
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe 2′-hydroxy group of flavin mononucleotide influences the catalytic function and promiscuity of the flavoprotein iodotyrosine dehalogenase†
Anton
Kozyryev
 ,
Petrina A.
Boucher
,
Carla M.
Quiñones-Jurgensen
and
Steven E.
Rokita
*
,
Petrina A.
Boucher
,
Carla M.
Quiñones-Jurgensen
and
Steven E.
Rokita
*
Department of Chemistry, Johns Hopkins University, 3400 N. Charles St., Maryland 21218, USA. E-mail: rokita@jhu.edu; Tel: +1-410-516-5793
First published on 4th August 2023
Abstract
The isoalloxazine ring system of the flavin cofactor is responsible for much of the catalytic power and diversity associated with flavoproteins. While the specificity of these enzymes is greatly influenced by the surrounding protein environment, the ribityl group of the cofactor may also participate in stabilizing transient intermediates formed by substrates and flavin. A conserved interaction between the phenolate oxygen of L-iodotyrosine and the 2′-hydroxy group of flavin mononucleotide (FMN) bound to iodotyrosine deiodianase (IYD) implied such a contribution to catalysis. Reconstitution of this deiodinase with 2′-deoxyflavin mononucleotide (2′-deoxyFMN) decreased the overall catalytic efficiency of L-iodotyrosine dehalogenation (kcat/Km) by more than 5-fold but increased kcat by over 2-fold. These affects are common to human IYD and its homolog from Thermotoga neapolitana and are best explained by an ability of the 2′-hydroxy group of FMN to stabilize association of the substrate in its phenolate form. Loss of this 2′-hydroxy group did not substantially affect the formation of the one electron reduced semiquinone form of FMN but its absence released constraints that otherwise suppresses the ability of IYD to promote hydride transfer as measured by a competing nitroreductase activity. Generation of IYD containing 2′-deoxyFMN also removed steric constraints that had previously limited the use of certain mechanistic probes. For example, L-O-methyl iodotyrosine could be accommodated in the active site lacking the 2′-hydroxy of FMN and shown to be inert to dehalogenation as predicted from a mechanism requiring ketonization of the phenolic oxygen. In the future, ancillary sites within a cofactor should now be considered when engineering new functions within existing protein architectures as demonstrated by the ability of IYD to promote nitroreduction after loss of the 2′-hydroxy group of FMN.
Introduction
Flavin-dependent enzymes are essential for life by catalyzing a broad range of reactions in both primary and secondary metabolism. While carbon–carbon and carbon–heteratom bond oxidation and reduction are most common, flavin is also associated with a number of other processes including activation of molecular oxygen, electron transfer, halogenation and certain non-redox transformations.1–4 The diversity of chemistry expressed by flavoproteins is directly attributed to the versatility of its core isoalloxazine ring system (Scheme 1). Typically, the surrounding protein environment is responsible for controlling the specific chemio-, regio- and stereospecificity of each enzyme by offering van der Waals interactions with the dimethylbenzene region, π-stacking over the aromatic system and polar interactions with the heteroatoms.1,5 The 5-phosphoribityl side chain extending from the N10 position for those enzymes utilizing flavin mononucleotide (FMN) and the additional phosphoadenosine for those utilizing flavin adenine dinucleotide (FAD) establish numerous protein interactions to secure cofactor binding but rarely are these implicated in catalysis.6–9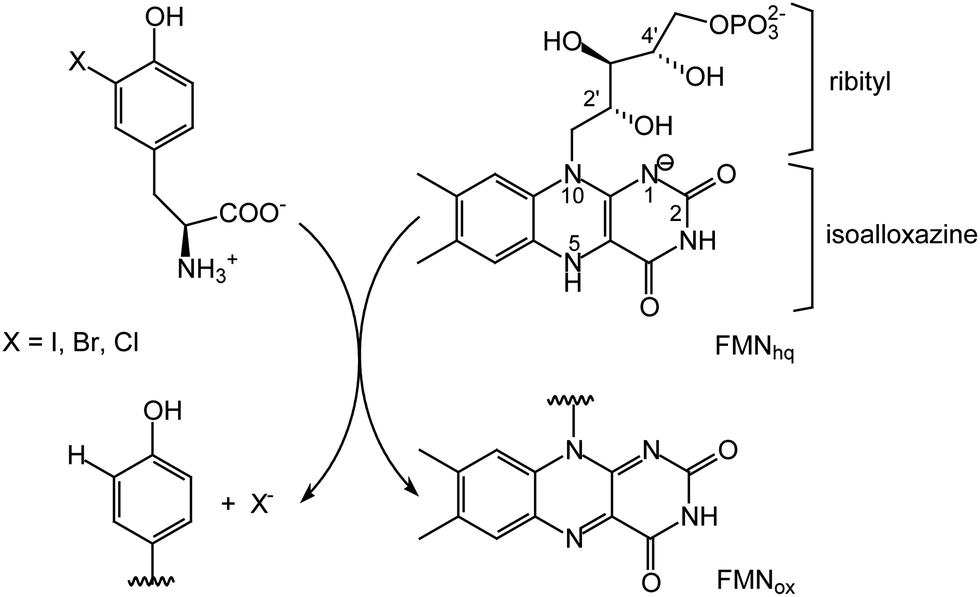 | ||
| Scheme 1 Flavin-dependent dehalogenation catalyzed by IYD. FMNox and FMNhq represent the oxidized and two electron reduced forms of flavin, respectively. | ||
Electron transfer flavoproteins (ETF) and acyl CoA dehydrogenases represent two exceptions in which the ribityl group intimately participates in enzyme turnover. The crystal structure of human ETF revealed a hydrogen bond between the 4′-hydroxy group of the ribityl side chain and the N1 of the isoalloxazine ring that is thought to stabilize the anionic semiquinone form of FAD.10 Reconstituting this ETF with 4′-deoxyFAD decreased the single electron reduction potential of oxidized FAD to its semiquinone by 116 mV while no perturbation of its two electron reduction from the oxidized to hydroquinone forms was detected.10 In an analogous example, the 2′-hydroxy of FAD was proposed to stabilize the increase in electron density associated with a two electron reduction of flavin in the berberine bridge enzyme.11 In contrast, acyl CoA dehydrogenases exploit their ribityl side chain for activating substrates. The 2′-hydroxy group of their FAD provides one of two hydrogen bonds to the thioester carbonyl oxygen of acyl CoAs.12,13 Loss of the hydrogen bond to FAD by reconstituting the enzyme with 2′-deoxyFAD decreased the rate of substrate oxidation by over seven orders of magnitude.14 This dramatic loss in activity is linked to a diminished ability of the catalyst to lower the pKa of the substrate's α-proton. Thus, the ribityl side chain has the potential to promote catalysis and does not solely function as an anchor for binding FMN and FAD to the active sites of enzymes.
Reductive dehalogenation catalyzed by the FMN-dependent enzyme iodotyrosine deiodinase (IYD) shares few similarities with the acyl CoA dehydrogenases yet the 2′-hydroxy of its FMN coordinates its halotyrosine substrates (Schemes 1 and 2).15–17 Studies to date suggest that the anionic phenolate rather than the neutral phenol form of the substrate binds to the active site and may be stabilized by the 2′-hydroxy group of FMN.16 Alternatively, the 2′-hydroxy group could promote catalysis by preferentially coordinating to the carbonyl of the electrophilic intermediate poised to accept an electron from the reduced FMN (Scheme 2).15,18 Additionally, the 2′-hydroxy group could serve as a proton donor to generate the electrophilic intermediate. The identity of this proton donor has remained elusive since none of the side chains proximal to the proposed site of protonation are ionizable and the acidity of the N5 proton of FMNhq does not affect catalytic turnover.16–18
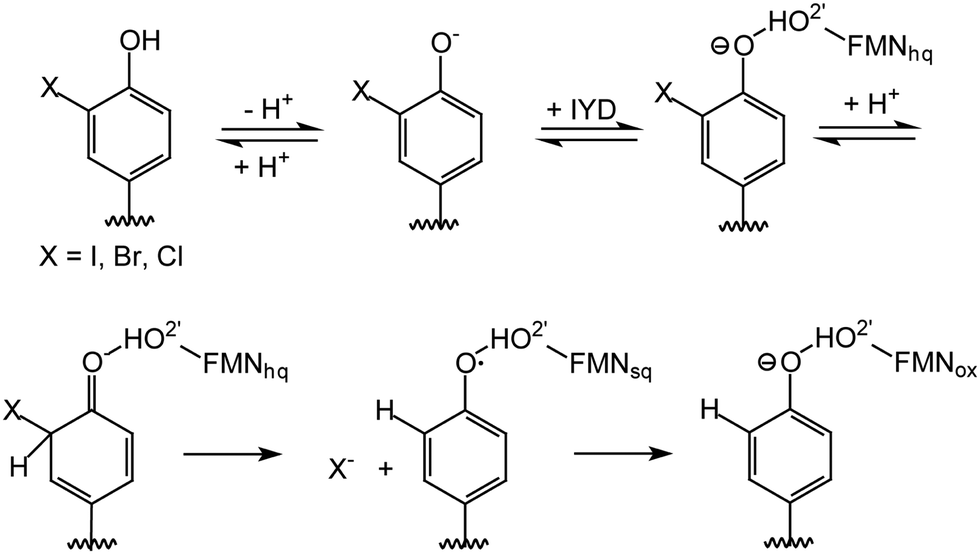 | ||
| Scheme 2 Likely mechanism for reductive dehalogenation by iodotyrosine deiodinase (IYD). FMNsq represents the one electron reduced form of flavin. | ||
To distinguish the role of the side chain hydroxy group, IYD has now been reconstituted with 2′-deoxyFMN and its properties are reported here. Binding and turnover studies indicate that the 2′-hydroxy group primarily affects substrate binding rather than catalytic intermediates. This represents a distinct function from the prior examples described above. Since 2′-deoxyFMN releases the steric constraints surrounding the phenolate, O-methyl iodotyrosine (O-Me I-Tyr) could now be studied with IYD to gain additional evidence for the proposed dearomatization of its substrate during dehalogenation. Surprisingly, loss of the 2′-hydroxy of FMN also released constraints that had limited substrate reduction to single electron transfer processes.
Results and discussion
Incorporating 2′-deoxyFMN in human IYD (HsIYD)
The role of the 2′-hydroxy group of FMN was first examined in HsIYD due to its prior mechanistic, redox and structural characterization.16,18–20 Rapid kinetics suggest that the chemistry of turnover was rate limiting since substrate binding is fast and the second order rate of FMNhq oxidation by I-Tyr is similar to a corresponding steady-state value of kcat/Km.19 However, neither proton nor electron transfer dominate turnover and instead the steady-state level of the electrophilic intermediate most affects the efficiency of deiodination.18 This enzyme is also amenable to reconstitution with a flavin analog as demonstrated earlier with 5-deazaFMN.21 An equivalent protocol based on guanidium hydrochloride treatment of HsIYD when bound to a nickel affinity column was used to remove native FMN and allow for reconstitution with 2′-deoxyFMN. In case the reconstitution process altered the general characteristics of the enzyme, HsIYD was also reconstituted with FMN for direct comparison with its 2′-deoxyFMN-containing derivative. Substrate binding to the resulting enzymes was conveniently measured by its ability to quench the fluorescence of the active site flavin in its oxidized form.22,23L-2-Iodotyrosine (I-Tyr) bound to the FMN-containing HsIYD with high affinity and a sub-micromolar dissociation constant similar to that observed previously with HsIYD characterized directly after purification (0.15 μM)16 (Table 1). Loss of the 2′-hydroxy group in HsIYD containing 2′-deoxyFMN diminished the affinity for I-Tyr by more than 1000-fold. Binding of the inert substrate analog L-2-fluorotyrosine (F-Tyr) was similarly weakened by over a 1000-fold. The analogous effect on Km for I-Tyr was more moderate but still an increase of 33-fold was measured (Table 1). In contrast to the diminished ability of the 2′-deoxyFMN enzyme to bind halotyrosines, the cofactor lacking the hydroxy group enhanced kcat by over 6-fold (Table 1).| HsIYD | X-Tyr | K D (μM) | k cat (×10−2 s−1) | K m (μM) | k cat/Km (×103 M−1 s−1) |
|---|---|---|---|---|---|
| a Data represent an average of two independent measurements. Error for the kinetic parameters represents the range of values determined from each trial (see ESI). b Values from ref. 18. c Values from ref. 16. d ND, not determined. | |||||
| FMN | I-Tyr | 0.42 ± 0.13 | 5.2 ± 0.2b | 10 ± 1b | 5.0 ± 0.7b |
| F-Tyr | 1.30 ± 0.4c | NDd | — | — | |
| deoxyFMN | I-Tyr | 1000 ± 60 | 32 ± 6 | 330 ± 100 | 0.97 ± 0.07 |
| F-Tyr | 1500 ± 70 | ND | — | — | |
These results are best rationalized by the ability of the 2′-hydroxy group of FMN to stabilize substrate as evident by its effect on enzyme affinity. In the absence of this stabilization, the energy of activation should then be diminished as manifest by the increase of kcat. If the 2′-hydroxy had instead been involved in protonating the substrate or stabilizing the non-aromatic intermediate, then kcat would have been expected to decrease significantly when FMN was replaced with 2′-deoxyFMN. Overall, the catalytic efficiency (kcat/Km) is lower in the absence of the 2′-hydroxy group of FMN. Thus, this group may optimize the association of the phenolate form of the substrate which is better poised than its neutral phenol form for subsequent carbon protonation (Scheme 2). Binding studies with the inert F-Tyr were a prelude to its use in testing the accumulation of the flavin semiquinone (FMNsq) during redox titration.16 However, the low affinity of F-Tyr would have required a concentration exceeding its solubility and hence another homolog of IYD was selected for reconstitution and further analysis.
Consequences of reconstituting IYD from a thermophilic organism with 2′-deoxyFMN
An IYD from Thermotoga neapolitana (TnIYD) had previously been expressed and characterized to identify the minimal features required for reductive dehalogenation.17 As expected, this homolog was highly thermostable and it was also found to bind halotyrosines with the highest affinity yet observed.17 Crystallographic analysis of TnIYD revealed the same close proximity of the phenolic oxygen of I-Tyr and the 2′-hydroxy group of FMN as that previously observed for HsIYD (Fig. 1).17 Accordingly, TnIYD was also subjected to the on-column reconstitution protocol used for HsIYD in anticipation of an enzyme derivative that retained sufficient binding to halotyrosines for redox characterization. This was confirmed by the sub-micromolar dissociation constant of I-Tyr with TnIYD containing 2′-deoxyFMN (Table 2). Even the substrate analog F-Tyr bound to this enzyme with a dissociation constant in the low micromolar range. Still, these values reflect a respective 25- and 50-fold decrease in affinity relative to those measured for TnIYD containing FMN (Table 2). Increases in both kcat and Km were also observed after exchanging FMN with 2′-deoxyFMN in TnIYD but the changes were less than half of those observed for HsIYD. Thus, the trends for IYD ranging from human to thermophile remain consistent and indicative of a role for the 2′-hydroxy group of FMN to stabilize the phenolate form of substrate. Alternative participation in proton transfer should have decreased rather than increased kcat.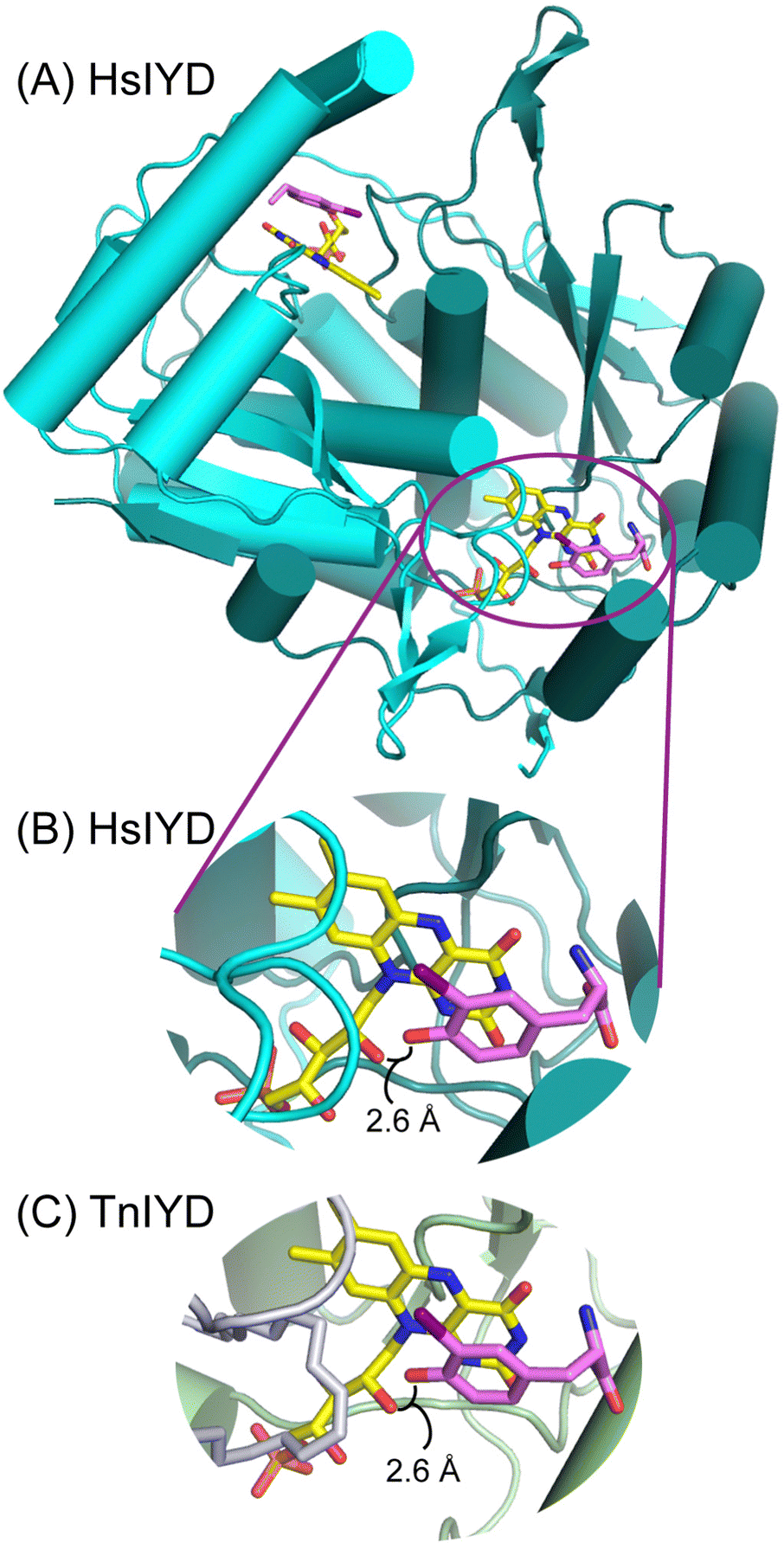 | ||
| Fig. 1 A conserved interaction between the phenolic oxygen of substrate and the 2′-hydroxy group of FMN in IYD. (A) The overall architecture of HsIYD illustrated in cyano and green represents the two polypeptides of the α2-homodimer (PDB: 4TTC).16 The carbon of FMN and I-Tyr are depicted in yellow and violet, respectively. (B) An individual active site from (A) is enlarged to highlight the close proximity of the phenolic oxygen and 2′-hydroxy group of FMN. (C) A corresponding view of the TnIYD and I-Tyr co-crystal reveals an equivalent association between these groups (PDB: 6Q1L).17 Its two polypeptides are rendered in gray and olive. | ||
| TnIYD | X-Tyr | K D (μM) | k cat (×10−2 s−1) | K m (μM) | k cat/Km (×103 M−1 s−1) |
|---|---|---|---|---|---|
| a Data represent an average of two independent measurements. Error for the kinetic parameter represents the range of values determined from each trial (see ESI). b Values from ref. 24. c Values from ref. 17. d ND, not determined. | |||||
| FMN | I-Tyr | 0.02 ± 0.1 | 2.7 ± 0.2b | 0.22 ± 0.07b | 120 ± 40b |
| F-Tyrc | 0.08 ± 0.03 | NDd | — | — | |
| deoxyFMN | I-Tyr | 0.5 ± 0.1 | 6.2 ± 0.8 | 24 ± 5 | 2.6 ± 0.6 |
| F-Tyr | 4.0 ± 0.4 | ND | — | — | |
Redox properties of TnIYD containing 2′-deoxyFMN
The range of reactions promoted by flavoproteins is attributed in part to the cofactor's ability to catalyze both single electron and hydride transfers.1 These can be differentially activated or suppressed by stabilizing or destabilizing the one electron reduced FMNsq. For example, nitroreductases from the nitroreductase superfamily promote hydride transfer during turnover and destabilize the potential formation of the one electron reduced FMNsq.25,26 In contrast, IYD is another member of this same superfamily but it stabilizes FMNsq for the single electron transfers necessary to dehalogenate its substrates.16 These different redox properties also correlate to the hydrogen bonding partner of the N5 position of FMN (Scheme 1). Enzymes of the superfamily ascribed to hydride transfer provide an amide proton proximal to the N5 whereas those members ascribed to single electron transfers provide a side chain hydroxy group.20,27,28 However, additional features of IYD likely contribute to the stabilization of FMNsq since loss of the side chain hydroxy next to the FMN N5 only decreased the efficiency of dehalogenation (kcat/Km) by 15-fold.27The potential for the 2′-hydroxy group of the ribityl side chain to offer additional stabilization to FMNsq was examined with the 2′-deoxyFMN containing TnIYD. Precedence for such a function was set by electron transfer flavoproteins that utilized the 4′-hydroxy group of the FMN side chain for this function.10 IYDs alone do not detectably accumulate FMNsq during reductive titration but this intermediate dominates the titration when an inert substrate analog F-Tyr binds to the active site, coordinates directly to the flavin and induced closure of an active site lid that sequesters the flavin and halotyrosine from solvent.16 TnIYD containing 2′-deoxyFMN behaved similarly when reduced by a xanthine/xanthine oxidase system under anaerobic conditions in the presence of excess F-Tyr (Fig. 2). Reduction of this enzyme was evident by a decrease in its absorbance at 450 nm and a concomitant increase in an absorbance at 575–600 nm that is typical of a neutral FMNsq.23,29 The ribityl hydroxy group within TnIYD consequently does not impact the single electron transfer mechanism and this is consistent with the ability of 2′-deoxyFMN to sustain the dehalogenation activity of IYD.
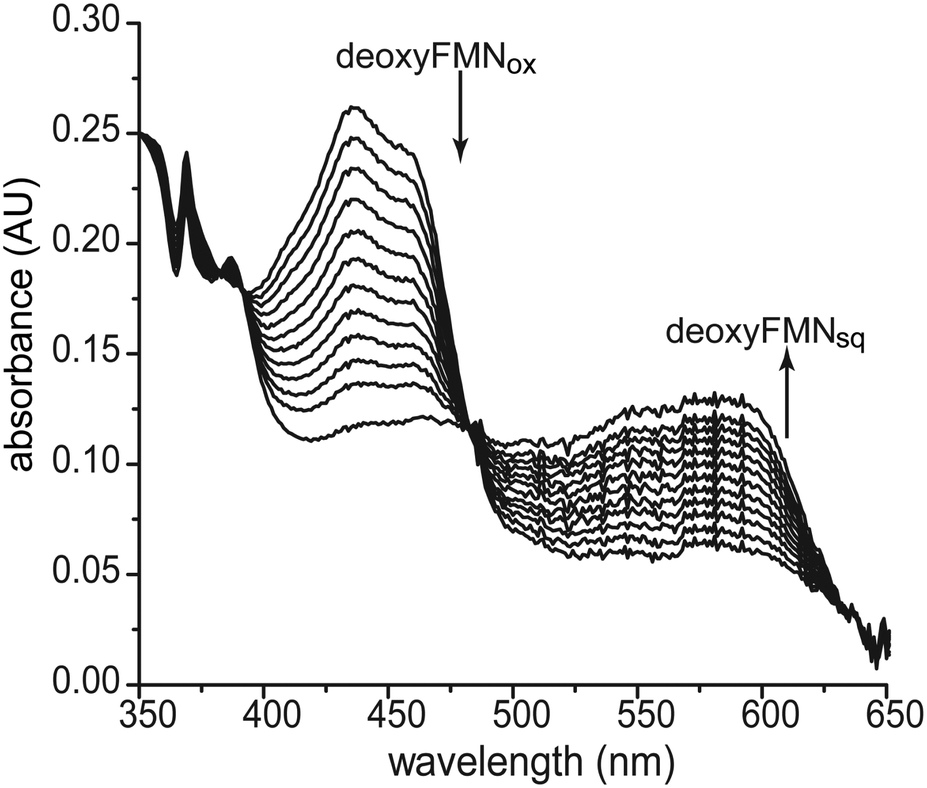 | ||
| Fig. 2 Reduction of TnIYD containing 2′-deoxyFMN. Xanthine oxidase was added to an anaerobic solution of TnIYD containing 2′-deoxyFMN (20 μM), xanthine, methyl viologen and F-Tyr (100 μM) in potassium phosphate pH 7.4 at 25 °C. The UV/Vis spectral changes were recorded every 2 min over 4 h and representative samples are included here. | ||
The environment surrounding FMN in IYD not only promotes single electron transfer but it also suppresses hydride transfer. For example, L-2-nitrotyrosine (O2N-Tyr) binds tightly to a bacteria IYD but participates in minimal redox chemistry with the reduced FMNhq form of the enzyme.27 However, loss of the hydrogen bond to the N5 position of FMN by mutation of a Thr to Ala in this enzyme relieves the suppression of hydride transfer and allows for full oxidation of FMNhq and concurrent reduction of nitrotyrosine.27 Similarly in the absence of active site lid closure and substrate coordination to flavin, a nascent hydride transfer activity is observed by HsIYD reduction of 2-nitrophenol.20 Conversely, single electron transfer is highly suppressed under these conditions since little reduction and dehalogenation of 2-iodophenol can be detected.20 Oxidation of the reduced form of IYD by nitroaromatics is consequently a convenient measure of its capacity to support hydride transfer in competition with single electron transfer.
As expected, after reducing TnIYD under anaerobic conditions to form FMNhq, it is not fully reoxidized by addition of even a 2-fold excess of O2N-Tyr (Fig. 3(A)). Similar to observations with HsIYD, addition of O2N-Tyr predominantly generates TnIYD containing FMNsq as observed by the formation of a species absorbing in the region of 575–600 nm that persists without change in the absence of molecular oxygen.20 No fully oxidized FMNox is apparent and little FMNhq remains. Surprisingly, TnIYD containing the reduced 2′-deoxyFMNhq is readily oxidized to its FMNox form by a stoichiometric concentration of O2N-Tyr (Fig. 3(B)). This ability to support both single electron and hydride transfer is remiscent of the Thr to Ala mutant in bacterial IYD that lacks the side chain hydrogen bond to the N5 position of flavin.27 However, the 2′-hydroxy group of FMN does not directly interact with the isoalloxazine ring. More likely, this polar group contributes to a network of interactions that are disrupted in its absence. Alternatively, loss of the 2′-hydroxy group of FMN may allow the substrate to reorient to a geometry favoring nitroreduction. This latter possibility seems less likely since the phenolate is still expected to remain similarly aligned to maintain its interaction with an amide in the protein backbone.17 The unexpected connection between the ribityl side chain and the redox processes promoted by IYD illustrates the intricate interplay between functional groups within the active sites of flavoproteins that may lead to unanticipated results when attempting to alter catalytic function or specificity by protein engineering.
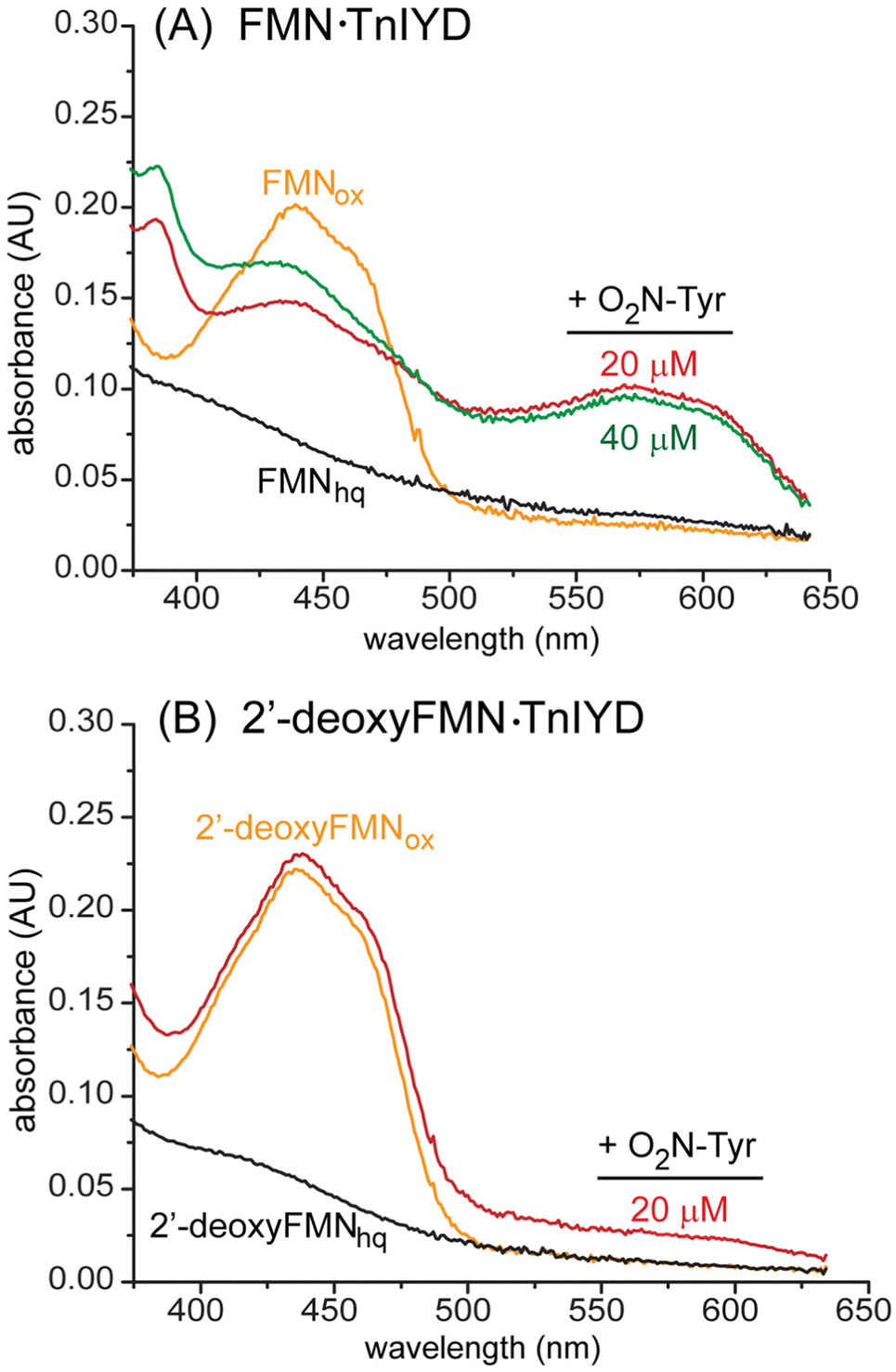 | ||
| Fig. 3 The contrasting efficiency of TnIYD to promote nitroreduction when alternatively containing FMN or 2′-deoxyFMN. (A) TnIYD (20 μM) containing FMNox (orange) was reduced to its FMNhq form (black) by dithionite under anaerobic conditions prior to an initial addition of 20 μM O2N-Tyr (red) and a second addition after 30 min for a total of 40 μM O2N-Tyr (green). Spectra were recorded 30 min after each addition and no changes were observed after extended incubation. (B) TnIYD containing 2′-deoxyFMNox (orange) was treated equivalently to generate its 2′-deoxyFMNhq form (black). Only one addition of 20 μM O2N-Tyr was necessary to regenerate the 2′-deoxyFMNox form (red) within 30 min. | ||
Mechanistic studies made possible by TnIYD containing 2′-deoxyFMN
The steric environment surrounding the substrate phenolate was thought to preclude substrate analogs that were previously envisioned to test the necessity of a non-aromatic tautomer in the dehalogenation process (Scheme 2). Removal of the 2′-hydroxy group of FMN released these constraints and allowed study of O-Me I-Tyr. An O-methyl (methoxy) group in conjugation with the aryl carbon–iodine bond maintains much of the electron-donating characteristics of a corresponding hydroxy group.30 Similarly, proton affinity for anisole is within 3% of that for phenol at least as measured in the gas-phase.31 However, methylation of the phenolic oxygen severely destabilizes carbon protonation to form the non-aromatic tautomer proposed in the catalytic mechanism and offers a poor mimic of a phenolate group (Scheme 2). In model studies using hydrogen donating solvents, 4-chlorophenol was dechlorinated 50-fold more efficiently than 4-chloroanisole.32 This difference has been correlated to the relative stability of the keto tautomer that is favored after addition of an electron or hydrogen to the aromatic system of phenol.32,33 Thus, dehalogenation of O-Me I-Tyr should be disfavored in the expected mechanism of IYD. This could not be confirmed with HsIYD containing 2′-deoxyFMN due to a lack of affinity of O-Me I-Tyr for this enzyme (Table 3). However, O-Me I-Tyr bound to the oxidized form of TnIYD containing 2′-deoxyFMN with a Kd that was only ∼12-fold higher than that of I-Tyr and still in the useful micromolar range (Tables 2 and 3). No deiodination of O-Me I-Tyr was detected by HPLC after more than doubling the standard incubation time and using a concentration of enzyme 10-fold greater than standard conditions. From the detection limit of these analyses, the kcat for O-Me I-Tyr is at least 2000-fold lower than that for I-Tyr (Tables 2 and 3). The lack of O-Me I-Tyr dehalogenation is consequently consistent with the proposed mechanism of flavin-catalyzed reductive dehalogenation (Scheme 2).| deoxyFMN | Ligand | K D (μM) | k cat (×10−2 s−1) |
|---|---|---|---|
| a See ESI for details. | |||
| HsIYD | O-Me I-Tyr | >5000 | <1.6 × 10−2 |
| TnIYD | O-Me I-Tyr | 6.3 ± 0.7 | <2.7 × 10−2 |
| I-aminoPhe | 57 ± 3 | <4.7 × 10−2 | |
Substituting the phenolic oxygen with nitrogen to form the aniline derivative of I-Tyr might be anticipated to support deiodination with IYD since the –NH2 group is highly electron donating and shares the potential for tautomerization by forming an imine equivalent to the keto derivative of the phenol.30 Furthermore, the efficiency of dehalogenation of 2-chloroaniline in hydrogen-donating solvents is only 2-fold less than that for 2-chlorophenol.32 However, the aniline derivative of I-Tyr (I-aminoPhe) was not dehalogenated by TnIYD containing 2′-deoxyFMN. Again from the detection limits set by the assay conditions and HPLC sensitivity, the kcat for I-aminoPhe is more than 1300-fold lower than that for I-Tyr (Tables 2 and 3). This lack of activity is not likely the result of a relatively low binding affinity of I-aminoPhe with TnIYD containing 2′-deoxyFMN (Table 3). Although its Kd is over 100-fold higher than that of I-Tyr, even weaker binding of I-Tyr to HsIYD containing 2′-deoxyFMN did not preclude turnover (Table 1). Most likely, the presence of the neutral –NH2 rather than the anionic phenolate perturbs the same network of interactions involving the 2′-hydroxy group of FMN that influences the stability of the bound substrate and the dynamics of the active site.
Conclusion
IYD joins the select number of flavoproteins that are known to rely on their ribityl side chain for enzyme turnover. Despite the decrease in kcat/Km for IYD after FMN is exchanged with 2′-deoxyFMN, its kcat increases in the absence of the 2′-hydroxy group. The close interaction between this 2′-hydroxy group and the substrate phenolate that was originally observed in crystal structures of IYD now appears to stabilize substrate association.16,17,34,35 Unlike electron transfer flavoproteins or acyl CoA dehydrogenases, the ribityl group of FMN in IYD is not necessary to stabilize intermediates of catalysis such as FMNsq or the non-aromatic keto form of a substrate.10,14 The ability of the 2′-hydroxy group of FMN to suppress hydride transfer by IYD was not anticipated and deserves further scrutiny. These results illustrate the potential for manipulating cofactor chemistry to create new catalysts by modulating a network of polar interactions surrounding but not directly associated with the isoalloxazine ring of FMN.Experimental
General materials
3-Fluoro-L-tyrosine (F-Tyr) was obtained from AstaTech, Inc (Bristol, PA). 3-Iodo-L-tyrosine (I-Tyr), O-methyl tyrosine, formic acid (88%) and trifluoroacetic acid were obtained from Acros Organics (Morris Plains, NJ). 3,4-Dimethylaniline, aniline, 2′-deoxy-D-ribose, NaBH3CN, xanthine oxidase from bovine milk, methyl viologen dichloride and sodium dithionite were obtained from Sigma-Aldrich (Madison, WI). SilicaFlash P60 silica (SiliCycle Inc., Quebec City, Quebec) was used for all column chromatography. Human IYD (HsIYD) lacking its N-terminal transmembrane sequence (residues 2–31) was expressed as a SUMO fusion, purified by Ni2+ affinity and size exclusion chromatography as described previously.16 IYD from Thermotoga neapolitana (TnIYD) was expressed with a C-terminal His6-tag and purified by Ni2+ affinity chromatography as described previously.17 Syntheses of 2′-deoxyFMN, O-Me I-Tyr and I-aminoPhe are described in the ESI.†General methods
NMR spectra were recorded on Brucker Avance-300 and Avance-400 spectrometers (1H resonances at 300 MHz and 400 MHz, respectively). Chemical shifts are reported in parts per million (ppm) and coupling constants (J) are reported in Hertz (Hz). The residual 1H and 13C signals from solvent were used as references. 19F and 31P signals were referenced indirectly from 1H data by the spectrometers. All UV measurements were performed with a Agilent 8453 spectrophotometer. All fluorescence measurements were performed with a Fluoromax-4 spectrofluorometer (Horiba Scientific). Mass analysis was performed with a Waters Acquity/Xevo-G2 UPLC-MS system with electrospray ionization (ESI) and q-TOF MS/MS analyzer. For all HPLC purification of synthetic materials, a Jasco PU-2080 system with a reverse phase preparative column (Econosphere C18 250 × 10 mm) was used.Reconstitution of HsIYD and TnIYD alternatively with FMN and 2′-deoxyFMN
Exchange of the FMN that copurifies with IYD was performed as described previously.21 For HsIYD, its SUMO-HsIYD fusion was bound to a Ni-affinity column and treated with increasing concentrations of guanidium hydrochloride to a maximum of 1.5 M in loading buffer (100 mM NaCl, 50 mM sodium phosphate pH 7.4, 10% glycerol, 20 mM imidazole and 0.05 mM tris(2-carboxethyl)phosphine (TCEP)) to release FMN. The gradient was then performed in reverse and the column was washed with either FMN or 2′-deoxyFMN (200 μM) in loading buffer for 4 h at 1 mL min−1 (4 °C). Excess cofactor was removed by an additional wash with loading buffer in the absence of added cofactor. The reconstituted SUMO-HsIYD fusion was eluted from the affinity column by addition of 250 mM imidazole to the loading buffer. The SUMO tag was removed by proteolysis and the reconstituted HsIYD was isolated by size exclusion chromatography under standard conditions.16 For TnIYD, an equivalent protocol was used without the need of a SUMO tag. Additionally, TnIYD was washed with loading buffer supplemented with sodium dithionite (2% final) to remove tyrosine bound in the active site prior to guanidium hydrochloride treatment.17Ligand binding to IYD23
Ligand was titrated into a solution of IYD (3 μM) in potassium phosphate (100 mM, pH 7.4) with gentle stirring (25 °C). Fluorescence (λex = 450 nm, λem = 516 nm) was measured 2 min after each addition. Dissociation constants were calculated by plotting the remaining fluorescence relative to the initial fluorescence (ΔF/F0) with respect to total enzyme ([E]T) and total ligand ([L]) using eqn (1) (Origin 9.1).22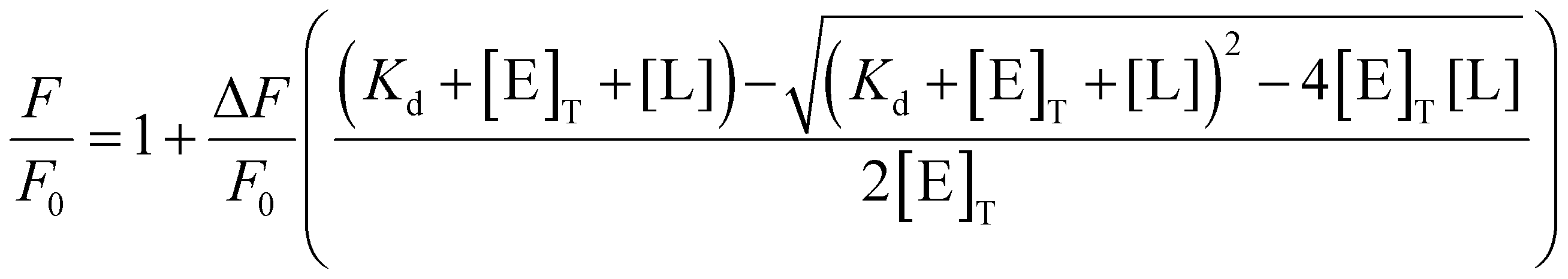 | (1) |
Catalytic deiodination
Enzyme-dependent deiodination was performed under standard conditions.36 Reaction was initiated by addition of 5% sodium dithionite in 5% sodium bicarbonate (100 μL) to a solution of substrate and enzyme (80 nM HsIYD or 100 nM TnIYD) in 111 mM potassium phosphate pH 7.4 (900 μL) at 25 °C. Reaction was quenched by 88% formic acid (50 μL) after sufficient time for product formation (40 min for 2′-deoxyFMN TnIYD, 15 min for 2′-deoxyFMN HsIYD) and an internal standard of Cl-Tyr was added (500 μM, 50 μL). The resulting mixture was centrifuged for 5 min at 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g and the supernatant (1 mL) was analyzed by reverse-phase C-18 HPLC using an Agilent 1200 series HPLC with a Varian Microsorb-MV 300-5 C18 250 × 4.6 mm column. HPLC solvent A was 0.44% aqueous formic acid and solvent B was 0.44% formic acid in acetonitrile. Elution began with 5% solvent B for 10 min followed by a linear gradient to 60% solvent B for 15 min and finally to 95% solvent B over 9 min with a flow rate of 1 mL min−1. O-Me I-Tyr (50 μM) and I-aminoPhe (50 μM) were individually incubated with 2′-deoxyFMN TnIYD (1 μM) for 2 h using equivalent conditions. m-Cresol (25 μM) was used as an internal standard for their chromatographic analyses. HPLC conditions used to monitor possible deiodination of O-Me I-Tyr followed the conditions used for I-Tyr. For possible deiodination of I-aminoPhe, a gradient of acetonitrile in buffer A (25 mM ammonium formate pH 6.4) was used as follows: 0% acetonitrile from 0–10 min, 0–10% acetonitrile from 10–17 min and a constant 10% acetonitrile from 17–22 min. For these assays, the disappearance of I-aminoPhe was used to monitor reaction because the deiodinated product coeluted with sodium dithionite.
000 g and the supernatant (1 mL) was analyzed by reverse-phase C-18 HPLC using an Agilent 1200 series HPLC with a Varian Microsorb-MV 300-5 C18 250 × 4.6 mm column. HPLC solvent A was 0.44% aqueous formic acid and solvent B was 0.44% formic acid in acetonitrile. Elution began with 5% solvent B for 10 min followed by a linear gradient to 60% solvent B for 15 min and finally to 95% solvent B over 9 min with a flow rate of 1 mL min−1. O-Me I-Tyr (50 μM) and I-aminoPhe (50 μM) were individually incubated with 2′-deoxyFMN TnIYD (1 μM) for 2 h using equivalent conditions. m-Cresol (25 μM) was used as an internal standard for their chromatographic analyses. HPLC conditions used to monitor possible deiodination of O-Me I-Tyr followed the conditions used for I-Tyr. For possible deiodination of I-aminoPhe, a gradient of acetonitrile in buffer A (25 mM ammonium formate pH 6.4) was used as follows: 0% acetonitrile from 0–10 min, 0–10% acetonitrile from 10–17 min and a constant 10% acetonitrile from 17–22 min. For these assays, the disappearance of I-aminoPhe was used to monitor reaction because the deiodinated product coeluted with sodium dithionite.
Reduction of reconstituted TnIYD using xanthine/xanthine oxidase
All assays were performed at 25 °C using xanthine/xanthine oxidase as described previously.16 Samples containing 1 mM xanthine, 12 μM methyl viologen and 100 μM F-Tyr in 100 mM potassium phosphate pH 7.4 were placed in sealable quartz cuvettes. Molecular oxygen was purged by bubbling the solution with argon for at least 20 min prior to addition of IYD (20 μM) and 2 min after addition. The head space was then purged with argon for 30 min and reduction was initiated by addition of xanthine oxidase (140 μg mL−1).Single-turnover of TnIYD with O2N-Tyr
Solutions of 100 mM 2-morpholinoethanesulfonate (MES) pH 6, 500 mM NaCl and 10% glycerol were flushed with a continuous flow of argon to remove dissolved molecular oxygen for 30 min. Enzyme was added to the solution with continuous flushing of argon for another 2 min. Additional flushing of the head space was extended for another 30 min before a minimal amount (∼2 μL) of 0.25% dithionite in 100 mM MES pH 6, 500 mM NaCl and 10% glycerol was added to fully reduce either 2′-deoxyFMNox or FMNox as monitored by loss of absorbance at 450 nm corresponding to their λmax. To confirm that no excess dithionite was present in these mixtures, between 10 and 20 μL of air saturated buffer was introduced into the mixture with a syringe. A small gain of absorbance at 450 nm was confirmed over 10 min. Reaction of the reduced enzyme (25 °C) was then initiated by addition of a solution of O2N-Tyr in 100 mM MES pH 6 that had been purged with argon for 1 h.Conflicts of interest
The authors do not have conflicts of interest in this work.Acknowledgements
This research was supported in part by a grant from the National Institutes of Health (RO1 GM130937, SR) and the National Science Foundation (BioREU, DBI-1262985, C. M. Q.-J.).References
- R. L. Fagan and B. A. Palfey, in Ch. 3 in Comprehensive Natural Products II, ed. T. P. Begley, Elsevier, Oxford, 2010, vol. 7, pp.37–114 Search PubMed.
- P. Macheroux, B. Kappes and S. E. Ealick, Flavogenomics – a genomic and structural view of flavin-dependent proteins, FEBS J., 2011, 278, 2625–2634 CrossRef CAS PubMed.
- V. Piano, B. A. Palfey and A. Mattevi, Flavins as covalent catalysts: new mechanisms emerge, Trend Biochem. Sci., 2017, 42, 457–469 CrossRef CAS PubMed.
- E. Romero, J. R. G. Castellanos, G. Gadda, M. W. Fraaije and A. Mattevi, Same substrate, many reactions: oxygen activation in flavoenzymes, Chem. Rev., 2018, 114, 1742–1769 CrossRef PubMed.
- M. W. Fraaije and A. Mattevi, Flavoenzymes: diverse catalysts with recurrent features, Trends Biochem. Sci., 2000, 25, 126–132 CrossRef CAS PubMed.
- A. Mattevi, A. J. Schierbeek and W. G. Hol, Refined crystal structure of lipoamide dehydrogenase from Azotobacter vinelandii at 2.2 Å resolution, J. Mol. Biol., 1991, 220, 975–994 CrossRef CAS PubMed.
- K. M. Fox and P. A. Karplus, Old yellow enzyme at 2 Å resolution: overall structure, ligand binding, and comparison with related flavoproteins, Structure, 1994, 2, 1089–1105 CrossRef CAS.
- G. N. Parkinson, J. V. Skelly and S. Neidle, Crystal structure of FMN-dependent nitroreductase from Escherichia coli B: A prodrug-activating enzyme, J. Med. Chem., 2000, 43, 3624–3631 CrossRef CAS PubMed.
- T. Oozeki, T. Nakai, K. Kozakai, K. Okamoto, S. Kuroda, K. Kobayashi, K. Tanizawa and T. Okajima, Functional and structural characterization of a flavoprotein monooxygenase essential for biogenesis of tryptophylquinone cofactor, Nat. Commun., 2021, 12, 993 CrossRef.
- T. M. Dwyer, S. Mortl, K. Kemter, A. Bacher, A. Fauq and F. E. Frerman, The intraflavin hydrogen bond in human electron transfer flavoprotein modulates redox potentials and may participate in electron transfer, Biochemistry, 1999, 38, 9735–9745 CrossRef CAS.
- S. Wallner, A. Winkler, S. Riedl, C. Dully, S. Horvath, K. Gruber and P. Macheroux, Catalytic and structural role of a conserved active site histidine in berberine bridge enzyme, Biochemistry, 2012, 51, 6139–6147 CrossRef CAS.
- J.-J. Kim, M. Wang and R. Paschke, Crystal structures of medium-chain acyl-CoA dehydrogenase from pig liver mitochondria with and without substrate, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 7523–7527 CrossRef CAS.
- S. Ghisla and C. Thorpe, Acyl-CoA dehydrogenases: a mechanistic overview, Eur. J. Biochem., 2004, 271, 494–508 CrossRef CAS PubMed.
- S. Engst, P. Vock, M. Wang, J.-J. Kim and S. Ghisla, Mechanism of activation of acyl-CoA substrates by medium chain acyl-CoA dehydrogenase: interaction of the thioester carbonyl with the flavin adenine dinucleotide ribityl side chain, Biochemistry, 1999, 38, 257–267 CrossRef CAS PubMed.
- Z. Sun, Q. Su and S. E. Rokita, The distribution and mechanism of iodotyrosine deiodinase defied expectations, Arch. Biochem. Biophys., 2017, 632, 77–87 CrossRef CAS.
- J. Hu, W. Chuenchor and S. E. Rokita, A switch between one- and two-electron chemistry of the human flavoprotein iodotyrosine deiodinase is controlled by substrate, J. Biol. Chem., 2015, 290, 590–600 CrossRef CAS.
- Z. Sun, B. Xu, S. Spisak, J. M. Kavran and S. E. Rokita, The minimal structure for iodotyrosine deiodinase function is defined by an outlier protein from thermophilic bacteria Thermotoga neapolitana, J. Biol. Chem., 2021, 297, 101385 CrossRef CAS PubMed.
- A. Kozyryev, D. Lemen, J. Dunn and S. E. Rokita, Substrate electronics dominate the rate of reductive dehalogenation promoted by the flavin-dependent iodotyrosine deiodinase, Biochemistry, 2023, 62, 1298–1306 CrossRef CAS PubMed.
- K. D. Bobyk, D. P. Ballou and S. E. Rokita, Rapid kinetics of dehalogenation promoted by iodotyrosine deiodinase from human thyroid, Biochemistry, 2015, 54, 4487–4494 CrossRef CAS PubMed.
- J. Hu, Q. Su, J. L. Schlessman and S. E. Rokita, Redox control of iodotyrosine deiodinase, Protein Sci., 2019, 28, 68–78 CrossRef CAS PubMed.
- Q. Su, P. A. Boucher and S. E. Rokita, Conversion of a dehalogenase to a nitroreductase by swapping its flavin cofactor with a 5-deazaflavin analog, Angew. Chem., Int. Ed., 2017, 56, 10862–10866 CrossRef CAS PubMed.
- J. R. Warner and S. D. Copley, Pre-steady-state kinetic studies of the reductive dehalogenation catalyzed by tetrachlorohydroquinone dehalogenase, Biochemistry, 2007, 46, 13211–13222 CrossRef CAS PubMed.
- P. M. McTamney and S. E. Rokita, A mammalian reductive deiodinase has broad power to dehalogenate chlorinated and brominated substrates, J. Am. Chem. Soc., 2009, 131, 14212–14213 CrossRef CAS PubMed.
- Z. Sun, PhD dissertation, Department of Chemistry, Johns Hopkins University, 2019.
- C. A. Haynes, R. L. Koder, A.-F. Miller and D. W. Rodgers, Structures of nitroreductase in three states, J. Biol. Chem., 2002, 277, 11513–11520 CrossRef CAS PubMed.
- R. L. Koder, C. A. Haynes, M. E. Rodgers, D. W. Rodgers and A.-F. Miller, Flavin thermodynamics explain the oxygen insensitivity of enteric nitroreductase, Biochemistry, 2002, 41, 14197–14205 CrossRef CAS PubMed.
- A. Mukherjee and S. E. Rokita, Single amino acid switch between a flavin-dependent dehalogenase and nitroreductase, J. Am. Chem. Soc., 2015, 137, 15342–15345 CrossRef CAS PubMed.
- E. Akiva, J. N. Copp, N. Tokuriki and P. C. Babbit, Evolutionary and molecular foundations of multiple contemporary functions of the nitroreductase superfamily, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E9549–E9558 CrossRef CAS PubMed.
- V. Massey, in Flavins Flavoproteins, Proceedings of the 10th International Symposium, Como, Italy, July 15–20, 1990, ed. B. Curti, S. Ronchi and G. Zanetti, Gruyter & Co., Berlin, 1991, pp.59–66 Search PubMed.
- C. Hansch, A. Leo and R. W. Taft, A survey of Hammett substituent constants and resonance and field parameters, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- E. P. L. Hunter and S. G. Lias, Evaluated gas phase basicities and proton affinities of molecules: an update, J. Phys. Chem. Ref. Data, 1998, 27, 413–656 CrossRef CAS.
- P. Mulder, I. W. C. E. Arends, D. Santoro and H.-G. Korth, The surprisingly facile thermal dehalogenation of chlorinated aromatics by a hydroaromatic donor solvent. Tautomerization of chlorinated phenols and anilines, J. Org. Chem., 2003, 68, 4247–4257 CrossRef CAS PubMed.
- E. D. Raczyńska, K. Kolczyńska and T. M. Stępniewski, Tautomeric preferences and π-electron delocalization for redox forms of phenol, Comput. Theor. Chem., 2011, 963, 176–184 CrossRef.
- S. R. Thomas, P. M. McTamney, J. M. Adler, N. LaRonde-LeBlanc and S. E. Rokita, Crystal structure of iodotyrosine deiodinase, a novel flavoprotein responsible for iodide salvage in thyroid glands, J. Biol. Chem., 2009, 284, 19659–19667 CrossRef CAS PubMed.
- N. Ingavat, J. M. Kavran, Z. Sun and S. E. Rokita, Active site binding is not sufficient for reductive deiodination by iodotyrosine deiodinase, Biochemistry, 2017, 56, 1130–1139 CrossRef CAS PubMed.
- A. Phatarphekar and S. E. Rokita, Functional analysis of iodotyrosine deiodinase from Drosophila melanogaster, Protein Sci., 2016, 25, 2187–2195 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cb00094j |
| This journal is © The Royal Society of Chemistry 2023 |