
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStitched peptides as potential cell permeable inhibitors of oncogenic DAXX protein†
Clare
Jelinska‡§
*abc,
Srinivasaraghavan
Kannan§
 d,
Yuri
Frosi
e,
Siti Radhiah
Ramlan
e,
Fernaldo
Winnerdy
a,
Rajamani
Lakshminarayanan
fgh,
Charles W
Johannes
e,
Christopher J
Brown
e,
Anh-Tuan
Phan
ai,
Daniela
Rhodes
abc and
Chandra S
Verma
*bdj
d,
Yuri
Frosi
e,
Siti Radhiah
Ramlan
e,
Fernaldo
Winnerdy
a,
Rajamani
Lakshminarayanan
fgh,
Charles W
Johannes
e,
Christopher J
Brown
e,
Anh-Tuan
Phan
ai,
Daniela
Rhodes
abc and
Chandra S
Verma
*bdj
aNTU Institute of Structural Biology, Experimental Medicine Building Level 06-01, 59 Nanyang Drive, 636921, Singapore. E-mail: csiclar@nus.edu.sg
bNTU School of Biological Sciences, 60 Nanyang Drive, 637551, Singapore. E-mail: chandra@bii.a-star.edu.sg
cNTU Lee Kong Chian School of Medicine, Experimental Medicine Building, 59 Nanyang Drive, 636921, Singapore
dBioinformatics institute (A*STAR), 30 Biopolis Street, Matrix Level 07-01, 138671, Singapore
eDITL, Institute of Cellular and Molecular Biology (A*STAR), 8a Biomedical Grove, 138648, Singapore
fOcular Infections and Anti-Microbials Research Group, Singapore Eye Research Institute, The Academia, 20 College Road, Singapore, 169856, Singapore
gDepartment of Pharmacy, National University of Singapore, Singapore, 117543, Singapore
hAcademic Clinical Program in Ophthalmology and Visual Sciences Academic Clinical Program, Duke-NUS Medical School, 169857, Singapore
iNTU School of Physical and Mathematical Sciences. 21 Nanyang link, 637371, Singapore
jDepartment of Biological Sciences, National University of Singapore, Block S3 #05-01, 16 Science Drive 4, 117558, Singapore
First published on 11th October 2023
Abstract
DAXX (Death Domain Associated Protein 6) is frequently upregulated in various common cancers, and its suppression has been linked to reduced tumor progression. Consequently, DAXX has gained significant interest as a therapeutic target in such cancers. DAXX is known to function in several critical biological pathways including chromatin remodelling, transcription regulation, and DNA repair. Leveraging structural information, we have designed and developed a novel set of stapled/stitched peptides that specifically target a surface on the N-terminal helical bundle domain of DAXX. This surface serves as the anchor point for binding to multiple interaction partners, such as Rassf1C, p53, Mdm2, and ATRX, as well as for the auto-regulation of the DAXX N-terminal SUMO interaction motif (SIM). Our experiments demonstrate that these peptides effectively bind to and inhibit DAXX with a higher affinity than the known interaction partners. Furthermore, these peptides release the auto-inhibited SIM, enabling it to interact with SUMO−1. Importantly, we have developed stitched peptides that can enter cells, maintaining their intracellular concentrations at nanomolar levels even after 24 hours, without causing any membrane perturbation. Collectively, our findings suggest that these stitched peptides not only serve as valuable tools for probing the molecular interactions of DAXX but also hold potential as precursors to the development of therapeutic interventions.
Introduction
DAXX (Death Domain Associated Protein 6), initially identified as a potentiator of apoptosis,1 is known to play crucial roles in several vital biological pathways, including chromatin remodeling, transcription regulation, DNA repair, and innate immunity. Its increased expression has been observed in diverse, epidemiologically prevalent cancers.2The diverse cellular functions of DAXX rely on its ability to shuttle between the cytoplasm and nucleus, forming a network of interactions facilitated by post-translational modifications such as phosphorylation and SUMOylation.3,4 Reversible SUMOylation is involved in the regulation of numerous proteins that interact with DAXX, and it serves as an important regulatory mechanism for DAXX itself, which can also undergo SUMOylation.5–7 For instance, as a co-repressor, DAXX has been shown to interact with multiple SUMOylated transcription factors that are derepressed upon increased association with DAXX.3,6 Additionally, DAXX can directly influence transcription by acting on chromatin through its histone chaperone activity.8 The ability of DAXX to impact transcription through multiple pathways may partly explain its seemingly diverse roles and its postulated involvement in oncogenic processes.9
One important nuclear pathway involves the well-studied interaction of DAXX as a partner of the chromatin remodeler ATRX, specifically in its role as an H3.3-specific chaperone.10,11 The histone chaperone function of DAXX is conferred by its DAXX histone binding domain (HBDDAXX),12,13 while the interaction between ATRX and DAXX is mediated by the DAXX-helical bundle domain (DHBDAXX).14 This domain of DAXX has also been reported to bind to multiple other protein partners (Rassf1C,15 Mdm2,16 HAUSP, and p5317) in a mutually exclusive ‘partner-switch’ mechanism.18,19 Interestingly, the interaction of SUMO with a SUMO interaction motif (SIM) located at the N-terminus of DAXX (NSIMDAXX)5 is thought to be allosterically regulated by self-interaction with a region on DHBDAXX that overlaps with the previously reported protein–protein interaction (PPI) surface of DAXX.20 These findings strongly suggest that a shared DHBDAXX PPI surface plays a central role in regulating a complex network of interactions that modulate DAXX function. Taken together, these observations suggest that the development of specific inhibitors targeting the promiscuous DHBDAXX PPI surface could serve as valuable tools for studying the intricate molecular interactions of DAXX with its partners in vitro, as well as potential therapeutics targeting DAXX.
Precise structural information regarding the interaction between DHBDAXX and its partner proteins is derived from atomic structures of apo DHBDAXX and complexes in which DHBDAXX is bound to peptides derived from either the Rassf1C tumor suppressor or the chromatin remodeler ATRX. The DHBDAXX domain comprises the antiparallel packing of four alpha helices (α1, α2, α4, and α5) and a short helix (α3) connecting helices α2 and α4.18,19,21 Both peptides from Rassf1C and ATRX bind between helices α2 and α5 of DHBDAXX. Despite their helical axes adopting nearly perpendicular orientations in the two complexes, the PPI surface occupied by the two peptides overlaps with a hydrophobic pocket on DHBDAXX, formed by residues V84, F87, Y124, V125, and I127. Consistent with this, NMR chemical shift perturbation data demonstrate significant changes in the amide cross-peak positions of these residues upon DHBDAXX binding to peptides derived from the tumor suppressor p53 and its negative regulator Mdm2, indicating that the same hydrophobic pocket also mediates these interactions.18 Overall, the structural information strongly supports the notion that the hydrophobic pocket on the DHBDAXX surface serves as an anchor point for a shared PPI surface. Given that this pocket is relatively shallow, it lends itself to inhibition by peptidomimetics rather than small molecules.22
To design peptidomimetic inhibitors targeting DHBDAXX binding, we exploit the well-established interaction between ATRX and DHBDAXX, which is significantly stronger than its interactions with other proteins (p53, Mdm2, Rassf1C, and NSIMDAXX).18,20 Through experiments and computations, we determined that the minimal interaction surface between ATRX and DAXX is characterized by ATRX-derived peptides adopting an alpha-helical conformation upon binding to DHBDAXX. Leveraging this information, we designed a series of stapled peptides,23–26 resulting in several nanomolar binders. Our most promising stapled peptide competitively binds to DHBDAXX with peptides derived from both ATRX and p53. Furthermore, it efficiently releases NSIMDAXX from auto-inhibition, enabling its interaction with SUMO−1. These results validate the utility of our peptidomimetic inhibitors in studying the interactions between DHBDAXX and its multiple interaction partners in vitro. To improve cell permeability, as our initial stapled peptides were unable to enter the cytoplasm, we generated ‘stitched’ peptides23 in which excessive negative charges were eliminated through mutations. These stitched peptides successfully entered cells and remained localized for over 24 hours without perturbing the outer membrane. In summary, our data demonstrate that ATRX-derived peptidomimetics targeting the DAXX protein binding interface serve as effective inhibitors of multiple DAXX interactions, providing potential tools for investigating DAXX interactions in cells and serving as potential therapeutic precursors.
Experimental
Protein expression and purification
The NSIM-DHBDAXX (residues 1-144 hDAXX), DHBDAXX (residues 55-144 hDAXX), and DIDATRX (residues 1190-1325 hATRX) coding sequences were cloned into a pNIC28-bsa4 plasmid vector with an N-terminal His6-tev purification tag. GST-DIDATRX and related fragments were cloned into a pNIC-GST plasmid vector with an N-terminal GST-His6-tev tag. All clones in pNIC vectors were prepared by the NTU protein production platform (https://proteins.sbs.ntu.edu.sg). The coding sequence for SUMO−1 (residues 1–97 hSUMO1) was synthesized and cloned into a pET28a plasmid with an N-terminal His6-tev purification tag (Genscript Ltd).All proteins, except SUMO−1 and 15N-NSIM-DHBDAXX, were expressed and purified by the NTU protein production platform following standardized protocols. SUMO−1 and 15N-NSIM-DHBDAXX were expressed in BL21(DE3) Rosetta T1R cells, cultured in either 2xTY media or M9 minimal media supplemented with 0.5 g L−1 15NH4Cl, respectively.
For all proteins, cell lysis was performed by sonication and initial purification was performed using Nickel NTA affinity chromatography. Cell lysates were loaded onto the column and washed with a standard lysis/Ni-NTA binding buffer (20 mM HEPES pH 7.5, 0.3 M NaCl, 10 mM Imidazole, 1 mM DTT, 10% glycerol) to remove unbound material. The bound proteins were then eluted with the same buffer containing 0.3 M Imidazole. The N-terminal His-tag of NSIM-DHBDAXX, DHBDAXX, or SUMO−1 proteins was cleaved off using tev protease during overnight dialysis against Ni-NTA binding buffer at 4 °C. The cleaved tag, uncleaved protein, tev protease, and other contaminants were removed using Nickel NTA resin, and the unbound protein was further purified using a HiLoad 16/60 Superdex 75 size exclusion column (GE Healthcare).
Except for proteins used in NMR experiments, a standard storage buffer (20 mM HEPES pH 7.5, 0.3 M NaCl, 1 mM DTT, 10% glycerol) was used for the final size-exclusion and concentration steps before flash freezing of purified proteins. For 15N NSIM-DHBDAXX and SUMO−1 proteins, a phosphate buffer (20 mM sodium phosphate pH 6.5, 0.5 M KCl, 0.1 mM EDTA, 10 mM DTT) was used in the final purification step. Protein concentrations were determined using the relevant molar extinction coefficient at 280 nm. Further characterization of purified proteins can be found in the ESI.†
Peptide synthesis and purification
The non-fluorescent linear and stapled peptides (SPEP1-7) were synthesized in-house or obtained from Mimotopes Pty Ltd. The FAM-SPEP7 stapled peptide was purchased from Synpeptide Co Ltd. FAM- and chloroalkane-labelled stitched peptides were synthesized at the Institute of Chemical & Engineering Science, (A*STAR). The purity of the peptides was confirmed using HPLC and mass spectrometry. Additional information on peptide purification can be found in the supplementary methods section. The initial stapled peptides (SPEP1-7) that were synthesised included N-terminal ‘SD’ residues (compared to ‘PEPI’), derived from 15N-labelled linear peptides used in NMR experiments not described in this manuscript. These residues were replaced by an N-terminal FAM-moeity for subsequent experiments and are demonstrably not important for binding in all our analyses, hence they are not discussed in the main text for clarity. Unless specified otherwise, concentrations of peptides without a FAM moiety or aromatic residues were determined by diluting from 10 mM peptide stocks in DMSO, prepared according to the synthesis report. The concentrations of FAM-labeled peptides were determined by measuring the absorbance at 495 nm, using an extinction coefficient of 75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm−1 M−1. The concentration of the p53 peptide was determined using the appropriate molar extinction coefficient at 280 nm. The sequences and chemical modifications of the peptides used are provided in the main figures and/or ESI.†
000 cm−1 M−1. The concentration of the p53 peptide was determined using the appropriate molar extinction coefficient at 280 nm. The sequences and chemical modifications of the peptides used are provided in the main figures and/or ESI.†
Co-immunoprecipitation
The purified GST-tagged DIDATRX and DIDATRX fragments were immobilized on Glutathione Sepharose 4B beads (GE Healthcare) using saturating conditions in binding buffer (20 mM HEPES, pH 7.5, 150 mM NaCl, 0.5 mM EDTA, 1 mM DTT, 10% Glycerol, 1% Triton). After washing off excess GST-tagged proteins HeLa nuclear extract (Ipracell Ltd), supplemented with protease inhibitors (Roche Ltd), was added to the beads. The samples were incubated at 4 °C for 4 hours, followed by washes (x4) with binding buffer. Bound proteins were eluted using 2x SDS PAGE sample loading buffer (Thermo Fisher) and heated at 95 °C for 5 minutes. For western blot analysis, the immunoprecipitated proteins were separated by SDS PAGE (NuPAGE 4–12% Bis-Tris, Thermo Fisher), transferred to a PVDF membrane, blocked with 10% BSA dissolved in PBS buffer containing 0.02% Tween 20, probed with a primary antibody (anti-DAXX, Sigma cat no. D7810, diluted 1:8000), and detected with horseradish peroxidase-conjugated anti-rabbit secondary antibodies (CST, cat no. 70745).
Isothermal titration calorimetry (ITC)
ITC experiments were performed using a MicroCal PEAQ-ITC calorimeter at 25 °C (Malvern). All peptides and proteins were dialysed overnight against ITC binding buffer (20 mM HEPES, pH 7.5, 150 mM NaCl, 10% glycerol, 1 mM DTT). The heat of binding was measured by 19 sequential 2 ml injections of peptide (or DIDATRX) at 0.1–1 mM into 0.2 mL DHBDAXX at 0.01–0.1 mM. Data analysis and curve fitting to a one-site binding model was performed using the PEAQ-ITC analysis software package provided with the calorimeter. In the absence of an absorbance signal to measure the concentrations of dialysed peptides, the concentration of DHBDAXX measured using A280nm and the stoichiometry (n = 1) were fixed during data fitting.
Circular dichroism spectropolarimetry (CD)
Far UV CD spectra between 190 and 260 nm were measured with a 2 nm bandwidth in a 2 mm cuvette at 25 °C using a Chiroscan spectropolarimeter (Applied PhotoPhysics). Peptides at 0.05 mM were dialysed against 10 mM sodium phosphate, pH 7.5, 150 mM NaF at room temperature overnight. The raw ellipticity measurements were converted to mean residue ellipticity (θMRE) for plotting (Fig. 4D and Fig. S3, ESI†) and values of θMRE at 222 nm were used to calculate percentage helicity (Fig. S4 and Table 2, ESI†) using established methods.Fluorescence polarisation
Fluorescence polarisation (FP) of the FAM-SPEP7 peptide with excitation and emission wavelengths of 485 nm and 535 nm respectively, bandwidths of 20 nm was measured using a SparkTM 10M instrument (Tecan) at 25 °C. A constant concentration of 10 nM FAM-SPEP7 was maintained throughout both saturation and competition binding experiments. In saturation binding experiments characterised by increasing FP upon binding, FAM-SPEP7 was titrated with varying amounts of either DHBDAXX or NSIM-DHBDAXX proteins. For competition binding experiments, a constant concentration of DHBDAXX (0.75 mM) was maintained to generate an 80% saturated FAM-SPEP7/DHBDAXX complex in the absence of competitor. Displacement of the FAM-SPEP7 peptide from this complex, characterised by decreasing FP was quantified by titration with increasing amounts of competitor ligand. All titrations were performed by making a 1:1 serial dilution of titrant into solutions of FAM-SPEP7 or FAM-SPEP7/DHBDAXX complex and allowing samples to equilibrate for 30 min at room temperature prior to measurement. FP data were fitted to a two-state binding model for saturation binding experiments or a competitive binding model for competition binding experiments.27
1H-15N HSQC NMR spectroscopy
1H-15N HSQC NMR spectra were recorded using a cryo-probe equipped Bruker Avance II 600 MHz spectrometer, processed and analysed using TopSpin4.0 software. Prior to titration, proteins were transferred into NMR buffer (10 mM potassium phosphate, pH 6.5, 100 mM KCl, 0.1 mM EDTA, 10 mM DTT, 10% D2O). We first titrated 15N DHBDAXX (100 mM in NMR buffer) with FAM-SPEP7 (10 mM in DMSO) to make a saturated complex. We observed discrete changes in the 1H–15N HSQC spectrum during the titration, where the transition between unbound and bound forms was in slow exchange on the NMR timescale (Fig. S9, ESI†). A small quantity of precipitated complex was formed during the titration. This was removed and the final concentration of soluble 15N DHBDAXX/FAM-SPEP7 complex was determined using the absorbance at 280 nm. This complex was titrated with unlabelled SUMO−1 (1.7 mM in NMR buffer), upon which we saw discrete changes in the 1H–15N HSQC spectrum, where affected cross peaks were in intermediate or fast exchange. We followed 9 fast exchange peaks during the titration to determine the saturation point of the observed transition. The chemical shift changes observed for each peak were plotted as a function of the ligand-substrate ratio and fitted to a 1:1 binding model to determine the individual dissociation constants (Fig. S9, ESI†), using the following equation:where, A = normalization constant, x = ligand-substrate molar ratio, KD = dissociation constant, V0 = initial volume (in μL), ni= initial amount of DHBDAXX—FAM-SPEP7 complex (in nmol), C = stock concentration of ligand (SUMO−1, in mM).
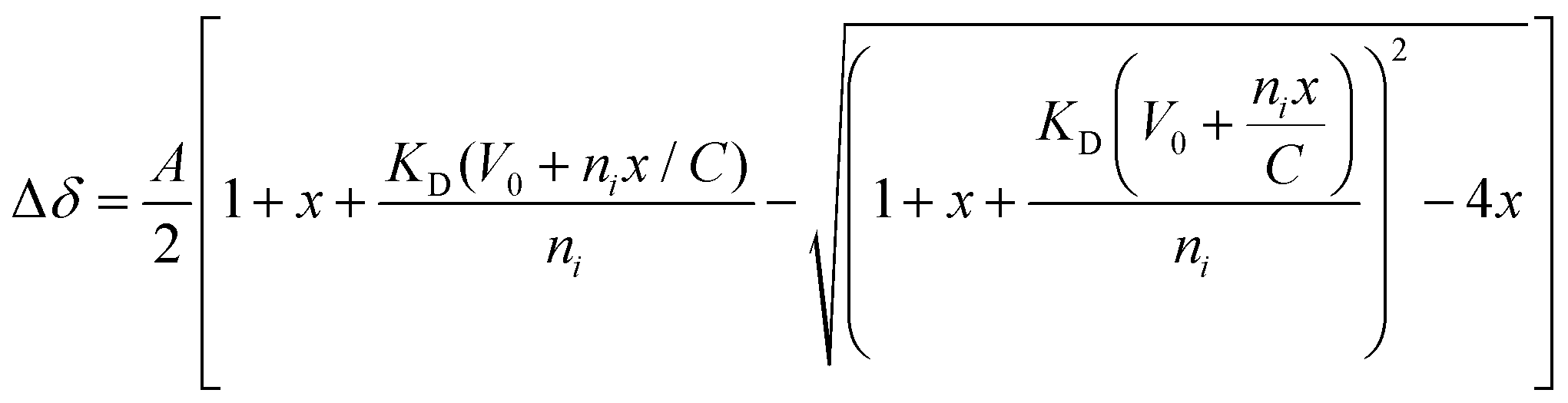
Lactate dehydrogenase (LDH) release assay
Cells were seeded into a 96-well plate in DMEM containing 10% FCS at a cell density of 5000 cells per well and incubated for 24 hours prior to replacement of cell media with 90 μL of culturing medium with 2% FCS. Cells were then treated with 10 μL of peptide for either 4 or 24 h. Final working concentration of DMSO was 1% v/v. Corresponding negative control wells with 1% DMSO only were also prepared. Cytosolic lactate dehydrogenase release was detected using the cytoTox 96® non-radioactive cytotoxicity Assay kit (Promega) as per manufacturer's instructions. Measurements were carried out using an Envision multiplate reader (PerkinElmer). Maximum LDH release was defined as the amount of LDH released when cells were lysed in the presence of 0.1% TRITON X-100 and was used to normalize the results.Live cell confocal microscopy
A total of 100000 U2OS cells were seeded in 3.5 cm glass-bottomed dishes using DMEM containing 10% FCS. After 24 hours, the media was replaced with DMEM containing either 0% or 2% FCS, and the cells were treated with 12.5 μM of the specified FAM-labelled peptides for either 5 hours or 24 hours. Following treatment, the cells were washed twice with PBS, and the media replaced with phenol red-free Optimem. Confocal images were captured at the designated time points using a Yokogawa CSU-22 spinning disk confocal system integrated with a motorized Nikon Eclipse Ti microscope. The microscope setup included a stage-top incubator and CO2 control system, a 100× 1.4 NA Plan Apo objective lens, a photometrics CoolSNAPHQ2 camera, and a 491 nm laser. MetaMorph software (Molecular Devices) was utilized to control acquisition parameters, shutters, filter positions, and focus.
NanoBRET assay
HCT116 cells stably expressing Nanoluc-Halotag fusion protein were plated at a density of 60000 cells per well in a 96-well white opaque tissue culture plate and incubated overnight. The following day, the medium was replaced with 90 μL of Optimem without red phenol, with or without 2% FCS (fetal calf serum). Chloroalkane-labelled stitched peptides were added to the cells at various concentrations and incubated for either 4 or 24 hours at 37 °C with 5% CO2. The final concentration of DMSO (dimethyl sulfoxide) was 1% v/v. To label unoccupied Nanoluc-Halotag molecules, 10 μL of 618 Nanobret-Ligand solution (12× concentration) was added to each well 30 minutes before each endpoint. To measure BRET (bioluminescence resonance energy transfer) signal inhibition, 10 μL of 12× Nano-Glo Substrate solution was added to each well, and the plates were immediately read on the Envision instrument.
Computational modelling
A three-dimensional atomistic model of DIDATRX (residues 1190–1325) was generated using the I-TASSER program.28 To construct the atomistic structure of the DHBDAXX-PEPATRX complex, the experimental structure of DHBDAXX and the structure of PEPATRX extracted from the DIDATRX model (this study) were utilized in the HADDOCK program (High Ambiguity Driven biomolecular DOCKing).29,30 Experimental structures of DHBDAXX in its apo form (PDB 2KZS), complexed with Rasf1c (PDB 2KZU), and in complex with PEPATRX (PDB 5GRQ, resolution 1.5 Å) were employed for the design of stapled and stitched peptide inhibitors targeting DAXX protein. The Amber18 program,31 utilising the Xleap module, was employed to prepare the system for Molecular Dynamics (MD) simulations. Staple and stitched linker parameters were taken from our previous study.32 The systems were neutralized with the appropriate number of counter ions and solvated in an octahedral box with TIP3P water molecules, ensuring a minimum distance of 10 Å between solute atoms and the box boundaries.33 MD simulations were performed using the pmemd.cuda module of the Amber18 package and the ff14SB force field, under explicit solvent conditions at 300 K, unless otherwise specified.34 Long-range electrostatic interactions were handled using the particle mesh Ewald method with a real space cut-off of 9 Å.35 Bond vibrations involving hydrogen atoms were constrained using the settle algorithm, enabling a time step of 2 fs.36 Energy minimization was carried out for solvent molecules and counter ions, initially with restraints (force constant: 50 kcal mol−1 Å−2) on protein and peptide atoms using the Steepest Descent minimizer (1000 steps), followed by unrestrained energy minimization to remove steric clashes. The system was gradually heated from 0 to 300 K through MD simulations with positional restraints (force constant: 50 kcal mol−1 Å−2) on protein and peptides over a period of 0.25 ns, allowing water molecules and ions to move freely. The positional restraints were gradually removed, followed by a 2 ns unrestrained equilibration at 300 K. The resulting structures were used as starting points for the production phase of the MD simulations. For each case, three independent MD simulations were performed with different initial random velocities, each running for 100 ns. Conformations were recorded every 4 ps. To enhance conformational sampling, Biasing Potential Replica Exchange MD (BP-REMD) simulations were applied to the peptides. BP-REMD is a Hamiltonian REMD technique that utilizes a biasing potential to promote dihedral transitions among replicas.37,38 Eight replicas, including a reference replica without any bias, were employed for BP-REMD simulations, with exchange attempts between neighbouring replicas made every 2 ps, accepted or rejected based on the metropolis criteria. Conformations sampled from the reference replica (without bias) were further analysed. Visualization of simulation trajectories was performed using VMD,39 and figures were generated using Pymol.39,40Binding energy calculations and energy decomposition analysis
The binding energies between the peptides and their partner proteins were determined using the molecular mechanics Poisson Boltzmann surface area (MMPBSA) method, which provides a reliable estimate of the binding strength.41,42 To calculate the binding energies, a subset of 250 conformations, taken at 200 ps intervals from the final 50 ns of the simulation trajectories, was used. It is important to note that due to the computational complexity and convergence challenges associated with entropy calculations, they were not included in the analysis.To gain further insights into the contributions of individual residues to the binding energy, the Generalized Born approximation to the MMPBSA method, known as MMGBSA, was employed. This energy decomposition scheme allows the decomposition of the overall binding energy into contributions from specific residues. The polar contribution to the solvation free energy was determined using the generalized born (GB) method with igb = 2, employing mbondi2 radii.31 Non-polar contributions were estimated using the ICOSA method, which considers the solvent-accessible surface area (SASA) and incorporates a surface tension proportionality constant of 0.0072 kcal mol−1 Å−2.
Additionally, in silico alanine scanning was performed to explore the impact of individual peptide residues on the binding energy. In this analysis, each peptide residue was mutated to alanine in each conformation of the MD simulation, and the change in binding energy relative to the wild-type peptide was calculated using MMPBSA. These energy changes were averaged over all conformations to provide a comprehensive assessment of the contribution of each residue to the binding affinity.
Results and discussion
Determination of the minimal interaction surface between ATRX and DAXX
To identify the minimal interaction surface between ATRX and DAXX, we first dissected the previously reported DAXX Interacting Domain of ATRX (DIDATRX, residues 1190–1325)14 using a combination of experimental and computational techniques. We observed a robust interaction between DIDATRX and the DAXX Helical Bundle (DHBDAXX, residues 55–144) using ITC (Kd 80 nM) (Fig. S1 and Table 1, ESI†). Since we couldn't identify a homologous structural template for the 136 amino acid long DIDATRX sequence using BLASTp we used the I-TASSER pipeline to predict its atomic structure.28,43 The prediction indicated a helical region spanning residues 1267 to 1284, which was consistent with secondary structure and disorder predictions (Fig. 1A).| Peptide | Kd (μM) | ΔH | −TΔS | ΔG |
|---|---|---|---|---|
| Calculated errors for fitted parameters are shown in parentheses. Units for changes in Enthalpy (ΔH), Free energy (ΔG) and Entropy (−TΔS) are kcal mol−1. | ||||
| 1255–1289 | 0.16 (0.01) | −12.2 (0.10) | 2.87 | −9.28 |
| 1255–1284 | 0.39 (0.03) | −10.6 (0.13) | 1.83 | −8.75 |
| 1261–1290 | 0.24 (0.02) | −13.8 (0.11) | 4.82 | −9.02 |
| 1263–1290 | 0.97 (0.09) | −13.4 (0.22) | 5.14 | −8.21 |
| 1265–1290 | 1.23 (0.08) | −13.3 (0.16) | 5.18 | −8.09 |
| 1267–1290 | 31.0 (7.64) | −11.9 (2.22) | 5.78 | −6.15 |
| 1267–1284 | 101 (162) | −9.3 (9.4) | 3.87 | −5.45 |
| 1264–1285 | 0.75 (0.07) | −13.3 (0.14) | 4.9 | −8.4 |
| 1190–1325 | 0.08 (0.01) | −11.7 (0.10) | 2.06 | −9.69 |
 | ||
| Fig. 1 Dissection of DIDATRX to define minimal sequence (PEPATRX) required for specific binding to DHBDAXX. (a) Results of per-residue disorder prediction (disEMBL https://dis.embl.de) for DIDATRX are plotted, with results from secondary structure prediction shown below. A helical region is predicted, spanning residues 1267–1284. (b) western blot analysis of pull-down assays used to determine which fragments of DIDATRX could pull down full-length DAXX protein from HeLa nuclear extracts.‘*’ denotes the shortest fragment (1255–1289) found to bind to DAXX and used as the basis to test a series of further peptide fragments in ITC binding experiments. (c) The sequence of the 1255–1289 fragment is shown with the predicted helical region highlighted in blue. Truncations around the helical region were made to identify critical binding residues. Plots of binding enthalpies determined using ITC upon titration of these peptide fragments against DHBDAXX are shown, where the identity of each peptide is indicated in the figure legend. Left panel: The results indicate that residues important for specific binding are contained within the N-terminal but not the C-terminal helix-flanking region. Right panel: The effect of systematically truncating the N-terminal flanking region, indicating that the two residues (N1265 and D1266) directly preceding the helix are critical for binding. | ||
To further understand the binding requirements, we tested purified GST-tagged DIDATRX fragments for interaction with full-length DAXX (Fig. 1B). Notably, DAXX was detected in the pulldowns of all fragments containing the predicted helix, except for GST-ATRX (1267–1325), suggesting the involvement of the region preceding the alpha helix in DIDATRX. To determine the essential residues for specific binding, we measured thermodynamic binding parameters using ITC for a series of DIDATRX peptide fragments (Table 1). The results (Fig. 1C and D) showed that the longest peptide in the series, ATRX (1255–1289), bound to DHBDAXX with an affinity similar to full-length DIDATRX (Kds 0.16 μM and 0.08 μM, respectively). This peptide included 12 residues flanking the N-terminal and 5 residues flanking the C-terminal ends of the predicted alpha helix (1267–1284). Assessing the contribution of these regions to binding (Fig. 1C and Table 1) revealed a negligible decrease in affinity (∼2-fold) upon deletion of the C-terminal flanking region. In contrast, deletion of the N-terminal flanking region dramatically reduced binding (∼200-fold), consistent with the pull-down data (Fig. 1B). The N-terminal flanking sequence contained a polyaspartate sequence (1259–1266) punctuated by a single asparagine (N1265), which we hypothesized to be important for binding to DHBDAXX due to electrostatics/H-bonds and/or additional specific interactions. Truncating this region revealed a ∼25-fold reduction in binding affinity and a significant change in enthalpy upon deleting the two amino acid residues (N1265 and D1266) directly preceding the alpha helix. This indicated that one or both residues are necessary for binding specificity (Fig. 1D and Table 1).
Based on these results, we defined a peptide fragment of ATRX (1264–1285) as PEPATRX, which was sufficient for specific interaction with DHBDAXX. The measured binding constant between PEPATRX and DHBDAXX (Kd 0.75 μM) was approximately 10-fold weaker than the corresponding interaction with the longer DIDATRX (Kd 80 nM) (Table 1).
Modelling the structure of the PEPATRX – DHBDAXX complex
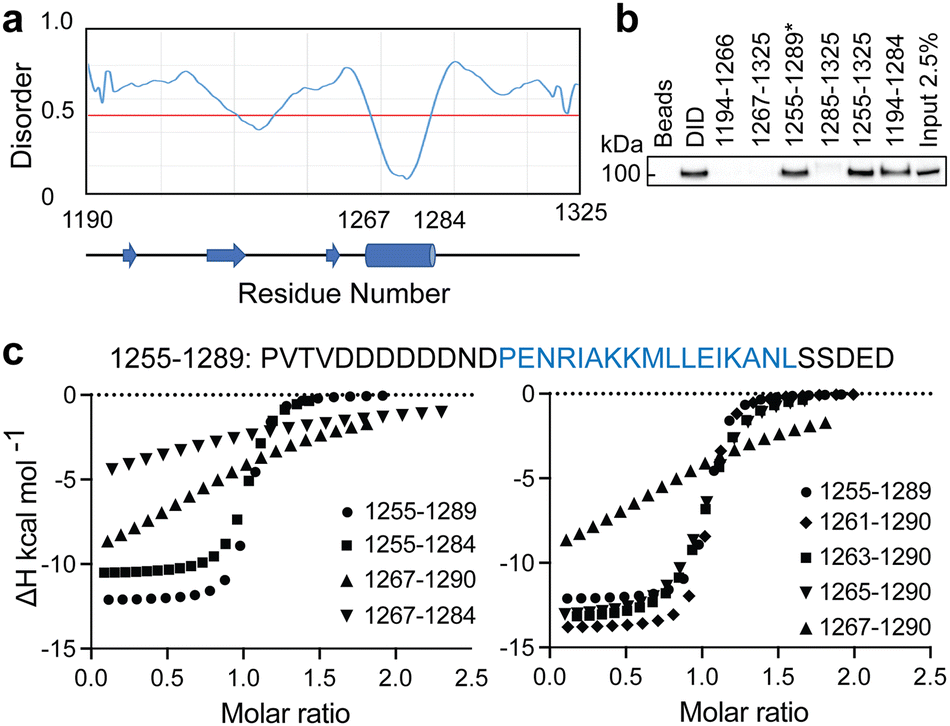
When we embarked on our inhibitor design studies, the only available structural information was the solution NMR structures of apo-DHBDAXX and DHBDAXX in complex with a peptide derived from the Rassf1C tumor suppressor (PEPRassf1C).18 It was observed that PEPRassf1C adopts a helical conformation upon binding to DHBDAXX, suggesting that PEPATRX, our peptide of interest, may also bind to DHBDAXX as an alpha helix.To investigate the 3-dimensional binding mode of PEPATRX, a helical model was generated using I-TASSER and then docked into the Rassf1C binding pocket of DHBDAXX using HADDOCK (High Ambiguity Driven biomolecular DOCKing).29,30 The docking protocol was established by successfully reproducing the bound conformation of the Rassf1C peptide from the NMR structure, with rmsd ∼1.5 Å between the top docking solution and the 1st conformation of the NMR ensemble (2KZU.pdb). Additional docking experiments of the PEPATRX helix (obtained from denovo modeling) against DHBDAXX were then carried out. The resulting poses of the peptide docked into the DHBDAXX pocket were organized into 7 major clusters (Fig. S2A, ESI†). To refine the bound poses, molecular dynamics (MD) simulations were performed on representative complexes from each cluster. In the MD simulations of DHBDAXX/PEPRassf1C, the peptide remained stably bound with no observed unbinding or protein unfolding events (rmsd ∼ 3 Å Fig. S2B, ESI†). However, in the case of DHBDAXX/PEPATRX, only 2 out of the 7 docked models remained stably bound, and these two poses exhibited overlapping conformations (Fig. S2C, ESI†). Remarkably, the positioning of the PEPATRX poses on the DHBDAXX surface differed significantly from the surface occupied by PEPRassf1C in the solution structure, with the helical axes juxtaposed by approximately 90 degrees (Fig. 2A).
 | ||
| Fig. 2 Comparison of the modelled DHBDAXX/PEPATRX structure with experimentally determined DHBDAXX/PEPRassf1C and DHBDAXX/PEPATRX structures. Cartoon representations comparing the 2 docked poses of PEPATRX (green and magenta) in the modeled DHBDAXX/PEPATRX structures with (a), the solution structure of DHBDAXX/PEPRassf1C (red) (pdb 2KZU) and (b), the co-crystal structures of DHBDAXX/PEPATRX (pdb 5Y18 (orange), 5GRQ (blue) and 5Y60 (cyan)). The structures were superimposed using the bound DHBDAXX (grey) backbone. | ||
During the course of our study, three X-ray crystal structures of DHBDAXX in complex with various ATRX peptides, which overlapped with PEPATRX, were published.19,21,44 In all of these crystal structures, residues 1267–1283 of ATRX adopted an alpha-helical conformation. The sidechain of D1266ATRX was observed to form a helix stabilizing hbond with the backbone amide of N1269ATRX and with the sidechain K122DAXX in structures 5GRQ.pdb and 5Y6O.pdb (visible only in some chains of the unit cell) that contained a flanking region preceding the helix, providing further support for the importance of residues preceding the helix in specific binding. Remarkably, the two stably docked poses from our in silico modelling aligned well with the bound ATRX peptide in the crystal structures, despite the initial juxtaposed orientation of PEPATRX in the model. This demonstrates the robustness of our de novo modelling approach (Fig. 2B). However, for subsequent design studies, we chose to utilize the structural information from the DHBDAXX/PEPATRX complex crystal structure (PDB, 5GRQ) due to the comprehensive insights it enables into the interaction. We designated the PEPATRX template sequence as ‘PEPI’ and renumbered the residues according to the peptide sequence (1–22). Detailed information on the specific interactions and surface charge complementarity between PEPI and DHBDAXX is shown in Fig. 3.
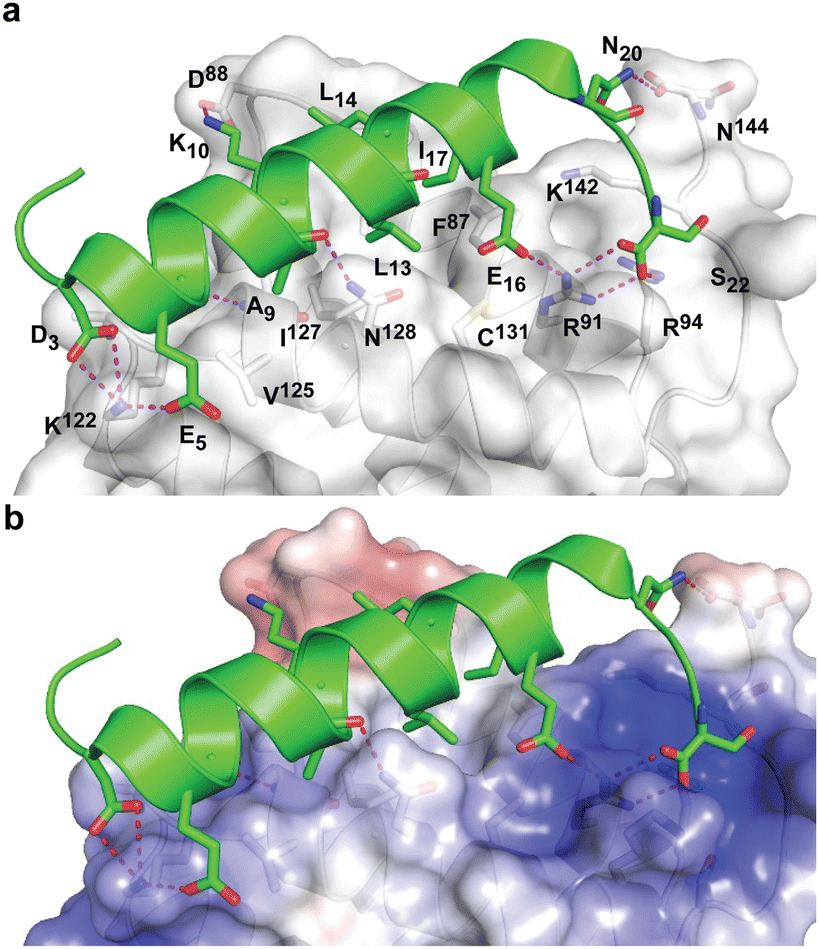 | ||
| Fig. 3 Structural views of DHBDAXX – PEPATRX interactions. (a) Cartoon representation of PEPATRX (green) bound to DHBDAXX surface (grey/white). The interacting protein–peptide residues are labelled and highlighted as sticks. (b) as in (a) but with labels removed and displaying the electrostatic surface potential of the DHBDAXX protein (red to blue colours indicating a range from −5 to +5 kcal mol−1). This highlights the charge complementary of the interaction surface to PEPATRX residues and locates the hydrophobic pocket on DHBDAXX that is important for binding both PEPRassf1C and PEPATRX. | ||
Conformational dynamics of PEPI and DHBDAXX in apo and bound states
To design tight binding analogues of PEPI it is essential to understand the behaviour of the peptide and the target in solution. This knowledge provides insights into the thermodynamic parameters that can be manipulated to enhance the affinity of the analogues. Therefore, we investigated the conformational behaviour of unbound PEPI in solution using Hamiltonian replica exchange molecular dynamics (HREMD). Simulations, initiated with the crystallographic helical structure, revealed that PEPI rapidly lost its secondary structure and became highly flexible within a short time frame of approximately 5 ns (Fig. S3A, ESI†). The conformational landscape of unbound PEPI was characterized by predominantly disordered states with transient helical motifs (∼15% helicity overall). Circular Dichroism spectropolarimetry (CD) measurements confirmed even lower helicity in solution (∼1.5%) (Fig. S3B and Table 2, ESI†). This emphasizes that the binding of PEPI to DHBDAXX in an alpha helical conformation is driven by strong enthalpic forces.| Peptide | K d | ΔH | −TΔS | ΔG | Helicity (%) |
|---|---|---|---|---|---|
| Calculated errors for fitted parameters are shown in parentheses. Units for changes in enthalpy (ΔH), free energy (ΔG) and entropy (−TΔS) are kcal mol−1. | |||||
| PEPI | 0.75 (0.07) | −13.3 (0.14) | 4.94 | −8.36 | 1.4 |
| SPEP1 | 0.09 (0.01) | −9.0 (0.17) | −0.57 | −9.63 | 17.4 |
| SPEP2 | 0.05 (0.01) | −10.2 (0.12) | 0.3 | −9.9 | 11.7 |
| SPEP3 | 0.18 (0.04) | −8.8 (0.20) | −0.39 | −9.19 | 17.9 |
| SPEP4 | 0.34 (0.05) | −7.6 (0.17) | −1.3 | −8.9 | 13.9 |
| SPEP5 | 0.55 (0.08) | −7.4 (0.25) | −1.1 | −8.5 | 8.8 |
| SPEP6 | 0.07 (0.01) | −8.5 (0.18) | −1.2 | −9.8 | 14.2 |
| SPEP7 | 0.08 (0.07) | −12.8 (0.11) | 3.1 | −9.7 | 23.4 |
We explored the structural dynamics of apo DHBDAXX in solution using classical MD simulations. Except for the N- and C-terminal residues (55–60 and 137–144, respectively), DHBDAXX was found to maintain stability throughout the simulations (rmsd < 2 Å) (Fig. S3C, ESI†) in accordance with the solution structure of apo DHBDAXX (2KZS.PDB).
In MD simulations of the DHBDAXX/PEPI complex, both DHBDAXX and PEPI exhibited stable conformations (rmsd ∼2.5 Å) (Fig. S3C, ESI†). The flexibility of the C-terminal region of apo DHBDAXX was reduced upon complexation due to the formation of salt-bridge and hydrogen bond interactions with PEPI. However, the flexibility at the N-terminal region of DHBDAXX remained unchanged. During the simulation, PEPI predominantly maintained its α-helical conformation while bound to DHBDAXX (∼70% α-helicity), with increased flexibility observed towards the C-terminal region of the peptide, which remained disordered throughout (Fig. S3C, ESI†). The hydrophobic and charged interactions observed in the co-crystal structure of DHBDAXX/PEPI (Fig. 3) were largely preserved during approximately 80% of the MD simulation. Collectively, these results indicate that the binding of PEPI to DHBDAXX is primarily driven by strong enthalpic forces, overcoming the entropic penalty associated with the folding of PEPI into the bound conformation.
Design of stapled peptide inhibitors of DHBDAXX
The low helicity of unbound PEPI in solution indicates that its binding to DHBDAXX occurs through an induced alpha helical conformation. To enhance the affinity for DHBDAXX by reducing the entropic cost of folding and binding, we aimed to constrain unbound PEPI into an alpha helical conformation resembling its bound state through hydrocarbon stapling. Our initial step was to identify suitable positions on PEPI that would, upon introduction of the hydrocarbon linkers, minimize perturbations to its interactions with DHBDAXX.The MD simulations of the DHBDAXX/PEPI complex were mined to decompose the contribution of each residue in PEPI to the overall binding energy (Fig. 4A). As anticipated, charged/polar residues (D3, E5, N6, K10, E16, and S22) and hydrophobic residues (A9, L13, L14, I17, and L21) at the DHBDAXX/PEPI interface, as observed in the crystal structure of the complex (Fig. 3), made favourable contributions to the binding.
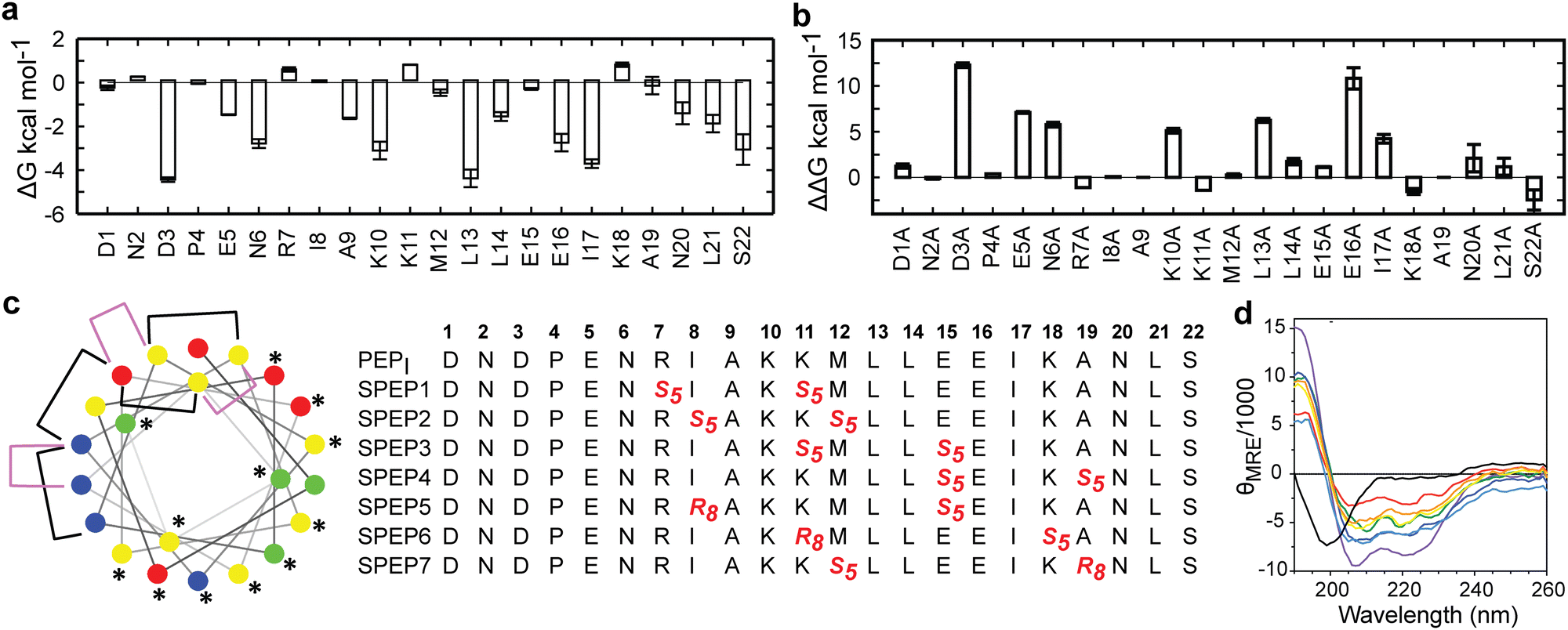 | ||
| Fig. 4 In silico analysis of DHBDAXX–PEPI key interacting residues and stapled peptide designs. (a) Per-residue binding free energy contributions of PEPI in the DHBDAXX/PEPI complex from MD simulations. (b) Computational alanine scanning of PEPI residues in the DHBDAXX/PEPATRX complex. (c) Helical wheel representation of the linear PEPATRX peptide sequence used for the design of stapled peptides. Colour-coded circles indicate polar/acidic (red), polar/basic (blue), polar/uncharged (green) and nonpolar (yellow) residues. Residues important for interaction with DHBDAXX are highlighted with asterisks. Positions bridged by hydrocarbon linkers i,i + 4 or i,i + 7 are indicated with magenta or black brackets, respectively. Sequences of stapled peptides (SPEP1–7) based on MD simulations are shown alongside using standard nomenclature with staple positions highlighted in red. Residue number is shown in bold above. (d) CD spectra of all stapled peptides (various colours) demonstrate a marked increase in helicity compared to the linear PEPI peptide (black). | ||
We performed in silico alanine scanning substitutions to explore the impact of each amino acid residue in PEPI on its interaction with DHBDAXX. The results (Fig. 4B) were consistent with the per-residue contributions obtained from the decomposition analyses. Major loss in affinity (with ΔΔG > 2 kcal mol−1) was observed for alanine mutations at residues D3, E5, N6, K10, E16, L13, and I17. Conversely, other positions displayed greater tolerance to alanine substitutions, with certain positively charged residues (R7, K11, and K18) even showing an increase in binding affinites. Taken together, these analyses suggested specific positions where staples could be incorporated without significantly affecting the interactions between DHBDAXX and PEPI.
Incorporating the staples also necessitated careful selection of sidechains with appropriate stereochemistry. We employed established strategies effective for stapling right-handed alpha-helices, such as S5 to S5 for (i, i + 4) linkages and R8 to S5 for (i, i + 7) linkages.45 Guided by these linkages and the insights from simulations, we designed seven stapled peptides referred to as SPEP 1–7 (Fig. 4C).
Peptide stapling increases helicity
To validate the design of our stapled peptides, we performed HREMD simulations on the SPEP 1–7 peptides in solution, initializing them from alpha helical conformations. As anticipated, all the stapled peptides exhibited higher overall helicities in solution, ranging from 26% to 58%, compared to the linear PEPI peptide with a helicity of approximately 15% (Fig. S4, ESI†). Analysis of per-residue helicity patterns also indicated that the stapled peptides tended to maintain helical structures in the central regions while exhibiting increased flexibility at the termini (Fig. S4, ESI†). Interestingly, no clear correlation emerged between changes in helicity and the specific positions of the staples, which is consistent with observations from previous studies on other systems.46,47To experimentally assess the helicity of both the linear PEPI and SPEP 1–7 peptides, we conducted CD spectropolarimetry (Fig. 4D and Fig. S4, ESI†). The experimental results confirmed a significant increase in helicity upon stapling, ranging from 9% to 23%, when compared to the linear PEPI peptide (1.5%). However, it is noteworthy that the experimental helicity values consistently appeared lower than the values obtained from the simulations (Fig. S4, ESI†).
Modelling of DHBDAXX – SPEP complexes
The binding of SPEP 1–7 peptides to DHBDAXX was modeled based on the co-crystal structure of the DHBDAXX/PEPATRX complex (5GRQ.pdb), where the ATRX residues 1264–1285 (PEPI) served as a starting point (Fig. 3). By individually incorporating the staples into the PEPI peptide and subjecting them to 100 ns MD simulations (triplicate runs), we observed that all SPEP 1–7 peptides maintained stability throughout the simulations, with rmsd values reaching approximately 2 Å from their respective starting conformations (Fig. S5, ESI†). The introduction of hydrocarbon linkers led to an increase in helicity within the bound peptides, which maintained a helical content of 75% to 85% throughout the simulations. Notably, flexibility was observed at both termini of the peptides, particularly at the C-terminal ends, which generally exhibited less helicity compared to the rest of the peptide. Exceptions to this trend were observed in SPEP6 and SPEP7, where the staple linker spanned the C-terminal region of PEPI. The bound conformations of DHBDAXX remained stable, with rmsd ∼3 Å from the starting conformation (Fig. S5, ESI†).Structural snapshots from the MD simulations of the complexes (Fig. 5) revealed that the hydrocarbon staples remained exposed to the solvent and did not directly engage with the DHBDAXX surface. Consistent with the crystal structure of DHBDAXX/PEPI (Fig. 3), the hydrophobic residues of SPEP 1–7, including A9, L13, and I7, remained buried, with L14 partially exposed in the hydrophobic pocket/binding site of DHBDAXX. In contrast, simulations of the unstapled DHBDAXX/PEPI complex showed L14 exposed to solvent (not shown). The salt bridges and hydrogen bond interactions observed in the crystal structure of the DHBDAXX/PEPI complex were well retained (∼90%) throughout the MD simulations of the DHBDAXX/SPEP complexes. Additionally, several new interactions were observed during the MD simulations of the DHBDAXX-SPEP complexes, although these interactions have not been experimentally verified and hence are not detailed here. Overall, analysis indicated that charged and polar residues (D3, E5, N6, K10, E16, S22) along with hydrophobic residues (A9, L13, L14, I17, L21) contributed significantly to the binding of the stapled peptides in the MD simulations (Fig. S6, ESI†). These analyses enabled the identification of several promising stapled peptides with satisfactory overall binding parameters (Table S1, ESI†).
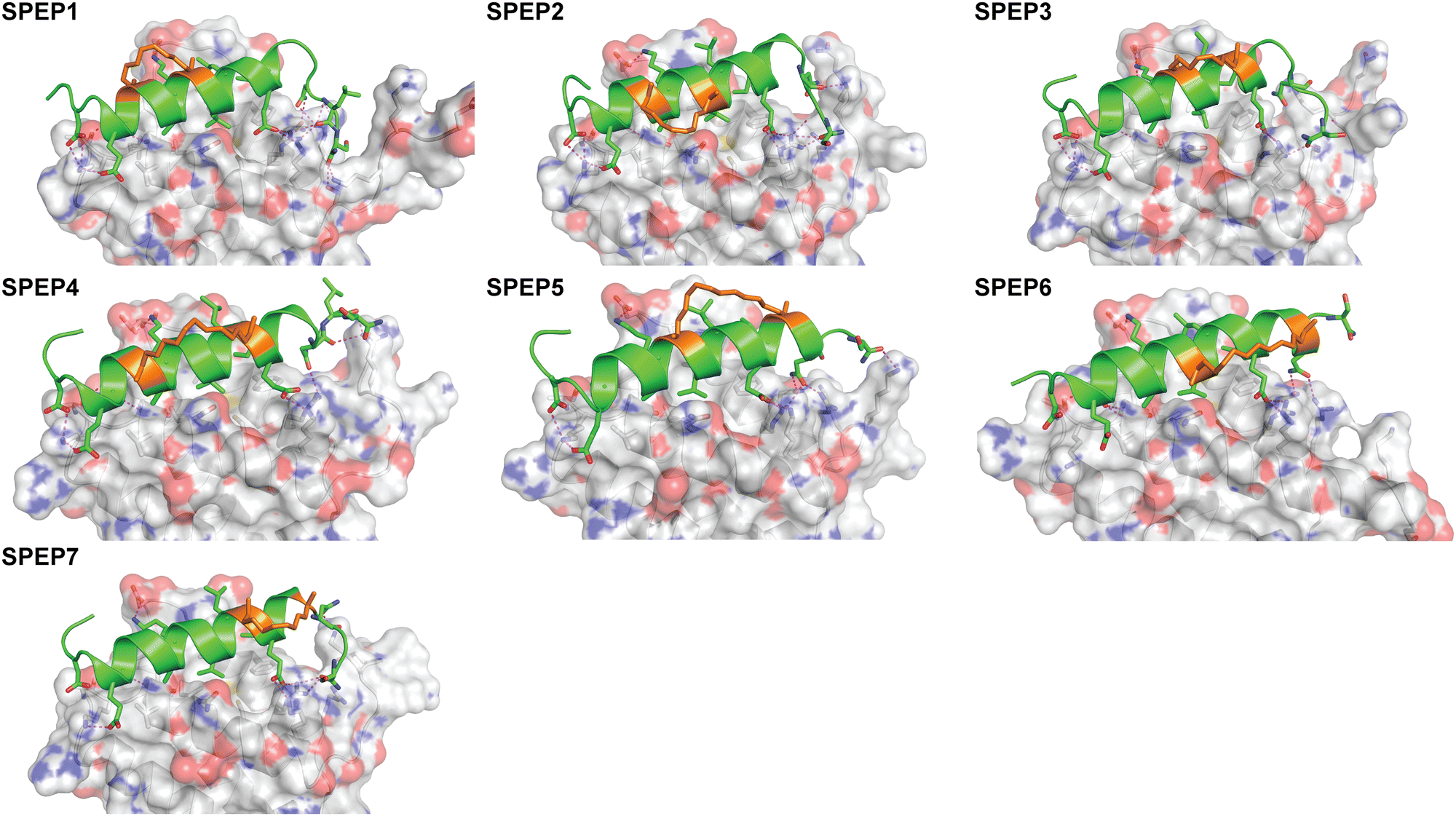 | ||
| Fig. 5 Structural representations of MD snapshots of DHBDAXX/SPEP complexes. Structural models for DHBDAXX complexes with SPEP1, SPEP2, SPEP3, SPEP4, SPEP5, SPEP6 and SPEP7 are shown. The DHBDAXX (white) and bound peptide (green) conformations are shown as surface and cartoon representations, respectively with interacting residues and staples (orange) highlighted in sticks. Hydrogen bonds are indicated by dotted lines (magenta). | ||
In conclusion, the MD analyses predict the formation of stable complexes between each of the SPEP peptides and DHBDAXX, with interface interactions closely resembling those observed in the DHBDAXX/linear PEPI complex crystal structure. Consequently, all seven SPEP designs were synthesized for experimental determination of their binding affinities.
Peptide stapling increases binding affinity to DHBDAXX
ITC measurements revealed that the binding affinity of SPEP 1–7 to DHBDAXX fell within the range of approximately 50 nM to 1 μM (Table 2 and Fig. S7, ESI†). All seven stapled peptides exhibited similar or higher affinities compared to the linear PEPI, with four peptides (SPEP 1, 2, 6, and 7) demonstrating binding strengths ∼10–20 times greater. While the stapled peptides yielded free energy gains of up to 1.5 kcal mol−1 upon binding, the linear PEPI exhibited more favorable enthalpic contributions (Table 2). It is worth noting that the linear peptide lacks structure in solution, necessitating a significant enthalpic gain to compensate for the entropic penalty incurred when the disordered peptide adopts an alpha helical conformation upon binding to DHBDAXX. In contrast, the stapled peptides are already alpha helical in solution, resulting in smaller entropic penalties during binding to DHBDAXX. However, the reduction in internal flexibility of the stapled peptides may result in fewer interactions with the target, leading to lower associated enthalpies. Although the computed distributions of contacts between the peptides and the target did not reveal a clear pattern, this is not surprising considering that most interactions are mediated by exposed sidechains. Additionally, the complexity of solvent interactions poses challenges for precise quantification. Surprisingly, experimental data indicated positive entropic contributions upon binding for five out of the seven peptides, suggesting increased disorder. While no direct correlation was observed between the flexibility of peptides in the bound state simulations and the experimentally measured order of entropies (6 > 4 > 5 > 1 > 3), our snapshots of the complexes suggested that peptides with higher entropy had staples exposed to the solvent (Fig. 5). It is plausible that this exposure of the hydrophobic region to a polar solvent generates frustration, resulting in higher entropies. Furthermore, the differential displacement of water molecules from the binding pocket of DHBDAXX may contribute to the observed increases in binding entropy. However, such increases in binding entropy could also indicate a loss of binding specificity, and indeed, this behaviour was consistently accompanied by decreases in binding enthalpy. For subsequent studies we ranked the four best binders based on binding free energies (ΔG) as follows (differences are very small, of the order of ∼1%): SPEP2 > SPEP6 > SPEP7 > SPEP1, but based on binding enthalpy, SPEP7 is the best binder; given that SPEP7 has a much larger binding enthalpy component, we chose this peptide for further explorations.Binding competition experiments
To assess the ability of our stapled peptides to inhibit multiple protein interaction partners that bind to DHBDAXX and disrupt DAXX function, we conducted competition experiments using a fluorescence polarization assay. Initially, we employed an N-terminally FAM-modified version of SPEP7 (FAM-SPEP7) to determine if the fluorescent label affected its binding to DHBDAXX. The results showed that FAM-SPEP7 exhibited comparable affinity to the non-fluorescent SPEP7 (Kd 46 nM vs. 80 nM, respectively), indicating that the FAM label did not interfere with binding (Fig. 6A). This validation allowed us to proceed with confidence using the fluorescence polarization assay for subsequent experiments. Furthermore, we demonstrated that FAM-SPEP7 could be displaced from DHBDAXX by the non-fluorescent SPEP1-7 peptides with similar affinities as measured by ITC, confirming that all peptides target the same interaction site (Fig. S8, ESI†). These findings support FAM-SPEP7 as an effective model peptide for probing DHBDAXX interactions with other binding partners (Fig. 6).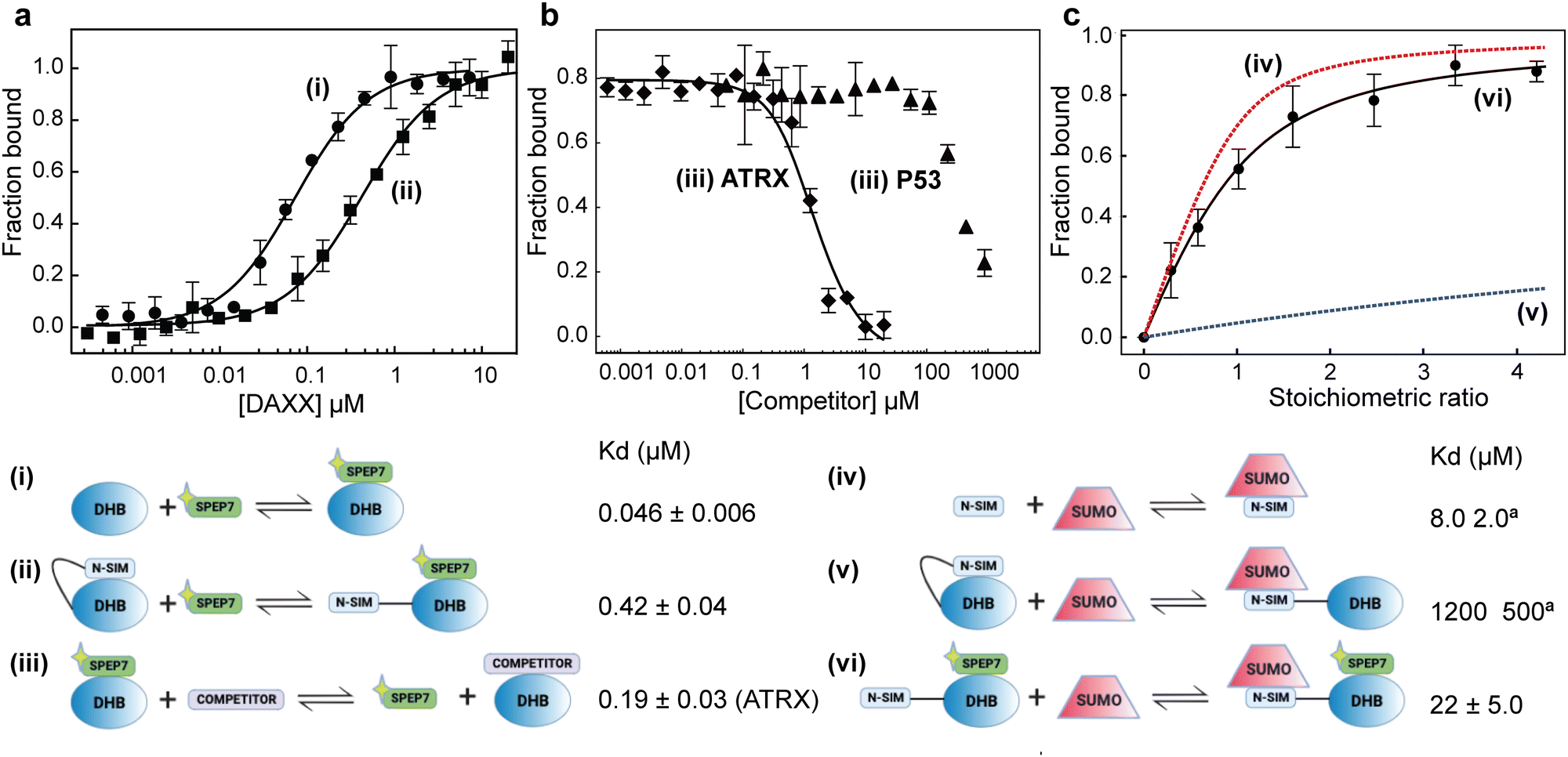 | ||
| Fig. 6 Multiple interactions and binding modes of DHBDAXX probed using FAM-SPEP7. Data depicted in (A)–(C) are labeled according to the relevant schematic ((i) to (vi)) that describes the corresponding binding interaction and associated Kd. A yellow star is used to indicate the FAM moiety. (a) Saturation binding of FAM-SPEP7 to DHBDAXX ((i), closed circles) or NSIM-DHBDAXX ((ii), closed squares). (b) (iii) Competitive displacement of FAM-SPEP7 from DHBDAXX by either DIDATRX (closed diamonds) or PEPP53 (closed triangles). The data points in (a) and (b), shown with standard error bars, are averages of three independent measurements. (c) Release of NSIMDAXX for binding to SUMO by FAM-SPEP7 ((vi) closed circles)). The data points with standard error bars are the normalized average chemical shift change observed for 7 cross-peaks in the 1H-15N HSQC spectrum of the 15N-NSIM-DHBDAXX/FAM-SPEP7 complex upon titration with SUMO1. An apparent Kd for the interaction was obtained by fitting the curve. Simulated binding curves (dashed lines) for NSIMDAXX/SUMO1 ((iv) red) and NSIM-DHBDAXX/SUMO1 ((v) blue dashed) interactions were calculated from published data. | ||
By conducting titration experiments with competitors such as DIDATRX or p53 peptide against the DHBDAXX/FAM-SPEP7 complex, we confirmed their ability to displace FAM-SPEP7 from DHBDAXX (Fig. 6B). The competitive binding affinity between DIDATRX and DHBDAXX obtained from the competition data was comparable to the direct binding affinity measured by ITC (Kd 193 nM vs. 80 nM, respectively), indicating a high level of complementarity between their interaction surfaces, as expected. However, we estimate ∼10-fold weaker competitive binding of the p53 peptide to DHBDAXX compared to its direct binding. This disparity may be attributed to the limited overlap between the binding sites of SPEP7 and the p53 peptide on DHBDAXX, which may primarily involve the hydrophobic pocket that characterizes the shared protein–protein interaction interface.21
In summary, our stapled peptides exhibited effective competition with multiple DAXX interaction partners in vitro, suggesting their potential to disrupt DAXX interactions in vivo. Additionally, protein interaction partners that bind to DHBDAXX with lower complementarity compared to the SPEP7/DHBDAXX interaction are expected to be more strongly inhibited.
Release of NSIMDAXX auto-inhibition
It has been proposed that the N-terminal SUMO Interaction Motif of DAXX (NSIMDAXX) can be sequestered from interaction with SUMO through self-interaction with DHBDAXX, serving as an important regulatory mechanism to prevent interactions of NSIMDAXX with non-cognate SUMOylated protein interaction partners.20 Release of NSIMDAXX could provide a mechanism by which DAXX could be sequestered (e.g. to PML bodies) and thereby provide a general means of inhibition in cells that aberrantly upregulate DAXX.3,5 We aimed to test whether our stapled peptide inhibitors could effectively release NSIMDAXX for interaction with SUMO−1 (NSIMDAXX has no binding preference for a particular homolog of SUMO20) using SPEP7 as a model.We initiated our investigation by quantifying the saturation binding of FAM-SPEP7 to NSIM-DHBDAXX using fluorescence polarization, and interestingly, we observed a ∼10-fold decrease in binding affinity compared to binding to DHBDAXX alone. This finding supports the notion of autoregulation (Fig. 6A). To confirm the release of NSIMDAXX from DHBDAXX upon SPEP7 binding, we conducted titration experiments using 1H-15N HSQC NMR spectroscopy to detect specific interactions. Titrating 15N-labeled NSIM-DHBDAXX with FAM-SPEP7 resulted in distinct changes in amide cross-peaks, indicating the formation of a specific complex (Fig. S9A, ESI†). Further titration of the pre-formed, saturated 15N-NSIM-DHBDAXX/FAM-SPEP7 complex with unlabeled SUMO−1 led to additional specific chemical shift perturbations, indicative of intermediate or fast exchange. We determined the binding affinity of SUMO−1 to NSIMDAXX within the NSIM-DHBDAXX/FAM-SPEP7 complex using 9 amide cross-peaks undergoing fast exchange (Fig. S9B, ESI†). Fitting the normalized, averaged data from these cross-peaks revealed an apparent Kd of ∼22 μM (Fig. 6C). This affinity is comparable to the reported direct interaction between SUMO−1 and isolated NSIMDAXX peptide (8 μM) and significantly stronger (approximately 50-fold) than the interaction observed between SUMO−1 and NSIM-DHBDAXX in the absence of a DHBDAXX binding inhibitor (1.2 mM).20 These results clearly demonstrate that SPEP7, the stapled peptide, effectively releases NSIMDAXX for binding to SUMO−1, thus enabling interactions with SUMOylated protein partners.
Strategy for enhancing cell permeability of SPEP Peptides
In our in vitro experiments, we observed strong binding of stapled peptides SPEP 1, 2, 6, and 7 to DHBDAXX. We also demonstrated that FAM-SPEP7 exhibited competitive binding against peptide ligands derived from cognate interaction partners and was able to release NSIMDAXX from auto-regulation. To further evaluate the potential of these peptides, we investigated their cell permeability. LDH release assays indicated no cytotoxicity in HCT116 cells treated with concentrations up to 50 μM of SPEP1-7 (Fig. S10A, ESI†). However, live cell imaging experiments with FAM-SPEP7 revealed its confinement in non-cytosolic compartments, suggesting limited access to endogenous DAXX (Fig. S10B, ESI†). To enhance the cellular uptake and target engagement of SPEP peptides, we explored key physicochemical properties known to be crucial for cellular permeability: charge, hydrophobicity, and amphipathicity.48,49 The PEPI template sequence, from which the SPEP1–7 peptides are derived, is highly charged, with an abundance of aspartate, glutamate, and lysine residues, resulting in an overall negative charge. The presence of charged residues can hinder cellular uptake due to the energy required to desolvate these residues for membrane traversal.50 Conversely, a net positive charge is generally beneficial for interacting with negative charges associated with cell membranes (phosphates, glycerols etc.). Therefore, we investigated whether substitutions or deletions of charged residues that are not involved in binding could improve permeability. Additionally, we explored the incorporation of an additional staple to increase the hydrophobic and amphipathic character of the peptides. These double-stapled peptides, known as “stitched” peptides, feature contiguous hydrocarbon staples anchored at a common attachment point. Recent studies have demonstrated that stitched peptides exhibit improved thermal and chemical stability, enhanced proteolytic resistance, increased helicity, and enhanced cell permeability.48,51,52Design of stitched peptides (STPEP) for improved cell permeability
The binding of SPEP1-7 peptides with DHBDAXX suggested the possibility of designing two stitched configurations by combining specific pairs of staples. These configurations, based on the SPEP1 + 6 and SPEP2 + 7 combinations, involved matching the positions of i, i + 4 staples in SPEPs 1 and 2 with the i, i + 7 staples in SPEPs 6 and 7, respectively. Additional deletions and mutations were incorporated to remove negative charges, resulting in the design of seven stitched peptides, STPEP 1–7 (Fig. 7).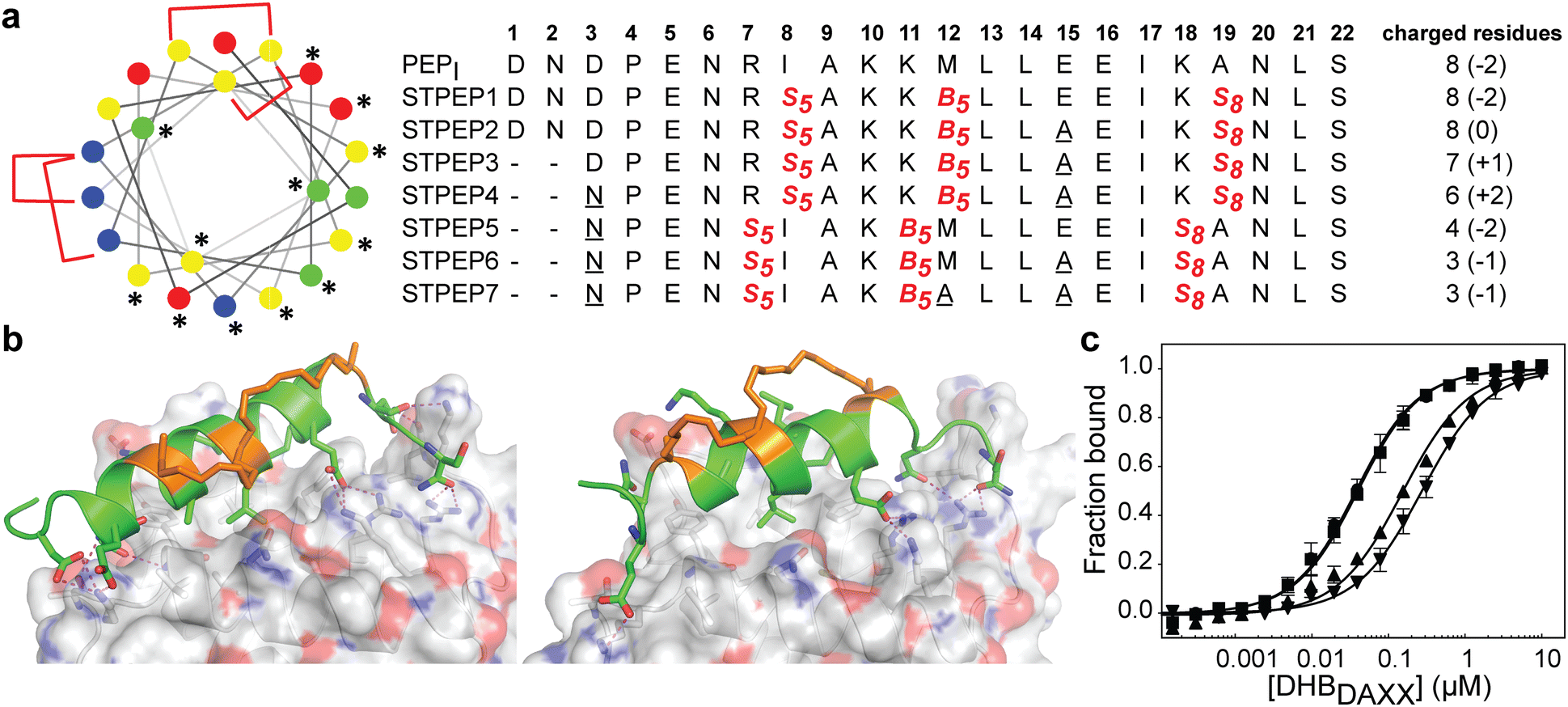 | ||
| Fig. 7 (a) Design of stitched peptide inhibitors of DHBDAXX PPI surface. (a) Helical wheel representation of the linear PEPI template sequence used for the design of stitched peptides. Annotations and colouring as in Fig. 4 except that positions bridged by contiguous hydrocarbon staples i, i,i + 4 plus i,i + 7 are highlighted with red brackets. Sequences of the stitched peptide (STPEP1-7) designs are shown with stitch positions or mutated residues highlighted in red or underlined, respectively. Deleted residues are denoted ‘−’. Residue number is shown in bold above. The number of charged residues, indicating the hydrophobicity of each peptide, is shown to the right with the net charge in parentheses. (b) Structural models generated from MD snapshots for DHBDAXX complexes with the two different stitch configurations. Structural representation as in Fig. 5. Left: Stitched configuration (used in STPEP1–4) resulting from combining SPEP2 and SPEP7 staples. Right: Stitched configuration (used in STPEP5–7) resulting from combining SPEP1 and SPEP6 staples. (c) Binding curves for the interaction of selected stitched peptides with DHBDAXX obtained using fluorescence polarisation. | ||
In HREMD simulations, the stitched peptides (STPEP1–7) showed comparable helicity to the parent singly stapled peptides, with greater helicity retained in the central regions and increased flexibility at the termini (Fig. S4, ESI†). The MD simulations of the STPEP/DHBDAXX complexes confirmed stable binding, with small rmsd values from the starting conformations of DHBDAXX or STPEPs (Fig. S11, ESI†). The hydrocarbon staples remained exposed to solvent without engaging the DHBDAXX surface. Structural snapshots from the simulations predicted that STPEP1, based on the SPEP2 + 7 stitch, retained all the interactions found in the individual stapled peptides, including those at the N- and C-termini, and exhibited similar binding affinity (Fig. 7B and Table S2, ESI†). Sequential introduction of mutations E15A (STPEP2) and ΔD1-N2 (STPEP3) did not affect binding. However, the D3N mutant (STPEP4), aimed at further reducing the negative charge, had a deleterious effect due to the loss of a side-chain interaction between D3 and K122 of DHBDAXX (Fig. 7 and Table S2, ESI†). MD simulations of the STPEP5-7 peptides, with a stitch based on SPEP1 + 6 staples, predicted a further decrease in binding affinity. Interestingly, reduced helicity was observed in both the bound and unbound forms of STPEP5-7, particularly towards the C-terminal region where several peptide-DHBDAXX interactions were lost. This behaviour deviated from the expected stability provided by the i, i + 7 staple incorporated into these peptides, which stabilizes helix formation in the singly stapled SPEP6 peptide (Fig. 5). These observations suggest that the stitched peptides STPEP5-7 may exhibit different behaviour than anticipated based on the individual staples.
To target endogenous DAXX, a compromise between cell permeability and binding affinity of the stitched peptides was considered. Four stitched peptides, STPEP2, STPEP3, STPEP4, and STPEP7, were thus selected for synthesis based on a range of properties predicted by computer simulations (Fig. 7A). These peptides were predicted to bind to DHBDAXX with decreasing affinity as follows: STPEP2 ≈ STPEP3 > STPEP4 > STPEP7 (Table S2, ESI†). In terms of hydrophobicity, the order was reversed, with STPEP7 being the most hydrophobic. Interestingly, the non-binding M12A mutation introduced in STPEP7 for ease of synthesis unexpectedly slightly increased affinity in the simulations. The exact cause of this effect remains unclear as to whether it resulted from the mutation or the staple position.
The selected stitched peptides were synthesized with an N-terminal FAM moiety to facilitate detection of binding to DHBDAXX, cell permeability, and localization. A noticeable decrease in solubility was observed compared to the stapled peptides, with solubility limits of 11, 4, 2, or 49 μM determined for STPEP2, STPEP3, STPEP4, or STPEP7, respectively. This reduced solubility prevented the use of ITC for determining binding thermodynamics or CD for quantifying helicity. However, dissociation constants could be determined using fluorescence anisotropy experiments.
The trends observed in the dissociation constants obtained were in excellent agreement with the MD simulations, validating the computational analyses. No change in affinity was measured for FAM-STPEP2 (E15A, Kd 32 nM) or STPEP3 (ΔD1-N2/E15A, Kd 35 nM) compared to FAM-SPEP7 (Kd 43 nM). However, a ∼4-fold decrease in binding was observed for STPEP4 (ΔD1-N2/D3N/E15A, Kd 148 nM), and a ∼7-fold decrease was observed for STPEP7 (ΔD1-N2/D3N/E15A, Kd 269 nM), which contained the destabilizing SPEP1 + 6 stitch (Fig. 7D and Table S2, ESI†).
Cell permeability of stitched peptides
To assess the cellular permeability and localization of the FAM-labeled stitched peptides, U2OS cells were selected due to their lack of ATRX expression and the presence of large DAXX-associated nuclear PML bodies, which could serve as indicators of peptide targeting to endogenous DAXX. When the cells were incubated with FAM-labeled stitched peptides, some degree of endosomal escape into the cell body was observed, particularly with FAM-STPEP2 and FAM-STPEP7 (Fig. 8A). This indicated an improvement in the cytosolic localization of the peptides. Although no punctate nuclear foci were observed, it was challenging to estimate whether the intracellular peptide concentration was sufficient for binding to DAXX due to the inability to measure absolute concentrations within the cell. To further estimate the extent of peptide intracellular delivery, we employed the NanoBRET assay.53 Chloroalkane versions of STPEP2 and STPEP7 were titrated in HT116 cells containing a cytosolic nanoluc-halotag sensor.53 Both peptides showed no significant cellular toxicity, and the Nanobret IC50 values determined after 24 hours of incubation were 7.1 nM for STPEP2 and 93 nM for STPEP7 (Fig. 8B). Although not directly measured, we assumed that the nuclear concentrations of the peptides were equal to those in the cytoplasm, as it is well-known that proteins smaller than 40 kDa can readily enter the nucleus via passive diffusion through nuclear pore complexes.54 While these results are promising, the intracellular concentrations of the stitched peptides do not reach the concentrations required for binding to endogenous DAXX as measured in vitro for STPEP2 and STPEP7 (32 nM and 269 nM, respectively) (Fig. 7D and Table S2, ESI†). In conclusion, stitched peptides have been successfully designed against DAXX and represent an improvement over the stapled analogues (also designed in this study). They also exhibit increased cytosolic localization, however the concentrations of peptides entering the cells appear to be too low to measure effective targeting of endogenous DAXX and the resulting changes in cellular activity. These findings suggest that further work is necessary to enhance cell permeability and determine whether the peptide concentration within the cell/nucleus is sufficient for effective target engagement.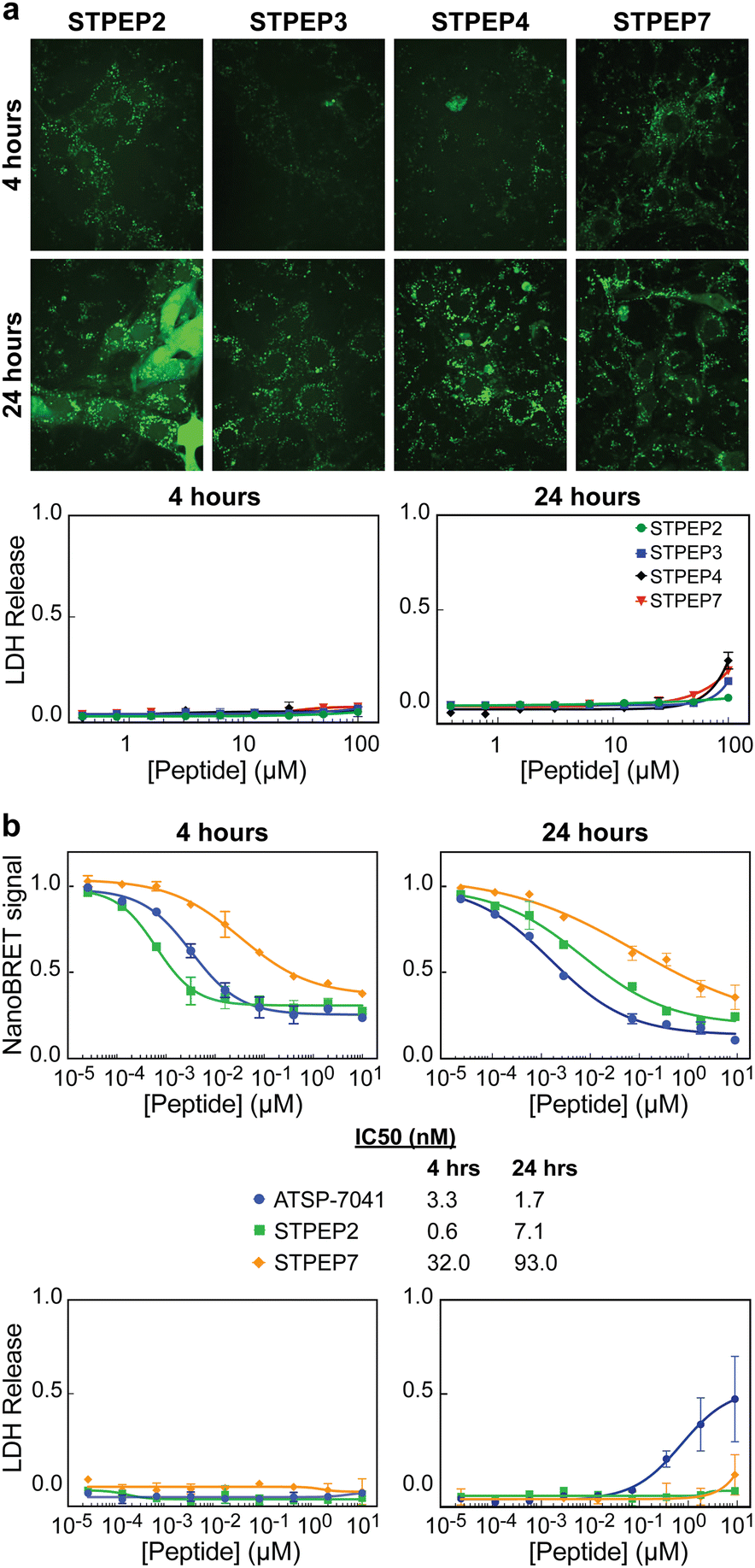 | ||
| Fig. 8 Cell permeability of stitched peptides. (a) Micrographs: Representative confocal images of U2OS cells treated with 25 μM of the indicated FAM-labelled stitched peptides for either 4 or 24 h. LDH assays: Titrations of stapled peptides were performed on U2OS cells in presence of 2% serum and LDH release was assessed at 4 and 24 h. (b) NanoBret assay: Titration of selected peptides vs Bret signal measured in HCT116 cells stably expressing the Nanoluc-Halotag fusion. The ATSP-7041 peptide was included as a positive control. Measurements were made 4 and 24 h after treatment with peptide and IC50s obtained from fitting the data are shown below the plots alongside the figure legends. LDH assays: Titrations of stitched peptides were performed on HCT116 cells in presence of 2% serum and LDH release was assessed at 4 and 24 h. All data points in plots correspond to the mean of three independent measurements with errors corresponding to ± 1 SD. | ||
Conclusions
DAXX, a multifunctional protein, is frequently overexpressed in human cancers, where it can promote cell proliferation (tumorigenesis) and chemoresistance.55,56 Understanding the intricate role of DAXX in cancer biology has been a challenge due to its numerous protein interaction partners.In this study, we aimed to explore the potential of targeting a promiscuous protein–protein interaction (PPI) surface of DAXX as a means to unravel its regulation and function and pave the way for therapeutic interventions. The PPI surface of interest is located in the DAXX Helical Bundle domain (DHBDAXX, residues 55-144), which interacts with multiple partners in a mutually exclusive manner, underscoring its importance as a regulatory domain.18,21 Additionally, the N-terminal SIM of DAXX is sequestered by this interaction surface, presumably preventing promiscuous interactions with non-cognate SUMOylated proteins.20 Releasing NSIMDAXX through competitive inhibition could potentially sequester DAXX when it is aberrantly upregulated in cancer, making our inhibitors promising candidates for therapeutic development.
Through systematic design and evaluation of stapled and stitched peptides based on the DAXX binding motif of ATRX, we successfully generated high-affinity binders that target the protein–protein interaction surface. The stapled peptides exhibit nM affinity for DHBDAXX, as demonstrated by binding assays (Table 2 and Fig. S7, ESI†) and competition experiments with ATRX and p53-derived peptides (Fig. 6B), highlighting their specificity. Furthermore, using a DAXX construct encompassing both the DHB and NSIM, we showed that the same stapled peptide can effectively relieve self-inhibition, allowing NSIMDAXX to interact with SUMO1 (Fig. 6 and Fig. S9, ESI†).
To enhance their cellular permeability, we further optimized the peptides through stitching, resulting in cell-permeable derivatives that displayed non-toxic behaviour. However, the observed cytosolic concentrations of the peptides fell below the dissociation constants measured for peptide binding to DHBDAXXin vitro. Therefore, future development of these peptidomimetic inhibitors may involve incorporating different staples, introducing mutations to modulate charge-hydrophobicity ratios, or conjugation with cyclic cell-penetrating peptides.57 These strategies are likely to yield more potent and cell-active peptide inhibitors for targeting endogenous DAXX, but fall outside of the scope of the current work. In conclusion, our work establishes a valuable foundation for the development of peptidomimetic tools to investigate DHBDAXX-mediated interactions within cells and as potential therapeutic agents.
Author contributions
C. J., S. K and C. S. V. conceptualized and designed the study. C. J. purified all protein reagents (unless otherwise stated) and performed all in vitro binding assays, with assistance from F.W for the collection of NMR data. C. J. and F. W. processed and analysed NMR spectra. C. J. B. and Y. F. performed live-cell imaging, LDH and NanoBret assays with assistance from SRR. Y. F. performed anaylsis of all cell-based assays. S.K performed all computational modelling and analysis. C. W. J. provided the stitched peptides. R. L. assisted with interpretation of CD data. C. J. wrote the paper with input from S. K., C. S. V and D. R. All authors contributed to the interpretation of the data as applicable to their technical contribution.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We thank staff at the NTU Institute of Structural Biology (NISB), Nanyang Technological University, for providing access to facilities; The NTU protein production facility for providing protein expression constructs and purified proteins; The Singapore Eye Research Institute for access to facilities; Rao Chang at the School of Biological Sciences, Nanyang Technological University for peptide synthesis; Charles W Johannes and staff at the Institute of Chemical & Engineering Science (A*STAR) for synthesis of stitched-peptides; The Bio-informatics Institute A*STAR and The National Supercomputing Centre (NSCC) for computational support. This work has been supported by NTU Lee Kong Chian School of Medicine and the Singapore Ministry of Education (MOE) Academic Research Fund (AcRF) Tier 3 (MOE2012-T3-1-001) grant.Notes and references
- X. Yang, R. Khosravi-Far, H. Y. Chang and D. Baltimore, Cell, 1997, 89, 1067–1076 CrossRef CAS PubMed.
- I. Mahmud and D. Liao, Nucleic Acids Res., 2019, 47, 7734–7752 CrossRef CAS PubMed.
- H. M. Shih, C. C. Chang, H. Y. Kuo and D. Y. Lin, Biochem. Soc. Trans., 2007, 35, 1397–1400 CrossRef CAS PubMed.
- J. J. Song and Y. J. Lee, J. Biol. Chem., 2004, 279, 30573–30578 CrossRef CAS PubMed.
- A. Santiago, A. C. Godsey, J. Hossain, L. Y. Zhao and D. Liao, Cell Cycle, 2009, 8, 76–87 CrossRef CAS PubMed.
- D. Y. Lin, Y. S. Huang, J. C. Jeng, H. Y. Kuo, C. C. Chang, T. T. Chao, C. C. Ho, Y. C. Chen, T. P. Lin, H. I. Fang, C. C. Hung, C. S. Suen, M. J. Hwang, K. S. Chang, G. G. Maul and H. M. Shih, Mol. Cell, 2006, 24, 341–354 CrossRef CAS PubMed.
- A. M. Ishov, A. G. Sotnikov, D. Negorev, O. V. Vladimirova, N. Neff, T. Kamitani, E. T. Yeh, J. F. Strauss, 3rd and G. G. Maul, J. Cell Biol., 1999, 147, 221–234 CrossRef CAS PubMed.
- S. J. Elsasser, K. M. Noh, N. Diaz, C. D. Allis and L. A. Banaszynski, Nature, 2015, 522, 240–244 CrossRef CAS PubMed.
- L. A. Puto, C. Benner and T. Hunter, Oncoscience, 2015, 2, 362–372 CrossRef PubMed.
- P. Drane, K. Ouararhni, A. Depaux, M. Shuaib and A. Hamiche, Genes Dev., 2010, 24, 1253–1265 CrossRef CAS PubMed.
- P. W. Lewis, S. J. Elsaesser, K. M. Noh, S. C. Stadler and C. D. Allis, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14075–14080 CrossRef CAS PubMed.
- S. J. Elsasser, H. Huang, P. W. Lewis, J. W. Chin, C. D. Allis and D. J. Patel, Nature, 2012, 491, 560–565 CrossRef PubMed.
- C. P. Liu, C. Xiong, M. Wang, Z. Yu, N. Yang, P. Chen, Z. Zhang, G. Li and R. M. Xu, Nat. Struct. Mol. Biol., 2012, 19, 1287–1292 CrossRef CAS PubMed.
- J. Tang, S. Wu, H. Liu, R. Stratt, O. G. Barak, R. Shiekhattar, D. J. Picketts and X. Yang, J. Biol. Chem., 2004, 279, 20369–20377 CrossRef CAS PubMed.
- D. Kitagawa, H. Kajiho, T. Negishi, S. Ura, T. Watanabe, T. Wada, H. Ichijo, T. Katada and H. Nishina, EMBO J., 2006, 25, 3286–3297 CrossRef CAS PubMed.
- J. Tang, L. K. Qu, J. Zhang, W. Wang, J. S. Michaelson, Y. Y. Degenhardt, W. S. El-Deiry and X. Yang, Nat. Cell Biol., 2006, 8, 855–862 CrossRef CAS PubMed.
- M. Gostissa, M. Morelli, F. Mantovani, E. Guida, S. Piazza, L. Collavin, C. Brancolini, C. Schneider and G. Del Sal, J. Biol. Chem., 2004, 279, 48013–48023 CrossRef CAS PubMed.
- E. Escobar-Cabrera, D. K. Lau, S. Giovinazzi, A. M. Ishov and L. P. McIntosh, Structure, 2010, 18, 1642–1653 CrossRef CAS PubMed.
- Z. Li, D. Zhao, B. Xiang and H. Li, Protein Cell, 2017, 8, 762–766 CrossRef CAS PubMed.
- E. Escobar-Cabrera, M. Okon, D. K. Lau, C. F. Dart, A. M. Bonvin and L. P. McIntosh, J. Biol. Chem., 2011, 286, 19816–19829 CrossRef CAS PubMed.
- X. Wang, Y. Zhao, J. Zhang and Y. Chen, Protein Cell, 2017, 8, 767–771 CrossRef CAS PubMed.
- H. Lu, Q. Zhou, J. He, Z. Jiang, C. Peng, R. Tong and J. Shi, Signal Transduction Targeted Ther., 2020, 5, 213 CrossRef PubMed.
- X. Li, S. Chen, W. D. Zhang and H. G. Hu, Chem. Rev., 2020, 120, 10079–10144 CrossRef CAS PubMed.
- P. M. Cromm, J. Spiegel and T. N. Grossmann, ACS Chem. Biol., 2015, 10, 1362–1375 CrossRef CAS PubMed.
- G. Fumagalli, R. J. Carbajo, J. W. M. Nissink, J. Tart, R. Dou, A. P. Thomas and D. R. Spring, J. Med. Chem., 2021, 64, 17287–17303 CrossRef CAS PubMed.
- M. T. J. Bluntzer, J. O’Connell, T. S. Baker, J. Michel and A. N. Hulme, Pept. Sci., 2021, 113, 24191 CrossRef.
- M. H. Roehrl, J. Y. Wang and G. Wagner, Biochemistry, 2004, 43, 16056–16066 CrossRef CAS PubMed.
- J. Yang, R. Yan, A. Roy, D. Xu, J. Poisson and Y. Zhang, Nat. Methods, 2015, 12, 7–8 CrossRef CAS PubMed.
- C. Dominguez, R. Boelens and A. M. Bonvin, J. Am. Chem. Soc., 2003, 125, 1731–1737 CrossRef CAS PubMed.
- G. C. P. van Zundert, J. Rodrigues, M. Trellet, C. Schmitz, P. L. Kastritis, E. Karaca, A. S. J. Melquiond, M. van Dijk, S. J. de Vries and A. Bonvin, J. Mol. Biol., 2016, 428, 720–725 CrossRef CAS PubMed.
- D. A. Case, I. Y. Ben-Shalom, S. R. Brozell, D. S. Cerutti, T. E. Cheatham, V. W. D. C. III, T. A. Darden, R. E. Duke, D. Ghoreishi, M. K. Gilson, H. Gohlke, A. W. Goetz, D. Greene, R. Harris, N. Homeyer, S. Izadi, A. Kovalenko, T. Kurtzman, T. S. Lee, S. LeGrand, P. Li, C. Lin, J. Liu, T. Luchko, R. Luo, D. J. Mermelstein, K. M. Merz, Y. Miao, G. Monard, C. Nguyen, H. Nguyen, I. Omelyan, A. Onufriev, F. Pan, R. Qi, D. R. Roe, A. Roitberg, C. Sagui, S. Schott-Verdugo, J. Shen, C. L. Simmerling, J. Smith, R. Salomon-Ferrer, J. Swails, R. C. Walker, J. Wang, H. Wei, R. M. Wolf, X. Wu, L. Xiao, D. M. York and P. A. Kollman, AMBER 2018, University of California, San Francisco, 2018.
- Y. S. Tan, J. Reeks, C. J. Brown, D. Thean, F. J. Ferrer Gago, T. Y. Yuen, E. T. Goh, X. E. Lee, C. E. Jennings, T. L. Joseph, R. Lakshminarayanan, D. P. Lane, M. E. Noble and C. S. Verma, J. Phys. Chem. Lett., 2016, 7, 3452–3457 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, J. Chem. Theory Comput., 2015, 11, 3696–3713 CrossRef CAS PubMed.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- S. Miyamoto and P. A. Kollman, J. Comput. Chem., 1992, 13, 952–962 CrossRef CAS.
- S. Kannan and M. Zacharias, Proteins, 2007, 66, 697–706 CrossRef CAS PubMed.
- K. Ostermeir and M. Zacharias, J. Comput. Chem., 2014, 35, 150–158 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14(33–38), 27–38 Search PubMed.
- W. De Lano, The PyMOL Molecular Graphics System, Version 1.2r3pre, Schrödinger, LLC.
- T. Hou, J. Wang, Y. Li and W. Wang, J. Comput. Chem., 2011, 32, 866–877 CrossRef CAS PubMed.
- N. Homeyer and H. Gohlke, Mol. Inform., 2012, 31, 114–122 CrossRef CAS PubMed.
- J. Zhang and T. L. Madden, Genome Res., 1997, 7, 649–656 CrossRef CAS PubMed.
- D. Hoelper, H. Huang, A. Y. Jain, D. J. Patel and P. W. Lewis, Nat. Commun., 2017, 8, 1193 CrossRef PubMed.
- L. D. Walensky and G. H. Bird, J. Med. Chem., 2014, 57, 6275–6288 CrossRef CAS PubMed.
- F. Bernal, A. F. Tyler, S. J. Korsmeyer, L. D. Walensky and G. L. Verdine, J. Am. Chem. Soc., 2007, 129, 2456–2457 CrossRef CAS PubMed.
- S. Kannan, P. G. A. Aronica, S. Ng, D. T. Gek Lian, Y. Frosi, S. Chee, J. Shimin, T. Y. Yuen, A. Sadruddin, H. Y. K. Kaan, A. Chandramohan, J. H. Wong, Y. S. Tan, Z. W. Chang, F. J. Ferrer-Gago, P. Arumugam, Y. Han, S. Chen, L. Renia, C. J. Brown, C. W. Johannes, B. Henry, D. P. Lane, T. K. Sawyer, C. S. Verma and A. W. Partridge, Chem. Sci., 2020, 11, 5577–5591 RSC.
- S. Kannan, P. G. A. Aronica, S. Ng, D. T. Gek Lian, Y. Frosi, S. Chee, J. Shimin, T. Y. Yuen, A. Sadruddin, H. Y. K. Kaan, A. Chandramohan, J. H. Wong, Y. S. Tan, Z. W. Chang, F. J. Ferrer-Gago, P. Arumugam, Y. Han, S. Chen, L. Renia, C. J. Brown, C. W. Johannes, B. Henry, D. P. Lane, T. K. Sawyer, C. S. Verma and A. W. Partridge, Chem. Sci., 2020, 11, 5577–5591 RSC.
- S. R. Perry, T. A. Hill, A. D. de Araujo, H. N. Hoang and D. P. Fairlie, Org. Biomol. Chem., 2018, 16, 367–371 RSC.
- P. G. Dougherty, A. Sahni and D. Pei, Chem. Rev., 2019, 119, 10241–10287 CrossRef CAS PubMed.
- G. J. Hilinski, Y. W. Kim, J. Hong, P. S. Kutchukian, C. M. Crenshaw, S. S. Berkovitch, A. Chang, S. Ham and G. L. Verdine, J. Am. Chem. Soc., 2015, 137, 8858 CrossRef CAS PubMed.
- G. H. Bird, N. Madani, A. F. Perry, A. M. Princiotto, J. G. Supko, X. He, E. Gavathiotis, J. G. Sodroski and L. D. Walensky, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14093–14098 CrossRef CAS PubMed.
- A. Peier, L. Ge, N. Boyer, J. Frost, R. Duggal, K. Biswas, S. Edmondson, J. D. Hermes, L. Yan, C. Zimprich, A. Sadruddin, H. Y. Kristal Kaan, A. Chandramohan, C. J. Brown, D. Thean, X. E. Lee, T. Y. Yuen, F. J. Ferrer-Gago, C. W. Johannes, D. P. Lane, B. Sherborne, C. Corona, M. B. Robers, T. K. Sawyer and A. W. Partridge, ACS Chem. Biol., 2021, 16, 293–309 CrossRef CAS PubMed.
- S. Shimozono, H. Tsutsui and A. Miyawaki, Biophys. J., 2009, 97, 1288–1294 CrossRef CAS PubMed.
- W. W. Pan, J. J. Zhou, X. M. Liu, Y. Xu, L. J. Guo, C. Yu, Q. H. Shi and H. Y. Fan, J. Biol. Chem., 2013, 288, 13620–13630 CrossRef CAS PubMed.
- S. B. Liu, X. P. Lin, Y. Xu, Z. F. Shen and W. W. Pan, J. Ovarian Res., 2018, 11, 90 CrossRef CAS PubMed.
- P. G. Dougherty, J. Wen, X. Pan, A. Koley, J. G. Ren, A. Sahni, R. Basu, H. Salim, G. Appiah Kubi, Z. Qian and D. Pei, J. Med. Chem., 2019, 62, 10098–10107 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cb00149k |
| ‡ Current address: Cancer Science Institute, National University of Singapore, MD6 level 07-01. |
| § These authors contributed equally to the work |
| This journal is © The Royal Society of Chemistry 2023 |