
Regioselective O-alkylation of 2-pyridones by TfOH-catalyzed carbenoid insertion†
Zhewei
Yan
ab,
Hangli
He
ab,
Dabiao
Yuan
ab,
Qiongjiao
Yan
a,
Wei
Wang
 a,
Haipeng
Jiang
a,
Haifeng
Wang
*ab and
Fener
Chen
*acd
a,
Haipeng
Jiang
a,
Haifeng
Wang
*ab and
Fener
Chen
*acd
aPharmaceutical Research Institute, Wuhan Institute of Technology, Wuhan, 430205, China. E-mail: skytacle@139.com
bSchool of Chemical Engineering & Pharmacy, Wuhan Institute of Technology, Wuhan, 430205, China
cEngineering Center of Catalysis and Synthesis for Chiral Molecules, Department of Chemistry, Fudan University, Shanghai, 200433, China
dShanghai Engineering Center of Industrial Catalysis for Chiral Drugs, Shanghai, 200433, China
First published on 7th December 2022
Abstract
Selective alkylation of 2-pyridone could solve a challenge in chemistry and streamline the synthesis of important molecules. Here we report the regioselective O-alkylation of 2-pyridones by TfOH-catalyzed carbenoid insertion. In the catalytic system, alkylation of 2-pyridone was achieved with unprecedented regioselectiviy (>99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). This protocol is characterized by mild reaction conditions, metal-free, and simplicity. Moreover, this method provides the desired products in good yield and demonstrates a broad substrate scope in this transformation.
:1). This protocol is characterized by mild reaction conditions, metal-free, and simplicity. Moreover, this method provides the desired products in good yield and demonstrates a broad substrate scope in this transformation.
The synthesis of substituted 2-pyridones is of great significance because these motifs are commonly found in biologically relevant molecules,1 drugs2 and natural products (Scheme 1).3 Accordingly, a variety of approaches have been developed to access this important motif.4 In principle, the direct alkylation of 2-pyridones can offer an appealing alternative, but the realization of this strategy has proven challenging and limits wider implementation due to the existence of two tautomeric forms of 2-pyridones (Scheme 2A).5 For instance, the ambident nucleophilic nature of 2-pyridone usually gives a mixture of N- and O-alkylated products in the presence of base (Scheme 2B).6
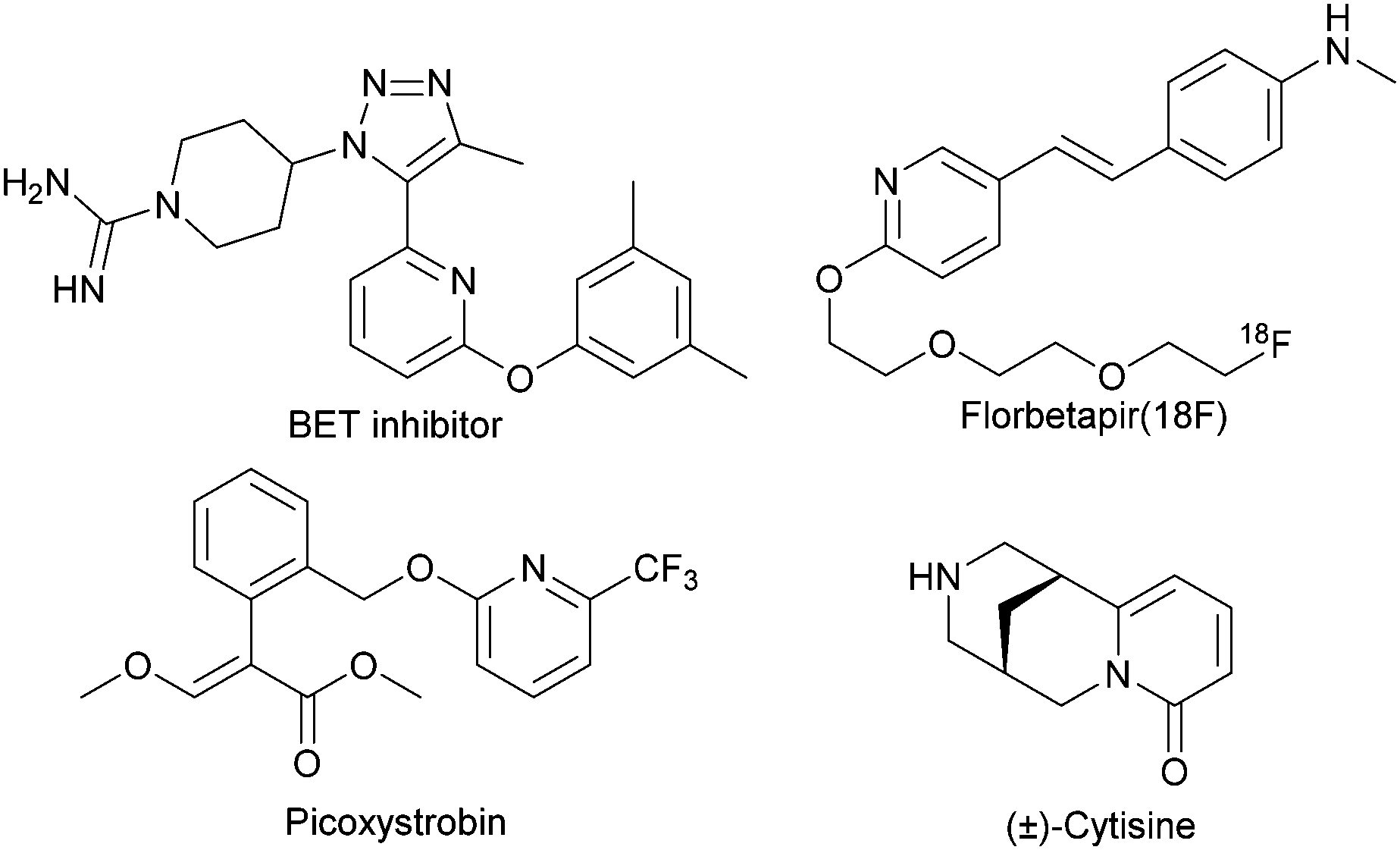 | ||
| Scheme 1 Representative biologically active compounds containing the 2-pyridone. | ||
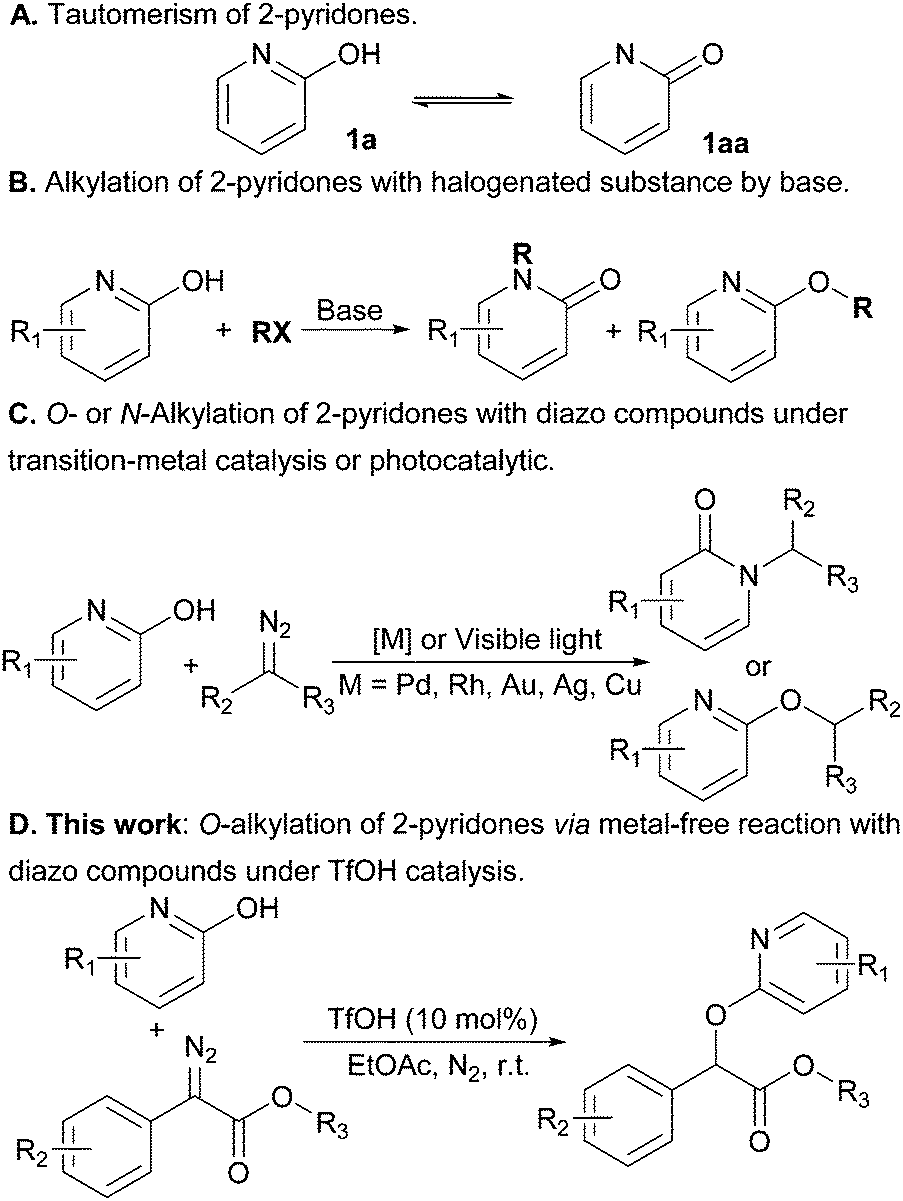 | ||
| Scheme 2 Site-selective alkylation of 2-pyridones. | ||
Towards the goal of the regioselectivity, various strategies ranging from photocatalytic methods,7 to transition-metals (palladium,8 rhodium,9 gold,10 silver11 and copper12) have been explored to achieve site-selective N- or O-functionalization (Scheme 2C). However, compared with the well documented photo and transition-metal catalytic protocols, studies on the acid-catalytic regioselective alkylation of 2-pyridones with diazo compounds remain underexplored. We became interested in this challenge as we envisioned that Brønsted acid catalyzed alkylation of diazo compounds would facilitate selective functionalization, providing a practical and scalable solution to the regioselective issues of the prevalent heterocycles. In fact, we recently demonstrated the regioselective N2-alkylation of indazoles in high regioselectivity by employing inexpensive TfOH as catalyst.13 This example reveals that Brønsted acids as catalysts in carbene transfer contribute to overcoming the challenge of selective alkylation. With this strategy in mind, we here wish to report our new discovery that TfOH-catalyzed regioselective O-alkylation of 2-pyridones with diazo compounds has been developed (Scheme 2D).
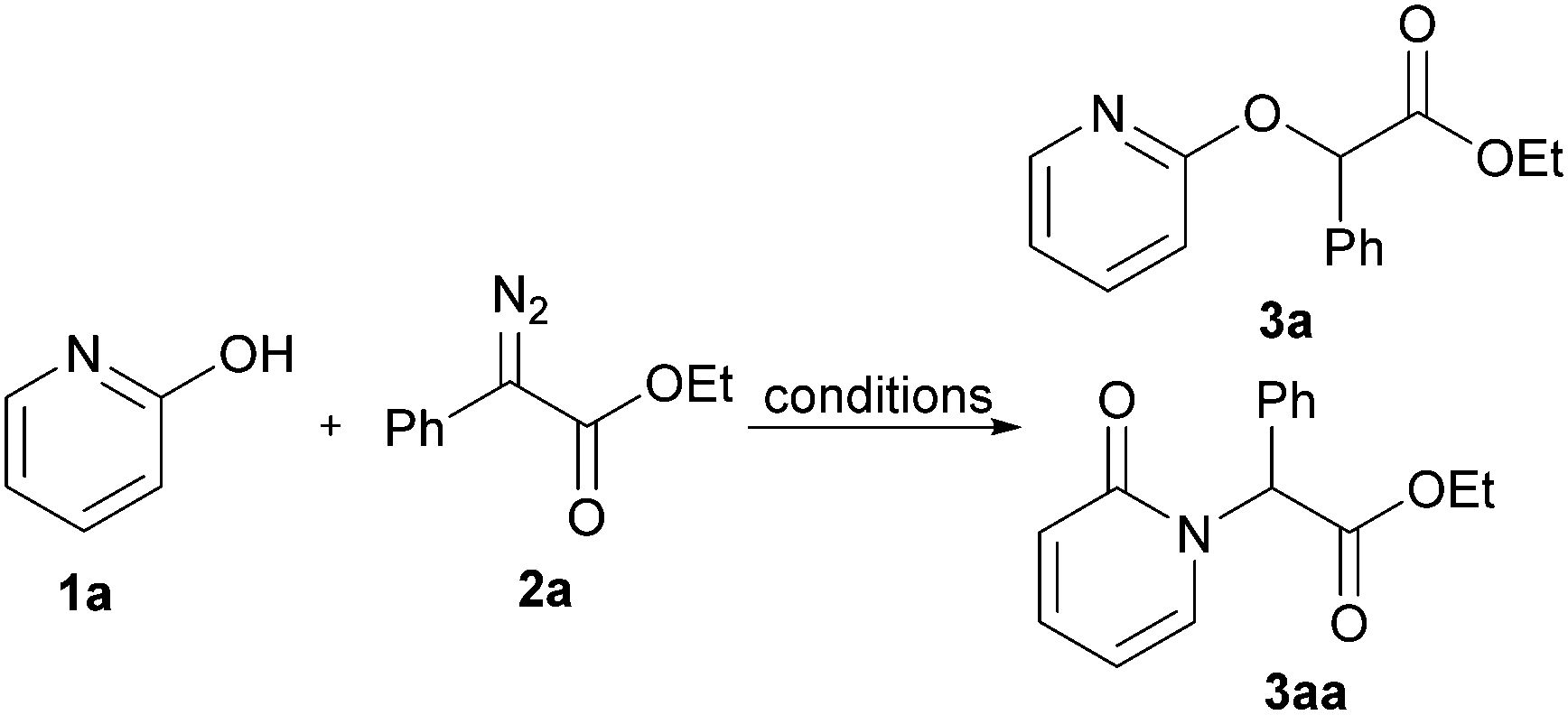
We commenced our study by employing 2-pyridone and ethyl 2-diazo-2-phenylacetate as model substrates to optimize the reaction conditions (Table 1). Various catalysts were first surveyed, including Cu(TfO)2, TFA, TsOH, H3PO4, TfOH, and the reaction yield was increased to 93% (>99:1 O-product: N-product ratio) when TfOH was chosen as an catalyst in 1,4-dioxane (Table 1, entries 1–5, respectively). Using others solvents, such as acetonitrile, toluene, methyl tert-butyl ether (MTBE) and dimethyl sulfoxide (DMSO), led to significant decreases in the yields (Table 1, entries 6–9). In contrast, when ethyl acetate was used, O-alkylated product 3a was obtained in 96% NMR yield, which was increased slightly than the yield in 1,4-dioxane (Table 1, entry 10). Increasing the amount of loading of TfOH led to lower yield (Table 1, entry 11). Notably, 1% of catalyst loading resulted in the N-alkylated product 3aa with high regioselectivity but poor yield (Table 1, entry 12). Unsurprisingly, no desired product was obtained when the TfOH catalyst was absent (Table 1, entry 13). Lowering the reaction temperature hindered the transformation and elevating the reaction temperature did not lead to any improvement (Table 1, entries 14 and 15). Subsequently, we further evaluated the equiv. of 2-diazo-2-phenylacetate, for which 1.5 equiv. of 2a was found to be optimal (Table 1, entries 16 and 17).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Cat. (mol%) | Solvent | T (°C) | Yieldb (%) | 3a/3aac |
| a Unless otherwise noted, all reactions were conducted with 2-pyridone 1a (0.10 mmol), diazo substrate 2a (0.15 mmol) and catalyst (0.01 mmol) in solvent (1.5 mL) under nitrogen for 3 hours. b Yield was determined by 1H NMR analysis of the crude mixture using 2-methylnaphthalene as an internal standard. c Determined by LC–MS of the crude products. d r.t. = room temperature. e The amount of 2a was 0.12 mmol. f The amount of 2a was 0.17 mmol. TFA: trifluoroacetic acid. TsOH: 4-methylbenzenesulfonic acid. TfOH: trifluoromethanesulfonic acid. MTBE: methyl tert-butyl ether. DMSO: dimethyl sulfoxide. | |||||
| 1 | Cu(TfO)2 | 1,4-Dioxane | r.t.d | 80 | 91:9 |
| 2 | TFA | 1,4-Dioxane | r.t. | — | — |
| 3 | TsOH | 1,4-Dioxane | r.t. | — | — |
| 4 | H3PO4 | 1,4-Dioxane | r.t. | — | — |
| 5 | TfOH | 1,4-Dioxane | r.t. | 93 | >99:1 |
| 6 | TfOH | CH3CN | r.t. | 81 | 94:6 |
| 7 | TfOH | Toluene | r.t. | 21 | 1:99 |
| 8 | TfOH | MTBE | r.t. | 90 | 92:8 |
| 9 | TfOH | DMSO | r.t. | — | — |
| 10 | TfOH | EtOAc | r.t. | 96 | >99:1 |
| 11 | TfOH (20) | EtOAc | r.t. | 93 | >99:1 |
| 12 | TfOH (1) | EtOAc | r.t. | 18 | 2:98 |
| 13 | TfOH (0) | EtOAc | r.t. | — | — |
| 14 | TfOH | EtOAc | 0 | 21 | 1:99 |
| 15 | TfOH | EtOAc | 50 | 93 | >99:1 |
| 16e | TfOH | EtOAc | r.t. | 87 | >99:1 |
| 17f | TfOH | EtOAc | r.t. | 96 | 91:9 |
With the optimized conditions in hand, we explored the generality of the reaction by first varying the scope of diazo compounds 2a–y using 2-pyridone 1a under standard conditions as shown in Scheme 3. Diazo compounds bearing methyl groups at the o-position or p-position of the phenyl rings were all well-tolerated to furnish the corresponding products 3b and 3c in good yield. But a para-methoxyl group substituted diazo ester gave s poor yield of 3d probably due to the high reactivity of the electron-donating group. In contrast, substituents with electron-withdrawing groups were all compatible with this reaction, giving the corresponding products 3f–i in moderate yields. Subsequently, various substituents bearing halogen atoms (–F, –Cl, –Br, –I) at ortho- and para-positions of the phenyl rings were also readily tolerated in this system, delivering the alkylated products 3j–p in good yields. Besides these examples, variation of the ester groups in 3q–t showed no steric effects and were compatible with this transformation. Notably, the presence of a tertiary butyl group on para-position of the phenyl ring and s naphthyl group were also able to deliver the products 3e and 3u in good yields. Beyond diazo esters, wild-type diazo compounds were also successful in the reaction, including a diazo ketone 2x, a diazo diphenylethan 2v and diazo acetic esters 2w, 2y, providing the respective products 3v–y in 35–82% yield.
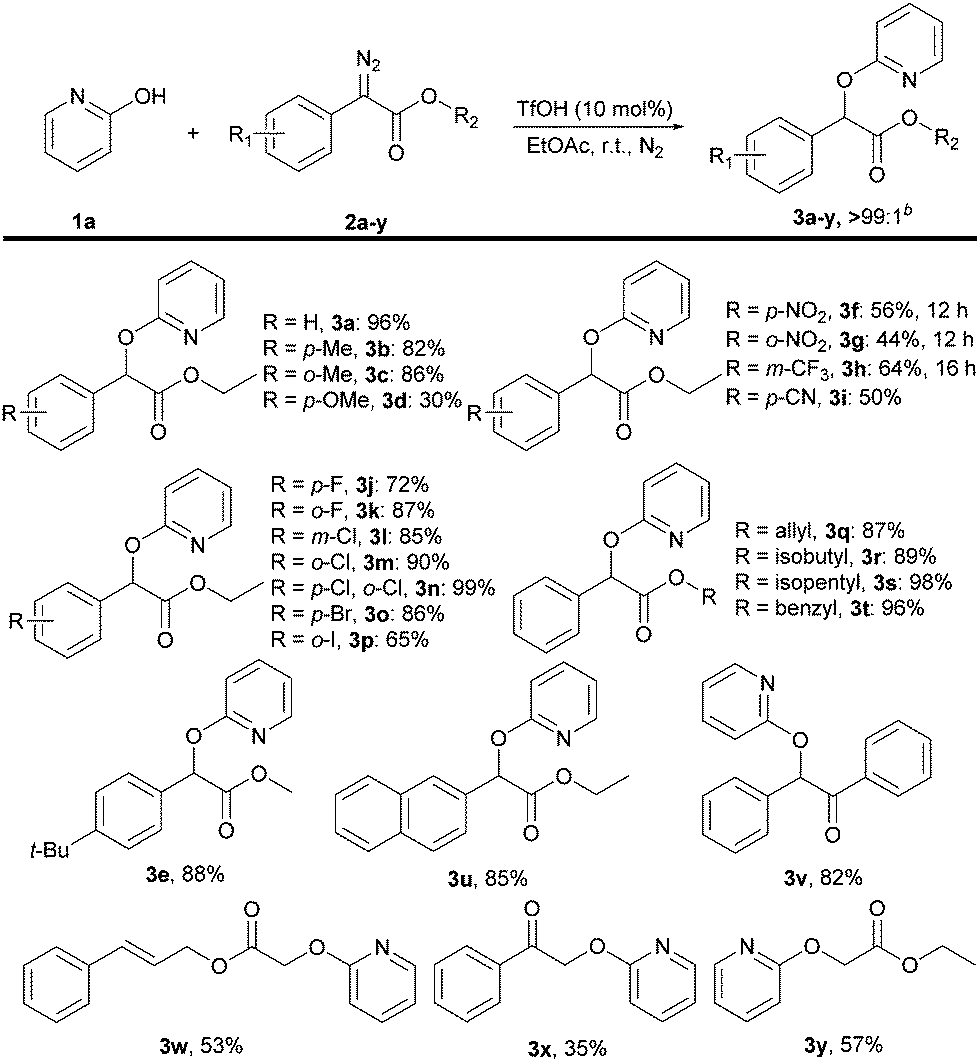 | ||
| Scheme 3 Substrate scope of diazo compoundsa. aReaction conditions: 1a (0.50 mmol), 2a–y (0.75 mmol) and TfOH (0.05 mmol) in EtOAc (3 mL) under nitrogen for 3–6 hours. Isolated yields are shown. bRatio of O- to N-product was determined by LC–MS of the crude products. | ||
We next evaluated the scope of 2-pyridones 1b–k transfer (Scheme 4). Halogen-substituted 2-pyridones at the 3-position and 5-position were well tolerated under the reaction conditions, giving the products 3ab–ae in good yields with excellent selectivity (>99:1). Pleasingly, electron-withdrawing groups at the 5-position of the phenyl rings all afforded the O–H insertion products 3af–ai in 31–70% yield. Quinolin-2(1H)-one 1j and isoquinolin-1(2H)-one 1k with ethyl 2-diazo-2-phenylacetate 2a also smoothly underwent selective O–H insertion to furnish 3aj and 3ak in 97% and 92% yield, respectively.
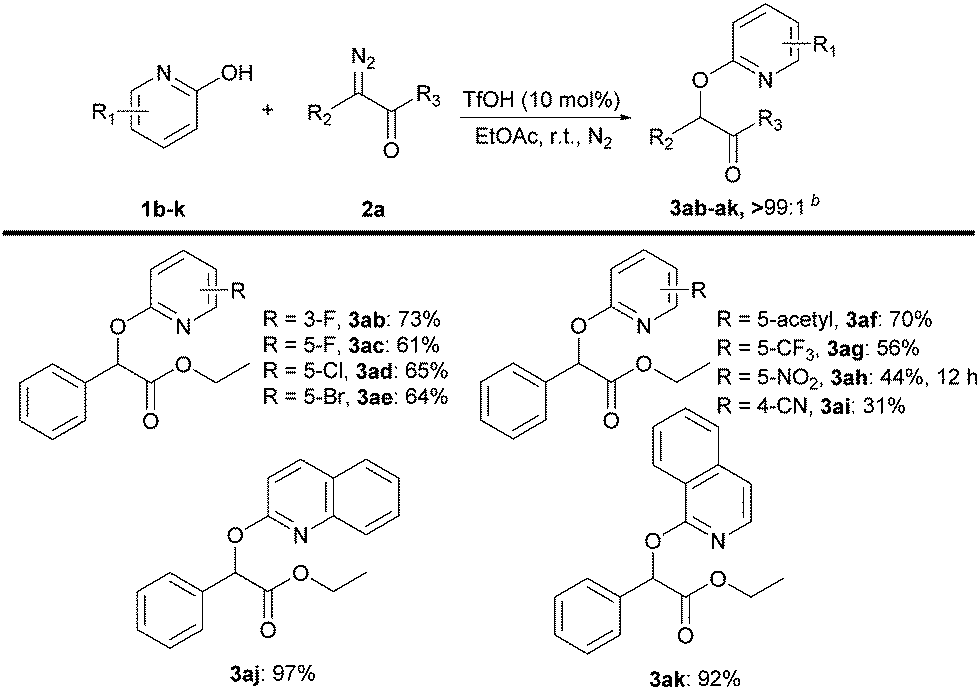 | ||
Scheme 4 Substrate scope of 2-pyridonesa. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) Reaction conditions: 1b–k (0.50 mmol), 2a (0.75 mmol) and TfOH (0.05 mmol) in EtOAc (3 mL) under nitrogen for 3–6 hours. Isolated yields are shown. bRatio of O- to N-product was determined by LC–MS of the crude products. Reaction conditions: 1b–k (0.50 mmol), 2a (0.75 mmol) and TfOH (0.05 mmol) in EtOAc (3 mL) under nitrogen for 3–6 hours. Isolated yields are shown. bRatio of O- to N-product was determined by LC–MS of the crude products. | ||
To evaluate the reliability and synthetic utility of this methodology, a gram scale synthesis of 3a was successfully achieved under optimal reaction conditions, providing product 1.28 g in 99% yield and >99:1 regioselectivity (Scheme 5A). To provide insight into the use of this method, we then engaged in subsequent studies to show its value for the facile synthesis of biological molecules in the treatment of type 2 diabetes mellitus.14 The diazo compound bearing a trifluoromethyl was readily attacked by substituted 2-pyridones under the standard conditions. Treatment of the alkylated product with sodium hydroxide in THF/H2O produced carboxylic acid 4 in 60% of total yield after acidification with hydrochloric acid (Scheme 5B).
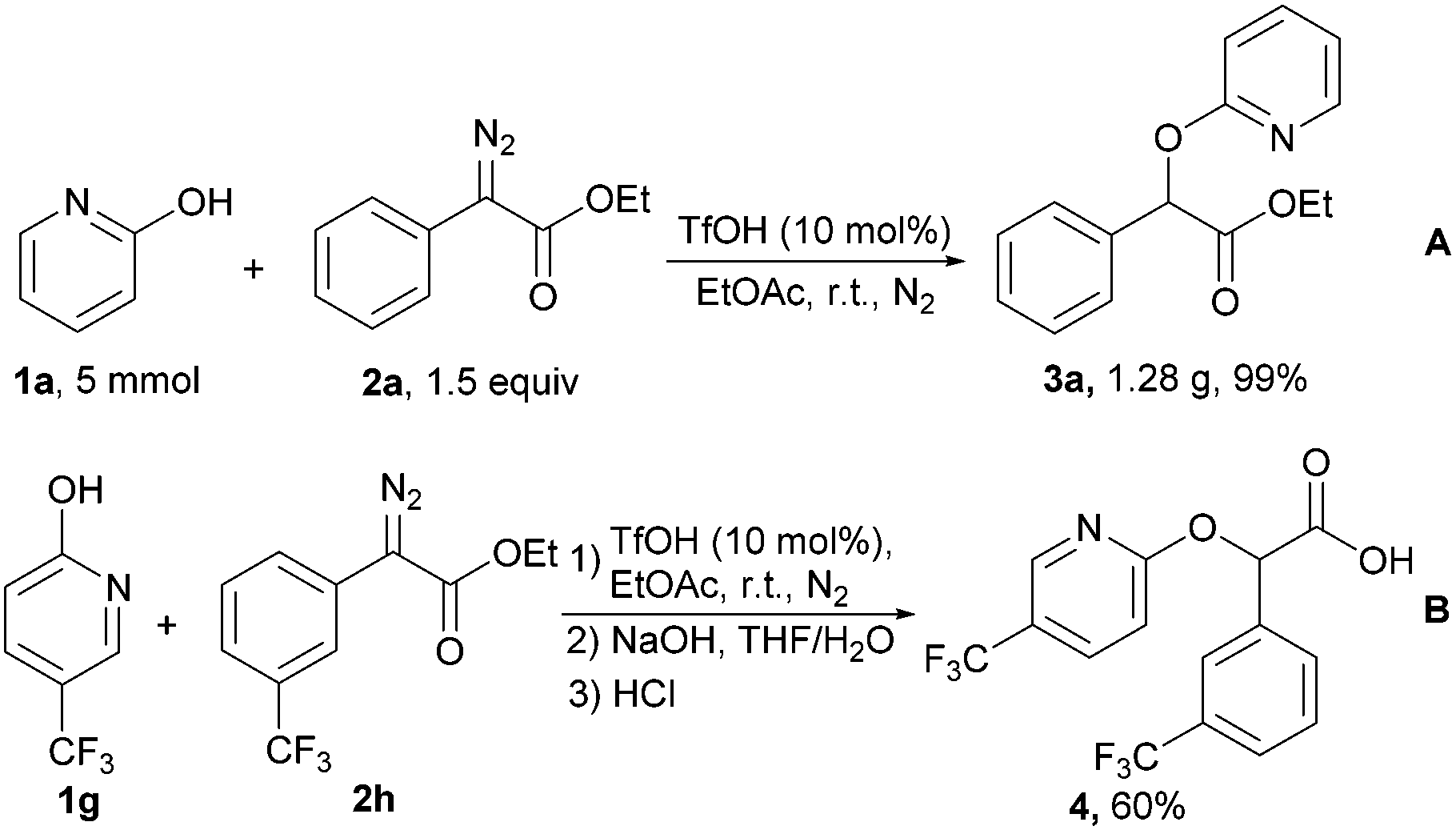 | ||
| Scheme 5 Gram-scale synthesis and applications of the O-alkylation reaction. | ||
To reveal the mechanism of the reaction, some control experiments were performed in Scheme 6. The anhydrous experiment proceeded smoothly under the standard reaction conditions and generated 3a in high yield (95%), indicating that a direct proton transfer from the 2-pyridone was involved in this transformation (Scheme 6A). Interestingly, treatment of 2a with TfOH led to the release of nitrogen, which was followed by the addition of 1a after 20 minutes, furnishing the corresponding product 3a in 94% yield (Scheme 6B). The results indicate that the in situ-generated intermediate could act as an effective coupling partner for this transformation. Deuterium-labeled 2-pyridone was utilized for the TfOH-catalyzed O-alkylation of 2-pyridones (Scheme 6C). The most conservation of deuterium was observed in the products 3d–3a despite proton transfer from TfOH and trace amount of water in the solvent, thus indicating that proton transfer from 2-pyridone was proven by the isotope labeling experiments.
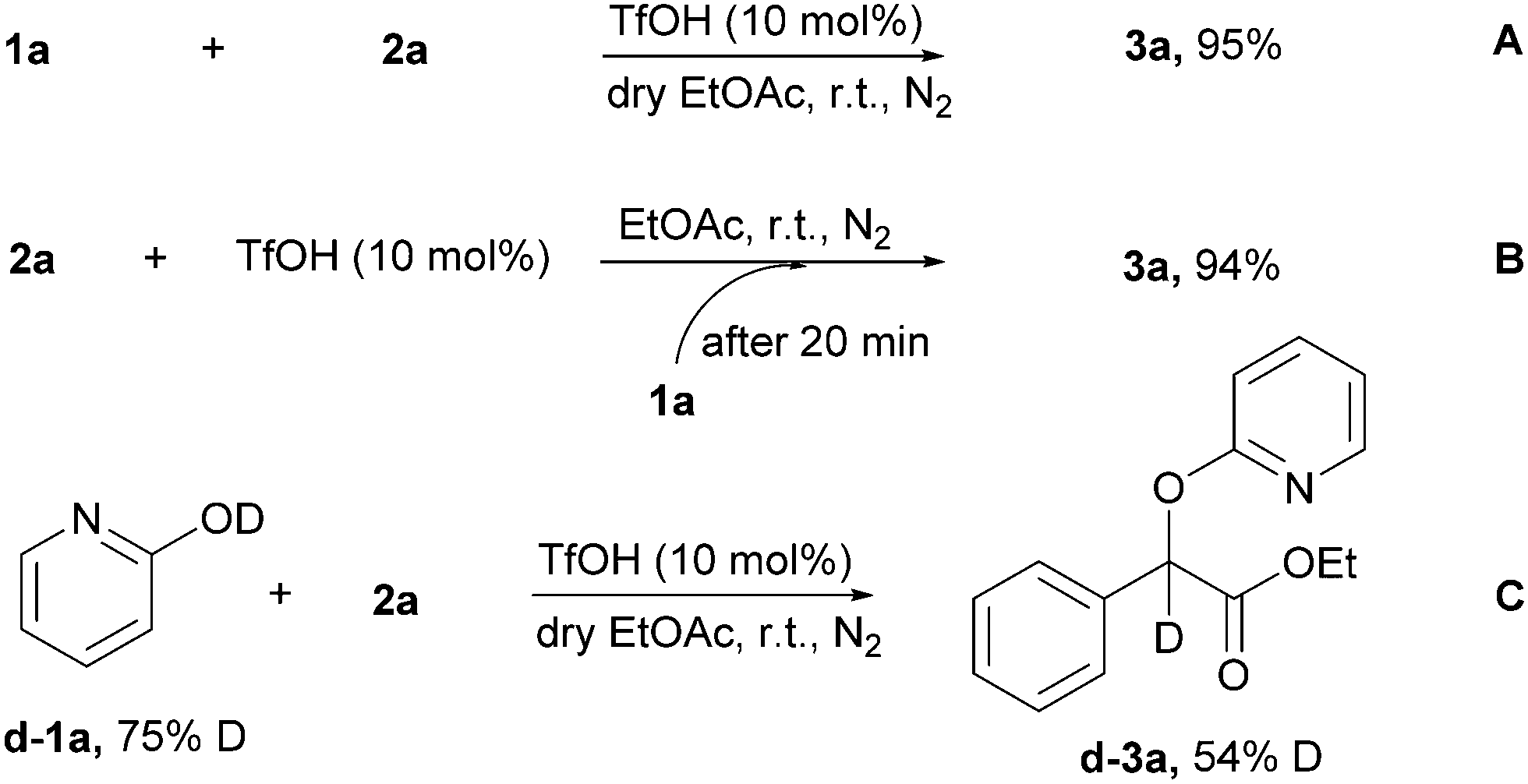 | ||
| Scheme 6 Control experiments. | ||
These findings allow us to propose a plausible reaction pathway depicted in Scheme 7. First, the diazo ester 2a reacts with TfOH to give an active intermediate I along with release of N2, which further is trapped by nucleophilic 2-pyridone 1a to generate intermediate II. Subsequently, the alkylated product 3a is afforded via further deprotonation. The high regioselectivity may be explained on the basis of the known density functional theory (DFT) computations, which show that bond dissociation energy (BDE) of O–H (1a) is lower than N–H (1aa).7d
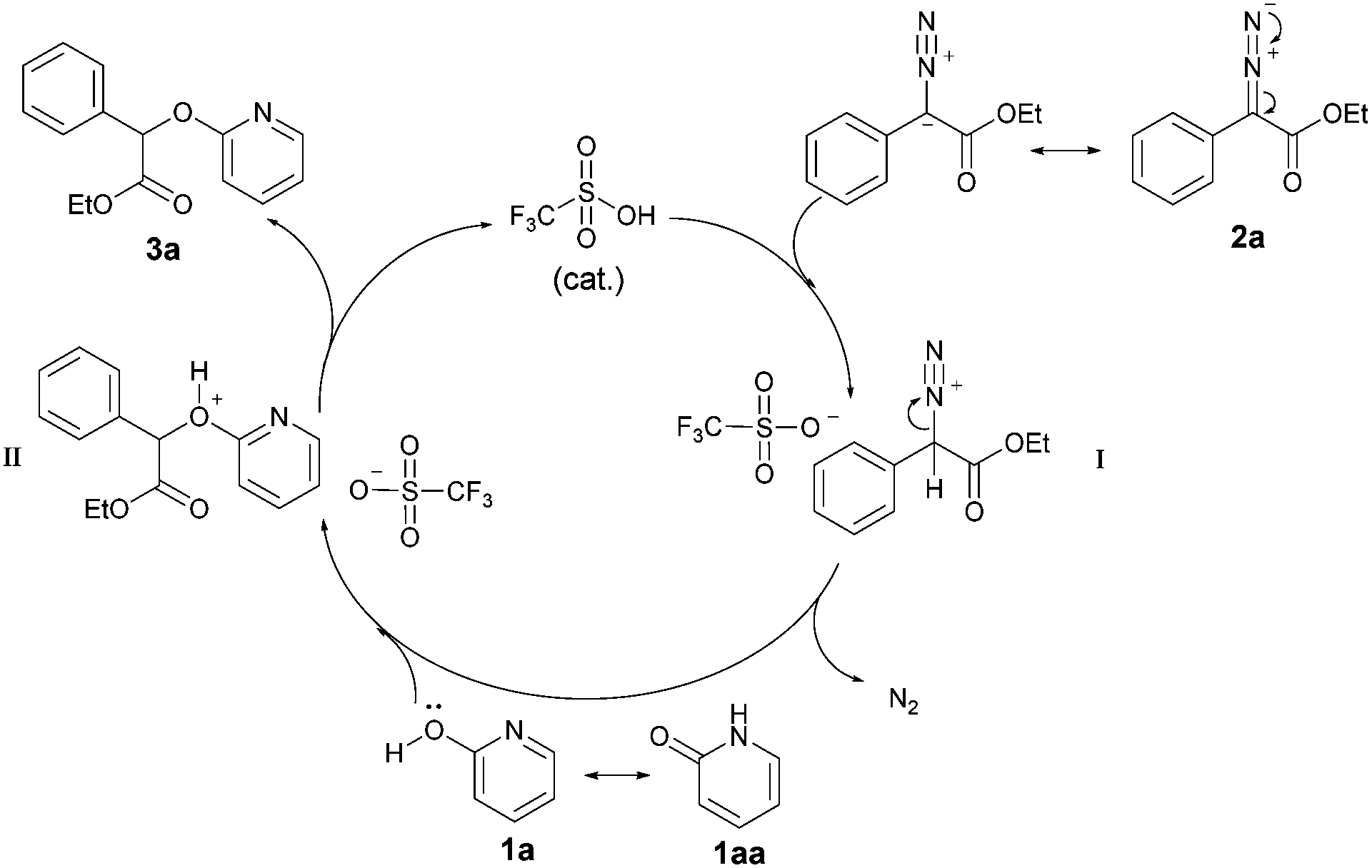 | ||
| Scheme 7 Plausible mechanism. | ||
In summary, we have developed a novel TfOH-catalyzed regioselective O-alkylation of 2-pyridones with diazo compounds in high regioselectivity (>99:1). This synthetic protocol employs readily available starting materials and reagents, proceeds under mild conditions, and tolerates a range of functional groups. The present work provides a new strategy for the regioselective formation of O-alkylated 2-pyridone. Further studies on Brønsted-acid-catalyzed alkylation reactions of diazo compounds are underway in our laboratory.
We thank the Scientific Research Project of Education Department of Hubei Province (B2020057) and the Open Fund of the Jiangxi Province Key Laboratory of Synthetic Chemistry (JXSC202002) for financial support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) A. S. Carlson, H. Cui, A. Divakaran, J. A. Johnson, R. M. Brunner, W. C. K. Pomerantz and J. J. Topczewski, ACS Med. Chem. Lett., 2019, 10, 1296–1301 CrossRef CAS PubMed; (b) T. Ogiyama, M. Yamaguchi, N. Kurikawa, S. Honzumi, Y. Yamamoto, D. Sugiyama, H. Takakusa and S. I. Inoue, Bioorg. Med. Chem., 2017, 25, 2234–2243 CrossRef CAS PubMed; (c) S. H. Reich, P. A. Sprengeler, G. G. Chiang, J. R. Appleman, J. Chen, J. Clarine, B. Eam, J. T. Ernst, Q. Han, V. K. Goel, E. Z. R. Han, V. Huang, I. N. J. Hung, A. Jemison, K. A. Jessen, J. Molter, D. Murphy, M. Neal, G. S. Parker, M. Shaghafi, S. Sperry, J. Staunton, C. R. Stumpf, P. A. Thompson, C. Tran, S. E. Webber, C. J. Wegerski, H. Zheng and K. R. Webster, J. Med. Chem., 2018, 61, 3516–3540 CrossRef CAS PubMed; (d) J. Yang, G. Su, Y. Ren and Y. Chen, Eur. J. Med. Chem., 2015, 101, 41–51 CrossRef CAS PubMed.
- (a) H. Duan, M. Ning, X. Chen, Q. Zou, L. Zhang, Y. Feng, L. Zhang, Y. Leng and J. Shen, J. Med. Chem., 2012, 55, 10475–10489 CrossRef CAS; (b) L. S. Lin, T. J. Lanza, J. P. Jewell, P. Liu, S. K. Shah, H. Qi, X. Tong, J. Wang, S. S. Xu, T. M. Fong, C. P. Shen, J. Lao, J. C. Xiao, L. P. Shearman, D. S. Stribling, K. Rosko, A. Strack, D. J. Marsh, Y. Feng, S. Kumar, K. Samuel, W. Yin, L. H. T. V. Ploeg, M. T. Goulet and W. K. Hagmann, J. Med. Chem., 2006, 49, 7584–7587 CrossRef CAS PubMed.
- (a) D. Gray and T. Gallagher, Angew. Chem., Int. Ed., 2006, 45, 2419–2423 CrossRef CAS PubMed; (b) D. Srtead, P. O'Brien and A. J. Sanderson, Org. Lett., 2005, 7, 4459–4462 CrossRef.
- (a) S. Ferrer, D. P. Naughton, I. Parveen and M. D. Threadgill, J. Chem. Soc., Perkin Trans. 1, 2002, 335–340, 10.1039/b109776h; (b) K. Hirano and M. Miura, Chem. Sci., 2018, 9, 22–32 RSC.
- M. Breugst and H. Mayr, J. Am. Chem. Soc., 2010, 132, 15380–15389 CrossRef CAS.
- (a) S. Chen, R. F. Graceffa and A. A. Boezio, Org. Lett., 2016, 18, 16–19 CrossRef CAS; (b) Y. Q. Fang, M. M. Bio, K. B. Hansen, M. S. Potter and A. Clausen, J. Am. Chem. Soc., 2010, 132, 15525–15527 CrossRef CAS; (c) X. Hao, Z. Xu, H. Lu, X. Dai, T. Yang, X. Lin and F. Ren, Org. Lett., 2015, 17, 3382–3385 CrossRef CAS; (d) A. Huang, K. Wo, S. Y. C. Lee, N. Kneitschel, J. Chang, K. Zhu, T. Mello, L. Bancroft, N. J. Norman and S. L. Zheng, J. Org. Chem., 2017, 82, 8864–8872 CrossRef CAS; (e) R. Islam, N. Ashida and T. Nagamatsu, Tetrahedron, 2008, 64, 9885–9894 CrossRef CAS; (f) D.-L. Mo, X.-H. Li, A.-H. Ye and C. Liang, Synthesis, 2018, 1699–1710 CrossRef.
- (a) R. D. Mandal, M. Saha and A. R. Das, Org. Biomol. Chem., 2022, 20, 2939–2963 RSC; (b) H. Ni, Y. Li, J. Deng, X. Shi and Q. Pan, New J. Chem., 2021, 45, 22432–22436 RSC; (c) J. Yang, J. Duan, G. Wang, H. Zhou, B. Ma, C. Wu and J. Xiao, Org. Lett., 2020, 22, 7284–7289 CrossRef CAS; (d) J. Yang, G. Wang, H. Zhou, Z. Li, B. Ma, M. Song, R. Sun and C. Huo, Org. Biomol. Chem., 2021, 19, 394–398 RSC.
- (a) J. A. Gurak, V. T. Tran, M. M. Sroda and K. M. Engle, Tetrahedron, 2017, 73, 3636–3642 CrossRef CAS; (b) Z. Lu, Y. Li, Y. Ru, S. Yang, C. Hao, M. Zuo, R. Jiao, W. Wu, Y. Zhou, H. Yao, N. Huang and Y. Fu, Chem. Commun., 2022, 58, 1215–1218 RSC.
- G. Xu, P. Chen, P. Liu, S. Tang, X. Zhang and J. Sun, Angew. Chem., Int. Ed., 2019, 58, 1980–1984 CrossRef CAS PubMed.
- (a) D. Huang, G. Xu, S. Peng and J. Sun, Chem. Commun., 2017, 53, 3197–3200 RSC; (b) G. Xu, K. Liu, Z. Dai and J. Sun, Org. Biomol. Chem., 2017, 15, 2345–2348 RSC.
- M. Gund, S. Mohana Roopan, F.-R. Nawaz Khan, J. S. Jin, K. Rajesh and A. Sudheer Kumar, Res. Chem. Intermed., 2011, 38, 1111–1118 CrossRef.
- Y. B. Wu, Y. Z. Wu, J. Wu, D. Xu, H. Jiang, W. W. Chang and C. Y. Ma, J. Org. Chem., 2021, 86, 6918–6926 CrossRef CAS.
- H. He, J. Yan, J. Jin, Z. Yan, Q. Yan, W. Wang, H. Jiang, H. Wang and F. Chen, Chem. Commun., 2022, 58, 6429–6432 RSC.
- (a) Z. Zhao, X. Chen, J. Wang, H. Sun and J. S. Liang, International Pat., WO2005/080340A1, 2005 Search PubMed; (b) M. E. Sanders and D. B. Karpf, International Pat., WO2006/102375A2, 2006 Search PubMed; (c) K. J. Tracey and M. P. Fink, International Pat., WO2006/002375A2, 2006 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc05676c |
| This journal is © The Royal Society of Chemistry 2023 |