
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile synthesis of sulfonyl fluorides from sulfonic acids†
Brodie J.
Thomson
a,
Samuel R.
Khasnavis
b,
Emma C.
Grigorian‡
b,
Rohun
Krishnan‡
b,
Theodore D.
Yassa‡
b,
Kelvin
Lee
b,
Glenn M.
Sammis
 *a and
Nicholas D.
Ball
*b
*a and
Nicholas D.
Ball
*b
aDepartment of Chemistry, University of British Columbia, 2036 Main Mall, Vancouver, British Columbia V6T 1Z1, Canada. E-mail: gsammis@chem.ubc.ca
bDepartment of Chemistry, Pomona College, 645 North College Avenue, Claremont, California 91711, USA. E-mail: nicholas.ball@pomona.edu
First published on 6th December 2022
Abstract
Herein, we demonstrate two complementary strategies for the syntheses of sulfonyl fluorides using sulfonic acids and their salts. One strategy involves the conversion of sulfonic acid sodium salts to sulfonyl fluorides using thionyl fluoride in 90–99% yields in one hour. Lessons learned from the mechanism of this reaction also have enabled a complementary deoxyfluorination of sulfonic acids using Xtalfluor-E® – a bench stable solid – allowing for the conversion of both aryl and alkyl sulfonic acids and salts to sulfonyl fluorides in 41–94% yields. Notably, using Xtalfluor-E® enabled milder conditions and the use of both sulfonic acids and their sodium salts.
Sulfonyl fluorides have been widely adopted as a novel set of click reagents since their introduction by Sharpless in 2014.1 The functional group is valuable due to its widespread applications in polymer chemistry, surface modifications, catalysis,2 and as precursors in the formation of additional sulfur(VI) species.1,3 In addition to its impact on synthesis, sulfonyl fluorides have found important applications as biological probes and protein inhibitors.4 Their high desirability across many scientific fields has instigated a robust development of synthetic strategies to access sulfonyl fluorides, including from sulfonamides,5 thiols and disulfides,6 sulfonyl hydrazides,7 sulfinic acids,7 Grignard reagents,8 arynes,9 aryl halides,10 carboxylic acids,11 and diazonium salts.12 Despite these innovations, an underexplored synthetic approach to sulfonyl fluorides has been to employ sulfonic acids. This strategy enables access to sulfonyl fluorides from stable, readily accessible S(VI) compounds without involving oxidation, reactive intermediates, or specialized compounds like sulfonamides or hydrazides.5,7 Current strategies toward the conversion of sulfonic acids to sulfonyl fluorides require two- step processes involving the in situ formation of sulfonyl chlorides (Scheme 1). Recent advances using this approach include a one-pot, two-step protocol, forming the sulfonyl chloride using trichloroacetonitrile13 or cyanuric chloride14 before Cl−/F− halogen exchange. While an improvement over the two-pot procedures, these strategies either are restricted to aryl sulfonic acids or require long reaction times (24 h). Furthermore, neither method was successful when using with electron poor substrates.
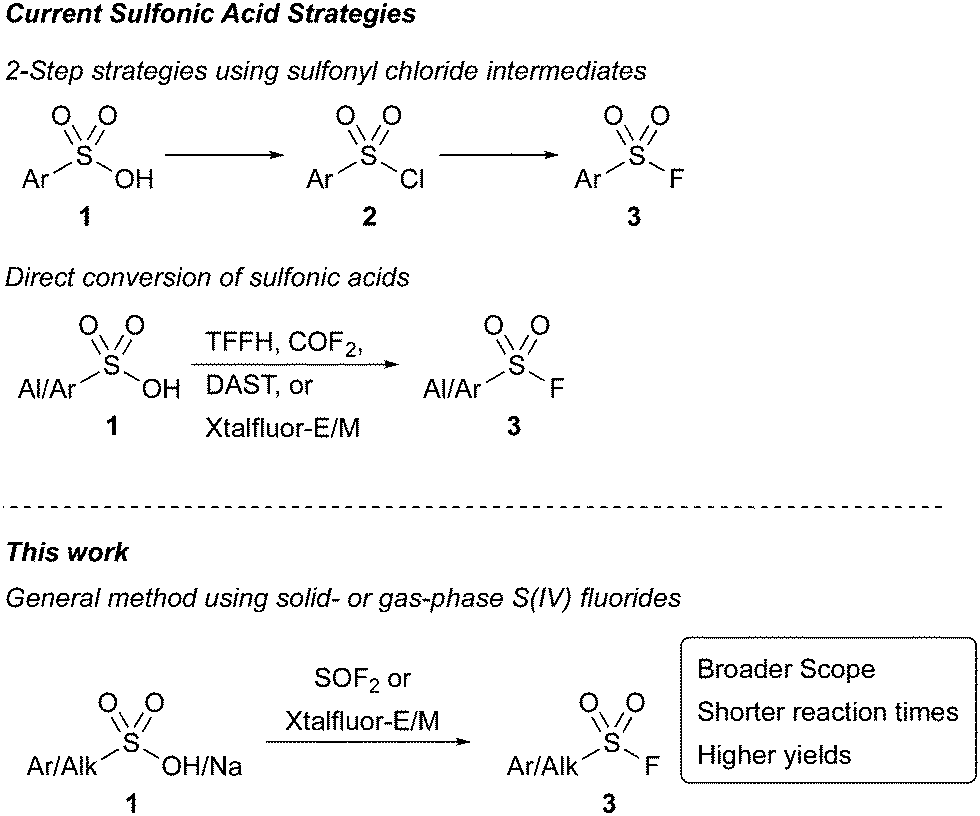 | ||
| Scheme 1 Strategies to access sulfonyl fluorides from sulfonic acids. | ||
An alternative deoxyfluorination approach that uses a reagent to both activate the sulfonic acid and provide a fluoride source would enable the direct access of sulfonyl fluorides from sulfonic acids and salts in a single step (Scheme 1). Efforts toward this direct deoxyfluorination strategy have been limited. Recently, 1,1,2,2-tetrafluoroethyl-N,N-dimethylamine15 has been employed with only a single substrate, and a method that uses carbonyl fluoride16 as the reactive intermediate was only demonstrated with five substrates. Liskamp and coworkers have also demonstrated the ability of amino sulfur(IV) fluorides, including DAST, Xtalfluor-E®, and Xtalfluor-M® to access a few sulfonyl fluoride inhibitors. However, yields were limited, and the reactions required heating reaction solutions at reflux containing HF and sulfonic acids over long reaction times.17 Collectively, these examples highlight the challenges of using sulfonic acids to make sulfonyl fluorides via deoxyfluorination, and the need for new methodologies. Herein, we demonstrate two complementary strategies toward sulfonyl fluorides directly from sulfonic acids and their salts using mild reaction conditions.
We began our investigation by exploring the use of thionyl fluoride – SOF2 – in the deoxyfluorination of sulfonate salts. Thionyl fluoride has recently been reported in a rapid deoxyfluorination of carboxylic acids to form the corresponding acyl fluorides.18 Notably, thionyl fluoride is believed to activate carboxylic acids via acyl fluorosulfinate intermediates, facilitating rapid conversion to the corresponding acyl fluoride. Importantly, thionyl fluoride gas can be generated in situ in the amounts needed using readily available bench-stable reagents. We hypothesized that thionyl fluoride could also be used to activate more challenging sulfonic acid derivatives toward sulfonyl fluorides.
Our studies began by treating pyridinium p-toluenesulfonic acid with a solution of thionyl fluoride in acetonitrile due to the solvent's previously reported effectiveness in the deoxyfluorination of carboxylic acids (Table 1, entry 1).18 No product was detected, and 4-methylbenzenesulfonic anhydride was observed as the exclusive product. Changing the reaction solvent to DMF in order to access similar intermediates reported in the COF2-mediated synthesis of sulfonic acids16 led to no reaction at 80 °C (entry 2), but the desired product was formed in 48% yield when the reaction was heated to 115 °C (entry 3). Elevating the temperature further to 130 °C increased the formation of 3b to 92% yield. Increasing the amount of thionyl fluoride to 3 equivalents afforded the desired sulfonyl fluoride (3a) in 98% yield in only 1 hour (entry 5). Sodium salt derivatives, which represent the most accessible source of sulfonic acids, generally afforded low yields (entry 6). To increase the yield, BF3·OEt2 was added as it has been shown to increase the electrophilicity of sulfur(IV) reagents.19 Gratifyingly, 3a was formed in quantitative yields by 19F NMR in 1 hour (entry 7).20
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | X | Additive | Solvent | T (°C) | SOF2 equiv.a | Yield (%) |
| Yields determined by 19F NMR spectroscopy at 0.25 mmol.a Equivalents of thionyl fluoride gas dissolved in DMF.b Only 4-methylbenzenesulfonic anhydride was detected by 1H NMR spectroscopy. | ||||||
| 1 | Pyridinium | — | MeCN | 80 | 2 | 0b |
| 2 | Pyridinium | — | DMF | 80 | 2 | 0 |
| 3 | Pyridinium | — | DMF | 115 | 2 | 48 |
| 4 | Pyridinium | — | DMF | 130 | 2 | 92 |
| 5 | Pyridinium | — | DMF | 130 | 3 | 98 |
| 6 | Na | — | DMF | 130 | 3 | 25 |
| 7 | Na | BF3·OEt2 | DMF | 130 | 3 | 99 |
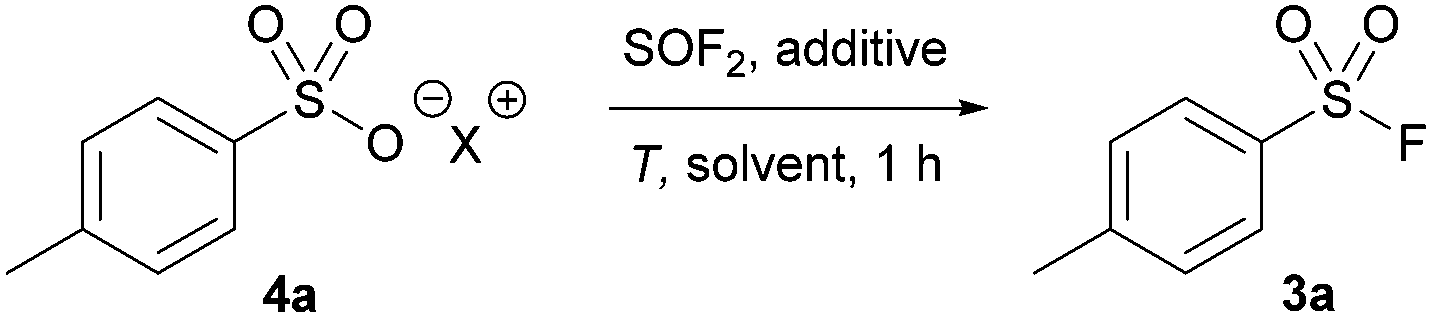
With our optimized protocol in hand, the substrate scope was investigated with a range of aromatic and aliphatic acids (Table 2). Alkyl substitution had no effect on the reaction, with 3a–3c all proceeding in near-quantitative yield. Similarly, the reaction was largely insensitive to electron rich and electron poor phenyl derivatives (3d–3g) Notably, this is the highest reported yield for 4-nitro derivative (3g) by any method without direct halogen exchange from sulfonyl chloride.21 Naphthylsulfonyl fluoride 3h was also synthesized in quantitative yield. Pyridine sulfuryl fluoride 3i was only synthesized in 14% 19F NMR yield, and significant quantities of the respective sulfonic acid anhydride were observed. Similarly, sulfuryl fluoride derivative 3j could not be synthesized by this method.
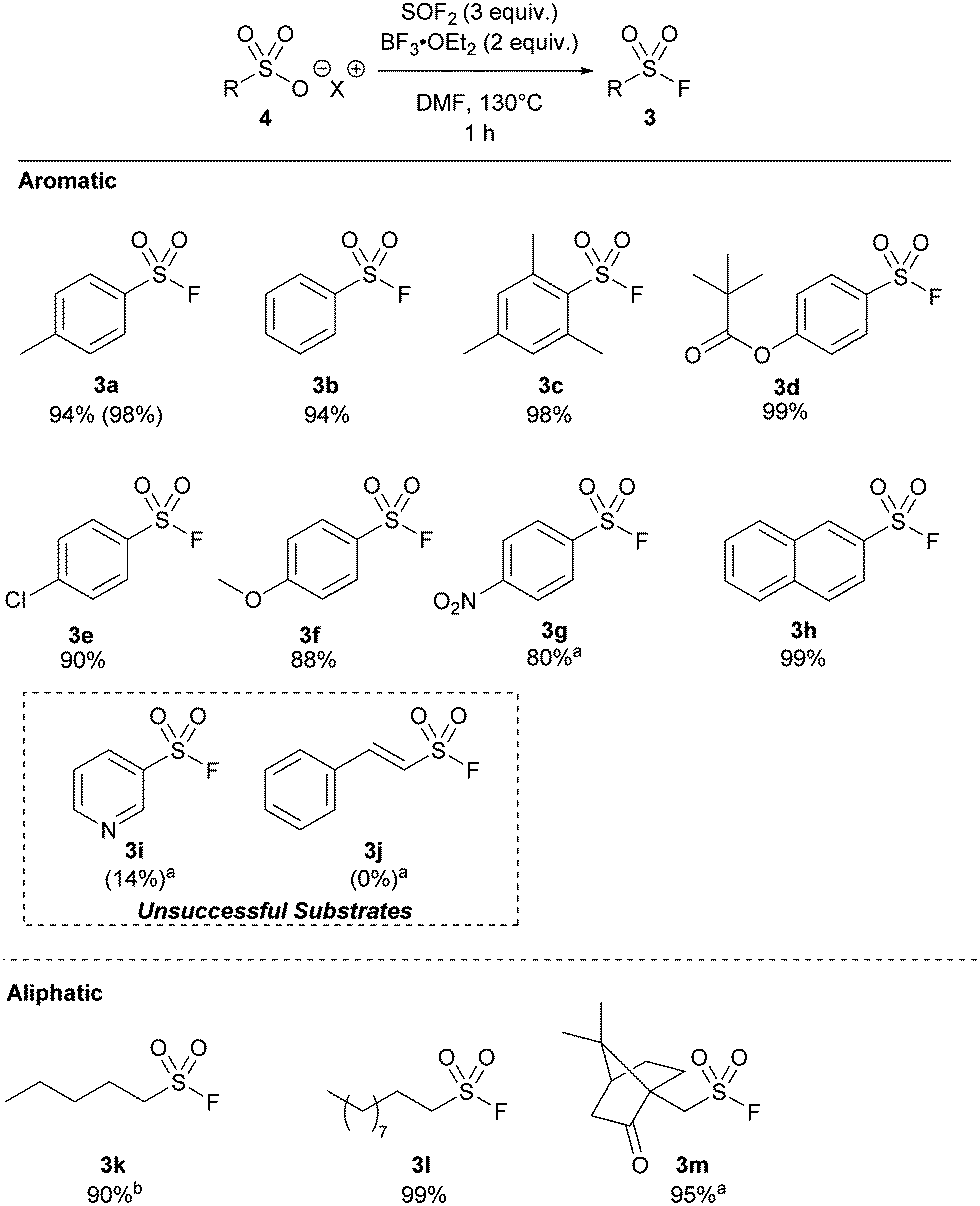
Significantly, we found our optimized protocol could also be applied to the synthesis of aliphatic substrates with products 3k–3l produced in ≥92% yield, as determined by 19F NMR spectroscopy. There are also few methods to make short chain sulfonyl fluorides with ease without going from a sulfonyl chloride. This method shows a higher yield for pentyl derivative 3k compared to the only existing method for this substrate.14
We next investigated the role of DMF in our optimized reaction. Heating a solution of thionyl fluoride dissolved in DMF to 130 °C resulted in the formation of FSO2−, indicated by a signal at +36 ppm by 19F NMR spectroscopy.22 The presence of FSO2− confirmed that deoxygenation of DMF was occurring within our reaction system forming either compound 5 or 7 (Scheme 2a).
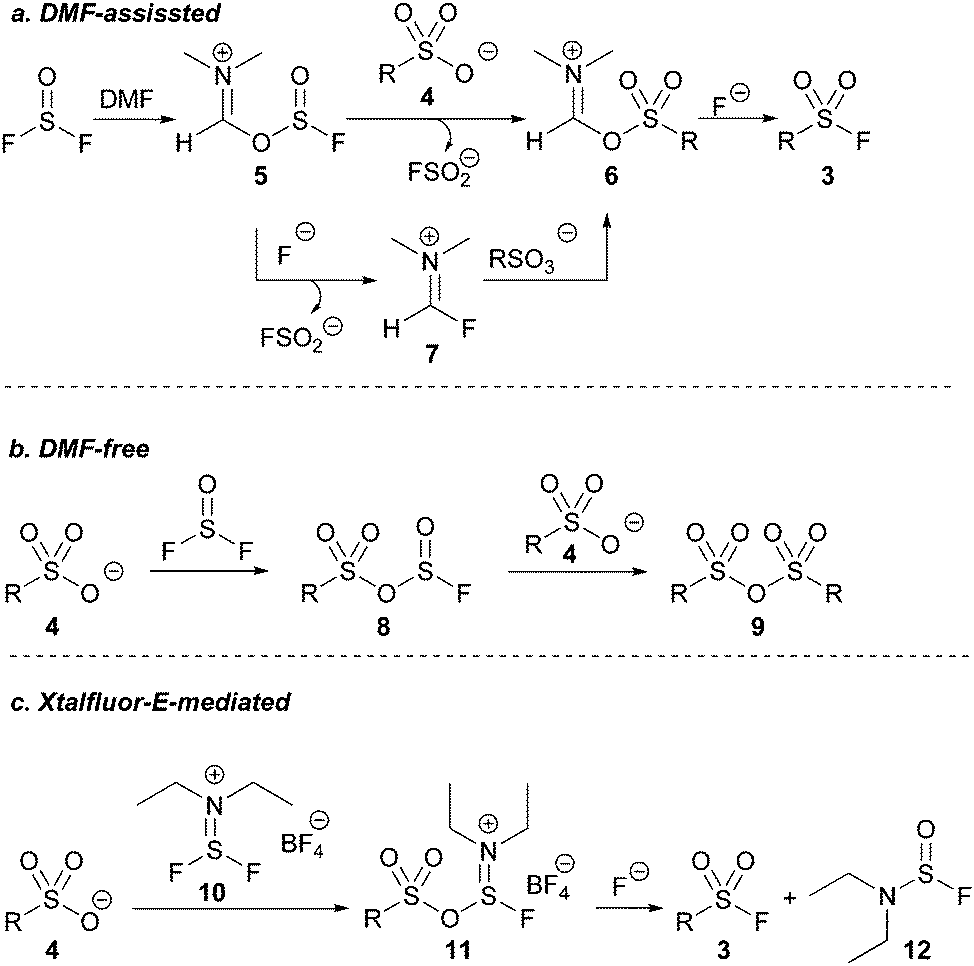 | ||
| Scheme 2 Working mechanistic models. | ||
Conversely, heating a solution of thionyl fluoride dissolved in chlorobenzene – a solvent with sufficiently high polarity and a boiling point comparable to DMF – to 130 °C in the presence of pyridinium p-toluene sulfonic acid afforded no detectable product by 19F NMR spectroscopy, and only anhydride 9 was detected (Scheme 2b). The similarly poor product yield using acetonitrile in the initial reaction optimisation (Table 1, entry 1), in which 9 was the exclusive product, suggest that intermediate 8 (Scheme 2b) is selectively formed in the absence of DMF. Another equivalent of a sulfonate salt can then react with the S(IV) center of intermediate 8, reforming 8, or it reacts with the S(VI) center to form sulfonic anhydride 9.
When DMF is used as a solvent, it reacts with SOF2 to presumingly form intermediate 5 (see ESI†). Intermediate 5, or Vilsmeier-like intermediate 7 may then react with the sulfonic acid in solution to form a DMF-activated sulfonic acid before subsequently reacting with free fluoride to afford the desired sulfonyl fluoride (Scheme 2a).
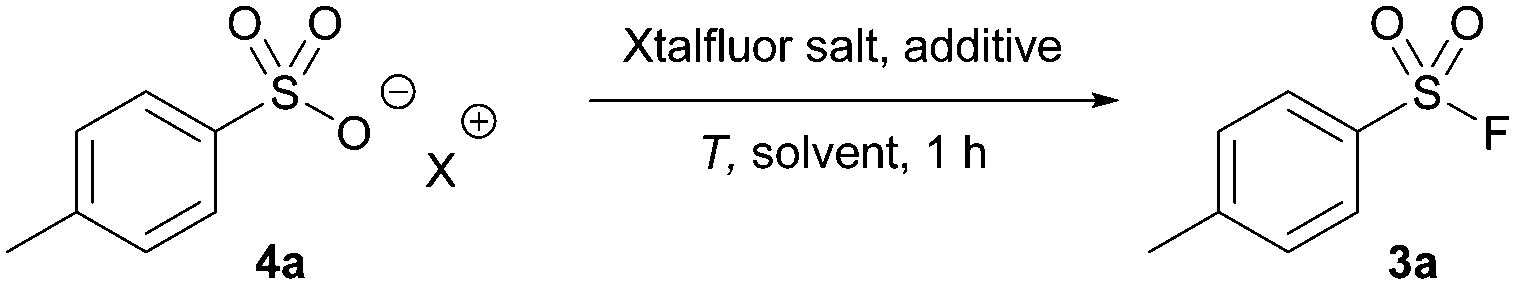
We were intrigued by intermediate 7 given its similarity in structure to the non-gaseous, deoxyfluorinating agents Xtalfluor-E® (10) and Xtalfluor-M® (a morpholine derivative of Xtalfluor-E®).23 We hypothesized that a sulfonate could add to Xtalfluor-E® (10) generating a species akin to 6 (intermediate 11) that could undergo addition of fluoride to give a sulfonyl fluoride 3. (Scheme 2c). Although the reagents were previously reported to form sulfonyl fluorides using harsh reaction conditions, we were optimistic that using the Xtalfluor salts under our optimized protocol may afford a more practical method.
Using our previous protocol with Xtalfluor-E®, sulfonyl fluoride (4a) was successfully formed in 80% yield using pyridinium acid and DMF as a solvent (Table 3, entry 1). Notably, the yields improved to 92% when the reaction was performed at room temperature in acetonitrile (entry 2), which offers a milder approach to previously reported methods.17 Similar to our reaction trials with thionyl fluoride, the yields of 4a were significantly reduced when sulfonic acid sodium salts were tested (entry 3); however, the addition of sodium fluoride and an increased temperature circumvented this issue and led to the formation of 3a in 92% yield in under 1 hour (entry 4). The reaction was equally efficient when Xtalfluor-M® was utilized (entry 5). However, we continued our study using Xtalfluor-E® due to its lower cost.24
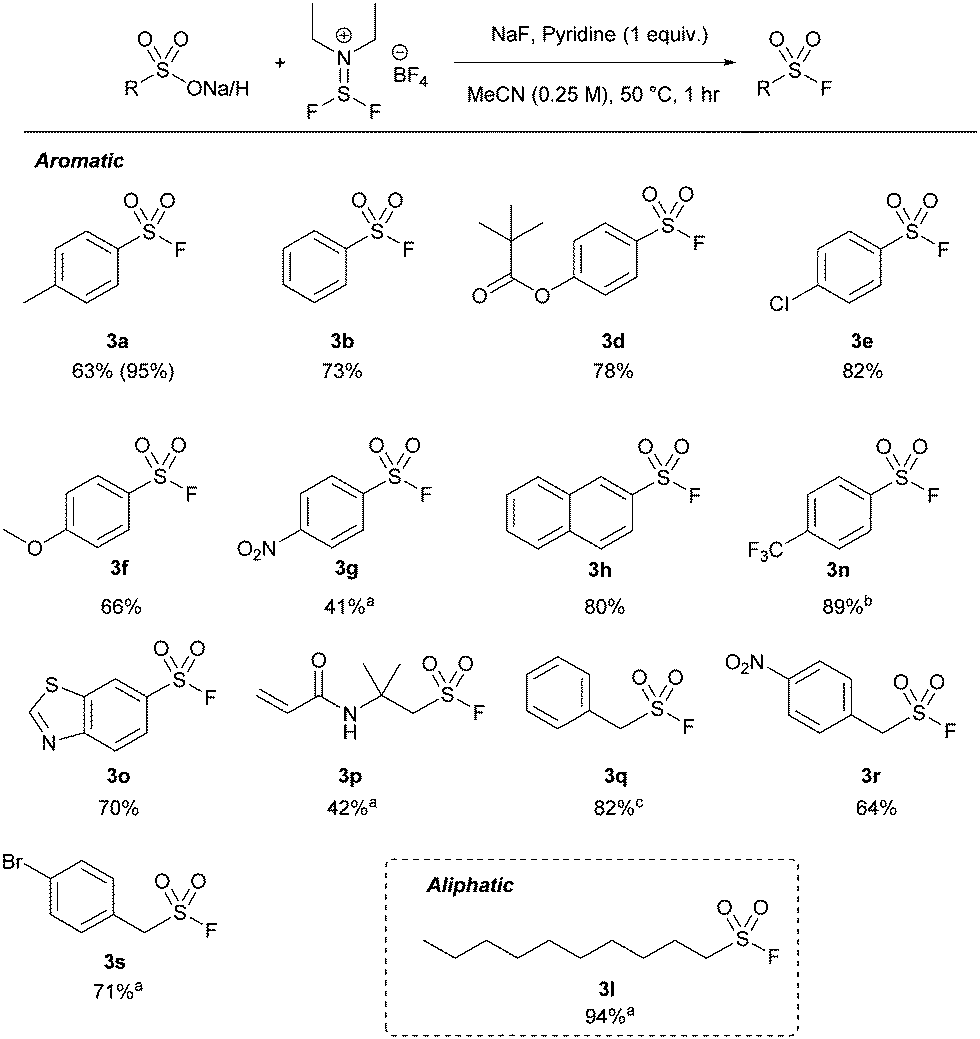
Xtalfluor-E® was successful with all the tested aromatic sulfonic acids, affording similar yields to thionyl fluoride (between 42 and 100%) by 19F NMR spectroscopy (Table 4). The reaction was more sensitive to electronic effects compared to the thionyl fluoride-mediated approach, where electronically rich systems (e.g.3a, f) afforded up to quantitative yields by 19F NMR spectroscopy. Conversely, electronically poor substrates, such as 3g and 3n, required longer reaction times. The comparatively lower isolated yield of 3g is due to its poor solubility in MeCN. Additionally, most alkyl and some benzyl substrates required extended reaction times. Unlike thionyl fluoride, heteroatomic substrate 3o was tolerated well under the optimized conditions, affording 71% isolated yield. We next tested aliphatic substrate pentanesulfonic acid (3k). However, pentene was identified as the major product.25 Longer-chain aliphatic substrates, such as 3l, prevented the formation of the discovered alkenes, affording excellent isolated yields in 94% with extended reaction times.
Isolated yields of sulfonyl fluorides are reported using a 0.250 mmol of sulfonic acid or sulfonate salt. Pyridine was used with all sulfonic acids and NaF was used with all sulfonate salts (see ESI). 19F NMR yields are reported in brackets. Some reactions were run for extended periods, either a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 16 hours, b3 hours, or c8 hours. 16 hours, b3 hours, or c8 hours. |
|---|
|
Compared to existing methods, the Xtalfluor-E® deoxyfluorination of aromatic sulfonic acids under our optimized conditions displayed milder reaction conditions in expedited timeframes. Furthermore, the salts are easy to access, store and handle. This method provides an excellent alternative route to access versatile sulfonyl fluoride moieties.
Overall, we have demonstrated two separate methods for accessing sulfonyl fluorides utilizing the sulfur (IV) oxidation in sulfur fluoride reagents thionyl fluoride and Xtalfluor-E®. Thionyl fluoride was found to be highly effective in the deoxyfluorination of both aliphatic and aromatic substrates, where further investigations found the successful reaction was a result of a novel DMF-activated intermediate. The use of Xtalfluor-E® was subsequently demonstrated to offer competitively high yields in the conversion of aromatic sulfonic acids across a diverse array of electronically rich and poor substrates. This alternative method offers an additional approach to access sulfonyl fluorides using a solid, bench-stable reagent with significantly milder conditions and ease-of-handling compared to thionyl fluoride and current reported methods.
N. D. B. thanks the National Institutes of General Medical Sciences of the National Institutes of Health for their funding (NIH-R15-GM134457-01A1). G. M. S. thank the University of British Columbia (UBC), and the Natural Sciences and Engineering Research Council of Canada (2020-RGPIN-04653) for funding. Student support was provided in the form of a MITACS Accelerate fellowships (B. J. T.).
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. Dong, L. Krasnova, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2014, 53, 9430–9448 CrossRef CAS PubMed.
- (a) P. K. Chinthakindi and P. I. Arvidsson, Eur. J. Org. Chem., 2018, 3648–3666 CrossRef CAS; (b) A. S. Barrow, C. S. Smedley, Q. Zheng, S. Li, J. Dong and J. E. Moses, Chem. Soc. Rev., 2019, 48, 4731–4758 RSC; (c) Z. Liu, J. Li, S. Li, G. Li, K. B. Sharpless and P. Wu, J. Am. Chem. Soc., 2018, 140, 2919–2925 CrossRef CAS PubMed; (d) R. Lekkala, R. Lekkala, B. Moku, K. P. Rakesh and H.-L. Qin, Org. Chem. Front., 2019, 6, 3490–3516 RSC; (e) C. Lee, A. J. Cook, J. E. Elisabeth, N. C. Friede, G. M. Sammis and N. D. Ball, ACS Catal., 2021, 11, 6578–6589 CrossRef CAS PubMed; (f) T. S.-B. Lou and M. C. Willis, Nat. Rev. Chem., 2022, 6, 146–162 CrossRef CAS.
- P. Mukherjee, C. P. Woroch, L. Cleary, M. Rusznak, R. W. Franzese, M. R. Reese, J. W. Tucker, J. M. Humphrey, S. M. Etuk, S. C. Kwan, C. W. Am Ende and N. D. Ball, Org. Lett., 2018, 20, 3943–3947 CrossRef CAS PubMed.
- A. Narayanan and L. H. Jones, Chem. Sci., 2015, 6, 2650–2659 RSC.
- M. Pérez-Palau and J. Cornella, Eur. J. Org. Chem., 2020, 17, 2497–2500 CrossRef.
- For representative examples, see: (a) G. Laudadio, A. A. Bartolomeu, L. M. H. M. Verwijlen, Y. Cao, K. T. de Oliveira and T. Noël, J. Am. Chem. Soc., 2019, 141, 11832–11836 CrossRef CAS PubMed; (b) S. W. Wright and K. N. Hallstrom, J. Org. Chem., 2006, 71, 1080–1084 CrossRef CAS PubMed.
- L. Tang, Y. Yang, L. Wen, X. Yang and Z. Wang, Green Chem., 2016, 18, 1224–1228 RSC.
- C. Lee, N. D. Ball and G. S. Sammis, Chem. Commun., 2019, 55, 14753–14756 RSC.
- J. Kwon and B. Moon Kim, Org. Lett., 2019, 21, 428–433 CrossRef CAS PubMed.
- (a) A. T. Davies, J. M. Curto, S. W. Bagley and M. C. Davies, Chem. Sci., 2017, 8, 1233–1237 RSC; (b) A. L. Tribby, I. Rodríguez, S. Sharffudin and N. D. Ball, J. Org. Chem., 2017, 82, 2294–2299 CrossRef CAS PubMed.
- Z. Ma, Y. Liu, X. Ma, X. Hu, Y. Guo, Q.-Y. Chen and C. Liu, Org. Chem. Front., 2022, 9, 1115–1120 RSC.
- For representative examples, see: (a) Z. Ma, L. Shan, X. Ma, X. Hu, Y. Guo, Q.-Y. Chen and C. Liu, J. Fluorine Chem., 2022, 254, 109948 CrossRef CAS; (b) T. Zhong, M.-K. Pang, Z.-D. Chen, B. Zhang, J. Weng and G. Lu, Org. Lett., 2020, 22, 3072–3078 CrossRef CAS PubMed; (c) Y. Liu, D. Yu, Y. Gup, J.-C. Xiao, Q.-Y. Chen and C. Liu, Org. Lett., 2020, 22, 2281–2286 CrossRef CAS PubMed.
- J.-G. Kim and D. O. Jang, Synlett, 2010, 20, 3049 Search PubMed.
- Y. Jiang, N. S. Alharbi, B. Sun and H.-L. Qin, RSC Adv., 2019, 9, 13863–13867 RSC.
- V. A. Petrov, S. Swearingen, W. Hong and W. C. Petersen, J. Fluorine Chem., 2001, 109, 25–31 CrossRef CAS.
- S. Zhao, Y. Guo, Z. Su, C. Wu, W. Chen and Q.-Y. Chen, Chin. J. Chem., 2021, 39, 1225–12320 CrossRef CAS.
- For representative examples, see: (a) R. Artschwager, D. J. Ward, S. Gannon, A. J. Brouwer, H. van de Langemheen, H. Kowalski and R. M. J. Liskamp, J. Med. Chem., 2018, 61, 5396–5411 CrossRef PubMed; (b) A. J. Brouwer, N. H. Álvarez, A. Ciaffoni, H. van de Langemheen and R. M. J. Liskamp, Bioorg. Med. Chem., 2016, 24, 3429–3435 CrossRef CAS PubMed; (c) A. J. Brouwer, T. Ceylan, A. M. Jonker, T. van der Linden and R. M. J. Liskamp, Bioorg. Med. Chem., 2011, 19, 2397–2406 CrossRef CAS PubMed.
- C. Lee, B. J. Thomson and G. M. Sammis, Chem. Sci., 2022, 13, 188–194 RSC.
- I. Ruppert, J. Fluorine Chem., 1982, 20, 79–84 CrossRef CAS.
- Sulfuryl fluoride also failed in yielding any desired product under similar conditions, suggesting the sulfur(IV) oxidation was required for the success of the reaction and highlighted the unique reactivity of thionyl fluoride.
- For representative examples, see: (a) L. Tang, X. Zhao, X. Yang, Y. Zhou and G. Zhou, CN Pat., 105198683, 2015 Search PubMed; (b) T. T. Bui, V. H. Tran and H.-K. Kim, Adv. Synth. Catal., 2022, 364, 341–347 CrossRef CAS; (c) D. Louvel, A. Chelagha, J. Rouillon, P.-A. Payard, L. Khrouz, C. Monnereau and A. Tlili, Chem. – Eur. J., 2021, 27, 8704–8707 CrossRef CAS PubMed.
- K. Gulbe, J. Luginina, E. Jansons, A. Kinens and M. Turks, Beilstein J. Org. Chem., 2021, 17, 964–976 CrossRef CAS PubMed.
- F. Beaulieu, L.-P. Beuregard, G. Courchesne, M. Couturier, F. LaFlamme and A. L’Heureux, Org. Lett., 2009, 11, 5050–5053 CrossRef CAS PubMed.
- Xtlalfluor-E® is currently priced $196.00/25g USD: Sigma-Aldrich, https://www.sigmaaldrich.com/CA/en/product/aldrich/719439, accessed October 2022. Xtalfluor-M® is currently priced at $302.00/25g USD: Sigma-Aldrich, https://www.sigmaaldrich.com/CA/en/product/aldrich/719447?gclsrc=aw.ds&gclsrc=ds&gclsrc=aw.ds, accessed October 2022.
- For a discussion on the formation of alkene side-products, see the ESI†.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc05781f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |