
Design strategies of covalent organic framework-based electrodes for supercapacitor application
Rao
Tao
,
Tianfu
Yang
,
Yan
Wang
,
Jingmin
Zhang
,
Zhengyi
Wu
and
Li
Qiu
 *
*
Yunnan Key Laboratory for Micro/Nano Materials & Technology, National Center for International Research on Photoelectric and Energy Materials, School of Materials and Energy, Yunnan University, Kunming 650091, P. R. China. E-mail: qiuli@ynu.edu.cn
First published on 30th January 2023
Abstract
Supercapacitors (SCs) have been recognized as a promising electrochemical energy storage (EES) device, thanks to their high-power density, long lifespan, fast charge–discharge capability, and eco-friendliness. The breakthrough of electrode materials that determine the electrochemical performance of SCs is urgently desired. Covalent organic frameworks (COFs), an emerging and burgeoning class of crystalline porous polymeric materials, have been found to have huge potential for application in EES devices by virtue of their unique properties including atomically adjustable structures, robust and tunable skeletons, well-defined and open channels, high surface areas, etc. In this feature article, we aim at summarizing the design strategies of COF-based electrode materials for SCs based on the representative advances. The current challenges and future perspectives of COFs for SC application are highlighted as well.
1. Introduction
As the environmental issues caused by the over-utilization of fossil energy have brought serious challenges to ecosystem health and social sustainability, the disparity between the looming energy crisis and growing energy requirements is among the greatest problems faced by our society today. The efficient development and utilization of renewable energy sources (e.g., wind, solar, geothermal and tidal energy) that cannot be used directly, is highly desirable. Currently, electrochemical energy storage (EES) is represented as a key technique for the development and utilization of these discontinuous clean power sources.1 Rechargeable supercapacitors (SCs) and batteries as widely utilized EES devices have received increasing attention thanks to their excellent electrochemical performance including high energy/power density, outstanding rate performance, long lifespan, and environmental friendliness.2,3 It should be noted that SCs integrate the merits of high energy density of batteries and superior power density of traditional capacitors.According to previous studies, SCs are traditionally classified into two categories, namely, electrochemical double layer capacitors (EDLCs) and pseudocapacitors, which store charge through electrostatic charge adsorption and reversible faradaic redox reactions at the electrolyte/electrode interface, respectively.4–6 Electrode materials, serving as the core component of SCs, determine the key performances including specific capacitance, rate performance, energy density, cycle stability, etc. For EDLCs, the charge–discharge process is of physical nature, and special attention has been paid to the design of electrode materials with excellent conductivity, high wettability, nanostructures, porous structures, etc.7 Their electrodes are mainly dominated by carbonaceous materials, including porous activated carbon, graphene, carbon nanotubes, and so forth, which generally lead to superior rate performance and high cycling stability.8–10 For pseudocapacitors, charge storage/separation is performed via reversible chemisorption and/or redox reactions, depending heavily on the chemical properties of the electrode materials.11 Transition metal compounds,12 conductive polymers,13 redox-active hybrids14 and porous organic materials15 have been demonstrated to be promising candidates for pseudocapacitive electrode materials, which usually showcase large capacitances and high energy densities. Actually, new hybrid supercapacitors that combine the electrode characteristics of EDLCs and pseudocapacitors have been developed recently, featuring one constituent as an EDLC type material and the other as a pseudocapacitive one.16 Overall, the energy storage mechanism of SC devices is determined from the electrode materials, and the currently reported electrode materials can be categorized into carbons, inorganics, organics and hybrids. As shown in Fig. 1, their representatives are listed as well.
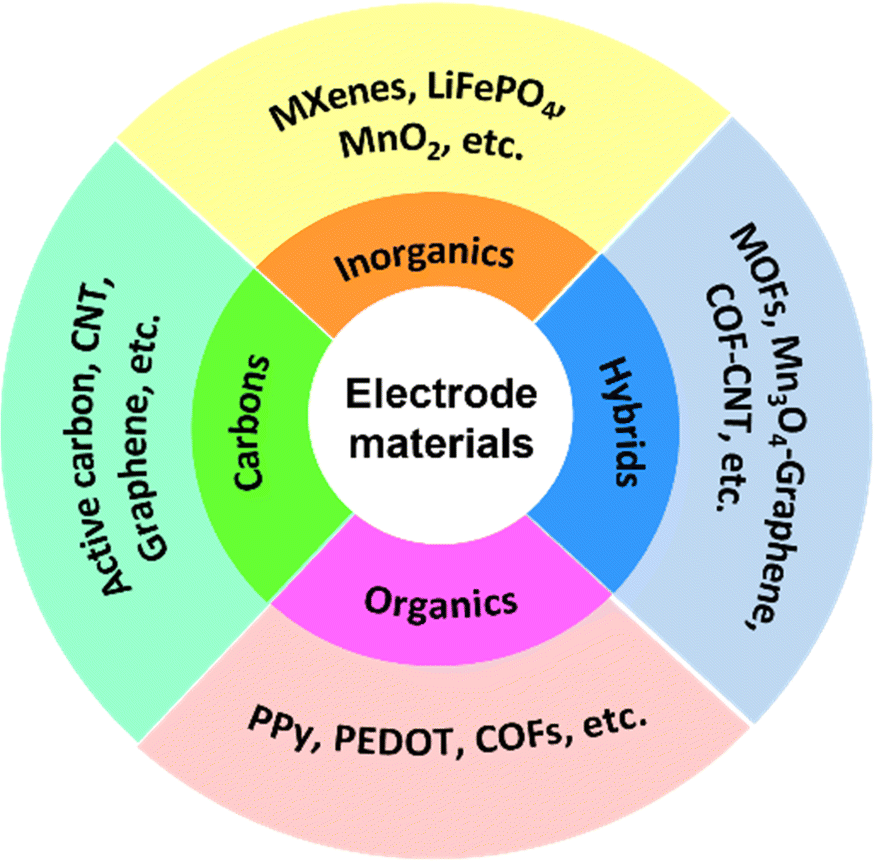 | ||
| Fig. 1 Classification of electrode materials for SC devices. | ||
To meet the higher application requirements of higher power/energy density, superior capacitance, better rate performance and longer service life, the breakthrough of novel SC electrode materials with high performance is highly desired. Nowadays, organic porous materials, especially covalent organic frameworks (COFs), have attracted great attention as electrode materials by virtue of their high specific surface area, adjustable functionalities, convenient electron/ion transport and designable redox sites.17–21 COFs are recognized as crystalline organic porous materials constructed entirely from light elements including C, H, O, N, B, etc., first reported by Yaghi and co-workers in 2005.22 Currently, one-dimensional (1D),23 two-dimensional (2D)24 and three-dimensional (3D)25 COFs have been designed and synthesized in accordance with the principle of reticular chemistry.26 The key step in constructing COFs is the formation of long-range order and porous structures, which relies heavily on the reversible equilibrium of dynamic covalent chemistry, manifesting that thermodynamically stable structures are assembled through “self-correction” between building blocks.27–29 With the increasing demand for harsh environment applications, many COFs synthesized via irreversible covalent reactions have sprung up.30–32 Nevertheless, COFs with the abovementioned reversible/irreversible linkages have similar requirements for the spatial configuration of building blocks, where the functional groups are obliged to undergo ring closure via covalent bonds. Besides, the formed extension layer generally needs to be stacked in an orderly and repetitive manner through mechanical interlocking and weak interactions such as hydrogen bonds, π–π interactions, etc. These structural features generally individualize COFs to have atomically precise structures, tailorable skeletons, open and regular channels, light weight, high surface areas, and facile built-in functions. Owing to their unique structural merits, COFs have been found to have tremendous potential in energy storage, gas storage and separation, optoelectronics, catalysis, sensing, drug delivery, etc.33–36 Since the initial development of COFs was dominated by the reversible boronate ester linkage that usually has poor hydrolytic and oxidative stability, it was not until 2013 that the SC application of COFs was first realized via Schiff base chemistry.37 Over the following 10 years, COFs have received growing attention for SC application. Nowadays, pristine bulk and nanostructured COFs, hybrids of COFs with other electrode materials (graphene, fullerene, conductive polymers, metal ions, etc.) and COF-based carbides are being developed for SC applications. Fig. 2 shows the number of articles published so far on COF-based materials for SC application and the corresponding citations (source: web of science, time: Dec. 31st, 2022). The growing trend in the number of articles published per year along with the outstanding number of citations indicate the prominence of next-generation COF-based SC technology.
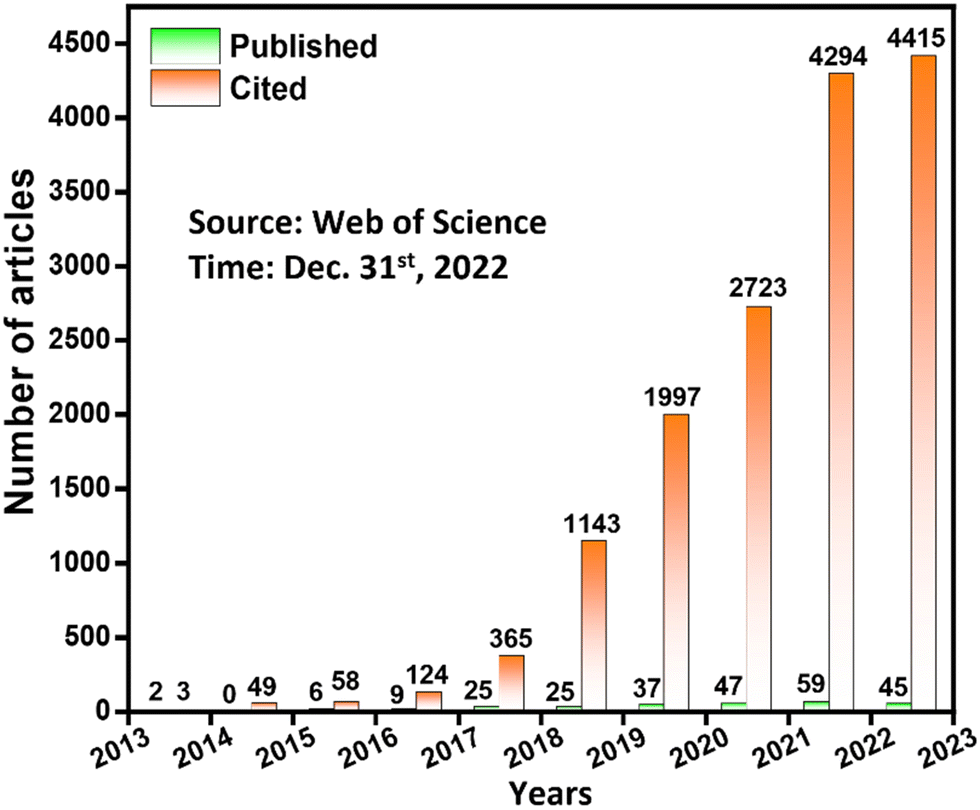 | ||
| Fig. 2 Bibliometric data on the number of papers published and the number of citations per year on COF-based SCs (the data were obtained by searching the following keywords on the web of science: (“covalent organic framework” OR “COF”) AND (“supercapacitor” OR “supercapacitors”)). | ||
So far, many good review articles on COF electrode materials are available,3,15,17,20,38–40 which provides us an excellent platform for tracking the relevant progress. In this feature article, we aim to give a complete picture on the nature of the recent design strategies of COF-based electrodes for SC application. The purpose is first to clarify the relationship between the structural properties and electrochemical performance of COF-based electrode materials. Then, the design strategies for COF-based SC electrode materials with high performance are systematically proposed. Besides, the current challenges and future perspectives for the design of COF-based SC electrodes are highlighted as well.
2. Electrode requirements for SCs
According to previous studies, specific capacitance, power/energy density, rate performance, and cycling stability have been considered as the main performance indicators to evaluate whether SC devices can be practically applied.41–43 Meanwhile, these main indicators are also the key technical parameters to guide the design of electrode materials. In this section, the core parameters are first described in detail, and the advantages of COF-based electrode materials for SCs oriented by the key parameters are then presented.2.1 Key performance indicators of SCs
2.2 Relationship between the structure of electrode materials and their SC performance
The electrochemical performance of SCs is closely related to the chemical structure of electrode materials, which might be notably different among various electrode materials. In summary, for COFs and other electrode materials, the specific surface area, type and accessibility of active sites, pore type and pore size, conjugation properties, crystalline structure and chemical linkage have significant effects on SC performance. The relationship between the structural properties of electrode materials and the electrochemical performance of SCs is summarized in Fig. 3.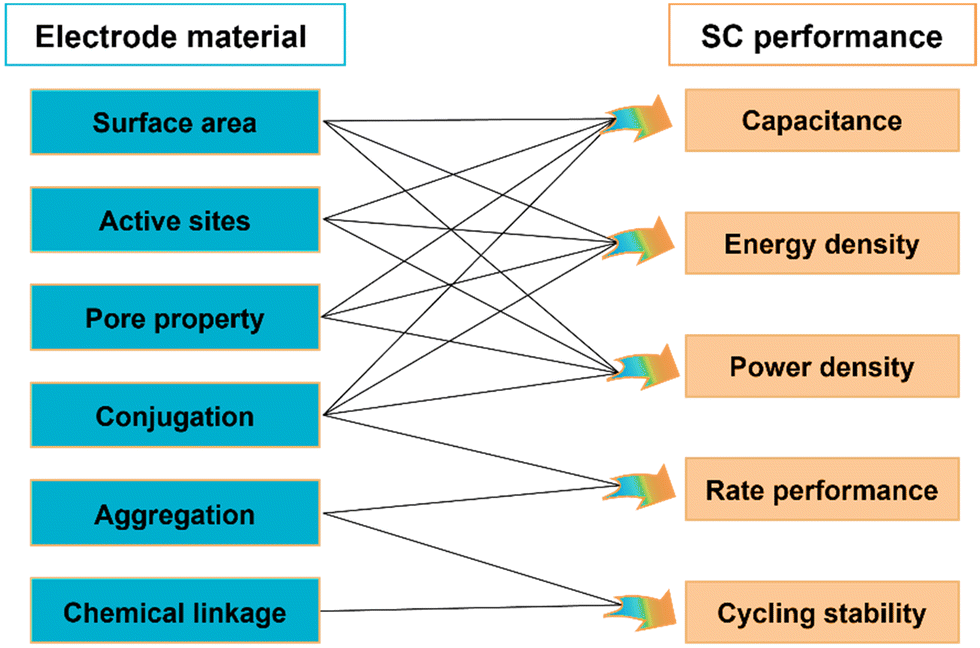 | ||
| Fig. 3 Relationship between the structural properties of electrode materials and the electrochemical performance of SCs. | ||
2.3 Advantages of COF-based electrode materials for SCs
Compared to carbon materials, transition metal oxides, amorphous porous materials, and metal–organic frameworks, COFs as a type of stable covalently bonded polymer are fully predesignable and synthetically controllable in structure. Accordingly, the application-oriented controllable construction based on the structure–performance relationship makes COF materials extremely charming. The structural properties including the pore size and distribution, specific surface area, degree of conjugation and surface functionalization determine the electrochemical performance of COFs. Specifically, the structure–performance characteristics of COF-based electrode materials in SC applications can be described in the following three aspects.(i) The covalent linkages and rigid skeletons of COFs endow them with outstanding thermal, chemical and electrochemical stability, and thus effectively suppressing the structural changes caused by ion deintercalation during the charge–discharge process and enhancing their cyclability.
(ii) The open and well-organized channels and high specific surface area facilitate the transfer of electrons/protons between COF electrodes and electrolytes, thereby improving the rate performance and power density.
(iii) The predesigned and abundant building blocks ensure that the COF platform shows great potential as electrodes for SCs: on one hand, tremendous electrochemically active organic moieties can be precisely organized into COF scaffolds, hurdling the obstacle of easy dissolution of organic molecules during the energy storage/conversion process and enabling high power density, remarkably enhanced energy density and outstanding cycle performance; on the other hand, feasible post-modification engineering allows electroactive units and other redox-active species (e.g., radicals, fullerenes, conducting polymers, metal oxides, etc.) to be decorated onto the walls or inside the channels of COFs, thereby endowing the resulting material with excellent capacitive properties.
3. Design strategies of COF-based electrodes for SCs
3.1 Introducing redox-active moieties into COFs
COFs are a versatile platform to tailor redox activities, thanks to their well-defined channels and walls, tunable skeletons, predesigned structures, and facile post-modification. In comparison to other electrode materials such as activated carbons, metal oxides, conducting polymers and amorphous porous organic materials, precise organization of electroactive units into COF skeletons is more feasible. Two approaches have been utilized to introduce redox-active moieties into COF skeletons: (i) designing redox-active building blocks for COF construction and (ii) introducing redox-active sites on the channel-walls of COF scaffolds linked by covalent bonds via post-modification. COFs with abundant redox-active sites enable not only high-speed mass transfer through their shape-preserving and orderly open channels, but also reversible high-throughput redox reactions through highly accessible redox-active centers on the channel walls, thus exhibiting large capacitance, excellent energy/power density, and high cyclability simultaneously. Herein, redox-active COFs are classified as the carbonyl/hydroxyl type, group 5A element-based type and free radical type based on the chemical structure of the redox moieties.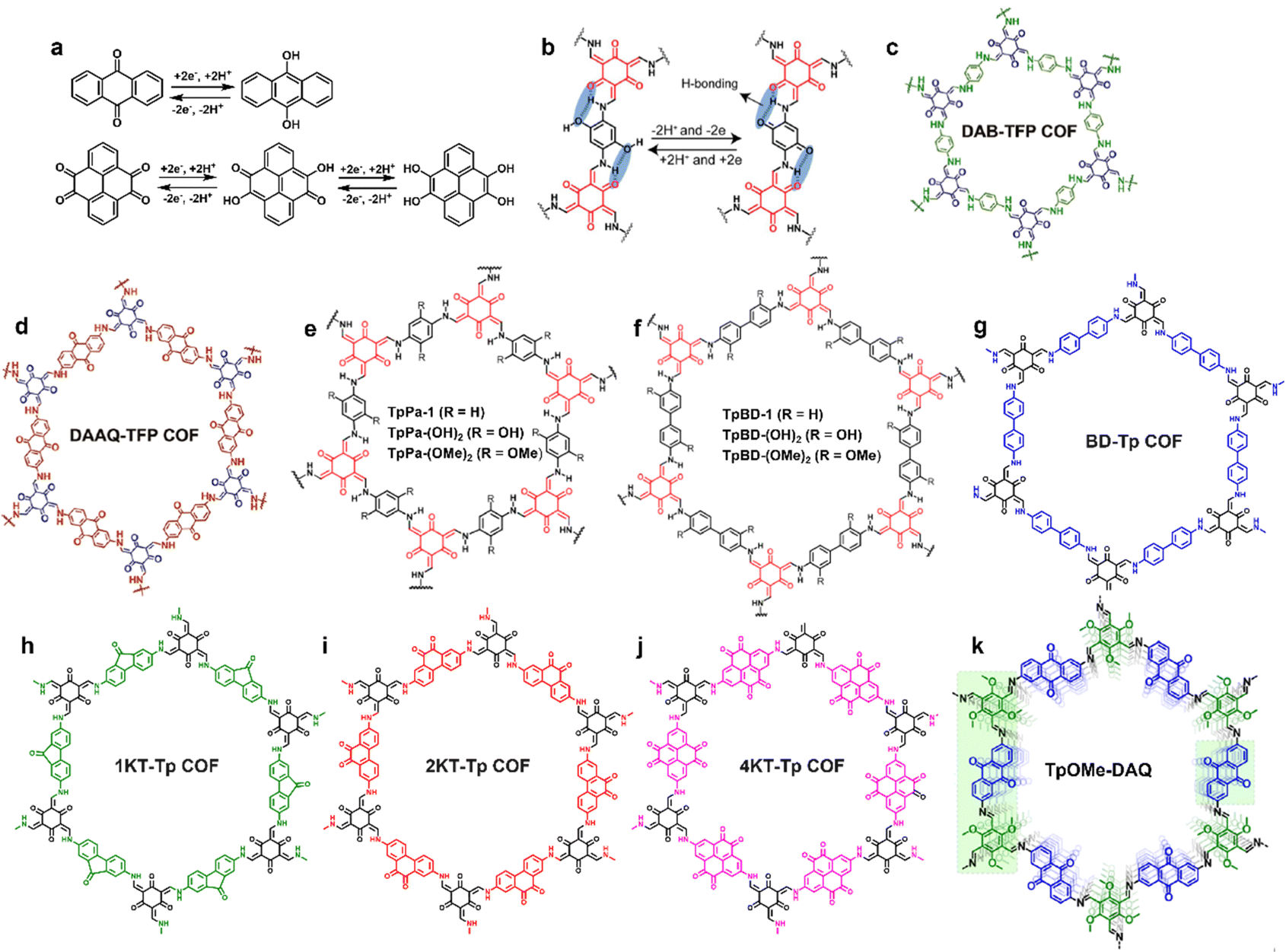 | ||
| Fig. 4 Energy storage process of carbonyl/hydroxyl redox-active groups through reversible transformation (a) between benzoquinone and hydroquinone, and (b) between H-bond-stabilized hydroquinone and benzoquinone. The chemical structure diagram of (c) DAB-TFP COFs, (d) DAAQ-TFP COFs (reproduced with permission from ref. 37 Copyright 2013, American Chemical Society), (e) TpPa-R [TpPa-1, TpPa-(OH)2, and TpPa-(OMe)2], and (f) TpBD-R [TpBD, TpBD-(OH)2, and TpBD-(OMe)2] (reproduced with permission from ref. 48 Copyright 2017, American Chemical Society). The chemical structure diagram of (g) BD-Tp COFs, (h) 1KT-Tp COFs, (i) 2KT-Tp COFs, (j) 4KT-Tp COFs (Reproduced with permission from ref. 49 Copyright 2020, Chinese Chemical Society), and (k) TpOMe-DAQ (reproduced with permission from ref. 50 Copyright 2018, American Chemical Society). | ||
For SC electrode application, both paraquinone- and orthoquinone-built-in COFs have been developed. Their representative chemical structures are shown in Fig. 4. Functional COFs with different carbonyl distribution densities were constructed as well. It is worth mentioning here that structural stability is always regarded as the most important parameter for the practical application of electrode materials. At the early stage of COF development, their construction was dominated by the reversible boronate ester linkage that exhibited poor hydrolytic and oxidative stability, which hindered the application of COFs to some extent. Until 2013, DAB-TFP COFs and DAAQ-TFP COFs were synthesized via combined Schiff-base condensation and enol–keto tautomerization (Fig. 4c and d) and first employed as electrode materials for SCs.37 As a control, DAB-TFP COFs without redox-active groups showed a capacitance of only 15 ± 6 F g−1 at 0.1 A g−1 in 1 M H2SO4 electrolyte, while DAAQ-TFP COFs decorated with redox-active paraquinone centers exhibited a higher specific capacitance of 48 ± 10 F g−1 and 83% capacitance retention over 5000 charge/discharge cycles. Under the same test conditions, the capacitance and capacitance retention of small DAAQ active units were 35 ± 7 F g−1 and 60%, respectively. This pioneering work demonstrated that introduction of redox-active centers into COF skeletons is a feasible design strategy to enhance the electrochemical properties of SCs.
The electro-transformation from hydroxyl groups to benzoquinone was also employed in the EES of SCs.48 In 2017, TpPa-(OH)2 and TpBD-(OH)2 (Fig. 4e and f) containing redox-active p-phenol functional moieties showcased specific capacitances of 396 and 86 F g−1 at 2 mV s−1 in 1 M phosphate buffer, respectively, in a three electrode configuration, while the initial specific capacitance values of contrastive TpPa-1, TpPa-(OMe)2, TpBD-1, and TpBD-(OMe)2 without phenolic OH groups were only 6, 13, 29, and 16 F g−1, respectively, under the same test conditions, which demonstrated the key role of intralayer hydrogen-bonds in determining the SC performance. The specific capacitance of TpBD-(OH)2 attributed to the irreversible redox transition of phenol/phenoxy radicals is merely one fifth of that of TpPa-(OH)2 ascribed to the reversible proton-coupled electron transfer (2H+/2e−) between hydroquinone and benzoquinone (Fig. 4b) along with 43% accessibility to active sites of the COF scaffold, and this different mechanism also accounted for the significant difference in capacitance retention in a three electrode configuration: 66% after 10000 cycles for TpPa-(OH)2 and only 2% after 1000 cycles for TpBD-(OH)2 as the phenoxy radical would quickly decompose.
The content of redox-active moieties in the COF skeleton has a remarkable impact on the electrochemical performance of SCs, which was exemplified using orthoquinone-based COFs systematically investigated in 2020.49 As shown in Fig. 4g–j, BD-Tp COFs, 1KT-Tp COFs, 2KT-Tp COFs and 4KT-Tp COFs containing none, one, two, and four ketone moieties in each linker units, respectively, were constructed via sequential Schiff-base condensation and enol–keto tautomerization. Compared with redox inactive groups in BD-Tp COFs or isolated carbonyl active sites in 1KT-Tp COFs, the orthoquinone moieties in 2KT-Tp COFs and 4KT-Tp COFs could not only provide denser redox-active sites but also enlarged conjugation upon reduction in the redox process. Therefore, 4KT-Tp COFs and 2KT-Tp COFs delivered higher capacitance values of 583 and 256 F g−1 at 0.2 A g−1 in 1 M H2SO4, respectively, than those of BD-Tp COF (20 F g−1) and 1KT-Tp COF (61 F g−1). Furthermore, all the three ketone containing COFs showed superb cycling stability with >92% capacitance retention over 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles.
000 cycles.
In 2018, reversible charge transfer between p-benzoquinone and hydroquinone for energy storage was reported.50 As shown in Fig. 4k, the chemical structure of TpOMe-DAQ is quite similar to that of DAAQ-TFP COFs,37 employing 2,4,6-trimethoxy-1,3,5-benzenetricarbaldehyde instead of 1,3,5-triformylpholoroglucinol as the trialdehyde building block. For TpOMe-DAQ, the interlayer hydrogen bond interaction between the N element of imine units (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) and the neighboring methoxyl group could effectively protect imine covalent linkages from hydrolysis, thus showing ultra-high cyclic performance with no significant capacitance degradation and Coulombic efficiency upon 10000 charge–discharge runs. The gravimetric and areal capacitance of TpOMe-DAQ was 169 F g−1 and 1600 mF cm−2 at 3.3 mA cm−2 in 3 M H2SO4, respectively. The superior areal capacitance was attributed to the thin sheet (200 μm) morphology. Besides, in the three electrode configuration, TpOMe-DAQ showcased excellent cyclic stability (>100000) with a capacitance increase during cycling originating from the improved accessibility to redox-active centers. Even in a solid-state SC device, 65% capacitance retention over 50000 cycles was achieved.
N) and the neighboring methoxyl group could effectively protect imine covalent linkages from hydrolysis, thus showing ultra-high cyclic performance with no significant capacitance degradation and Coulombic efficiency upon 10000 charge–discharge runs. The gravimetric and areal capacitance of TpOMe-DAQ was 169 F g−1 and 1600 mF cm−2 at 3.3 mA cm−2 in 3 M H2SO4, respectively. The superior areal capacitance was attributed to the thin sheet (200 μm) morphology. Besides, in the three electrode configuration, TpOMe-DAQ showcased excellent cyclic stability (>100000) with a capacitance increase during cycling originating from the improved accessibility to redox-active centers. Even in a solid-state SC device, 65% capacitance retention over 50000 cycles was achieved.
Although benzoquinone functionalized COFs have been widely utilized in SC energy storage devices, it should be noted that not all COFs containing benzoquinone exhibit pseudocapacitive behaviour. For instance, electrochemical measurements of DTFP-1 COFs did not exhibit characteristic redox curves, but rather charges were stored primarily through an EDLC mechanism.51 Generally, with extended π conjugation, inherent microporosity, and uniform channels, the vast majority of redox-active COFs store energy through both the reversible faradaic process and the EDLC mechanism, but the former dominates. Extending conjugation to improve conductivity and increasing the content and accessibility of redox moieties to amplify the faradaic redox effect are powerful approaches to improve the electrochemical performance of COF-based SCs.
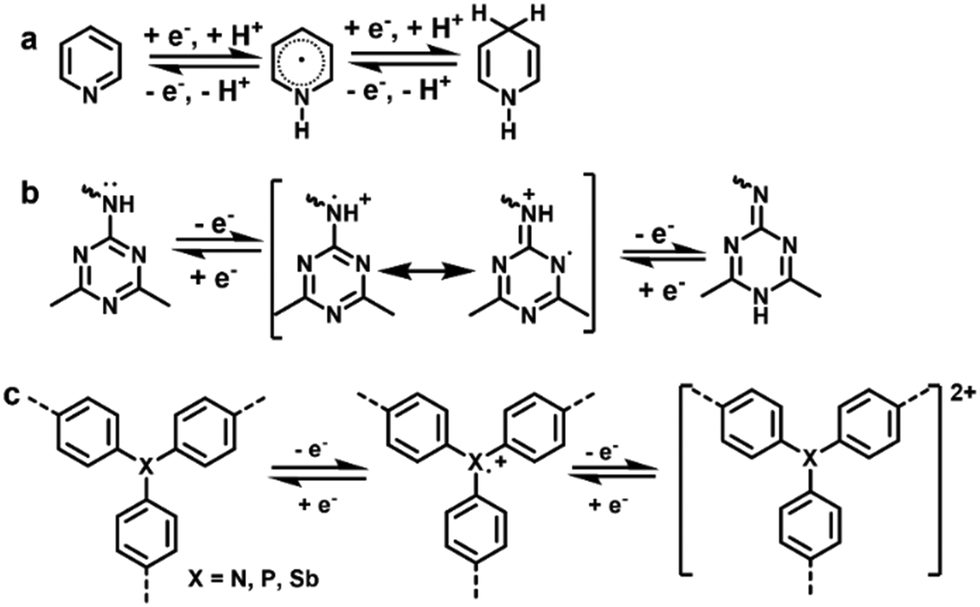 | ||
| Fig. 5 Energy storage mechanisms of (a) pyridinic N-, (b) triazinyl N-, and (c) triphenyl N/P/Sb-based redox-active moieties. | ||
A large number of COFs containing N-based moieties have been constructed, but electrochemically active N centers are extremely rare. In 2016, a pyridine-based COF (TaPa-Py COF, Fig. 6a) was reported to exhibit electrochemical activities, which was the pioneering work on heteroatom-based COFs exploited in SCs.53 CV measurement illustrated that the TaPa-Py COF showed a reversible faradaic process ascribed to the proton-coupled electron transfer of pyridine moieties originating from the intercalation of H+ into the COF (Fig. 5a). To demonstrate the pseudocapacitive behavior of pyridine moieties, the electrochemical behavior of the contrastive DAB-TFP COF based of the p-phenylenediamine linker instead of diaminopyridine (DAP) was also tested, and only an EDLC behavior was observed. Benefiting from a combination of pseudocapacitance provided by pyridine moieties and EDLC capacitance originating from the ordered porous structure, TaPa-Py COFs delivered a capacitance of 209 F g−1, which was much higher than that of redox-inactive DAB-TFP COFs (98 F g−1) and DAP-monomers (47 F g−1) in a three electrode configuration. A similar advantage of TaPa-Py COFs in comparison with DAB-TFP COFs was also observed in a two electrode configuration. Due to a stable chemically linked crystalline structure, TaPa-Py COF based devices exhibited a high cyclability with 92% capacitance retention at 2 A g−1 over 6000 charge–discharge cycles.
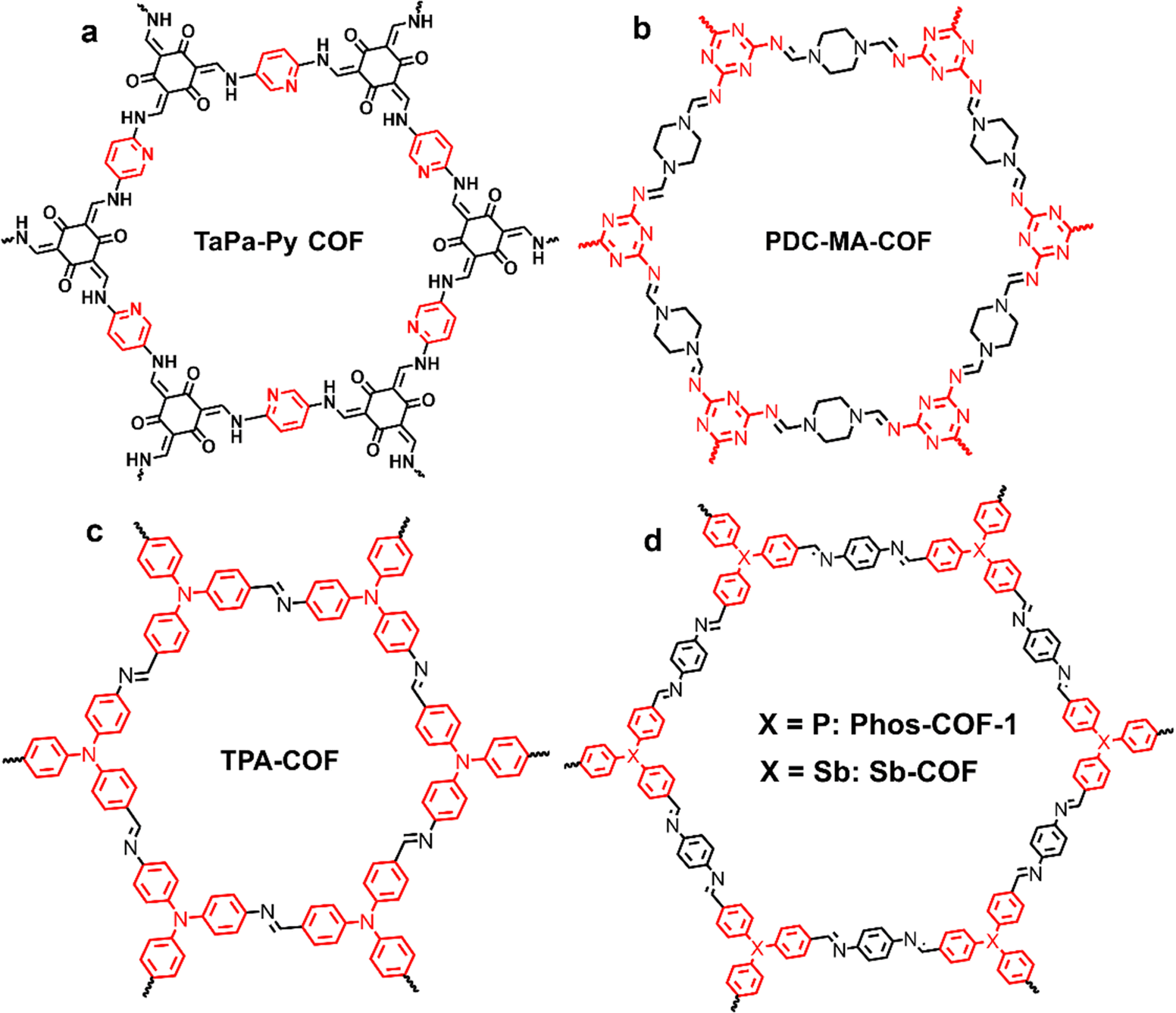 | ||
| Fig. 6 Chemical structure representation of redox-active group 5A element N/P/Sb-based COFs. | ||
In 2019, a series of pyridine-rich COFs (termed IISERP-COF10-12) were developed as SC electrode materials based on the pyridinal reversible faradaic process.54 As shown in Fig. 7a–c, the three IISERP-COFs share high structural similarities, where the keto–enol tautomerism along with hydrogen bonding interactions led to high stability. The abundant pyridyl and triazine groups ensure reversible interactions with H+ from the acidic electrolyte, enabling rapid and immense charge storage. Generally, the high surface area contributes primarily toward the EDLC behavior, while redox-active functional sites store energy in a pseudocapacitive way. In a three electrode configuration, IISERP-COF 10 delivered the highest specific capacitance of 546 F g−1, while IISERP-COF11 and IISERP-COF12 showed capacitance values of 310 and 400 F g−1, respectively, which correspond to their high specific surface area (IISERP-COF10, 1233 m2 g−1; IISERP-COF11, 921 m2 g−1; and IISERP-COF12, 1067 m2 g−1). Meanwhile, IISERP-COF12 with a greater number of OH groups predominantly adopts the keto form that shows higher affinity toward a protic electrolyte than the enolic form, thus exhibiting the highest pseudocapacitive contribution ratio among the three IISERP-COFs (Fig. 7d–f). Their capacitance retention follows the order of IISERP-COF10 (70%) < IISERP-COF11 (75%) < IISERP-COF12 (82%), which was due to the increased number of keto–enol stabilized hydroxyl groups as well. In solid-state devices, IISERP-COF10 yielded a record high areal capacitance of 92 mF cm−2 at 0.5 mA cm−2 thus far among all the COF-based solid-state SCs.
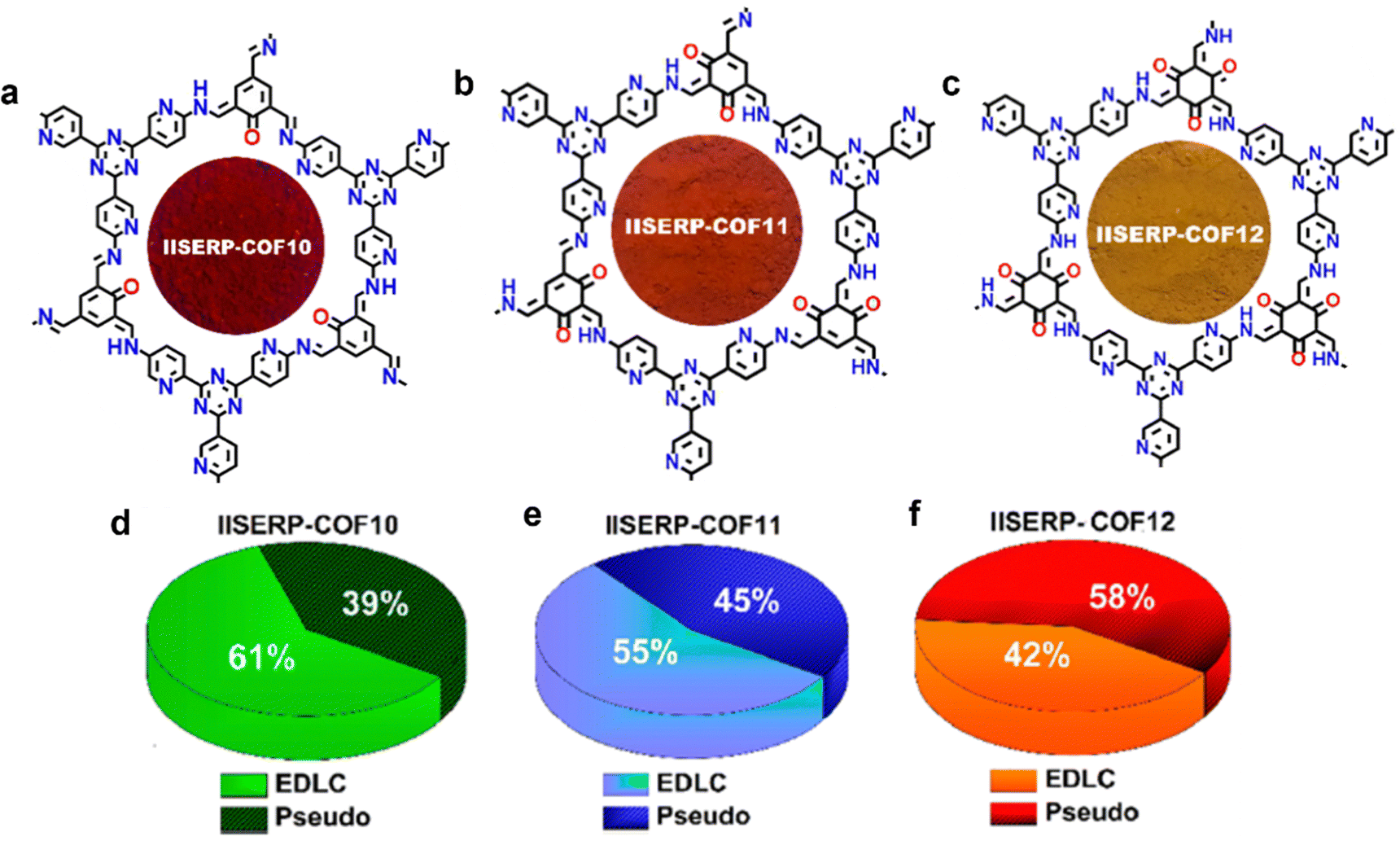 | ||
| Fig. 7 Chemical structure diagram of (a) IISERP-COF 10, (b) IISERP-COF 11, and (c) IISERP-COF 12, and (d–f) graphical representation of their capacitance contribution coming from the EDLC and pseudocapacitance in 1 M H2SO4. Reproduced with permission from ref. 54 Copyright 2019, American Chemical Society. | ||
Triazine was found to be a novel type of redox active center as well. In 2019, pseudocapacitive characteristics of PDC-MA-COFs with a high nitrogen content of 47.87% (Fig. 6b) synthesized from piperazinedicarboxaldehyde (PDC) and melamine (MA) were reported.55 The high nitrogen content and specific surface area, and abundant pores endow PDC-MA-COFs with outstanding electrochemical characteristics, while the interlayer hydrogen bonding provides it excellent stability. As illustrated in Fig. 5b, the triazine active moiety could experience a redox transition between aromatic and quinone structures. PDC-MA-COFs thus delivered a high specific capacitance of 335 and 94 F g−1 at 1.0 A g−1 in 6 M NaOH in three- and two-electrode configurations, respectively, and 88% capacitance retention after 20000 charge–discharge cycles was achieved in the two electrode configuration.
In 2020, our group reported the pioneering work on phosphine-based COFs (Phos-COF-1)60,61 for the controlled synthesis of metal nanoparticles. Meanwhile, its supercapacitive performance as an electrode was investigated where group 5A triphenyl moieties (i.e., triphenylphosphine) acted as redox centers. Phos-COF-1 delivered a decent capacitance of 100 F g−1 at 1 A g−1 in 3 M Na2SO4 with 90% capacitance retention over 5000 charge–discharge cycles in a three electrode configuration.58 Later, we developed Sb-COFs with triphenylantimony active sites in 2022.59 Sb-COFs demonstrate a similar PXRD pattern with Phos-COF-1, and the atomically structural difference is only that the Sb atoms in Sb-COFs take the place of P atoms in Phos-COF-1 (Fig. 6d). In electrochemical experiments, Sb-COFs showed a higher specific capacitance of 260 F g−1 at 2 A g−1, a high Coulombic efficiency of 99%, and 72% capacitance retention over 10000 cycles in a three electrode configuration, manifesting its high energy transfer efficiency and good electrochemical stability. CV measurements demonstrated that Phos-COF-1 and Sb-COFs exhibited similar redox behavior, but the latter had a superior electrochemical performance, which was due to the higher triphenylphosphine site utilization and better conductivity associated with its micromorphology (see Section 3.2.1). The electrochemical properties of Phos-COF-1 were further improved by compositing with graphene aerogels, which will be summarized in Section 3.2.2.
The redox activity of triphenylamine-based COFs (TPA-COF) was also exploited for electrochemical energy storage of SCs in 2021.57 The chemical structure of TPA-COFs is shown in Fig. 6c. TPA-COFs exhibited a uniform nanofiber-like structure along with a high specific surface area of 398.59 m2 g−1. Due to the presence of redox active triphenylamine units, the TPA-COF electrode exhibited pseudo-capacitance characteristics and showed a high specific capacitance of 263.1 F g−1 at 0.1 A g−1 in 1 M H2SO4 in a three electrode configuration. Its capacitance retention was 111% over 5000 charge–discharge cycles, which was attributed to the pore expansion originating from the shuttling of ions during the continuous charge–discharge process.
If hydrogen bonds are formed between heteroatoms and hydrogen atoms in the COF skeleton, electron transfer is expected to accelerate, thereby enabling materials with enhanced or even nascent electrochemical properties, which was recently realized in COF scaffolds containing heteroatoms N, S, etc.48,62 As presented in section 3.1.1 (Fig. 4b), intramolecular hydrogen bonding promoted redox processes.48 In 2020, the effect of intramolecular hydrogen bonds on electrochemical performance of PG-BBT COFs was demonstrated (Fig. 8a).62 PG-BBT COFs were constructed via sequential reversible Schiff base condensation and irreversible oxidation. When PG-BBT COFs underwent a redox process, each proton of the phenol group was transferred to the adjacent N atom to form a hydrogen bond, and therefore 6e−/6H+ transfer for each unit (Fig. 5b). The capacitance of PG-BBT was 724 F g−1 at 1 A g−1 along with 96% capacitance retention over 10000 charge–discharge cycles, indicating outstanding electrochemical performance.
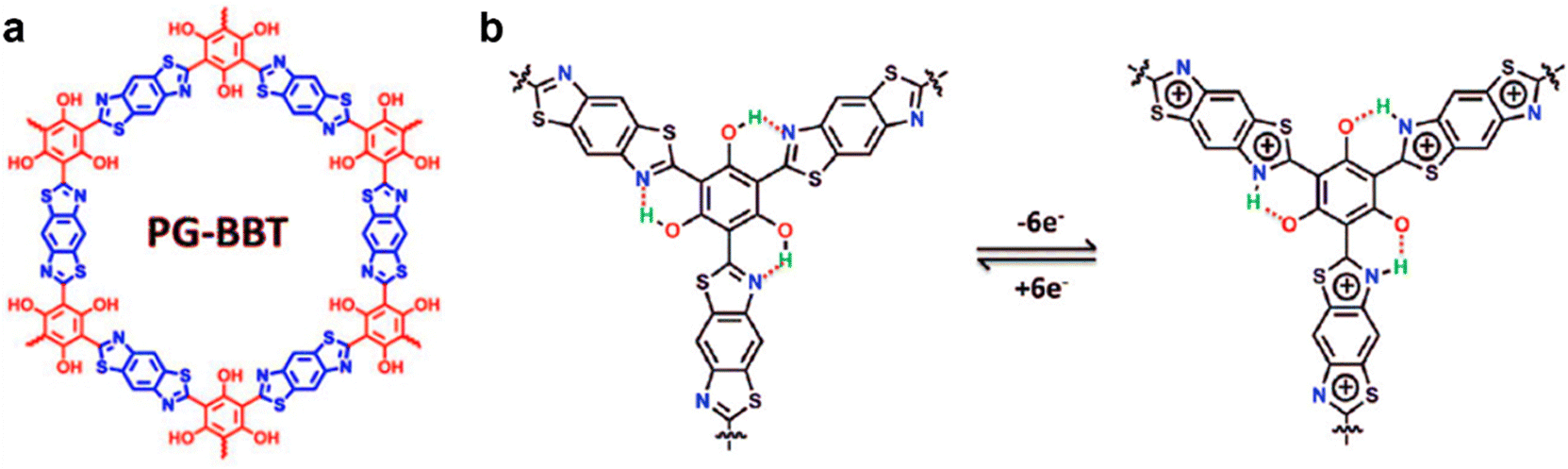 | ||
| Fig. 8 (a) Chemical structure diagram, and (b) energy storage mechanism via the intramolecular hydrogen bond process of PG-BBT. Reproduced with permission from ref. 62 Copyright 2020, The Royal Society of Chemistry. | ||
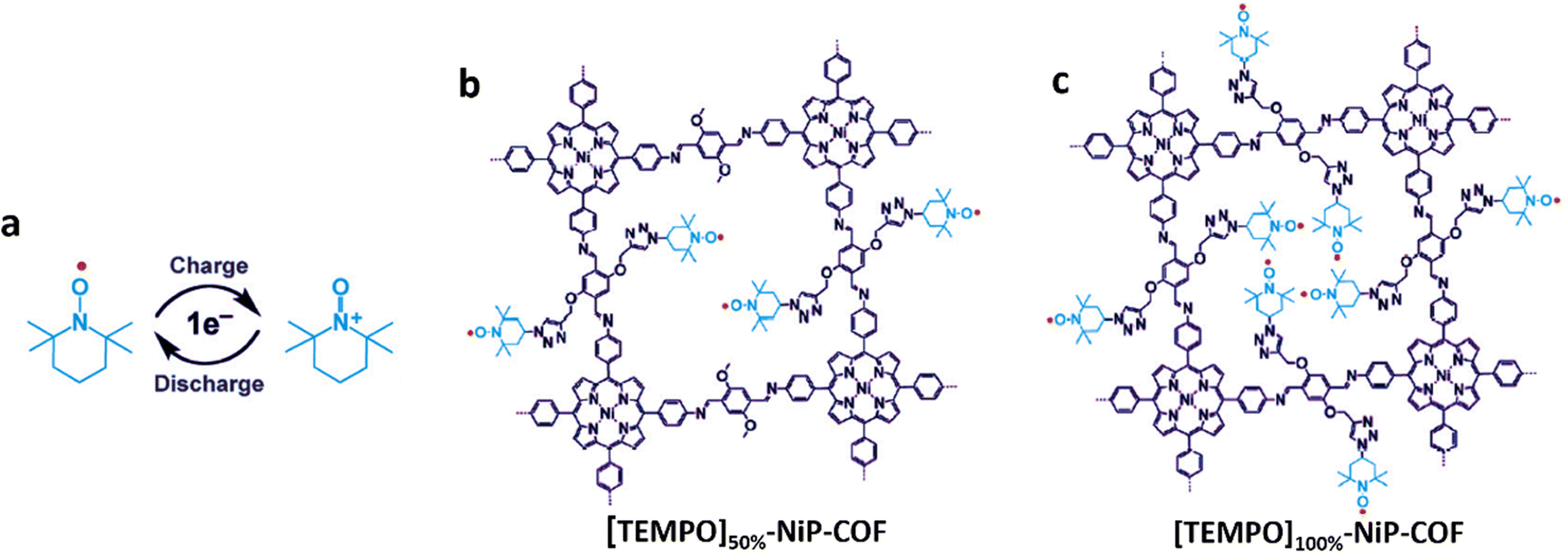 | ||
| Fig. 9 (a) Energy storage mechanism of TEMPO based on a one-electron redox reaction, and the chemical structure diagram of (b) [TEMPO]50%-NiP-COFs and (c) [TEMPO]100%-NiP-COFs. Reproduced with permission from ref. 63 Copyright 2015, Wiley-VCH. | ||
3.2 Improving the accessibility of redox-active moieties
Thanks to the outstanding performance-oriented designability, ordered and well-established channels and easily functionalized walls, COFs have exhibited tremendous application potential in many fields. However, the poor processability, low electrochemical accessibility to the active sites and inferior electrical conductivity severely limit their practical applications. For SC electrodes, the long-range columnar channels of bulk COFs formed by stacked atomic layers and edge defects usually have a negative influence on efficient ion transport and high accessibility to the redox-active sites. In this section, the strategy via morphology control including nanosheets standing on a substrate, free standing thin films, and nanostructures for enhancing the accessibility of redox-active moieties will be summarized and analyzed.Nanosheet-like COFs with a thickness of single- or few-atom layers and nanostructured COFs with various shapes can shorten the length of ion transport channels and increase the exposure rate of active sites, thus enhancing capacitive properties. Pristine powder COFs usually need to be mixed with carbon black and a binder before studying their electrochemical performance. As shown in Fig. 10a, the grain boundaries and random orientation remain large obstacles to achieve high conductivity and fast mass transfer. The first COF (DAAQ-TFP COF) developed as a SC electrode delivered a low capacitance of 48 ± 10 F g−1, with only ∼3.0% utilization rate of redox-active DAAQ sites, which was due to random orientation of microcrystalline DAAQ-TFP COF powder.37 In 2015, Dichtel and co-workers demonstrated that the oriented COF thin films could improve the accessibility to redox-active sites and shorten the ion transport path, thus enhancing the electrochemical performance.66 DAAQ-TFP COF thin films were prepared on a Au substrate electrode (Fig. 10b). The thickness could be modulated by varying the initial monomer concentration. A much higher utilization efficiency (80–99%) of DAAQ moieties in thin films was obtained in comparison to the randomly oriented polycrystalline powder (∼3.0%). Consequently, the oriented COF thin films exhibited a 4-fold increase in areal capacitance in comparison to the powder COF electrode. 3D graphene has also been employed as a support for the growth of electrochemically active COF films.67 In 2015, a π-conjugated surface COF (COFDAAQ-BTA) with anthraquinone moieties was incorporated into 3D graphene through a noncovalent way. The layered COFDAAQ-BTA-3DG hybrid delivered an areal capacitance of 31.7 mF cm−2 with an enhanced accessibility of 13.45% (vs. 2.5% for 2D COF powder) of the DAAQ moieties in 1 M KOH.
 | ||
| Fig. 10 Schematic diagram of the existing morphology of (a) microcrystalline COF powder and (b) oriented COF thin film on the electrode surface. Reproduced with permission from ref. 66 Copyright 2015, American Chemical Society. | ||
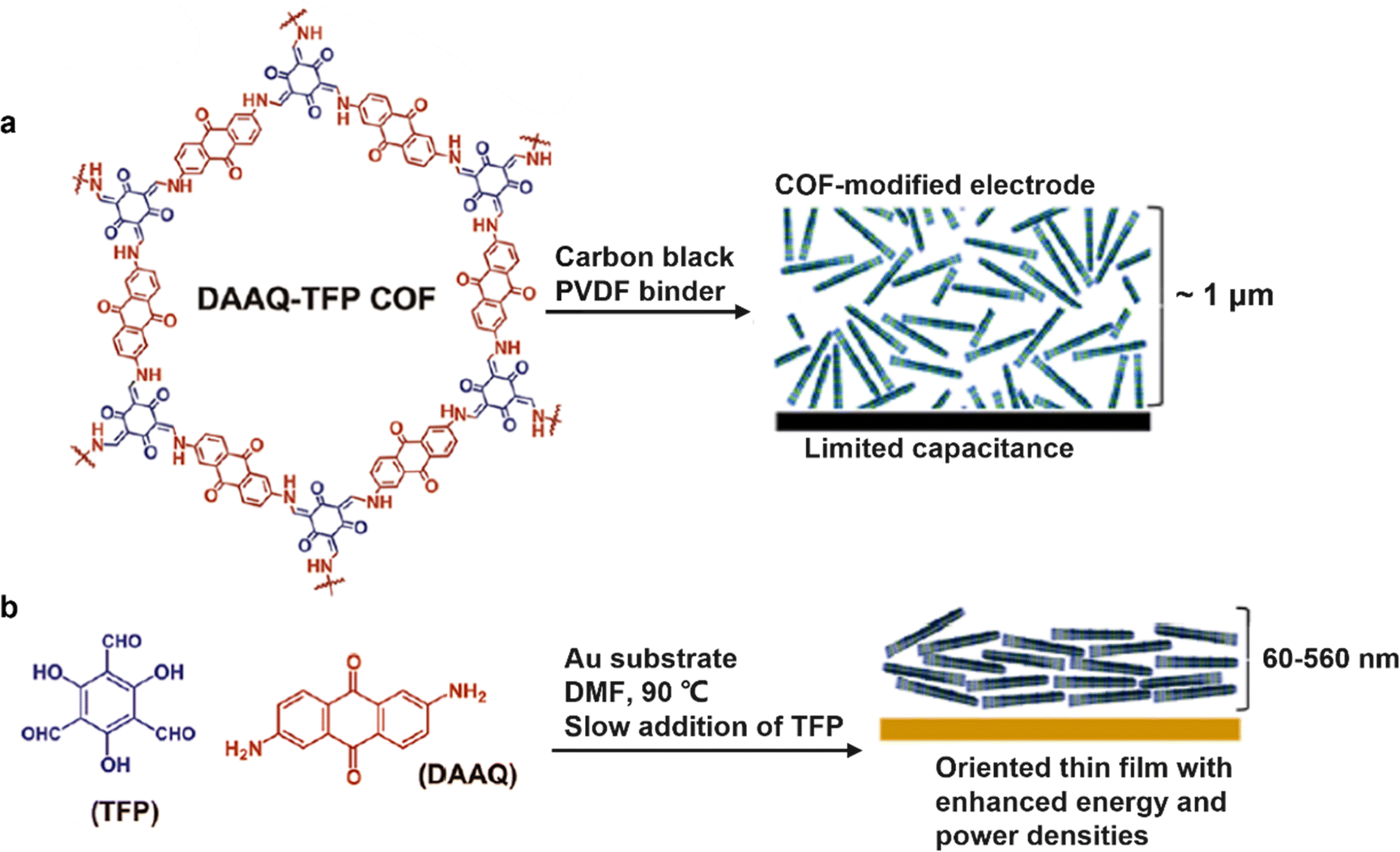
Free-standing thin films were also prepared for SC electrodes. So far, substantial efforts have been devoted to DAAQ-functioned COFs as redox-active materials. Very representatively, the TpOMe-DAQ COF (chemical structure shown in Fig. 4k) was constructed via a p-toluenesulfonic acid (PTSA) catalyzed mechanochemical grinding approach (Fig. 11a).50 It presented an independent electrochemical behavior without any substrate, support, or additives (Fig. 11b), and delivered a gravimetric capacitance of 169 F g−1 and an areal capacitance of 1600 mF cm−2, thanks to the thin flake morphology (∼200 μm). Slightly earlier, using a similar film-forming strategy, another flexible DAAQ-based COF electrode was prepared, where two different linkers including π-electron-rich anthracene (Da) and redox active anthraquinone (Dq) were simultaneously constructed into a β-ketoenamine linked COF.68 The effect of the ratio of the two employed linkers on the electrochemical and mechanical properties of COFs was further investigated. The larger the molar ratio of Dq: Da in diamine building blocks, the better the capacitive performance and the lower the mechanical strength of the COF thin film.
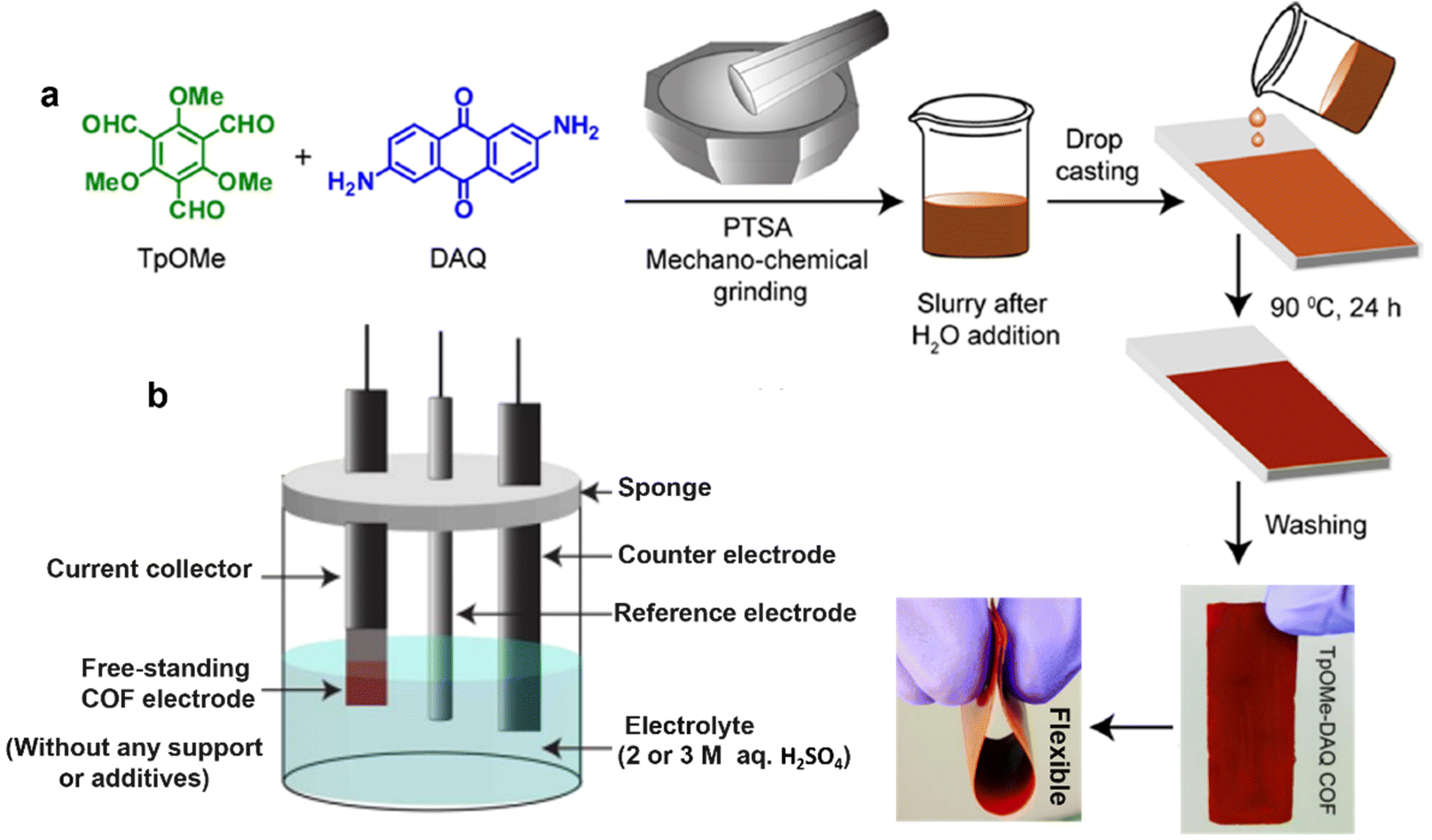 | ||
| Fig. 11 (a) Fabrication procedure diagram of the TpOMe-QAQ COF thin film and its flexible nature, and (b) free-standing TpOMe-QAQ COFs as the working electrode without any support or additives. Reproduced with permission from ref. 50 Copyright 2018, American Chemical Society. | ||
Constructing nanostructured COFs is a useful way to enhance the accessibility of redox-active groups, although its development is still in infancy. Currently, the growth and morphology of COFs are mainly controlled by the thermodynamics of the reaction system or the added template agent. In terms of SC electrode materials, the study on COF nanostructures is still relatively rare. As summarized in Section 3.1.2, the chemical structures of Sb-COFs and Phos-COF-1 are slightly different. Their pore size distribution, specific surface area, and electrochemical behavior were all quite similar as well. However, the accessibility to redox-active sites (42% vs. 12%) and specific capacitance (260 F g−1 at 2 A g−1vs. 100 F g−1 at 1 A g−1) differed significantly, which was largely due to the COF particle size.58,59 Sb-COFs had a nanostructured morphology with an average diameter of 100 ± 20 nm and a length of ∼200 nm statistically derived from SEM and TEM images (Fig. 12a and b), while Phos-COF-1 existed in an aggregated bulk formed by a great deal of particles with an average size of ∼10 μm (Fig. 12c and d). Besides, nanostructured Sb-COFs with intrinsic opening channels could enable effective electrolyte infiltration, and the small particle size can shorten the transport and diffusion path lengths for electrolyte ions, thus providing fast kinetics and high charge–discharge rates. Moreover, a stable crystalline structure could endure strain/stress in the electrode matrix, ensuring enhanced cyclability.69 The superior capacitive performance of Sb-COFs indicated that the nanostructured COFs possess great potential in SC applications.
 | ||
| Fig. 12 (a) SEM and (b) TEM images of Sb-COFs. Reproduced with permission from ref. 59 Copyright 2022, The Royal Society of Chemistry. (c) SEM and (d) TEM images of Phos-COF-1. Reproduced with permission from ref. 58 Copyright 2021, Springer. | ||
3.3 Enhancing the electrical conductivity
Combining high surface areas and notably abundant redox-active sites, COFs can synergize with energy storage principles of EDLCs and pseudocapacitors in SC application. However, their electrochemical performance is severely limited by the low charge/discharge rates, which are fundamentally caused by the inferior electrical conductivity of COF architectures. To overcome the obstacle, compositing conductive additives with COFs was implemented. It is noteworthy that the enhanced electrical conductivity would be beneficial for a better accessibility to redox-active sites as well. The most classical electrochemically active DAAQ-COF has been intensively studied as a model pristine COF material.In 2016, encapsulating poly(3,4-ethylenedioxythiophene) (PEDOT), the most widely studied conducting polymer with excellent conductivity and high stability, inside the nanochannels of thick DAAQ-COF films to accelerate the charge–discharge rate was investigated.70 As shown in Fig. 13a, employing 3,4-ethylenedioxythiophene as a monomer, PEDOT was in situ electropolymerized inside the pores of COF films that was grown on a Au substrate. The preparation procedure of COF films was similar to the abovementioned work.66 The obtained PEDOT-modified DAAQ-TFP film exhibited 10-fold higher current response, a 12-fold increase in volumetric power density and 30-fold higher volumetric energy density than those of the pristine DAAQ-COF film. Besides, the charge transfer rate of the PEDOT-modified DAAQ-TFP film could be up to 1600 C without deteriorating the performance. However, the PEDOT chains that penetrated deep into the channels of the DAAQ-TFP significantly weakened the intrinsic porous property, with an ultralow BET surface area of 6 cm2 cm−2 after nine electropolymerization cycles, which was detrimental to the surface capacitance. In 2019, via in situ solid-state polymerization, PEDOT linear chains were controlled to grow inside the DAAQ-TFP channels and retained a high porosity with a BET specific surface area of 131 m2 g−1.71 The PEDOT@AQ-COF electrode exhibited an ultrarapid charge–discharge rate (998 F g−1 at 500 A g−1), an superb conductivity of 1.1 S cm−1, as well as a high capacitance of 1663 F g−1 at 1 A g−1. The above two studies demonstrated that the engineered introduction of conducting polymers into the cavities of COFs can efficiently improve the conductivity of COF-based electrodes for high performance SCs.
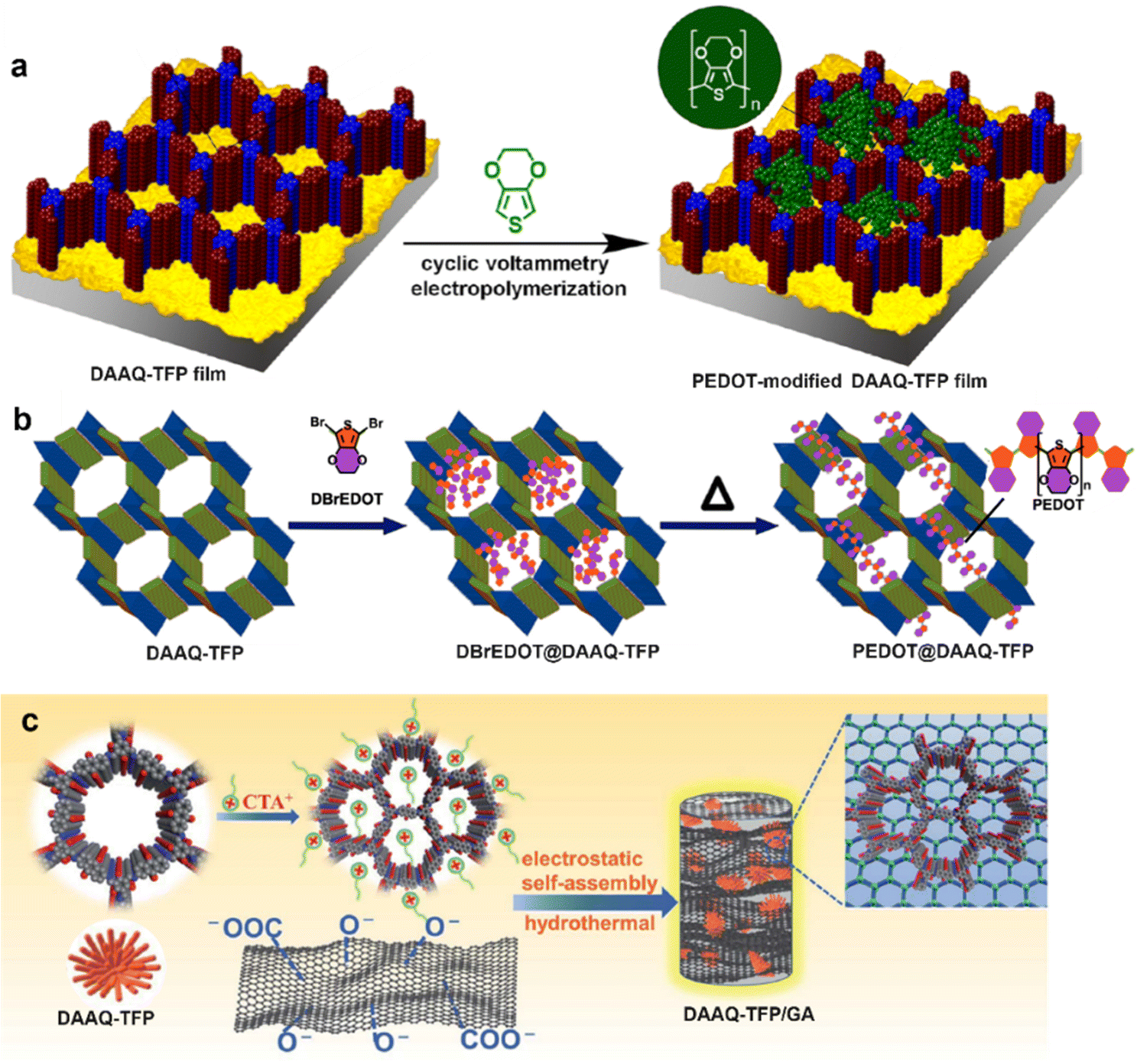 | ||
| Fig. 13 Synthetic scheme of (a) PEDOT-modified DAAQ-TFP films, and (b) PEDOT@DAAQ-TFP powder. Reproduced with permission from ref. 70 Copyright 2016 and ref. 71 Copyright 2019, American Chemical Society, respectively; (c) Preparation procedure of DAAQ-TFP/GA. Reproduced with permission from ref. 72 Copyright 2021, the Royal Society of Chemistry. | ||
Besides conducting polymers, 3D graphene aerogels (GAs) have also attracted a great deal of attention in energy storage application due to their excellent electrical conductivity and unique mechanics characteristics. Currently, a few COF/graphene composites have been fabricated. In 2021, DAAQ-TFP was first modified with the cationic surfactant CTAB (cetyltrimethylammonium bromide) followed by self-assembly with the negatively charged GA via electrostatic interaction (Fig. 13c).72 The 3D cross-linking conductive network in the obtained free-standing DAAQ-TFP/GA composite could efficiently surmount the limitations of slow electron transfer and low accessibility to the active sites in the COF skeleton. The as-obtained DAAQ-TFP/GA electrode exhibited a high specific capacitance of 378 F g−1 at 1 A g−1 and fast kinetics with ∼93.4% capacitive contribution at 3 mV s−1. Besides, the asymmetric SC assembled with DAAQ-COFs/GA and pristine GA electrodes exhibited an energy density of 30.5 W h kg−1. In early 2021, our group also made similar progress in the SC electrode application of graphene/COF composites. We chose reduced graphene oxide (rGO) aerogel as a conducting network support, and four Phos-COF-1/rGO composites (PPrGO-1, PPrGO-2, PPrGO-3, and PPrGO-4) with different mol% rGO were synthesized via a solvothermal method where the polymer was formed in the presence of rGO.73 Thanks to the well-established interfacial interaction and high specific surface area (690 m2 g−1), PPrGO-2 exhibited the lowest charge transfer resistance (3.85 Ω cm−2) and series resistance (1.72 Ω cm−2) in 1 M KOH. Correspondingly, the as-assembled PPrGO-2//AC (active carbon) could be operated in a wide potential window of 1.6 V, displaying a high energy density of 33.3 W h kg−1 with a power density of 8370 W kg−1 and superb cyclability with 88% capacitance retention after 12000 successive charge/discharge cycles. The excellent electrochemical performance of PPrGO-2 was ascribed to the conjugated structure, highly enhanced conductivity, and easily accessible redox sites. Meanwhile, Phos-COF-1 was also hydrothermally composited with rGO to give a hierarchical nanostructured PP/rGO which then served as the anode for high-performance SCs. Employing α-MnO2 nanowires as the cathode, a high-voltage aqueous asymmetric SC was successfully assembled, which had an extended potential window of 2 V, and exhibited a significantly enhanced capacitance of 380.2 F g−1 at 2 A g−1 with a good rate capability of 165.0 F g−1 at 4 A g−1. A high specific energy of 39 W h kg−1 with extraordinary cyclability with 96% capacitance retention over 10000 cycles was achieved as well.74
Enhancing conductivity via a metal ion assisted strategy has also been carried out to improve the capacitive performance of COF-based SCs. In 2019, Ni-COFs were solvothermally synthesized via choosing 1,2,4,5-benzenetetraamine (BTA), 2,5-dihydroxy-1,4-benzenedicarboxaldehyde (HBC) as building blocks and Ni(OAc)2·4H2O as an electrochemically active center.75 The chemical structure and charge–discharge mechanism of Ni-COFs are presented in Fig. 14. The reversible transformation between hydroquinone and benzoquinone occurred during the energy storage/conversion process. Benefiting from the 2D highly conjugated structure and the incorporation of Ni(II) centers, Ni-COFs exhibited high electrical conductivity of 1.2 and 1.3 × 10−2 S cm−1 for thin film and powder, respectively. Besides, Ni-COFs showed an ultrahigh specific capacitance of 1257 F g−1 at 1 A g−1 with 94% capacitance retention over 10000 cycles. The as-assembled activated carbon//Ni-COF showed a high capacitance of 417 F g−1, and an excellent energy density of 130 W h kg−1. This work demonstrated a feasible approach to enhance the electrochemical performance through introducing metal ion centers.
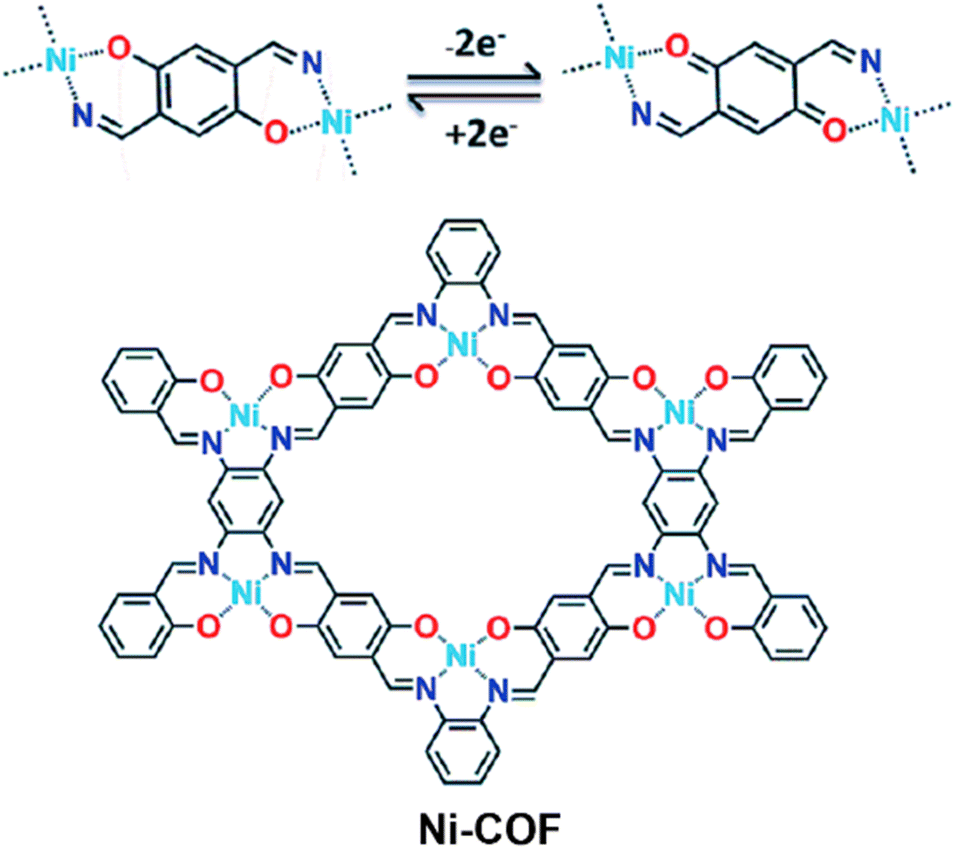 | ||
| Fig. 14 Energy storage mechanism (top) and chemical structure (bottom) of Ni-COFs. Reproduced with permission from ref. 75 Copyright 2019, The Royal Society of Chemistry. | ||
3.4 COFs as supports for other electrode materials
COFs feature with ordered channels and good structural tailorability, which are considered to be an outstanding porous support for other electrochemically active species.76 For one thing, the easily functionalized construction of the channel-wall composition can provide a chemical platform for the controllable introduction of guest molecules; for another thing, their uniform channels can effectively confine guest species inside the channel without aggregation or loss during application. Therefore, by incorporating highly electrochemically active species such as metal oxides, hydroxides, fullerenes into the channels of COFs, high electrochemical activity and cycle performance are expected. However, the studies on COF-dispersed and stabilized electrochemically active metal oxides/hydroxides have been rarely reported. Therefore, numerous opportunities exist in the field, which will be discussed in detail in the last section.The unique π-electron conjugated system and excellent electron affinity of fullerenes make them attractive candidates for electrode materials in EES devices. However, fullerene materials usually lack a certain degree of order on the nanoscale, resulting in low electrochemical utilization and a limited ion transport rate, thus greatly limiting their application in energy storage. In 2021, we designed an azide group-functionalized COF to hang the fullerene carbonyl/hydroxyl type into the ordered pores through “click” chemistry to create ordered nonporous fullerenes. (Fig. 15).77 Powder X-ray diffraction and N2 adsorption–desorption test results showed that the obtained material ([C60]X-COF, X = 0.025; 0.05; 0.09; 0.016) could retain the crystallinity and porosity of the COF support, but the pore volume decreased with the increase in C60 loading. Among the four [C60]X-COFs with different C60 contents, [C60]0.05-COF electrode material showed the best electrochemical performance, which was because an appropriate C60 loading can endow sufficient active sites without blocking the pores that serve as mass transfer paths. All [C60]X-COFs exhibited much higher capacitance values than those (<14 F g−1) of pristine COF and C60 in a three electrode configuration, among which [C60]0.05-COFs gave the maximum specific capacitance of 63.1 F g−1 at 0.7 A g−1. The assembled [C60]0.05-COF//rGO delivered an energy density of up to 21.4 W h kg−1 at a power density of 900 W kg−1 in an operating voltage window of 1.8 V with 99% capacitance retention after 5000 cycles. This work discloses that constructing ordered nanoporous C60 templated by a COF material is an efficient strategy to enhance its electrochemical accessibility and thus capacitive performance.
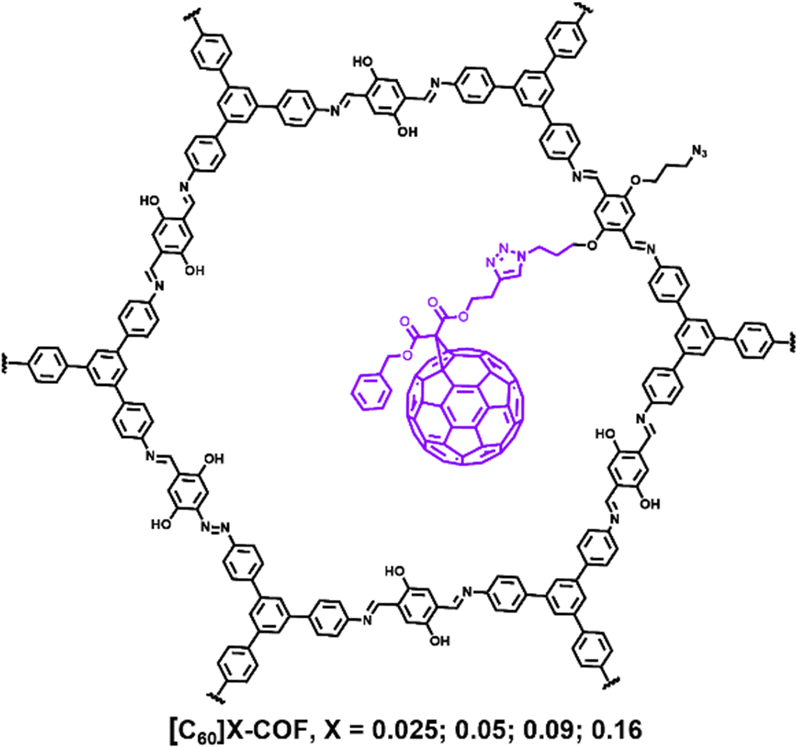 | ||
| Fig. 15 Chemical structure of [C60]X-COFs. Reproduced with permission from ref. 77 Copyright 2021, Elsevier. | ||
3.5 COFs as precursors for porous carbon electrode materials
Porous carbon materials have been drawing intensive attention with regard to metal-free electrode application thanks to their pore structure, high specific surface areas, and high chemical and mechanical stability. Doping heteroatoms (N, B, S, P, etc.) into sp2 carbon networks is a feasible approach to modify the electronic properties and provide electroactive sites.78–80 Actually, as aforementioned, the vast majority of pristine COFs deliver weak electrical conductivity, which severely restricts the electrochemical applications of COFs. Carbonization at high temperature has been considered as an effective approach to enhance their electrical conductivities. Compared with amorphous organic porous material precursors, carbonizing crystalline COFs is more beneficial to obtain porous carbon materials with uniform pore size and controlled introduction of heteroatoms.81,82 Although the carbonization treatment might destroy the periodical features of pristine COFs, many investigations have demonstrated that the carbides derived from COF precursors showcased enhanced electrochemical performance compared to original COFs.83,84 Hitherto, both approaches to carbonize COFs were developed, including template-free direct carbonization and templated pyrolysis, while the former has a wider range of applications.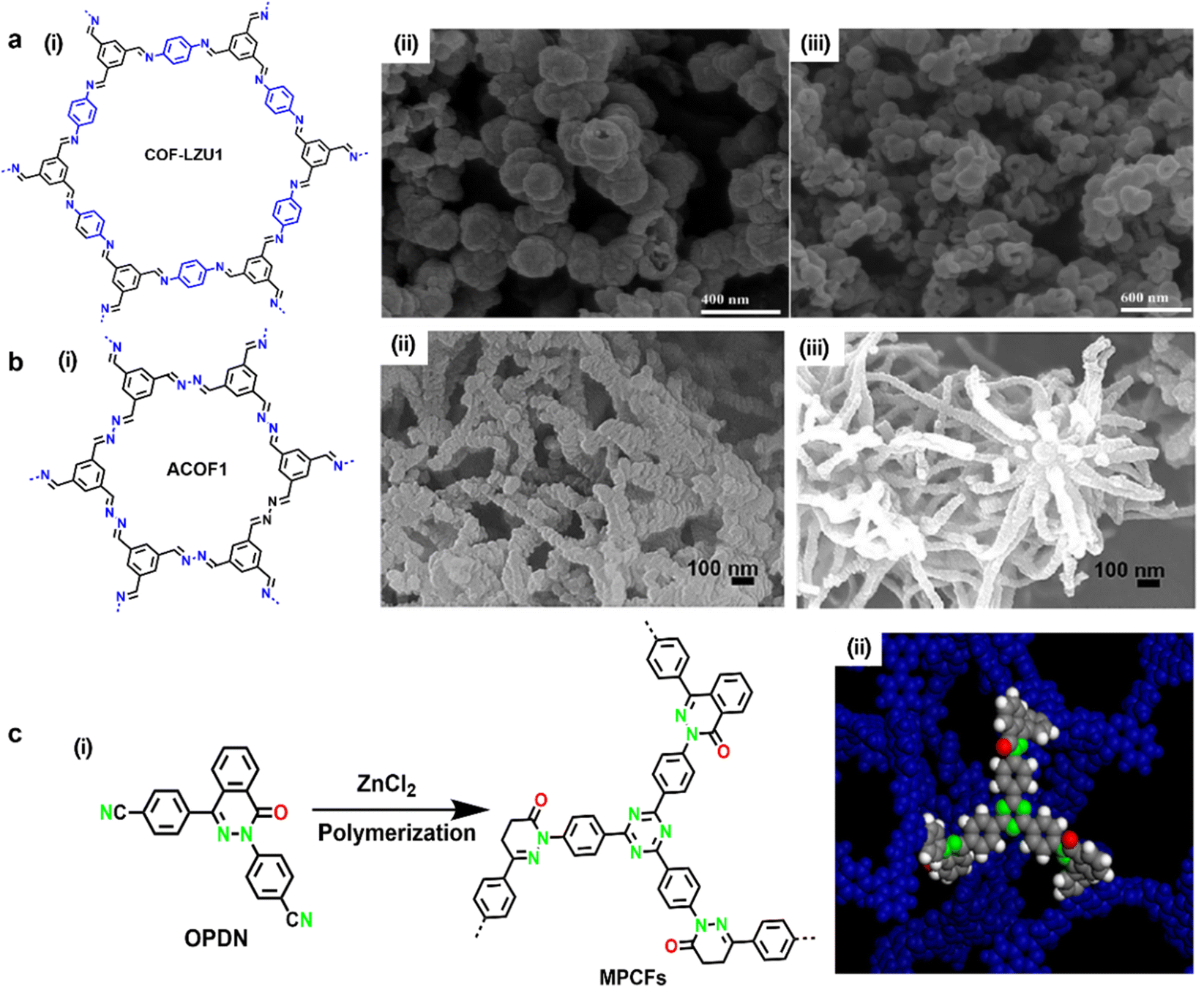 | ||
| Fig. 16 (a-i) Chemical structure of COF-LZU1, and SEM image of COF-LZU1 (a-ii) before and (a-iii) after carbonization. Reproduced with permission from ref. 85 Copyright 2013, American Chemical Society. (b-i) Chemical structure of ACOF1 and SEM image of ACOF1 (b-ii) before and (b-iii) after carbonization. Reproduced with permission from ref. 87 Copyright 2017, Elsevier. (c-i) Synthesis schematic illustration of MPCFs and (c-ii) its simulated fragments. Reproduced with permission from ref. 88 Copyright 2017, American Chemical Society. | ||
The N and O atomically homogeneously doped porous carbon frameworks (MPCFs) were polymerized from 4,4′-(4-oxophthalazine-1,3(4H)-diyl)dibenzonitrile through the trimerization of cyano groups and subsequent ionothermal synthesis catalyzed by ZnCl2 at 600 °C, 650 °C, 700 °C, respectively (Fig. 16c).88 After pyrolysis at high temperature, the black carbonized powder (MPCFs@700) obtained at 700 °C had the highest specific surface area of 1638 m2 g−1 and a pore diameter of ∼2 nm, thus affording a high specific capacitance of 302 F g−1 in a three electrode configuration. When 1-butyl-3-methylimidazolium tetrafluoroborate was employed as an electrolyte to broaden the special window (up to 3.5 V), an SC device based on MPCFs@700 exhibited a specific capacitance of 155 F g−1 and an energy density of 65 W h kg−1 at 0.1 A g−1. Furthermore, the as-prepared MPCFs@700 showed excellent cyclability in aqueous, organic and ionic liquid electrolyte respectively, demonstrating its long term durability.
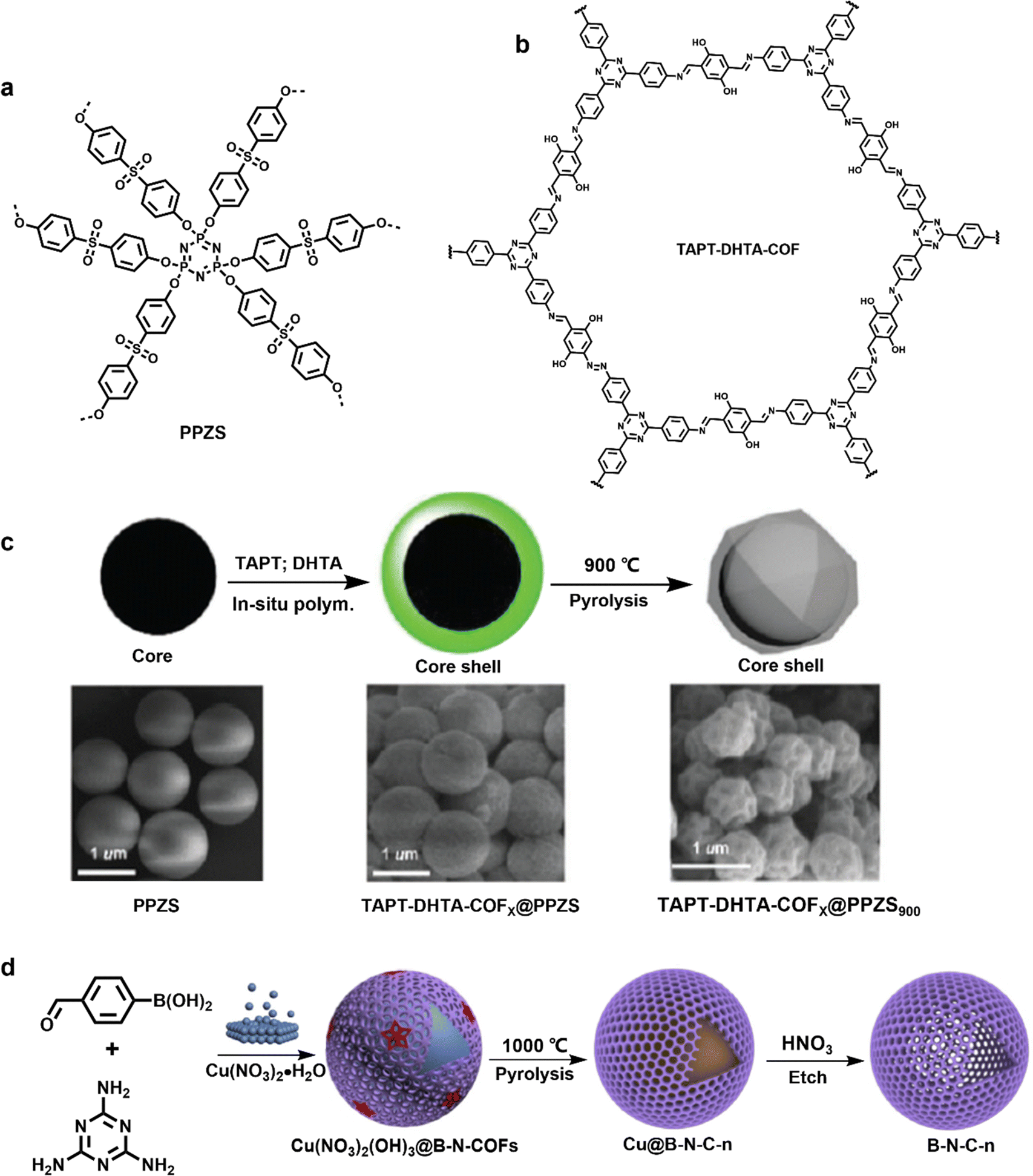 | ||
| Fig. 17 Chemical structure of (a) PPZS and (b) TAPT–DHTA-COFs, and (c) preparation schematic illustration of TAPT-DHTA-COFX@PPZS and TAPTDHTA-COFX@PPZS900. Reproduced with permission from ref. 89 Copyright 2017, The Royal Society of Chemistry. (d) Synthesis schematic illustration of B-N-C capsules. Reproduced with permission from ref. 90 Copyright 2019, Elsevier. | ||
B, N-codoped carbon (B-N-C) was successfully derived via thermal conversion of COFs by using metallic Cu particles as the template core in 2019.90 As shown in the Fig. 17d, Cu(NO3)2 served as both a chemical equilibrium control agent and a catalyst for Schiff-base reaction between melamine and 4-formylphenylboronic acid to synthesize Cu(NO3)2(OH)3@B-N-COFs in one step. Core-capsule-like Cu@B-N-Cs with a thin carbon shell was then obtained by pyrolysis at 900 °C, 1000 °C, and 1100 °C. Hollow B-N-C capsules with ∼15 nm thickness were finally formed after removing the Cu core with HNO3. The resulting B-N-C-1000 showcased a high specific capacitance of 230 F g−1 at 5 A g−1. The storage of B-N-C was mainly dominated by EDLC mechanism via adsorption of electrolyte ions onto the surface of electrode materials.
It has been confirmed that K2CO3 can be reduced to emit K and CO gases by carbon at a high temperature of >627 °C in an inert atmosphere.91 In 2019, O and N co-doped carbons (ONC-T1s) were obtained from K2CO3-assisted pyrolysis of AQ-COF.92 During the carbonization process, K2CO3 inside the channels of AQ-COFs serves not only as an activator to create pores and cause swelling of the carbon layers, but also as a template to prevent the skeleton collapse. Among the resulting ONC-T1s (carbonized at 550, 700 and 850 °C, respectively), ONC-T1-700 and ONC-T1-850 exhibited hierarchical pore size distributions from micropores to mesopores along with ultrahigh BET surface areas of 3451 and 1518 m2 g−1, respectively. In 1 M H2SO4, ONC-T1-700 and ONCT1-850 achieved high specific capacitance values of 768 and 1711 F g−1 at 1 A g−1, respectively, accompanied with a fast charge–discharge rate and superb cyclability.
3.6 Pros and cons of COF-based electrode materials
As summarized above, pristine COFs, hybrid COFs and carbonized COFs have been developed as electrode materials for SCs. Each COF-based electrode material has its pros and cons in terms of chemical synthesis and structural properties.The electrochemical properties of pristine COFs are usually determined from the chemical structure and aggregation morphology. Their SC performances are regulated through the precise predesign of the chemical component, which is superior to other electrode materials. Correspondingly, due to the high requirements for the constructing conditions of COFs, the development of high-performance novel pristine COF electrode materials is also very challenging, usually requiring a reasonable combination of conjugated skeletons and highly active-redox centers.
Hybrid COFs provide a broad development prospect for SC applications of COF-based electrode materials. The SC performance of COFs could be further optimized and the cost could be easily reduced in combination with electrochemically active units such as activated carbon, metal oxides, conductive polymers, etc. However, the analysis of the structure–property relationship and the function-oriented construction of COF-based hybrids are generally difficult.
The morphology of carbonized COFs depends largely on carbonization temperature and the thermal stability of pristine COFs. The purpose of carbonization is to improve the conductivity of COFs and retain the regular pore structure in the meantime. It may also increase the accessibility to active sites and even introduce new sites through templated carbonization. However, the controllability of template-free carbonized COFs is usually poor. Although the templated carbonization can improve the controllability to some extent, the process is usually complex and the cost is usually high, which is not conducive to the commercial application.
4. Conclusions and outlook
COFs are an emerging class of organic porous materials with ordered porous properties. Due to their adjustable redox centers, high specific surface areas, ordered open channels, COFs have drawn great attention in the application of electrode materials. For SC devices, to fabricate electrodes with high specific capacitance, high energy/power density and excellent cycling stability, constructing COFs with high specific surface areas, high conductivity and abundant accessible redox active sites is highly desired. In the past few decades, a series of electrochemically active COFs exhibiting excellent SC performance have been developed, and they have demonstrated tremendous potential for SC applications. In this feature article, design strategies of COF-based electrode materials for SCs, including (i) introducing redox-active moieties into COFs, (ii) improving the accessibility of redox-active moieties, (iii) enhancing the electrical conductivity, (iv) COFs as supports for other electrode materials, and (v) COFs as precursors for porous carbon electrode materials, have been emphatically discussed via summarizing representative research studies. Furthermore, the challenges as well as perspectives in the future are highlighted as follows:(1) Low-cost preparation of COFs on a large scale is one of the key challenges for them to achieve practical applications. Besides the traditional solvothermal synthesis, some high-throughput approaches have been developed, such as liquid crystal-directed synthesis93 and an ultrasound-assisted process.94 Batch preparation of COFs can be achieved through these high-throughput synthesis strategies. Of course, other preparation methods, such as photocatalytic synthesis, electrocatalytic synthesis, and microwave synthesis, are also expected to result in large-scale production of COFs.
(2) The design and synthesis of COFs with high electrochemical activity is a very important research direction, which is related to whether COF-based electrodes can be applied into our daily life and industry applications. Here are three suggested strategies to obtain COFs with high capacitive performance: (i) to develop new building blocks and new linkage types with electrochemical activity; (ii) to chemically anchor the electrochemical active moieties on the backbone of COFs; and (iii) to physically composite electrochemical active centers on the COF supports. Although some COFs with good capacitive performance have been obtained using these strategies, more types of COFs with better performance need to be developed.
(3) The type, number and accessibility of redox active centers are the key to obtain COF materials with high specific capacitance, excellent energy density and cycling stability. Multi-type redox centers need to be developed and exploited. These novel COFs can be constructed via the rational design of small organic molecules with high redox activity as building blocks of COFs.
(4) More preparation strategies for free-standing thin sheets and more available COF sources are needed to meet commercial applications.
(5) Superior capacitive performances for the composites of COF scaffolds and classical electrochemically active species (i.e., conducting polymers and metal oxides/hydroxides) are reasonably expected. Although a few studies on conductive polymer functionalized COFs have been reported, bridging the vast gap in development is of great importance. Metal oxides/hydroxides featuring abundance, low cost, and satisfying electrochemical properties have also been studied as potential electrodes for SCs. However, the relatively low electrical conductivity and inferior structural stability hinder their practical application. Compositing COFs and metal oxides/hydroxides is a feasible strategy to integrate their merits: (i) the porous nature of COFs facilitate good interaction between the electrolyte and electrochemical active metal oxides/hydroxides; (ii) the ultrafine metal oxide/hydroxide nanoparticles dispersed on the surface or dwelled inside the channel of the COFs could be almost fully accessible; (iii) COF carriers with high superiority in designability can overcome the poor conductivity of metal oxides/hydroxides; and (iv) the synergistic effects of electrochemically active centers of COFs and metal oxides/hydroxides lead to a super-excellent capacitive performance.
Author contributions
Rao Tao: writing the original draft, reviewing, and editing. Tianfu Yang and Yan Wang: reviewing, and editing. Jingmin Zhang and Zhengyi Wu: making figures. Li Qiu: reviewing, editing, and supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant 51962036), the Key Project of Natural Science Foundation of Yunnan (grant 202201AS070011), High-Level Talents Introduction in Yunnan Province (grant C619300A025), the Major Science and Technology Project of Precious Metal Materials Genetic Engineering in Yunnan Province (grants 2019ZE001-1 and 202002AB080001), and Yunnan University (grant C176220100005).References
- D. Zhu, G. Xu, M. Barnes, Y. Li, C. P. Tseng, Z. Zhang, J. J. Zhang, Y. Zhu, S. Khalil, M. M. Rahman, R. Verduzco and P. M. Ajayan, Adv. Funct. Mater., 2021, 31, 2100505 CrossRef CAS.
- T. Sun, J. Xie, W. Guo, D. S. Li and Q. Zhang, Adv. Energy Mater., 2020, 10, 1904199 CrossRef CAS.
- Y. Dai, C. Liu, Y. Bai, Q. Kong and H. Pang, Nanotechnol. Rev., 2022, 11, 1005–1046 CrossRef CAS.
- G. Wang, L. Zhang and J. Zhang, Chem. Soc. Rev., 2012, 41, 797–828 RSC.
- D. P. Dubal, N. R. Chodankar, D. H. Kim and P. Gomez-Romero, Chem. Soc. Rev., 2018, 47, 2065–2129 RSC.
- A. González, E. Goikolea, J. A. Barrena and R. Mysyk, Renew. Sust. Energ. Rev., 2016, 58, 1189–1206 CrossRef.
- C. Merlet, C. Péan, B. Rotenberg, P. A. Madden, B. Daffos, P. L. Taberna, P. Simon and M. Salanne, Nat. Commun., 2013, 4, 2701 CrossRef CAS.
- F. Béguin, V. Presser, A. Balducci and E. Frackowiak, Adv. Mater., 2014, 26, 2219–2251 CrossRef.
- Y. Zhai, Y. Dou, D. Zhao, P. F. Fulvio, R. T. Mayes and S. Dai, Adv. Mater., 2011, 23, 4828–4850 CrossRef CAS.
- L. Hao, X. Li and L. Zhi, Adv. Mater., 2013, 25, 3899–3904 CrossRef CAS.
- R. Chen, M. Yu, R. P. Sahu, I. K. Puri and I. Zhitomirsky, Adv. Energy Mater., 2020, 10, 1903848 CrossRef CAS.
- G. Zhang, X. Xiao, B. Li, P. Gu, H. Xue and H. Pang, J. Mater. Chem. A, 2017, 5, 8155–8186 RSC.
- J. Kim, J. Lee, J. You, M. S. Park, M. S. Al Hossain, Y. Yamauchi and J. H. Kim, Mater. Horiz., 2016, 3, 517–535 RSC.
- X. Xiao, L. Zou, H. Pang and Q. Xu, Chem. Soc. Rev., 2020, 49, 301–331 RSC.
- X. Liu, C. F. Liu, S. Xu, T. Cheng, S. Wang, W. Y. Lai and W. Huang, Chem. Soc. Rev., 2022, 51, 3181–3225 RSC.
- D. P. Chatterjee and A. K. Nandi, J. Mater. Chem. A, 2021, 9, 15880–15918 RSC.
- J. Lin, Y. Zhong, L. Tang, L. Wang, M. Yang and H. Xia, Nano Select, 2022, 3, 320–347 CrossRef CAS.
- S. Jin, O. Allam, S. S. Jang and S. W. Lee, InfoMat, 2022, 4, e12277 CrossRef CAS.
- L. Yang and N. Huang, J. Polym. Sci., 2022, 60, 2225–2238 CrossRef CAS.
- M. Sajjad and W. Lu, J. Energy Storage, 2021, 39, 102618 CrossRef.
- K. Zhang, K. O. Kirlikovali, R. S. Varma, Z. Jin, H. W. Jang, O. K. Farha and M. Shokouhimehr, ACS Appl. Mater. Interfaces, 2020, 12, 27821–27852 CrossRef CAS.
- A. P. Cote, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef CAS.
- Z. Chen, K. Wang, X. Hu, P. Shi, Z. Guo and H. Zhan, ACS Appl. Mater. Interfaces, 2020, 13, 1145–1151 CrossRef.
- S. B. Alahakoon, S. D. Diwakara, C. M. Thompson and R. A. Smaldone, Chem. Soc. Rev., 2020, 49, 1344–1356 RSC.
- X. Guan, F. Chen, S. Qiu and Q. Fang, Angew. Chem., Int. Ed., 2022, 62, e202213203 Search PubMed.
- O. M. Yaghi, M. J. Kalmutzki and C. S. Diercks, Introduction to reticular chemistry: metal-organic frameworks and covalent organic frameworks, John Wiley & Sons, 2019 Search PubMed.
- Y. Jin, C. Yu, R. J. Denman and W. Zhang, Chem. Soc. Rev., 2013, 42, 6634–6654 RSC.
- X. Chen, K. Geng, R. Liu, K. T. Tan, Y. Gong, Z. Li, S. Tao, Q. Jiang and D. Jiang, Angew. Chem., Int. Ed., 2020, 59, 5050–5091 CrossRef CAS.
- S. Kandambeth, K. Dey and R. Banerjee, J. Am. Chem. Soc., 2019, 141, 1807–1822 CrossRef CAS.
- Y. Su, Y. Wan, H. Xu, K. Otake, X. Tang, L. Huang, S. Kitagawa and C. Gu, J. Am. Chem. Soc., 2020, 142, 13316–13321 CrossRef CAS.
- H. L. Qian, F. L. Meng, C. X. Yang and X. P. Yan, Angew. Chem., Int. Ed., 2020, 59, 17607–17613 CrossRef CAS.
- B. Zhang, M. Wei, H. Mao, X. Pei, S. A. Alshmimri, J. A. Reimer and O. M. Yaghi, J. Am. Chem. Soc., 2018, 140, 12715–12719 CrossRef CAS.
- X. Li, K. Kawai, M. Fujitsuka and Y. Osakada, Surf. Interfaces, 2021, 25, 101249 CrossRef CAS.
- N. Keller and T. Bein, Chem. Soc. Rev., 2021, 50, 1813–1845 RSC.
- M. Wu and Y. Yang, Chin. Chem. Lett., 2017, 28, 1135–1143 CrossRef CAS.
- X. Liu, D. Huang, C. Lai, G. Zeng, L. Qin, H. Wang, H. Yi, B. Li, S. Liu and M. Zhang, Chem. Soc. Rev., 2019, 48, 5266–5302 RSC.
- C. R. DeBlase, K. E. Silberstein, T. T. Truong, H. D. Abruña and W. R. Dichtel, J. Am. Chem. Soc., 2013, 135, 16821–16824 CrossRef CAS.
- J. Li, X. Jing, Q. Li, S. Li, X. Gao, X. Feng and B. J. C. S. R. Wang, Chem. Soc. Rev., 2020, 49, 3565–3604 RSC.
- M. Li, J. Liu, T. Zhang, X. Song, W. Chen and L. Chen, Small, 2021, 17, e2005073 CrossRef.
- S. Wang, Y. Guo, F. Wang, S. Zhou, T. Zeng and Y. Dong, New Carbon Mater., 2022, 37, 109–135 CrossRef CAS.
- C. Gao, J. Huang, Y. Xiao, G. Zhang, C. Dai, Z. Li, Y. Zhao, L. Jiang and L. Qu, Nat. Commun., 2021, 12, 2647 CrossRef CAS.
- J. C. Russell, V. A. Posey, J. Gray, R. May, D. A. Reed, H. Zhang, L. E. Marbella, M. L. Steigerwald, Y. Yang, X. Roy, C. Nuckolls and S. R. Peurifoy, Nat. Mater., 2021, 20, 1136–1141 CrossRef CAS.
- X. Chen, R. Paul and L. Dai, Natl. Sci. Rev., 2017, 4, 453–489 CrossRef CAS.
- Q. Wu, T. He, Y. Zhang, J. Zhang, Z. Wang, Y. Liu, L. Zhao, Y. Wu and F. Ran, J. Mater. Chem. A, 2021, 9, 24094–24147 RSC.
- B. Häupler, A. Wild and U. S. Schubert, Adv. Energy Mater., 2015, 5, 1402034 CrossRef.
- Y. Liang, P. Zhang and J. Chen, Chem. Sci., 2013, 4, 1330–1337 RSC.
- P. Poizot, J. Gaubicher, S. Renault, L. Dubois, Y. Liang and Y. Yao, Chem. Rev., 2020, 120, 6490–6557 CrossRef CAS PubMed.
- S. Chandra, D. Roy Chowdhury, M. Addicoat, T. Heine, A. Paul and R. Banerjee, Chem. Mater., 2017, 29, 2074–2080 CrossRef CAS.
- M. Li, J. Liu, Y. Li, G. Xing, X. Yu, C. Peng and L. Chen, CCS Chem., 2020, 2, 696–706 Search PubMed.
- A. Halder, M. Ghosh, M. A. Khayum, S. Bera, M. Addicoat, H. S. Sasmal, S. Karak, S. Kurungot and R. Banerjee, J. Am. Chem. Soc., 2018, 140, 10941–10945 CrossRef CAS.
- P. Bhanja, K. Bhunia, S. K. Das, D. Pradhan, R. Kimura, Y. Hijikata, S. Irle and A. Bhaumik, ChemSusChem, 2017, 10, 921–929 CrossRef CAS.
- A. F. M. El-Mahdy, Y. H. Hung, T. H. Mansoure, H. H. Yu, T. Chen and S. W. Kuo, Chem. – Asian J., 2019, 14, 1429–1435 CrossRef CAS.
- A. M. Khattak, Z. A. Ghazi, B. Liang, N. A. Khan, A. Iqbal, L. Li and Z. Tang, J. Mater. Chem. A, 2016, 4, 16312–16317 RSC.
- S. Haldar, R. Kushwaha, R. Maity and R. Vaidhyanathan, ACS Materials Lett., 2019, 1, 490–497 CrossRef CAS.
- L. Li, F. Lu, R. Xue, B. Ma, Q. Li, N. Wu, H. Liu, W. Yao, H. Guo and W. Yang, ACS Appl. Mater. Interfaces, 2019, 11, 26355–26363 CrossRef CAS.
- R. Xue, Y. Zheng, D. Qian, D. Xu, Y. Liu, S. Huang and G. Yang, Mater. Lett., 2022, 308, 131229 CrossRef CAS.
- S. Xiong, J. Liu, Y. Wang, X. Wang, J. Chu, R. Zhang, M. Gong and B. Wu, J. Appl. Polym. Sci., 2022, 139, 51510 CrossRef CAS.
- M. Sajjad, R. Tao and L. Qiu, J. Mater. Sci.: Mater. Electron., 2021, 32, 1602–1615 CrossRef CAS.
- K. Kang, Z. Wu, M. Zhao, Z. Li, Y. Ma, J. Zhang, Y. Wang, M. Sajjad, R. Tao and L. Qiu, Chem. Commun., 2022, 58, 3649–3652 RSC.
- R. Tao, X. Shen, Y. Hu, K. Kang, Y. Zheng, S. Luo, S. Yang, W. Li, S. Lu, Y. Jin, L. Qiu and W. Zhang, Small, 2020, 16, e1906005 CrossRef.
- Y. Liu, A. Dikhtiarenko, N. Xu, J. Sun, J. Tang, K. Wang, B. Xu, Q. Tong, H. J. Heeres, S. He, J. Gascon and Y. Fan, Chem. – Eur. J., 2020, 26, 12134–12139 CrossRef CAS.
- T. Li, X. Yan, Y. Liu, W. Zhang, Q. Fu, H. Zhu, Z. Li and Z. Gu, Polym. Chem., 2020, 11, 47–52 RSC.
- F. Xu, H. Xu, X. Chen, D. Wu, Y. Wu, H. Liu, C. Gu, R. Fu and D. Jiang, Angew. Chem., Int. Ed., 2015, 54, 6814–6818 CrossRef CAS.
- X. Zhang, Z. Xiao, X. Liu, P. Mei and Y. Yang, Renew. Sust. Energ. Rev., 2021, 147, 111247 CrossRef CAS.
- J. Lee, S. Hong, Y. Heo, H. Kang and M. Kim, Dalton Trans., 2021, 50, 14081–14090 RSC.
- C. R. DeBlase, K. Hernández-Burgos, K. E. Silberstein, G. G. Rodríguez-Calero, R. P. Bisbey, H. D. Abruña and W. R. J. A. N. Dichtel, ACS Nano, 2015, 9, 3178–3183 CrossRef CAS.
- Z. Zha, L. Xu, Z. Wang, X. Li, Q. Pan, P. Hu and S. Lei, ACS Appl. Mater. Interfaces, 2015, 7, 17837–17843 CrossRef CAS.
- M. A. Khayum, V. Vijayakumar, S. Karak, S. Kandambeth, M. Bhadra, K. Suresh, N. Acharambath, S. Kurungot and R. Banerjee, ACS Appl. Mater. Interfaces, 2018, 10, 28139–28146 CrossRef.
- S. Chen, W. Xing, J. Duan, X. Hu and S. Z. Qiao, J. Mater. Chem. A, 2013, 1, 2941–2954 RSC.
- C. R. Mulzer, L. Shen, R. P. Bisbey, J. R. McKone, N. Zhang, H. D. Abruna and W. R. Dichtel, ACS Cent. Sci., 2016, 2, 667–673 CrossRef CAS.
- Y. Wu, D. Yan, Z. Zhang, M. M. Matsushita and K. Awaga, ACS Appl. Mater. Interfaces, 2019, 11, 7661–7665 CrossRef CAS.
- N. An, Z. Guo, J. Xin, Y. He, K. Xie, D. Sun, X. Dong and Z. Hu, J. Mater. Chem. A, 2021, 9, 16824–16833 RSC.
- M. Sajjad, R. Tao, K. Kang, S. Luo and L. Qiu, ACS Appl. Energy Mater., 2021, 4, 828–838 CrossRef CAS.
- M. Sajjad, K. Kang, Z. Wu, Y. Ma, R. Tao and L. Qiu, J. Energy Storage, 2021, 40, 102772 CrossRef.
- T. Li, W. Zhang, Y. Liu, Y. Li, C. Cheng, H. Zhu, X. Yan, Z. Li and Z. Gu, J. Mater. Chem. A, 2019, 7, 19676–19681 RSC.
- R. Tao, X. Ma, X. Wei, Y. Jin, L. Qiu and W. Zhang, J. Mater. Chem. A, 2020, 8, 17360–17391 RSC.
- X. Zhao, M. Sajjad, Y. Zheng, M. Zhao, Z. Li, Z. Wu, K. Kang and L. Qiu, Carbon, 2021, 182, 144–154 CrossRef CAS.
- J. P. Paraknowitsch and A. Thomas, Energy Environ. Sci., 2013, 6, 2839–2855 RSC.
- Y. Deng, Y. Xie, K. Zou and X. Ji, J. Mater. Chem. A, 2016, 4, 1144–1173 RSC.
- L. Miao, H. Duan, M. Liu, W. Lu, D. Zhu, T. Chen, L. Li and L. Gan, Chem. Eng. J., 2017, 317, 651–659 CrossRef CAS.
- Q. Xu, Y. Tang, X. Zhang, Y. Oshima, Q. Chen and D. Jiang, Adv. Mater., 2018, 30, e1706330 CrossRef.
- X. Hu, Y. Long, M. Fan, M. Yuan, H. Zhao, J. Ma and Z. Dong, Appl. Catal., B, 2019, 244, 25–35 CrossRef CAS.
- J. K. Sun and Q. Xu, Energy Environ. Sci., 2014, 7, 2071–2100 RSC.
- X. Zhang, G. Zhu, M. Wang, J. Li, T. Lu and L. Pan, Carbon, 2017, 116, 686–694 CrossRef CAS.
- X. Liu, L. Zhou, Y. Zhao, L. Bian, X. Feng and Q. Pu, ACS Appl. Mater. Interfaces, 2013, 5, 10280–10287 CrossRef CAS.
- S. Ding, J. Gao, Q. Wang, Y. Zhang, W. Song, C. Su and W. Wang, J. Am. Chem. Soc., 2011, 133, 19816–19822 CrossRef CAS.
- G. Kim, J. Yang, N. Nakashima and T. Shiraki, Chem. – Eur. J., 2017, 23, 17504–17510 CrossRef CAS.
- F. Hu, J. Wang, S. Hu, L. Li, W. Shao, J. Qiu, Z. Lei, W. Deng and X. Jian, ACS Appl. Mater. Interfaces, 2017, 9, 31940–31949 CrossRef CAS.
- Q. Xu, Y. Tang, L. Zhai, Q. Chen and D. Jiang, Chem. Commun., 2017, 53, 11690–11693 RSC.
- Z. Zhou, X. Zhang, L. Xing, J. Liu, A. Kong and Y. Shan, Electrochim. Acta, 2019, 298, 210–218 CrossRef CAS.
- D. W. McKee and D. Chatterji, Carbon, 1978, 16, 53–57 CrossRef CAS.
- D. Yan, Y. Wu, R. Kitaura and K. Awaga, J. Mater. Chem. A, 2019, 7, 26829–26837 RSC.
- F. Tan, Y. Zheng, Z. Zhou, H. Wang, X. Dong, J. Yang, Z. Ou, H. Qi, W. Liu, Z. Zheng and X. Chen, CCS Chem., 2022, 4, 3751–3761 CrossRef CAS.
- W. Zhao, P. Yan, B. Li, M. Bahri, L. Liu, X. Zhou, R. Clowes, N. D. Browning, Y. Wu, J. W. Ward and A. I. Cooper, J. Am. Chem. Soc., 2022, 144, 9902–9909 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |