
Lignin C–C bond cleavage induced by consecutive two-photon excitation of a metal-free photocatalyst†
Pengju
Li
,
Rong
Liu
,
Zijian
Zhao
,
Fushuang
Niu
and
Ke
Hu
 *
*
Department of Chemistry and Shanghai Key Laboratory of Molecular Catalysis and Innovative Materials, Fudan University, 220 Handan Road, Shanghai 200433, P. R. China. E-mail: khu@fudan.edu.cn
First published on 17th January 2023
Abstract
Photocatalytic lignin valorization has caught widespread attention; yet the reaction systems often employ noble metal complexes, hydrogen atom transfer (HAT) agents, and/or sacrificial electron donors/acceptors that do not comply with atom economy or environmental friendliness. Herein, we discovered that N-phenylphenothiazine (PTH) as a metal-free photocatalyst induced the cleavage of the lignin Cα–Cβ bond under ambient conditions free of those additional agents with a high yield and selectivity toward benzoic acid. Transient spectroscopic investigations revealed that the energy-demanding Cα–Cβ bond cleavage was induced by the potent oxidant, 2PTH˙+*, that was derived from consecutive two-photon excitation of PTH.
Lignin, rich in aromatic carbon units, is a renewable energy source to replace the use of fossil fuels on a large scale. Yet breaking the profuse C–O and C–C bonds selectively into small value-added compounds is challenging. Currently, various strategies have been reported for the valorization of lignin, including pyrolysis, gasification, acid or base treatment, hydrogenolysis, etc.1–5 These methods often require harsh reaction conditions such as high temperature, high pressure, corrosive solvents, etc., so better methods are urgently needed. Photocatalysis, typically considered as a green and mild method for organic redox synthesis, has gained increasing popularity for biomass valorization.6–14 However, cleavage of the high energy C–C or C–O bonds requires potent photooxidants or photoreductants, among which noble-metal-containing ruthenium or iridium coordination compounds are often used.15–19 Furthermore, hydrogen atom transfer (HAT) agents and sacrificial electron or hole donors are often used as necessary additives in an inert atmosphere to complete the catalytic cycle.15,16,18,20–25 These additives generate unproductive waste to the extent that the value of the obtained small aromatic compounds may not offset it. Therefore, it is highly desirable to find a photocatalytic strategy that can selectively cleave lignin C–C bonds into high-value-added aromatic compounds in an ambient environment without metal-containing photocatalysts, sacrificial reagents or other additives.
Herein, we report that N-phenylphenothiazine (PTH) as a metal-free photocatalyst surprisingly induces the selective cleavage of lignin Cα–Cβ bonds under ambient conditions without any other additives. Though a mild photooxidant in its singlet excited state, the doublet excited state of the radical cation (2PTH˙+*) is a potent photooxidant to induce the oxidative cleavage of the Cα–Cβ bond in lignin models through consecutive two-photon excitation (Scheme 1). Two-color two-pulse laser flash photolysis experiments revealed a rapid subnanosecond electron transfer process likely through a static quenching mechanism.26–28 A wide scope of major lignin units were successfully cleaved in high yields through simple steady state illumination of PTH, demonstrating the potential utility of this simple, commercially-available, metal-free photocatalyst for lignin valorization in an environmentally-friendly manner.
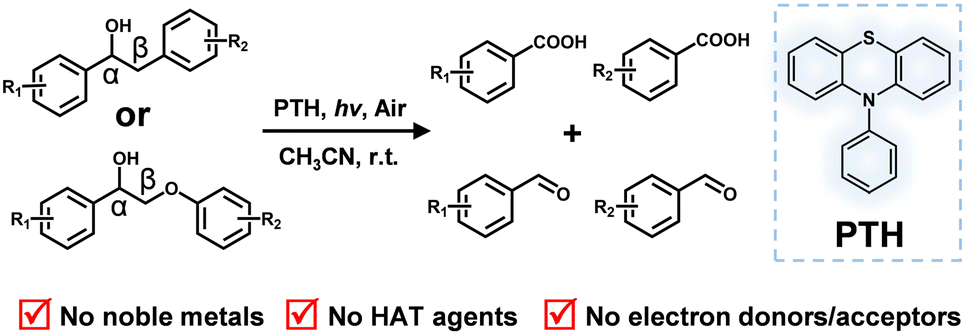 | ||
| Scheme 1 Photocatalytic cleavage of the Cα–Cβ bond of lignin model substrates. | ||
PTH showed mainly UV absorption in the ground state (Fig. 1a). Electrochemical oxidation of PTH in CH3CN resulted in PTH˙+ formation showing three distinctive visible to near IR absorption bands (λmax = 514 nm, 772 nm, and 864 nm) (Fig. S1, ESI†) that were consistent with the literature.29 PTH˙+ was air-stable as the absorption was retained upon solution exposure to the air. When the PTH solution was irradiated with 390 nm light in ambient air, a similar absorption change attributed to the formation of PTH˙+ was also observed (Fig. S2, ESI†). Molecular oxygen dissolved in CH3CN was presumably the electron acceptor responsible for quenching the PTH excited state (Fig. S3, ESI†).30
 | ||
| Fig. 1 (a) The UV-vis-NIR absorption spectra of PTH and PTH˙+ in CH3CN. (b) Full TA spectra of PTH obtained after 355 nm pulsed-laser excitation in the presence of 100 mM 1a in aerated CH3CN at the indicated time delays. (c) Absorption difference spectra of PTH in the presence of 1000 mM 1a in a two-color two-pulse TA experiment at 500 ns time delay before the second laser pulse (blue) and after the second laser pulse (green). Inset: Transient kinetic data monitored at the probe wavelength of 510 nm for PTH˙+ with a two-color two-pulse laser excitation sequence shown in the laser pulse scheme above (kinetics in green) or with only the first 355 nm pulsed-laser excitation (kinetics in blue). (d) Stern–Volmer-type plot based on the two-color two-pulse TA experiment at different concentrations of 1a. The subscript PP stands for pump-probe and PPP pump–pump–probe. The inset shows the meaning of ΔAbsPP and ΔAbsPPP. | ||
Cyclic voltammetry (CV) was performed to evaluate the redox properties of PTH and the lignin model substrate 1,2-diphenylethanol (1a). PTH showed a reversible redox wave at E°(PTH˙+/0) = 0.92 V vs. NHE (Fig. S4, ESI†) and an irreversible peak at Ep/2 = 1.59 V attributed to PTH2+/˙+. The oxidation of 1a was also irreversible with Ep/2 = 1.9 V, which was more positive than Ep/2(PTH2+/˙+). CV of the PTH and 1a mixture did not show any catalytic current for the oxidation of 1a as expected due to unfavourable thermodynamics (Fig. S4, ESI†).
A steady-state fluorescence quenching experiment was carried out to test whether the PTH excited state could oxidize 1a. Unsurprisingly, titration of 1a up to 100 mM did not lead to any measurable PTH fluorescent quenching (Fig. S5, ESI†). The excited state of PTH apparently lacked the oxidative power to initiate the oxidation of 1a.
Nanosecond transient absorption (TA) spectroscopy was employed to explore the electron transfer dynamics between PTH and 1a. Fig. 1b shows the full TA spectra of PTH in the presence of 1a in aerated CH3CN after nanosecond pulsed-laser excitation (λex = 355 nm). The positive absorption bands from the visible to near infrared region corresponded to the absorption of PTH˙+, which was consistent with the oxidation of PTH from both the spectroelectrochemistry and photolysis data (Fig. S2 and S6, ESI†).
The visible to NIR absorption of PTH˙+ allowed it to absorb a second photon to reach the doublet excited state, PTH˙+*. Prior studies have shown that PTH˙+* is a potent photooxidant with E°(PTH˙+*/0) = 2.31 V.27 It was of great interest to study the PTH˙+* reactivity towards the degradation of lignin models with redox potential around 2.0 V. Nanosecond two-color two-pulse TA experiments were carried out to investigate the quenching of PTH˙+* by 1a. The first laser pulse (λex = 355 nm) generated PTH* that subsequently transferred an electron to O2 to form the long-lived PTH˙+ as the spectral simulation showed in Fig. 1b. The second laser pulse (λex = 532 nm) was delayed by 500 ns relative to the first and selectively excited the transiently formed PTH˙+. The second laser pulse resulted in significant bleaching of the PTH˙+ absorption signal monitored at 510 nm in the presence of 1000 mM 1a (Fig. 1c inset). Absorption difference spectra of PTH recorded from 390 nm – 950 nm in oxygen-saturated CH3CN after two pulsed-laser excitations are displayed in Fig. 1c. Upon titration of increasing concentrations of 1a, the bleach signal of delta absorbance monitored at 510 nm gradually increased right after the second laser pulse excitation. For the control sample that was in the absence of 1a, the second laser pulse did not result in the bleaching of the PTH˙+ absorption (Fig. S7, ESI†). A Stern–Volmer-type plot for the quenching of PTH˙+* by 1a was obtained by plotting ΔAbsPP/ΔAbsPPP as a function of increasing concentrations of 1a (Fig. 1d). The plot showed a linear regression, from which the slope was equal to 0.15 M−1. Considering that the lifetime of PTH˙+* was only 36 ps,29 it was highly unlikely that the reductive quenching approached through bimolecular diffusion, but rather ground state association of PTH˙+* and 1a with the equilibrium constant of 0.15 M−1.
Steady-state photolysis of PTH and 1a was carried out in CH3CN under ambient air conditions. Deviations from the standard condition along with the reaction conversions and product yields are also listed in Table 1. First, we optimized the photocatalyst PTH concentration and chose 15 mol% as the standard reaction concentration since concentration as high as 20 mol% did not result in a significant increase in the product yield (Table 1, entries 1–3). Three major products were detected including benzoic acid (1b), benzaldehyde (1c), and diphenylethanone (1d). The formation of 1b and 1c clearly demonstrated that PTH under light promoted oxidative cleavage of the Cα–Cβ bond in appreciable yields (entry 1). Note that 100% selective Cα–Cβ bond cleavage in 1a would theoretically result in 200% yield of 1b and 1c combined. Nevertheless, the combined yield of 96% was still among the average of lignin Cα–Cβ bond cleavage in the literature;18,31 yet our photocatalytic system only contained PTH photocatalyst without other HAT or sacrificial agents. It is also noteworthy that when a white light LED source was added in combination with the 390 nm light, the rate of product formation was significantly faster than using 390 nm light irradiation alone (Fig. S8, ESI†). Greater visible light absorption by PTH˙+ was indeed helpful for the generation of PTH˙+*.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Deviation from standard conditions | Con. (%) | Yield of products (%) | ||
| 1b | 1c | 1d | |||
| Standard reaction conditions: 0.1 mmol 1a, 15 mol% PTH, 2 mL CH3CN, 390 nm LED (15 mW·cm−2), air, 48 h, room temperature. Yield of 1b, 1c, 1d = moles of product 1b, 1c, 1d formed/moles of 1a × 100%. n.r. = no reaction. n.d. = not detected. All yields were determined by HPLC analysis. Electron, hole, or radical scavengers: 200 mM AgNO3, 200 mM BQ, 200 mM TEOA, 200 mM TEMPO. | |||||
| 1 | None | 96 | 82 | 14 | 23 |
| 2 | 10 mol% PTH | 93 | 66 | 16 | 24 |
| 3 | 20 mol% PTH | 96 | 87 | 16 | 24 |
| 4 | Ar instead of air | n.r. | n.d | n.d. | n.d. |
| 5 | In the dark | n.r. | n.d | n.d. | n.d. |
| 6 | No PTH | 33 | 12 | 8 | 12 |
| 7 | 2,6-Lutidine (4 equiv.) | 30 | 5 | 1 | 5 |
| 8 | HCOOH (4 equiv.) | 85 | 43 | 18 | 22 |
| 9 | 10% water | 76 | 31 | 16 | 28 |
| 10 | AgNO3 | 71 | 27 | 11 | 39 |
| 11 | BQ | 66 | 11 | 18 | 36 |
| 12 | TEOA | 24 | 1 | 1 | 1 |
| 13 | TEMPO | 25 | 1 | 3 | 1 |
| 14 | MeOH instead of CH3CN | 36 | 5 | 3 | 12 |
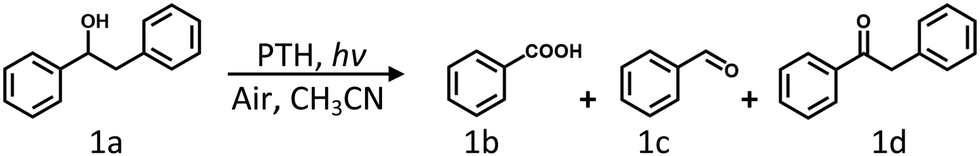
When the ambient air environment was replaced by Ar, there was almost no conversion of 1a and no product was detected (entry 4). The solution dissolved molecular oxygen was the default electron acceptor for PTH*, while PTH* by itself was unable to perform photoredox chemistry towards 1a as suggested by the previous fluorescence quenching experiment. When the reaction was placed in the dark, no conversion of 1a was found, which eliminated the possibility of thermoreactions (entry 5). Conversion of 1a in the absence of PTH was observed but in a much slower rate (entry 6). The observation was not unprecedented though when lignin substrates were exposed to UV light.32,33 The above variations of reaction conditions confirmed that light, photocatalyst, and O2 were all essential factors to achieve efficient Cα–Cβ bond cleavage for 1a. It is noteworthy that the addition of other environmental factors such as acid, base, or water dramatically suppressed the yields of Cα–Cβ bond cleavage products (entries 7–9). As we emphasized, the PTH photocatalyzed Cα–Cβ bond cleavage for 1a proceeded under mild conditions without the need for additional additives.
The scope of the lignin model substrates that included β–1 and β–O–4 linkages (2a–6a) was also investigated for PTH photocatalyzed Cα–Cβ bond cleavage reactions. The oxidations of 2a–6a were all irreversible with Ep/2 spanning between 1.59 V and 1.92 V (Fig. S9–S13, ESI†), which were well below E°(PTH˙+*/0). The photocatalytic reaction conversions were all close to 100% across the substrate scope under the standard reaction conditions (Table S1, ESI†). Substituents to the aromatic rings of 1a resulted in comparatively high yields and selectivity as well (Table S1, entries 2 and 3, ESI†). Similar to the β–1 model, the Cα–Cβ bond of the β–O–4 linkage model was also cleaved with moderate yields under the standard reaction conditions (Table S1, entries 4 and 5, ESI†).
Although 1a conversion approached unity after 48 hours of photocatalysis, the uncleaved product 1d was still present in a significant amount. Therefore, we set out to monitor the product yields by sampling the reaction solution every 24 hours (Fig. 2). The conversion of 1a climbed close to unity within 48 hours and stayed almost invariant in the next 120 hours, but the ratio of the product yields kept changing over time. Initially, all three products were present but the yields of 1c and the uncleaved product 1d gradually decreased to almost 0 while that of 1b increased to almost the only product. The yields of products over time suggested that 1d might be the kinetically competitive product but was subject to further photoredox induced cleavage to the final thermodynamically stable 1b. The long-time photolysis result was encouraging in terms of selectively obtaining high purity of the Cα–Cβ-bond-cleaved photoproduct.
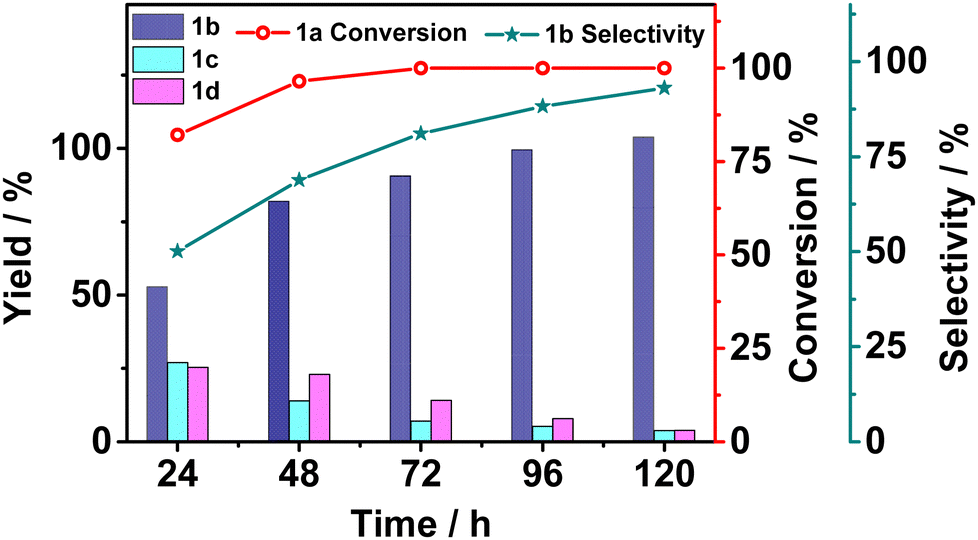 | ||
| Fig. 2 The product yields from photocatalytic Cα–Cβ bond cleavage of 1a over the elapsed time. 1b selectivity = moles of product 1b/moles of product (1b + 1c + 1d) × 100%. | ||
Electron, hole, and radical scavengers including AgNO3, 1,4-benzoquinone (BQ), triethanolamine (TEOA), and 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) were used to test possible reactive intermediates for elucidation of the reaction mechanism (Table 1, entries 10–13). The addition of TEOA significantly decreased the product yields, suggesting that PTH˙+* was a necessity. The formation of O2˙![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) − was also critical to the reaction conversion because BQ as a superoxide scavenger and AgNO3 as a competitive electron acceptor to O2 could both significantly decrease the product yields. The addition of TEMPO dramatically reduced the efficiency of the reaction, indicating that the catalytic process involved free radical intermediates.
− was also critical to the reaction conversion because BQ as a superoxide scavenger and AgNO3 as a competitive electron acceptor to O2 could both significantly decrease the product yields. The addition of TEMPO dramatically reduced the efficiency of the reaction, indicating that the catalytic process involved free radical intermediates.
Based on the above control tests and transient spectroscopic data, the following photocatalytic mechanism was proposed for lignin Cα–Cβ bond cleavage (Scheme 2). Under the first photon irradiation, the photoexcited PTH was oxidatively quenched by O2 with PTH˙+ and O2˙− as the primary photoproducts. PTH˙+ was insufficient to oxidize 1a, but with its absorption of the second photon, the doublet excited state PTH˙+* was a super-oxidant (E°(PTH˙+*/0) > 2.3 V) that abstracted an electron to generate a Cβ-centered radical and released a proton (H+). Subsequently, the Cβ-centered radical of 1a reacted with O2˙− and H+ to produce a peroxide intermediate. Next, the unstable peroxide intermediate induced cleavage of the Cα–Cβ bond to form benzaldehyde and water.34 Control experiments showed that benzaldehyde was further oxidized to produce benzoic acid.
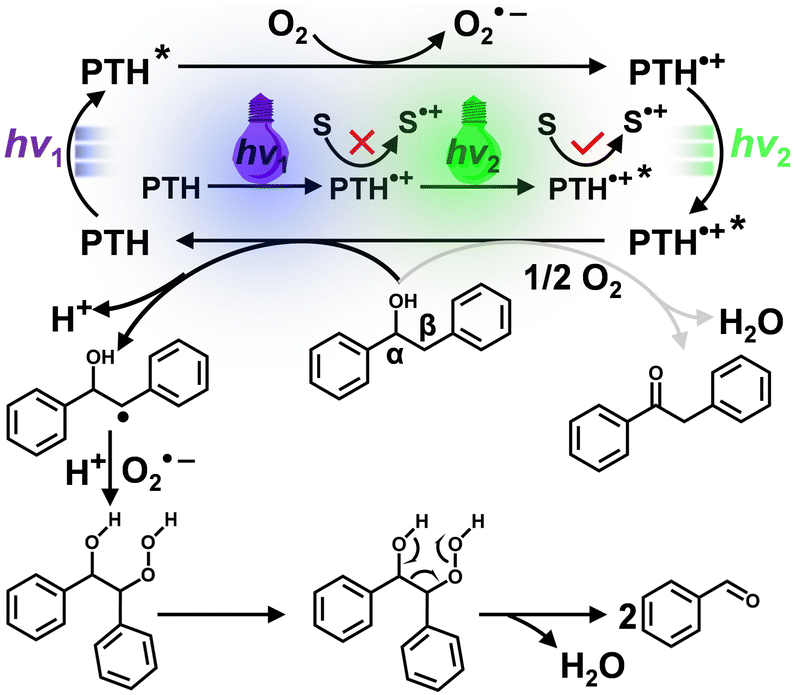 | ||
| Scheme 2 Proposed mechanism for Cα–Cβ bond cleavage of 1avia consecutive photo-excitation of the PTH photocatalyst. | ||
In summary, we proposed a new photocatalytic strategy where a commercially available, metal-free small organic photocatalyst, PTH, can be used to valorize lignin under ambient air conditions through photocatalysis. The doublet excited state PTH˙+* formed from consecutive two-photon excitation was a potent oxidant to activate the C(sp3)–H bond of lignin models possibly through ground state association with the substrate and subsequently induced Cα–Cβ bond cleavage. The conversion of a variety of major lignin models was close to unity with a benzoic acid moiety as the major photocleaved product. The reaction conditions were mild without any additional HAT agents and sacrificial electron/hole donors. Therefore, the photocatalytic strategy is a green approach to value-added carbon sources from lignin waste.
This work is supported by the National Natural Science Foundation of China (22173022) and the Natural Science Foundation of Shanghai (21ZR1404400, 19DZ2270100).
Conflicts of interest
There are no conflicts to declare.Notes and references
- Y. Liao, S.-F. Koelewijn, G. Van den Bossche, J. Van Aelst, S. Van den Bosch, T. Renders, K. Navare, T. Nicolaï, K. Van Aelst and M. Maesen, Science, 2020, 367, 1385–1390 CrossRef CAS PubMed.
- W.-J. Liu, W.-W. Li, H. Jiang and H.-Q. Yu, Chem. Rev., 2017, 117, 6367–6398 CrossRef CAS PubMed.
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS PubMed.
- Q. Meng, J. Yan, R. Wu, H. Liu, Y. Sun, N. Wu, J. Xiang, L. Zheng, J. Zhang and B. Han, Nat. Commun., 2021, 12, 1–12 CrossRef PubMed.
- Z. Liu, H. Li, X. Gao, X. Guo, S. Wang, Y. Fang and G. Song, Nat. Commun., 2022, 13, 1–11 Search PubMed.
- X. Wu, X. Fan, S. Xie, J. Lin, J. Cheng, Q. Zhang, L. Chen and Y. Wang, Nat. Catal., 2018, 1, 772–780 CrossRef CAS.
- N. Luo, T. Montini, J. Zhang, P. Fornasiero, E. Fonda, T. Hou, W. Nie, J. Lu, J. Liu and M. Heggen, Nat. Energy, 2019, 4, 575–584 CrossRef CAS.
- H. Liu, H. Li, J. Lu, S. Zeng, M. Wang, N. Luo, S. Xu and F. Wang, ACS Catal., 2018, 8, 4761–4771 CrossRef CAS.
- X. Wu, S. Xie, C. Liu, C. Zhou, J. Lin, J. Kang, Q. Zhang, Z. Wang and Y. Wang, ACS Catal., 2019, 9, 8443–8451 CrossRef CAS.
- G. Han, T. Yan, W. Zhang, Y. C. Zhang, D. Y. Lee, Z. Cao and Y. Sun, ACS Catal., 2019, 9, 11341–11349 CrossRef CAS.
- X. Wu, N. Luo, S. Xie, H. Zhang, Q. Zhang, F. Wang and Y. Wang, Chem. Soc. Rev., 2020, 49, 6198–6223 RSC.
- S.-H. Li, S. Liu, J. C. Colmenares and Y.-J. Xu, Green Chem., 2016, 18, 594–607 RSC.
- M.-Y. Qi, M. Conte, M. Anpo, Z.-R. Tang and Y.-J. Xu, Chem. Rev., 2021, 121, 13051–13085 CrossRef CAS PubMed.
- M.-Y. Qi, M. Conte, Z.-R. Tang and Y.-J. Xu, ACS Nano, 2022, 16, 17444–17453 CrossRef CAS PubMed.
- K. Chen, J. Schwarz, T. A. Karl, A. Chatterjee and B. König, Chem. Commun., 2019, 55, 13144–13147 RSC.
- G. Magallanes, M. D. Karkas, I. Bosque, S. Lee, S. Maldonado and C. R. Stephenson, ACS Catal., 2019, 9, 2252–2260 CrossRef CAS.
- Q. Zhu and D. G. Nocera, ACS Catal., 2021, 11, 14181–14187 CrossRef CAS.
- E. Ota, H. Wang, N. L. Frye and R. R. Knowles, J. Am. Chem. Soc., 2019, 141, 1457–1462 CrossRef CAS PubMed.
- S. Li, S. Kim, A. H. Davis, J. Zhuang, E. W. Shuler, D. Willinger, J.-J. Lee, W. Zheng, B. D. Sherman and C. G. Yoo, ACS Catal., 2021, 11, 3771–3781 CrossRef CAS.
- I. Bosque, G. Magallanes, M. Rigoulet, M. D. Kärkäs and C. R. Stephenson, ACS Cent. Sci., 2017, 3, 621–628 CrossRef CAS PubMed.
- S. T. Nguyen, P. R. Murray and R. R. Knowles, ACS Catal., 2019, 10, 800–805 CrossRef.
- J. D. Nguyen, B. S. Matsuura and C. R. Stephenson, J. Am. Chem. Soc., 2014, 136, 1218–1221 CrossRef CAS PubMed.
- C. Yang, M. D. Karkas, G. Magallanes, K. Chan and C. R. Stephenson, Org. Lett., 2020, 22, 8082–8085 CrossRef CAS PubMed.
- H. Li and O. S. Wenger, Angew. Chem., Int. Ed., 2022, 61, e202110491 CAS.
- S. Li, Z. Hao, K. Wang, M. Tong, Y. Yang, H. Jiang, Y. Xiao and F. Zhang, Chem. Commun., 2020, 56, 11243–11246 RSC.
- F. Glaser and O. S. Wenger, JACS Au, 2022, 2, 1488–1503 CrossRef CAS PubMed.
- P. Li, A. M. Deetz, J. Hu, G. J. Meyer and K. Hu, J. Am. Chem. Soc., 2022, 144, 17604–17610 CrossRef CAS PubMed.
- S. Wu, J. Žurauskas, M. Domański, P. S. Hitzfeld, V. Butera, D. J. Scott, J. Rehbein, A. Kumar, E. Thyrhaug and J. Hauer, Org. Chem. Front., 2021, 8, 1132–1142 RSC.
- J. A. Christensen, B. T. Phelan, S. Chaudhuri, A. Acharya, V. S. Batista and M. R. Wasielewski, J. Am. Chem. Soc., 2018, 140, 5290–5299 CrossRef CAS PubMed.
- S. Alkaitis, G. Beck and M. Grätzel, J. Am. Chem. Soc., 1975, 97, 5723–5729 CrossRef CAS.
- Y. Wang, Y. Liu, J. He and Y. Zhang, Sci. Bull., 2019, 64, 1658–1666 CrossRef CAS PubMed.
- Y. Kang, X. Yao, Y. Yang, J. Xu, J. Xin, Q. Zhou, M. Li, X. Lu and S. Zhang, Green Chem., 2021, 23, 5524–5534 RSC.
- A. Temiz, U. C. Yildiz, I. Aydin, M. Eikenes, G. Alfredsen and G. Çolakoglu, Appl. Surf. Sci., 2005, 250, 35–42 CrossRef CAS.
- T. Hou, N. Luo, H. Li, M. Heggen, J. Lu, Y. Wang and F. Wang, ACS Catal., 2017, 7, 3850–3859 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc06730g |
| This journal is © The Royal Society of Chemistry 2023 |