
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Contemporary photoelectrochemical strategies and reactions in organic synthesis
Ling
Qian
 a and
Min
Shi
*ab
a and
Min
Shi
*ab
aKey Laboratory for Advanced Materials and Institute of Fine Chemicals, School of Chemistry & Molecular Engineering, East China University of Science and Technology, 130 Meilong Road, Shanghai, 200237, P. R. China
bState Key Laboratory of Organometallic Chemistry, Center for Excellence in Molecular Synthesis, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Science, Chinese Academy of Sciences, 345 Lingling Road, Shanghai, 200032, P. R. China. E-mail: mshi@mail.sioc.ac.cn
First published on 21st February 2023
Abstract
In recent years, with the development of organic synthetic chemistry, a variety of organic synthetic methods have been discovered and applied in practical production. Photochemistry and electrochemistry have been widely used in organic synthesis recently due to their advantages such as mild conditions and green and environmental protection and have now been developed into two of the most massive synthetic strategies in the field of organic synthesis. In order to further enhance the potential of photochemistry and electrochemistry and to overcome the limitations of each, organic synthetic chemists have worked to combine the two synthetic strategies together to develop photoelectrochemistry as a new synthetic method. Photoelectrochemistry achieves the complementary advantages and disadvantages of photochemistry and electrochemistry, avoids the problem of using stoichiometric oxidants or reductants in photochemistry and easy dimerization in electrochemistry, generates highly reactive reaction intermediates under mild conditions, and achieves reactions that are difficult to accomplish by single photochemistry or electrochemistry. This review summarizes the research progress in the field of photoelectrochemistry from the perspective of photoelectro-chemical catalysts in recent years, analyzes the catalytic mechanism of various catalysts in detail, and gives a brief outlook on the research direction and development prospects in this field.
 Ling Qian | Ling Qian was born in 1996 in Anhui (P. R. China). He received his BS degree in 2019 from the Hengyang Normal University, before joining a master's program in the East China University of Science & Technology. He is currently a postgraduate under the direction of Professor Min Shi. |
 Min Shi | Dr Min Shi was born in 1963 in Shanghai, China. He received his BS in 1984 (Institute of Chemical Engineering of East China) and PhD in 1991 (Osaka University, Japan). He had his postdoctoral research experience with Prof. Kenneth M. Nicholas at the University of Oklahoma (1995–6) and worked as an ERATO Researcher in Japan Science and Technology Corporation (JST) (1996–8). He is currently a group leader of the State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry (SIOC). |
1. Introduction
With the rapid development of photochemistry1 and electrochemistry,2 a large number of remarkable chemical reactions have been discovered by chemists, significantly driving the development of chemical research. However, both photochemistry and electrochemistry have certain limitations that make some reactions not well suited to photocatalysis or electrocatalysis to obtain the expected products. Net redox reactions in photocatalysis often require the addition of large quantities of stoichiometric or superstoichiometric oxidants or reductants, which may complicate the chemical reactions and produce unwanted byproducts, and the range of accessible substrates for photocatalysis is generally limited by the redox potential of the excited state, which relates to the limited energy of single visible photons. Although electrochemistry does not require the use of stoichiometric oxidants or reductants and can use electric current to achieve redox reactions and electron transfer at the electrode to obtain reactive intermediates, the redox potential of many reactive intermediates is often more accessible than that of the precursors, and secondary oxidation or radical dimerization continues to occur at the electrode surface before they can be transferred to solution, making the reaction difficult due to increased side reactions and electrode passivation. In order to overcome these drawbacks, researchers have thought of combining the two strategies in a single reaction system and developed a photoelectrochemical strategy based on photochemistry and electrochemistry. The photoelectrochemical strategy allows the cyclic regeneration of catalysts to be accomplished by selecting the appropriate potential without the need to search for redox reagents with suitable redox potentials, requiring the use of smaller stoichiometric oxidants/reductants compared to photocatalysis, or milder oxidants/reductants and being able to separate them by space from the half reaction of interest, and photoelectrochemistry also achieves the activation of some substrates that are challenging in photocatalysis by controlling the potential. It has been favored by researchers in recent years and has developed rapidly, and the number of literature reports has exploded. There is also a proliferation of reviews in this emerging field. In the review by Barham and König,3a photoelectrochemistry was systematically classified into three main forms: (1) electrochemically mediated photo-redox catalysis (e-PRC), (2) decoupled photoelectrochemistry (dPEC), and (3) interfacial photoelectrochemistry (iPEC), and some special terms in photoelectrochemistry are elaborated. Recently, Lambert's group3b has also summarized the research results of recent years according to the redox type of photoelectro-catalytic reactions. These reviews help chemists to get a more detailed understanding of the latest research results in this field, as well as explain some of the terminology used in this field. To a certain extent, they are excellent inspiration for researchers working in this field.In this review, we first give a brief statement of the terms used in photoelectrochemistry, and a simple description and classification of the mechanism of photoelectrochemical catalyst action, which is basically included in the catalytic process of the examples that follow, and then briefly introduces the development of photochemistry and electrochemistry and their accepted mechanisms so that the readers can have a brief background knowledge before understanding the photoelectrochemistry. Finally, we introduce the beginning and development of photoelectrochemistry, mainly from photoelectrochemical catalysts and their catalytic mechanisms to introduce some recent research achievements in this field, and in addition, we also give a brief summary and outlook on the development and future work in this field.
2. Brief statement
In the IUPAC definition, photoelectrochemistry refers to “term applied to a hybrid field of chemistry employing techniques which combine photochemical and electrochemical methods for the study of the oxidation–reduction chemistry of the ground or excited states of molecules or ions. In general, it is the chemistry resulting from the interaction of light with electrochemical systems.” Therefore, we believe that the research results contained in this review can be collectively referred to as photoelectrochemistry, and the catalysts used in these reactions are collectively referred to as photoelectro-chemical catalysts.In photoelectrochemistry, the cyclic turnover of the photo-electrochemical catalyst is an important part of the completion of the reaction. In combined photochemical and electro-chemical catalysis, the electrochemical process generally has two modes of action. One is that the catalyst is first activated into an intermediate with stronger oxidizing or reducing properties at the electrode via an electrocatalytic process, and then subsequently irradiated by light to generate an excited state catalyst, which has stronger redox properties. Then the excited state catalyst undergoes an electron transfer process with the substrate, and the catalyst returns to the ground state and then participates in the next catalytic cycle. In the other the terminal redox agent is replaced by an electrochemical process, which allows the catalyst to cycle and regenerate. In this review, we classify photoelectrochemical catalysts into three types according to these two mechanisms of action: (1) organic electrochemically recycled photocatalysts, (2) organic electrochemically activated photocatalysts, and (3) inorganic electrochemically recycled photocatalysts.
3. Photoredox catalytic reactions
In the 1970s, Kellogg4 found a photoreduction reaction in which the use of N-methyl-1,4-dihydropyridine compounds as the terminal reducing agent, and the catalytic amount of Ru(bipy)3Cl2 as the photocatalyst to absorb visible light, can accelerate the decomposition of sulfide ions to obtain the corresponding alkane and sulfur ether (Scheme 1). Photocatalytic redox reactions began to enter the vision of chemists, but this strategy has been ignored for a long time and has developed very slowly.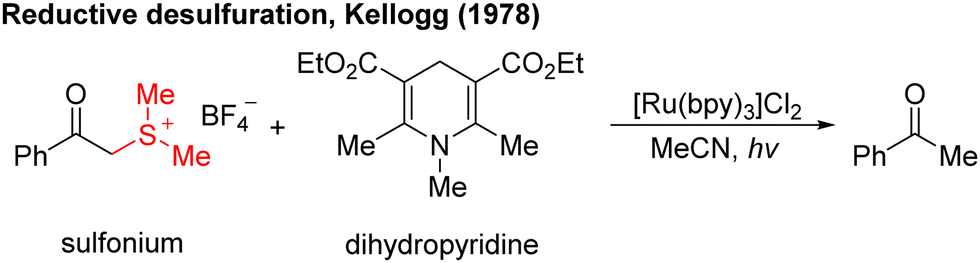 | ||
| Scheme 1 The first reaction of photocatalysis. | ||
By 2008, Yoon's group developed an effective intramolecular [2+2] cycloaddition reaction of olefin by using the photocatalyst Ru(bipy)3Cl2 (Scheme 2a).5 In the same year, MacMillan's group also used the Ru(bipy)3Cl2 photocatalyst to achieve asymmetric alkylation of aldehydes at the α-site (Scheme 2b),6 for the first time to achieve the combination of photocatalysis and organic small molecule catalysis. The next year, Stephenson's group developed a photoredox-catalyzed dehalogenation reduction reaction of alkyl halides (Scheme 2c).7 Notably, this reaction could be performed without the use of toxic tin reagents and was environmentally friendly. These reports have aroused great interest and attention of organic synthetic chemists. Since then, the number of reports on photocatalysis has increased significantly and the research in this field has been fully developed. Compared with traditional redox reaction, photocatalytic redox reaction uses much cheaper, cleaner, and environmentally friendly light energy, and also uses a catalytic amount of photoredox catalysts instead of the stoichiometric chemical oxidants or reductants, saving resources. Moreover, these reactions can be carried out under milder reaction conditions in most cases, using a simple device, and with easy operation.
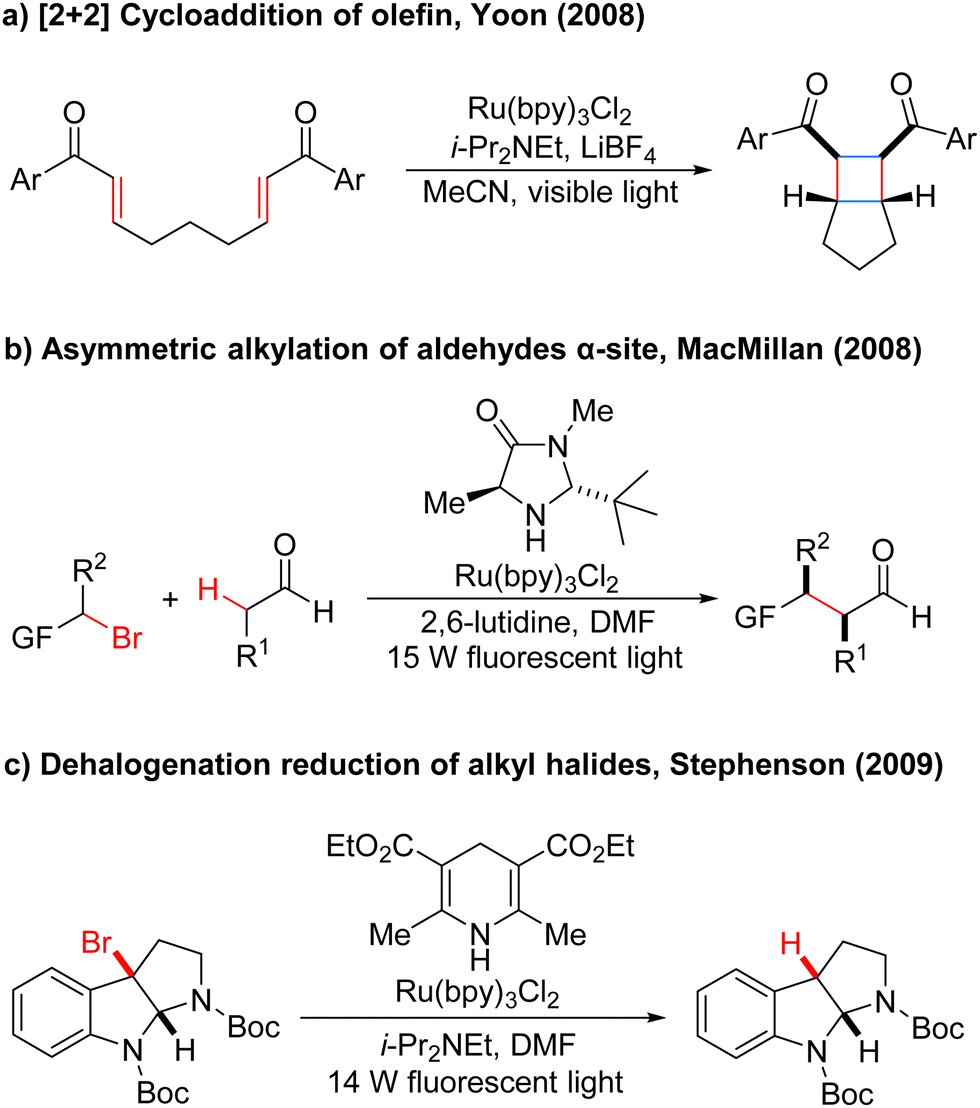 | ||
| Scheme 2 Classic examples of photocatalysis. | ||
As the research on photocatalytic reactions has intensified, significant attention has been paid to the study of the reaction mechanism of photocatalysis. Since most of the organic compounds cannot absorb visible light very well by themselves, they need to absorb light energy with the help of photocatalysts, so that the light energy can be converted into chemical energy. In photoredox catalytic reactions, the photocatalyst acts as an oxidizing or reducing agent in the chemical reaction. After absorbing visible light, the photocatalyst transitions from the ground state to the excited state, which has stronger oxidation and reduction properties, and in most cases, it interacts with substrates or reagents through the single electron transfer (SET) pathway to return to the ground state and obtain a highly active intermediate, which is generally a radical species. In the single electron transfer process, the excited photocatalyst can be returned to the ground state by oxidation quenching and reduction quenching (Scheme 3). In addition to the single electron transfer pathway, the excited state photocatalyst can also exchange energy with the substrate through the energy transfer (EnT) pathway to directly excite the substrate into the active intermediate (Scheme 3).
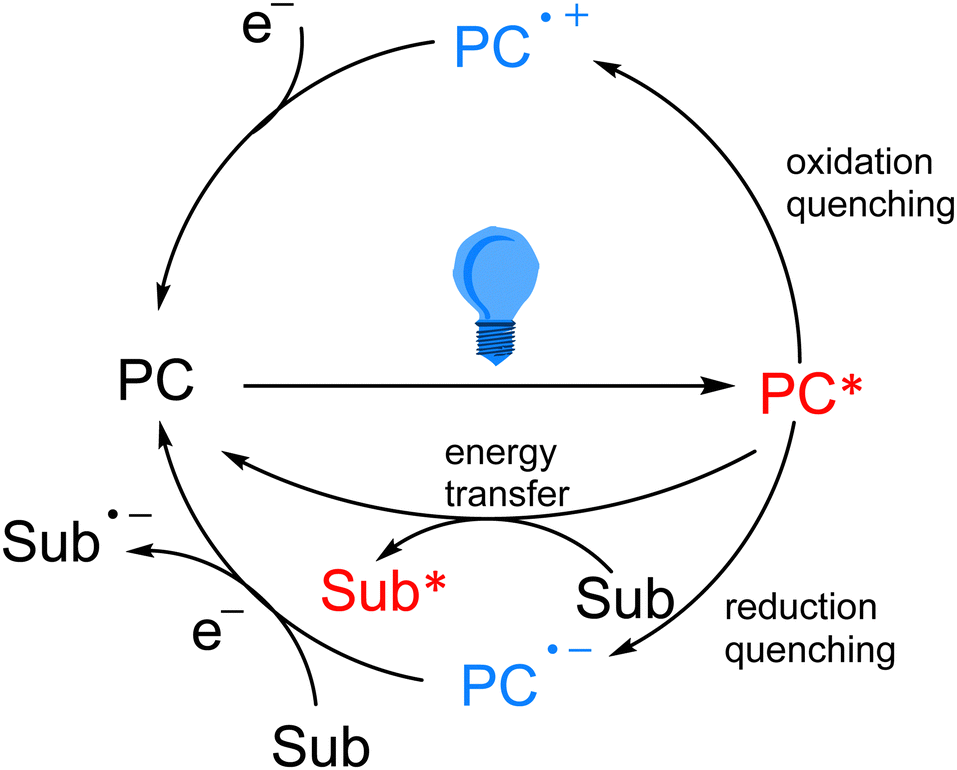 | ||
| Scheme 3 Typical quenching types of photocatalysis. | ||
Photocatalytic reaction, as a synthetic strategy developed rapidly in the last decade or so, can be used to synthesize various C–C, C–N, C–O, and C–P bonds with high efficiency and selectivity due to its unique mechanism. However, the scope of photoredox catalysis is limited by the redox potential of its substrate and the excitation potential of the photocatalyst. In addition, to achieve a net oxidation or reduction reaction an additional terminal oxidant or reductant is required as a sacrificial reagent to enable the catalytic cycle to be turned around. These sacrificial reagents may react with catalysts, substrates, reagents or active intermediates to produce unwanted byproducts, reducing the efficiency of the main reaction or making it impossible to proceed, complicating the reaction system, and rendering the addition of sacrificial reagents uneconomical. For example, in 2010, Stephenson and co-workers developed a photocatalytic protocol of photoredox-mediated direct inter-molecular C–H functionalization of heterocyclic arenes with diethyl bromomalonate.8 When they used Et3N as a sacrificial reducing agent, the yield of the target product was only 25% after 24 hours, even though a large amount of substrate was employed. If using 4-methoxy-N,N-diphenylaniline, the yield of the target product increased to 82%. This is due to the fact that amino radical cation of the former can donate a hydrogen atom to the target substrate radical species, which was produced by dehalogenation of diethyl bromomalonate, leading to a substantial amount of reduced diethyl malonate.
4. Electrocatalytic reactions
Electrocatalysis has a very long history as photocatalysis and has been used as an efficient and highly selective synthetic strategy in modern organic synthesis. Since the invention of the cell by Volta9 in 1800, synthetic organic electrochemistry has been slowly developing. It was not until the 1830s that Faraday's10 pioneering scheme for the electrolysis of acetic acid to produce alkanes attracted the chemists’ interest in this non-spontaneous organic reaction driven by electric currents (Scheme 4a). In 1845, Schoenbein11 discovered the reaction of reduction and dehalogenation of trichloromethyl sulfonic acid at the cathode, which realized the electrocatalytic chemical reaction of cathodic reduction for the first time (Scheme 4b). Inspired by Faraday, in 1874, Kolbe12 achieved the oxidative decarboxylation of carboxylic acids at the anode to generate alkyl radicals for dimerization into alkanes (Scheme 4c). This strategy provides a convenient means to obtain alkyl radicals. Since then, organic synthetic chemists began to realize the powerful potential of this new strategy and the trend of future development of organic synthesis and created the field of electrochemical organic synthesis. | ||
| Scheme 4 The beginning reactions of electrochemistry. | ||
A simple electrochemical catalytic reaction device requires a power supply connected to an anode electrode and a cathode electrode, which are inserted into an electrolyte solution to form a closed-loop pathway. Mechanistically, electrocatalytic reactions occur by applying an electrical potential to cause molecules to gain or lose electrons at an electrode or in an electrolyte, resulting in a corresponding reduction or oxidation reaction. Electrocatalytic reactions can be divided into direct electrolysis and indirect electrolysis depending on whether the substrate gains or loses electrons at the electrode. In a direct electrolysis method, the substrate oxidizes by losing electrons at the anode or reduces by gaining electrons at the cathode to obtain a highly reactive intermediate before proceeding to the subsequent functionalization, which is a heterogeneous reaction. In contrast, indirect electrolysis requires the addition of a redox mediator to indirectly transfer electrons between the substrate and the electrode, so the redox mediator is also called an electron transfer medium. The redox mediator oxidizes by losing electrons at the anode or reduces by gaining electrons at the cathode to produce an active redox mediator, which makes the substrate undergo the corresponding electron reduction or oxidation by electron transfer in contact with the substrate, and then get functionalized by the subsequent reaction.
Compared with traditional organic synthetic strategies for redox reactions, electrochemistry decreases the use of chemical reagents by using electrical energy, has milder conditions, consumes less energy overall, decreases environmental pollution, and is more in line with the tenets of green chemistry. Moreover, the course of the reaction can be precisely controlled by controlling the electrode potential to improve the selectivity of the main reaction, and the functional groups are well tolerated. However, electrochemical methods also have certain limitations, such as direct electrolysis is a heterogeneous reaction, the radical active species generated at the electrode may not be able to migrate to the solution before the secondary oxidation or radical dimerization and other side reactions occur to affect the yield, and the electrode surface is contaminated by intermediates from starting material decomposition, leading to passivation. The radical-active species generated by indirect electrolysis are prone to over-oxidation because they have a lower oxidation potential than the precursors. In addition, some commonly used solvents (e.g., tetrahydrofuran, toluene) have poor electrical conductivity, and the choice of solvent is a factor to be considered in electrochemical synthesis; alternatively, a supporting electrolyte may be added to the solvent to improve the conductivity of the solvent, but some supporting electrolytes are expensive, toxic and may lead to the risk of explosion, such as LiClO4 and NaClO4. In a complex chemical reaction, a high potential may be required to produce an active intermediate, and at such extreme potentials the reaction will not be well controlled, and the reaction selectivity will be greatly decreased.
5. Early work on photoelectrochemical reactions
Although photochemistry and electrochemistry have a long and rich history of development, and after more than ten years of development, the two strategies have been well complemented and improved to achieve many reactions that traditional redox reactions cannot accomplish, but because of the inherent limitations of their unique models, for many reactions still cannot achieve the ideal solution for researchers. The emergence of photoelectrochemistry, which perfectly complements the advantages and disadvantages of these two strategies, is a very young and challenging field, and early reports on photoelectro-chemistry did not clearly recognize it as an emerging synthetic method with great development potential.It is generally accepted in the field of photochemistry that the first reaction related to photoelectrochemistry was reported by Moutet and Reverdy in 1979 (Scheme 5a).13 1,1-Diphenylethene (DPE) 1 was irradiated using a mercury lamp and phenothiazine (PTZ) acted as a photoelectrochemical catalyst and cyclization occurred at a potential of 0.79 V to form the product 2. In this reaction, the photoelectrochemical catalyst phenothiazine belongs to the class of organic electrochemically activated photocatalysts. Three years later, Moutet and Reverdy published another report of photoelectro-chemistry, in which the N,N,N′,N′-tetraphenyl-p-phenylenediamine (TPPD) was used as a photoelectrochemical catalyst and under UV irradiation. Benzyl alcohol 3 is oxidized to benzaldehyde 4 by two successive electron transfer processes (Scheme 5b).14 The TPPD catalyst in this protocol belongs to the same class of organic electrochemically activated photocatalysts as the above PTZ catalyst. In 1983, Scheffold also published a report on the photoelectrochemical one-step synthesis of 4-oxo aldehydes, ketones, esters and nitriles. Anhydride 5 reacts with electron-deficient olefin 6 catalyzed by vitamin B12 and Co(III) species by visible light irradiation at 500 W using a divided cell and a mercury cell cathode to produce the addition compounds 7 (Scheme 5c).15 The vitamin B12 in this protocol is mainly used to reduce the Co(III) species to Co(I) at the beginning of the reaction, and the catalyst that undertakes the catalytic cycle of the reaction is Co(III), which mechanistically belongs to the type of inorganic electrochemically recycled photocatalysts. The specific reaction processes and mechanisms of the three works mentioned here are summarized in the reviews by Lambert et al.3 and are available for readers to review on their own, and thus it will not be repeated here.
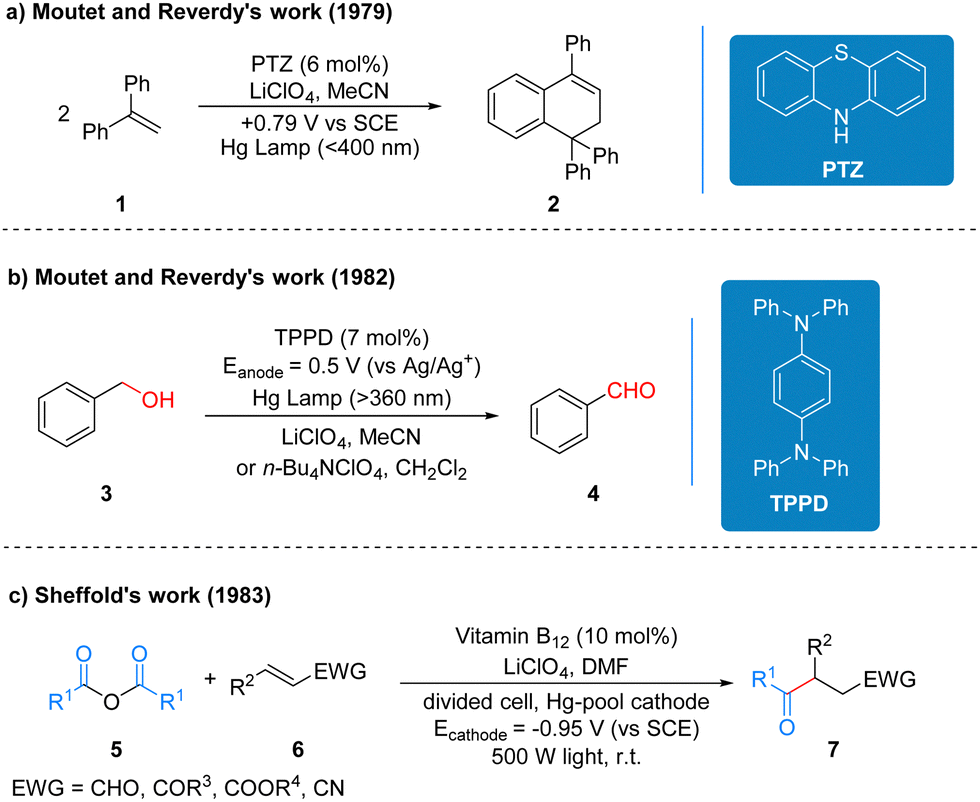 | ||
| Scheme 5 Relevant example of early work on photoelectrochemistry. | ||
From a certain perspective, the three previously mentioned works were the beginning of photoelectrochemistry, but because the great potential of this combined photochemical and electrochemical catalytic reaction strategy was not realized initially, the strategy did not receive much attention from organic synthetic chemists during the first two decades of its development. In the last five years, the photoelectrochemical strategy has been gradually came to the attention of organic chemists. Since 2019, there has been a large increase in the number of reactions related to photoelectrochemistry, various types of photoelectrochemical methods have appeared, and a variety of efficient photoelectro-chemical catalysts have been used, and the research in this field has become more and more in-depth. Among them, various efficient photocatalysts can be roughly classified into three categories according to their mechanism of action: organic electrochemically recycled photocatalysts, organic electrochemically activated photo-catalysts, and inorganic electrochemically recycled photocatalysts. The following introduces some research achievements in the field of photoelectrochemistry in recent years from the perspective of these three types of photoelectrochemical catalysts.
6. The prosperous development of modern photoelectrochemical reactions
6.1 Organic electrochemically recycled photocatalysts
 | ||
| Scheme 6 Photoelectrochemistry of heteroarene alkylation. | ||
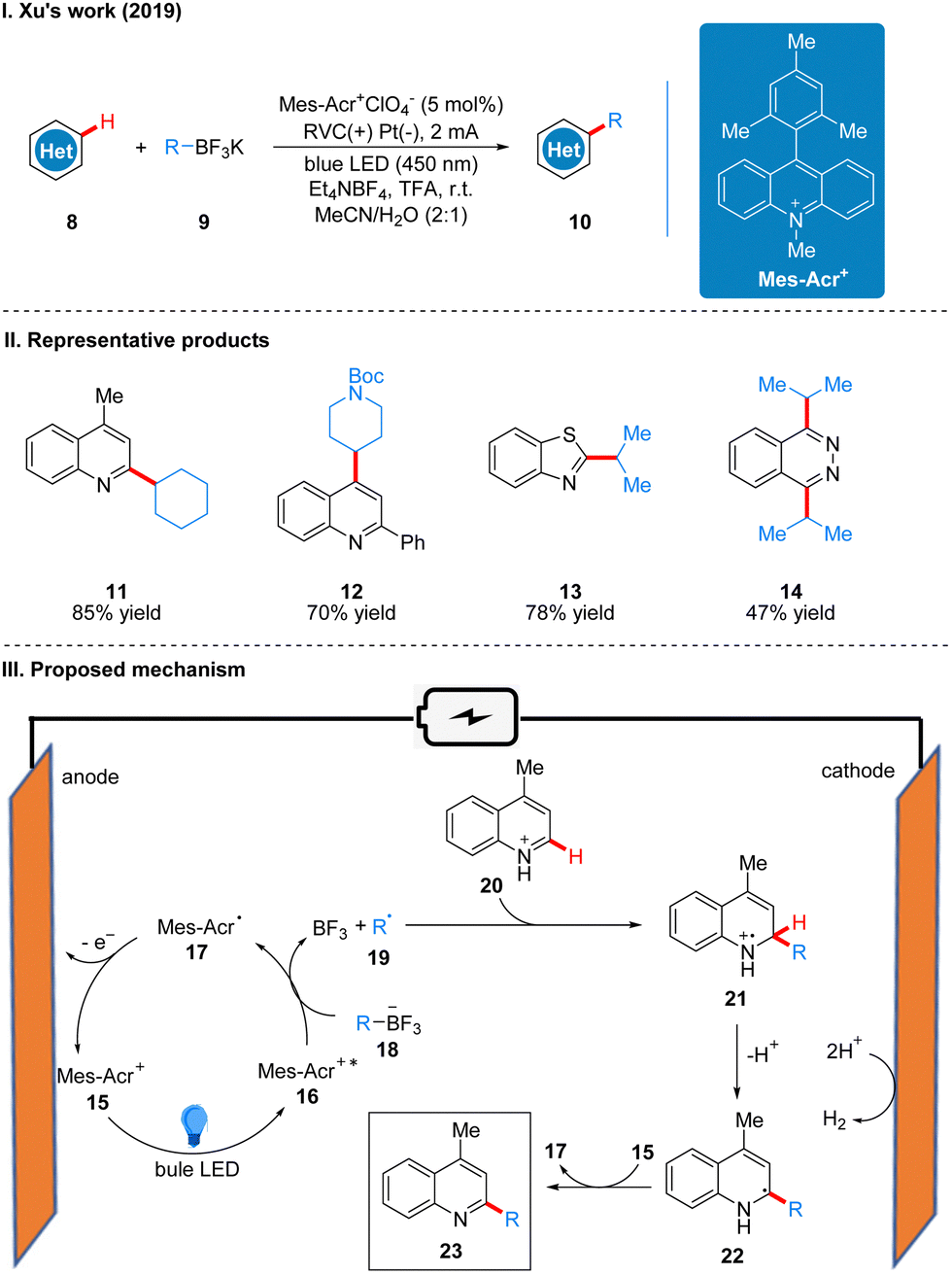
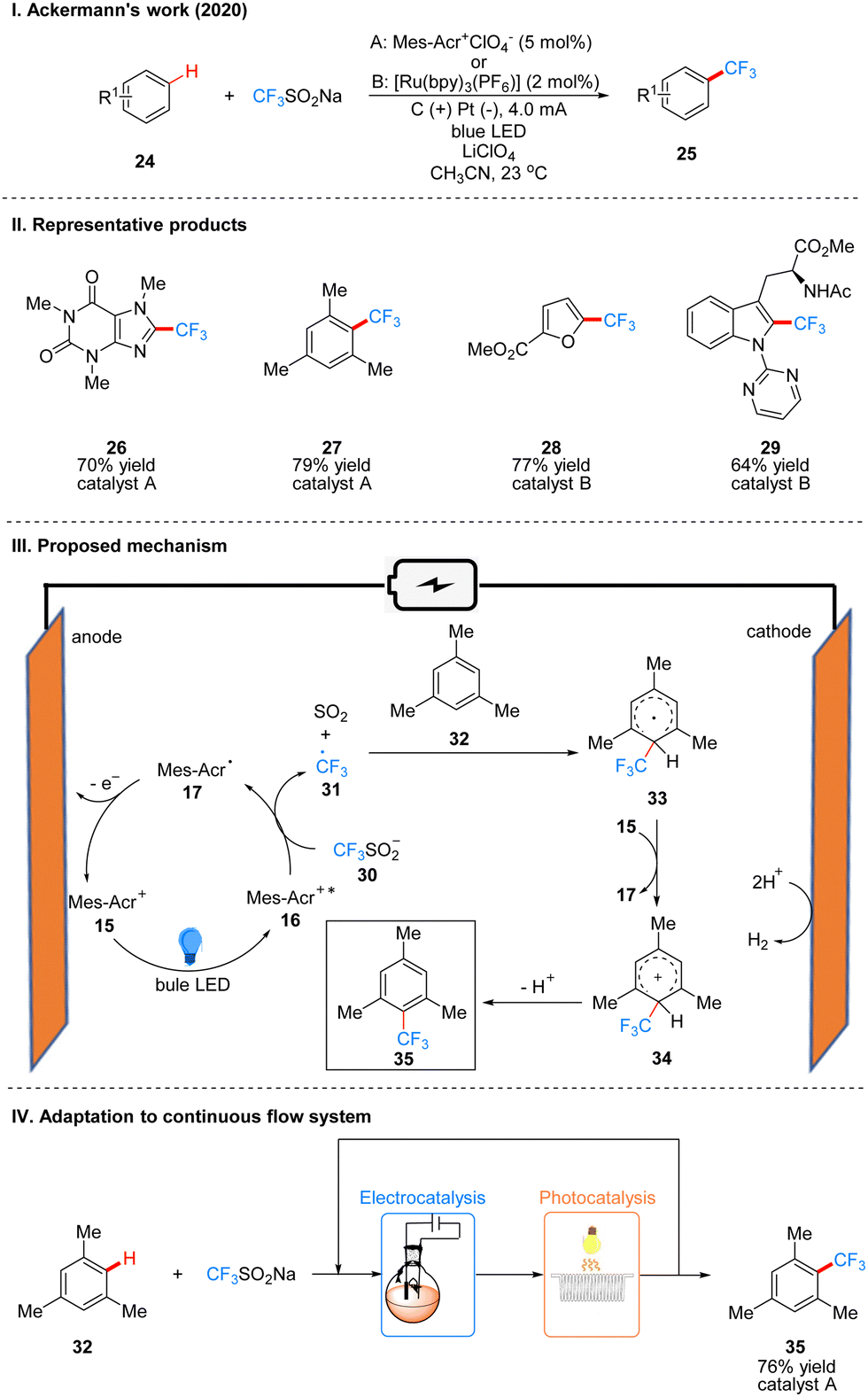
In 2020, Ackerman and co-workers reported a photoelectro-catalyzed C–H trifluoromethylation reaction of (het)arenes 24 to produce 25, which was the first trifluoromethylation reaction to utilize photoelectron co-catalysis (Scheme 7(I)).18 This reaction utilized Mes-Acr+ or [Ru(bpy)3](PF6)2 as the photoelectrochemical catalyst, and the trifluoromethyl source reagent used was the Langlois reagent (CF3SO2Na). The reaction has high chemoselectivity and functional group tolerance (including esters and amides), and the trifluoro-methylation products are also well obtained for arenes containing groups with high site resistance. The reaction proceeds very well to trifluoromethylation with good yields for a wide range of heteroarenes, such as furans, thiophenes, benzothiophenes, indoles, quinolines and pyrimidines. The part of the products is displayed in Scheme 7(II) (26–29). The mechanism of the Mes-Acr+ catalyst is the same as that reported above, where Mes-Acr+15 generates the excited state of Mes-Acr+ (Mes-Acr+*) 16 under light irradiation, and Mes-Acr+* 16 undergoes a single electron transfer (SET) process with the Langlois reagent 30 to obtain trifluoromethyl radical 31 and Mes-Acr˙ 17, which was oxidized to regenerate Mes-Acr+ in the anode, completing the catalyst cycle. Subsequently, the trifluoromethyl radical 31 was captured by aromatics 32 to generate radical intermediate 33, which was oxidized by Mes-Acr+ to obtain the cationic intermediate 34. Finally 34 deprotonated to obtain trifluoro-methylated product 35 (Scheme 7(III)). The reaction can also be applied in fluidic systems and can be used for gram scale synthesis (Scheme 7(IV)).
 | ||
| Scheme 7 Photoelectrochemistry of C–H trifluoromethylation of arenes. | ||
 | ||
| Scheme 8 Photoelectrochemistry of SNAr reaction using DDQ. | ||
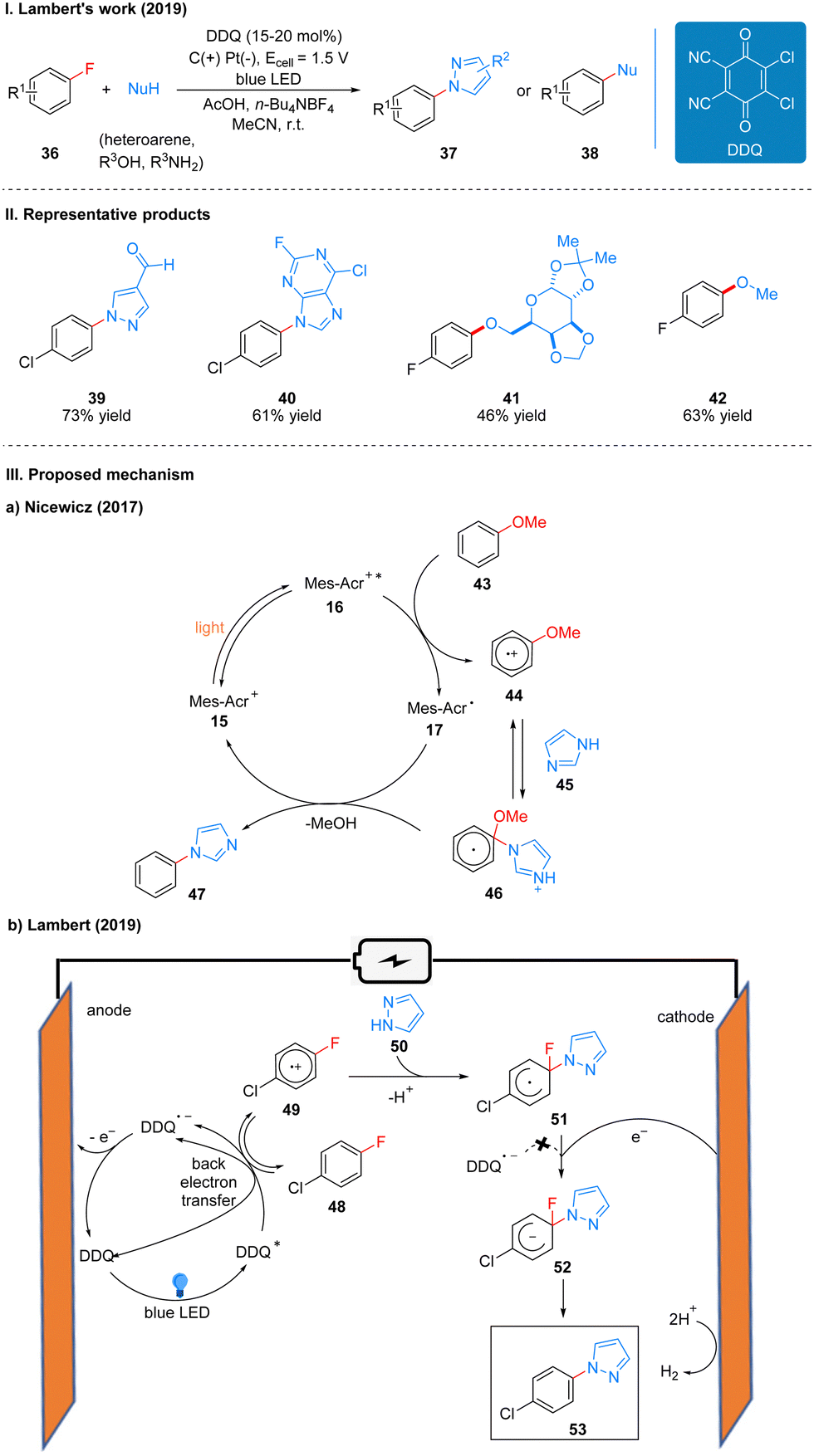
Previously, Nicewicz and co-workers20 reported the SNAr reaction of an inactive alkoxy arenes catalyzed by the photocatalyst N-methyl-9-mesitylacridinium (Mes-Acr). Mechanistically, the key to the reaction is the generation of an aryl ether radical cation 44 by the alkoxy arene 43 catalyzed by Mes-Acr+*, which is easy for nucleophilic attack to obtain the radical intermediate 46, followed by the transfer of electrons from the reduced photocatalyst 17 to the radical intermediate 46, and finally the alkoxy substituent is removed to complete the aromatization to get product 47 (Scheme 8(IIIa)). Because the electron-donating alkoxy substituent is necessary to achieve the initial oxidation event, the substrate scope is only applicable to alkoxy-type inactive aromatics. To achieve the SNAr reaction for less electron-rich arenes (e.g., fluorobenzene), a more oxidizing photocatalyst needs to be found to complete the initial oxidation of the inactive arenes, while the reduced catalyst can also transfer electrons to the radical intermediate to generate the initial photocatalyst to complete the catalytic cycle. DDQ is an ideal oxidant with a strong oxidizing power (Ered = 3.18 V vs. SCE), the excited state DDQ generated under light can undergo a single electron transfer (SET) process with p-chlorofluorobenzene 48 to obtain a radical cation 49, which is subsequently attacked by a nucleophilic reagent 50 to afford a radical intermediate 51. However, the reduced DDQ radical anion cannot transfer electrons to the radical intermediate 51, reducing it to a radical anion 52, at which point a cathode is required to give electrons to the radical intermediate 51, and reduced it to the radical anion 52, and finally the fluoride ion is removed to re-aromatize the product 53. The previously reduced DDQ radical anion is oxidized at the anode to regenerate DDQ, completing the catalytic cycle (Scheme 8(IIIb)). Notably, the alkoxybenzene used in the Nicewicz's work did not observe the expected product in this scheme, and the authors suggest that while single-electron oxidation events readily occur, the anti-electron transfer is also very rapid, overtaking the nucleophilic reagent to attack the radical cation intermediate 68, leading to the re-generation of the radical cation 68 into an inactive aromatic.
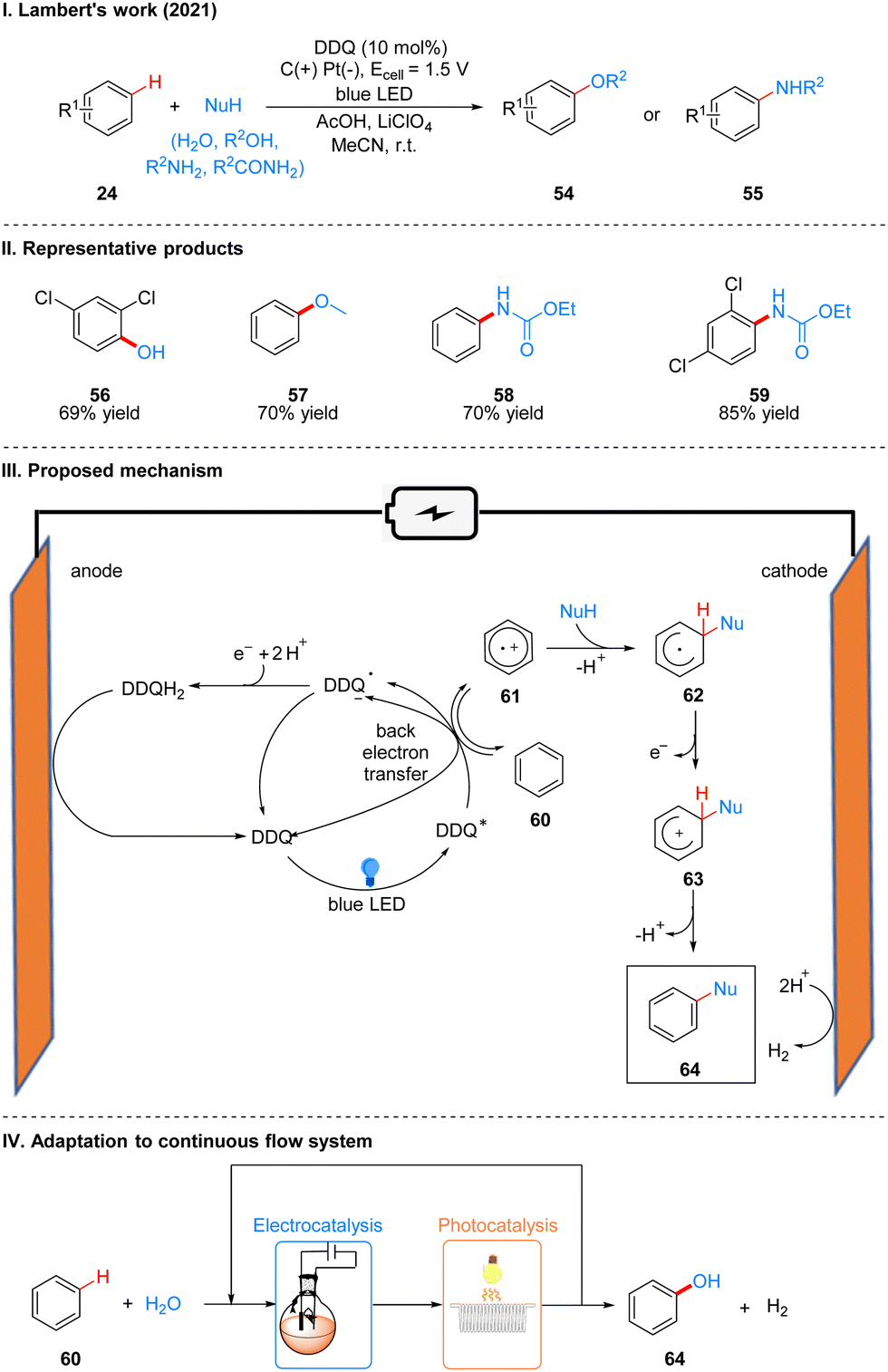
In 2021, Lambert's group further discovered the photoelectro-chemically catalyzed direct C–H heterofunctionalization reaction of aromatic hydrocarbons.21 Using DDQ as a photoelectrochemical catalyst, under a mild electrical potential with blue LEDs irradiation, aromatics 24 can directly react with nucleophilic reagents, such as alcohols, water, carboxylic acids, aminocarbonates, and amides, to produce functionalized products 54 or 55 (Scheme 9(I)). The part of products is displayed in Scheme 9(II) (56–59).
 | ||
| Scheme 9 Photoelectrochemistry of arene heterofunctionalization using DDQ. | ||
Direct C–H functionalization of aromatics is an effective strategy for the synthesis of substituted aromatics. Both transition metal-catalyzed and direct electrolytic oxidation methods have long been known, but these methods usually have low yields, poor selectivity, and a limited range of substrates. For the rapidly developing photoredox methods, the C–H bond functionalization of aromatics is often applicable to electron-rich arenes because they have low redox potentials, while for electron-deficient arenes the redox potentials are higher and not easily oxidized to aryl radical cations, so it is a great challenge for electron-deficient and electron-neutral aromatics. Based on the previous studies19,24 and the work of Fukuzumi22 and König,23 the researchers concluded that DDQ would be a suitable catalyst for this photoelectrochemical reaction. Mechanistically, DDQ is irradiated with blue LEDs to obtain the excited state DDQ (DDQ*), and then undergoes a single electron transfer (SET) process with benzene 60 to form the benzene radical cation 61, followed by the attack of the phenyl radical cation 61 with nucleophilic reagents to give the radical intermediate 62, which then undergoes single electron oxidation to form 63, and deprotonation to afford the final product 64. The DDQ radical anion (DDQ˙−), after getting one molecule of electrons and two molecules of protons to form DDQH2, which is immediately followed by the regeneration of the DDQ catalyst at the anode oxidation (Scheme 9(III)). Hydrogen protons are reduced at the cathode to form H2. To further scale up the process, they applied a continuous flow system to the reaction, with electrochemical modules and multiple photochemical reaction channels in series, which not only effectively increased the reaction yield, but also successfully achieved a 15 mmol scale conversion of benzene to phenol (Scheme 9(IV)).
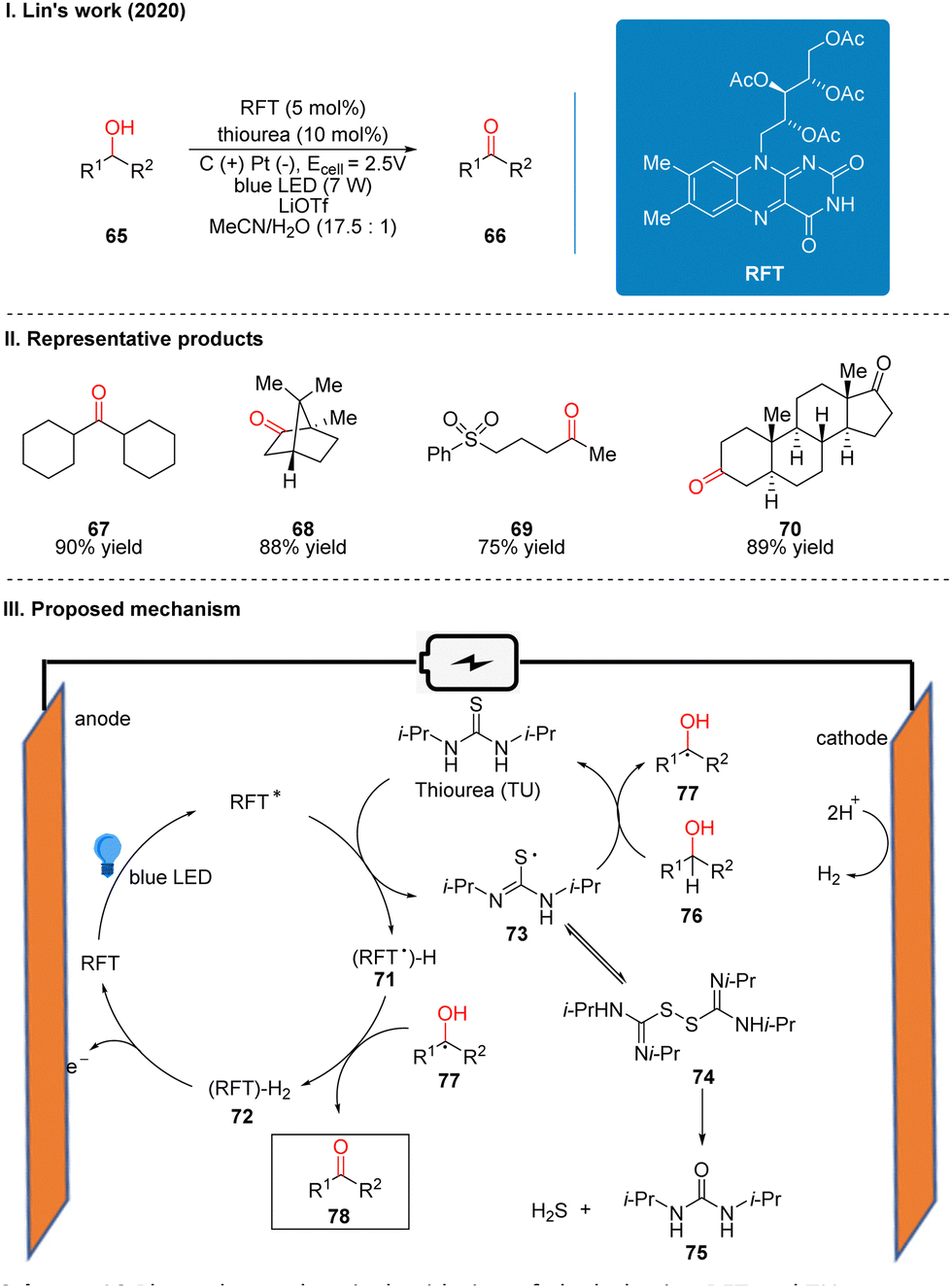 | ||
| Scheme 10 Photoelectrochemical oxidation of alcohol using RFT and TU. | ||
In 2020, Lei's group reported a generalized Mn-catalyzed direct C(sp3)–H bond azidation reaction to obtain 80 under photoelectron-chemical catalytic conditions (Scheme 11(I)).28 The reaction is applicable to various benzyl-site C(sp3)–H bonds, conventional alkyl and remote C(sp3)–H bonds, and has high regioselectivity, with greater reactivity for tertiary C(sp3)–H than for primary/secondary C(sp3)–H, such as product 82. The part of the products is displayed in Scheme 11(II) (81–84). In this reaction, which requires the use of a manganese catalyst and an organic HAT photocatalyst (e.g., DDQ, 9-fluorenone, 4,4′-dimethoxybenzo-phenone) to complete the catalysis, the authors suggest that HAT photocatalyst 88 generates excited state 89 under light irradiation, which subsequently reacts with alkanes 90 to obtain alkyl radical 91 and radical intermediate 90, which is oxidized to 88 at the anode. In another catalytic cycle, the Mn(II) 85 and N3− coordination to get 86, which is anodically oxidized to intermediate 87, and 87 subsequently reacted with alkyl radical 91 generated by HAT photocatalysts to give the azidation product 92 (Scheme 11(III)).
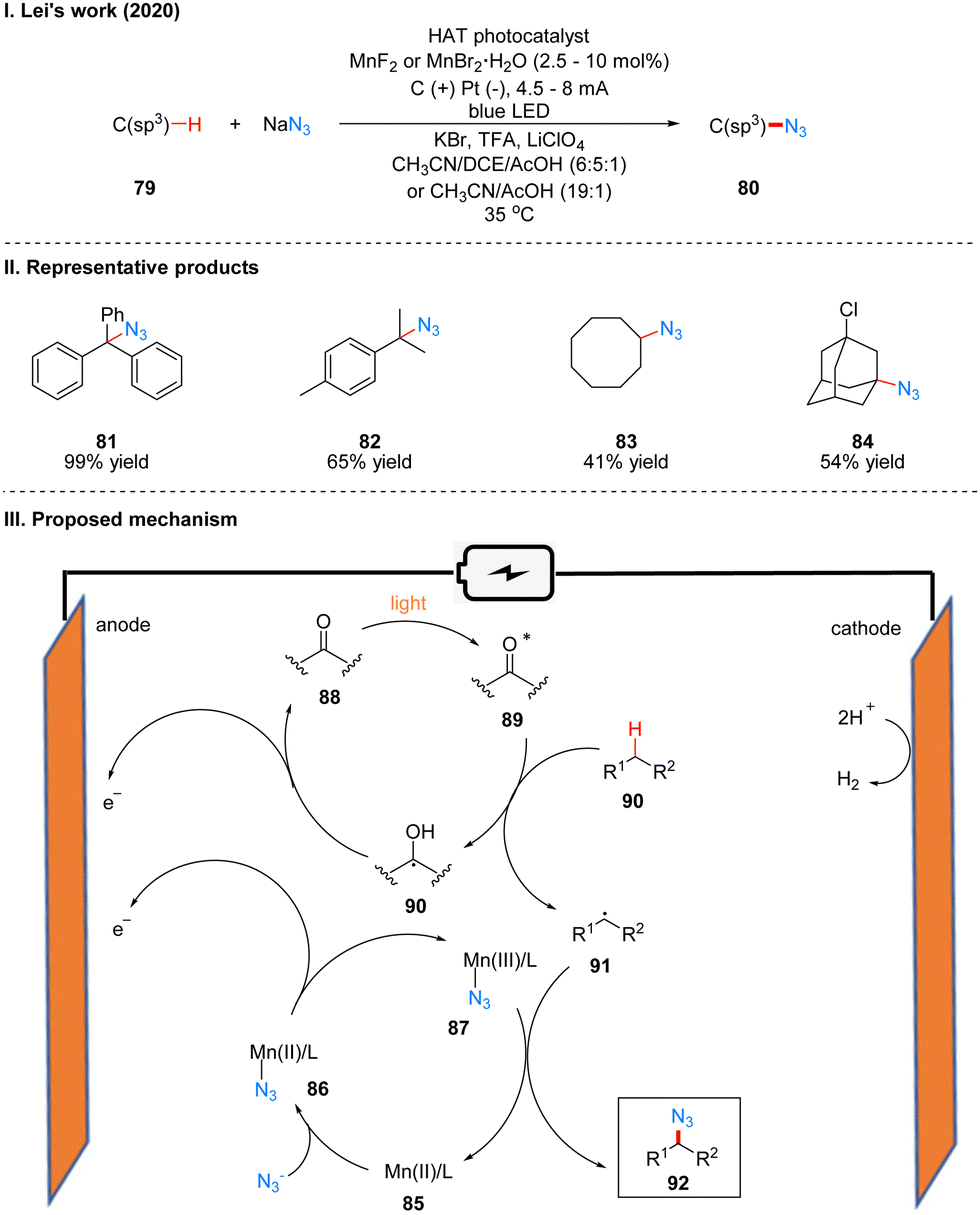 | ||
| Scheme 11 Photoelectrochemistry of oxidative azidation. | ||
In 2021, Ravelli and co-workers reported photoelectrochemically catalyzed dehydrogenation cross-coupling reactions of benzothiophene 93 and alkane 79 with C(sp3)–H bonds to obtain 94 by using tetrabutylammonium decatungstate (TBADT) as the photo-electrochemical catalyst (Scheme 12(I)).29 While previous work30 by this author has used K2S2O8 as a terminal oxidant to achieve the photocatalytic Minisci-type reaction of heteroarenes, this strategy utilizes electrochemical oxidation instead of the K2S2O8 oxidant, which is more favorable in terms of safety, economy and environment. This reaction has a good range of substrate applicability for various substituents of benzothiazole and chain alkanes, cyclic alkanes. A part of the products is displayed in Scheme 12(II) (95–98). The mechanism of the reaction is shown in Scheme 12(III). TBADT is excited by light to obtain the excited state of TBADT (TBADT*), which is responsible for the activation of aliphatic C(sp3)–H bonds, and alkanes 99 are oxidized by the single electron transfer (SET) of TBADT* to obtain alkyl radicals 100, and 100 then undergoes addition with benzothiazole 101 to obtain nitrogen radical intermediates 102. It is confirmed that intermediates 102 undergo a proton-mediated spin-center shift (SCS) to give the tertiary carbon alkyl radical 103, and finally the product 104 was obtained under the oxidation of TBADT and deprotonation. The reduced TBADT was regenerated by anodic oxidation.
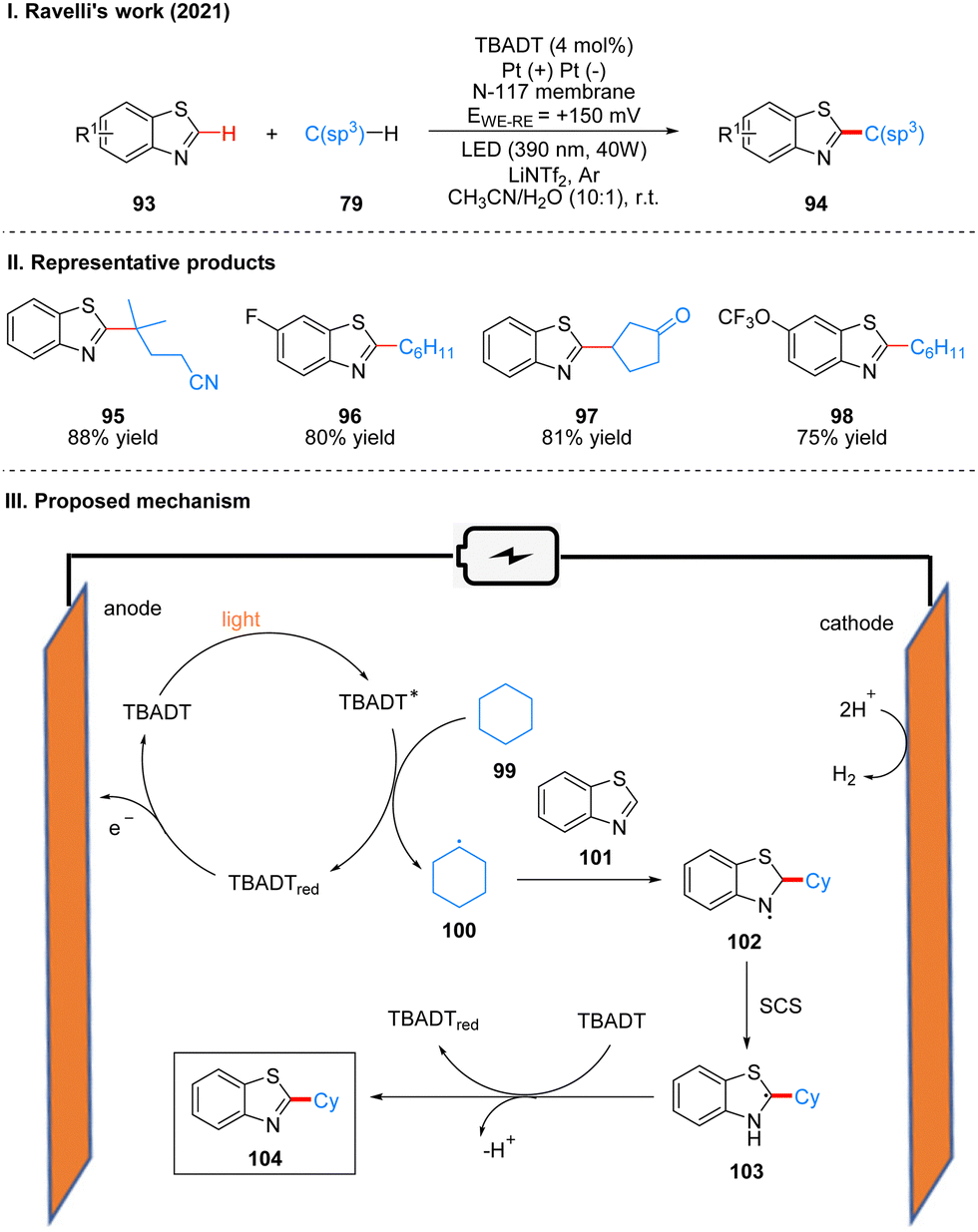 | ||
| Scheme 12 Photoelectrochemical alkylation of benzothiazole. | ||
6.2 Organic electrochemically activated photocatalysts
 , 3-cyano-1-methylquinolinium
, 3-cyano-1-methylquinolinium 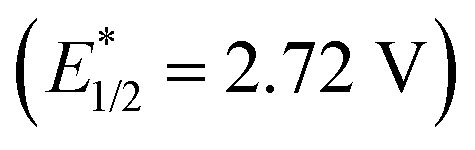 , and even 2,6-dichloro-3,5-dicyanoquinone (DDQ;
, and even 2,6-dichloro-3,5-dicyanoquinone (DDQ;  ). Thus, they concluded that the TAC ion could serve as a powerful new photoelectron-chemical catalyst that could well oxidize a variety of challenging substrates, which were unlikely to succeed in the works of Nicewicz,35 Lei,36 Grätzel and Hu.37
). Thus, they concluded that the TAC ion could serve as a powerful new photoelectron-chemical catalyst that could well oxidize a variety of challenging substrates, which were unlikely to succeed in the works of Nicewicz,35 Lei,36 Grätzel and Hu.37
 | ||
| Scheme 13 Photoelectrochemistry of the C–H/N–H coupling reaction. | ||
The most critical part of the whole reaction process is that the TAC ion (TAC+) 111 produces an open-shell TAC radical dication (TAC˙2+) 112 at the anode, which produces an excited TAC radical dication (TAC˙2+*) 113 with strong oxidation properties under the irradiation of CFL. Then the substrate benzene 114 and TAC2+* undergo a single electron transfer (SET) process to obtain benzene radical cation 115, which reduces TAC˙2+* to TAC+ to continue to participate in the next catalytic cycle at the anode. The coupling of benzene radical cation 115 and azoles 116 occurs to obtain radical intermediate 117, which are subsequently oxidized and diprotonated in the presence of TAC radical dications to form product 118 (Scheme 13(III)).
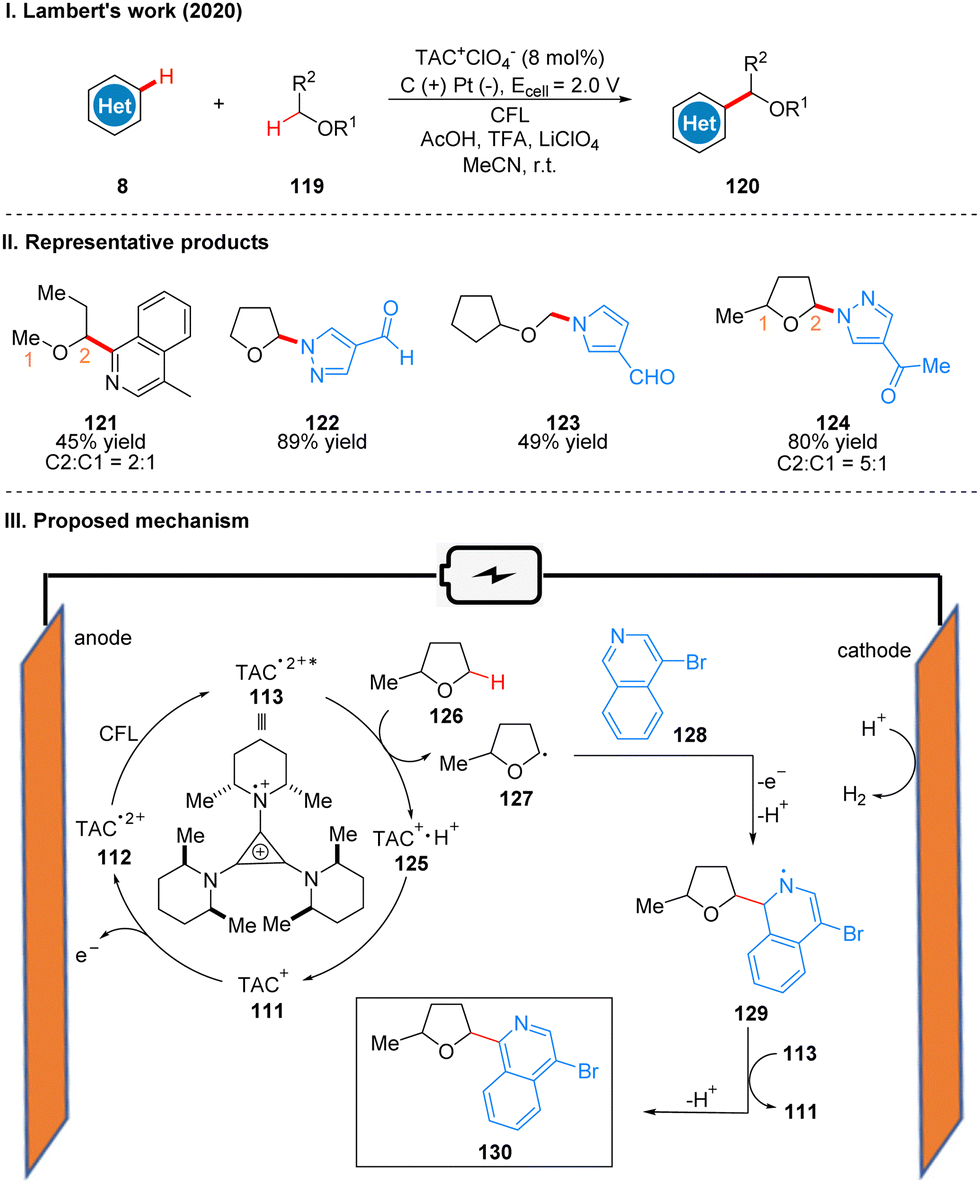
In 2020, Lambert's group reported a photoelectroncatalyzed Minisci-type reaction for the highly selective C–H functionalization of ethers 119 at the α-position (Scheme 14(I)).38 Minisci-type reactions for the addition of radicals to heterocyclic arenes are an important tool for building molecular complexity, and the strategy is highly effective for building biologically active molecules, but many examples of the strategy require the use of stoichiometric peroxides and have poor regioselectivity. From their previous study,31 it is known that the photoexcited state of the TAC radical dication has amine radical cationic properties on one of the nitrogen substituents, and thus they envisioned the excited state TAC radical dication intermediate 113 to be an effective acceptor for hydrogen atom transfer that would accept well the hydrogen atom given by the ether 126 to form radical 127 while producing protonated 125. Next the radical 127 reacts with the substituted isoquinoline 128 to form the radical intermediate 129, which may undergo a second oxidation by TAC˙2+*, and another proton is removed to give the product 130. Simultaneous 125 deprotonation regenerates the TAC+ catalyst 111 (Scheme 14(III)). It is worth mentioning that the reaction has a special regioselectivity. For either the pri/ter or sec/ter C–H bond, the products obtained are regional isomers with small site resistance due to the large site resistance of the tertiary C–H bond (Scheme 14(II), 124); whereas for the pri/sec C–H bond, the products obtained are second C–H bond functionalized products (Scheme 14(II), 121). The reaction is also applicable to other radical acceptors such as alkenes, alkynes, and azoles. A part of the products is displayed in Scheme 14(II) (121–124).
 | ||
| Scheme 14 Photoelectrochemical Minisci ether functionalization. | ||
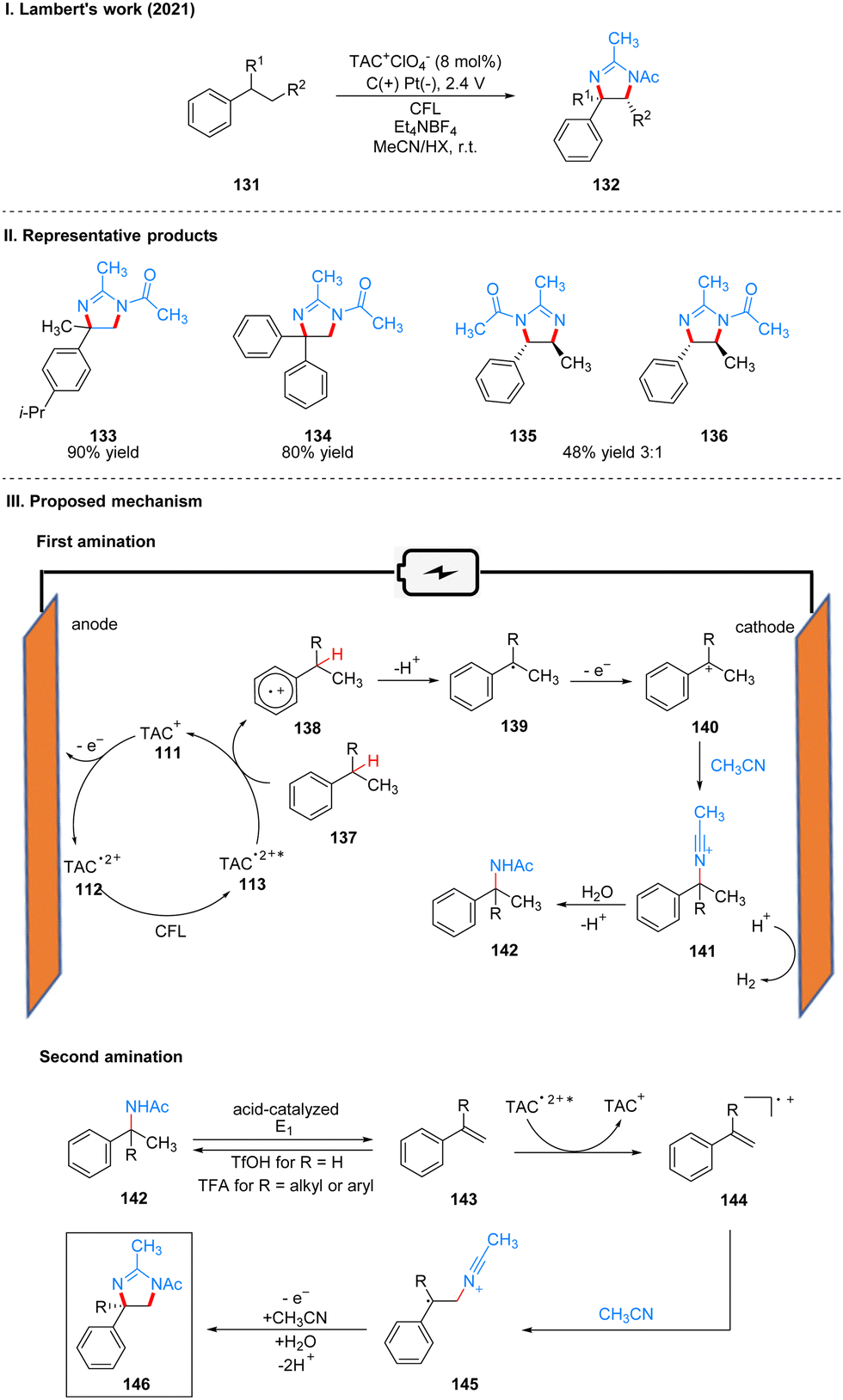
In 2021, Lambert's group reported another example of a photoelectrochemical reaction for Ritter-type C–H bond di-amination with TAC ion photoelectrochemical catalyst. Under the irradiation of CFL and 2.4 V potential using carbon positive and platinum negative electrodes, the diamination product 132 can be obtained by the reaction of substrate 131 at room temperature catalyzed by TAC+ClO4− (Scheme 15(I)).39 From the classical Hofmann–Löffler–Freytag (HLF) reaction to the modern development of transition metal photoredox reactions, C–N bond construction methods have been continuously developed, and although all these strategies have their powerful efficiency and substrate applicability, the vast majority of them can only construct a single C–N bond due to the alteration of the surrounding C–H bond activity caused by the installation of hetero-functionalization. Considering that nitrogen heterocycles are common and essential in bio-molecules and drug-like molecules, researchers have attempted to achieve vicinal C–H diaminization reactions. From previous work, it is known that the photoexcited TAC radical dication is a very strong oxidant for the oxidative functionalization of electron-deficient aromatic hydrocarbons, and they speculate that this oxidant can also be well accomplished for the C–H monoamination at the benzyl position to obtain amide 142, and then the C–H bond at the neighboring position can undergo the secondary amination reaction again. For the mechanism, they suggest that the reaction starts with a process consistent with an electrocatalytic Ritter-type reaction, where the substrate 137 undergoes a single electron transfer (SET) process with TAC˙2+* 113 to form a radical cationic intermediate 138, followed by deprotonation and secondary oxidation to give the benzyl carboncation 140, and then reacts with the solvent acetonitrile to obtain 141, which then form the Ritter-type product 142. The secondary amination process may be caused by the E1 elimination of amide 142 under the catalysis of acid to generate the corresponding styrene derivative 143, which was subsequently oxidized again by TAC˙2+* to form the radical cationic intermediate 144, and then underwent a series of solvent capture and oxidation events to give the final product dihydroimidazolium compounds 146 (Scheme 15(III)). The mechanistic process of the styrene intermediates was demonstrated by the trace amounts of styrene products obtained in the experiments, but the key to diamination is the slow generation of styrene, otherwise too much styrene will dimerize, but no dimerization products were detected under these conditions. This strategy of adjacent C–H bond diamination is not only applicable to styrene substrates with branched chains at the benzylic position, but also to unbranched substrates, which can be achieved simply by changing the solvent. Interestingly, the product generated by changing the electrolyte from Et4NBF4 to LiClO4 changed from dihydroimidazole to 2-oxazoline. A part of the products is displayed in Scheme 15(II) (133–136).
 | ||
| Scheme 15 Photoelectrochemical reaction of vicinal C–H diamination. | ||
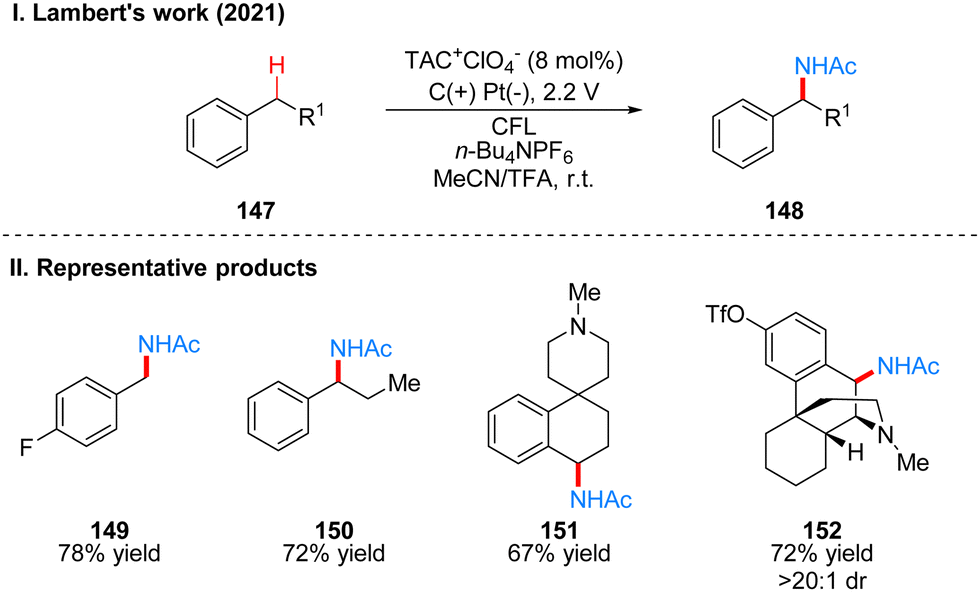
Subsequently, Lambert's group reported another work on photoelectrochemical catalytic Ritter-type C–H monoamination to produce benzylacetamide 148 by substrate 147 (Scheme 16(I)).40 The conditions of this reaction are almost the same as the previous work, also using TAC ions as photoelectrochemical catalyst, except that the reaction is only applicable to substrates without branched chains in the benzyl position and using trifluoroacetic acid (TFA) as co-solvent, if the substrate has branched chains to obtain the above-mentioned imidazoles of diamination products. For the above diamination reaction in the benzyl position without branched substrate also the diamination product can be obtained because of the use of trifluoromethanesulfonic acid (TfOH) as the co-solvent. This strategy also justifies the mechanism of styrene formation by E1 elimination in the diamination process, because the E1 elimination reaction of a branched substrate with TFA at the benzyl position can easily occur, so that the diamination product is obtained, but for a substrate without a branched chain, the E1 elimination reaction is more difficult and requires a stronger acid, such as TfOH. Therefore, only monoamination products can be obtained when TFA is used as a co-solvent in this reaction. The mechanism of the process is the same as that described above for the pre-diamination part and is not repeated here (Scheme 15(III), First amination). The part of products is displayed in Scheme 16(II) (149–152).
 | ||
| Scheme 16 Photoelectrochemical reaction of C–H amination. | ||
The most widely used strategy for the double oxidation of olefins, which is an important part of organic synthesis, is to use transition metal catalysis, especially osmium, but it involves high cost and high toxicity. In 2012, Yoshida's group41 discovered an electrochemical catalytic method to form a bis(alkoxysulfonium) intermediate by electrochemical oxidation of olefins in DMSO, which is then hydrolyzed to produce 1,2-diol. The problem of cost and toxicity is well solved by this method. However, because the “cation pool” strategy42 requires a high oxidation potential, the range of substrates is very limited, and only five substrates are shown in this paper, which leads to the inability of this method to be widely applied. In 2021, Lambert's group used the TAC ion photoelectrochemical catalyst to achieve a highly chemoselective and diastereoselective acetoxyhydroxylation of olefins 156 to obtain 157 (Scheme 17(I)).43 The substrate applicability of this strategy was greatly extended, with good yields for intracyclic olefins, chain olefins, heterocyclic olefins, and 1,3-conjugated diolefins. Carboxylic acids are also not limited to acetic acid, but benzoic acid, cyclopropanecarboxylic acid, thiophene-carboxylic acid, furoic acid, etc. can be obtained as dioxy-genation products. The method can also be used to achieve multigram synthesis of monoester products using a continuous flow system. The part of products is displayed in Scheme 17(II) (158–161). Mechanistically, under an undivided cell with an applied potential of 2.0 V and the irradiation of visible light, the TAC ion is converted to TAC˙2+*, which subsequently oxidizes the olefinic substrate 162 to the radical cation 163. It is then captured by the nucleophilic reagent acetic acid to form radical 164, which was further oxidized to the oxocarbenium intermediate 165, and was finally hydrolyzed to give the dioxygen product 166 (Scheme 17(III)).
 | ||
| Scheme 17 Photoelectrochemical acetoxyhydroxylation of aryl olefin. | ||
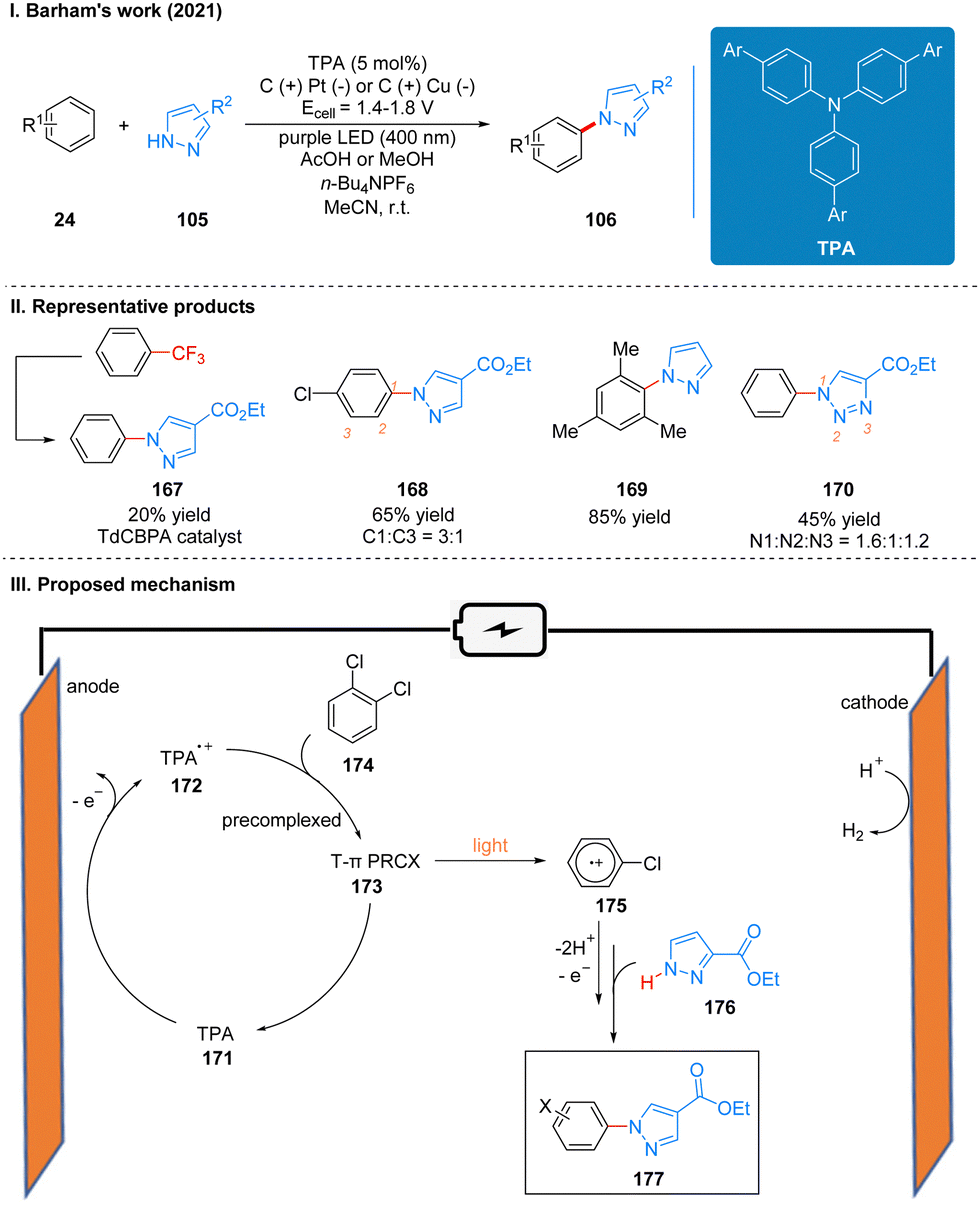 | ||
| Scheme 18 Photoelectrochemical C–H/N–H coupling reaction using TPA. | ||
The reaction efficiency can be optimized for different electron-deficient levels of aromatics by adjusting the aryl group of the TPA in this reaction, and the expected products of very electron-deficient substrates can be observed by using more electron-deficient TPA variants as well. It is noteworthy that the extremely electron-deficient 1,2,4-trifluorobenzene and trifluorotoluene substrates produce not the product of C–H/N–H coupling but the product of SNAr (Scheme 18(II), 167). This is the first time that a chemistry reaction of C(sp2)–CF3 to C(sp2)–N(Het-Ar) substitution has been achieved, and it highlights the potential for using high energy excited states by the precomplex. The part of the products is displayed in Scheme 18(II) (167–170). The report also used a large number of experiments to address the connection between the structure and reactivity of TPA. In contrast to the previous mechanism, the catalyst is oxidized to form TPA radical cations 172 at anode, which are subsequently precomplexed with the substrate 174 to obtain a reactive T-π precomplex (PRCX) 173, and then PRCX is reductively quenched via a single electron transfer (SET) process under the excitation of light to generate aryl radical cations 175 and regenerated TPA catalysts. The aryl radical cation 175 is trapped by the N-heteroarenes 176 to form the product 177 after losing protons and electrons (Scheme 18(III)).
In 2020, Lambert's group also reported a photoelectrochemically catalyzed functionalization reaction of aryl halide reduction in collaboration with Lin (Scheme 19(I)).47 Based on previous work on TAC ions, we can know that the TAC ion (TAC+) was electrochemically oxidized and photoexcited to obtain an open-shell excited state TAC radical dication (TAC˙2+*), which is super oxidizing and can oxidize many substances that are not easily oxidized. They wanted to use electrochemical cathodic reduction and photo-catalysis to obtain an open-shell strongly reductive species for the reduction functionalization of aryl halides with a high reduction potential.
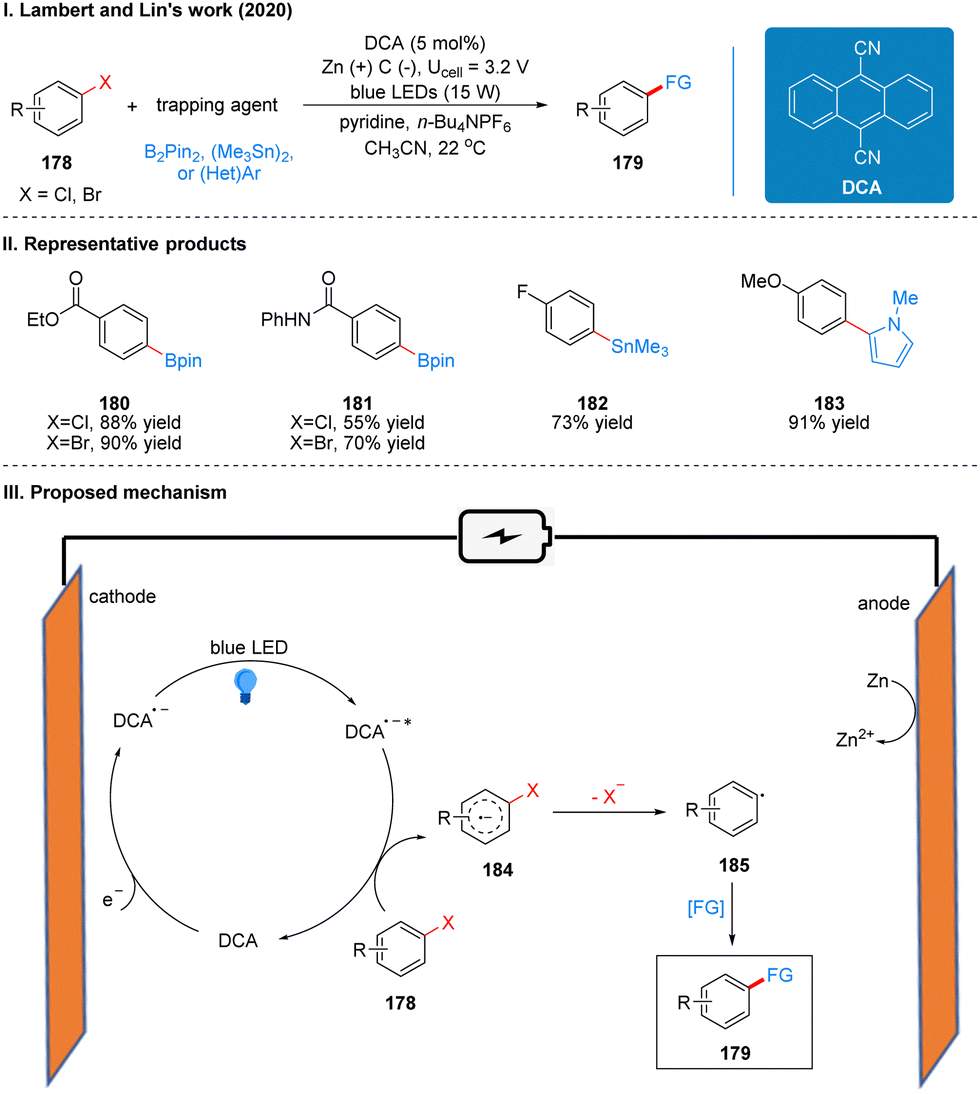 | ||
| Scheme 19 Photoelectrochemical reductive coupling reaction of aryl halide using DCA. | ||
The reaction uses dicyanoanthracene (DCA) as a photoelectro-catalyst, which gets an electron at the cathode to reduce to DCA radical anion (DCA˙−), which is a deep-orange solution. This solution is irradiated with blue LEDs at 15 W to form a strongly reduced excited DCA radical anion (DCA˙−*) (Ered = −3.3 V vs. SCE), which is comparable to many metal monomers (e.g., lithium). The DCA˙−* then undergoes a single electron transfer (SET) process with the aryl halide 178 to obtain the aryl halide radical anion 184, which then undergoes mesolytic cleavage to form the aryl radical 185 and finally completes the functionalization to obtain the product 179 (Scheme 19(III)). To prevent the formation of zinc bridges between the cathode and anode to decrease the yield, the reaction is carried out in a divided cell. Due to the strong reducing properties of DCA˙−*, this catalytic system can be used for the reductive functionalization of some challenging electron-rich aromatics. For example, the reductive borylation of 4-chloroanisole (Ered = −2.90 V vs. SCE), a substrate that cannot be reductively functionalized in a high yield using photoredox catalysis. However, the reactivity was not satisfactory for some chlorinated aromatics, while the brominated aromatics reacted quite well. The intramolecular competition reaction also showed that the C–Br bond breakage was faster than the C–Cl bond breakage, probably due to the fact that the back electron transfer rate from the radical anion intermediate 165 to DCA was much faster than the C–Cl bond breakage. It is worth noting that the system, although it has a strongly reductive properties, is well tolerated for many functional groups, such as ketones, esters, carbamates, amides and other sensitive functional groups. The reaction can also be reductively functionalized using various radical trapping agents as radical acceptors, such as bis(pinacolato)diboron (B2pin2), hexamethylditin, 1,4-difluorobenzene, and N-methyl-pyrrole, extending the photoelectrochemical protocol further to the synthesis of C–C, C–Sn, and C–B bonds. The part of the products is displayed in Scheme 19(II) (180–183).
Coincidentally, Wickens was also inspired by the work of Lambert's group on the photoelectrochemical catalytic generation of the superoxide TAC˙2+*. A photoelectric reaction using N-arylmaleimide (NpMI) as a photoelectrochemical catalyst for the reductive functionalization of aryl halides 186 was reported almost simultaneously (Scheme 20(I)).48 The authors concluded that the open-shell NpMI radical anion (NpMI˙−*), obtained after electrochemical and photochemical excitation of the NpMI catalyst, is highly reductive (Ered = −3.3 V vs. SCE) and its ability is also comparable to that of metallic monolithic sodium lithium. The mechanistic process is similar to the work described above and will not be described here. The difference is that the reaction can be well applied to aryl halides where the previous work does not apply, and the radical trapping agent is replaced with N-methylpyrrole and triethyl phosphite, thus achieving efficient construction of C–P, C–C bonds. A part of the products is displayed in Scheme 20(II) (187–190). Wu and co-workers developed a strategy for the functionalization of aryl halide reduction using perylene diimide (PDI) as a photocatalyst in 2021 (Scheme 21(I)).49 This strategy uses the photoelectrode as the cathode, which uses in interfacial photoelectrochemistry, so we think this protocol is out of our review's scope, and herein we do not make too much introduction, and interested readers can get a closer look at this work through Lambert's review3a or Wu's study.49 The part of the products is displayed in Scheme 21(II) (191–194).
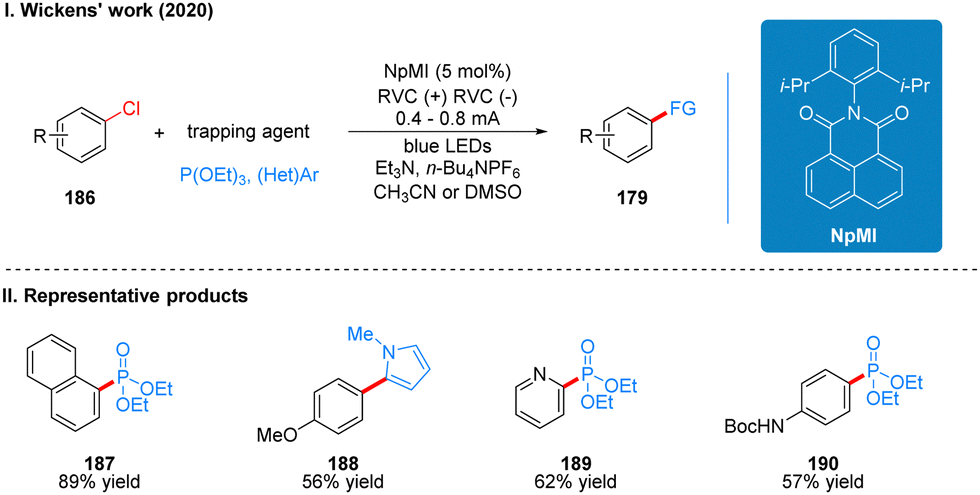 | ||
| Scheme 20 Photoelectrochemical reductive coupling reaction of aryl halide using NpMI. | ||
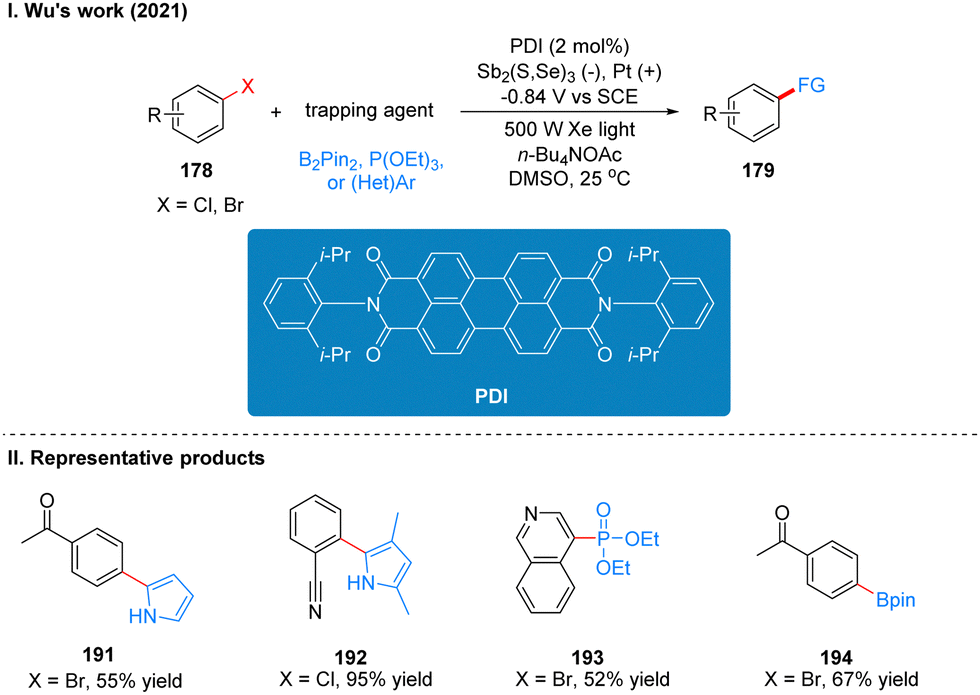 | ||
| Scheme 21 Functionalization of aryl halide reduction by iPEC. | ||
In 2022, Wu's group reported a photoelectrochemically catalyzed transition-metal-free CAr–F arylation reaction. The unique properties of fluorinated organic molecules give them great potential for applications in biomedicine, materials science, catalysts, pesticide and other fields. In the past decades, the introduction of fluorinated organic molecule fractions has mostly relied on transition metal-catalyzed approaches, such as Ni- and Pd-catalyzed cross-coupling of polyfluorinated aromatic hydrocarbons and highly reactive aromatic metals, but the strong metal–F bonds generated in these approaches usually hinder catalyst turnover and thus always require specialized ligands or high temperatures, and toxicity and heavy metal residues are inevitable in these methods. Wu and co-workers utilizes PDI as a photoelectrochemical catalyst to achieve the arylation of polyfluorinated arenes under a current of 1 mA and blue light irradiation (Scheme 22(I)).50 This scheme overcomes the problems of transition metal catalysis mentioned above by combining photoelectrochemical catalytic reduction catalyzed by PDI and radical oxidation of nitrogen–oxygen in a photoelectrochemical catalytic cell, and is also well suited for fluorinated arenes that are inactive in photocatalysis. This protocol has a wide range of substrate applicability and is well tolerated for many functional groups such as ester groups, aldehyde groups, and cyano groups, and it has a high regioselectivity, yields are comparable to those of transition metal catalysts. The late functionalization and gram-scale synthesis of the drug compounds were also achieved smoothly, which shows the value of this protocol in drug synthesis. A part of the products is displayed in Scheme 22(II) (197–200).
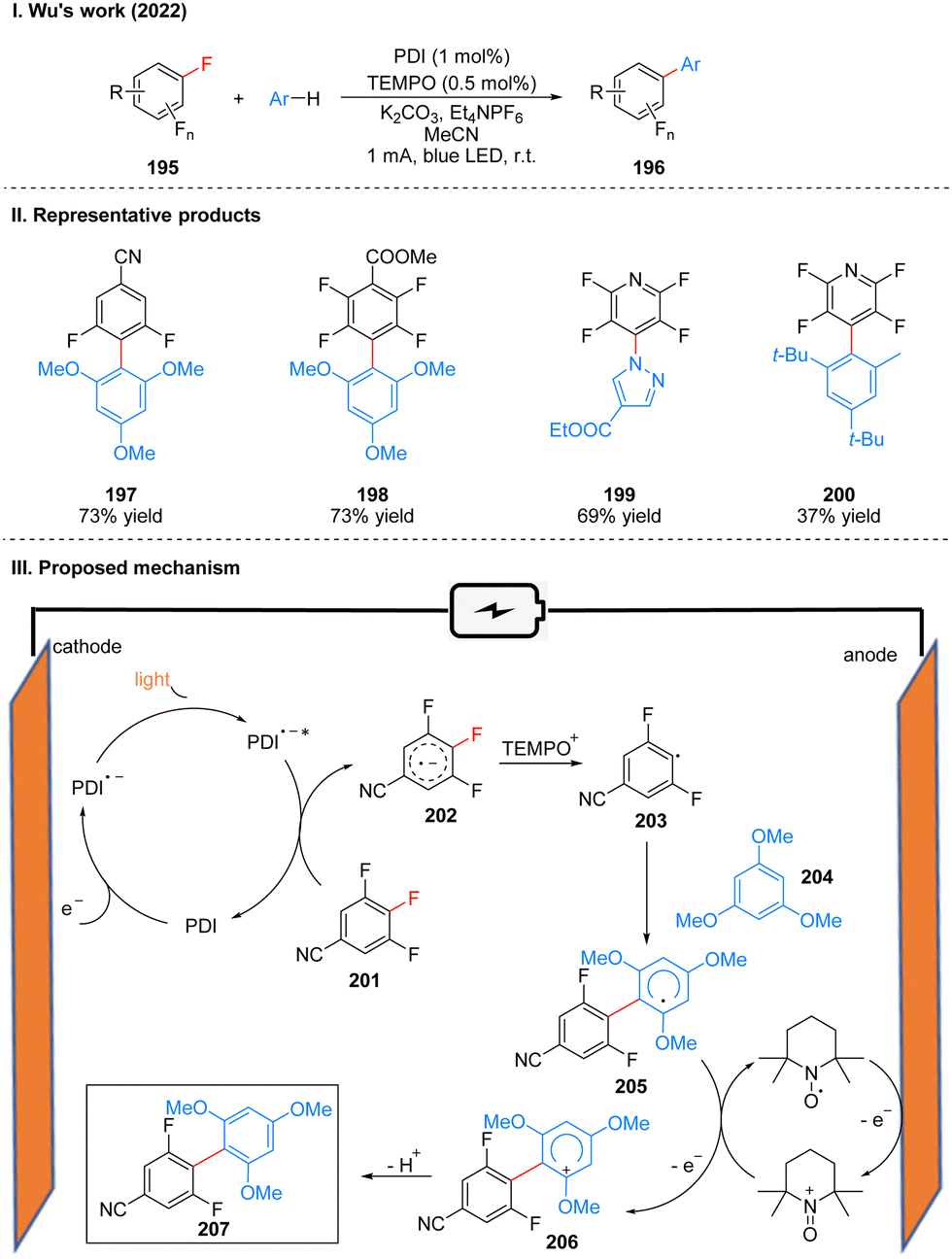 | ||
| Scheme 22 Photoelectrochemistry of CAr–F arylation reaction. | ||
Mechanistically, PDI is first reduced to PDI˙− on the cathode, and then under light irradiation, excited state PDI (PDI˙−*) is formed, which is a catalyst with strong reducing properties. PDI˙−* and the polyfluorinated arene 201 undergo a single electron transfer (SET) process to obtain the radical anion 202, and regenerate the PDI catalyst, which participates in the next catalytic cycle. At the same time, TEMPO is oxidized to TEMPO+ at the anode, which captures the F− in the radical anion 202 to generate the radical 203. 203 is then captured by the aromatics 204 to generate the stable poly-fluorobiphenyl radical 205, which is subsequently oxidized again by TEMPO+ to generate the more stable cationic intermediate 206, and finally undergoes deprotonation to obtain the aromatization product 207 (Scheme 22(III)). It is noteworthy that TEMPO can be oxidized to TEMPO+ at the anode, which can stabilize the highly reactive radical anion 202 and generate the radical 203. TEMPO+ can also oxidize the radical 205 to the cationic intermediate 206.
In 2021, after Lambert,47 Wickens48 reported, Barham and König also reported a photoelectrochemical catalytic reduction of phosphinated alcohols 209 to olefins 210 or saturated alkanes 211 using a n-BuO-NpMI photoelectrochemical catalytic reductant (Scheme 23(I)).51 The authors, influenced by the work of Lambert and Wickens, also tried using DCA and NpMI as photoelectrochemical catalytic reductants, but neither worked well and many phosphinated alcohol substrates failed to react. Thus, the researchers modified the NpMI catalyst to obtain the n-BuO-NpMI catalyst, which can selectively break the C(sp3)–O bond well to obtain the target product. This strategy has a good range of substrate suitability and functional group tolerance, with good yields for esters, ethers, amides, aryl halides and halide analogues. Notably, aryl halides are well tolerated in this scheme compared to Scheme 20,48 due to the fact that the n-BuO-NpMI catalyst allows its radical anions to form a pre-assembly with the phosphate substrate, thus promoting the C(sp3)–O bond cleavage that determines reactivity. The part of the products is displayed in Scheme 23(II) (212–215). As with the previous photoelectrochemical catalytic reductants, under the irradiation of light, n-BuO-NpMI gets an electron to form the excited state of the n-BuO-NpMI radical anion (n-BuO-NpMI˙−*) at the cathode, which reduced the phosphinated alcohol 209 to the radical anion 216. Then the C–O bond is cleaved to obtain the alkyl radical 217, and then a molecule of electrons was obtained to form the carbon anion 218. Subsequently, another molecule of proton is obtained to generate the saturated product 211, or the elimination of the leaving group at the α-position to generate the olefin product 210 (Scheme 23(III)).
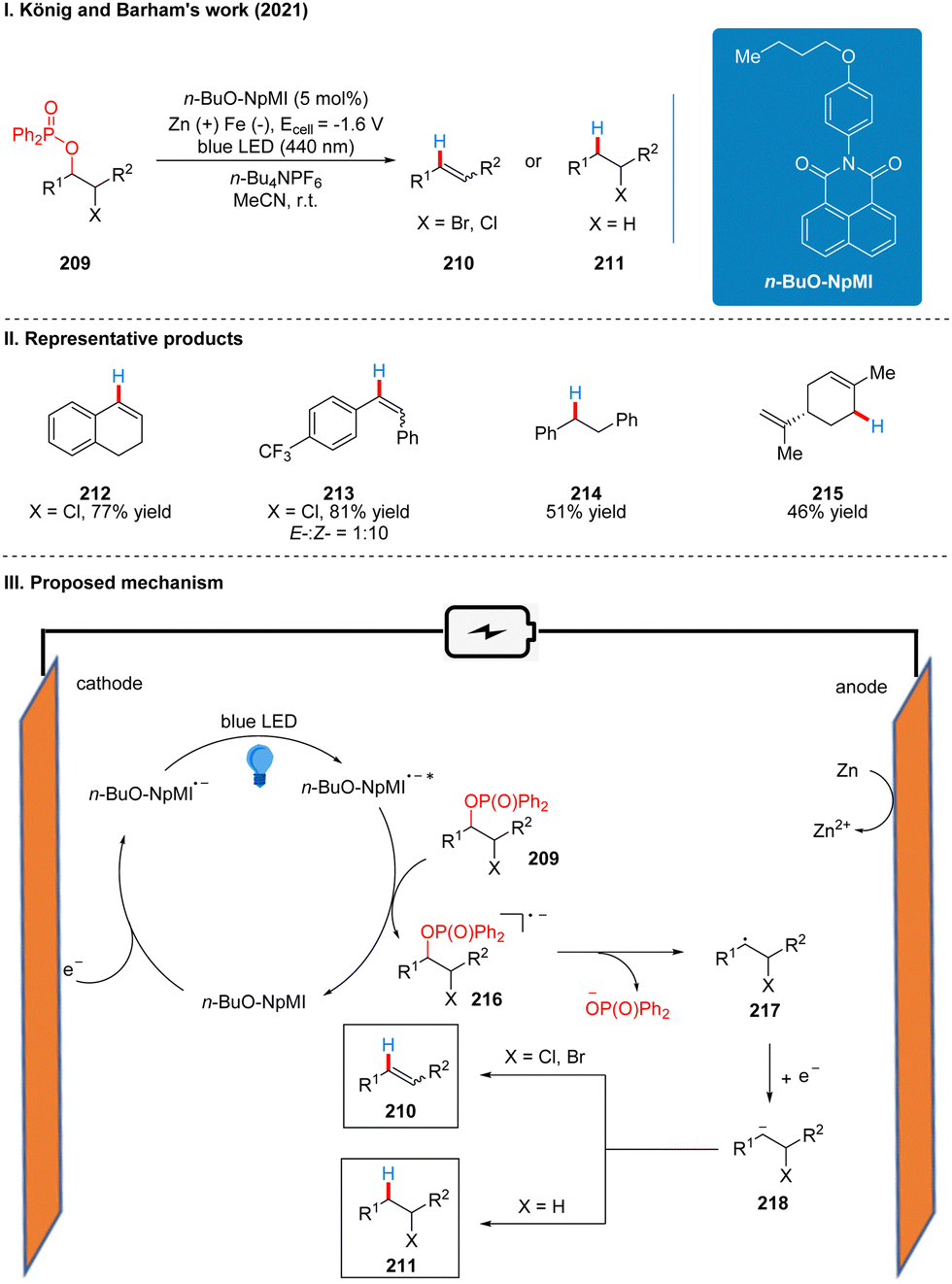 | ||
| Scheme 23 Photoelectrochemical reduction of phosphinated alcohol. | ||
In 2021, Wickens and co-workers discovered another new photoelectrochemical catalytic reductant for the reductive functionalization of strong C(sp2)–N bonds and C(sp2)–O bond cleavage. The reaction utilizes the newly developed 4-DPAIPN as a photoelectrochemical catalyst for the reductive functionalization of trialkyl phenylamine salts or aryl phosphites under the irradiation of 405 nm light in a divided cell with −1.6 V (vs. SCE) potential (Scheme 24(I)).52 In the initial test, the authors used several common photoelectrochemical catalyst reductants, which are used in the above strategy, but none of these catalysts could achieve the desired catalytic effect. Through continuous modification of the catalysts, it was finally found that the 4-DPAIPN catalyst could achieve a very good catalytic effect. The method can be used not only to reduce C(sp2)–N and C(sp2)–O bonds to C(sp2)–H bonds, but also to obtain various heteroaromatic, phosphorylated and boronized products containing C–C, C–P and C–B bonds after the addition of radical trapping reagents. The catalytic approach can also tolerate a range of functional groups such as ethers, esters, amides, free alcohols, and heterocycles. The part of the products is displayed in Scheme 24(II) (220–222).
 | ||
| Scheme 24 Photoelectrochemical reductive functionalization. | ||
6.3 Inorganic electrochemically recycled photocatalysts
In 2020, Xu's group reported a Minisci-type photoelectro-chemical reaction by using CeCl3·7H2O as the catalyst, in which carboxylic acids are decarboxylated under photoelectro-chemical conditions to produce alkyl radicals or carbamoyl radicals to achieve C–H bond coupling to obtain C–H alkylation products 223 or carbamoylation products 224 with hetero-arenes (Scheme 25(I)).53 This reaction is suitable for a wide range of substrates, and good yields can be obtained for various primary, secondary and tertiary aliphatic acids, and good tolerance for functional groups such as ester groups, ether bonds, hydroxyl groups and N-Boc. The part of the products is displayed in Scheme 25(II) (225–228). It is worth noting that C–H alkylation using trivalent cerium as the catalyst requires external strong acid, such as HCl, but the catalytic effect of trivalent cerium in C–H carbamoylation is not satisfactory and requires the use of 4-CzIPN as catalyst instead, and no additional acid is required, probably because the acidity of ammonium oxalate is sufficient as the carbamoylation reagent and proton source.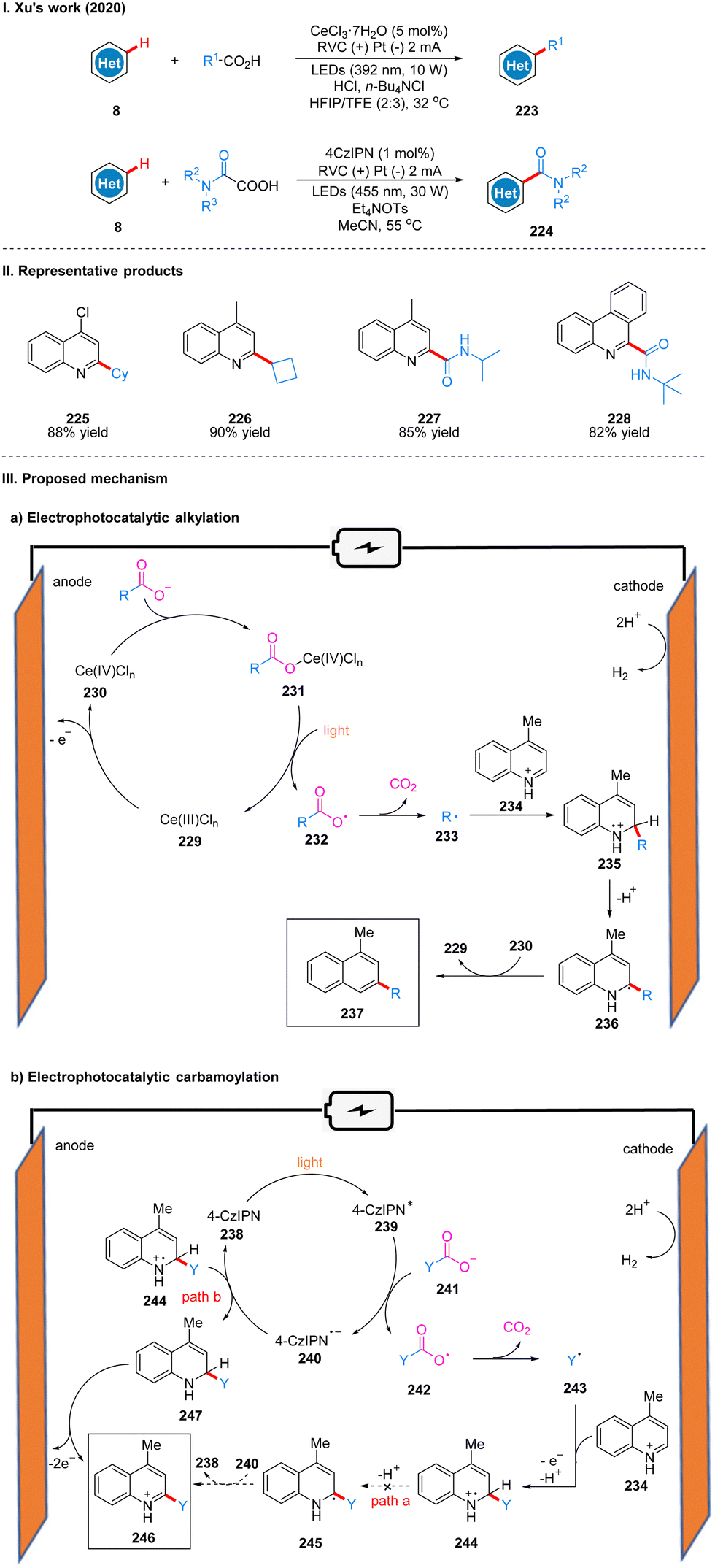 | ||
| Scheme 25 Photoelectrochemical alkylation of heteroarene. | ||
In the reactions of these two different catalysts, the mechanisms are also slightly different. In the mechanism catalyzed by cerium, Ce(III) 229 is first oxidized to Ce(IV) 230 at the anode, which subsequently coordinated with carboxylic acid to form a complex 231, producing a carboxyl radical 232 under light irradiation, then decarboxylation gives an alkyl radical 233, and the alkyl radical 233 is coupled to a protonated lepidine 234 to produce a radical cation 235. Then it undergoes deprotonation to form a radical intermediate 236, and oxidation by Ce(IV) to form an alkylation product 237 (Scheme 25(IIIa)). In the mechanism of 4-CzIPN catalysis, 4-CzIPN 238 is irradiated by light to obtain the excited state of 4-CzIPN (4-CzIPN*) 239, followed by a single electron transfer (SET) process with the oxalate ion 241 to obtain the carboxyl radical 242 and 4-CzIPN radical anion (4-CzIPN˙−) 240, then 242 undergoes decarboxylation to obtain the carbamoyl radical 243. The coupling of the carbamoyl radical 243 and the protonated lepidine yields the radical cation 244 in this step because the reduction potential of 4-CzIPN (Ep/2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) red = −1.22 V vs. SCE) is lower than that of the protonated product 244 (Ep/2red = −0.72 V vs. SCE), thus 4-CzIPN cannot successfully oxidize the intermediate 245, but can only accept an electron to obtain dihydroquinoline intermediate 247 by highly reduced 4-CzIPN radical anion through route B, and finally 247 was oxidized at the anode to obtain the carbamoylated product 246 (Scheme 25(IIIb)). In addition, the production of dihydroquinoline was also detected in the experiment to justify the above mechanism. The strategy can be easily scaled up and successfully achieved the synthesis of alkylation and carbamoylation products on the decagram scale.
red = −1.22 V vs. SCE) is lower than that of the protonated product 244 (Ep/2red = −0.72 V vs. SCE), thus 4-CzIPN cannot successfully oxidize the intermediate 245, but can only accept an electron to obtain dihydroquinoline intermediate 247 by highly reduced 4-CzIPN radical anion through route B, and finally 247 was oxidized at the anode to obtain the carbamoylated product 246 (Scheme 25(IIIb)). In addition, the production of dihydroquinoline was also detected in the experiment to justify the above mechanism. The strategy can be easily scaled up and successfully achieved the synthesis of alkylation and carbamoylation products on the decagram scale.
In 2022, Lei's group developed a photoelectrochemical efficient and highly selective ring-opening functionalization reaction of cyclic alcohols. Using Ce(III) as a photoelectrochemical catalyst, the ring-opening functionalization reaction of the cyclic alcohol 248 or 249 underwent a constant current of 2 mA and blue LEDs irradiation to obtain the remote functionalized product ketone 250 or 251 (Scheme 26(I)).54 The reaction is not only suitable for ring-opening alkylation, thioetherification and oxime etherification of cycloalcohols, but also ring-opening chlorination, alkenylation and arylation by slightly modifying the reaction conditions, showing an unprecedented range of substrates for the synthesis of compounds with remote C–CN, C–C, C–S, C–Cl, C–oxime bonds, etc. The part of the products is displayed in Scheme 26(II) (252–255). Mechanistically, the researchers used a CeIIICl3 precursor dissolved in acetonitrile with the help of Et4NCl to obtain [CeIIICl6]3−256, which was subsequently oxidized to Ce(IV) species at the anode. As evidenced by UV-vis and X-ray absorption spectroscopy, ligand exchange with the alcohol 258 in the presence of base generates Ce(IV)–alcohol salt intermediate 257. Subsequently, under the irradiation of light, intermediate 257 undergoes homogeneous photolysis by ligand metal charge transfer (LMCT) to form the corresponding alkoxy radical 259 and regenerated [CeIIICl6]3−. Due to the instability of the oxygen-centered radical, 259 occurs β-cleavage to get a carbonyl-containing remote alkyl radical 260, and the aryl sulfonyl compound acts as a radical acceptor to receive the radical 260, finally giving the functionalized product 261, with the sulfonyl group being reduced to benzenesulfonate at the cathode (Scheme 26(III)). The experiments showed that the ring tension of the cycloalkyl alcohols had a small effect on the reaction rate, and the substituents on the aryl ring alcohols had no obvious effect on the reaction yield.
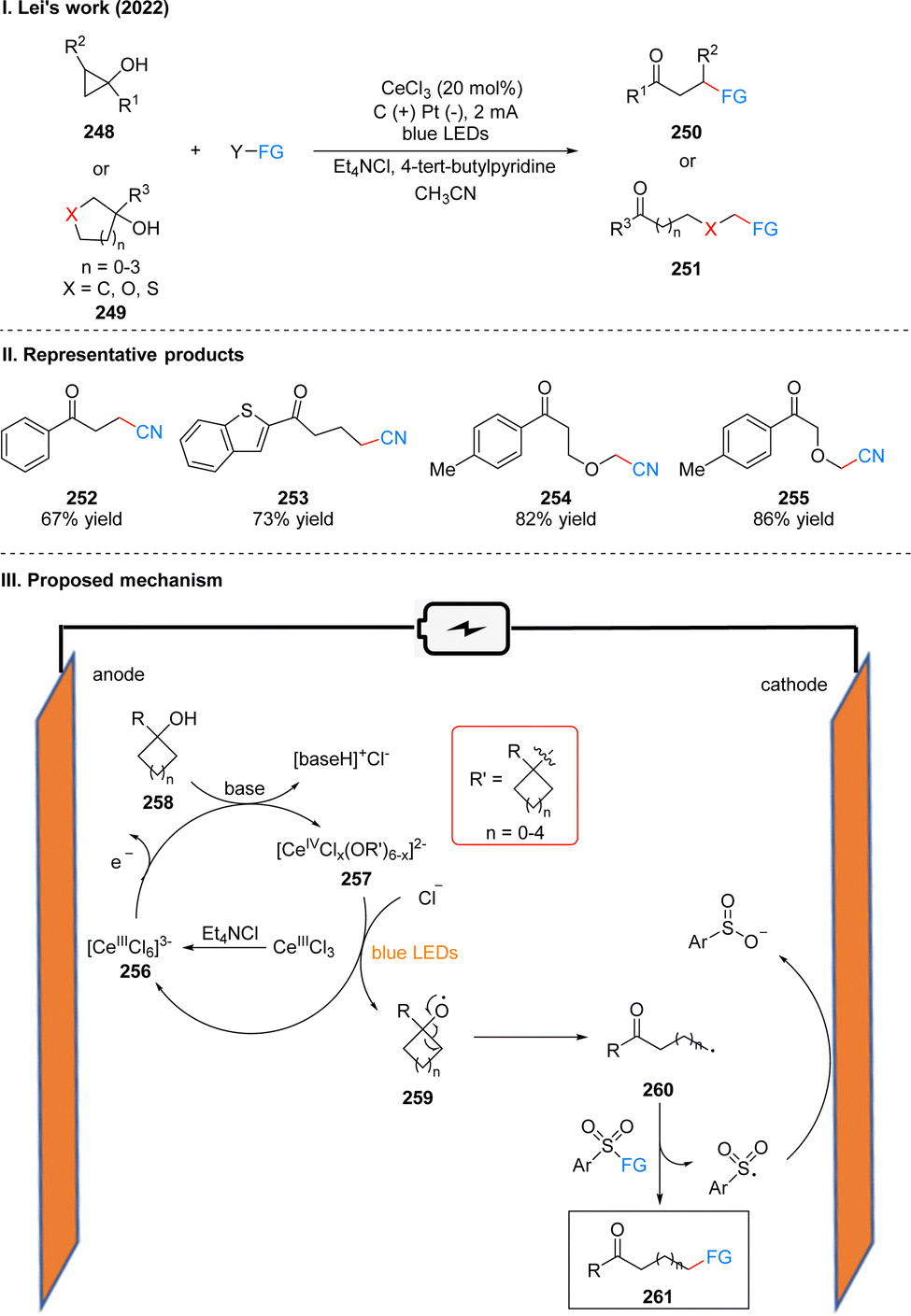 | ||
| Scheme 26 Photoelectrochemical ring-opening functionalization of cyclic alcohol. | ||
In 2022, Fu's group reported a photoelectrochemical reaction of carboxylic acid 262 decarboxylative azidation catalyzed by the combination of manganese and cerium metal, using TMS-N3263 as the nitrogen source (Scheme 27(I)).55 The reaction showed good tolerance for functional groups such as ketones, cyano, aminocarbonates, and heteroarenes. The part of the products is displayed in Scheme 27(II) (265–268). Mechanistically, Mn(II) catalyst and N3− coordinate to produce intermediate 87 by anodic oxidation. In another metal-catalytic cycle, Ce(III) was oxidized to Ce(IV) at the anode, and coordinated with carboxylate 269 to obtain Ce(IV)–carboxylate complex intermediate 270. And the intermediate 270 was decarboxylated under blue LED irradiation to form the alkyl radical 271 and regenerated Ce(III) catalyst. The alkyl radical 271 is captured by the Mn(III)–N3 intermediate to produce the final azide product 264 (Scheme 27(III)). This protocol is a good decarboxylative azidation method for some electron-rich molecules that are prone to oxidative decomposition under conventional chemical conditions, researchers have used several arylacetic acids for reactions of decarboxylative azidation under conventional oxidative conditions, without obtaining desirable yields that could be used in a synthetic sense, and this reaction also allows the application of separate photochemical and electrochemical parts for large-scale synthesis using a flow device.
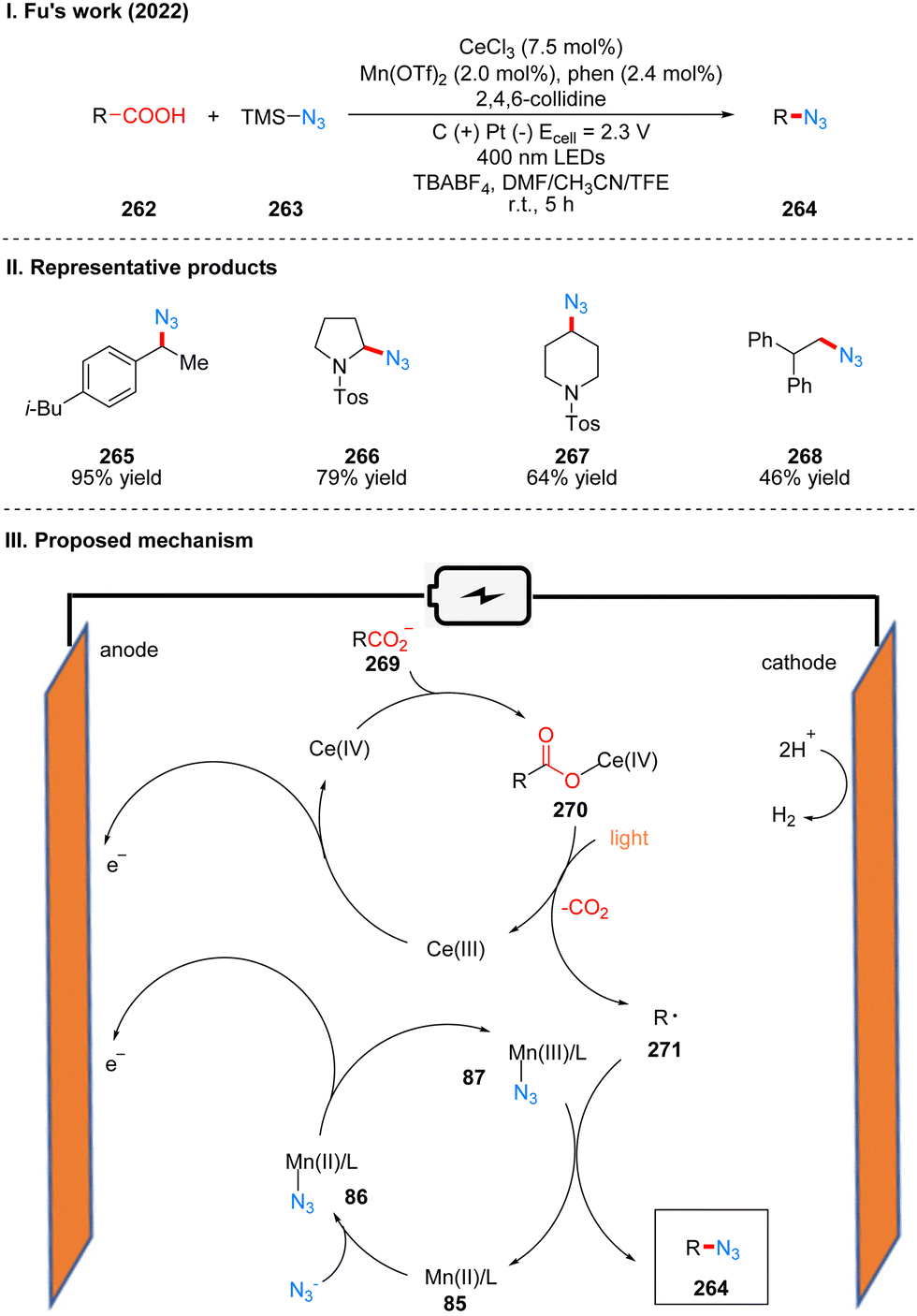 | ||
| Scheme 27 Photoelectrochemical decarboxylative azidation of carboxylic acid. | ||
In 2022, Chen, Liu, and Li collaborated to report an electro-photocatalysis promote alkyl radical cycling in situ for the alkylation of heteroarenes (Scheme 28(I)).56 Using a reticulated vitreous carbon electrode at a potential of −1.4 V (vs. Fc/Fc+) and blue LEDs irradiation, heteroarene and Katritzky salts were alkylated under the photoelectrochemical conditions in the presence of catalytic Ru(bpy)3Cl2·6H2O to obtain heteroarene alkylation products 10. The reaction exhibits a surprisingly wide range of substrate suitability, including a wide variety of cycloalkanes, heterocyclic alkanes, straight chain alkanes and heteroarenes, and can also be used for ligand modification and late functionalization of bioactive scaffolds, which will have great potential for applications in the fields of pharmaceuticals, natural products and materials science. The part of the products is displayed in Scheme 28(II) (266–269). The key to the success of this reaction is the use of the photocatalyst to promote the cycle of alkyl radical in situ. The reduction of Katritzky salts 270 at the cathode forms the persistent radical intermediate 271, followed by the release of energy to undergo N–C bond breaking to give the alkyl radical 272. The alkyl radical 272via energy release drives the cross-coupling reaction of the alkyl radical 272 and the radical intermediate 271 to form the intermediate 273, which is a relatively fast process. Subsequently, the intermediate 273 was oxidized to form alkyl radical 272 and Katritzky salt 270 by Ru(III), completing the radical cycle. The alkyl radical 272 can also undergo a high-energy radical addition reaction with the quinoline to form the nitrogen radical cation 274, and then 274 undergoes a SET process with excited Ru(II) to produce 2-alkylquinoline 275 and Ru(III), which is used in the in situ radical cycle. 2-Alkylquinoline 275 is re-aromatized by oxidation at the anode to give the product 276 (Scheme 28(III)).
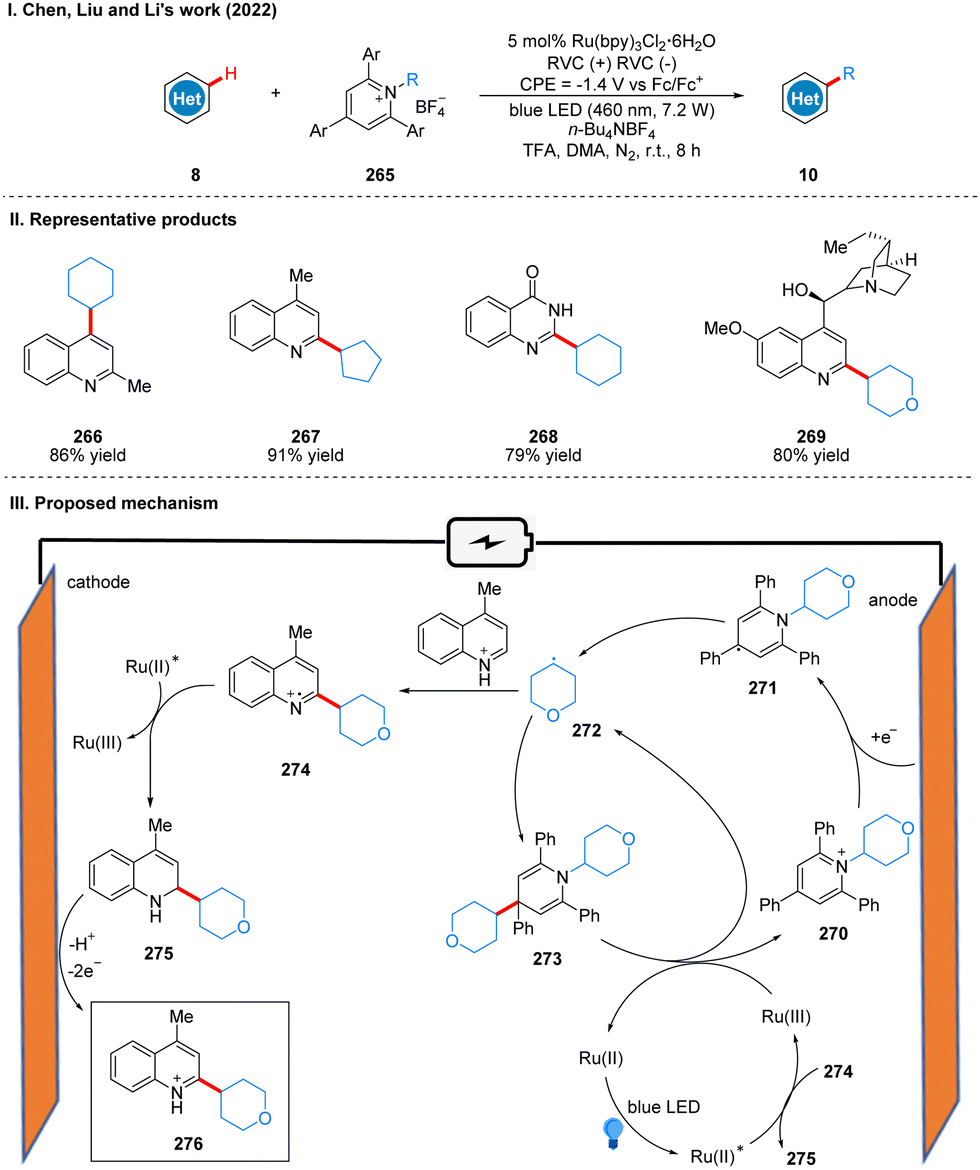 | ||
| Scheme 28 Photoelectrochemical alkylation of heteroarene using Ru catalyst. | ||
7. Conclusions and prospects
Photocatalysis and electrocatalysis are two important strategies in organic synthesis, but because of their respective limitations, chemists have developed a method to combine the two together, namely the photoelectrochemical strategy, which effectively overcomes the disadvantages of the net redox reaction in photocatalysis requiring the addition of terminal oxidants and the easy peroxidation and dimerization in electrocatalysis are eliminated, and the advantages of the two are perfectly combined to become a more effective organic synthetic strategy. Moreover, the preassembly technique involved in Scheme 18 can overcome the short lifetime of photoexcited anion/cation doublet states and achieve the photochemistry of higher-order doublet (Dn) excited states, breaking through the extremely high redox potential. Thus, preassembly is very important in the design and modification of photoelectrochemical catalysts to help us achieve hi-power redox reactions. Because of these advantages, photoelectrochemical strategy is expected to: (1) widen the “redox window” of SET chemistry; (2) provide milder conditions allowing significant functional group tolerance and chemoselectivity; and (3) improve energy conservation and atom economy.Although the photoelectrochemical strategy has many advantages, there are some areas that need to be improved. First, the experimental setups and reaction operations used in photoelectrochemistry have a high degree of complexity, which is a great challenge for some researchers who are new to the field or some non-experts. Second, the photoelectrochemical strategy does not completely eliminate redox additives, stoichiometric redox additives are present in reactions, and due to the use of electrochemical potentials there is an inevitable faradaic efficiency problem in the reactions, so the field needs to seek solutions to address the issue of limited faradaic efficiency. Finally, most of the photoelectrochemical catalysts currently used are selected from those used in photochemistry or electrochemistry, and in this review, many researchers in the work presented use the almost same photocatalysts with similar catalytic mechanisms, and therefore there is a paucity of photoelectrochemical catalysts available for the development. We can strengthen the research on catalysts to discover more efficient catalysts for various types of photoelectrochemical reactions and to expand the scope of photoelectrochemical reactions so that the redox functionalization of more challenging substrates can be accomplished.
Photoelectrochemistry, as a rapidly developing and emerging research field in recent years, is developing rapidly, and many classical reactions can be improved by photoelectrochemistry to upgrade the mild reaction conditions and the applicable scope of substrates, thus expanding the application scope of the reaction. Although there are still many shortcomings and areas that can be improved, the authors believe that in the future, with the further development of photoelectrochemical strategy, it will become another important synthetic strategy after photocatalysis and electrocatalysis.
Author contributions
Shi, M. directed the perspective and revised the manuscript. Qian, L. carried out the literature collection, organization, and wrote the manuscript. Qian, L drew the schemes and checked them.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from the National Key R & D Program of China (2022YFC2303100), the National Natural Science Foundation of China (21372250, 21121062, 21302203, 20732008, 21772037, 21772226, 21861132014, 91956115 and 22171078) and the Fundamental Research Funds for the Central Universities (222201717003).Notes and references
- (a) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC; (b) J.-P. Goddard, C. Ollivier and L. Fensterbank, Acc. Chem. Res., 2016, 49, 1924–1936 CrossRef CAS PubMed; (c) M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed; (d) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed; (e) S. Crespi and M. Fagnoni, Chem. Rev., 2020, 120, 9790–9833 CrossRef CAS PubMed; (f) A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. F. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Chem. Rev., 2022, 122, 1485–1542 CrossRef CAS; (g) N. Holmberg-Douglas and D. A. Nicewicz, Chem. Rev., 2022, 122, 1925–2016 CrossRef CAS PubMed.
- (a) J.-i Yoshida, K. Kataoka, R. Horcajada and A. Nagaki, Chem. Rev., 2008, 108, 2265–2299 CrossRef CAS PubMed; (b) R. Francke and R. D. Little, Chem. Soc. Rev., 2014, 43, 2492–2521 RSC; (c) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed; (d) K. D. Moeller, Chem. Rev., 2018, 118, 4817–4833 CrossRef CAS PubMed; (e) J. Liu, L. Lu, D. Wood and S. Lin, ACS Cent. Sci., 2020, 6, 1317–1340 CrossRef CAS PubMed; (f) J. C. Siu, N. K. Fu and S. Lin, Acc. Chem. Res., 2020, 53, 547–560 CrossRef CAS PubMed; (g) C. J. Zhu, N. W. J. Ang, T. H. Meyer, Y. A. Qiu and L. Ackermann, ACS Cent. Sci., 2021, 7, 415–431 CrossRef CAS; (h) L. F. T. Novaes, J. J. Liu, Y. F. Shen, L. X. Lu, J. M. Meinhardt and S. Lin, Chem. Soc. Rev., 2021, 50, 7941–8002 RSC.
- (a) H. Huang, K. A. Steiniger and T. H. Lambert, J. Am. Chem. Soc., 2022, 144, 12567–12583 CrossRef CAS PubMed; (b) J. P. Barham and B. König, Angew. Chem., Int. Ed., 2020, 59, 11732–11747 CrossRef CAS PubMed; (c) S. Z. Wu, J. Kaur, T. A. Karl, X. H. Tian and J. P. Barham, Angew. Chem., Int. Ed., 2022, 61, e202107811 CAS.
- (a) D. M. Hedstrand, W. M. Kruizinga and R. M. Kellogg, Tetrahedron Lett., 1978, 19, 1255–1258 CrossRef; (b) T. J. van Bergen, D. M. Hedstrand, W. M. Kruizinga and R. M. Kellogg, J. Org. Chem., 1979, 44, 4953–4962 CrossRef CAS.
- M. A. Ischay, M. E. Anzovino, J. Du and T. P. Yoon, J. Am. Chem. Soc., 2008, 130, 12886–12887 CrossRef CAS PubMed.
- D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77–80 CrossRef CAS PubMed.
- J. M. R. Narayanam, J. W. Tucker and C. R. J. Stephenson, J. Am. Chem. Soc., 2009, 131, 8756–8757 CrossRef CAS PubMed.
- L. Furst, B. S. Matsuura, J. M. R. Narayanam, J. W. Tucker and C. R. J. Stephenson, Org. Lett., 2010, 12, 3104–3107 CrossRef CAS PubMed.
- A. G. A. Volta, Nat. Philos. Chem. Arts, 1800, 4, 179–187 Search PubMed.
- M. Faraday, Ann. Phys., 1834, 47, 438 Search PubMed.
- C. F. Schoenbein, Liebigs Ann. Chem., 1845, 54, 164 Search PubMed.
- H. J. Kolbe, Prakt. Chem., 1847, 41, 137 CrossRef.
- J.-C. Moutet and G. Reverdy, Tetrahedron Lett., 1979, 20, 2389–2392 CrossRef.
- J.-C. Moutet and G. Reverdy, J. Chem. Soc., Chem. Commun., 1982, 654–655 RSC.
- R. Scheffold and R. Orlinski, J. Am. Chem. Soc., 1983, 105, 7200–7202 CrossRef CAS.
- H. Yan, Z. W. Hou and H. C. Xu, Angew. Chem., Int. Ed., 2019, 58, 4592–4595 CrossRef CAS PubMed.
- J. K. Matsui, D. N. Primer and G. A. Molander, Chem. Sci., 2017, 8, 3512–3522 RSC.
- Y. Qiu, A. Scheremetjew, L. H. Finger and L. Ackermann, Chem, 2020, 26, 3241–3246 CrossRef CAS PubMed.
- H. Huang and T. H. Lambert, Angew. Chem., Int. Ed., 2020, 59, 658–662 CrossRef CAS PubMed.
- N. E. S. Tay and D. A. Nicewicz, J. Am. Chem. Soc., 2017, 139, 16100–16104 CrossRef CAS PubMed.
- H. Huang and T. H. Lambert, Angew. Chem., Int. Ed., 2021, 60, 11163–11167 CrossRef CAS PubMed.
- (a) K. Ohkubo, A. Fujimoto and S. Fukuzumi, J. Am. Chem. Soc., 2013, 135, 5368–5371 CrossRef CAS PubMed; (b) K. Ohkubo, K. Hirose and S. Fukuzumi, Chem. – Eur. J., 2015, 21, 2855–2861 CrossRef CAS PubMed.
- (a) S. Das, P. Natarajan and B. König, Chem. – Eur. J., 2017, 23, 18161–18165 CrossRef CAS PubMed . For a recent review, see:; (b) P. Natarajan and B. König, Eur. J. Org. Chem., 2021, 2145–2161 CrossRef CAS.
- A related photocatalytic arene hydroxylation/amination reaction that requires UV light has also been reported: Y.-W. Zheng, B. Chen, P. Ye, K. Feng, W. Wang, Q.-Y. Meng, L.-Z. Wu and C.-H. Tung, J. Am. Chem. Soc., 2016, 138, 10080–10083 CrossRef CAS PubMed.
- W. Zhang, K. L. Carpenter and S. Lin, Angew. Chem., Int. Ed., 2020, 59, 409–417 CrossRef CAS PubMed.
- C. Feldmeier, H. Bartling, K. Magerl and R. M. Gschwind, Angew. Chem., Int. Ed., 2015, 54, 1347–1351 CrossRef CAS PubMed.
- G. de Gonzalo and M. W. Fraaije, ChemCatChem, 2013, 5, 403–415 CrossRef CAS.
- L. Niu, C. Jiang, Y. Liang, D. Liu, F. Bu, R. Shi, H. Chen, A. D. Chowdhury and A. Lei, J. Am. Chem. Soc., 2020, 142, 17693–17702 CrossRef CAS PubMed.
- L. Capaldo, L. L. Quadri, D. Merli and D. Ravelli, Chem. Commun., 2021, 57, 4424–4427 RSC.
- M. C. Quattrini, S. Fujii, K. Yamada, T. Fukuyama, D. Ravelli, M. Fagnoni and I. Ryu, Chem. Commun., 2017, 53, 2335–2338 RSC.
- H. Huang, Z. M. Strater, M. Rauch, J. Shee, T. J. Sisto, C. Nuckolls and T. H. Lambert, Angew. Chem., Int. Ed., 2019, 58, 13318–13322 CrossRef CAS PubMed.
- F. Gerson, G. Plattner and Z. Yoshida, Mol. Phys., 1971, 21, 1027–1032 CrossRef CAS.
- R. Weiss and K. Schloter, Tetrahedron Lett., 1975, 16, 3491–3494 CrossRef.
- R. W. Johnson, Tetrahedron Lett., 1976, 17, 589–592 CrossRef.
- N. A. Romero, K. A. Margrey, N. E. Tay and D. A. Nicewicz, Science, 2015, 349, 1326–1330 CrossRef CAS PubMed.
- L. Niu, H. Yi, S. Wang, T. Liu, J. Liu and A. Lei, Nat. Commun., 2017, 8, 14226–14232 CrossRef PubMed.
- L. Zhang, L. Liardet, J.-S. Luo, D. Ren, M. Grätzel and X. L. Hu, Nat. Catal., 2019, 2, 366–373 CrossRef CAS PubMed.
- H. Huang, Z. M. Strater and T. H. Lambert, J. Am. Chem. Soc., 2020, 142, 1698–1703 CrossRef CAS PubMed.
- T. Shen and T. H. Lambert, Science, 2021, 371, 620–626 CrossRef CAS PubMed.
- T. Shen and T. H. Lambert, J. Am. Chem. Soc., 2021, 143, 8597–8602 CrossRef CAS PubMed.
- Y. Ashikari, T. Nokami and J.-I. Yoshida, Org. Lett., 2012, 14, 938–941 CrossRef CAS PubMed.
- J.-I. Yoshida, A. Shimizu and R. Hayashi, Chem. Rev., 2018, 118, 4701–4730 CrossRef PubMed.
- H. Huang and T. H. Lambert, J. Am. Chem. Soc., 2021, 143, 7247–7252 CrossRef CAS.
- S. Wu, J. Žurauskas, M. Domański, P. S. Hitzfeld, V. Butera, D. J. Scott, J. Rehbein, A. Kumar, E. Thyrhaug, J. Hauer and J. P. Barham, Org. Chem. Front., 2021, 8, 1132–1142 RSC.
- D. Y. Jeong, D. S. Lee, H. L. Lee, S. Nah, J. Y. Lee, E. J. Cho and Y. You, ACS Catal., 2022, 12, 6047–6059 CrossRef CAS.
- J. S. Beckwith, A. Aster and E. Vauthey, Phys. Chem. Chem. Phys., 2022, 24, 568–577 RSC.
- H. Kim, H. Kim, T. H. Lambert and S. Lin, J. Am. Chem. Soc., 2020, 142, 2087–2092 CrossRef CAS.
- N. G. W. Cowper, C. P. Chernowsky, O. P. Williams and Z. K. Wickens, J. Am. Chem. Soc., 2020, 142, 2093–2099 CrossRef CAS PubMed.
- Y.-J. Chen, T. Lei, H.-L. Hu, H.-L. Wu, S. Zhou, X.-B. Li, B. Chen, C.-H. Tung and L.-Z. Wu, Matter, 2021, 4, 2354–2366 CrossRef CAS.
- Y. J. Chen, W. H. Deng, J. D. Guo, R. N. Ci, C. Zhou, B. Chen, X. B. Li, X. N. Guo, R. Z. Liao, C. H. Tung and L. Z. Wu, J. Am. Chem. Soc., 2022, 144, 17261–17268 CrossRef CAS PubMed.
- X. Tian, T. A. Karl, S. Reiter, S. Yakubov, R. de Vivie-Riedle, B. König and J. P. Barham, Angew. Chem., Int. Ed., 2021, 60, 20817–20825 CrossRef CAS PubMed.
- C. P. Chernowsky, A. F. Chmiel and Z. K. Wickens, Angew. Chem., Int. Ed., 2021, 60, 21418–21425 CrossRef CAS PubMed.
- X. L. Lai, X. M. Shu, J. Song and H. C. Xu, Angew. Chem., Int. Ed., 2020, 59, 10626–10632 CrossRef CAS.
- Z. Yang, D. Yang, J. Zhang, C. Tan, J. Li, S. Wang, H. Zhang, Z. Huang and A. Lei, J. Am. Chem. Soc., 2022, 144, 13895–13902 CrossRef CAS.
- Y. Wang, L. Li and N. Fu, ACS Catal., 2022, 12, 10661–10667 CrossRef CAS.
- K. Wang, X. Liu, S. Yang, Y. Tian, M. Zhou, J. Zhou, X. Jia, B. Li, S. Liu and J. Chen, Org. Lett., 2022, 24, 3471–3476 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |