
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFirst-principles study of interfacial features and charge dynamics between spiro-MeOTAD and photoactive lead halide perovskites†
Adriana
Pecoraro
 a,
Francesca
Fasulo
b,
Michele
Pavone
bc and
Ana B.
Muñoz-García
*ac
a,
Francesca
Fasulo
b,
Michele
Pavone
bc and
Ana B.
Muñoz-García
*ac
aDepartment of Physics “E. Pancini”, University of Naples Federico II, Napoli, Italy. E-mail: anabelen.munozgarcia@unina.it
bDepartment of Chemical Sciences, University of Naples Federico II, Napoli, Italy
cNational Reference Center for Electrochemical Energy Storage (GISEL)-INSTM, Florence, Italy
First published on 3rd April 2023
Abstract
The main stability and performance issues of perovskite solar cells arise from the interfaces between the perovskite and the hole transport material. Here we address these interface issues by means of state-of-the-art first-principles calculations, providing new insights into charge transfer times and mechanisms and how they depend on the perovskite chemical composition and local interfacial environment.
Solar cells with hybrid organic–inorganic lead halide perovskites (LHPs) as photoactive moieties revolutionized the photovoltaic scenario, thanks to their intrinsic properties such as low exciton binding energies, high absorption coefficients, and long charge carrier lifetimes.1 The efficiencies of perovskite solar cells (PSCs) rapidly increased when moving from the original methylammonium lead triiodide (MAPbI3, MAPI) to LHPs with mixed cationic and/or anionic compositions.2 In particular, the triple cation/dual anion Cs0.05(FA0.83MA0.17)0.95Pb(I0.83Br0.17)3 stoichiometry with MA = CH3NH3+, FA = HC(NH2)+ (called triLHP in this work) has become a standard in PSCs due to its high efficiency, improved stability and device reproducibility.3 This improvement has been ascribed to an entropy-induced major structural stability of the LHP, diminished defect content and better electronic properties.4 Here, we aim at assessing if and to what extent compositional changes from MAPI to triLHP also affect charge transfer (CT) properties at the LHP/spiro-MeOTAD interface, being spiro-MeOTAD the archetypal hole transport material (HTM) used in start-of-the-art PSCs.5,6 In particular, we use hybrid density functional theory (DFT)-based calculations to study interfacial structural and electronic features, and, for the first time, we outline the charge dynamics features of such interfaces and estimate hole injection rates with the projection-operator diabatization (POD) approach,7 as recently applied for the MAPI/TiO2 interface.8
The computational details and structural models of the bulk LHP (Fig. S1, ESI†) and interfaces studied in this work are collected in the ESI.† A comprehensive discussion on the thermodynamic stability of PSC interfaces is provided in a recent review.9 To build LHP/spiro-MeOTAD interfaces, we considered the thermodynamically favored (010) surface10 with both AX terminations (A = MA in MAPI or Cs, FA, MA in triLHP) and PbX2 terminations (X = I in MAPI or I, Br in triLHP) since surface terminations have been found to influence the energy of spiro-MeOTAD frontier orbitals11 and because we are interested in relating the CT features to changes in chemical interactions. For these reasons, we also considered different local compositions of both terminations in triLHP (Fig. S2, ESI†): AX terminations exposing or not the Cs atom (CsFAMAX and FAMAX in the text, respectively), plus an additional CsFAMAX layer with a maximized interaction of Cs with the most active methoxy groups of spiro-MeOTAD,12,13 CsFAMAX(O–Cs). Similarly, we considered two PbX2-terminated surfaces, lacking or containing a Cs atom at the subsurface layer, PbX2 and PbX2(Cs), respectively. The minimum-energy structures of all these interfaces are shown in Fig. 1a. The most stable interfaces have been determined in terms of binding energies (Eb) between the LHP and spiro-MeOTAD moieties computed as Eb = ELHP/Spiro − ELHP − ESpiro, where ELHP/Spiro, ELHP and ESpiro are the electronic energies of the fully relaxed interfaces, isolated LHP surfaces, and isolated spiro-MeOTAD molecule, respectively. Total Eb values have been further decomposed into three main contributions:
| Eb = Ea + ESpirod + ELHPd = (ELHP/Spiro − ELHP* − ESpiro*) + (ESpiro* − ESpiro) + (ELHP* − ELHP) | (1) |
 | ||
| Fig. 1 (a) Lateral views of the relaxed LHP/spiro-MeOTAD (LHP = MAPI or triLHP) interfaces with AX- and PbX2-type terminations showing electron density difference isosurfaces upon interface formation. Only the two outermost layers of each LHP are shown for the ease of viewing. Yellow and blue regions denote charge accumulation and depletion zones, respectively. Isosurface value = 0.001 e− bohr−3. (b) Decompositions of the binding energies of spiro-MeOTAD on different LHP terminations and chemical environments according to eqn (1). | ||
The first contribution is the adhesion energy (Ea) and stems from the electronic interaction between the moieties and it is calculated from the LHP and spiro-MeOTAD energies at the interface geometries (ELHP* and ESpiro*). These energetic terms allow us to also evaluate the distortion energies of spiro-MeOTAD (ESpirod) and LHP (ELHPd), accounting for energetic penalties due to conformational changes upon interface formation. Defined in this way, Ea is an attractive (negative) term, while distortion energies are positive quantities that partially compensate Ea to deliver the final Eb. This analysis is displayed in Fig. 1b, while the corresponding numerical values are listed in Table S1 (ESI†).
Our results reveal an opposite affinity of the spiro-MeOTAD molecule for either termination depending on LHP composition: while adsorption on the MAI surface is preferred for MAPI, PbX2 is preferred for triLHP. Moreover, Cs appears to have a stabilizing effect in the MA terminated triLHP when linked to the methoxy group of spiro-MeOTAD: FAMAX and CsFAMAX(O–Cs) deliver similar Eb, with binding in CsFAMAX being less convenient.
Regarding PbX2 termination in LHP, the presence of Cs in the subsurface layer reinforces the PbX2(Cs)/spiro-MeOTAD binding with respect to PbX2/spiro-MeOTAD by ∼1 eV, making the PbX2(Cs)/spiro-MeOTAD the most stable interface among all explored interfaces (∼−4.5 eV). The binding energy trends reflect both the structural and electronic features of the interface. On one hand, more negative binding energies correspond to the closest molecule/surface distances, as discussed in the ESI† (Fig. S3 and Table S1). On the other hand, the dominant term in Eb is the adhesion energy, which suggests a mostly electronic interaction, with low distortion energies for both components, especially for the surface, with a low conformational freedom involving only cations of the topmost layer (Fig. S4, ESI†). A more in-depth structural analysis of structural rearrangements on spiro-MeOTAD is reported in Fig. S5 (ESI†). At all LHP interfaces, the angles of the main fluorene moiety and the -methoxyphenyl residues vary by 2° and 4°, respectively, compared to the free spiro-MeOTAD molecule. These structural variations occur with an elongation of the methoxy CH3–O bond of ∼0.02/0.03 Å at the most stable triLHP(PbX2(Cs))/spiro-MeOTAD interface. The electronic nature of the LHP/spiro-MeOTAD interaction is depicted by the charge density difference plots (Fig. 1a). Most of the charge rearrangement at the interface involves the fluorene ring and the methoxy groups of spiro-MeOTAD, which interact predominantly with the A cation and with the Pb atoms of AX and PbX2 terminations, in agreement with previous investigations.12,13 This analysis points out also a direct role of the Cs atom in stabilizing the interaction with spiro-MeOTAD. In the CsFAMAX(O–Cs) system, the increased charge density along the methoxy O–Cs bond and the O–Cs distance (∼3.25 Å) suggests the formation of direct bonding.14 Also the strongest binding in the PbX2(Cs) system is accompanied by a pronounced charge rearrangement involving the whole interface.
On these interfaces, we computed the atom-projected density of states (pDOS; see Fig. S6, ESI†) to obtain a first qualitative indication of frontier orbital energetics and the thermodynamic feasibility of CT. In all the cases, the spiro-MeOTAD highest occupied molecular orbital (HOMO) lies at higher energy than the LHP valence band maximum (VBM), providing a convenient driving force for hole injection. The energy difference between the donor and the acceptor band edges is the largest for the PbX2 terminations of both LHPs, suggesting a more effective transfer process than that in AX ones, as also pointed out by Wang et al.15 Nevertheless, a driving force of ∼0.2 eV or higher is sufficient for efficient CT, so all the considered interfaces are in principle suitable.16 The pDOS plots also indicate the presence of several HOMO−X (X = 1, 2, 3…) states, lying at higher energies than the LHP VBM, which can behave as additional CT channels. Motivated by this finding, we calculated the donor–acceptor coupling matrix elements for transitions occurring between both the HOMO and HOMO−1 molecular orbitals (MOs) of spiro-MeOTAD and the LHP VBM. Since the hole transfer process from the LHP VBM to the spiro-MeOTAD HOMO is conceptually equivalent to an electron transfer in the opposite direction, we consider spiro-MeOTAD as the electron donor, while the LHP acts as the acceptor. We calculated the spectral function Γd(E) from electronic couplings:
 | (2) |
 | ||
| Fig. 2 Left panels: coupling matrix elements |Vad| between the spiro-MeOTAD HOMO and HOMO−1 donor states and LHP VB acceptor states and spectral function Γ(E) calculated according to eqn (2) for (a) AX-type and (b) PbX2-type interfaces. The spiro-MeOTAD donor state is represented by red vertical lines in |Vad| and Γ(E) plots. Right panels: Atom-projected density of states (pDOS) computed at the HSE06 level of theory. | ||
The actual LHP states involved in the CT process can be extracted from the atom-projected pDOS, as shown in Fig. 2 (right panels). The low energy region with the highest couplings at AX-interfaces is composed of spiro-MeOTAD oxygen p states and MA/FA cations. While in CsFAMAX Cs states are not present in the −6 to −9 eV region, such states appear in CsFAMAX-OCs, where couplings are much higher. All pDOS for PbX2 derived interfaces feature mixed Pb and halide states interacting with spiro-MeOTAD oxygen p states in the −2 to −4 eV region. Also in this case, Cs states correlated to higher couplings appear for triLHP-PbX2(Cs). To gain insight on the nature of donor states and clarify the differences between HOMO and HOMO−1 in terms of couplings, we show interfacial spiro-MeOTAD MOs and the corresponding energies in Fig. 3. For comparison, the same orbitals for isolated spiro-MeOTAD are plotted in Fig. S7 (ESI†).
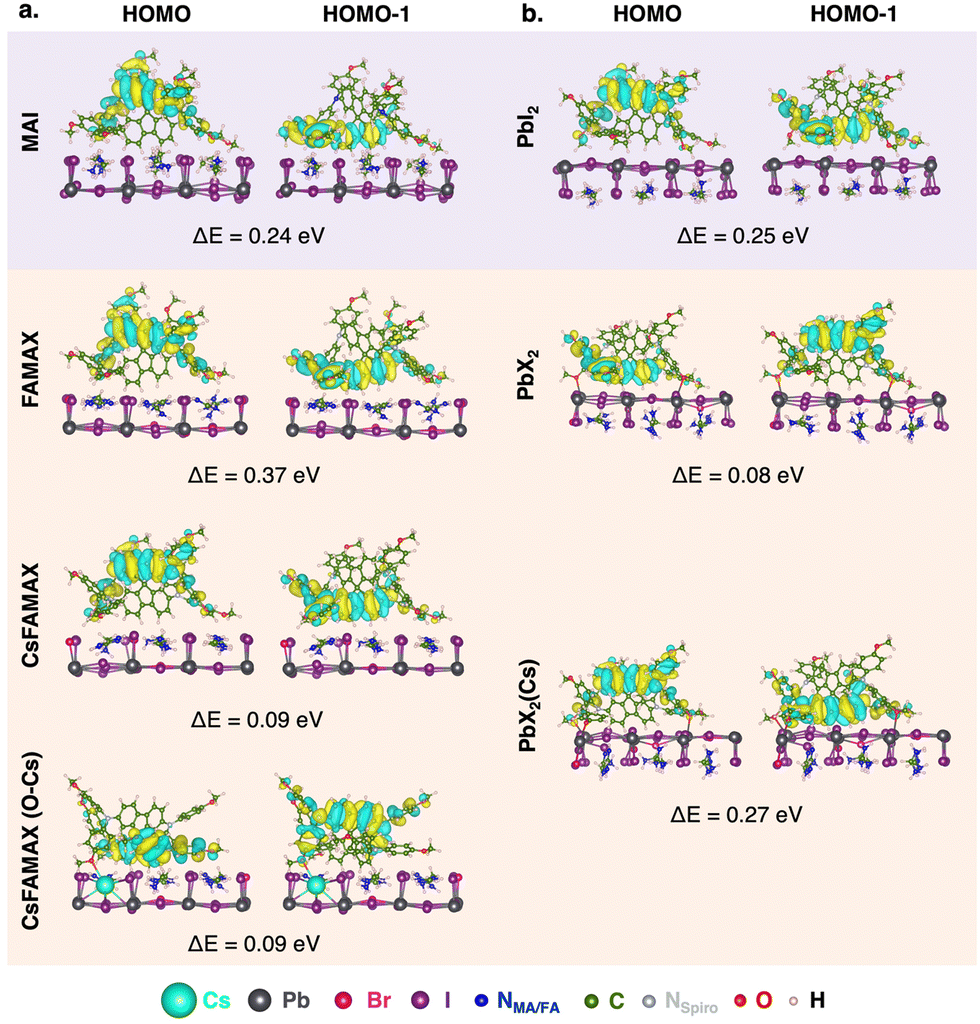 | ||
| Fig. 3 Spiro-MeOTAD HOMO and HOMO−1 MOs and difference in energy between HOMO and HOMO−1 (ΔE = EHOMO − EHOMO−1) for all LHP/spiro-MeOTAD interfaces. | ||
As suggested in other works,18 our calculations predict a surface-induced light-to-moderate degeneracy loss between HOMO and HOMO−1 (separated by only 0.04 eV in the isolated molecule) for interfacial spiro-MeOTAD with energy differences from 0.08 to 0.37 eV. As shown in Fig. 3, either the HOMO−1 or HOMO orbital points to the surface, while the other MO points to the vacuum region. For all surfaces explored, the highest couplings occur through such MO oriented towards the surface, i.e. HOMO−1 for MAPI-MAI and PbI2, triLHP-FAMAX and PbX2(Cs) and HOMO for triLHP-PbX2. An exception, and apparent contradiction, occurs for triLHP-CsFAMAX and CsFAMAX(O–Cs) cases, where more advantageous couplings occur through HOMO and HOMO−1 MOs, respectively, which are not spatially located towards the perovskite surface. Still, a closer look at these MOs (Fig. S8, ESI†) can explain this finding: they comprise the methoxy group directly linked to the Cs-rich surface at 2.43 Å and 3.25 Å for CsFAMAX and CsFAMAX(O–Cs), respectively. Particularly for the CsFAMAX(O–Cs)/HOMO−1 case, strong couplings in the −6 to −9 eV region that also involve Cs states suggest that Cs acts as a direct hole transfer booster.
The computed hole injection times, corresponding to the coupling between the LHP and spiro-MeOTAD states described above and extracted from Γ(E), are reported in Table 1. All the obtained values are of the order of picoseconds (ps), in agreement with other experimental works,19–22 with triLHP presenting the fastest injections, in particular for the FAMAX/spiro-MeOTAD (HOMO−1) (1.65 ps), CsFAMAX(O–Cs)/spiro-MeOTAD (HOMO−1) (2.50 ps) and PbX2(Cs)/spiro-MeOTAD (HOMO−1) (2.68 ps) interfaces. These results show that (i) triLHP is always more efficient for CT than MAPI; (ii) MAPI is only efficient at the MAI termination (1.80 ps versus 4.93 ps at the PbI2 one); and (iii) Cs acts as an additional CT booster not only when directly linked to spiro-MeOTAD, but also when it is present as subsurface species.
| MAPI | triLHP | |||
|---|---|---|---|---|
| τ (ps): AX-type | MAI | FAMAX | CsFAMAX | CSFAMAX(O–Cs) |
| HOMO → VB | 11.7 | 38.7 | 5.70 | 16.7 |
| HOMO−1 → VB | 1.80 | 1.65 | 12.9 | 2.50 |
| τ (ps): PbX2-type | PbI2 | PbX2 | PbX2(Cs) |
|---|---|---|---|
| HOMO → VB | 31.6 | 5.24 | 14.3 |
| HOMO−1 → VB | 4.93 | 13.1 | 2.68 |
In conclusion, our DFT study addresses the interfaces of the spiro-MeOTAD HTM with the well-studied MAPI and with the much less explored triple cation LHP formulation, providing new insights on the chemical features leading to favorable binding and enhanced CT. In agreement with the recent literature, electronic interactions are crucial for interface stability, with FA and Cs atoms strengthening the binding to spiro-MeOTAD methoxy groups. Regarding CT, we find a thermodynamically favourable scenario for all interfaces, PbX2 terminations presenting the highest driving force. Being close in energy and well above the LHP VBM, we include both the HOMO and HOMO−1 orbitals of spiro-MeOTAD in computing couplings and injection times. Cross-analysis of coupling matrix elements and pDOS shows the specific LHP states acting as CT channels, while analysis of MOs highlights the correlation between the best couplings and orbital localization towards the LHP surface. Our estimates of injection times predict that the CT occurs on the ps timescale, in agreement with experimental works. The donor state is the spiro-MeOTAD HOMO−1 orbital in most cases, which injects the electron to states in the deep LHP VB region or at higher energy for MAX and PbX2 terminations, respectively. Interestingly, our results also highlight a new role for the Cs cation at the triLHP surface: besides its beneficial effects on the structural stability of LHP, Cs offers a convenient anchoring point to the HTM molecule and provides effective CT channels for hole transport. Overall, our analysis offers a novel atomic-scale perspective for a comprehensive rationalization of CT mechanisms and times for some among the most representative systems of current state-of-the-art PSCs, paving the route to further investigation on the effects of dopants, defects or buffer interlayers on key processes as interfacial CT.
This work was supported by the Italian MISE-ENEA Research Agreement on the Electric System, ENEA-CRESCO for computing facilities, and PON R&I 2014–2020-Asse IV “Istruzione e ricerca per il recupero-REACT-EU”.
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. Y. Kim, J.-W. Lee, H. S. Jung, H. Shin and N.-G. Park, Chem. Rev., 2020, 120, 7867–7918 CrossRef CAS PubMed
.
- H.-S. Kim, A. Hagfeldt and N.-G. Park, Chem. Commun., 2019, 55, 1192–1200 RSC
- M. Saliba, T. Matsui, J.-Y. Seo, K. Domanski, J.-P. Correa-Baena, M. K. Nazeeruddin, S. M. Zakeeruddin, W. Tress, A. Abate, A. Hagfeldt and M. Grätzel, Energy Environ. Sci., 2016, 9, 1989–1997 RSC
- C. Yi, J. Luo, S. Meloni, A. Boziki, N. Ashari-Astani, C. Grätzel, S. M. Zakeeruddin, U. Röthlisberger and M. Grätzel, Energy Environ. Sci., 2016, 9, 656–662 RSC
- Z. Hawash, L. K. Ono and Y. Qi, Adv. Mater. Interfaces, 2018, 5, 1700623 CrossRef
- Y. Saygili, H.-S. Kim, B. Yang, J. Suo, A. B. Muñoz-Garcia, M. Pavone and A. Hagfeldt, ACS Energy Lett., 2020, 5, 1271–1277 CrossRef CAS
- I. Kondov, M. Čížek, C. Benesch, H. Wang and M. Thoss, J. Phys. Chem. C, 2007, 111, 11970–11981 CrossRef CAS
- J. Haruyama, K. Sodeyama, I. Hamada, L. Han and Y. Tateyama, J. Phys. Chem. Lett., 2017, 8, 5840–5847 CrossRef CAS PubMed
- P. Schulz, D. Cahen and A. Kahn, Chem. Rev., 2019, 119, 3349 CrossRef CAS PubMed
- C. Ma, M. Grätzel and N.-G. Park, ACS Energy Lett., 2022, 7, 3120–3128 CrossRef CAS
- C. Quarti, F. De Angelis and D. Beljonne, Chem. Mater., 2017, 29, 958–968 CrossRef CAS
- A. Torres and L. G. C. Rego, J. Phys. Chem. C, 2014, 118, 26947–26954 CrossRef CAS
- C. Coppola, A. Pecoraro, A. B. Muñoz-García, R. Infantino, A. Dessì, G. Reginato, R. Basosi, A. Sinicropi and M. Pavone, Phys. Chem. Chem. Phys., 2022, 24, 14993–15002 RSC
- A. Leclaire, J. Solid State Chem., 2008, 181, 2338–2345 CrossRef CAS
- Q. Wang, E. Mosconi, C. Wolff, J. Li, D. Neher, F. De Angelis, G. P. Suranna, R. Grisorio and A. Abate, Adv. Energy Mater., 2019, 9, 1900990 CrossRef
- A. Pecoraro, A. Di Maria, P. Delli Veneri, M. Pavone and A. B. Muñoz-García, Phys. Chem. Chem. Phys., 2020, 22, 28401–28413 RSC
- N. Droseros, B. Dänekamp, D. Tsokkou, P. P. Boix and N. Banerji, APL Mater., 2019, 7, 041115 CrossRef
- D. Shi, X. Qin, Y. Li, Y. He, C. Zhong, J. Pan, H. Dong, W. Xu, T. Li, W. Hu, J.-L. Brédas and O. M. Bakr, Sci. Adv., 2016, 2, e1501491 CrossRef PubMed
- P. Piatkowski, B. Cohen, F. J. Ramos, M. D. Nunzio, M. K. Nazeeruddin, M. Grätzel, S. Ahmad and A. Douhal, Phys. Chem. Chem. Phys., 2015, 17, 14674–14684 RSC
- C. S. Ponseca Jr., E. M. Hutter, P. Piatkowski, B. Cohen, T. Pascher, A. Douhal, A. Yartsev, V. Sundström and T. J. Savenije, J. Am. Chem. Soc., 2015, 137, 16043–16048 CrossRef PubMed
- J. Leng, J. Liu, J. Zhang and S. Jin, J. Phys. Chem. Lett., 2016, 7, 5056–5061 CrossRef CAS PubMed
- K. Pydzińska-Białek, V. Drushliak, E. Coy, K. Załļski, J. Flach, J. Idígoras, L. Contreras-Bernal, A. Hagfeldt, J. A. Anta and M. Ziółek, ACS Appl. Mater. Interfaces, 2020, 12, 30399–30410 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Computational details and supporting results. See DOI: https://doi.org/10.1039/d3cc00960b |
| This journal is © The Royal Society of Chemistry 2023 |