
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
ortho-Functionalization of azobenzenes via hypervalent iodine reagents†
Ester Maria
Di Tommaso
 a,
Melanie
Walther
bc,
Anne
Staubitz
*bc and
Berit
Olofsson
*a
a,
Melanie
Walther
bc,
Anne
Staubitz
*bc and
Berit
Olofsson
*a
aDepartment of Organic Chemistry, Arrhenius Laboratory, Stockholm University, Stockholm, Sweden. E-mail: berit.olofsson@su.se
bUniversity of Bremen, Institute for Analytical and Organic Chemistry, Bremen, Germany. E-mail: staubitz@uni-bremen.de
cUniversity of Bremen, MAPEX Center for Materials and Processes, Bremen, Germany
First published on 31st March 2023
Abstract
ortho-Functionalized azobenzenes are much sought after molecular switches, as they may be tuned to absorb in the visible range of light and the (Z)-isomers can have high thermal half-lives. To enable straightforward access to these targets, we have developed a synthetic route via novel ortho-substituted azobenzene-functionalized diaryliodonium salts. Selective transfer of the azobenzene moiety to O-, N-, C- and S-nucleophiles under mild, transition metal-free conditions gives access to an unprecedented range of ortho-substituted azobenzenes. The photoswitching properties of the reagents were investigated and the structure was determined by X-ray crystallography.
Azobenzenes are important molecular switches that can be photochemically switched between the thermodynamically stable (E)-isomer and the metastable (Z)-isomer.1 Exceptions exist, in which the stabilities are reversed.2 They show a high thermal3 and photochemical stability,1 and the switching mechanisms are well understood.4 Using targeted syntheses, it is often possible to obtain certain properties. Hence, there is an increasing number of applications, e.g. in switchable polymers with diverse purposes,5 biomedical applications,6 or as solar thermal fuels.7 For each task, the azobenzene has to be finely adjusted due to two main issues.8 The thermal stability of the less stable isomer can cause an unavoidable background isomerization9 and the overlap of the isomers’ absorption spectra results in photostationary states (PSSs) of low selectivity for either isomer at a particular wavelength.10
Current research focuses on ortho-substituted azobenzenes to reach thermally stable (Z)-isomers or an absorption in the visible range of light.11 Switching occurs with wavelengths as long as 720 nm,12 and highly selective PSS (tetra-ortho-fluorinated azobenzene: PSS(Z) 91%/PSS(E) 86%).10 However, there are few synthetic methods available for their synthesis (Scheme 1a).3,13 They can be prepared from appropriately substituted anilines11a,14 and further functionalized via nucleophilic substitution,15 or through transition metal-catalyzed C–H activation.16 While azobenzenes decorated with carbon,17 oxygen,17a,18 nitrogen19 and halide16,19b substituents in the ortho-position have been synthesized, synthetic drawbacks include the need for transition metal catalysts, long reaction times at elevated temperatures and scope limitations. Surprisingly, the literature is void of cross-coupling methods in the ortho-position, irrespective of whether the azobenzene moiety serves as an organometallic nucleophilic component or as an electrophilic halogenated cross-coupling partner,13 despite access to ortho-metalated azobenzenes.20
 | ||
| Scheme 1 Synthesis of ortho-substituted azobenzenes. (a) State of the art. (b) This work. | ||
Hypervalent iodine(III) compounds are efficient reagents for a wide range of transformations under mild reaction conditions.21 Diaryliodonium salts have been recognized as highly reactive electrophilic arylating reagents with a variety of carbon and heteroatom nucleophiles under both metal-free and metal-catalyzed conditions.22 Furthermore, they are relatively non-toxic, bench-stable and easily synthesized through one-pot reactions of iodoarenes and arenes or arylboronic acids.23
We envisioned that the combination of azobenzenes with iodine(III) chemistry could overcome the limitations in the synthesis of ortho-functionalized azobenzenes. Herein, we showcase the successful synthesis and applications of ortho-azobenzene diaryliodonium salts in metal-free arylations to access hitherto unobtainable products (Scheme 1b).
The study commenced by investigating suitable conditions for the synthesis of diaryliodonium salts 3 from the corresponding ortho-iodoazobenzenes 1.24 Our standard one-pot conditions with mCPBA and triflic or tosic acid and a suitable arene failed,23b,d as did one-pot reactions of 1 with arylboronic acids.23c To our delight, a sequential one-pot reaction25 with oxidation of iodoarene 1 using mCPBA and tosic acid at 40 °C, followed by the addition of arene 2 at room temperature was successful for the synthesis of the novel iodonium salts 3 (Scheme 2). In this way, the otherwise unsubstituted ortho-azobenzene iodonium salt 3a was obtained in 84% isolated yield from 1a. A stepwise synthesis of 3a with isolation of the corresponding [hydroxy(tosyloxy)iodo]arene before treatment with anisole gave 3a in 78% overall yield.24 Anion exchange to the corresponding triflate salt 3a′ could be performed by in situ treatment with triflic acid or workup with NaOTf.24,25b
 | ||
| Scheme 2 Scope of ortho-azobenzene(aryl)iodonium salts 3 (isolated yields after precipitation). [a] After workup with NaOTf. [b] TFE as the only solvent. [c] TsOH·H2O (2.2 equiv.), mCPBA (2.0 equiv.) and anisole (2.0 equiv.). | ||
The scope investigations were focused on the synthesis of unsymmetric diaryliodonium salts (Ar1Ar2IX) to avoid wasting a precious azobenzene moiety in subsequent arylations. The anisyl moiety is often an efficient “dummy group” in chemoselective arylations with Ar1Ar2IX under metal-free conditions.26 Anisole was hence utilized as the standard arene in reactions with substituted ortho-iodoazobenzenes 1b–d to provide iodonium salts 3a–d in good to high yields. Substituents were well tolerated on the para-azo aryl moiety, with bromo-substituted salt 3b well positioned for further late-stage functionalizations. Further substituents on the iodo-substituted aryl group were also tolerated, as shown by methyl-substituted product 3d. Mesitylene and trimethoxybenzene could be utilized to provide iodonium salts 3e and 3f bearing a mesityl or trimethoxyphenyl (TMP) dummy group, which are reported to give chemoselective arylations with certain nucleophiles.26 Bis(diaryliodonium) salt 3g was obtained in good yield through difunctionalization of the corresponding diiodo-azobenzene 1e.
It was possible to grow single crystals of the (E)-isomer of salt 3a, suitable for X-ray diffraction analysis (Fig. 1, CCDC number: 2215795). Diaryliodonium salts generally have the typical T-shape of iodine(III) compounds, where one aryl group and the counterion reside in the hypervalent bond.21 Interestingly, the X-ray structure of 3a displays an N–I-aryl hypervalent bond with an N–I interaction of 2.616 Å, whereas the tosylate anion coordinates perpendicular to the hypervalent bond (bond angles: C(13)–I(1)–O(3) 81.84(10)°; O(3)–I(1)–C(1) 90.45(9)°). While azo-coordination to iodine(III) is unknown, Nachtsheim and coworkers have recently reported on related N–I interactions in iodine(III) reagents.27 Preliminary calculations indicate that the pseudocyclic structure is favored for the (E)-isomer, whereas the (Z)-isomer lacks the N–I stabilizing interaction and the expected I–OTs coordination is favored.24
 | ||
| Fig. 1 X-ray crystal structure of the (E)-isomer of 3a with selected bond lengths. Selected angles: C(13)–I(1)–O(3) 81.84(10)°; O(3)–I(1)–C(1) 90.45(9)°; N(1)–I(1)–O(3) 107.47°; C(1)–I(1)–N(1) 69.2(1)°; C(13)–I(1)–C(1) 94.56(12)°; N(1)–I(1)–C(13) 8.54(5). | ||
The influence of the hypervalent iodine moiety on the photoswitching behavior of the azobenzene was examined by 1H NMR and UV/vis spectroscopy in CDCl3/CHCl3, comparing iodonium salt 3a and the parent azobenzene 1a. Under ambient conditions, both molecules existed exclusively as the (E)-isomer. Upon irradiation with UV light (340 nm), both molecules switched to the corresponding (Z)-isomer ((E)/(Z) ratio: 32/68 (1a), 29/71 (3a)). Re-isomerization occurred upon irradiation with visible light (450 nm) ((E)/(Z) ratio: 90/10 (1a), 72/28 (3a)). A complete conversion to the (E)-isomer could not be achieved photochemically but no significant photodegradation was detected for 1a and 3a upon repeated switching. The incorporation of the hypervalent moiety had almost no influence on the absorption maxima of the azobenzene switch: (π–π*: 325 nm (1a), 338 nm (3a); n–π*: 434 nm (1a), 436 nm (3a)). However, the thermal half-life was significantly decreased (t1/2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :124.6 h (1a), 5.83 h (3a)). This can be attributed to the strong electron acceptor character of the iodine(III) moiety, and the N–I stabilizing interaction in the (E)-isomer of 3a.
:124.6 h (1a), 5.83 h (3a)). This can be attributed to the strong electron acceptor character of the iodine(III) moiety, and the N–I stabilizing interaction in the (E)-isomer of 3a.
To demonstrate the utility of the novel reagents, well-established methods for transition metal-free arylation of various nucleophiles with diaryliodonium salt 3a were utilized to provide a range of ortho-substituted azobenzene products. The transformations proceeded with complete chemoselectivity and retained (E)-configuration (Scheme 3). O-Arylation of phenol and carboxylic acids28 delivered the azobenzene-functionalized diaryl ether 4 and aryl esters 5 and 6 in good yields. The aryl ester 6 could be hydrolyzed under mild conditions into 2-hydroxyazobenzene 7 in 81% yield, which has excellent features as a structural motif in pharmaceutical and materials science.18b The synthesis of product 7 was also feasible through the hydroxylation of 3a with hydrogen peroxide29 in a moderate yield. As Pd-catalyzed ortho-acyloxylation and hydroxylation of azobenzenes is reported,17a,18 we were eager to evaluate the reactivity of 3a with other types of nucleophiles.
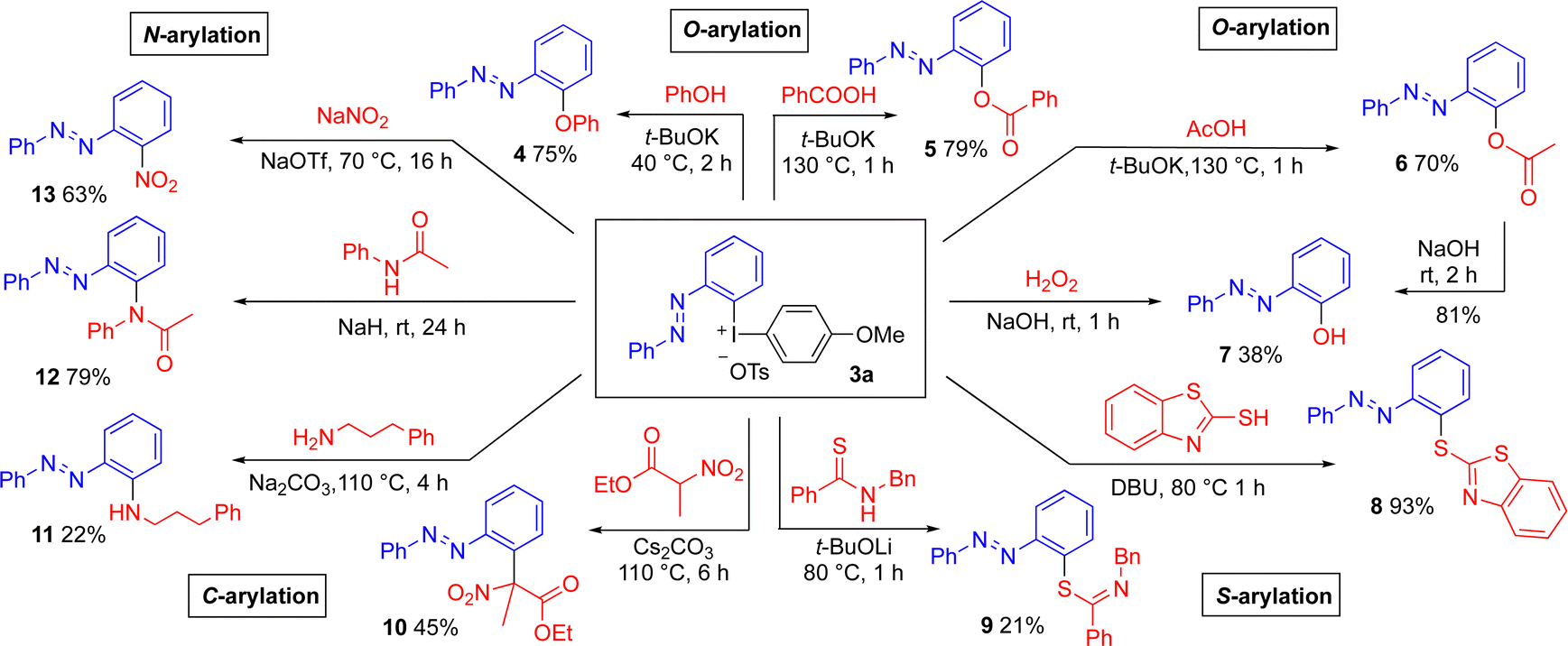 | ||
| Scheme 3 Metal-free arylations of C, N, O and S-nucleophiles with reagent 3a. | ||
To our delight, S-arylation with a mercaptothiazole30 delivered the novel, heterocyclic azobenzene product 8 in excellent yield (93%). The arylation of thioamide31 to thioimidates 9 was more difficult. Even the C-arylation of a substituted nitroester32 could be performed to provide the sterically congested product 10. Methods for N-arylation were next explored, and the arylation of a primary amine33 proved to be challenging. The novel, amino-substituted product 11 was obtained in 22% yield due to partial decomposition of 3a under the reaction conditions. On the other hand, the arylation of acetanilide34 proceeded smoothly to give the tertiary amide 12 in 79% yield. Furthermore, arylation of sodium nitrite35 delivered ortho-nitro azobenzene (13) in 63% yield.
The O-arylation methodology28 was subsequently employed to achieve more advanced ortho-functionalized azobenzene products that should be relevant in supramolecular chemistry, materials science14,36 and biological chemistry37 (Scheme 4). The reaction of bis(diaryl)iodonium salt 3g with phenol gave the ortho-diphenoxylated azobenzene 14 and the steroid estrone was arylated with reagent 3a to diaryl ether 15 in 60% yield. Moreover, the O-arylation of protected ribose38 delivered the arylated derivative 16. Arylation of acetyl-protected cholic acid produced ester 17 in moderate yield, as the basic conditions caused partial hydrolysis of the acetate groups, with competing arylation to give byproduct 6 in 20% yield.24 Finally, the O-arylation of oleic acid produced the functionalized ester 18. It should be noted that 8–11 and 15–18 are novel ortho-azobenzenes, illustrating the utility of the method to quickly reach a variety of targets. In comparison with literature methods to reach the known products 4–7 and 12–14, our method often has milder conditions and shorter reaction times.24
 | ||
| Scheme 4 O-Arylations to provide complex ortho-substituted azobenzenes. Conditions: 3 (1.0 equiv.), nucleophile and tBuOK (1.2-2.1 equiv.), in toluene. | ||
In conclusion, the synthesis of ortho-azobenzene-derived diaryliodonium salts was achieved in high yields. The novel iodonium reagents were demonstrated as chemoselective arylation reagents with a range of C, N, O and S-nucleophiles under mild and metal-free conditions. The X-ray crystal structure revealed that the nitrogen is involved in hypervalent bonding with the iodine(III) center. The photoswitching properties of the azobenzene were retained in the hypervalent azobenzene derivatives but the incorporation of the hypervalent iodine bond influenced the position of the absorption maxima and drastically reduced the half-life time.
Financial support for this study was provided through the Swedish Research Council (2015-04404 and 2019–04232). Dr Marian Olaru is acknowledged for measuring the crystal structure and Malte Pankau for initial contributions to the project.
A.S. and B.O. conceived the project; E.M.D.T. and M.W. performed the experiments and analyzed the data; all authors contributed to writing the manuscript; A.S. and B.O. supervised the project and acquired the funding. All authors have read and agreed to the published version of the manuscript.
Conflicts of interest
There are no conflicts to declare.Notes and references
- H. M. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- R. Siewertsen, H. Neumann, B. Buchheim-Stehn, R. Herges, C. Näther, F. Renth and F. Temps, J. Am. Chem. Soc., 2009, 131, 15594–15595 CrossRef CAS PubMed.
- E. Merino, Chem. Soc. Rev., 2011, 40, 3835–3853 RSC.
- T. Kumpulainen, B. Lang, A. Rosspeintner and E. Vauthey, Chem. Rev., 2017, 117, 10826–10939 CrossRef CAS PubMed.
- (a) K. M. Herbert, H. E. Fowler, J. M. McCracken, K. R. Schlafmann, J. A. Koch and T. J. White, Nat. Rev. Mater., 2022, 7, 23–38 CrossRef CAS; (b) A. Natansohn and P. Rochon, Chem. Rev., 2002, 102, 4139–4175 CrossRef CAS PubMed; (c) T. Ikeda and O. Tsutsumi, Science, 1995, 268, 1873–1875 CrossRef CAS PubMed.
- (a) N. Yasuike, K. M. Blacklock, H. Lu, A. S. I. Jaikaran, S. McDonald, M. Uppalapati, S. D. Khare and G. A. Woolley, ChemPhotoChem, 2019, 3, 431–440 CrossRef CAS PubMed; (b) A. C. Impastato, A. Shemet, N. A. Veprek, G. Saper, H. Hess, L. Rao, A. Gennerich and D. Trauner, Angew. Chem., Int. Ed., 2022, 61, e202115846 CrossRef CAS PubMed.
- (a) A. Kunz, A. H. Heindl, A. Dreos, Z. Wang, K. Moth-Poulsen, J. Becker and H. A. Wegner, ChemPlusChem, 2019, 84, 1145–1148 CrossRef CAS PubMed; (b) Z. Wang, P. Erhart, T. Li, Z.-Y. Zhang, D. Sampedro, Z. Hu, H. A. Wegner, O. Brummel, J. Libuda, M. B. Nielsen and K. Moth-Poulsen, Joule, 2021, 5, 3116–3136 CrossRef CAS.
- L. N. Lameijer, S. Budzak, N. A. Simeth, M. J. Hansen, B. L. Feringa, D. Jacquemin and W. Szymanski, Angew. Chem., Int. Ed., 2020, 59, 21663–21670 CrossRef CAS PubMed.
- H. A. Wegner, Angew. Chem., Int. Ed., 2012, 51, 4787–4788 CrossRef CAS PubMed.
- D. Bléger, J. Schwarz, A. M. Brouwer and S. Hecht, J. Am. Chem. Soc., 2012, 134, 20597–20600 CrossRef PubMed.
- (a) C. Knie, M. Utecht, F. Zhao, H. Kulla, S. Kovalenko, A. M. Brouwer, P. Saalfrank, S. Hecht and D. Bleger, Chem. – Eur. J., 2014, 20, 16492–16501 CrossRef CAS PubMed; (b) A. A. Beharry, O. Sadovski and G. A. Woolley, J. Am. Chem. Soc., 2011, 133, 19684–19687 CrossRef CAS PubMed; (c) M. J. Hansen, M. M. Lerch, W. Szymanski and B. L. Feringa, Angew. Chem., Int. Ed., 2016, 128, 13713–13716 Search PubMed.
- M. Dong, A. Babalhavaeji, C. V. Collins, K. Jarrah, O. Sadovski, Q. Dai and G. A. Woolley, J. Am. Chem. Soc., 2017, 139, 13483–13486 CrossRef CAS PubMed.
- M. Walther, W. Kipke, S. Schultzke, S. Ghosh and A. Staubitz, Synthesis, 2021, 1213–1228 CAS.
- S. Mehrparvar, Z. N. Scheller, C. Wölper and G. Haberhauer, J. Am. Chem. Soc., 2021, 143, 19856–19864 CrossRef CAS PubMed.
- K. Kuntze, J. Viljakka, E. Titov, Z. Ahmed, E. Kalenius, P. Saalfrank and A. Priimagi, Photochem. Photobiol. Sci., 2022, 21, 159–173 CrossRef CAS PubMed.
- D. B. Konrad, J. A. Frank and D. Trauner, Chem. – Eur. J., 2016, 22, 4364–4368 CrossRef CAS PubMed.
- (a) C. Qian, D. Lin, Y. Deng, X.-Q. Zhang, H. Jiang, G. Miao, X. Tang and W. Zeng, Org. Biomol. Chem., 2014, 12, 5866–5875 RSC; (b) M. Li and Y. Ye, ChemCatChem, 2015, 7, 4137–4142 CrossRef CAS; (c) Z. Liu, Y. Xian, J. Lan, Y. Luo, W. Ma and J. You, Org. Lett., 2019, 21, 1037–1041 CrossRef CAS PubMed.
- (a) T. H. L. Nguyen, N. Gigant, S. Delarue-Cochin and D. Joseph, J. Org. Chem., 2016, 81, 1850–1857 CrossRef CAS PubMed; (b) X. Fu, Z. Wei, C. Xia, C. Shen, J. Xu, Y. Yang, K. Wang and P. Zhang, Catal. Lett., 2017, 147, 400–406 CrossRef CAS.
- (a) Y.-F. Liang, X. Li, X. Wang, Y. Yan, P. Feng and N. Jiao, ACS Catal., 2015, 5, 1956–1963 CrossRef CAS; (b) Z. Ahmed, A. Siiskonen, M. Virkki and A. Priimagi, Chem. Commun., 2017, 53, 12520–12523 RSC.
- (a) J. Hoffmann, T. J. Kuczmera, E. Lork and A. Staubitz, Molecules, 2019, 24, 303 CrossRef PubMed; (b) M. Juribašić, I. Halasz, D. Babić, D. Cinčić, J. Plavec and M. Ćurić, Organometallics, 2014, 33, 1227–1234 CrossRef.
- (a) A. Yoshimura and V. V. Zhdankin, Chem. Rev., 2016, 116, 3328–3435 CrossRef CAS PubMed; (b) Patai's Chemistry of Functional Groups: The Chemistry of Hypervalent Halogen Compounds, Wiley, ed. B. Olofsson, I. Marek and Z. Rappoport, Chichester, 2019 Search PubMed; (c) Hypervalent Iodine Chemistry, ed. T. Wirth, Springer International Publishing, Cham, 2016 Search PubMed.
- (a) E. A. Merritt and B. Olofsson, Angew. Chem., Int. Ed., 2009, 48, 9052–9070 CrossRef CAS PubMed; (b) K. Aradi, B. L. Tóth, G. L. Tolnai and Z. Novák, Synlett, 2016, 1456–1485 CAS; (c) P. Villo and B. Olofsson, Patai's Chemistry of Functional Groups, Wiley, Chichester, 2019, pp. 461–522 Search PubMed.
- (a) M. D. Hossain, Y. Ikegami and T. Kitamura, J. Org. Chem., 2006, 71, 9903–9905 CrossRef CAS PubMed; (b) M. Bielawski, M. Zhu and B. Olofsson, Adv. Synth. Catal., 2007, 349, 2610–2618 CrossRef CAS; (c) M. Bielawski, D. Aili and B. Olofsson, J. Org. Chem., 2008, 73, 4602–4607 CrossRef CAS PubMed; (d) M. Zhu, N. Jalalian and B. Olofsson, Synlett, 2008, 592–596 CAS.
- See the ESI† for details.
- (a) T. L. Seidl, S. K. Sundalam, B. McCullough and D. R. Stuart, J. Org. Chem., 2016, 81, 1998–2009 CrossRef CAS PubMed; (b) M. D. Hossain and T. Kitamura, Tetrahedron, 2006, 62, 6955–6960 CrossRef CAS; (c) E. Lindstedt, M. Reitti and B. Olofsson, J. Org. Chem., 2017, 82, 11909–11914 CrossRef CAS PubMed.
- (a) C. H. Oh, J. S. Kim and H. H. Jung, J. Org. Chem., 1999, 64, 1338–1340 CrossRef CAS; (b) J. Malmgren, S. Santoro, N. Jalalian, F. Himo and B. Olofsson, Chem. – Eur. J., 2013, 19, 10334–10342 CrossRef CAS PubMed; (c) D. R. Stuart, Chem. – Eur. J., 2017, 23, 15852–15863 CrossRef CAS PubMed.
- (a) K. Aertker, R. J. Rama, J. Opalach and K. Muñiz, Adv. Synth. Catal., 2017, 359, 1290–1294 CrossRef CAS; (b) A. Boelke, E. Lork and B. J. Nachtsheim, Chem. – Eur. J., 2018, 24, 18653–18657 CrossRef CAS PubMed; (c) A. Boelke, S. Sadat, E. Lork and B. J. Nachtsheim, Chem. Commun., 2021, 57, 7434–7437 RSC.
- N. Jalalian, T. B. Petersen and B. Olofsson, Chem. – Eur. J., 2012, 18, 14140–14149 CrossRef CAS PubMed.
- M. Reitti, R. Gurubrahamam, M. Walther, E. Lindstedt and B. Olofsson, Org. Lett., 2018, 20, 1785–1788 CrossRef CAS PubMed.
- S. Sarkar, N. Wojciechowska, A. A. Rajkiewicz and M. Kalek, Eur. J. Org. Chem., 2022, e202101408 CAS.
- P. Villo, G. Kervefors and B. Olofsson, Chem. Commun., 2018, 54, 8810–8813 RSC.
- C. Dey, E. Lindstedt and B. Olofsson, Org. Lett., 2015, 17, 4554–4557 CrossRef CAS PubMed.
- N. Purkait, G. Kervefors, E. Linde and B. Olofsson, Angew. Chem., Int. Ed., 2018, 57, 11427–11431 CrossRef CAS PubMed.
- F. Tinnis, E. Stridfeldt, H. Lundberg, H. Adolfsson and B. Olofsson, Org. Lett., 2015, 17, 2688–2691 CrossRef CAS PubMed.
- M. Reitti, P. Villo and B. Olofsson, Angew. Chem., Int. Ed., 2016, 55, 8928–8932 CrossRef CAS PubMed.
- (a) X. Yao, T. Li, J. Wang, X. Ma and H. Tian, Adv. Opt. Mater., 2016, 4, 1322–1349 CrossRef CAS; (b) E. Fuentes, M. Gerth, J. A. Berrocal, C. Matera, P. Gorostiza, I. K. Voets, S. Pujals and L. Albertazzi, J. Am. Chem. Soc., 2020, 142, 10069–10078 CrossRef CAS PubMed.
- Y. M. Osornio, P. Uebelhart, S. Bosshard, F. Konrad, J. S. Siegel and E. M. Landau, J. Org. Chem., 2012, 77, 10583–10595 CrossRef CAS PubMed.
- G. L. Tolnai, U. J. Nilsson and B. Olofsson, Angew. Chem., Int. Ed., 2016, 55, 11226–11230 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2215795. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc01060k |
| This journal is © The Royal Society of Chemistry 2023 |