
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licenceα-Synuclein and biological membranes: the danger of loving too much
Silvia
Mansueto
 a,
Giuliana
Fusco
*b and
Alfonso
De Simone
*a
a,
Giuliana
Fusco
*b and
Alfonso
De Simone
*a
aDepartment of Pharmacy, University of Naples, Via Montesano 49, 80131 Naples, Italy. E-mail: alfonso.desimone@unina.it
bCentre for Misfolding Diseases, Department of Chemistry, University of Cambridge, Lensfield Road, CB2 1EW Cambridge, UK. E-mail: gf203@cam.ac.uk
First published on 15th June 2023
Abstract
The aberrant aggregation of α-Synuclein (αS), a disordered protein primarily localised at the neuronal synapses, is associated with a number of neurodegenerative disorders including Parkinson's disease (PD). The biological properties of αS are strictly connected with its ability to bind synaptic membranes under both physiological and pathological conditions. Here we overview the recent studies on the structural and biological properties of the membrane interaction by αS. The characterisation of this state is particularly challenging as the membrane binding of αS is weak, transient and features a considerable degree of conformational disorder. Advancements in this area have been achieved through combinations of nuclear magnetic resonance (NMR), super-resolution microscopy, cryo-EM and cellular biophysics. Current data clarified the central role of the equilibrium between ordered and disordered states of αS at the membrane surface, which regulates the membrane affinity, the aggregation into amyloid fibrils and the promotion of vesicle clustering. Recent results on toxic oligomeric species of αS also revealed common features in the membrane interaction of functional and aberrant forms of this protein. These findings therefore evidence the challenging nature of identifying suitable therapeutics to target the aberrant aggregation of αS in PD while leaving its normal physiological form unperturbed.
1. Introduction
αS is a 14 kDa protein that is located predominantly at the presynaptic terminals and whose aggregation is associated with Parkinson's disease (PD).1–8 Fibrillar aggregates of αS have been identified as the major constituents of intraneuronal inclusions, known as Lewy bodies, that form in dopaminergic neurons of PD patients. Genetic traits also exist linking αS and familial forms of early onset PD, including missense mutations as well as duplications or triplications of the αS encoding gene (SNCA).9,10 The aberrant aggregation of αS is also associated with other neurodegenerative disorders such as dementia with Lewy bodies and multiple system atrophy,2,6 whereas aggregates composed of the non-amyloid-β component (NAC) region of the protein (residues 61–95) have also been found in association with Alzheimer's disease (AD).11Despite the general consensus on the pathological relevance of αS aggregation, the functional role of this protein remains highly debated.12 Its prevalence at the presynaptic terminals suggests an involvement in the process of neurotransmission, with evidences pointing at a possible involvement in synaptic plasticity13 and learning.14 A consensus is gradually emerging on a putative role of αS in the regulation of the homeostasis of synaptic vesicles (SVs),15–20 particularly in contexts where intense neuronal activity is required.21 Additional studies have indicated the possibility of other functions of αS, including a regulator of pools of SVs,22,23 an inhibitor of the ER-to-Golgi vesicle trafficking,19,24 and a binder of mitochondria that mitigates oxidative stress.25–28 While these biochemical processes are fundamentally different, they share a common characteristic, namely the binding to cellular membranes as a critical step for αS to attain biological activity. It is indeed becoming increasingly evident that the interaction with membranes is critical for the physiological behaviour of αS.29 Membrane binding of αS appears to be tightly regulated in vivo30 and has a relevant role also in the context of αS aggregation and for the toxicity of its aggregates.1,31,32
In this feature article, we first summarize the current structural knowledge about the mechanism of membrane binding by αS. We then discuss the current understanding of the biological relevance of the membrane interaction in αS monomers and the aberrant aggregates (Fig. 1).
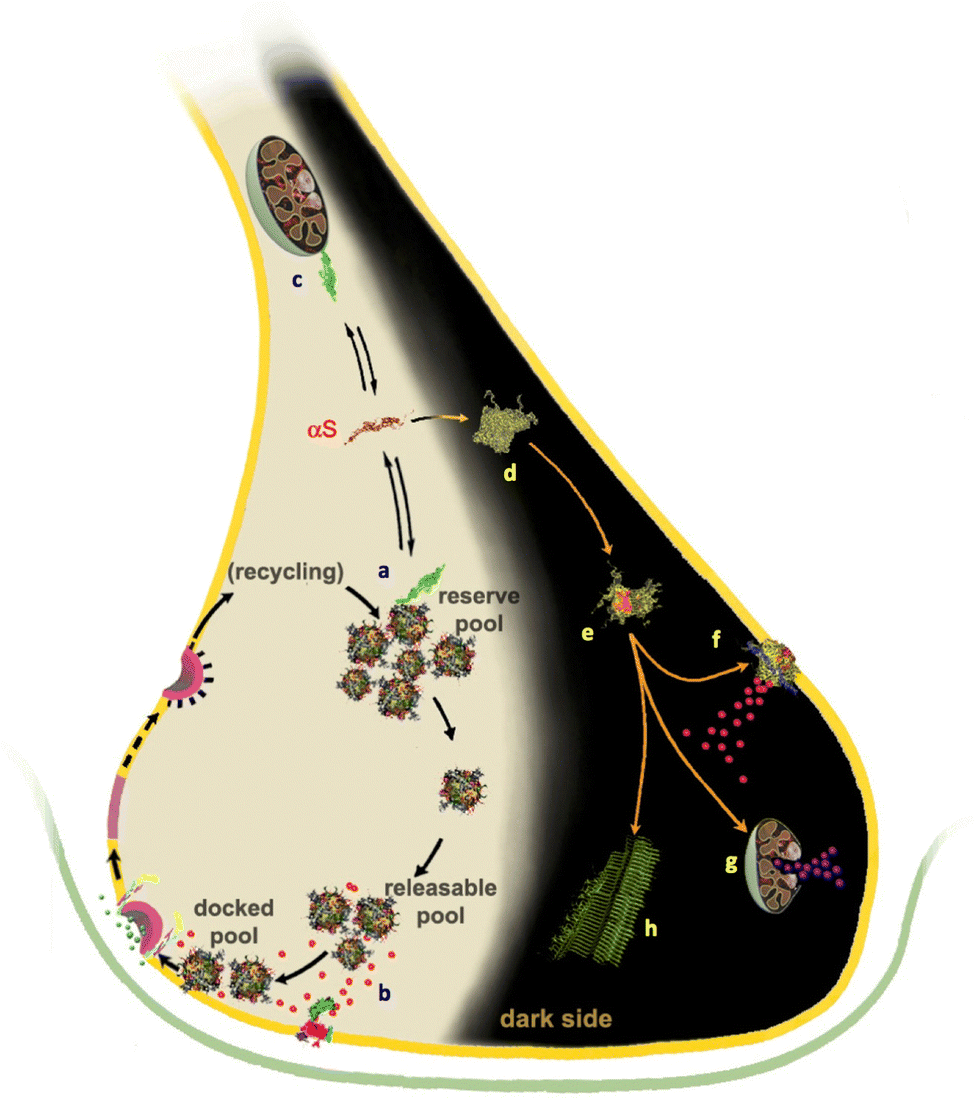 | ||
| Fig. 1 Membrane interactions of αS in functional and pathological contexts. Under physiological conditions (left side of the synapse), αS has been shown to interact with SVs and promote their clustering18–20 into SV pools forming at the synaptic termini17,22–24 (a). In addition, in the active zone αS has been associated to a chaperone role of the SNARE formation15,16 (b). αS also binds to mitochondria where it has been associated with the mitigation of oxidative stress (c). Under aggregation-prone conditions (right “dark side” of the synapse), αS is believed to aggregate first in amorphous early aggregates (d). Subsequently, these evolve into pre-fibrillar oligomeric species (e) that diffuse in the cellular milieu and establish aberrant interactions affecting the cellular viability.1 The toxic oligomers are able to bind and disrupt the plasma membrane (f), causing calcium influx and metal dishomeostasis, and to disrupt the mitochondrial integrity (g) by provoking the release of cytochrome C. These oligomers ultimately evolve into mature fibrils, which have been shown to be the major constituents of Lewy bodies. Several literature evidences indicate that αS fibrils are considerably less neurotoxic than oligomeric species,1 however, mature amyloids can still act as pathogenic agents by acting as reservoirs of toxic oligomers that are released as a result of fragmentation.139 | ||
2. Conformational pathways of αS-membrane interaction
In its cytosolic form, αS is monomeric and disordered.33,34 Nuclear magnetic resonance (NMR) studies in aqueous solution35,36 and in the cellular milieu34 indicated that this protein lacks any significant secondary and tertiary structure in its cytosolic state. Despite featuring a general structural disorder, αS is unusually compact with respect to other intrinsically disordered proteins (IDP) as shown in NMR,37 EPR38 and MD39 studies. This observation reflects the peculiar sequence of αS, where only the C-terminal region (residues 98–140) presents the typical characteristics of IDPs, whereas the N-terminal 97 residues show some levels of propensity toward structural order.29 This asymmetry in the αS sequence is maintained in the membrane binding mechanism, which is driven by a disorder-to-order transition in the N-terminal region acquiring a character of an amphipathic α-helix at the surface of lipid bilayers. The conformational switch from a purely disordered cytosolic monomer to a partially helical state is promoted by 7 imperfect sequence repeats of 11 residues located in the N-terminal region of αS and encoding for amphipathic class A2 lipid-binding segments.35,40–42 It was shown that these acquired helical segments of αS lay on the membrane surface upon binding (Fig. 2).43 The presence of multiple repeat modules in the membrane binding domain confers αS the plasticity to interact with a variety of assemblies, including detergent micelles, small vesicles and planar lipid bilayers.40,41,44,45 Upon interaction with detergent micelles, αS was found to fold into a broken α-helix composed of two helical segments (residues 3–37 and 45–92), with the C-terminal region remaining essentially disordered and not associated with the detergents. By contrast, EPR studies identified a single extended helix spanning the first 97 residues upon membrane binding.46 Both topologies have in fact been found in association with detergent micelles or lipid membranes.44,47–50 Using solid-state NMR (ssNMR) we clarified that, beyond the topological and structural properties, it is the dynamical nature of membrane-bound αS to play a critical biological role.51 Even upon membrane binding, αS indeed retains a significant level of structural disorder with some regions existing in equilibrium between ordered-bound and disordered-unbound conformations. More specifically, the αS sequence can be divided into three regions having distinct structural and dynamical properties when bound to the surface of lipid bilayers51 (Fig. 2A). These regions have different roles for the membrane interaction, and influence in individual ways the thermodynamics and kinetics of the binding.52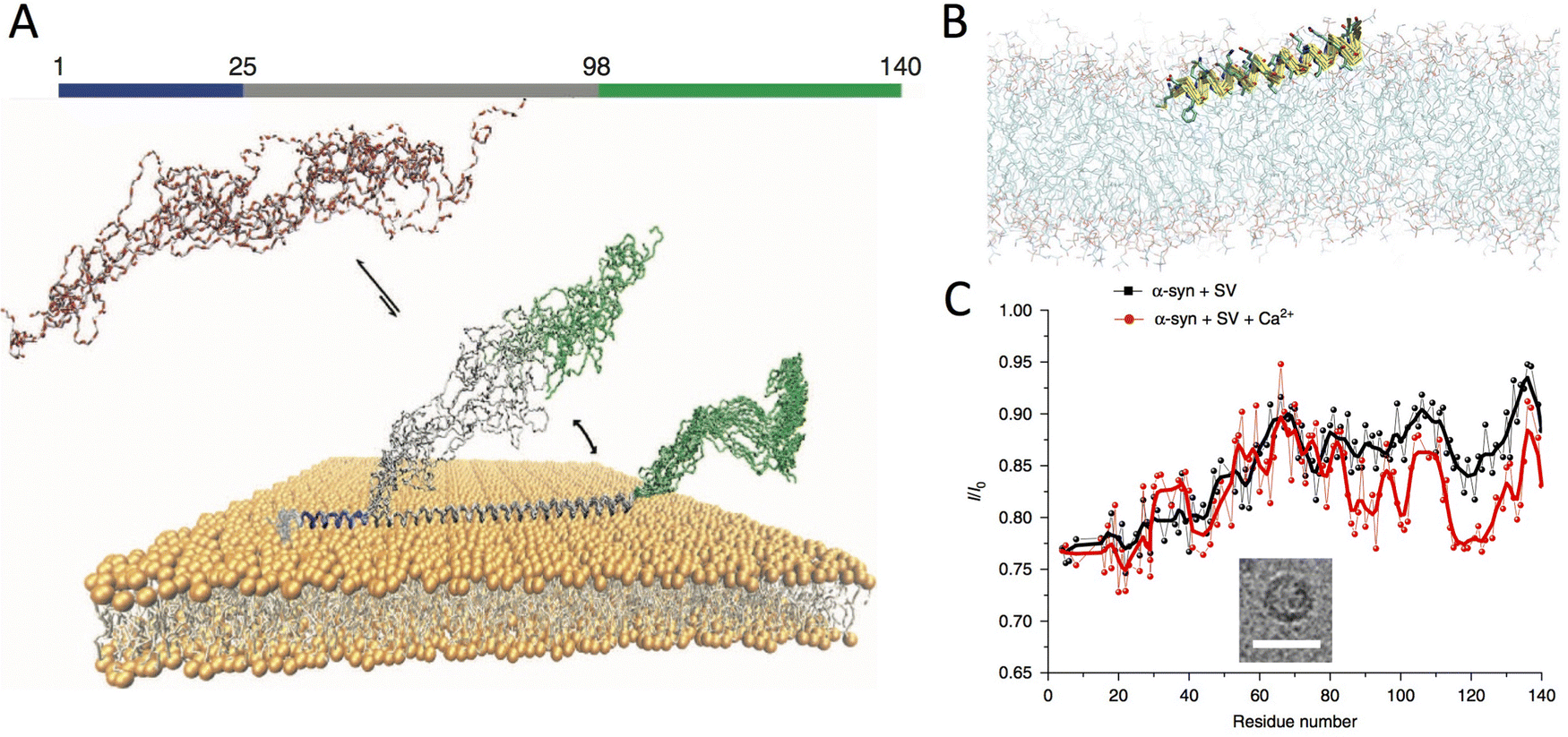 | ||
| Fig. 2 Structural bases of αS-membrane interactions. (A) The membrane bound state of αS features three regions having distinctive structural and dynamical properties. These include a rigid N-terminal anchor (residues 1–25, blue), adopting the structure of an amphipathic helix, a central region (residues 26–98, grey) being in conformational equilibrium between membrane-tethered and membrane-detached states, and a C-terminal region (residues 99–140, green) remaining substantially disordered and poorly associated with the membrane. Adapted from ref. 51, with permission from Nature Publishing Group, copyright 2014. (B) The membrane-anchor was shown to adopt a topology where the amphipathic α-helix lays parallel on the membrane surface by partially inserting the first 12 residues in the hydrophobic interior of the lipid bilayer. Adapted from ref. 43, with permission from Nature Publishing Group, copyright 2016. (C) Binding of calcium ions was shown to alter the modes of interaction with ex vivo SVs, as probed by NMR. A cryo-EM image of the SV is inserted in the CEST NMR plot (scale bar 50 nm). Adapted from ref. 60, with permission from Nature Publishing Group, copyright 2018. | ||
The primary region of αS to foster membrane binding is the N-terminus (residues 1 to 25), which acts as an anchor promoting the interaction with lipid bilayers (Fig. 2B).51 In association with membranes, this region folds into an ideal amphipathic α-helix characterised by a hydrophobic surface opposed to a lysine-rich hydrophilic surface. The ability to strongly anchor αS to the membrane was associated to a partial insertion of the N-terminal 12 residues into the hydrophobic region of the lipid bilayer (Fig. 2B).43 Moreover, the N-terminal acetylation of αS has been shown to considerably increase the local membrane affinity of the protein,35 and our studies clarified that this effect is not associated with topological-structural alterations of the membrane-bound state,53 but likely arises from an enhanced helical character in the cytosolic state.35 Following the N-terminal anchor, the central region (residues 26–97) was found to modulate the overall membrane affinity of αS by acting as a “sensor” of the properties of the lipid bilayer.51 This region, which includes the aggregation-prone non amyloid-β component (NAC, residues 60–95), was found to exist in equilibrium between detached and membrane bound states, with the latter adopting an helical structure as indicated by EPR44,46 and NMR40 data. Enhanced coarse-grained simulations revealed that N-terminal and central regions of αS have different modes of membrane binding. In the N-terminal region, a binding step through tethered-extended conformations is energetically possible, however, it is the folding into an amphipathic α-helical structure to critically strengthen and “lock” the membrane interaction.54 By contrast, in the central region the energetics of disordered-tethered and helical-locked conformations are more similar, particularly in the segment 87–97 where these two binding modes have the same energy.55 These fundamental characteristics have implications for the ability of a single αS molecule to simultaneously bind multiple membranes (described below). Finally, the last portion of the αS sequence, the highly negatively charged and proline-rich C-terminal region (residues 98–140), remains primarily disordered and poorly associated with lipid bilayers51 or detergent micelles.41,56 Using ssNMR in conjunction with paramagnetic relaxation enhancement (PRE), we observed that the C-terminal region establishes weak and transient interactions with the membrane surface,57 a property that might have functional relevance in influencing protein–protein interactions, such as for example those established with synaptobrevin-2 upon SV binding.16,20
3. αS binding to synaptic vesicles
The localisation of αS at presynaptic terminals, and a number of in vivo evidences, strongly indicate an involvement in SV trafficking.12 In this context, it is generally considered that αS binds SVs, despite initial proteomic studies did not found this protein in association to SVs.58 Recently, ultra-definition experiments have, however, identified αS as a “SV visitor” protein,59 in line with the overall body of evidences supporting the weak and transient association of αS with SVs. Super-resolution imaging of synaptosomes showed that αS strongly colocalizes with SVs at the pre-synaptic terminals, and that this process is regulated by calcium ions that weakly bind to the C-terminus of the protein (Fig. 2C).60 In addition to this evidence for SV binding, an emerging idea is forming about a role for αS as a chaperone of the assembly of the SNARE complex through direct interaction with synaptobrevin-2 on SV surface.15,16 Indeed triple knock out mice lacking α-, β-, and γ-synucleins were found to develop neuropathological phenotypes associated with impaired SNARE activity with ageing.61 It was also shown that, as a result of the binding to SV, αS is able to rescue the SNARE formation in mice lacking CSPα.62Upon SV binding, αS has the ability to promote their clustering,18–20 a mechanism involved with the maintenance of SV pools at the synaptic termini.17,22–24 We obtained experimental evidence for an underlying structural mechanism for the promotion of SV clustering, which is based on the subtle equilibrium between structured and disordered conformations adopted by αS when bound to the surface of SVs. In particular, the study of the pathological αS variants A30P and E46K revealed that the N-terminal (residues 1 to 25) and central (residues 65 to 97) regions of the protein are somewhat independent in their binding to the membrane surface.57 This observation suggested that, in addition to interact with the same membrane, the two regions could bind simultaneously across two different membranes. The resulting “double-anchor” (Fig. 3A) provides a mechanism for the αS promotion of indirect interactions between two separate vesicles. In aqueous solutions, this process can be quantified with assays monitoring the fusion of liposomes (Fig. 3C) and the clustering of ex vivo SVs (Fig. 3B).57 By characterising these processes, it was found that the efficiency by which the double-anchor mechanism promotes vesicle-vesicle interactions depends on the amount of detachment of the central region of αS from the membrane surface. Indeed the bound–unbound equilibrium can be altered by several factors such as the characteristics of the lipid bilayers, including curvature, charge,51 levels of cholesterol,63 packing defects and surface hydrophobicity,41,44,64–66 as well as the properties of αS, including mutations57 and post-translational modifications.67 The double-anchor mechanism clarifies a number of experimental observations where mutational variants affecting the membrane affinity of the two individual anchors impairs vesicle clustering, as observed in S. cerevisiae18 or in aqueous solutions.20 These investigations also identify a new link between functional and aberrant behaviour of membrane-bound αS.57 In particular, the evolutionary pressure toward sequences of αS that promote the detachment of the central region from the membrane surface may have a dual effect of (i) improving the efficiency of the double-anchor mechanism in vesicle clustering and (ii) enhancing the exposure of the amyloidogenic NAC region. The latter effect, however, may favour unwanted αS self-assembly thereby enhancing its propensity to aggregate at the membrane surface.17,68–71
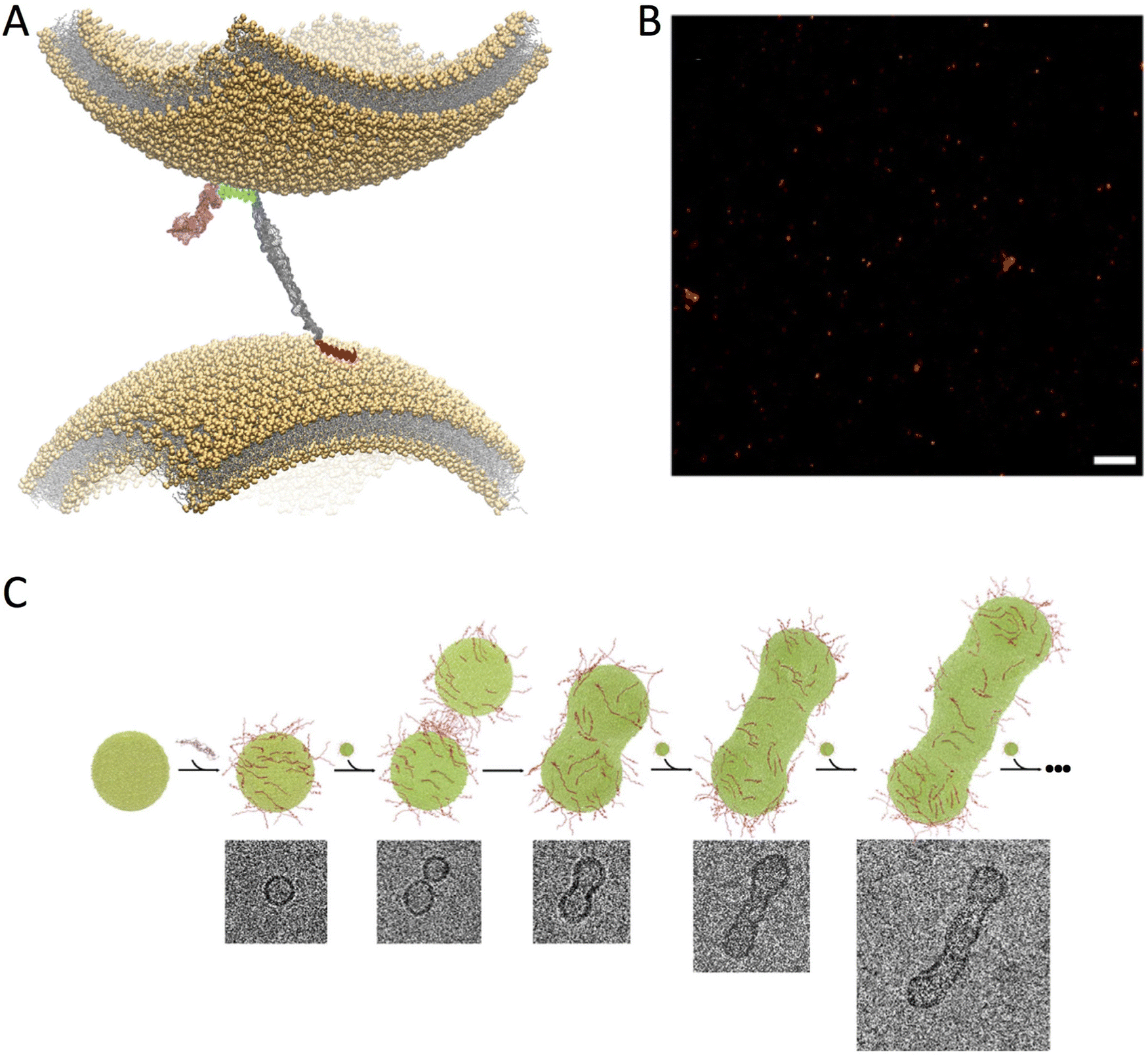 | ||
| Fig. 3 Double-anchor mechanism. (A) Structural model of the double-anchor mechanism by which a single αS molecule binds simultaneously two lipid vesicles that are up to 150 Å apart, thereby promoting their indirect interaction. A first anchor, which spans residues 1 to 25 in N-terminal region (red), binds a vesicle in an amphipathic helical conformation while a second anchor, spanning the region 65 to 97 (cyan) of αS, binds a second vesicle. The C-terminal region (residues 99 to 140) and the linker (residues 26 to 59) are shown in pink and grey, respectively. (B) dSTORM imaging of the clustering of ex vivo SVs upon incubation in vitro with αS. Scale bar 1 μm. C) Stepwise representation of vesicle fusion promoted by αS in vitro as probed by cryo-EM. αS molecules (red) bind dynamically the surface of lipid vesicles (green) by promoting their fusion in a “worm-like” assembly. All images adapted from ref. 57, with permission from Nature Publishing Group, copyright 2016. | ||
4. Binding to other membranes and organelles
In addition to SV binding, αS has been associated with a variety of other synaptic membranes. In particular, experimental evidences have been obtained for αS binding to mitochondrial membranes,72 mitochondrial-associated membranes,73 and presynaptic membranes (PM).74 In the specific case of binding to the PM, monomeric forms of αS have been shown to localise intracellularly near the PM,75,76 while interaction with the outer PM leaflet has been associated with the cellular uptake of αS monomers77 or pathological aggregates.1,78 We studied the modes of binding of αS with the PM and found that the interactions with inner and outer leaflets (IPM and OPM) are significantly different (Fig. 4A). In particular, αS was found to have poor affinity for OPM, although alterations of the content of gangliosides (GM) in the lipid bilayer, particularly GM1 and GM3, enhanced the affinity for this membrane. This finding has relevance in the context some neurodegenerative conditions where increased GM content occurs in the PM79 as well as for lipid rafts,80 where GM are highly abundant.81 By contrast we found that αS has considerable affinity for IPM, and that the structural properties of this binding make it ideal to promote a double-anchor mechanism between the IPM and SVs (Fig. 4B), which is in line with some literature hypotheses.21,82,83 Using model vesicles, we also found that αS stabilizes thermodynamically and kinetically the docking of SVs on the IPM in a concentration-dependent manner.81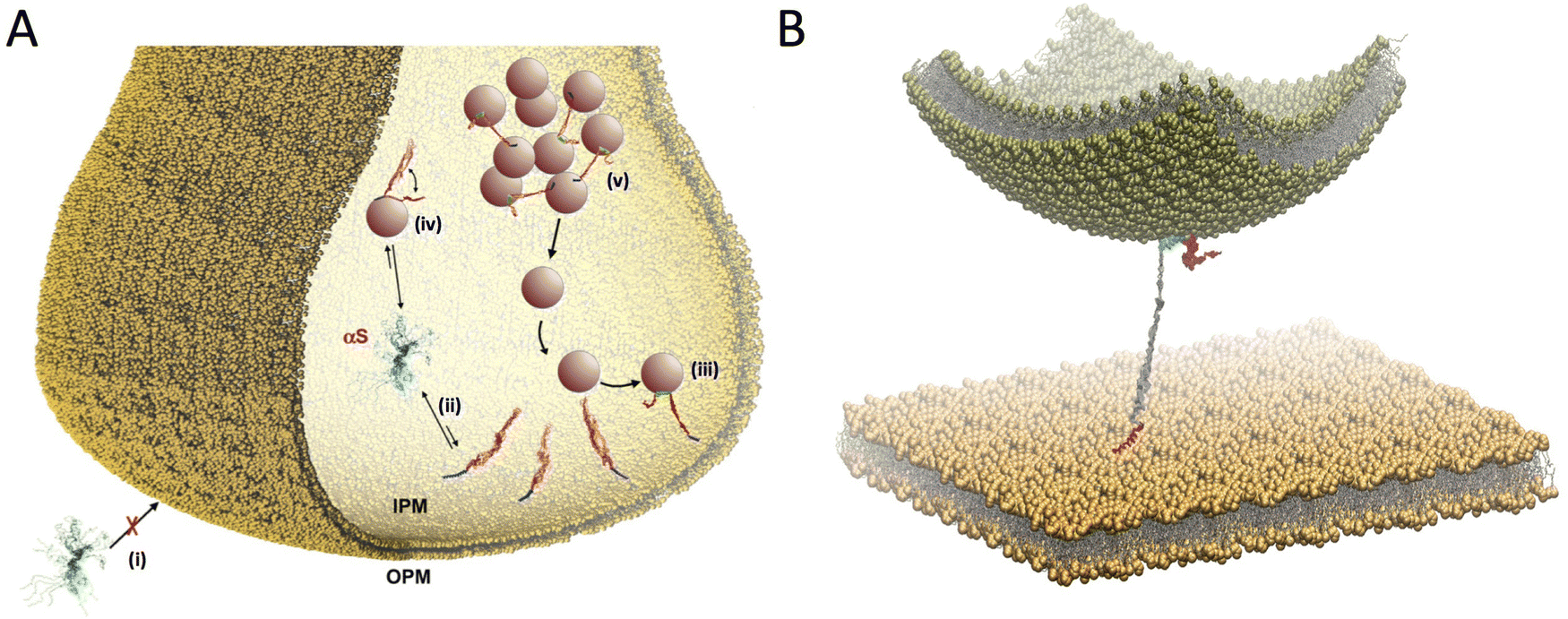 | ||
| Fig. 4 Multiple membrane interactions by αS at the synaptic termini. (A) Scheme of competing interactions between αS and biological membranes at the presynaptic terminal. αS has negligible affinity to bind the outer PM (OPM) (i) but significantly higher affinity for the internal PM (IPM) (ii). The IPM bound state includes primarily the binding through the N-terminal anchor of αS (blue), with the region 65–140 (red) being mostly dissociated from the IPM surface. This conformation is optimal to promote a double-anchor mechanism (first anchor in blue and second anchor spanning residues 65–97 in green) to stabilise the SV docking on the IPM surface (iii). Competing with this process, the binding to SVs (iv) promotes the clustering of SV in pools (v) slowing down the SV diffusion toward the active zone. (B) Double anchor mechanism by which αS stabilizes SVs (green vesicle) on the IPM (flat yellow membrane) in a concentration dependent manner. The N-terminal region (red) binds the IPM while the central region (residues 65–97, cyan) interacts with the SV. The C-terminal region (residues 98–140) and the linker between the anchors (residues 26–64) are drawn in magenta and gray, respectively. Adapted from ref. 81, with permission from Nature Publishing Group, copyright 2021. | ||
The ability of αS to induce both SV–IPM and SV–SV interactions explains, at least in part, some literature controversies about the promotion15,16 or inhibition84–86 of the SNARE activity. In particular, the promotion of SV clusters20 downregulates the flow of vesicles toward the active zone17 thereby negatively contributing to SV exocytosis. By contrast, the stabilisation of the SV docking on the IPM surface81 as well as the role of chaperone for the SNARE formation16 are expected to favour the exocytosis of SVs. These literature evidences, therefore, delineate a subtle equilibrium between different roles of αS in competing processes within the overall SV exocytosis (Fig. 4A). This balance can be perturbed by external factors such as for example the lipid dyshomeostasis leading to an increase of GM content in the PM79 as well as calcium bursts,60 post-translational modifications67 (particularly Ser 87,87 Ser 12988 and Tyr 3982) or αS aggregation. These recent data thereby provide a link between functional and pathological properties of αS in the context of SV trafficking.
In addition to the PM, αS binding to mitochondrial membranes is attracting considerable interest. Mitochondrial dysfunction is a hallmark of the pathogenesis of PD and other neurodegenerative disorders,89–91 and alterations of the ATP and ROS production have been reported in the context of PD in addition to changes in the mitochondrial morphology and integrity. Studies have demonstrated that αS localisation in mitochondria has an effect on their functions and integrity92–97 and experimental evidences exist about the binding of αS to both synthetic models of mitochondrial membranes and ex vivo mitochondria. It has been shown that αS co-localizes with both outer and inner mitochondrial membranes, with some studies pointing to an exclusive interaction with OMM92 or IMM,96,97 and others indicating binding to both types of membranes.93,94 In addition, αS appears to interact also with mitochondrial associated membranes (MAMs).73 Pathological point mutations of αS, including A53T and A30P, have been shown to alter its mitochondrial interactions and abundance in isolated mitochondria.73,93 It remains to be clarified whether the mitochondrial co-localization of αS is promoted by protein–protein interactions, including chaperones98 or membrane translocases such as TOM40,93 TOM2099 and VDAC100 or only by its intrinsic affinity for membrane binding. Studies of αS binding to model synthetic mitochondrial membranes showed the key role of the N-terminal region of the protein as the deletion of the first 11 residues impaired this interaction.96
There is a debate about the role of αS in mitochondrial morphology and dynamics,101 with studies indicating that αS induces mitochondrial fragmentation92,102,103 particularly when in the form of the PD variant A53T.104 The underlying mechanisms of αS induced mitochondrial fragmentation are still unknown, with studies pointing to a direct involvement of the αS-membrane interaction.102 Other investigations, however, have revealed that αS inhibits mitochondrial fusion92 thereby posing new questions on its mitochondrial role in functional and pathological contexts. It is also now clear that in addition to the physiological monomeric form, aggregates of αS are also involved in mitochondrial interactions under pathological conditions. A key question arises whether αS aggregates induce mitochondrial dysfunction or it is the loss of mitochondrial function and integrity to trigger the formation of oligomeric αS species. In this context, the increase of cardiolipin on the OMM has been shown to modulate the accumulation of αS aggregates,105 whereas αS oligomers, but not monomeric or fibrillar species, were shown to damage isolated mitochondria106 and mitochondrial-mimicking membranes107 where αS oligomers generated pores in the membranes. These and other studies also flagged the importance of cardiolipin in promoting the interaction of αS with mitochondrial membranes97 and in enhancing the pore-forming activity of its oligomeric species.
5. Pathological membrane binding by αS aggregates
The pathological relevance of αS aggregation into Lewy bodies is now established and recent structural works have revealed that lipids represent also a significant component of these intraneuronal depositions forming in conjunction with PD.108 A key focus therefore exists on the role of the membrane interaction in the process of αS aggregation. Many studies have been performed on the role of membranes in altering the kinetics and pathways of αS aggregation, with different results showing either inhibition or acceleration of this process depending on the experimental conditions and the membrane properties.17,71,109–111 Literature data indicate that the membrane interaction has dramatic effects on the conformational ensemble of αS, including the expansion from a compact state as observed in the cytosol34,112 and the consequent exposure of the amyloidogenic NAC region at the membrane surface.113 The mechanism by which membrane binding enhances αS aggregation in vitro is indeed of particular interest in the context of synucleinopathies. Solid-state NMR studies have revealed that membrane-induced αS aggregation passes through an α-helical intermediate, which is not observed when the protein aggregates in aqueous solution.69 By combining kinetics and structural studies, prefibrillar and early fibrillar intermediates of αS forming on the surface of anionic vesicles were isolated and characterised at high resolution using ssNMR.114 This study showed a segmental folding process that enables the gradual build-up of the structural elements composing membrane-induced αS fibrils. Moreover, structural studies of membrane-induced αS aggregates have shown the role of lipids in directing the protofilament fold and in mediating the arrangement of protofilaments.115 The involvement of membranes in the fibrillization of αS may also explain the different polymorphic amyloids structures in post mortem analyses of patients affected by synucleinopathies.108,116Despite the progress in the characterisation of fibrillar structures of αS,117–122 including WT and pathological mutants123,124 as well as ex vivo aggregates,108,116 the properties of oligomeric species of this protein remain still elusive. Understanding the nature of αS oligomers is of critical importance as it has become evident both in vitro125–128 and in vivo32,129 that these small and diffusible aggregates are likely to be the key aggregates inducing neurotoxicity in PD.
To clarify the mechanism by which toxic αS oligomers induce membrane disruption, we characterised the structural properties and membrane interactions of two types oligomers of exhibiting significantly different toxicity levels.1 In particular, type-B* αS oligomers, in contrast to the type-A* species, were found to induce toxicity when incubated with neuroblastoma cells and primary cortical neurons, including the generation of intracellular ROS, the increase in the basal calcium levels and the impairment of the mitochondrial activity.1 When unilaterally infused into the substantia nigra of rats, type-B* αS oligomers triggered the deposition in neurons and microglia of fibrils of αS with significant levels of S129 phosphorylation, as well as persistent neuroinflammatory response, early mitochondrial loss and a gradual nigrostriatal dopaminergic loss, associated with motor and cognitive impairment.130 Further studies found that bilateral intracerebral infusion of the type-B* αS oligomers induced memory deficits in rats whereas spreading of toxic αS was detected within anatomically interconnected areas of the brain.131 The analyses showed neuroinflammation in distant cognition-relevant regions of the brain as well as a proinflammatory phenotype in microglia, as shown by increased levels of microglial tumor necrosis factor alpha (TNF-α).131
ssNMR investigations of the type-B* αS oligomers clarified the critical role two elements of the aggregates in the process of binding and disruption of neuronal membranes (Fig. 5A and B).1 The first element is the highly lipophilic N-terminal region of αS, which is exposed at the surface of the type-B* oligomers. This region promotes strong binding with neuronal membranes whereas a second region, the fibrillar core of the oligomers that is formed by a portion of the NAC region, inserts into the lipid bilayer thereby disrupting the membrane integrity. Mutations reducing the membrane affinity of the N-terminal region of αS were found to suppress the toxicity of type-B* oligomers when incubated with neuroblastoma cells and primary cortical neurons. Collectively these data suggest that targeting the membrane interaction of αS aggregates provides a critical therapeutic opportunity to suppress their toxicity. This hypothesis was tested using an antibody that specifically binds the N-terminal region of αS protomers within the type-B* oligomers,132 showing a potent inhibition of the toxicity of the aggregates when incubated with neuronal cells. The antibody was also shown to rescue the pathological phenotype of C. elegans strains overexpressing αS in muscles (Fig. 5C).132 In addition to suppressing the toxicity derived from αS aggregation in C. elegans, the antibody was shown to reduce the overall aggregation of the protein in vivo. In similar contexts, the use of small molecules such as Trodusquemine to displace toxic protein oligomers from cellular membranes has shown promising results.133
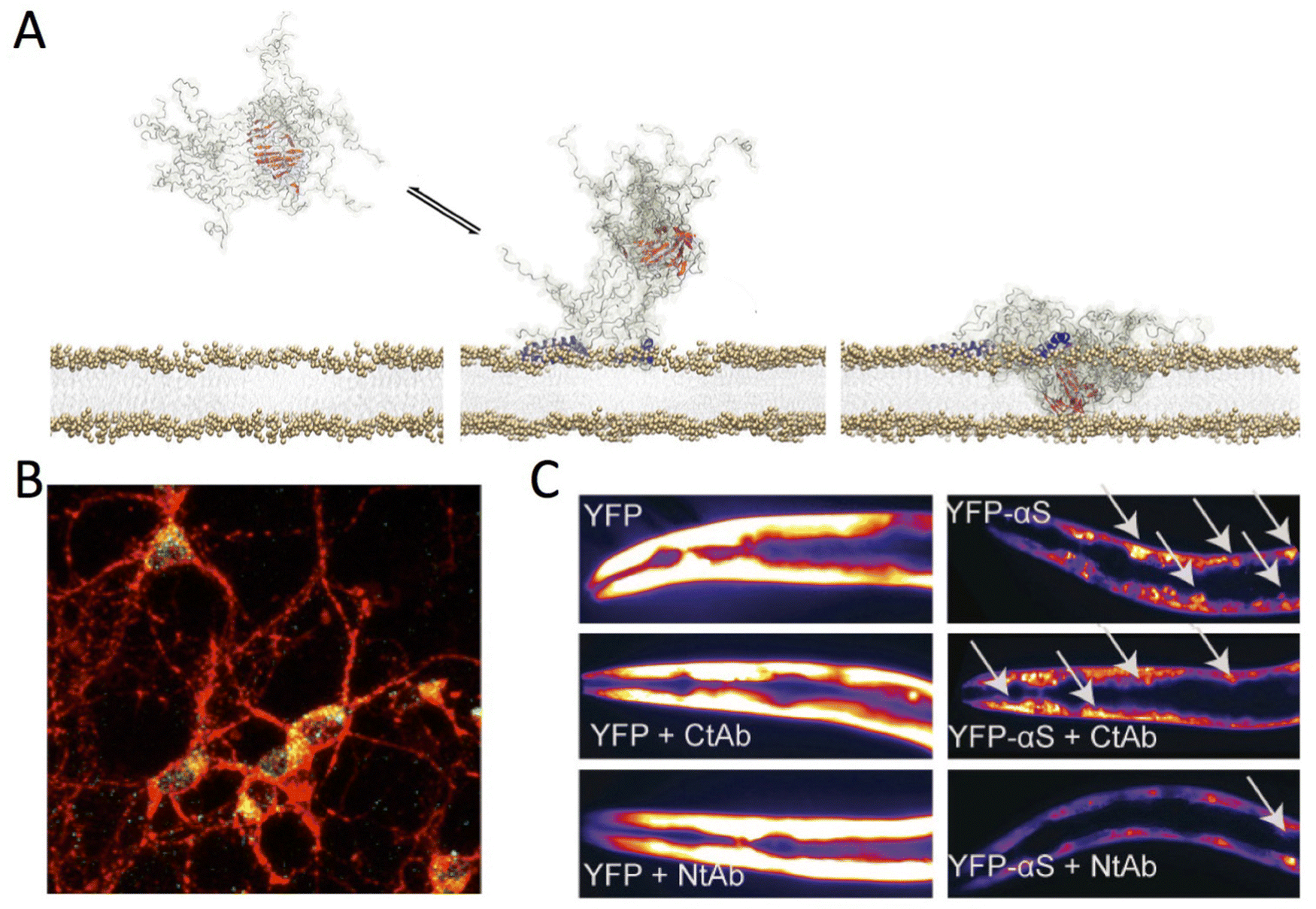 | ||
| Fig. 5 Aberrant membrane disruption by αS oligomers. (A) Toxic type-B* αS oligomers were found to possess both structured (red) and disordered (grey) regions. The oligomers bind the membrane surface using the N-terminal regions of αS protomers in the assemblies (blue). The structured rigid core of the oligomers was found to insert into the lipid bilayer thereby disrupting its integrity. Adapted from ref. 1, with permission from the American Association for the Advancement of Science (licence number 5567500159295), copyright 2017. (B) Membrane interaction of type-B* αS oligomers with primary cortical neurons imaged using confocal scanning microscopy. Red and green fluorescence indicates the cell membranes and the αS oligomers, respectively. Adapted from ref. 132, with permission from American Chemical Society, copyright 2019. (C) Effect antibodies targeting the N- and C-terminal regions of αS (Nt-Ab and Ct-Ab) in C. elegans models of αS toxicity. These worms overexpress YFP-tagged αS in the muscles and the aggregation of the protein generates toxicity. Representative fluorescence microscopy images of worms at day 11 of adulthood expressing YFP (left) or YFP-αS (right) is shown in the absence (top) or presence of 0.4 μM of Ct-Ab (middle) or Nt-Ab (bottom). The inclusions are indicated by white arrows. Adapted from ref. 132, with permission from American Chemical Society, copyright 2019. | ||
6. Conclusions
We are now beginning to uncover the connection between the normal and pathological forms of αS and how specific structural elements of this protein, normally employed for its functional membrane interaction, can be recruited in processes associated with aberrant αS aggregation. It has been demonstrated that toxic oligomeric species of αS exploit the lipophilic nature of the N-terminal membrane anchor to interact with biological membranes and promote unphysiologically strong binding that irreversibly disrupts the membrane integrity. These recent studies therefore revealed that the membrane interactions of physiological monomers and pathological aggregates of αS, despite being involved in dramatically different cellular processes, share common features, including the mechanism of membrane anchoring promoted by the N-terminal region. The similarities in the interaction propensity of monomeric and aggregated species of αS may be at the origin of the synaptic dysfunction observed in some pathological contexts. αS oligomers were indeed shown to impair the SNARE formation by establishing aberrant interactions with the N-terminal region of synaptobrevin-2.134 The concomitance of all PD familial variants in the membrane-binding region of αS also indicate alterations in the membrane binding, as demonstrated in vitro,57,67,135 which may in turn affect the biological behaviour of the protein in the context of SV trafficking.136,137 Finally, a link between dysfunction and aggregation has been proposed whereby the fibrils growth induces toxic effects by depleting αS monomers, leading to a loss of function of the protein.138Taken together these recent literature studies uncover the nature of the tremendous challenges ahead in identifying suitable molecular strategies to target pathological aggregates of αS in PD without interfering with its physiological monomeric form, as the biological properties of both αS species are inextricably linked with similar strategies of membrane interaction. To achieve this critical milestone in PD research, it will be fundamentally important to advance our understanding of the physiological function of αS at the neuronal synapses.
Conflicts of interest
There are no conflicts to declare.References
- G. Fusco, S. W. Chen, P. T. F. Williamson, R. Cascella, M. Perni, J. A. Jarvis, C. Cecchi, M. Vendruscolo, F. Chiti, N. Cremades, L. Ying, C. M. Dobson and A. De Simone, Science, 2017, 358, 1440–1443 CrossRef CAS PubMed.
- H. A. Lashuel, C. R. Overk, A. Oueslati and E. Masliah, Nat. Rev. Neurosci., 2013, 14, 38–48 CrossRef CAS PubMed.
- M. G. Spillantini, M. L. Schmidt, V. M. Lee, J. Q. Trojanowski, R. Jakes and M. Goedert, Nature, 1997, 388, 839–840 CrossRef CAS PubMed.
- R. W. Newberry, J. T. Leong, E. D. Chow, M. Kampmann and W. F. DeGrado, Nat. Chem. Biol., 2020, 16, 653–659 CrossRef CAS PubMed.
- K. C. Luk, V. Kehm, J. Carroll, B. Zhang, P. O'Brien, J. Q. Trojanowski and V. M. Lee, Science, 2012, 338, 949–953 CrossRef CAS PubMed.
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2017, 86, 27–68 CrossRef CAS PubMed.
- V. N. Uversky and D. Eliezer, Curr. Protein Pept. Sci., 2009, 10, 483–499 CrossRef CAS PubMed.
- S. J. Lee and E. Masliah, Nature, 2015, 522, 296–297 CrossRef CAS PubMed.
- M. C. Chartier-Harlin, J. Kachergus, C. Roumier, V. Mouroux, X. Douay, S. Lincoln, C. Levecque, L. Larvor, J. Andrieux, M. Hulihan, N. Waucquier, L. Defebvre, P. Amouyel, M. Farrer and A. Destee, Lancet, 2004, 364, 1167–1169 CrossRef CAS PubMed.
- A. B. Singleton, M. Farrer, J. Johnson, A. Singleton, S. Hague, J. Kachergus, M. Hulihan, T. Peuralinna, A. Dutra, R. Nussbaum, S. Lincoln, A. Crawley, M. Hanson, D. Maraganore, C. Adler, M. R. Cookson, M. Muenter, M. Baptista, D. Miller, J. Blancato, J. Hardy and K. Gwinn-Hardy, Science, 2003, 302, 841 CrossRef CAS PubMed.
- K. Ueda, H. Fukushima, E. Masliah, Y. Xia, A. Iwai, M. Yoshimoto, D. A. Otero, J. Kondo, Y. Ihara and T. Saitoh, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 11282–11286 CrossRef CAS PubMed.
- J. Burre, J. Parkinson's Dis., 2015, 5, 699–713 CAS.
- D. D. Murphy, S. M. Rueter, J. Q. Trojanowski and V. M. Lee, J. Neurosci., 2000, 20, 3214–3220 CrossRef CAS PubMed.
- J. M. George, H. Jin, W. S. Woods and D. F. Clayton, Neuron, 1995, 15, 361–372 CrossRef CAS PubMed.
- J. Burre, M. Sharma and T. C. Sudhof, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E4274–4283 CrossRef CAS PubMed.
- J. Burre, M. Sharma, T. Tsetsenis, V. Buchman, M. R. Etherton and T. C. Sudhof, Science, 2010, 329, 1663–1667 CrossRef CAS PubMed.
- P. K. Auluck, G. Caraveo and S. Lindquist, Annu. Rev. Cell Dev. Biol., 2010, 26, 211–233 CrossRef CAS PubMed.
- J. H. Soper, S. Roy, A. Stieber, E. Lee, R. B. Wilson, J. Q. Trojanowski, C. G. Burd and V. M. Lee, Mol. Biol. Cell, 2008, 19, 1093–1103 CrossRef CAS PubMed.
- A. D. Gitler, B. J. Bevis, J. Shorter, K. E. Strathearn, S. Hamamichi, L. J. Su, K. A. Caldwell, G. A. Caldwell, J. C. Rochet, J. M. McCaffery, C. Barlowe and S. Lindquist, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 145–150 CrossRef CAS PubMed.
- J. Diao, J. Burre, S. Vivona, D. J. Cipriano, M. Sharma, M. Kyoung, T. C. Sudhof and A. T. Brunger, eLife, 2013, 2, e00592 CrossRef PubMed.
- J. Lautenschlager, C. F. Kaminski and G. S. Kaminski Schierle, Trends Cell Biol., 2017, 27, 468–479 CrossRef CAS PubMed.
- K. J. Vargas, S. Makani, T. Davis, C. H. Westphal, P. E. Castillo and S. S. Chandra, J. Neurosci., 2014, 34, 9364–9376 CrossRef PubMed.
- V. M. Nemani, W. Lu, V. Berge, K. Nakamura, B. Onoa, M. K. Lee, F. A. Chaudhry, R. A. Nicoll and R. H. Edwards, Neuron, 2010, 65, 66–79 CrossRef CAS PubMed.
- A. A. Cooper, A. D. Gitler, A. Cashikar, C. M. Haynes, K. J. Hill, B. Bhullar, K. Liu, K. Xu, K. E. Strathearn, F. Liu, S. Cao, K. A. Caldwell, G. A. Caldwell, G. Marsischky, R. D. Kolodner, J. Labaer, J. C. Rochet, N. M. Bonini and S. Lindquist, Science, 2006, 313, 324–328 CrossRef CAS PubMed.
- N. Plotegher, E. Gratton and L. Bubacco, Biochim. Biophys. Acta, 2014, 1840, 2014–2024 CrossRef CAS PubMed.
- M. Ramezani, M. M. Wilkes, T. Das, D. Holowka, D. Eliezer and B. Baird, NPJ Parkinsons Dis., 2019, 5, 12 CrossRef PubMed.
- S. Menges, G. Minakaki, P. M. Schaefer, H. Meixner, I. Prots, U. Schlotzer-Schrehardt, K. Friedland, B. Winner, T. F. Outeiro, K. F. Winklhofer, C. A. von Arnim, W. Xiang, J. Winkler and J. Klucken, Sci. Rep., 2017, 7, 42942 CrossRef CAS PubMed.
- A. S. Maltsev, J. Chen, R. L. Levine and A. Bax, J. Am. Chem. Soc., 2013, 135, 2943–2946 CrossRef CAS PubMed.
- G. Fusco, M. Sanz-Hernandez and A. De Simone, Curr. Opin. Struct. Biol., 2018, 48, 49–57 CrossRef CAS PubMed.
- H. J. Lee and S. J. Lee, J. Biol. Chem., 2002, 277, 48976–48983 CrossRef CAS PubMed.
- N. Lorenzen, L. Lemminger, J. N. Pedersen, S. B. Nielsen and D. E. Otzen, FEBS Lett., 2014, 588, 497–502 CrossRef CAS PubMed.
- B. Winner, R. Jappelli, S. K. Maji, P. A. Desplats, L. Boyer, S. Aigner, C. Hetzer, T. Loher, M. Vilar, S. Campioni, C. Tzitzilonis, A. Soragni, S. Jessberger, H. Mira, A. Consiglio, E. Pham, E. Masliah, F. H. Gage and R. Riek, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 4194–4199 CrossRef CAS PubMed.
- C. A. Waudby, C. Camilloni, A. W. Fitzpatrick, L. D. Cabrita, C. M. Dobson, M. Vendruscolo and J. Christodoulou, PLoS One, 2013, 8, e72286 CrossRef CAS PubMed.
- F. X. Theillet, A. Binolfi, B. Bekei, A. Martorana, H. M. Rose, M. Stuiver, S. Verzini, D. Lorenz, M. van Rossum, D. Goldfarb and P. Selenko, Nature, 2016, 530, 45–50 CrossRef CAS PubMed.
- A. S. Maltsev, J. Ying and A. Bax, Biochemistry, 2012, 51, 5004–5013 CrossRef CAS PubMed.
- S. Gil, T. Hosek, Z. Solyom, R. Kummerle, B. Brutscher, R. Pierattelli and I. C. Felli, Angew. Chem., 2013, 52, 11808–11812 CrossRef CAS PubMed.
- J. H. Lee, F. Li, A. Grishaev and A. Bax, J. Am. Chem. Soc., 2015, 137, 1432–1435 CrossRef CAS PubMed.
- M. Drescher, Top. Curr. Chem., 2012, 321, 91–119 CrossRef CAS PubMed.
- M. M. Dedmon, K. Lindorff-Larsen, J. Christodoulou, M. Vendruscolo and C. M. Dobson, J. Am. Chem. Soc., 2005, 127, 476–477 CrossRef CAS PubMed.
- C. R. Bodner, C. M. Dobson and A. Bax, J. Mol. Biol., 2009, 390, 775–790 CrossRef CAS PubMed.
- T. S. Ulmer, A. Bax, N. B. Cole and R. L. Nussbaum, J. Biol. Chem., 2005, 280, 9595–9603 CrossRef CAS PubMed.
- D. Eliezer, E. Kutluay, R. Bussell, Jr. and G. Browne, J. Mol. Biol., 2001, 307, 1061–1073 CrossRef CAS PubMed.
- G. Fusco, A. De Simone, P. Arosio, M. Vendruscolo, G. Veglia and C. M. Dobson, Sci. Rep., 2016, 6, 27125 CrossRef CAS PubMed.
- C. C. Jao, B. G. Hegde, J. Chen, I. S. Haworth and R. Langen, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 19666–19671 CrossRef CAS PubMed.
- H. T. Kratochvil, R. W. Newberry, B. Mensa, M. Mravic and W. F. DeGrado, Faraday Discuss., 2021, 232, 9–48 RSC.
- C. Y. Cheng, J. Varkey, M. R. Ambroso, R. Langen and S. Han, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 16838–16843 CrossRef CAS PubMed.
- S. Chandra, X. Chen, J. Rizo, R. Jahn and T. C. Sudhof, J. Biol. Chem., 2003, 278, 15313–15318 CrossRef CAS PubMed.
- S. B. Lokappa and T. S. Ulmer, J. Biol. Chem., 2011, 286, 21450–21457 CrossRef CAS PubMed.
- E. R. Georgieva, T. F. Ramlall, P. P. Borbat, J. H. Freed and D. Eliezer, J. Am. Chem. Soc., 2008, 130, 12856–12857 CrossRef CAS PubMed.
- E. R. Georgieva, T. F. Ramlall, P. P. Borbat, J. H. Freed and D. Eliezer, J. Biol. Chem., 2010, 285, 28261–28274 CrossRef CAS PubMed.
- G. Fusco, A. De Simone, T. Gopinath, V. Vostrikov, M. Vendruscolo, C. M. Dobson and G. Veglia, Nat. Commun., 2014, 5, 3827 CrossRef CAS PubMed.
- C. R. Bodner, A. S. Maltsev, C. M. Dobson and A. Bax, Biochemistry, 2010, 49, 862–871 CrossRef CAS PubMed.
- M. Runfola, A. De Simone, M. Vendruscolo, C. M. Dobson and G. Fusco, Sci. Rep., 2020, 10, 204 CrossRef CAS PubMed.
- C. Navarro-Paya, M. Sanz-Hernandez and A. De Simone, Life, 2020, 10, 98 CrossRef CAS PubMed.
- C. Navarro-Paya, M. Sanz-Hernandez and A. De Simone, Front. Mol. Biosci., 2022, 9, 857217 CrossRef CAS PubMed.
- T. S. Ulmer and A. Bax, J. Biol. Chem., 2005, 280, 43179–43187 CrossRef CAS PubMed.
- G. Fusco, T. Pape, A. D. Stephens, P. Mahou, A. R. Costa, C. F. Kaminski, G. S. Kaminski Schierle, M. Vendruscolo, G. Veglia, C. M. Dobson and A. De Simone, Nat. Commun., 2016, 7, 12563 CrossRef PubMed.
- S. Takamori, M. Holt, K. Stenius, E. A. Lemke, M. Gronborg, D. Riedel, H. Urlaub, S. Schenck, B. Brugger, P. Ringler, S. A. Muller, B. Rammner, F. Grater, J. S. Hub, B. L. De Groot, G. Mieskes, Y. Moriyama, J. Klingauf, H. Grubmuller, J. Heuser, F. Wieland and R. Jahn, Cell, 2006, 127, 831–846 CrossRef CAS PubMed.
- Z. Taoufiq, M. Ninov, A. Villar-Briones, H. Wang, T. Sasaki, M. Roy, F. Beauchain, Y. Mori, T. Yoshida, S. Takamori, R. Jahn and T. Takahashi, Proc. Natl. Acad. Sci. U. S. A., 2019, 117, 33586–33596 CrossRef PubMed.
- J. Lautenschlager, A. D. Stephens, G. Fusco, F. Strohl, N. Curry, M. Zacharopoulou, C. H. Michel, R. Laine, N. Nespovitaya, M. Fantham, D. Pinotsi, W. Zago, P. Fraser, A. Tandon, P. S. George-Hyslop, E. Rees, J. J. Phillips, A. De Simone, C. F. Kaminski and G. S. K. Schierle, Nat. Commun., 2018, 9, 712 CrossRef PubMed.
- A. Pelkonen and L. Yavich, Neurosci. Lett., 2011, 487, 350–353 CrossRef CAS PubMed.
- S. Chandra, G. Gallardo, R. Fernandez-Chacon, O. M. Schluter and T. C. Sudhof, Cell, 2005, 123, 383–396 CrossRef CAS PubMed.
- W. K. Man, A. De Simone, J. D. Barritt, M. Vendruscolo, C. M. Dobson and G. Fusco, Front. Neurosci., 2020, 14, 18 CrossRef PubMed.
- J. N. Rao, C. C. Jao, B. G. Hegde, R. Langen and T. S. Ulmer, J. Am. Chem. Soc., 2010, 132, 8657–8668 CrossRef CAS PubMed.
- M. M. Ouberai, J. Wang, M. J. Swann, C. Galvagnion, T. Guilliams, C. M. Dobson and M. E. Welland, J. Biol. Chem., 2013, 288, 20883–20895 CrossRef CAS PubMed.
- G. F. Wang, C. Li and G. J. Pielak, Protein Sci., 2010, 19, 1686–1691 CrossRef CAS PubMed.
- L. Reimer, H. Gram, N. M. Jensen, C. Betzer, L. Yang, L. Jin, M. Shi, D. Boudeffa, G. Fusco, A. De Simone, D. Kirik, H. A. Lashuel, J. Zhang and P. H. Jensen, PNAS Nexus, 2022, 1, pgac259 CrossRef PubMed.
- M. Grey, S. Linse, H. Nilsson, P. Brundin and E. Sparr, J. Parkinson's Dis., 2011, 1, 359–371 CAS.
- G. Comellas, L. R. Lemkau, D. H. Zhou, J. M. George and C. M. Rienstra, J. Am. Chem. Soc., 2012, 134, 5090–5099 CrossRef CAS PubMed.
- M. Grey, C. J. Dunning, R. Gaspar, C. Grey, P. Brundin, E. Sparr and S. Linse, J. Biol. Chem., 2015, 290, 2969–2982 CrossRef CAS PubMed.
- C. Galvagnion, A. K. Buell, G. Meisl, T. C. Michaels, M. Vendruscolo, T. P. Knowles and C. M. Dobson, Nat. Chem. Biol., 2015, 11, 229–234 CrossRef CAS PubMed.
- M. Vicario, D. Cieri, M. Brini and T. Calì, Front. Neurosci., 2018, 12, 388 CrossRef PubMed.
- C. Guardia-Laguarta, E. Area-Gomez, C. Rub, Y. Liu, J. Magrane, D. Becker, W. Voos, E. A. Schon and S. Przedborski, J. Neurosci., 2014, 34, 249–259 CrossRef CAS PubMed.
- D. Snead and D. Eliezer, Exp. Neurobiol., 2014, 23, 292–313 CrossRef PubMed.
- D. L. Fortin, M. D. Troyer, K. Nakamura, S. Kubo, M. D. Anthony and R. H. Edwards, J. Neurosci., 2004, 24, 6715–6723 CrossRef CAS PubMed.
- T. F. Outeiro and S. Lindquist, Science, 2003, 302, 1772–1775 CrossRef CAS PubMed.
- J. Y. Park, K. S. Kim, S. B. Lee, J. S. Ryu, K. C. Chung, Y. K. Choo, I. Jou, J. Kim and S. M. Park, J. Neurochem., 2009, 110, 400–411 CrossRef CAS PubMed.
- C. Masaracchia, M. Hnida, E. Gerhardt, T. Lopes da Fonseca, A. Villar-Pique, T. Branco, M. A. Stahlberg, C. Dean, C. O. Fernandez, I. Milosevic and T. F. Outeiro, Acta Neuropathol. Commun., 2018, 6, 79 CrossRef PubMed.
- N. Yamamoto, T. Matsubara, T. Sato and K. Yanagisawa, Biochem. Biophys. Acta, 2008, 1778, 2717–2726 CAS.
- L. Liu, K. Zhang, L. Tan, Y. H. Chen and Y. P. Cao, Alzheimer Dis. Assoc. Disord., 2015, 29, 63–69 CrossRef CAS PubMed.
- W. K. Man, B. Tahirbegi, M. D. Vrettas, S. Preet, L. Ying, M. Vendruscolo, A. De Simone and G. Fusco, Nat. Commun., 2021, 12, 927 CrossRef CAS PubMed.
- I. Dikiy, B. Fauvet, A. Jovicic, A. L. Mahul-Mellier, C. Desobry, F. El-Turk, A. D. Gitler, H. A. Lashuel and D. Eliezer, ACS Chem. Biol., 2016, 11, 2428–2437 CrossRef CAS PubMed.
- I. Dikiy and D. Eliezer, Biochem. Biophys. Acta, 2012, 1818, 1013–1018 CAS.
- N. Thayanidhi, J. R. Helm, D. C. Nycz, M. Bentley, Y. Liang and J. C. Hay, Mol. Biol. Cell, 2010, 21, 1850–1863 CrossRef CAS PubMed.
- Y. Lai, S. Kim, J. Varkey, X. Lou, J. K. Song, J. Diao, R. Langen and Y. K. Shin, Biochemistry, 2014, 53, 3889–3896 CrossRef CAS PubMed.
- D. C. DeWitt and E. Rhoades, Biochemistry, 2013, 52, 2385–2387 CrossRef CAS PubMed.
- A. Oueslati, K. E. Paleologou, B. L. Schneider, P. Aebischer and H. A. Lashuel, J. Neurosci., 2012, 32, 1536–1544 CrossRef CAS PubMed.
- A. D. Stephens, M. Zacharopoulou, R. Moons, G. Fusco, N. Seetaloo, A. Chiki, P. J. Hooper, I. Mela, H. Lashuel, J. J. Philips, A. De Simone, F. Sobott and G. S. Kaminski Schierle, Nat. Commun., 2020, 10, 1038 Search PubMed.
- M. Nguyen, Y. C. Wong, D. Ysselstein, A. Severino and D. Krainc, Trends Neurosci., 2019, 42, 140–149 CrossRef CAS PubMed.
- G. Cenini, A. Lloret and R. Cascella, Oxid. Med. Cell. Longevity, 2019, 2019, 2105607 Search PubMed.
- M. T. Lin and M. F. Beal, Nature, 2006, 443, 787–795 CrossRef CAS PubMed.
- F. Kamp, N. Exner, A. K. Lutz, N. Wender, J. Hegermann, B. Brunner, B. Nuscher, T. Bartels, A. Giese, K. Beyer, S. Eimer, K. F. Winklhofer and C. Haass, EMBO J., 2010, 29, 3571–3589 CrossRef CAS PubMed.
- L. Devi, V. Raghavendran, B. M. Prabhu, N. G. Avadhani and H. K. Anandatheerthavarada, J. Biol. Chem., 2008, 283, 9089–9100 CrossRef CAS PubMed.
- K. Nakamura, V. M. Nemani, E. K. Wallender, K. Kaehlcke, M. Ott and R. H. Edwards, J. Neurosci., 2008, 28, 12305–12317 CrossRef CAS PubMed.
- M. S. Parihar, A. Parihar, M. Fujita, M. Hashimoto and P. Ghafourifar, Cell. Mol. Life Sci., 2008, 65, 1272–1284 CrossRef CAS PubMed.
- M. Robotta, H. R. Gerding, A. Vogel, K. Hauser, S. Schildknecht, C. Karreman, M. Leist, V. Subramaniam and M. Drescher, ChemBioChem, 2014, 15, 2499–2502 CrossRef CAS PubMed.
- I. G. Zigoneanu, Y. J. Yang, A. S. Krois, E. Haque and G. J. Pielak, Biochem. Biophys. Acta, 2012, 1818, 512–519 CAS.
- B. M. Burmann, J. A. Gerez, I. Matecko-Burmann, S. Campioni, P. Kumari, D. Ghosh, A. Mazur, E. E. Aspholm, D. Sulskis, M. Wawrzyniuk, T. Bock, A. Schmidt, S. G. D. Rudiger, R. Riek and S. Hiller, Nature, 2020, 577, 127–132 CrossRef CAS PubMed.
- R. Di Maio, P. J. Barrett, E. K. Hoffman, C. W. Barrett, A. Zharikov, A. Borah, X. Hu, J. McCoy, C. T. Chu, E. A. Burton, T. G. Hastings and J. T. Greenamyre, Sci. Transl. Med., 2016, 8, 342ra378 Search PubMed.
- L. J. Martin, S. Semenkow, A. Hanaford and M. Wong, Neurobiol. Aging, 2014, 35, 1132–1152 CrossRef CAS PubMed.
- N. J. Thorne and D. A. Tumbarello, Front. Mol. Neurosci., 2022, 15, 947191 CrossRef CAS PubMed.
- K. Nakamura, V. M. Nemani, F. Azarbal, G. Skibinski, J. M. Levy, K. Egami, L. Munishkina, J. Zhang, B. Gardner, J. Wakabayashi, H. Sesaki, Y. Cheng, S. Finkbeiner, R. L. Nussbaum, E. Masliah and R. H. Edwards, J. Biol. Chem., 2011, 286, 20710–20726 CrossRef CAS PubMed.
- E. K. Butler, A. Voigt, A. K. Lutz, J. P. Toegel, E. Gerhardt, P. Karsten, B. Falkenburger, A. Reinartz, K. F. Winklhofer and J. B. Schulz, PLoS Genet., 2012, 8, e1002488 CrossRef CAS PubMed.
- V. M. Pozo Devoto and T. L. Falzone, Dis. Models & Mech., 2017, 10, 1075–1087 Search PubMed.
- T. Ryan, V. V. Bamm, M. G. Stykel, C. L. Coackley, K. M. Humphries, R. Jamieson-Williams, R. Ambasudhan, D. D. Mosser, S. A. Lipton, G. Harauz and S. D. Ryan, Nat. Commun., 2018, 9, 817 CrossRef PubMed.
- E. S. Luth, I. G. Stavrovskaya, T. Bartels, B. S. Kristal and D. J. Selkoe, J. Biol. Chem., 2014, 289, 21490–21507 CrossRef PubMed.
- S. Ghio, A. Camilleri, M. Caruana, V. C. Ruf, F. Schmidt, A. Leonov, S. Rayazanov, C. Griesinger, R. Cauchi, F. Kamp, A. Giese and N. Vassallo, ACS Chem. Neurosci., 2019, 10, 3815–3829 CrossRef CAS PubMed.
- Y. Yang, Y. Shi, M. Schweighauser, X. Zhang, A. Kotecha, A. G. Murzin, H. J. Garringer, P. W. Cullinane, Y. Saito, T. Foroud, T. T. Warner, K. Hasegawa, R. Vidal, S. Murayama, T. Revesz, B. Ghetti, M. Hasegawa, T. Lashley, S. H. W. Scheres and M. Goedert, Nature, 2022, 610, 791–795 CrossRef CAS PubMed.
- M. Zhu and A. L. Fink, J. Biol. Chem., 2003, 278, 16873–16877 CrossRef CAS PubMed.
- R. J. Perrin, W. S. Woods, D. F. Clayton and J. M. George, J. Biol. Chem., 2001, 276, 41958–41962 CrossRef CAS PubMed.
- E. Jo, J. McLaurin, C. M. Yip, P. S. George-Hyslop and P. E. Fraser, J. Biol. Chem., 2000, 275, 34328–34334 CrossRef CAS PubMed.
- D. Bhattacharyya, R. Kumar, S. Mehra, A. Ghosh, S. K. Maji and A. Bhunia, Chem. Commun., 2018, 54, 3605–3608 RSC.
- R. Pariary, D. Bhattacharyya and A. Bhunia, Gene Rep., 2019, 16, 100423 CrossRef.
- L. Antonschmidt, R. Dervişoǧlu, V. Sant, K. T. Movellan, I. Mey, D. Riedel, C. Steinem, S. Becker, L. B. Andreas and C. Griesinger, Sci. Adv., 2021, 7, eabg2174 CrossRef CAS PubMed.
- B. Frieg, L. Antonschmidt, C. Dienemann, J. A. Geraets, E. E. Najbauer, D. Matthes, B. L. de Groot, L. B. Andreas, S. Becker, C. Griesinger and G. F. Schröder, Nat. Commun., 2022, 13, 6810 CrossRef CAS PubMed.
- M. Schweighauser, Y. Shi, A. Tarutani, F. Kametani, A. G. Murzin, B. Ghetti, E. Matsubara, T. Tomita, T. Ando, K. Hasegawa, S. Murayama, M. Yoshida, M. Hasegawa, S. H. Scheres and M. Goedert, Nature, 2020, 585, 464–469 CrossRef CAS PubMed.
- M. D. Tuttle, G. Comellas, A. J. Nieuwkoop, D. J. Covell, D. A. Berthold, K. D. Kloepper, J. M. Courtney, J. K. Kim, A. M. Barclay, A. Kendall, W. Wan, G. Stubbs, C. D. Schwieters, V. M. Lee, J. M. George and C. M. Rienstra, Nat. Struct. Mol. Biol., 2016, 23, 409–415 CrossRef CAS PubMed.
- L. Bousset, L. Pieri, G. Ruiz-Arlandis, J. Gath, P. H. Jensen, B. Habenstein, K. Madiona, V. Olieric, A. Bockmann, B. H. Meier and R. Melki, Nat. Commun., 2013, 4, 2575 CrossRef PubMed.
- J. Gath, L. Bousset, B. Habenstein, R. Melki, B. H. Meier and A. Bockmann, Biomol. NMR Assignments, 2014, 8, 395–404 CrossRef CAS PubMed.
- H. Heise, W. Hoyer, S. Becker, O. C. Andronesi, D. Riedel and M. Baldus, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 15871–15876 CrossRef CAS PubMed.
- B. Li, P. Ge, K. A. Murray, P. Sheth, M. Zhang, G. Nair, M. R. Sawaya, W. S. Shin, D. R. Boyer, S. Ye, D. S. Eisenberg, Z. H. Zhou and L. Jiang, Nat. Commun., 2018, 9, 3609 CrossRef PubMed.
- R. Guerrero-Ferreira, N. M. Taylor, A. A. Arteni, P. Kumari, D. Mona, P. Ringler, M. Britschgi, M. E. Lauer, A. Makky, J. Verasdonck, R. Riek, R. Melki, B. H. Meier, A. Böckmann, L. Bousset and H. Stahlberg, eLife, 2019, 8, e36402 CrossRef PubMed.
- D. R. Boyer, B. Li, C. Sun, W. Fan, K. Zhou, M. P. Hughes, M. R. Sawaya, L. Jiang and D. S. Eisenberg, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 3592–3602 CrossRef CAS PubMed.
- D. R. Boyer, B. Li, C. Sun, W. Fan, M. R. Sawaya, L. Jiang and D. S. Eisenberg, Nat. Struct. Mol. Biol., 2019, 26, 1044–1052 CrossRef CAS PubMed.
- N. Cremades, S. I. Cohen, E. Deas, A. Y. Abramov, A. Y. Chen, A. Orte, M. Sandal, R. W. Clarke, P. Dunne, F. A. Aprile, C. W. Bertoncini, N. W. Wood, T. P. Knowles, C. M. Dobson and D. Klenerman, Cell, 2012, 149, 1048–1059 CrossRef CAS PubMed.
- S. W. Chen, S. Drakulic, E. Deas, M. Ouberai, F. A. Aprile, R. Arranz, S. Ness, C. Roodveldt, T. Guilliams, E. J. De-Genst, D. Klenerman, N. W. Wood, T. P. Knowles, C. Alfonso, G. Rivas, A. Y. Abramov, J. M. Valpuesta, C. M. Dobson and N. Cremades, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E1994–2003 CAS.
- P. R. Angelova, M. H. Ludtmann, M. H. Horrocks, A. Negoda, N. Cremades, D. Klenerman, C. M. Dobson, N. W. Wood, E. V. Pavlov, S. Gandhi and A. Y. Abramov, J. Cell Sci., 2016, 129, 1792–1801 CAS.
- H. L. Roberts and D. R. Brown, Biomolecules, 2015, 5, 282–305 CrossRef CAS PubMed.
- O. W. Wan and K. K. Chung, PLoS One, 2012, 7, e38545 CrossRef CAS PubMed.
- L. Boi, A. Pisanu, M. F. Palmas, G. Fusco, E. Carboni, M. A. Casu, V. Satta, M. Scherma, E. Janda, I. Mocci, G. Mulas, A. Ena, S. Spiga, P. Fadda, A. De Simone and A. R. Carta, Int. J. Mol. Sci., 2020, 21, 8535 CrossRef CAS PubMed.
- M. F. Palmas, M. Etzi, A. Pisanu, C. Camoglio, C. Sagheddu, M. Santoni, M. F. Manchinu, M. Pala, G. Fusco, A. De Simone, L. Picci, G. Mulas, S. Spiga, M. Scherma, P. Fadda, M. Pistis, N. Simola, E. Carboni and A. R. Carta, Cells, 2022, 11, 2628 CrossRef CAS PubMed.
- R. Cascella, M. Perni, S. W. Chen, G. Fusco, C. Cecchi, M. Vendruscolo, F. Chiti, C. M. Dobson and A. De Simone, ACS Chem. Biol., 2019, 14, 1352–1362 CrossRef CAS PubMed.
- R. Limbocker, B. Mannini, F. S. Ruggeri, R. Cascella, C. K. Xu, M. Perni, S. Chia, S. W. Chen, J. Habchi, A. Bigi, R. P. Kreiser, A. K. Wright, J. A. Albright, T. Kartanas, J. R. Kumita, N. Cremades, M. Zasloff, C. Cecchi, T. P. J. Knowles, F. Chiti, M. Vendruscolo and C. M. Dobson, Commun. Biol., 2020, 3, 435 CrossRef CAS PubMed.
- B. K. Choi, M. G. Choi, J. Y. Kim, Y. Yang, Y. Lai, D. H. Kweon, N. K. Lee and Y. K. Shin, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 4087–4092 CrossRef CAS PubMed.
- M. B. Fares, N. Ait-Bouziad, I. Dikiy, M. K. Mbefo, A. Jovicic, A. Kiely, J. L. Holton, S. J. Lee, A. D. Gitler, D. Eliezer and H. A. Lashuel, Hum. Mol. Genet., 2014, 23, 4491–4509 CrossRef CAS PubMed.
- M. Sharma and J. Burré, Trends Neurosci., 2023, 46, 153–166 CrossRef CAS PubMed.
- P. Calabresi, A. Mechelli, G. Natale, L. Volpicelli-Daley, G. Di Lazzaro and V. Ghiglieri, Cell Death Dis., 2023, 14, 176 CrossRef PubMed.
- N. M. Kanaan and F. P. Manfredsson, J. Parkinson's Dis., 2012, 2, 249–267 CAS.
- A. Bigi, R. Cascella, F. Chiti and C. Cecchi, BioEssays, 2022, 44, e2200086 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2023 |