
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntense absorption of azulene realized by molecular orbital inversion†
Takahiro
Tsuchiya
 *a,
Tomohiro
Hamano
a,
Masahiro
Inoue
a,
Tomoya
Nakamura
b,
Atsushi
Wakamiya
b and
Yasuhiro
Mazaki
*a
*a,
Tomohiro
Hamano
a,
Masahiro
Inoue
a,
Tomoya
Nakamura
b,
Atsushi
Wakamiya
b and
Yasuhiro
Mazaki
*a
aDepartment of Chemistry, Kitasato University Kitasato 1-15-1, Sagamihara, Kanagawa 252-0373, Japan. E-mail: ttsuchi@kitasato-u.ac.jp
bInstitute for Chemical Research, Kyoto University Uji, Kyoto 611-0011, Japan
First published on 26th July 2023
Abstract
The introduction of diarylamino groups at the 2- and 6-positions of azulene was found to invert the order of the orbital energy levels and allowed the HOMO−LUMO transition, resulting in a substantial increase in absorbance in the visible region. In addition, the stability of their one-electron oxidised species was improved by introducing bromine or methoxy groups at the 1- and 3-positions.
Benzene and acenes, such as naphthalene and anthracene, display excellent electron-donating properties when they contain substituted bis(diarylamino) groups1–10 and have garnered interest due to their potential applications such as organic semiconductors,1 multi-electron redox materials2–4 for rechargeable batteries,5,6 chromophore linkers for nonlinear optically active metal–organic framework (MOF) materials,7 and dopants in the emitting layers for organic light-emitting devices (OLEDs).8 These substituted acenes are also attracting attention because of their ability to absorb visible light, which enables them to be used as photoreduction catalysts.9,10 Furthermore, azulene11–20 is a structural isomer of naphthalene, with relatively high HOMO and low LUMO energy levels due to its unique polarisation structure (Fig. 1a), and it is also known as a redox-active molecule compared to naphthalene, anthracene, pyrene and so on.11–15 In this context, hole-transporting materials with a two-dimensionally expanded π-system around the azulene core for efficient perovskite solar cells have also recently attracted interest.21,22 In addition, 1,3-bis(diarylamino)azulene 1 (Scheme 1a) is reported to exhibit high electron-donating properties.23 However, we are not aware of a system in which diarylamino groups have been introduced at the 2- and 6-positions of azulene. The 1- and 3-positions of azulene have large HOMO coefficients, while the 2- and 6-positions have large HOMO−1 and LUMO coefficients (Fig. 1b).11–15 Hence, it is conceivable that the electronic structure of azulene changes significantly depending on the positions where the diarylamino groups are introduced.
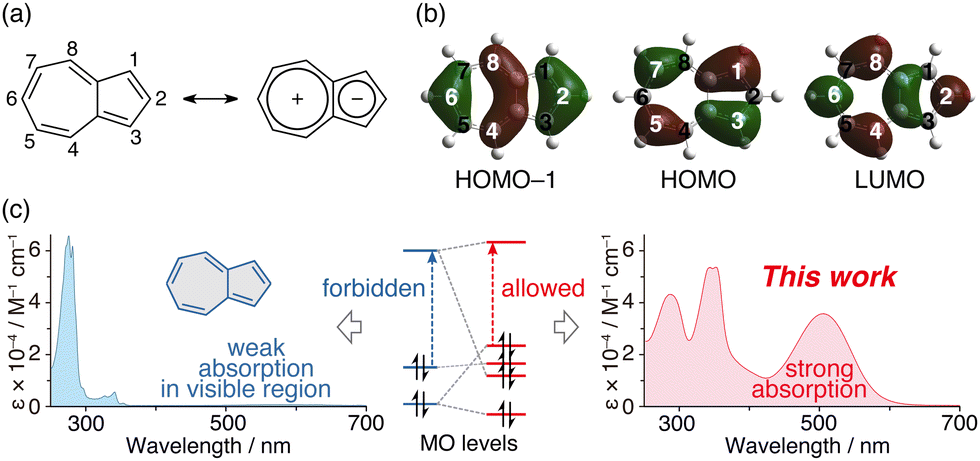 | ||
| Fig. 1 (a) Numbering of atoms in azulene and its polarized resonance structure. (b) HOMO–1, HOMO and LUMO of azulene. (c) Concept for molecular-orbital inverted azulene possessing strong absorption of visible light. | ||
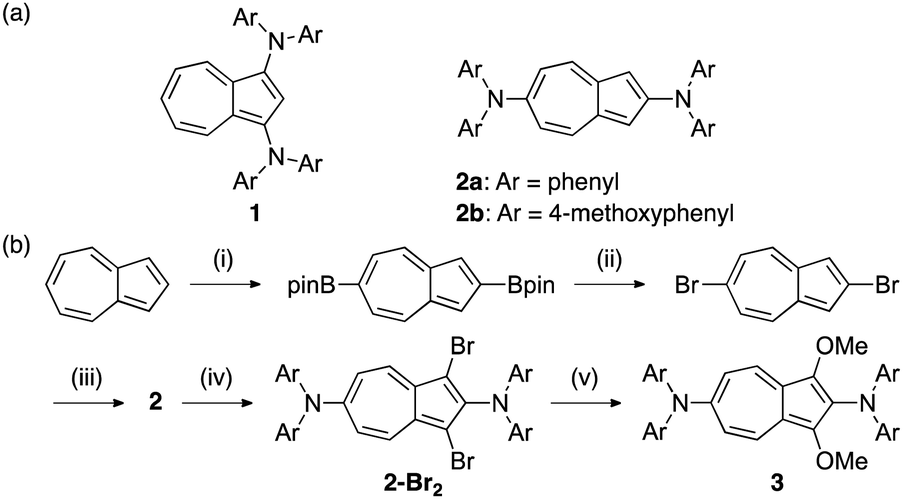 | ||
Scheme 1 (a) Structural formulae of 1 and 2. (b) Synthesis of 2, 2-Br2 and 3. Reagents and conditions: (i) pin2B2, [IrCl(cod)]2, bpy, cyclohexane, reflux, 48 h, 45%; (ii) CuBr, DMF, 80 °C, 2 h, 88%; (iii) Ar2NH, Pd(OAc)2, tBuOK, tBu3PHBF4, toluene, reflux, 2 h, 2a: 80%, 2b: 82%; (iv) NBS, CHCl3, −78 °C, 2 h, 2a-Br2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :82%, 2b-Br2:84%; (v) MeONa, CuBr, MeOH, DMF, 100 °C, 5 d, 3a: 74%, 3b: 58%. :82%, 2b-Br2:84%; (v) MeONa, CuBr, MeOH, DMF, 100 °C, 5 d, 3a: 74%, 3b: 58%. | ||
Azulene exhibits a bright-blue colour and shows absorption derived from the S0–S1 transition at approximately 580 nm.24 However, its molar-absorption coefficient is approximately 350 M−1 cm−1, which is very weak for π–π* transition absorption (Fig. 1c, left).24 This is due to the forbidden HOMO–LUMO transition; the HOMO of azulene is distributed across its odd-positioned carbon atoms, while the coefficients of the LUMO and HOMO−1 are greater in its even-positioned carbon atoms. Therefore, the ability to control the molecular-orbital (MO) energy levels of azulene will lead to the creation of a material having new optical and electronic characteristics (Fig. 1c, right).
In this study, we introduced diarylamino groups at the azulene 2- and 6-positions of compound 2, and then investigated its optical properties and redox behaviour. Moreover, we evaluated the stabilisation of the one-electron oxidised species of 3, in which methoxy groups were introduced at the 1- and 3-positions of azulene.
The synthesis of 1 through the reaction between 1,3-dibromoazulene25 and diarylamine has previously been reported.23 Although 1,3-dibromoazulene can be obtained by the reaction of azulene with N-bromosuccinimide (NBS),25 a precursor of 2, 2,6-dibromoazulene, has been conventionally synthesised from α-tropolone in five steps (14% overall yield).26,27 One of the factors that made the synthesis of 2 difficult is the complicated route of raw material synthesis. However, recent reports have shown the successful borylation of the 2- and 6-positions of azulene through its iridium-catalysed reaction with bis(pinacolato)diboron (B2pin2).28 Hence, in the present study, we synthesised 2,6-dibromoazulene through bromination of 2,6-diborylazulene (Scheme 1b).
The reaction of the obtained diborylazulene with copper bromide gave dibromoazulene with a yield of 92%. Subsequently, 2 was formed by a coupling reaction between dibromoazulene and diarylamine in toluene, in the presence of a Pd-catalyst. Bromination with NBS was used to introduce substituents at positions 1 and 3 for compound 2. Subsequently, 1,3-dimethoxy compound 3 was obtained by reacting the dibromide compound with sodium methoxide in methanol in the presence of copper bromide.
Compounds 2, 2-Br2, and 3 were characterized by NMR measurements (Fig. S1–S9, ESI†). Interestingly, in the 1H NMR spectrum of 2a in CDCl3, the ortho- and meta-position phenyl signals of the diphenylamino group at the 2-position of azulene were observed to be equivalent (Fig. S2a, S3, and S4, ESI†). This is a peculiar phenomenon in CDCl3, and when the solvent was changed to acetone-d6, the signals did not appear to be equivalent (Fig. S2b–S2e, ESI†). It is possible that the signals overlapped to produce something that resembles this multiplet in CDCl3.
Single crystal X-ray structure analysis was conducted on compounds 2a, 2a-Br2, 2b, and 2b-Br2 (Fig. S10–S13, ESI†). The asymmetric unit of 2-Br2 was one molecule, whereas for 2 there was disorder, where the five-membered and seven-membered rings of azulene were inverted, and the asymmetric unit contained half a molecule. Each amino group nitrogen of 2 and 2-Br2 had an almost planar structure, and the dihedral angle between the azulene and amino-group planes was 24° in 2a and 28° in 2b, while in 2-Br2, the dihedral angles were significantly different between the 5-membered- and 7-membered-ring sides, 65° and 13° in 2a-Br2 and 67° and 13° in 2b-Br2, respectively. It was concluded that the dihedral angle on the 2-position side was large because of the steric repulsion between the bromines at the 1- and 3-positions and the phenyl groups. The carbon–carbon bonds of the azulene skeleton tended to elongate slightly around the introduction position of the diarylamino group and bromine.
Compounds 2, 2-Br2, and 3 exhibit brown or reddish-orange colours, which are significantly different from that of the raw material azulene (blue colour). All ultraviolet-visible (UV-Vis) absorption spectra showed absorption bands near 400 to 700 nm (2a: λmax 480 nm, ε 31500 M−1 cm−1; 2b: λmax 490 nm, ε 31700; 2a-Br2: λmax 492 nm, ε 28300, 2b-Br2: λmax 516 nm, ε 34600; 3a: λmax 494 nm, ε 31000; and 3b: λmax 506 nm, ε 37300) in tetrahydrofuran (THF) as shown in Fig. 2a. Absorption in these visible light regions (ε 28300–37300 M−1 cm−1) is much stronger than by azulene (S0–S2, λmax 340 nm, ε 4000 M−1 cm−1; S0–S1, λmax 580 nm, ε 350)24 and also the previously reported 1,3-bis(diphenylamino)azulene (1) (λmax 420 nm, ε 10000 M−1 cm−1; λmax 667 nm, ε 300).23 These absorption bands of 2, 2-Br2, and 3 are distributed at similar or longer wavelengths than those of oligothiophenes (P3HT: 455 nm;29 MK-2: λmax 480 nm, ε 38400 M−1 cm−1;30 MK-3: λmax 485 nm, ε 40100;30 and MK-4: λmax 474 nm, ε 3310030), which are commercially available as organic dyes for dye-sensitised solar cells, and 1,4-bis(diphenylamino)naphthalene (λmax 375 nm, ε 9200 M−1 cm−1),10 which has been used as a photoreduction catalyst under visible light. The absorption spectra of 2, 2-Br2, and 3 showed relatively good agreement with the results of calculations thus using time-dependent density-functional theory (TDDFT) (Fig. 2b), indicating that the strong absorption in the visible light region is derived from the allowed HOMO–LUMO transition (Fig. 3).
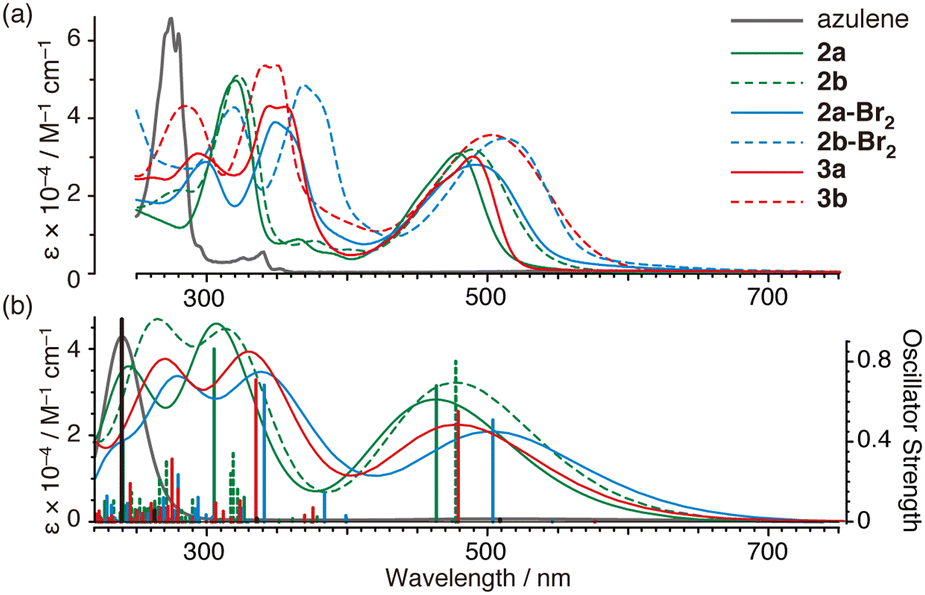 | ||
| Fig. 2 (a) UV-Vis absorption spectra of azulene (black line), 2 (green line), 2-Br2 (blue line) and 3 (red line) in THF. (b) Simulated absorption spectra at the TD-B3LYP/6-31G(d)//B3LYP/6-31G(d) level. | ||
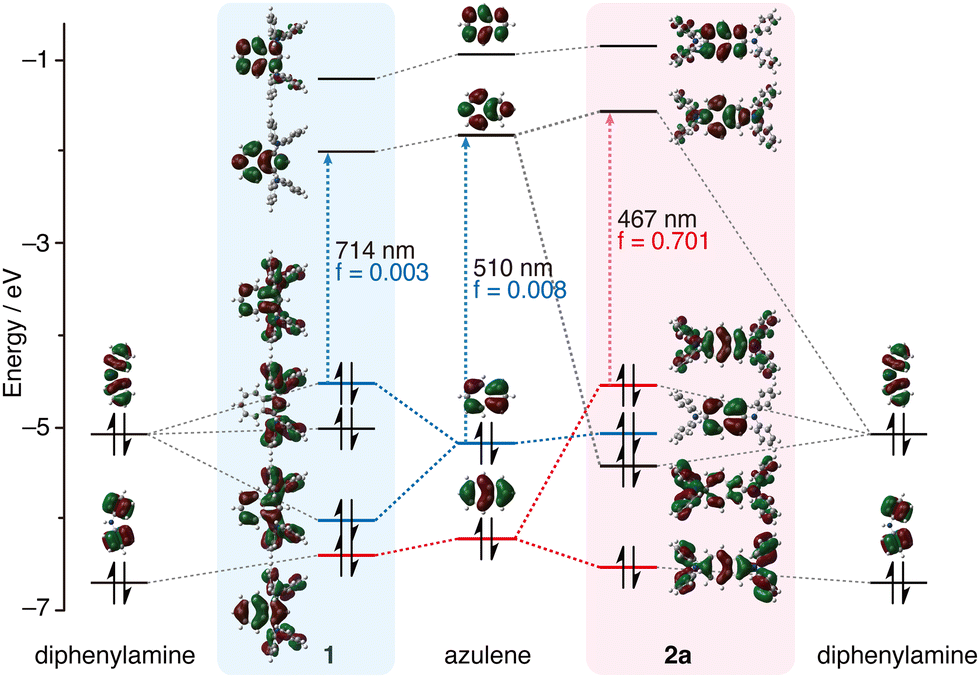 | ||
| Fig. 3 MO diagrams of azulene, diphenylamine, 1 and 2a calculated at the B3LYP/6-31(d) level. | ||
Fig. 3 shows the MO diagram of azulene, diphenylamine, 1, and 2a. Since the 2- and 6-positions of azulene are located in the HOMO node, the introduction of diphenylamino groups at the 2- and 6-positions does not affect the HOMO; however, it is shown that the HOMO−1 and LUMO of azulene strongly form a linear combination with the HOMO of diphenylamine. Consequently, the HOMO of 2a is distributed in both the azulene skeleton and the diphenylamino group, and is formed by the antibonding interaction between the HOMO−1 of azulene and the HOMO of diphenylamine. And the HOMO of 2a is located at a higher level (destabilised) than that of the original azulene. This means that the order of the MO energy levels is inverted between azulene and 2a. As a result, it is conceivable that the HOMO–LUMO transition of 2a is allowed because it is a transition between orbitals with a large coefficient on even-numbered carbons, resulting in stronger absorption in the long-wavelength region. Introduction of one diphenylamino group at the 2- or 6-position of azulene also forms a linear combination of the HOMO−1 of azulene with the HOMO of diphenylamine (Fig. S14, ESI†). However, the interactions are weaker than when doubly substituted, and the LUMO of azulene does not form a linear combination with the HOMO of diphenylamine unlike 2. As a result, the HOMO energy levels of 2- and 6-(diphenylamino)azulene are lower than that of 2.
In 1, the HOMO of the azulene forms a linear combination with the HOMO of diphenylamine (Fig. 3). The HOMO of 1 is also higher in energy than that of the original azulene, thus reducing the HOMO–LUMO gap. Despite this, the HOMO–LUMO transition of 1 remains forbidden. On the other hand, the order of the MO energy levels is consistent among 2, 2-Br2 and 3, and the HOMO–LUMO transitions of the compounds are all allowed (Fig. S15, ESI†). Introduction of methoxy groups at the para-positions of the diphenylamino groups leads to an overall energy increase in the MO levels except for HOMO−1 (Fig. S16, ESI†).
Redox potential measurements were taken to examine the electronic properties in detail (Fig. 4 and Table S8, ESI†). The first reduction potential of 2a (redE1: −2.16 V vs. Fc/Fc+) showed a value similar to that of azulene (redE1: −2.13 V), whereas the first oxidation potential of 2a (oxE1: 0.03 V) had a cathodic shift of about 500 mV vs. Fc/Fc+ relative to that of azulene (oxE1: 0.56 V). The oxidation potential of 2,6-bis(diphenylamino)naphthalene is reportedly 0.24 V,8 indicating that 2a has a higher electron donating capacity. Moreover, compared with 2a, 2b (oxE1: −0.12 V, redE1: −2.28 V) had a cathodic shift of about 100 mV for both oxidation and reduction potentials, and the electron donating ability was further improved. These results are consistent with the results of DFT calculations.
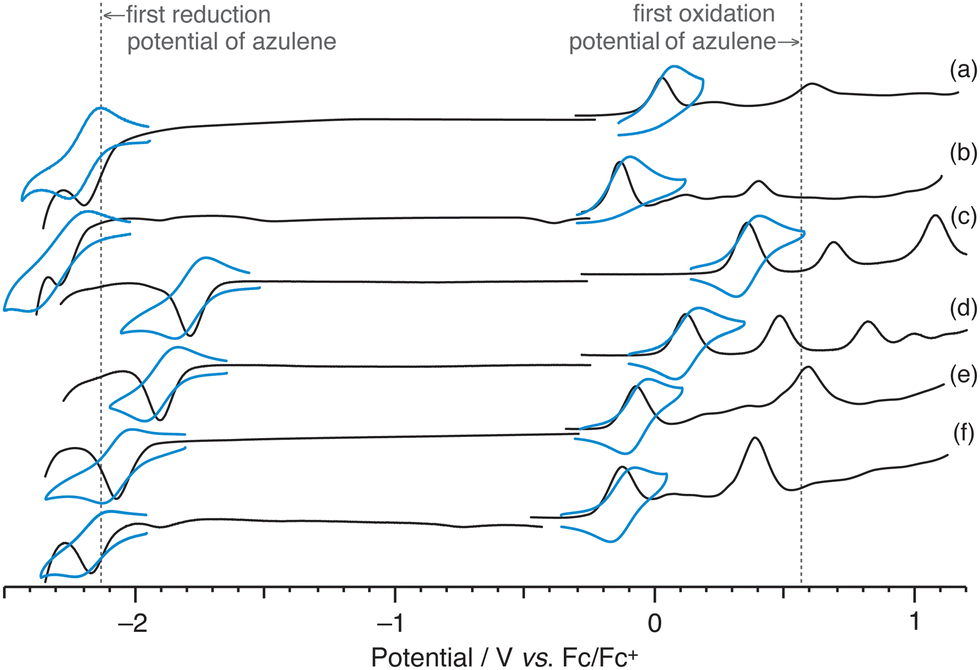 | ||
| Fig. 4 Square-wave voltammogram (SWV, black line) and cyclic voltammogram (CV, blue line) of (a) 2a, (b) 2b, (c) 2a-Br2, (d) 2b-Br2, (e) 3a and (f) 3b. | ||
Although the first reduction waves of 2 showed reversible behaviour in cyclic voltammetry (CV) (Fig. 4a and b, blue lines), the reversibility of the first oxidation processes was not observed. In contrast, it was found that the first oxidation processes of 2-Br2 (2a-Br2:0.36 V, 2b-Br2:0.10 V) and 3 (3a: −0.07 V, 3b: −0.14 V), which possess the functional groups at 1,3-positions, were reversible, and the electron donating ability of 3 was more significant than that of 2. It is known that the irreversible behaviour of the first oxidation process is characteristic of azulene, and that the radical cations of azulene polymerise at the 1,3-positions where the HOMO coefficient is large.31 Although the spin density of the radical cation of 2 is spread throughout the molecule (Fig. S17, ESI†), it is shown that the spin density at the 1,3-positions is still relatively high. We believe that by introducing substituents at the 1,3-positions like 2-Br2 and 3, the polymerisation at these positions was suppressed and reversibility appeared.
The morphology and electronic characteristics of the azulene derivatives in the thin film were also investigated. Atomic force microscopy (AFM) measurements for the spin-coated samples showed a relatively uniform and flat surface (Fig. S18, ESI†). The UV-Vis absorption spectra of 2 and 2a-Br2 in the spin-coated films exhibited a shape similar to those in solution (Fig. 5a), but the absorption in the visible region was shifted by about 13 to 24 nm to longer wavelengths than those in solution. Since the mixed film with poly(methyl methacrylate) (PMMA) showed absorption at almost the same position as that in the solution, it was concluded that the long wavelength shift was caused by intermolecular interactions between the azulene derivatives in the solid state.
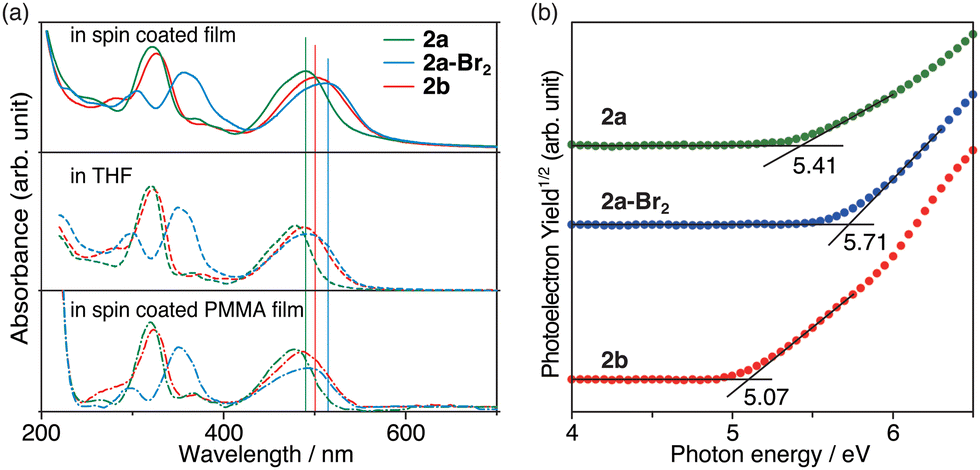 | ||
| Fig. 5 (a) UV-Vis absorption spectra in spin coated film, THF, and spin coated PMMA film and (b) PYS spectra of 2a (green), 2a-Br2 (blue), and 2b (red). | ||
The ionic potentials of 2 in the spin-coated thin-film state were determined by photoelectron yield spectroscopy (PYS) measurements under vacuum (Fig. 5b). The HOMO energies of the thin films of 2a, 2a-Br2 and 2b were found to be −5.41, −5.71 and −5.07 eV, respectively. These values are comparable to those of prevalently used hole transporting materials [2,2′,7,7′-tetrakis(N,N-di-p-methoxyphenylamine)-9,9′-spirobifluorene (spiro-OMeTAD): −5.05 eV;32 poly[bis(4-phenyl)(2,4,6-trimethylphenyl)amine] (PTAA): −5.1 eV;33 poly(3,4-ethylenedioxythiophene)polystyrene sulfonate (PEDOT:PSS): −5.1 eV;34 and azulene-core-based two-dimensionally expanded π-systems: −4.96 to −5.00 eV21] for solid-state dye-sensitized solar cells (ssDSSCs) and perovskite solar cells (PSCs).
In conclusion, we successfully synthesised azulene-based organic electron donors that strongly absorb visible light. This was achieved by introducing electron-donating substituents at the 2- and 6-positions of azulene, resulting in an inversion of the orbital energy level order of original azulene and permitting its previously forbidden HOMO–LUMO transition. Furthermore, it was confirmed that their one-electron oxidised species could be stabilised by introducing a substituent at the 1- and 3-positions. Compounds 2 and 3 have higher HOMOs than metal porphyrins and thiophene trimers and tetramers and exhibit strong absorption properties over a wide range of wavelengths. Therefore, we believe that these findings provide a rational approach to constructing a wide variety of azulene-based π-conjugated molecules for photonic, optoelectronic, and photocatalytic applications.
This work was supported by the International Collaborative Research Program of Institute for Chemical Research, Kyoto University (grant # 2022-22).
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. Freudenberg, D. Jansch, F. Hinkel and U. H. F. Bunz, Chem. Rev., 2018, 118, 5598 CrossRef CAS PubMed.
- M. Roy, J. H. Walton, J. C. Fettinger and A. L. Balch, Chem. – Eur. J., 2022, 28, e202104631 CrossRef CAS PubMed.
- J. Zhang, Z. Chen, L. Yang, F.-F. Pan, G.-A. Yu, J. Yin and S. H. Liu, Sci. Rep., 2016, 6, 1 CrossRef PubMed.
- X. Wang, Z. Zhang, Y. Song, Y. Su and X. Wang, Chem. Commun., 2015, 51, 11822 RSC.
- G. Wang, E. Dmitrieva, B. Kohn, U. Scheler, Y. Liu, V. Tkachova, L. Yang, Y. Fu, J. Ma and P. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202116194 CrossRef CAS PubMed.
- Y. Chen, X. Gao, L. R. Johnson and P. G. Bruce, Nat. Commun., 2018, 9, 1 CrossRef PubMed.
- D. C. Mayer, A. Manzi, R. Medishetty, B. Winkler, C. Schneider, G. Kieslich, A. Pöthig, J. Feldmann and R. A. Fischer, J. Am. Chem. Soc., 2019, 141, 11594 CrossRef CAS PubMed.
- C. Wu, P. I. Djurovich and M. E. Thompson, Adv. Funct. Mater., 2009, 19, 3157 CrossRef CAS.
- R. Taniguchi, N. Noto, S. Tanaka, K. Takahashi, S. K. Sarkar, R. Oyama, M. Abe, T. Koike and M. Akita, Chem. Commun., 2021, 57, 2609 RSC.
- N. Noto, T. Koike and M. Akita, ACS Catal., 2019, 9, 4382 CrossRef CAS.
- T. Tsuchiya, M. Higashibeppu and Y. Mazaki, ChemistryOpen, 2023, 12, e202100298, DOI:10.1002/open.202100298.
- T. Tsuchiya, Y. Katsuoka, K. Yoza, H. Sato and Y. Mazaki, ChemPlusChem, 2019, 84, 1659 CrossRef CAS PubMed.
- T. Tsuchiya, R. Umemura, M. Kaminaga, S. Kushida, K. Ohkubo, S. I. Noro and Y. Mazaki, ChemPlusChem, 2019, 84, 655 CrossRef CAS PubMed.
- A. Konishi and M. Yasuda, Chem. Lett., 2021, 50, 195 CrossRef CAS.
- H. Xin, B. Hou and X. Gao, Acc. Chem. Res., 2021, 54, 1737 CrossRef CAS PubMed.
- A. H. Elwahy and K. Hafner, Asian J. Org. Chem., 2021, 10, 2010 CrossRef CAS.
- P. Bakun, B. Czarczynska-Goslinska, T. Goslinski and S. Lijewski, Med. Chem. Res., 2021, 30, 834 CrossRef CAS PubMed.
- A. G. Lvov and A. Bredihhin, Org. Biomol. Chem., 2021, 19, 4460 RSC.
- L. C. Murfin and S. E. Lewis, Molecules, 2021, 26, 353 CrossRef CAS PubMed.
- T. Shoji, S. Ito and M. Yasunami, Int. J. Mol. Sci., 2021, 22, 10686 CrossRef CAS PubMed.
- M. A. Truong, J. Lee, T. Nakamura, J. Y. Seo, M. Jung, M. Ozaki, A. Shimazaki, N. Shioya, T. Hasegawa, Y. Murata, S. M. Zakeeruddin, M. Grätzel, R. Murdey and A. Wakamiya, Chem. – Eur. J., 2019, 25, 6741 CrossRef CAS PubMed.
- H. Nishimura, N. Ishida, A. Shimazaki, A. Wakamiya, A. Saeki, L. T. Scott and Y. Murata, J. Am. Chem. Soc., 2015, 137, 15656 CrossRef CAS PubMed.
- G. Nöll, S. Amthor, M. Avola, C. Lambert and J. Daub, J. Phys. Chem. C, 2007, 111, 3512 CrossRef.
- S. V. Shevyakov, H. Li, R. Muthyala, A. E. Asato, J. C. Croney, D. M. Jameson and R. S. Liu, J. Phys. Chem. A, 2003, 107, 3295 CrossRef CAS.
- A. Ito, M. Watanabe, A. Ishii, R. Yamasaki and I. Okamoto, Tetrahedron Lett., 2021, 86, 153523 CrossRef CAS.
- Q. Fan, D. Martin-Jimenez, D. Ebeling, C. K. Krug, L. Brechmann, C. Kohlmeyer, G. Hilt, W. Hieringer, A. Schirmeisen and J. M. Gottfried, J. Am. Chem. Soc., 2019, 141, 17713 CrossRef CAS PubMed.
- F. Schwarz, M. Koch, G. Kastlunger, H. Berke, R. Stadler, K. Venkatesan and E. Lörtscher, Angew. Chem., Int. Ed., 2016, 55, 11781 CrossRef CAS PubMed.
- M. Narita, T. Murafuji, S. Yamashita, M. Fujinaga, K. Hiyama, Y. Oka, F. Tani, S. Kamijo and K. Ishiguro, J. Org. Chem., 2018, 83, 1298 CrossRef CAS PubMed.
- K. Rahimi, I. Botiz, J. O. Agumba, S. Motamen, N. Stingelin and G. Reiter, RSC Adv., 2014, 4, 11121 RSC.
- Z.-S. Wang, N. Koumura, Y. Cui, M. Takahashi, H. Sekiguchi, A. Mori, T. Kubo, A. Furube and K. Hara, Chem. Mater., 2008, 20, 3993 CrossRef CAS.
- M. Saitoh, J. Yano, T. Nakazawa, Y. Sugihara and K. Hashimoto, J. Electroanal. Chem., 1996, 418, 139 CrossRef CAS.
- W. H. Nguyen, C. D. Bailie, E. L. Unger and M. D. McGehee, J. Am. Chem. Soc., 2014, 136, 10996 CrossRef CAS PubMed.
- S. Zhang, M. Stolterfoht, A. Armin, Q. Lin, F. Zu, J. Sobus, H. Jin, N. Koch, P. Meredith and P. L. Burn, ACS Appl. Mater. Interfaces, 2018, 10, 21681 CrossRef CAS PubMed.
- F. Hermerschmidt, F. Mathies, V. R. Schröder, C. Rehermann, N. Z. Morales, E. L. Unger and E. J. List-Kratochvil, Mater. Horiz., 2020, 7, 1773 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2254119, 2254124, 2254133, and 2254134. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc02311g |
| This journal is © The Royal Society of Chemistry 2023 |