
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Glycosylation of vicinal di- and trifluorinated glucose and galactose donors†
Kler
Huonnic
a and
Bruno
Linclau
 *ab
*ab
aSchool of Chemistry, University of Southampton, Southampton SO17 1BJ, UK
bDepartment of Organic and Macromolecular Chemistry, Ghent University, Ghent 9000, Belgium. E-mail: Bruno.linclau@ugent.be
First published on 28th June 2023
Abstract
The acid-catalysed formation of glycosidic bonds is more difficult when glycosyl donors are fluorinated, especially at the 2-position. Here we report high-yielding glycosidation and glycosylation reactions of 2,3-difluorinated- and 2,3,4-trifluorinated gluco- and galactopyranoside donors with a variety of acceptors under conventional trichloroacetimidate/TMSOTf activation in moderate to high anomeric selectivities. This methodology allows access to highly fluorinated glycans, illustrated with the synthesis of a pentafluorinated disaccharide.
Glycans are a large class of biomolecules that have important roles in diverse areas such as human health and biomaterials.1,2 The study of how glycans interact with proteins and how glycan structure determines materials properties is therefore of great importance. Selective fluorination of glycans is one of the strategies that enable such studies. It can be used for epitope-mapping studies, for example by comparing binding affinities3,4 or by using 19F NMR based experiments,5 for 18F based diagnostic applications,6 or fluorine can be introduced to deliberately modify protein-carbohydrate interactions as its small size, high electronegativity, and non-polarizable electron pairs affect most of the interaction mechanisms.7 Finally, carbohydrate fluorination influences their hydrolytic and enzymatic stability, and has received attention for property optimisation in a drug development context.8,9 Insights in glycan-based materials properties can be obtained by fluorination, which disrupts local hydrogen bonding networks.10,11
In the vast majority of cases, monofluorinated sugar units are employed. Site-selective fluorination, even of smaller oligosaccharides, is not trivial, and glycosylation involving fluorinated monosaccharide building blocks is usually the only practical way to obtain fluorinated glycans. Enzymatic glycosylations are possible,12,13 but so far only work with monofluorinated donors.14 Fluorination has a negative effect on glycosylation rates because its strong electron withdrawing effect destabilizes glycosylation transition states.7 Chemical synthesis of glycans involving one or more monofluorinated sugar building blocks has been successfully demonstrated.15,16
Glycosylation involving polyfluorinated donors is less common,17 and demonstrated with 2,6-18,19 and 3,6-20 difluorinated donors (Fig. 1A). With fluorination at successive carbon atoms next to the anomeric centre, only glycosidation reactions have been reported (Fig. 1B). With a hexafluorinated donor, DiMagno notably achieved glycosidation using the very reactive triflate leaving group.21 Our group reported an anomeric alkylation approach for the related tetrafluorinated donors.22,23 Both strategies were successfully employed by the Giguère group for the first glycosidations of 2,3,4-trifluorinated donors, using an in situ generated glycosyl bromide with phenolate nucleophiles,24,25 or using Ag2O induced anomeric alkylation with alkyl iodides.26 Unfortunately, this method was shown not to work for glycosylation with a 6-iodinated galactose acceptor. Using the same donor, BF3·OEt2-mediated SN1-type anomeric alkylation also led to good results (not shown).26,27
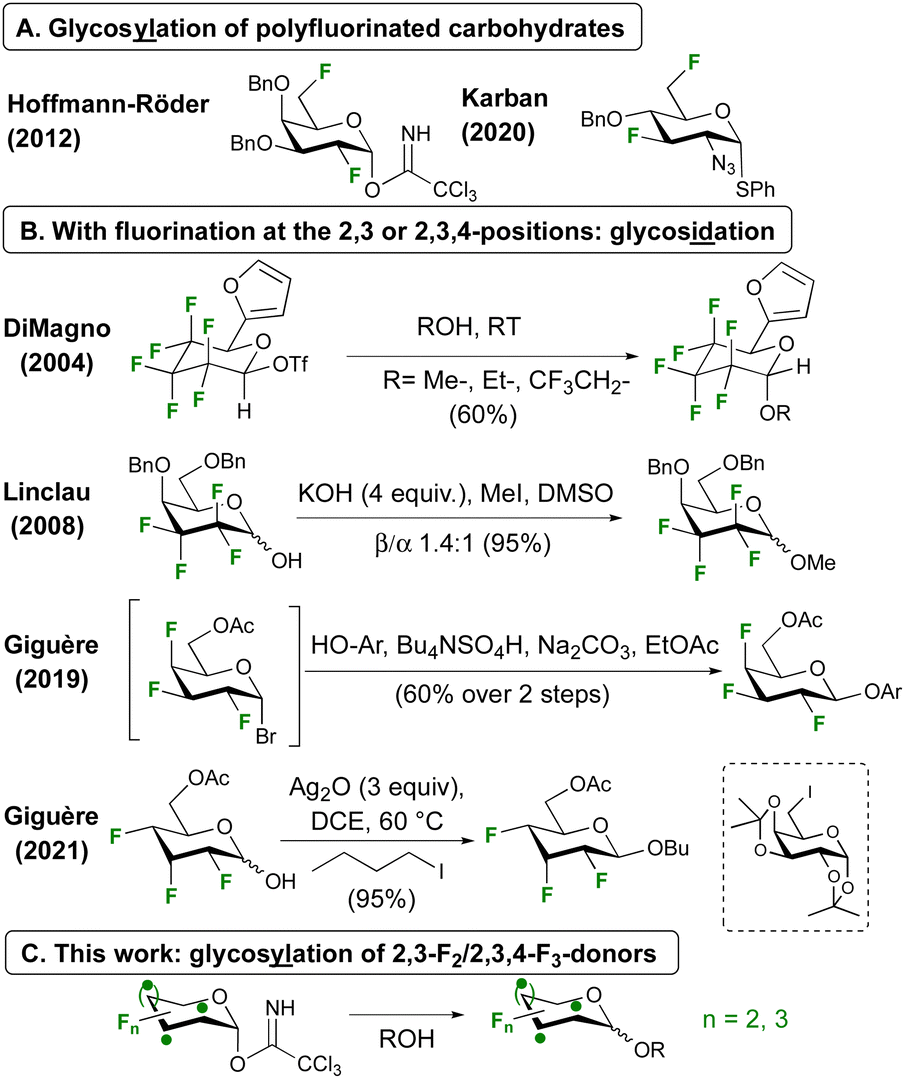 | ||
| Fig. 1 Proposed polyfluorosugar glycosylation, with precedent. | ||
Incorporation of polyfluorinated sugars into glycans will enable novel opportunities for the study of protein-carbohydrate interactions and glycan-based biomaterials. As anomeric alkylation is inherently less attractive for this purpose, not least due to the inevitable inversion of configuration at an acceptor ring position, a conventional glycosylation approach is desired. Here we report our first results of glycosidation and glycosylation of 2,3-difluoro- and 2,3,4-trifluorinated glucose and galactose donors (Fig. 1C).
Anticipating a low donor reactivity, our attention went first to the use of glycosyl triflates as donors, which were synthesised by treatment of the reducing sugar with Tf2O. However, they proved too reactive to be isolated, and their reaction with excess methanol as acceptor led to a complex reaction mixture. The corresponding tosylate donor, which was synthesised by reaction of the hemiacetal with Ts2O, was stable enough to be isolated, but glycosidation reactions also led to complex reaction mixtures. To our delight, attempts to use the conventional trichloroacetimidate group as donor proved successful, prompting their further investigation.
The syntheses of the trichloroacetimidate donors was achieved according to the procedure developed by Schmidt et al28 using the corresponding reducing sugar, 10 equiv. of trichloroacetonitrile, and 0.1 equiv. of DBU (Scheme 1). The reaction was stirred overnight to maximise the formation of the α-anomer, and high yields and selectivities were obtained. Deactivating ester protecting groups were selected in the first instance as a ‘worst case’ reactivity scenario.
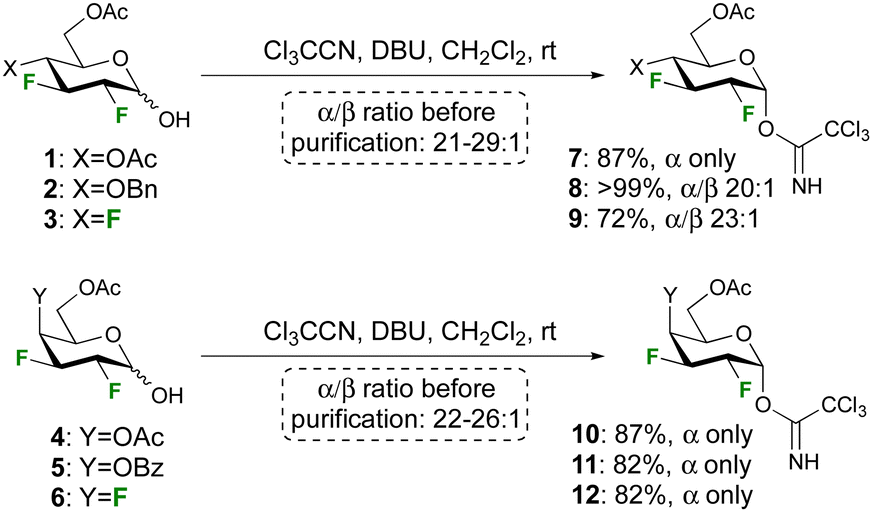 | ||
| Scheme 1 Syntheses of the trichloroacetimidate donors. | ||
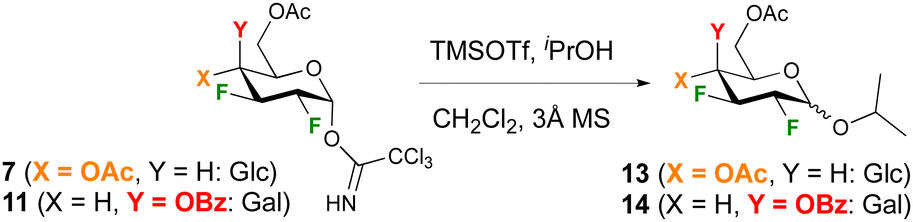
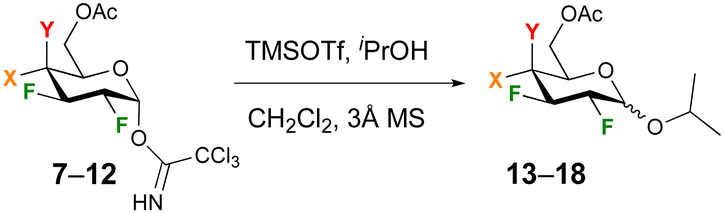
To enable comparison with Gilmour's seminal work on 2-F glucose and -galactose donors,29 glycosidation with 2-propanol under TMSOTf catalysis was investigated first. In our hands, increasing the equivalents of TMSOTf from 0.1 to 0.2 and using rigorously dry conditions including distilled dichloromethane solvent over CaH2 was required to achieve reproducible reactions. In order to ensure quantitative integration of anomeric ratios by 19F{1H} NMR analysis of the crude reaction mixture, a delay time of 3 s was used. The glucose and galactose donors 7 and 11 were investigated first (Table 1). In general, reactions proceeded in excellent yields with the glucose donor displaying a lower reactivity compared to the galactose donor, for which almost quantitative yields were obtained even at −50 °C. This was a surprising finding, given the corresponding peracetylated 2-deoxy-2-fluoro galactose trichloroacetimidate was reported not to react below 0° (leading to 45% yield of the isopropyl glycoside).16,30 The anomeric selectivity is enhanced in favour of the β-anomer at lower temperature, with high β-selectivities obtained for the galactose donor. Reaction of the glucose donor 7 at room temperature or higher resulted in the α-glycoside as the major anomer in a 1.2/1 ratio.
|
|
||
|---|---|---|
| Temp. | Yielda (α/β ratiob) | |
| 13 (Glc) | 14 (Gal) | |
| a Isolated yield. b Determined by 19F{1H} NMR spectroscopy of the crude reaction mixture. c Not tried. d Calculated from isolated mixture of SM and P. | ||
| −60 °C | c | 32% (1/17) |
| −50 °C | c | 97% (1/8.6) |
| −40 °C | 52%d (1/5.6) | 98% (1/5.4) |
| −30 °C | >99% (1/3.2) | 90% (1/4.0) |
| −20 °C | 93% (1/2.5) | >99% (1/2.2) |
| 0 °C | >99% (1/1.3) | |
| Rt | >99% (1.2/1) | >99% (1/1.1) |
| 40 °C | >99% (1.2/1) | |
The glycosidation was then investigated with the different donor substrates (Table 2), all at temperatures of −30 °C. For the glucose donors, the change of an acetate for a benzyl group (8) led to a slight decrease in yield and anomer ratio, which was unexpected based on Gilmour's report of pronounced β-selectivity when using benzyl protecting groups.29 With 2,3,4-trifluorination (9), a low 34% yield of 16 was obtained, in moderate ratio. For the galactose donors, the presence of a smaller acetate (10) instead of benzoate (11) slightly increased the anomeric product ratio, while reaction of the 2,3,4- trifluorinated galactose donor 12 yielded the glycoside 18 in quantitative yield with a much higher 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 β-selectivity.
:9 β-selectivity.
Next, glycosylation using sugar-based acceptors was investigated. Such glycosylations are more challenging, given the reduced reactivities of sugar alcohol groups. The six donors 7–12 were reacted with galactoside diacetonide (Table 3). To enable comparison with the data in Table 2, reactions were also carried out at −30 °C. For both the glucose and galactose donors, excellent yields were obtained, which were superior (in the case of glucose-based donors 7–9) or similar (galactose-based 10–12) compared to the isopropanol glycosidation yields. In contrast, in all cases significant erosion of the anomeric selectivities was observed compared to the results in Table 2.
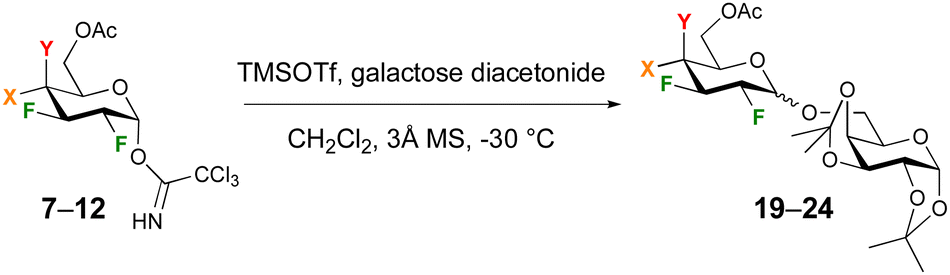
The glycosylation scope of 11 and 12 was further investigated (Fig. 2). Glycosylation at the 4-position of a rhamnoside acceptor led to disaccharides 25 and 26 in excellent yield. In contrast, glycosylation at the 6-position of a 2-deoxy-2-azidoglucoside acceptor was low-yielding (27, 28), although raising the temperature to 0 °C was sufficient to obtain 28 in 89% yield. Glycosylation with a difluorinated acceptor at −30 °C to give the tetra- and pentafluorinated disaccharides 29 and 30 proceeded only in good yield with donor 10, but increasing the temperature to 0 °C significantly enhanced the yield. The anomeric selectivities were much reduced, with glycosylations to give 27 and 29 even displaying α-selectivity.
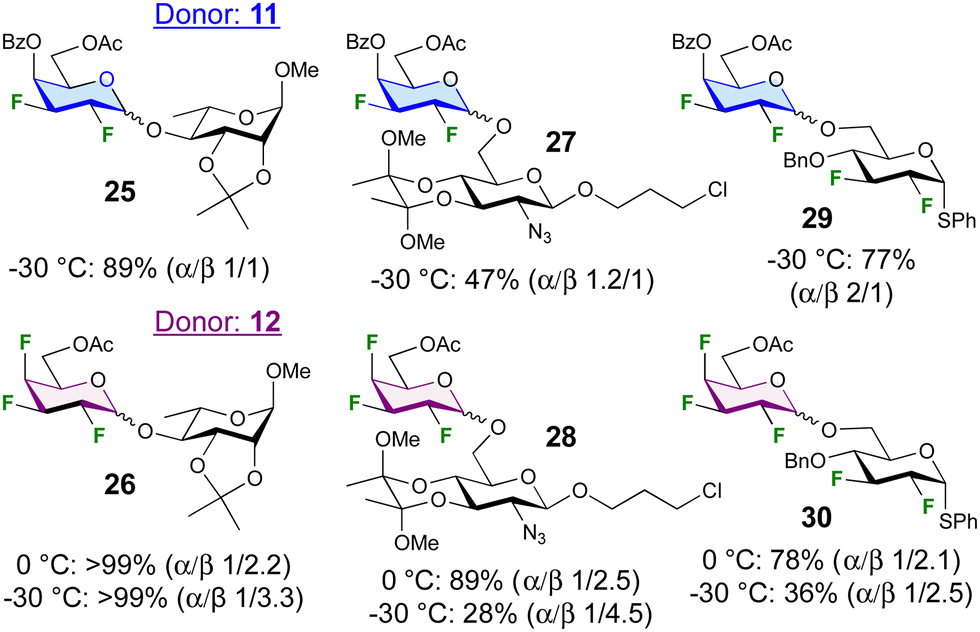 | ||
| Fig. 2 Glycosylation results with other acceptors. | ||
A control experiment was conducted to investigate the possibility of anomerisation under the reaction conditions (Scheme 2). A mixture of trichloroacetimidate 7 (1.0 equiv.) and isopropyl galactopyranoside 17 (1.0 equiv., α/β 1:5.2) were subjected to glycosylation conditions, with 1-propanol (2.4 equiv.) as acceptor at 0 °C. 19F{1H} NMR analysis of the crude reaction mixture indicated no change in the anomeric ratio of 17, or any evidence for the formation of cross-glycosylation products such as 13 or 32. Hence, the reaction is not reversible under the employed reaction conditions (Scheme 2). Incidentally, the higher anomeric ratio of the glycosidation product 31 compared to that of the isopropyl glycoside (1:1.3 α/β, Table 1) is expected given the lower steric hindrance of propanol.30
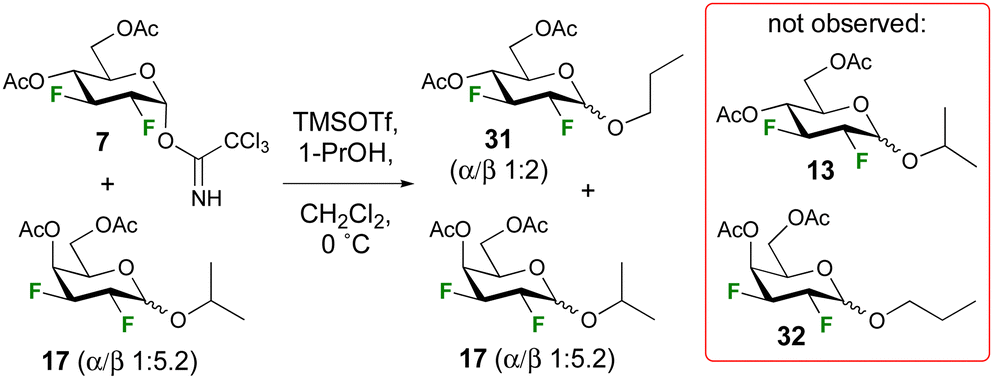 | ||
| Scheme 2 Control experiment regarding in-situ anomerisation. | ||
A tentative model for the observed anomeric selectivities of the peracetylated donors, limited to the 4H3 and 3H4 halfchair conformations, is shown in Fig. 3. The reactivity of the acetylated polyfluorinated donors is clearly similar compared to the peracetylated 2-deoxy-2-fluoroGlc/Gal trichloroacetimida-tes investigated by Gilmour,29,30 While this was not expected, fluorine and acetate groups do have similar ‘field inductive effect parameters’.31 Hence, a similar “SN1-like”29 transition state (TS) is assumed as well as the preponderance of the Anh-Eisenstein 1,2-induction model placing F2 in axial position.29 This leads to the 3H4 halfchair as lower-energy transition state, in accordance with the generally observed β-selectivity. When glucose-configured, substituents at the 3- and 4-position are axial, which has been reported to individually lead to stabilisation of the TS,32 but as part of a glucose framework also undergo destabilising steric interactions,32,33 leading to lower levels of β-selectivities. This could be the reason why 13, with a small fluorine at C3, is formed with higher selectivity compared to 32, which has a larger acetate group at C3. There may also be unfavourable 1,3-diaxial interactions between the substituents at the 2- and 4-positions, which will be larger with a 4F substituent, consistent with a lower β-selectivity for 16. For the galactoses, the substituent at the 4-position is equatorial, which is also expected to destabilise the positive charge at the reacting centre. This effect may lead to a shift in reaction mechanism away from SN1, which would favour β-selectivity. Glycosylation with the less reactive sugar acceptors is expected to give increased SN1 character.34 Larger nucleophiles also experience repulsion from the axial substituent at C3,35 which reduces β-selectivities, as observed (Table 3 and Fig. 2). These data show that this reduction in β-selectivity is much more pronounced with a 4-OBz substituent compared to 4F, indicating that the effect of a 4-OBz group further increases the TS energy difference in favour of the 4H3 halfchair, for which it is tempting to invoke remote ester participation.36 However, there are other effects at play, including involvement of covalent glycosyl triflates and hyperconjugative stabilisations, as well as the presence of other oxonium ion conformations.33,37,38
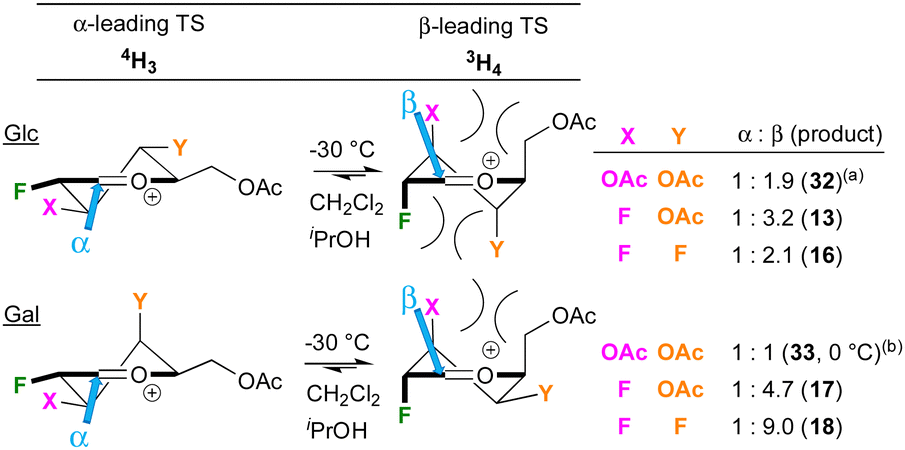 | ||
| Fig. 3 Simplified model to describe the anomeric selectivities. (a) 32 = isopropyl 3,4,6-tri-O-acetyl-2-deoxy-2-fluoroglucoside, See ref. 29. (b) 33 = isopropyl 3,4,6-tri-O-acetyl-2-deoxy-2-fluorogalactoside, See ref. 30. | ||
In summary, a series of novel 2,3-difluorinated and 2,3,4-trifluorinated glucose and galactose trichloroacetimidates are excellent donors for both glycosidation and glycosylation under typical Lewis-acid catalysed conditions, despite their high fluorination content. Excellent anomeric selectivities were obtained for glycosidation reactions with isopropanol, but glycosylation reactions using less reactive alcohol acceptors generally proceeded with low selectivities. As expected, galactose configured donors are more reactive. The glycosylation allows the synthesis of polyfluorinated disaccharides with multiple fluorination in each ring. Further optimisations to increase anomeric selectivities are ongoing. This convenient glycosylation of polyfluorinated glycosyl donors opens up new avenues in glycan-based glycobiology and materials research.
K. H. and B. L. thank Southampton University for funding. B. L. acknowledges the Research Foundation Flanders (FWO, Belgium) for an Odysseus Type I grant (G0F5621N).
Conflicts of interest
There are no conflicts to declare.Notes and references
- A. Varki, Glycobiology, 2017, 27, 3–49 CrossRef CAS PubMed.
- L. Su, Y. Feng, K. Wei, X. Xu, R. Liu and G. Chen, Chem. Rev., 2021, 121, 10950–11029 CrossRef CAS PubMed.
- C. P. J. Glaudemans, Chem. Rev., 1991, 91, 25–33 CrossRef CAS.
- I. P. Street, C. R. Armstrong and S. G. Withers, Biochemistry, 1986, 25, 6021–6027 CrossRef CAS PubMed.
- T. Diercks, A. S. Infantino, L. Unione, J. Jimenez-Barbero, S. Oscarson and H. J. Gabius, Chem. – Eur. J., 2018, 24, 15761–15765 CrossRef CAS PubMed.
- E. Campbell, C. Jordan and R. Gilmour, Chem. Soc. Rev., 2023, 52, 3599–3626 RSC.
- B. Linclau, A. Arda, N. C. Reichardt, M. Sollogoub, L. Unione, S. P. Vincent and J. Jimenez-Barbero, Chem. Soc. Rev., 2020, 49, 3863–3888 RSC.
- R. Hevey, Pharmaceuticals, 2019, 12, 55 CrossRef CAS PubMed.
- R. Hevey, Chem. – Eur. J., 2021, 27, 2240–2253 CrossRef CAS PubMed.
- G. Fittolani, S. Djalali, M. A. Chaube, T. Tyrikos-Ergas, M. C. S. Dal Colle, A. Grafmüller, P. H. Seeberger and M. Delbianco, Org. Biomol. Chem., 2022, 20, 8228–8235 RSC.
- K. Anggara, Y. Zhu, G. Fittolani, Y. Yu, T. Tyrikos-Ergas, M. Delbianco, S. Rauschenbach, S. Abb, P. H. Seeberger and K. Kern, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2102168118 CrossRef CAS PubMed.
- S.-J. Richards, T. Keenan, J.-B. Vendeville, D. E. Wheatley, H. Chidwick, D. Budhadev, C. E. Council, C. S. Webster, H. Ledru, A. N. Baker, M. Walker, M. C. Galan, B. Linclau, M. A. Fascione and M. I. Gibson, Chem. Sci., 2021, 12, 905–910 RSC.
- J. Orwenyo, W. Huang and L.-X. Wang, Biorg. Med. Chem., 2013, 21, 4768–4777 CrossRef CAS PubMed.
- C. E. Council, K. J. Kilpin, J. S. Gusthart, S. A. Allman, B. Linclau and S. S. Lee, Org. Biomol. Chem., 2020, 18, 3423–3451 RSC.
- A. S. Infantino, D. Bengtsson and S. Oscarson, Carbohydr. Res., 2022, 512, 108515 CrossRef CAS PubMed.
- A. H. Viuff, J. C. Hansen, A. B. Christiansen and H. H. Jensen, Synth. Commun., 2013, 43, 1557–1562 CrossRef CAS.
- K. Huonnic and B. Linclau, Chem. Rev., 2022, 122, 15503–15602 CrossRef CAS PubMed.
- T. Oberbillig, H. Löwe and A. Hoffmann-Röder, J. Flow Chem., 2012, 2, 83–86 CrossRef CAS.
- A. Baumann, S. Marchner, M. Daum and A. Hoffmann-Röder, Eur. J. Org. Chem., 2018, 3803–3815 CrossRef CAS.
- V. Hamala, L. Cervenkova Stastna, M. Kurfirt, P. Curinova, M. Dracinsky and J. Karban, Org. Biomol. Chem., 2020, 18, 5427–5434 RSC.
- J. C. Biffinger, H. W. Kim and S. G. DiMagno, ChemBioChem, 2004, 5, 622–627 CrossRef CAS PubMed.
- R. S. Timofte and B. Linclau, Org. Lett., 2008, 10, 3673–3676 CrossRef CAS PubMed.
- S. Golten, C. Q. Fontenelle, R. S. Timofte, L. Bailac, M. Light, M. Sebban, H. Oulyadi and B. Linclau, J. Org. Chem., 2016, 81, 4434–4453 CrossRef CAS PubMed.
- V. Denavit, D. Laine, C. Bouzriba, E. Shanina, E. Gillon, S. Fortin, C. Rademacher, A. Imberty and D. Giguère, Chem. – Eur. J., 2019, 25, 4478–4490 CrossRef CAS PubMed.
- V. Denavit, D. Lainé, J. St-Gelais, P. A. Johnson and D. Giguère, Nat. Commun., 2018, 9, 4721 CrossRef PubMed.
- T. Tremblay, J. St-Gelais, M. Houde and D. Giguère, J. Org. Chem., 2021, 86, 4812–4824 CrossRef CAS PubMed.
- V. Denavit, J. St-Gelais, T. Tremblay and D. Giguère, Chem. – Eur. J., 2019, 25, 9272–9279 CrossRef CAS PubMed.
- R. R. Schmidt, Angew. Chem., Int. Ed. Engl., 1986, 25, 212–235 CrossRef.
- C. Bucher and R. Gilmour, Angew. Chem., Int. Ed., 2010, 49, 8724–8728 CrossRef CAS PubMed.
- E. Durantie, C. Bucher and R. Gilmour, Chem. – Eur. J., 2012, 18, 8208–8215 CrossRef CAS PubMed.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- D. M. Smith and K. A. Woerpel, Org. Biomol. Chem., 2006, 4, 1195–1201 RSC.
- P. O. Adero, H. Amarasekara, P. Wen, L. Bohé and D. Crich, Chem. Rev., 2018, 118, 8242–8284 CrossRef CAS PubMed.
- S. van der Vorm, T. Hansen, H. S. Overkleeft, G. A. van der Marel and J. D. C. Codée, Chem. Sci., 2017, 8, 1867–1875 RSC.
- L. Ayala, C. G. Lucero, J. A. C. Romero, S. A. Tabacco and K. A. Woerpel, J. Am. Chem. Soc., 2003, 125, 15521–15528 CrossRef CAS PubMed.
- J. Kalikanda and Z. Li, J. Org. Chem., 2011, 76, 5207–5218 CrossRef CAS PubMed.
- T. Hansen, L. Lebedel, W. A. Remmerswaal, S. van der Vorm, D. P. A. Wander, M. Somers, H. S. Overkleeft, D. V. Filippov, J. Désiré, A. Mingot, Y. Bleriot, G. A. van der Marel, S. Thibaudeau and J. D. C. Codée, ACS Cent. Sci., 2019, 5, 781–788 CrossRef CAS PubMed.
- A. A. Hettikankanamalage, R. Lassfolk, F. S. Ekholm, R. Leino and D. Crich, Chem. Rev., 2020, 120, 7104–7151 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc02518g |
| ‡ Using our optimised conditions, glycosidation with acetylated 2-fluorinated glucose trichloroacetimidate proceeded only at −20 °C or higher. See ESI.† |
| This journal is © The Royal Society of Chemistry 2023 |