
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mono-cyclononatetraenyl lanthanide complexes†
Luca
Münzfeld
 ,
Adrian
Hauser
,
Michael T.
Gamer
and
Peter W.
Roesky
*
,
Adrian
Hauser
,
Michael T.
Gamer
and
Peter W.
Roesky
*
Institute of Inorganic Chemistry, Karlsruhe Institute of Technology (KIT), Engesserstraße 15, Karlsruhe 76131, Germany. E-mail: roesky@kit.edu
First published on 6th July 2023
Abstract
The synthesis of the first half-sandwich complexes based on the cyclononatetraenyl (Cnt = C9H9−) ligand ([LnIII(η9-Cnt)(η3-BH4)2(thf)] (Ln = La, Ce)) is reported. The title compounds were obtained from the reaction of [Ln(BH4)3(thf)3] and [K(Cnt)]. Further solvation of [LnIII(η9-Cnt)(η3-BH4)2(thf)] with tetrahydrofuran (THF) resulted in a reversible decoordination of the Cnt ring and the formation of the ionic species [LnIII(η3-BH4)2(thf)5][Cnt]. Removal of THF from [LaIII(η9-Cnt)(η3-BH4)2(thf)] gave the polymeric compound [LaIII(μ-η2:η2-BH4)2(η3-BH4)(η9-Cnt)]n.
The ground-breaking discovery of ferrocene paved the way towards modern organometallic chemistry.1 The arising family of metallocene and sandwich compounds, i.e. complexes where a metal ion or atom is complexed by two cyclic and aromatic ligand moieties, was quickly expanded from d- to f-elements.2 Although not literally being sandwich compounds, trivalent tris-cyclopentadienyl complexes of the type [LnIII(η5-C5H5)3] reported by Wilkinson and Birmingham3 are generally considered to be the starting point of lanthanidocene as well as organolanthanide chemistry.4 While tris-cyclopentadienyl complexes are rarely used for further derivatization, the rapid development of organolanthanide complexes started when bis(cyclopentadienyl)lanthanide halides became available. Already in 1963 Dubeck et al. reported on the synthesis of a variety of thermally stable bis(cyclopentadienyl)lanthanide chlorides.5 A significant advance was the stabilization of highly reactive organolanthanide species with the pentamethylcyclopentadienyl ligand in the early 1980s.4a,6 The resulting compounds, which show high solubility in nonpolar solvents, crystallize well, and are stable towards ligand redistribution due to the steric properties of the pentamethylcyclopentadienyl ligand.4a In the form of bis(pentamethylcyclopentadienyl) lanthanide σ-alkyl and hydride complexes such as [(η5-C5Me5)2LnIIICH(SiMe3)2] and [(η5-C5Me5)2LnIII(μ-H)]2, this compound class plays a key role in the catalytic σ-bond metathesis.7
Similarly, the pentamethylcyclopentadienyl ligand and related systems facilitated the selective synthesis of mono-(cyclopentadienyl) lanthanide complexes (half-sandwich complexes). A number of mono-(cyclopentadienyl) lanthanide bisborohydride compounds were prepared,8 in which different substitution patterns on the five membered ring have been achieved.9 The synthesis of the analogue species with an unsubstituted cyclopentadienyl ligand is very difficult because a rearrangement to the thermodynamic more stable bis(cyclopentadienyl) compound is often observed.10
Recently, the “bigger brother” of cyclopentadienyl, which is the cyclononatetraenyl anion (Cnt = C9H9−), a flat and monoanionic 10π-electron aromatic ligand,11 was introduced in coordination chemistry. Inspired by work of Sitzmann et al. on barium compounds,12 Nakajima et al.13 and Nocton et al.14 introduced the Cnt ligand in the coordination sphere of divalent lanthanides forming homoleptic sandwich compounds [LnII(η9-Cnt)2] (Ln = Eu, Sm, Tm, Yb). The groups of Nocton and us disclosed the heteroleptic trivalent sandwich compounds [(Cnt)LnIII(η8-COT)] (COT = cyclooctatetraendiid, C8H82−)15 We showed that the Er complex [(Cnt)ErIII(η8-COT)] showed interesting single molecular magnet behaviour.15a Solvation of [(Cnt)LnIII(η8-COT)] (Ln = La, Ce, Nd, Tb, Er) with THF resulted in neutral [(η4-Cnt)LnIII(thf)2(η8-COT)] (Ln = La, Ce) and ionic [LnIII(thf)x(η8-COT)][Cnt] (x = 4 (Ce, Nd, Tb), 3 (Er)) species in a solid-to-solid transformation. The resulting compounds act as switchable luminophores and single-molecule magnets.16 Nocton and co-workers also reported on the tris(cyclononatetraenyl) rare earth complexes [LnIII(Cnt)3] (Ln = Y, Gd, Tb, Dy, Ho, Er, Tm).17 These compounds were obtained by salt metathesis form LnI3 and [K(Cnt)].11 In contrast to [(η5-C5H5)LnIII], not all carbon atoms are bound to the lanthanide metal. Instead, the Cnt ligand is significantly bent in order to adapt to the size of the metal ion. For the smaller metals the ligand does not switch away but swings over the metal ion, maximizing electrostatic interactions.17
For the intended synthesis of the target compounds the trisborohydrides of the rare earth elements [LnIII(BH4)3(thf)3]18 were employed as suitable precursors. The BH4− ligand exhibits many advantages over other anionic ligands, especially over chloride because it is much more electron-donating and better soluble in common organic solvents.19 Although BH4− is isosteric with Cl−, the electronic properties allow the isolation of otherwise unsaturated and inaccessible species. Moreover, halide derivatives have a significant larger tendency to form ate complexes or bridged derivatives, which are less soluble. The BH4− ligand can also be identified spectroscopically and thus is a suitable probe to monitor reactions by 1H and/or 11B NMR as well as by IR spectroscopy.8 [LnIII(BH4)3(thf)3] is conveniently accessible from LnCl3 and NaBH4.18,20
Reaction of [LnIII(BH4)3(thf)3] with [K(Cnt)] in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio at 60 °C in toluene led to the anticipated half-sandwich complexes [LnIII(η9-Cnt)(η3-BH4)2(thf)] (Ln = La (1), Ce (2)).21 Crystals of these compounds were isolated from toluene at −10 °C in good yields of 68% and 72%, respectively, after filtration (Scheme 1). Interestingly, in the analogous reaction starting from [SmIII(BH4)3(thf)3], only the earlier reported divalent sandwich complex [SmII(η9-Cnt)2],14 was isolated (Scheme 1). A similar reduction was observed by reacting SmIIII3 with three equivalents of [K(Cnt)].17 Attempts to synthesize corresponding compounds of the smaller lanthanides ErIII and DyIII were unsuccessful. Here, no reaction was observed at moderate temperatures, probably due to the smaller ionic radius. Only from about 150 °C in xylene a clear color change and the formation of a colorless precipitate was observable. However, it was not possible to isolate characterizable products from the reaction mixtures.
:1 ratio at 60 °C in toluene led to the anticipated half-sandwich complexes [LnIII(η9-Cnt)(η3-BH4)2(thf)] (Ln = La (1), Ce (2)).21 Crystals of these compounds were isolated from toluene at −10 °C in good yields of 68% and 72%, respectively, after filtration (Scheme 1). Interestingly, in the analogous reaction starting from [SmIII(BH4)3(thf)3], only the earlier reported divalent sandwich complex [SmII(η9-Cnt)2],14 was isolated (Scheme 1). A similar reduction was observed by reacting SmIIII3 with three equivalents of [K(Cnt)].17 Attempts to synthesize corresponding compounds of the smaller lanthanides ErIII and DyIII were unsuccessful. Here, no reaction was observed at moderate temperatures, probably due to the smaller ionic radius. Only from about 150 °C in xylene a clear color change and the formation of a colorless precipitate was observable. However, it was not possible to isolate characterizable products from the reaction mixtures.
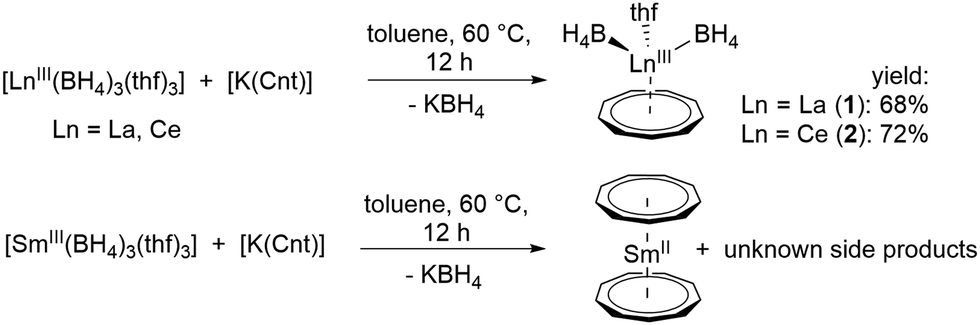 | ||
| Scheme 1 Synthesis of 1 and 2 (top), and reaction of [K(Cnt)] with [SmIII(BH4)3(thf)3] (bottom). | ||
Both compounds 1 and 2 crystallize in the monoclinic space groups P21/n (1) and P21/c (2), respectively and show the expected piano-chair-like coordination motif consisting of an η9-coordinated Cnt ligand, two terminal η3-coordinated BH4 anions and a coordinated THF molecule (Fig. 1). Thus, compounds 1 and 2 represent the first half-sandwich complexes of the Cnt ligand at all. At first glance, this structural motif resembles the silylated cyclooctatetraendiid complexes [LnIII(η8-COTTIPS)BH4] (Ln = La, Ce, Sm, Er; COTTIPS = 1,4-bis-triisopropylsilyl-cyclooctatetraendiid)).22 Only the number of borohydride and THF ligands is inverted for charge balance.
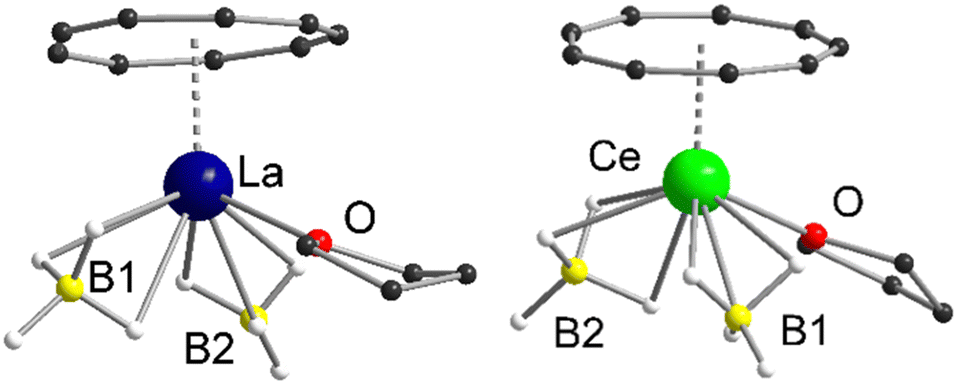 | ||
| Fig. 1 Left: Molecular structure of [LaIII(η9-Cnt)(η3-BH4)2(thf)] (1) in the solid-state. Right: Molecular structure of [CeIII(η9-Cnt)(η3-BH4)2(thf)] (2) in the solid-state. Carbon bound hydrogen atoms are omitted for clarity. For detailed bond lengths and angles see Fig. S15 and S16 (ESI†). | ||
Within 1 and 2 the Ln–Ct (Ct = ring centroid) distances account for 2.1149(2) Å (1) and 2.0739(4) Å (2), with Ln–C distances from 2.906(4)-2.985(3) Å (1) and 2.869(7)-2.929(7) Å (2), respectively. There is a tiny shift of the Cnt-ring from the ideal η9-coordination in 2, that however is still within the error range. The positions of all hydride atom could be localized and freely refined, confirming beyond doubt the coordination mode of the borohydride ligands in the solid state (Fig. 1). Moreover, Raman spectra of the two compounds show bands at 2231 cm−1 and 2453 cm−1 for 1 and 2234 cm−1 and 2452 cm−1 for 2, which are characteristic for this binding mode.23 As described earlier Raman spectroscopy is a valuable tool for the characterization of Cnt complexes.15a The characteristic ring vibrations of the Cnt ring (νasym(η9-Cnt) = 1520 cm−1 (1), 1522 cm−1 (2); νsym(η9-Cnt) = 678 cm−1 (1), 679 cm−1 (2)) and Ln–Cnt vibration (ν(Ln-Cnt) = 145 cm−1 (1), 140 cm−1 (2)), can be clearly assigned. The remaining, less intense bands between 800 cm−1 and 1500 cm−1 were attributed to the THF ligand, respectively.24
As expected, the 1H NMR spectra of 1 and 2 in solution show a singlet that can be assigned to the nine isochronous aromatic protons of the η9-Cnt ligand. In the case of the diamagnetic species 1, the signal is localized at δ = 7.01 ppm in THF-d8. In the case of the paramagnetic compound 2, the corresponding signal is shifted as expected and is located at δ = 5.41 ppm. At δ = 1.26–0.61 ppm for 1 in THF-d8 and δ = 39.4 ppm for 2, the broad resonances of the hydridic BH4 protons were detected. The corresponding signals in the 11B NMR spectra were observed at δ = 19.8 ppm for 1 and δ = 35.2 ppm for 2. However, it should be noted here that in some cases the resonances in the proton spectra of the diamagnetic compound 1 did not show the expected integral ratio. In these cases, the integrals over the borohydride resonances were too large. A contamination of the samples with the precursor [LnIII(BH4)3(THF)3] is unlikely since the precursor signals have different chemical shifts. Therefore, we suggest that solubility equilibria may lead to a changed chemical composition of the species in solution, because the deviations changes with concentration.
Therefore, we investigated this issue in more detail by performing solvation experiments of compounds 1 and 2 in THF. Both 1 and 2 are readily soluble in warm THF, and single crystals can be isolated after separation of insoluble residues by filtration and slow cooling to room temperature (Scheme 2) (note compounds 1 and 2 were crystallized from toluene). Single crystal X-ray structural analysis revealed the formation of ionic compounds [LnIII(η3-BH4)2(thf)5][Cnt] (Ln = La (3), Ce (4)) featuring structural motifs with fully displaced Cnt ligands in both cases (Fig. 2).21 A similar displacement of the Cnt ligand by THF was observed for the sandwich complexes [(Cnt)LnIII(η8-COT)] (Ln = Ce, Nd, Tb, Er) with THF.16 Among others the ionic species [LnIII(thf)x(η8-COT)][Cnt] (x = 4 (Ce, Nd, Tb), 3 (Er)) with a non-coordinated Cnt anion were isolated. This clearly shows that the Cnt anion is significantly weaker bound to the lanthanide ions than the smaller cyclopentadienyl ligand.
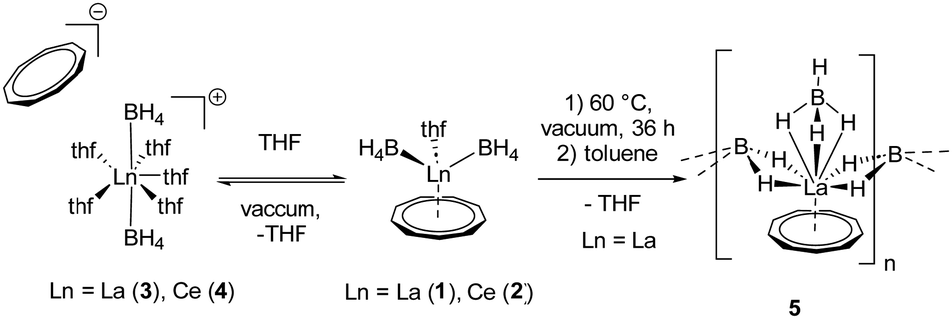 | ||
| Scheme 2 Possible structural transformations of compounds 1 and 2 by varying crystallization conditions. Left: Ionic complexes 3 and 4 after THF solvation and crystallization. Middle: Neutral parent compounds 1 and 2 after crystallization from toluene. Right: Polymeric and solvent-free species 5 after drying in high vacuum and crystallization from toluene. | ||
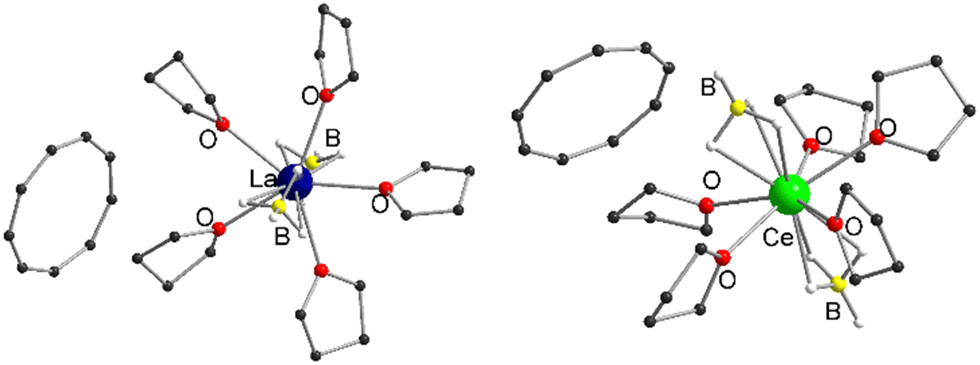 | ||
| Fig. 2 Left: Molecular structure of [LaIII(η3-BH4)2(thf)5][Cnt] (3) in the solid-state. Left: Molecular structure of [CeIII(η3-BH4)2(thf)5][Cnt] (4) in the solid-state. B For detailed bond lengths and angles see Fig. S17 and S18 (ESI†). | ||
Compounds 3 and 4 crystallize with one cation, two half-occupied anions and two half-occupied non-coordinating THF molecules in the asymmetric unit. Furthermore, one of the THF ligands of 4 is statistically disordered. Since crystals of the two compounds were reproducibly twinned, structural refinement was performed after twin integration with X-area.25 The cationic units in both cases are pentagonal-bipyramidal coordinated [LnIII(η3-BH4)2(thf)5]+ fragments, with the axial positions occupied by two BH4− anions and the equatorial positions by five neutral THF ligands each. A similar structural motif is already known in the form of [NdIII(η3-BH4)2(thf)5][BPh4], which was prepared by protolysis of a borohydride ligand in [NdIII(BH4)3(thf)3] with [NH(Et)3][BPh4] in THF.26 Since the binding metric of the central cationic fragment of 3 and 4 are almost identical to that [NdIII(η3-BH4)2(thf)5][BPh4], a detailed discussion will not be given here (see Fig. S17 and S18 for selected bond lengths and angles, ESI†).
Since the related full displacement of the Cnt ring with THF in the ionic compounds [LnIII(thf)x(η8-COT)][CNT] (x = 4 (Ce, Nd, Tb), 3 (Er)) is fully reversible,16 we were interested to study the back reaction of 3 and 4 to compounds 1 and 2. After drying the crystals of the THF adducts 3 and 4 in high vacuum, Raman spectra of the isolated solids were obtained analogous to those of the unsolvated compounds 1 and 2. In particular, the Ln-Cnt vibrational bands are clearly observable (see above). As seen for [(η9-CNT)LnIII(η8-COT)] (Ln = Ce, Nd, Tb, Er),16 this effect offers some potential with respect to switchable materials.
Besides coordinating more THF molecules to the metal atom in compounds 1 and 2, it is also possible to remove the THF ligand from the coordination sphere in compound 1 by drying this substance in high vacuum at 60 °C for several days. As result a polymeric species [LaIII(μ-η2:η2-BH4)2(η3-BH4)(η9-Cnt)]n (5) (Scheme 2) is formed.21 It should be noted, however, that much of the originally used compound 1 seems to be decomposed in this process, or the polymeric species is only very poorly soluble in toluene and benzene. Thus, only a few single crystals of 5 could be isolated after extraction of the dried compound 1 with hot toluene and evaporation of the solvent at room temperature. In case of compound 2 no crystalline product could be obtained.
In the 1H NMR and 13C{1H} NMR spectra of compound 5, the expected peak for the CNT ligand is seen at δ = 7.44 ppm (1H) and δ = 113.4 ppm (13C{1H}). The integral of the BH4 groups in the 1H NMR spectrum (δ = 0.96–0.38 ppm) is higher than expected (vide supra). Compound 5 crystallizes with three units of the polymeric chain and two molecules of toluene in the asymmetric unit. With respect to the monomeric units, a piano-chair-like structural motif with an η9-coordinated Cnt ligand is formed similar to the parent compound 1, with the THF ligand of 1 replaced by a bridging μ-η2:η2-BH4 ligand of one of the neighboring molecules (Fig. 3). The second borohydride ligand remains in the original terminal η3-coordination mode. The assignment of the binding modes is based on freely refined BH4 hydrogen atoms. The polymeric structure as a whole can be described as a coordination polymer of the type [LaIII(μ-η2:η2-BH4)2(η3-BH4)(η9-Cnt)]n with trigonal prism as chain links. The average La-CtCnt and La-B(η3-BH4) distances are in good agreement with those of the monomeric compound 1, averaging to 2.03 Å and 2.74 Å, respectively. The La-B(η2-BH4) distances of the bridging borohydride ligands average 2.97 Å, slightly longer than those of the terminal η3-BH4 ligands, as expected. In general, μ-η2:η2-bridging BH4 groups are very rare and to the best of our knowledge not reported for lanthanum. However, bridging BH4 groups with different coordination modes have been previously observed for lanthanum,27e.g., in [LaIII(Htp)2(μ-BH4)]2 (Htp = 2,5-di-tert-butylphospholyl)28 and [LaIII(Cpttt)(BH4)2(thf)2] (Cpttt = C5H2tBu3-1,2,4).29
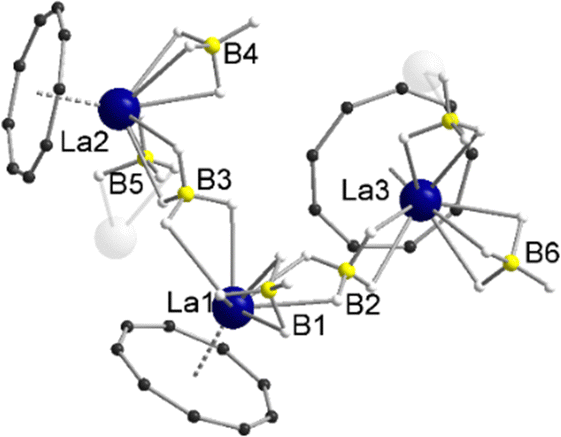 | ||
| Fig. 3 Molecular structure of the asymmetric unit of [LaIII(μ-η2:η2-BH4)2(η3-BH4)(η9-Cnt)]n (5) in the solid-state. Carbon bound hydrogen atoms and toluene molecules are omitted for clarity. For detailed bond lengths and angles see Fig. S19 (ESI†). | ||
In summary, we report the synthesis of the first Cnt half sandwich complexes. The use of the borohydrides [LnIII(BH4)3(thf)3] of the early lanthanides La and Ce allowed the access to the desired complexes [LnIII(η9-Cnt)(η3-BH4)2(thf)] (Ln = La (1), Ce (2)) in good yields. Both compounds form piano stool coordination polyhedra in the solid state. Depending on amount of coordinated THF molecules a rich diversity of compounds could be obtained, ranging from ionic pentagonal-bipyramidal species (3 and 4) with a non-coordinated Cnt ligand over monomeric half-sandwich complexes (1 and 2) to a trigonal-prismatic coordination polymer (5).
AH and PR gratefully acknowledge financial support from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) through the Collaborative Research Centre “4f for Future” (CRC 1573, project number 471424360) project C1.
Conflicts of interest
There are no conflicts to declare.References
- (a) T. J. Kealy and P. L. Pauson, Nature, 1951, 168, 1039 CrossRef CAS; (b) G. Wilkinson, M. Rosenblum, M. C. Whiting and R. B. Woodward, J. Am. Chem. Soc., 1952, 74, 2125 CrossRef CAS; (c) E. O. Fischer and W. Pfab, Z. Naturforsch., B: Chem. Sci., 1952, 7, 377 CrossRef; (d) P. Štěpnička, Ferrocenes: Ligands, Materials and Biomolecules, John Wiley & Sons, Ltd, Chichester, 2008 CrossRef; (e) P. Štěpnička, Dalton Trans., 2022, 51, 8085 RSC.
- C. Elschenbroich, Organometallics, Wiley-VCH, Weinheim, 3 edn, 2006 Search PubMed.
- (a) G. Wilkinson and J. M. Birmingham, J. Am. Chem. Soc., 1954, 76, 6210 CrossRef CAS; (b) J. M. Birmingham and G. Wilkinson, J. Am. Chem. Soc., 1956, 78, 42 CrossRef CAS.
- (a) H. Schumann, J. A. Meese-Marktscheffel and L. Esser, Chem. Rev., 1995, 95, 865 CrossRef CAS; (b) H. Schumann, Angew. Chem., Int. Ed. Engl., 1984, 23, 474 CrossRef.
- R. E. Maginn, S. Manastyrskyj and M. Dubeck, J. Am. Chem. Soc., 1963, 85, 672 CrossRef CAS.
- (a) P. L. Watson, J. F. Whitney and R. L. Harlow, Inorg. Chem., 1981, 20, 3271 CrossRef CAS; (b) L. Gong, A. Streitwieser and A. Zalkin, J. Chem. Soc., Chem. Commun., 1987, 460 RSC; (c) S. Schäfer, S. Kaufmann, E. S. Rösch and P. W. Roesky, Chem. Soc. Rev., 2023, 52, 4006 RSC.
- (a) F. T. Edelmann, Organolanthoid Chemistry: Synthesis, Structure, Catalysis, Springer Berlin Heidelberg, Berlin, Heidelberg, 1996, p. 247 Search PubMed; (b) P. L. Watson and G. W. Parshall, Acc. Chem. Res., 1985, 18, 51 CrossRef CAS; (c) W. J. Evans, I. Bloom, W. E. Hunter and J. L. Atwood, J. Am. Chem. Soc., 1983, 105, 1401 CrossRef CAS; (d) G. Jeske, H. Lauke, H. Mauermann, H. Schumann and T. J. Marks, J. Am. Chem. Soc., 1985, 107, 8111 CrossRef CAS; (e) G. A. Molander and J. O. Hoberg, J. Org. Chem., 1992, 57, 3266 CrossRef CAS; (f) M. A. Giardello, V. P. Conticello, L. Brard, M. R. Gagne and T. J. Marks, J. Am. Chem. Soc., 1994, 116, 10241 CrossRef CAS; (g) P. L. Watson, J. Am. Chem. Soc., 1982, 104, 337 CrossRef CAS; (h) P. L. Watson and D. C. Roe, J. Am. Chem. Soc., 1982, 104, 6471 CrossRef CAS; (i) M. R. Gagne and T. J. Marks, J. Am. Chem. Soc., 1989, 111, 4108 CrossRef CAS; (j) M. R. Gagne, C. L. Stern and T. J. Marks, J. Am. Chem. Soc., 1992, 114, 275 CrossRef CAS; (k) M. R. Gagne, S. P. Nolan and T. J. Marks, Organometallics, 1990, 9, 1716 CrossRef CAS; (l) G. A. Molander and M. Julius, J. Org. Chem., 1992, 57, 6347 CrossRef CAS; (m) P.-F. Fu and T. J. Marks, J. Am. Chem. Soc., 1995, 117, 10747 CrossRef CAS; (n) K. N. Harrison and T. J. Marks, J. Am. Chem. Soc., 1992, 114, 9220 CrossRef CAS; (o) E. A. Bijpost, R. Duchateau and J. H. Teuben, J. Mol. Catal. A: Chem., 1995, 95, 121 CrossRef CAS; (p) G. A. Molander and J. O. Hoberg, J. Am. Chem. Soc., 1992, 114, 3123 CrossRef CAS.
- S. M. Guillaume, L. Maron and P. W. Roesky, in Handbook on the Physics and Chemistry of Rare Earths, eds. J.-C. G. Bünzli and V. K. Pecharsky, Elsevier, 2014, vol. 44, pp. 1 Search PubMed.
- (a) D. Barbier-Baudry, F. Bouyer, A. S. Madureira Bruno and M. Visseaux, Appl. Organomet. Chem., 2006, 20, 24 CrossRef CAS; (b) F. Bonnet, M. Visseaux, D. Barbier-Baudry, A. Hafid, E. Vigier and M. M. Kubicki, Inorg. Chem., 2004, 43, 3682 CrossRef CAS PubMed.
- S. Arndt and J. Okuda, Chem. Rev., 2002, 102, 1953 CrossRef CAS PubMed.
- T. J. Katz and P. J. Garratt, J. Am. Chem. Soc., 1964, 86, 5194 CrossRef CAS.
- M. D. Walter, G. Wolmershäuser and H. Sitzmann, J. Am. Chem. Soc., 2005, 127, 17494 CrossRef CAS PubMed.
- K. Kawasaki, R. Sugiyama, T. Tsuji, T. Iwasa, H. Tsunoyama, Y. Mizuhata, N. Tokitoh and A. Nakajima, Chem. Commun., 2017, 53, 6557 RSC.
- M. Xémard, S. Zimmer, M. Cordier, V. Goudy, L. Ricard, C. Clavaguéra and G. Nocton, J. Am. Chem. Soc., 2018, 140, 14433 CrossRef PubMed.
- (a) L. Münzfeld, C. Schoo, S. Bestgen, E. Moreno-Pineda, R. Köppe, M. Ruben and P. W. Roesky, Nat. Commun., 2019, 10, 3135 CrossRef PubMed; (b) M. Tricoire, L. Münzfeld, J. Moutet, N. Mahieu, L. La Droitte, E. Moreno-Pineda, F. Gendron, J. D. Hilgar, J. D. Rinehart, M. Ruben, O. Cador, B. Le Guennic, P. W. Roesky and G. Nocton, Chem. – Eur. J., 2021, 13558 CrossRef CAS PubMed.
- L. Münzfeld, M. Dahlen, A. Hauser, N. Mahieu, S. K. Kuppusamy, J. Moutet, M. Tricoire, R. Köppe, L. La Droitte, O. Cador, B. Le Guennic, G. Nocton, E. Moreno-Pineda, M. Ruben and P. W. Roesky, Angew. Chem., Int. Ed., 2023, 62, e202218107 CrossRef PubMed.
- O. Stetsiuk, L. La Droitte, V. Goudy, B. Le Guennic, O. Cador and G. Nocton, Organometallics, 2022, 41, 133 CrossRef CAS.
- S. M. Cendrowski-Guillaume, M. Nierlich, M. Lance and M. Ephritikhine, Organometallics, 1998, 17, 786 CrossRef CAS.
- Z. Xu and Z. Lin, Coord. Chem. Rev., 1996, 156, 139 CrossRef CAS.
- (a) U. Mirsaidov, G. N. Boiko, A. Kurbonbekov and A. Rakhimova, Dokl. Akad. Nauk Tadzh. SSR, 1986, 29, 608 CAS; (b) U. Mirsaidov and A. Kurbonbekov, Dokl. Akad. Nauk Tadzh. SSR, 1985, 28, 219 CAS; (c) U. Mirsaidov, I. B. Shaimuradov and M. Khikmatov, Zh. Neorg. Khim., 1986, 31, 1321 CAS; (d) S. M. Cendrowski-Guillaume, G. Le Gland, M. Nierlich and M. Ephritikhine, Organometallics, 2000, 19, 5654 CrossRef CAS.
- L. Münzfeld, Dissertation, Karlsruher Institut für Technologie, 2022.
- L. Münzfeld, X. Sun, S. Schlittenhardt, C. Schoo, A. Hauser, S. Gillhuber, F. Weigend, M. Ruben and P. W. Roesky, Chem. Sci., 2022, 13, 945 RSC.
- M. Ephritikhine, Chem. Rev., 1997, 97, 2193 CrossRef CAS PubMed.
- B. Cadioli, E. Gallinella, C. Coulombeau, H. Jobic and G. Berthier, J. Phys. Chem., 1993, 97, 7844 CrossRef CAS.
- X-AREA: Program for the Acquisition and Analysis of Data, Stoe & Cie GmbH, Darmstadt, German, 2018 Search PubMed.
- (a) D. Robert, M. Kondracka and J. Okuda, Dalton Trans., 2008, 2667 RSC; (b) T. Arliguie, L. Belkhiri, S.-E. Bouaoud, P. Thuéry, C. Villiers, A. Boucekkine and M. Ephritikhine, Inorg. Chem., 2009, 48, 221 CrossRef CAS PubMed.
- F. Ortu, Chem. Rev., 2022, 122, 6040 CrossRef CAS PubMed.
- J. Liu, L. E. Nodaraki, P. J. Cobb, M. J. Giansiracusa, F. Ortu, F. Tuna and D. P. Mills, Dalton Trans., 2020, 49, 6504 RSC.
- F. Ortu, D. Packer, J. Liu, M. Burton, A. Formanuik and D. P. Mills, J. Organomet. Chem., 2018, 857, 45 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization, NMR and Raman spectra, single X-ray diffraction data. CCDC 2267628–2267632. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc02717a |
| This journal is © The Royal Society of Chemistry 2023 |