
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Na2Fe2Se2O: a double anti-perovskite with prevalence of anionic redox activity in Na-ion batteries†
Mikhail V.
Gorbunov
 *a,
Thomas
Doert
b and
Daria
Mikhailova
a
*a,
Thomas
Doert
b and
Daria
Mikhailova
a
aInstitute for Materials Chemistry, IFW Dresden, Helmholtzstraße 20, 01069, Dresden, Germany. E-mail: m.gorbunov@ifw-dresden.de
bFaculty of Chemistry and Food Chemistry, TUD Dresden University of Technology, Helmholtzstr. 10, 01069, Dresden, Germany
First published on 25th October 2023
Abstract
Na2Fe2Se2O with the I4/mmm space group was studied in sodium-ion batteries, delivering a reversible specific capacity of more than 140 mA h g−1. Operando XRD and XAS studies disclosed bifunctional redox behaviour with the prevalence of anionic electrochemical activity and a likely partial decomposition of the material, which, however, does not influence the electrochemical behaviour of the system.
Although lithium-ion batteries are nowadays dominating the market of energy solutions for portative electronics and electric vehicles, the development of alternative systems is imperative, as the amount of lithium in the Earth's crust is insufficient to fulfil the rapidly growing demand for electrochemical energy sources.1 Some of the promising post-lithium systems overcoming the limited raw material availability are Na-ion batteries.2 However, many of them suffer from the sluggish kinetics of sodium diffusion in the solid state; thus, it is necessary to search and develop novel materials with higher efficiency in terms of enhanced specific electrochemical characteristics and longer cycle life.1
One of the novel material classes, which was recently tested, is double anti-perovskites (also classified as anti-Ruddlesden–Popper type structures3–5). Unlike single cubic anti-perovskites, they are not suffering from the disadvantage of alkali metals and electrochemically active elements sharing the same crystallographic position,6 which is known to impede the (de)intercalation kinetics and to cause relatively rapid structural degradation.7 One example of a double anti-perovskite structure is Na2Fe2S2O, which was recently tested in sodium-ion batteries. It was reported to exhibit promising electrochemical characteristics. In particular, it was shown to deliver a specific capacity of about 150 mA h g−1 in half-cells vs. sodium with an average discharge voltage around 2.0 V, and about 125 mA h g−1 in full-cells with a pre-cycled hard carbon at a rate of 0.1C (1 Na+ per Na2Fe2OS2 formula in 10 h).5 However, the impressive capacity values decay relatively fast with each subsequent cycle.
Nevertheless, the discussed results stimulated us to test the Se-based analogue Na2Fe2Se2O, known for its 2D magnetic properties,8 electrochemically. In the current work, we present our systemized studies.
The crystal structure of Na2Fe2Se2O with two cationic and two anionic sites is shown in Fig. 1a, and the space group is I4/mmm. Both iron and sodium in Na2Fe2Se2O are coordinated in distorted octahedra: iron with four selenium atoms forming the square and two oxygen atoms forming the apices, and sodium with five selenium atoms and one oxygen along the four-fold axis. Sodium and iron layers are stacked alternatingly along the c-axis. Oxygen and selenium layers are also stacked alternatingly, making the structure promising for reversible sodium extraction. Although Na2Fe2Se2O has a higher formula weight compared to Na2Fe2S2O (331.59 vs. 237.81 g mol−1), one can expect similar capacity values for both materials due to higher structural stability of the desodiated Se-containing phase, similarly to Li2FeS1−xSexO.11 A powder X-ray diffractogram of the synthesized material is shown in Fig. 1b, as well as the respective Rietveld fit. The results of the Rietveld refinement are available in Table S1 (ESI†). As one can see, besides the main phase, the sample also contains small amounts of FeSe12 and γ-Fe2O3 (maghemite)13 as admixtures. The volumetric fraction of the target compound was estimated to be more than 90%, which is acceptable for further studies. The chemical composition determined by ICP-OES corresponds to Na1.96(3)Fe1.97(3)Se1.98(3)O1.14(9) (see Table S1, ESI†), which is in good agreement with the target stoichiometry.
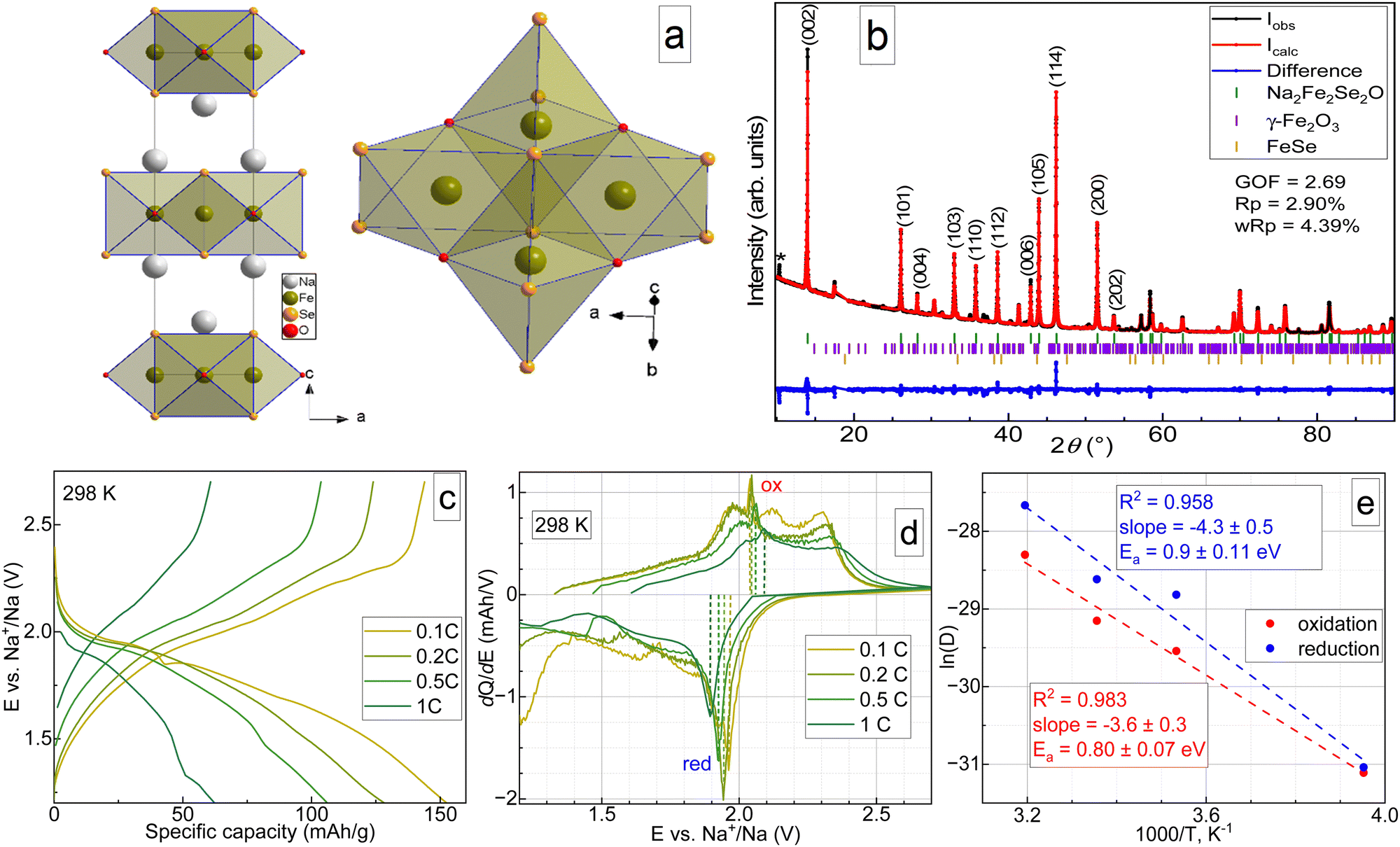 | ||
| Fig. 1 Preliminary physicochemical characterization of Na2Fe2Se2O and its electrochemical tests. (a) Polyhedral view of Na2Fe2Se2O crystal structure. (b) Powder X-ray diffractogram of the synthesized material. The wavelength was 1.78896 Å, the refinement by the Rietveld method9 was done in Jana2006.10 * stands either for an unidentified phase or a detector artefact. (c) Typical galvanostatic charge–discharge curves for Na2Fe2Se2O half-cells at 298 K at various current densities. (d) Corresponding differential capacity plots. (e) Arrhenius plots for diffusion coefficients at 0.2C calculated at four different temperatures using Randles–Sevcik routine from differential capacity plots and evaluated activation energies for sodium migration in Na2Fe2Se2O during charge (red) and discharge (blue). | ||
Galvanostatic cycling with potential limitations was done at four temperatures to evaluate the electrochemical kinetics. Typical charge–discharge curves of Na2Fe2Se2O-based half-cells at 298 K are presented in Fig. 1, together with differential capacity plots (panel d). The data for elevated and lowered temperatures and the results of rate capability tests are available in the supplementary file (Fig. S2, ESI†). At a low current density of 0.1C, Na2Fe2Se2O delivers a specific capacity of up to 150 mA h g−1 with an average discharge voltage of about 1.8 V, but the rate capability is moderate, as it delivers only 60 mA h g−1 at 1C. As visible from the differential capacity plots, electrochemical removal and insertion of sodium both represent a multi-stage process, which seems to change with the temperature, current, and cycle number. At 298 K, the charge process of Na2Fe2Se2O can be characterized by four stages between 1.8 and 2.4 V in the first cycle.
With subsequent cycles, the number of oxidative peaks in the differential curve reduces to three, indicating a possible alteration of the charge compensation mechanism. Upon discharge, two stages are observed. However, the peak at slightly lower than 2.0 V is ascribed to the main process of Na+ intercalation. Even at room temperature, the coulombic efficiency during the formation cycle deviates significantly from 100% (Fig. S2k, ESI†), reflecting a higher discharge capacity, which may be related to either parasitic reactions in the electrolyte or stepwise amorphization of the material at deep states of (dis)charge. Temperature elevation leads to an even higher deviation of the coulombic efficiency from 100% and faster merging of the peaks on dQ/dE curves, which allows assuming a faster degradation of the material and requires slight changes in the potential cut-off for the cycling. At 283 K, Na2Fe2Se2O showed the best rate capability (Fig. S2l, ESI†), and its capacity returned to almost 100% of the initial capacity after 25 cycles at various current densities, which may be probably due to inhibited decomposition. At 283 K, the charge process looks the same as at room temperature for lower current densities; however, oxidation turns into a single-step process at higher currents of 0.5 and 1C (Fig. S2b and e, ESI†). The discharge part of the curve does not change its shape, but the lower voltage peak becomes more resolved. Further reduction of the temperature to 253 K leads to drastic decay of the electrochemical performance (Fig. S2c and f, ESI†), which can be attributed to a significant decrease of the electrolyte conductivity. Differential capacity plots were used to evaluate the chemical diffusion coefficients for sodium ions using the Randles–Ševčík equation,14 and the activation barrier for their migration in the crystal structure of Na2Fe2Se2O. The latter was done using an Arrhenius plot14 and the obtained values of Ea are 0.9 ± 0.11 and 0.80 ± 0.07 eV for sodium extraction and insertion, respectively. It should be noted that only one oxidation peak and one reduction peak were considered for these calculations, as they showed acceptable data reliability: the natural logarithm of the diffusion coefficient value vs. the inverse temperature 1000/T showed linear behaviour with R2 ≥ 0.95. The changes in the shapes in the upper part of differential capacity plots may be evidence for the partial decomposition of Na2Fe2Se2O during cycling. This phenomenon required further advanced physicochemical studies.
Structural and redox behaviours of Na2Fe2Se2O upon reversible sodium extraction were evaluated by conducting operando XRD and XAS experiments using synchrotron radiation facilities (beamlines P02.1 and P64 of PETRA III, DESY, Hamburg, Germany15,16). The results are shown in Fig. 2.
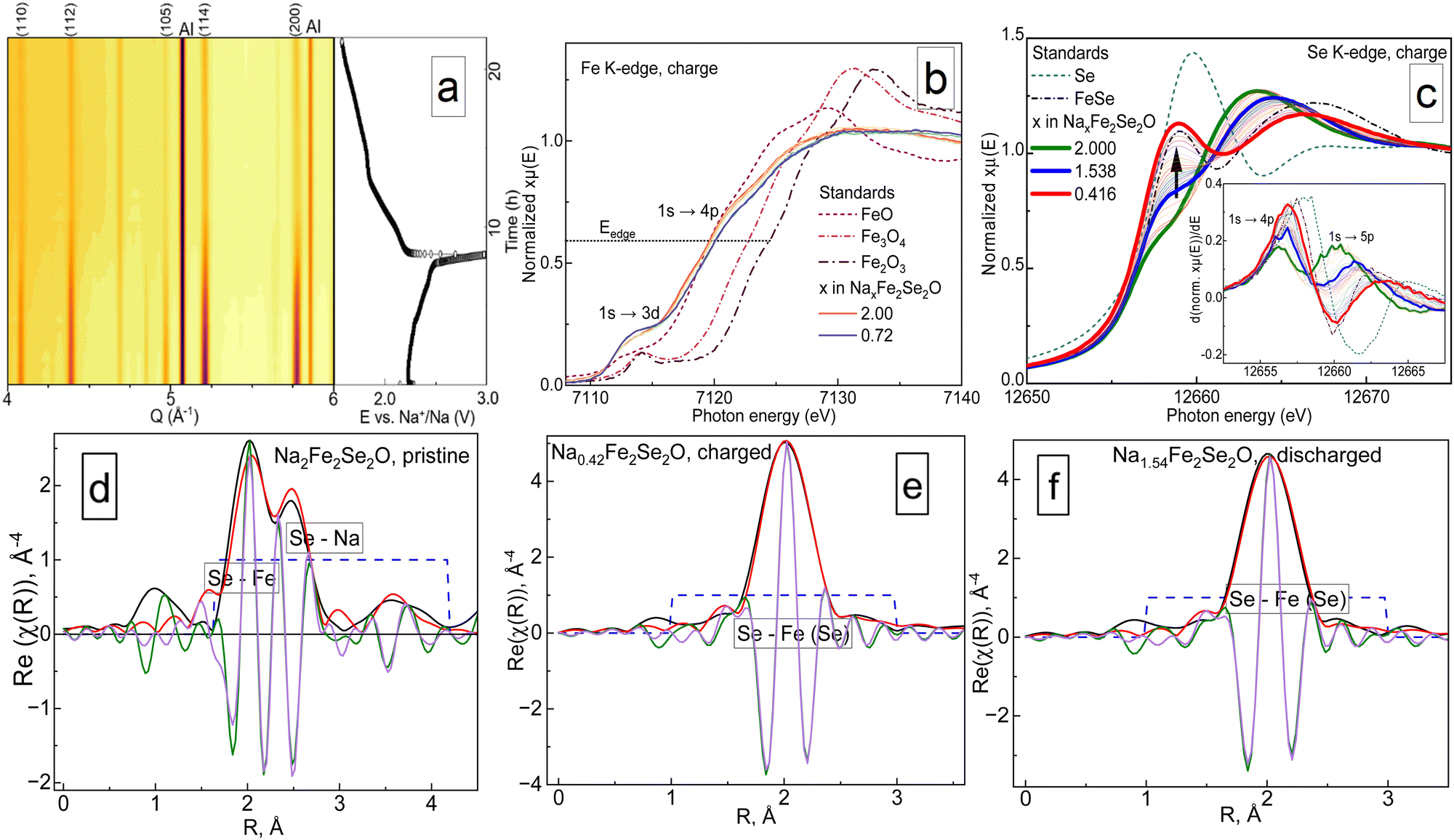 | ||
| Fig. 2 The results of operando for Na2Fe2Se2O. (a) Contour plots of X-ray diffractograms together with the corresponding charge–discharge curve. (b) and (c) X-ray absorption spectra in Fe and Se K-edge regions during the cell charge process, (d)–(f) XAFS calculations for the pristine, charged and discharged states. XAS data processing was done using the Demeter software package.17 It should be noted that in panels (b) and (c), only the points corresponding to pristine, fully charged and discharged material are highlighted and shown in the legend. | ||
As visible from panel a, the material suffers from a serious degree of amorphization, and the crystallinity is not regained after discharge. However, the structural symmetry of the crystalline phase remains tetragonal, even when a significant amount of sodium is removed. Similar results were reported earlier for Na2Fe2S2O, which showed even higher crystallinity loss.5 The cell volume of Na1.2Fe2Se2O reduces from 248.5 Å3 in the initial state to 247.3 Å3, equivalent to a relative volume change of about 0.48% (Fig. S3d, ESI†). The contour plot clearly shows that there is no broadening of reflections, but only decreasing intensity. The decrease of the Bragg reflection intensities (Fig. S3e, ESI†) without reflection broadening may come from the partial decomposition of the material, which was suggested from the electrochemical analysis. Most likely, the remaining X-ray intensities belong to the electrochemically inactive part of Na2Fe2Se2O inside the grains, surrounded by a layer of decomposition products. A small value of 0.48% relative to the volume change can be rationalised by a narrow range of x in Na2−xFe2Se2O under which the material remains stable. The structure collapses when a significant amount of sodium is removed.
In panels (b)–(f) of Fig. 2, the results of X-ray absorption spectroscopy are visualised. A 3d–4p orbital hybridization allows the dipole-forbidden 1s → 3d feature to be observed near the iron K-edge, as for the cubic anti-perovskites.7,11,18–20 In contrast to the latter, iron is almost electrochemically inactive in the title compound since the energy position of the K-edge only shifts a little with cycling, whereas the K-edge of selenium changes its energy position and shape significantly when sodium is removed. Our previous studies confirmed that anionic electrochemical activity plays an important role for cubic anti-perovskites in batteries: it causes amorphization and may be the main factor for the structural collapse in this family of materials. Current results are the first experimental evidence for the prevalence of Se2−/Se0 redox couples over Fe3+/Fe2+ ones in anti-perovskites during galvanostatic cycling. It is noteworthy though that, unlike for 3d metals, the energy position of the Se K-edge does not depend linearly on the oxidation state.21,22 Nevertheless, a relative shift from the initial point, which was determined from the first energy derivative (see the insets of panels (c) and (d)) of the normalised spectra,22 can be considered as an acceptable measure for the degree of oxidation. As shown in Fig. S3c (ESI†), the position of the Se K-edge changes much stronger during charging than that of the Fe K-edge. Moreover, this change is reversible during discharge. Another interesting point to notice is the behaviour of the peak at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 657 eV corresponding to a 1s → 4p electron transition. Generally, this type of transition is forbidden,21 as the Se2− anion has a completely occupied 4p-orbital. Nevertheless, it is present in the experimental spectra, since the Fe–Se bond should not be classified as ionic, but rather as a covalent polar one, both from the Fe–Se distance in Na2Fe2Se2O and from the difference in their electronegativity values,23 which does not exceed the value of 1.7, an empirical criterion for an ionic bond.24 The peak corresponding to the 1s → 4p transition grows with charge and decays on discharge; however, it does not regain its initial shape, when the same amount of sodium is incorporated into the structure. Note that this feature represents a superposition of contributions from five Se–Na and four Se–Fe bonds, meaning that even at deep states of charge, it would not reach the shape corresponding to pure selenium. This is demonstrated by highlighting the spectra recorded for the Na1.54Fe2Se2O composition on charge and discharge in panel (b) of Fig. 2 and in panel (b) of Fig. S3 (ESI†) by a thick blue line. One can clearly see that the intensity of the 1s → 4p electron transition for the same Na1.54Fe2Se2O composition is higher on the discharge part of the measurement, and its shape is resolved much better. Since this kind of transition is a characteristic for covalently bonded selenium, we can assume that during the charge process of Na2Fe2Se2O, selenium oxidizes to its elemental state, and intercalation of sodium ions into electrochemically formed Se may occur during discharge.
657 eV corresponding to a 1s → 4p electron transition. Generally, this type of transition is forbidden,21 as the Se2− anion has a completely occupied 4p-orbital. Nevertheless, it is present in the experimental spectra, since the Fe–Se bond should not be classified as ionic, but rather as a covalent polar one, both from the Fe–Se distance in Na2Fe2Se2O and from the difference in their electronegativity values,23 which does not exceed the value of 1.7, an empirical criterion for an ionic bond.24 The peak corresponding to the 1s → 4p transition grows with charge and decays on discharge; however, it does not regain its initial shape, when the same amount of sodium is incorporated into the structure. Note that this feature represents a superposition of contributions from five Se–Na and four Se–Fe bonds, meaning that even at deep states of charge, it would not reach the shape corresponding to pure selenium. This is demonstrated by highlighting the spectra recorded for the Na1.54Fe2Se2O composition on charge and discharge in panel (b) of Fig. 2 and in panel (b) of Fig. S3 (ESI†) by a thick blue line. One can clearly see that the intensity of the 1s → 4p electron transition for the same Na1.54Fe2Se2O composition is higher on the discharge part of the measurement, and its shape is resolved much better. Since this kind of transition is a characteristic for covalently bonded selenium, we can assume that during the charge process of Na2Fe2Se2O, selenium oxidizes to its elemental state, and intercalation of sodium ions into electrochemically formed Se may occur during discharge.
The XAFS analysis on the Se K-edge (panels (d)–(f) of Fig. 2, and Fig. S3c, ESI†) shows an irreversible local structural transformation in its second coordination sphere when x in Na2−xFe2Se2O reaches a value below 1. For an appropriate fit, an artificially “short” Se–Fe distance was first introduced, as has been done before for the Fe–S distance in Na2Fe2S2O5. Interestingly, similar results may be achieved by introducing α-Se25 with a Se–Se distance of 2.33 Å into the structural model. The ability of selenium to intercalate sodium ions at an average voltage of 1.5 V is well-known.26 We consider the second option as more reasonable, since the shape of the Se K-edge for Na2−xFe2Se2O at deep states of charge (x ≥ 1) looks like a superposition of those for FeSe and elemental selenium. Therefore, while discussing the electrochemical performance, we must define the material as a composite, consisting of the initial phase and electrochemically formed compounds, most likely elemental selenium and binary selenides of sodium and iron.
In the end, the hypothesis of a composite formation instead of pure Na2Fe2Se2O during charge and discharge appears to be more reasonable, based on the results of three advanced characterisation techniques applied. Our suggestion is that elemental selenium is formed gradually during each subsequent charging cycle, especially when a significant amount of sodium is extracted, and continuously replaces the initial phase in the electrode material. A similar assumption may be proposed for Na2Fe2S2O5; however, for this material, more precise evaluation of the redox activity of sulphur may be required. Activation barriers for sodium diffusion (for the stabilised system after 3 cycles) estimated from Arrhenius plots are comparable to the values calculated theoretically for sodium diffusion in double anti-perovskite superionic conductors (0.12–0.44 eV).27
Improvement of the batteries based on anti-Ruddlesden–Popper phases could perhaps be achieved by tuning the cationic or (and) anionic compositions, as it was earlier done for cubic anti-perovskites.11,18,19 However, the structural instability of these systems upon electrochemical cycling may be a critical limitation. Even though the practical application of Na2Fe2Se2O in batteries can hardly be realized, it may be considered as an interesting model system for tuning various properties of layered materials, like electrochemical activity of different ionic species.
M. V. Gorbunov thanks the IFW excellence program for the financial support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- J.-Y. Hwang, S.-T. Myung and Y.-K. Sun, Chem. Soc. Rev., 2017, 46, 3529 RSC.
- M. D. Slater, D. Kim and E. Lee, et al. , Adv. Funct. Mater., 2013, 23, 947 CrossRef CAS.
- S. N. Ruddlesden and P. Popper, Acta Crystallogr., 1957, 10, 538 CrossRef CAS.
- S. N. Ruddlesden and P. Popper, Acta Crystallogr., 1958, 11, 54 CrossRef CAS.
- J. Gamon, A. J. Perez and L. A. Jones, et al. , J. Mater. Chem. A, 2020, 8, 20553 RSC.
- K.-T. Lai, I. Antonyshin and Y. Prots, et al. , J. Am. Chem. Soc., 2017, 139, 28 CrossRef PubMed.
- D. Mikhailova, L. Giebeler and S. Maletti, et al. , ACS Appl. Energy Mater., 2018, 1, 6593 CrossRef CAS.
- J. B. He, D. M. Wang and H. L. Shi, et al. , Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 205212 CrossRef.
- H. M. Rietveld, J. Appl. Crystallogr., 1969, 2, 65 CrossRef CAS.
- V. Petricek, M. Dusek and L. Palatinus, Z. Kristallogr., 2014, 229, 345 CAS.
- M. A. A. Mohamed, M. V. Gorbunov and M. Valldor, et al. , J. Mater. Chem. A, 2021, 9, 23095 RSC.
- W. Schuster, H. Mikler and K. L. Komarek, Chem. Monthly, 1979, 110, 1153 CrossRef CAS.
- C. Pecharroman, T. Gonzalez-Carreno and J. Iglesias, Phys. Chem. Miner., 1995, 22, 21 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, John Wiley & Sons, 2nd edn, 2001 Search PubMed.
- A.-C. Dippel, H.-P. Liermann and J. T. Walter, et al. , J. Synchrotron Radiat., 2015, 22, 675 CrossRef CAS PubMed.
- W. A. Caliebe, V. Murzin and A. Kalinko, et al. , AIP Conf. Proc., 2019, 2054, 060031 CrossRef.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537 CrossRef CAS PubMed.
- M. V. Gorbunov, S. Carocci and S. Maletti, et al. , Inorg. Chem., 2020, 59, 15626 CrossRef CAS PubMed.
- M. V. Gorbunov, S. Carocci and I. G. Gonzalez Martinez, et al. , Front. Energy Res., 2021, 9, 657962 CrossRef.
- A. L. Ryser, D. G. Strawn and M. A. Marcus, et al. , Environ. Sci. Technol., 2006, 40, 46 CrossRef PubMed.
- J. T. Henthorn, R. J. Arias and S. Koroidov, et al. , J. Am. Chem. Soc., 2019, 141, 34 CrossRef PubMed.
- M. Newhille, Rev. Mineral. Geochem., 2014, 78, 33 CrossRef.
- Handbook of Chemistry and Physics, ed. W. M. Haynes, D. R. Lide, T. J. Bruno, Boca Raton, London, New York, 2015 Search PubMed.
- P. Atkins and L. Jones, Chemistry: Molecules, Matter and Change, W.H. Freeman & Co, New York, 1997 Search PubMed.
- P. Cherin and P. Unger, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1972, 28, 313 CrossRef CAS.
- X. L. Huang, C. Zhou and W. He, et al. , ACS Nano, 2021, 15, 4 Search PubMed.
- Z. Zu, Y. Wang and G. Shao, J. Mater. Chem. A, 2019, 7, 10483 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed description of the experiments and additional data. See DOI: https://doi.org/10.1039/d3cc04722a |
| This journal is © The Royal Society of Chemistry 2023 |