
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBoundary research between organic conductors and transistors: new trends for functional molecular crystals
Tomofumi
Kadoya
 *a and
Toshiki
Higashino
*b
*a and
Toshiki
Higashino
*b
aDepartment of Chemistry, Konan University, 8-9-1 Okamoto, Higashinada, Kobe 658-8501, Japan. E-mail: tkadoya@konan-u.ac.jp
bResearch Institute for Advanced Electronics and Photonics, National Institute of Advanced Industrial Science and Technology (AIST), 1-1-1 Higashi, Tsukuba, Ibaraki 305-8565, Japan. E-mail: t-higashino@aist.go.jp
First published on 2nd June 2023
Abstract
In organic transistors, the contact resistance at the interface between the organic semiconductor (OSC) and electrode reduces the device performance. To solve this problem, a buffer layer is inserted between the electrode and OSC layer to decrease the depletion layer that forms at the OSC surface. Especially in bottom-contact transistors, the effects of interfacial potentials and carrier traps can be suppressed using high-conductivity organic-conductors and carbon pastes as electrodes. Accordingly, a “self-contact” transistor was proposed based on chemically doping OSCs. This technique enables us to fabricate organic conductor electrodes specific to each OSC, resulting in transistors with low contact resistance. However, organic conductors based on OSCs are essential to expand the fabrication of these devices using the doping method. Consequently, OSCs have been used to develop a promising new class of organic conductors with high conductivity and excellent thermoelectric properties. Although developing organic conductors based on OSCs remains relatively unexplored, it is expected to become a new trend in functional molecular compounds.
1. Introduction
Organic semiconductors (OSCs) were ushered in by the discovery of electrical conduction in molecular crystals such as phthalocyanines and violanthrones in around 1950.1,2 The room-temperature conductivity of these materials was approximately 10−10 S cm−1, and such π-extended aromatic molecules were pointed out as intrinsic semiconductors.3 The electrical conductivity of organic materials was greatly enhanced by the advent of charge-transfer (CT) complexes/salts (e.g. perylene–bromine salts showed a conductivity of 0.1–1 S cm−1).4 The partial CT state between the constituent electron donor and acceptor acts as a high-concentration chemical doping. This doping method is quite efficient and reliable for generating conduction carriers in molecular materials, and has led to a number of CT complexes/salts exhibiting metallic and even superconducting states, thus greatly expanding the field of organic conductors.5–7 Needless to say, the progress in the field has been largely driven by the evolution of tetrathiafulvalene (TTF), a remarkably excellent donor.8–12 Note that although these CT complexes/salts are, in many cases, formed as two- or multiple-component systems involving donors, acceptors, or inorganic ions, single-component organic conductors have also been developed through sophisticated molecular design and crystal engineering.13–30Steady progress has been made with conventional OSCs over the years based on their design versatility, as well as their intriguing properties such as low-environmental-impact processability, cost-effective applications, inherent light weight, and flexibility. These OSCs features, in combination with physical methods for generating currents or voltages through photoexcitation, thermal gradients, and electric field effects, have led to the development of next-generation organic electronic devices such as organic photovoltaics (OPVs),31–36 organic thermoelectrics (OTEs),37–40 and organic field-effect transistors (OFETs).41–47 Among them, OFETs provide a platform for investigating the intrinsic carrier transport properties of organic materials, as well as their application in switching devices. Typically, carrier injection into the organic active layer of OFETs produces semiconducting resistivity, but metallic states can also be achieved by tuning the carrier density using high-κ dielectrics such as ionic liquids.48–52 Superconducting states can also be driven by controlling the band filling of Mott insulators.53–55 In this case, field-effect carrier injection causes only the band filling of the active layer (target material) to change without any influence on the chemical composition. Therefore, this is a suitable method for the rapid and precise control of the carrier density near the Fermi level in strongly correlated electron systems.
The performance of OFETs has been limited by the contact resistance (carrier-injection barrier) at the semiconductor–metal interface.56–59 OSCs do not generally form Ohmic junctions, in which the carrier-injection barrier is approximately zero, with metal electrodes, in contrast to inorganic semiconductors. This can be attributed to the degree of morphological disorder of OSC molecules and charge transfer at the metal–organic junctions, which create interfacial carrier traps and potential shifts.43,46,60,61 In other words, the interface between the OSC and the metal electrode tends to be a Schottky junction where charge injection resistance exists. The details of Schottky junctions are complicated, for which various theoretical models have been proposed based on photoelectron spectroscopic (spectroscopy) evaluation.61–64 Therefore, to improve device performance, various methods have been developed to minimize the contact resistance.
In this highlight, we first focus on the fundamental issue of contact resistance at the OSC–electrode interface in organic transistors, and introduce techniques to reduce the contact resistance, such as the insertion of buffer layers and use of CT-complex electrodes. Next, to fabricate CT-complex electrodes, we introduce a new type of OFET, which can be described as a “self-contact transistor”, in which a part of the semiconductor is converted into a conducting material and directly used as an electrode. We then highlight recent studies on non-TTF-type organic conductors derived from high-performance OSCs.
2. Contact resistance at the organic semiconductor/metal electrode
2.1. Laminated buffer layers into semiconductor/electrode interfaces
The energy level between an OSC and an electrode is important when considering contact resistance. The simplest model is the Mott–Schottky model, which takes the difference between the work function EW of the electrode and the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) levels of the OSC.59,65 However, photoelectron spectroscopic experiments under ultrahigh vacuum have reported incorporating the value of the vacuum level shift into this Mott–Schottky model equation.60–64,66 Nevertheless, deciding which model to apply can be challenging depending on the OSC/metal junction combination. In general, numerous carrier traps exist at the interface between the OSCs and metals. Therefore, even in the combination of OSCs and metal electrodes, which was originally expected to form an Ohmic junction, the carrier density becomes low owing to charge trapping, and the region behaves like a depletion layer. Such junctions often become Schottky junctions. At the Schottky interface, charge injection from the electrode to the semiconductor occurs via carrier migration through the depletion layer owing to the tunnelling effect. Therefore, increasing the carrier concentration at the electrode/semiconductor interface reduces the depletion layer thickness and enables effective tunnel injection.As described above, the energy-level difference between the EW of the electrode and the HOMO/LUMO levels of OSCs is a major factor in contact resistance. Thus, the OSC/electrode interface has been tuned by chemical modifications to align the energy levels. For example, some electron-donating/accepting materials are often used as n- or p-type dopants at the OSC/electrode interface. N-(p-) dopants must have a HOMO (LUMO) level shallower (deeper) than (or close to) the LUMO (HOMO) level of the matrix materials. The n-/p-dopants inject electrons/holes into the OSC surface. The resultant high charge density at the electrode/OSC interface improves the contact resistance by reducing of the width of the depletion layer and enhancing the tunnel injection.
As an example of OSC doping, Minari et al. demonstrated that contact p-doping using iron(III) chloride (FeCl3) enables the improvement of the transistor characteristics of dioctylbenzothieno[3,2-b][1]benzothiophene (C8-BTBT) OFETs (Fig. 1).67 Contact doping results in the formation of acceptor levels in the forbidden band and generates holes in the HOMO level edge of C8-BTBT. The induced holes significantly increase the charge density and passivate active traps at the contacts, significantly reducing the contact resistance from 200 to 8.8 kΩ cm. A strong organic acceptor, 2,3,5,6,-tetrafluoro-7,7,8,8-tetracyanoquinodimethane (F4TCNQ), also acts as a good p-type dopant. Abe et al. developed a method of controlling the threshold voltage of vacuum-deposited pentacene through F4TCNQ doping while keeping a high field-effect mobility and on/off ratio.68 Soeda et al. reported that the F4TCNQ doping of solution-crystallised C8-BTBT produces high-mobility OFETs with a low threshold voltage and high on/off ratio in air.69
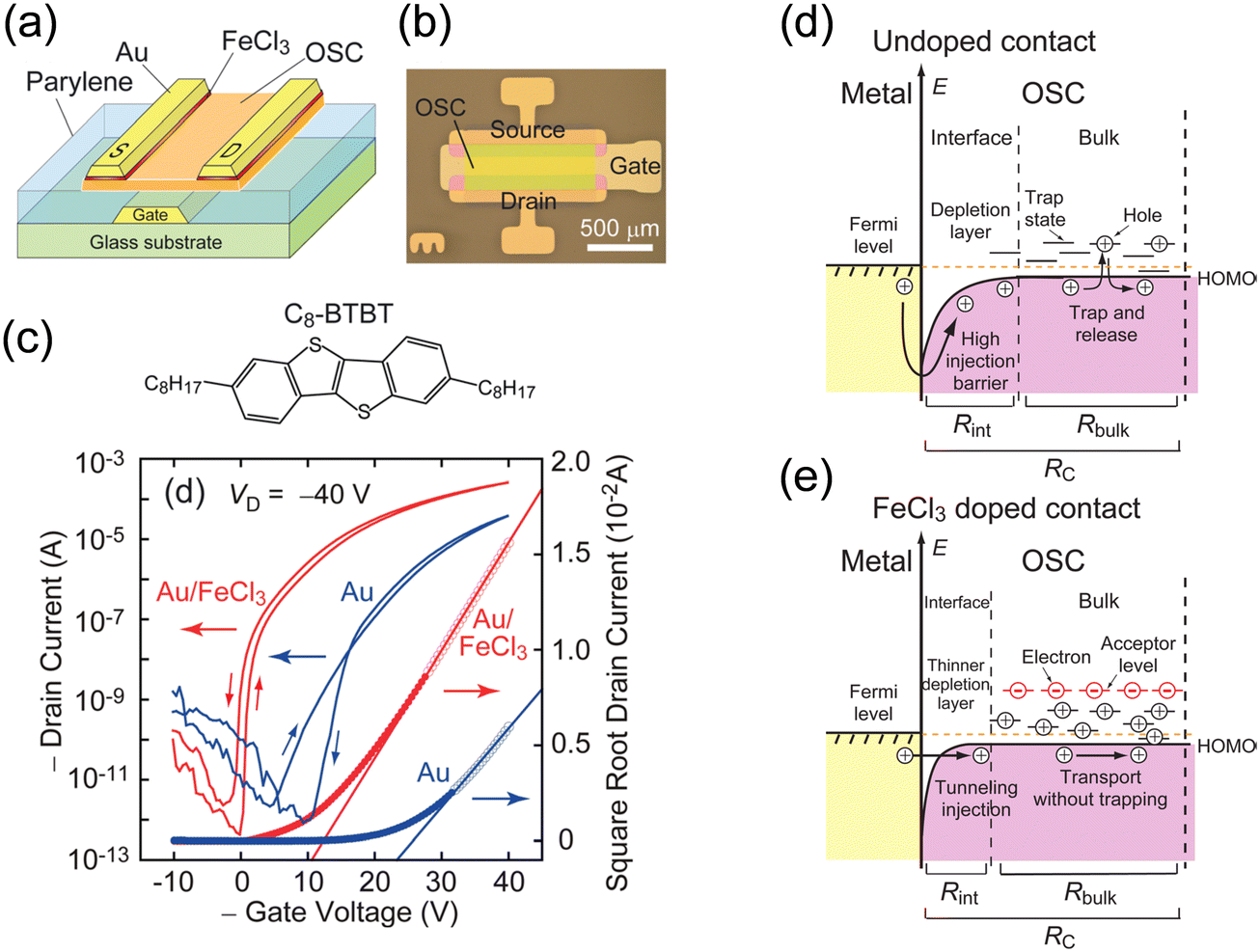 | ||
| Fig. 1 (a) Schematic of OFETs based on C8-BTBT with a chemically doped contact. (b) Optical micrograph of an OFET device. (c) Molecular structure of C8-BTBT and typical transfer characteristics of the devices with doped and undoped contacts. The channel length and width of these devices are 100 and 1000 μm, respectively. (d) Energy diagram of the OSC device contact without acceptor doping. (e) Energy diagram of the OFET contact showing the reduction of Schottky barrier thickness and the occupation of trap states in the access region by charge carriers generated by acceptor doping at the contact. Reprinted with permission from ref. 67. Copyright 2012, AIP Publishing. | ||
There are fewer n-type dopants than the p-type ones. Pyronin B chloride, TTF derivatives, 1,3-dimethyl-2-aryl-2,3-dihydro-1H-benzoimidazole (DMBI) derivatives, pentacene derivatives and organometallics have been reported as n-type dopants capable of electron injection.70–75 In particular, the mechanism of DMBI dopants is complicated; a reaction between the dopant and host, which begins with either hydride or hydrogen atom transfer and ultimately leads to the formation of host radical anions is responsible for the n-doping effect.
Surface modifications of the source-drain electrodes are also effective for energy-level alignment. Self-assembled monolayers (SAMs) based on fluorinated molecules are used for bottom-contact devices with Au and Ag electrodes.76–79 The EW of the modified electrodes shifts more deeply to accurately match the HOMO level of p-channel OSCs, significantly improving the carrier injection and stability of the OTFTs. Not only do SAMs increase the carrier concentration, but they also reduce the disturbance of the thin-film morphology at the electrode–semiconductor interface due to the lower surface energy. In the case of Ag and Cu electrodes, dimethyldicyanoquinonediimine (DMDCNQI) becomes a good candidate for electrode dopants, because it forms CT salts X(DMDCNQI)2 (X = Cu and Ag), which act as a buffer layer.80 The effect of the buffer layer can be interpreted as a reduction in the unfavourable interfacial potential for the charge injection between the semiconductor and the electrode. Note that the EW of the Ag electrode shifts more deeply to approximately 5.3 eV by covering the electrode surface with a few nanometres of MoO3 layer, leading to efficient hole injection into OSCs.81
2.2. Conducting organic materials as a low-contact-resistance electrodes
Contact resistance depends on the device structure of OFETs, as well as the energy level at the OSC/electrode junction. The electronic state at the junction differs between the bottom- and top-contact configurations, even in the same combination of OSCs and metals. This is closely related to the morphology of OSC molecules in the active layer. Generally, the morphology of OSC layers is strongly affected by the physically adsorbed substrate, to afford distinct molecular packing/orientation with those of the bulk crystal, which is known as thin-film phase. The morphological change largely depends on the surface energy of the substrate. Thus, it becomes more drastic when the OSC layer forms on the boundary region between the electrode and dielectric in bottom-contact OFETs, as seen in vacuum-deposited pentacene with the edge-on/face-on orientations. This morphological disorder causes a large contact resistance and significant degradation of the transistor performances (Fig. 2).82,83 Thus, the contact resistance tends to be lower for top-contact devices, where the electrode is created later on top of the OSC layer. Even for bottom-contact devices, this morphological problem can be mitigated by treating the electrode surfaces with SAMs. However, a particularly simple and effective method is to use high-conductivity organic materials instead of metal electrodes. Because the surface energies of organic materials are almost the same with each other, the morphological change of OSCs are considerably suppressed.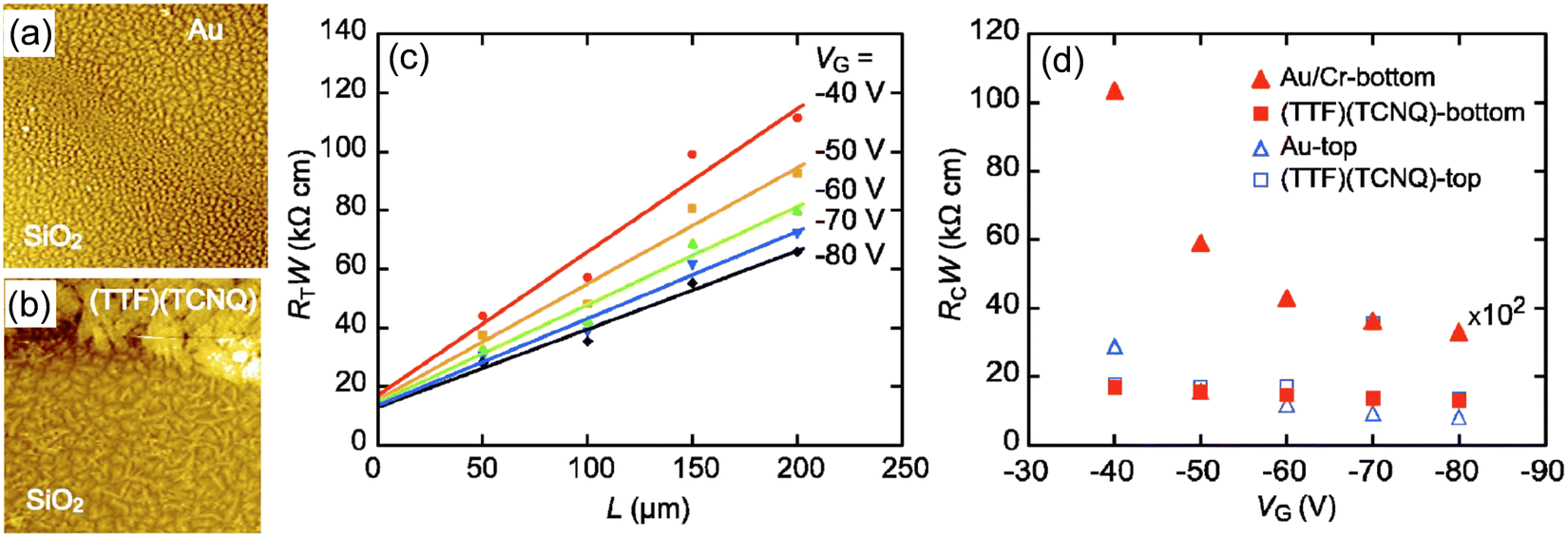 | ||
| Fig. 2 AFM images of pentacene-based bottom-contact transistors, (a) near the channel electrode boundary for an Au-electrode transistor and (b) near the channel-electrode boundary for a (TTF)(TCNQ)-electrode transistor. (c) Analysis of contact resistance for bottom-contact (TTF)(TCNQ) transistors. (d) Gate voltage (VG) dependence of the contact resistance RcW. Contact resistance for bottom-contact Au transistors is multiplied by 1/100. Reprinted with permission from ref. 83. Copyright 2007, AIP Publishing. | ||
For example, (TTF)(TCNQ), a well-studied CT complex,84–87 can be used as an organic electrode, because of the high conductivity with metallic behaviour and the film-forming capability through vacuum deposition. When (TTF)(TCNQ) thin films are used as the electrode, the contact resistance is significantly reduced even with the bottom-contact device. The performance reaches nearly that of a top-contact Au-electrode device.83 Moreover, the Fermi level of the CT complex can be controlled by changing the combination of the donor and the acceptor. Despite restrictions on the number of combinations, electron/hole injection into the OSC layer can be arbitrarily controlled.88 Thus, organic electrodes not only eliminate the various factors that create depletion layers at the electrode-semiconductor interface, but also allow to control of the EW of the electrodes. In addition to CT complexes, CT salts can become organic electrodes by utilizing the nanoparticles formed with ionic liquids or dispersant polymers, instead of the vacuum deposition method.89,90 The nanoparticles are prepared as inks that disperse homogeneously in polar solvents, such as ethanol and water, enabling the use of simple solution processes to form conductive organic films. Alternatively, conductive carbon paste can be adapted to similar low-contact-resistance electrodes using a solution method (Fig. 3(a)).91–93 Carbon paste is also attractive because it is less expensive than metal paste. High-resolution carbon electrodes can be created from carbon paste by laser sintering. This technique provides a short-channel transistor with a channel length of approximately 2.5 μm, without using photolithography. The resulting carbon films are as thin as 60 nm and has high transparency, it is a promising candidate for an electrode material to replace indium tin oxide.
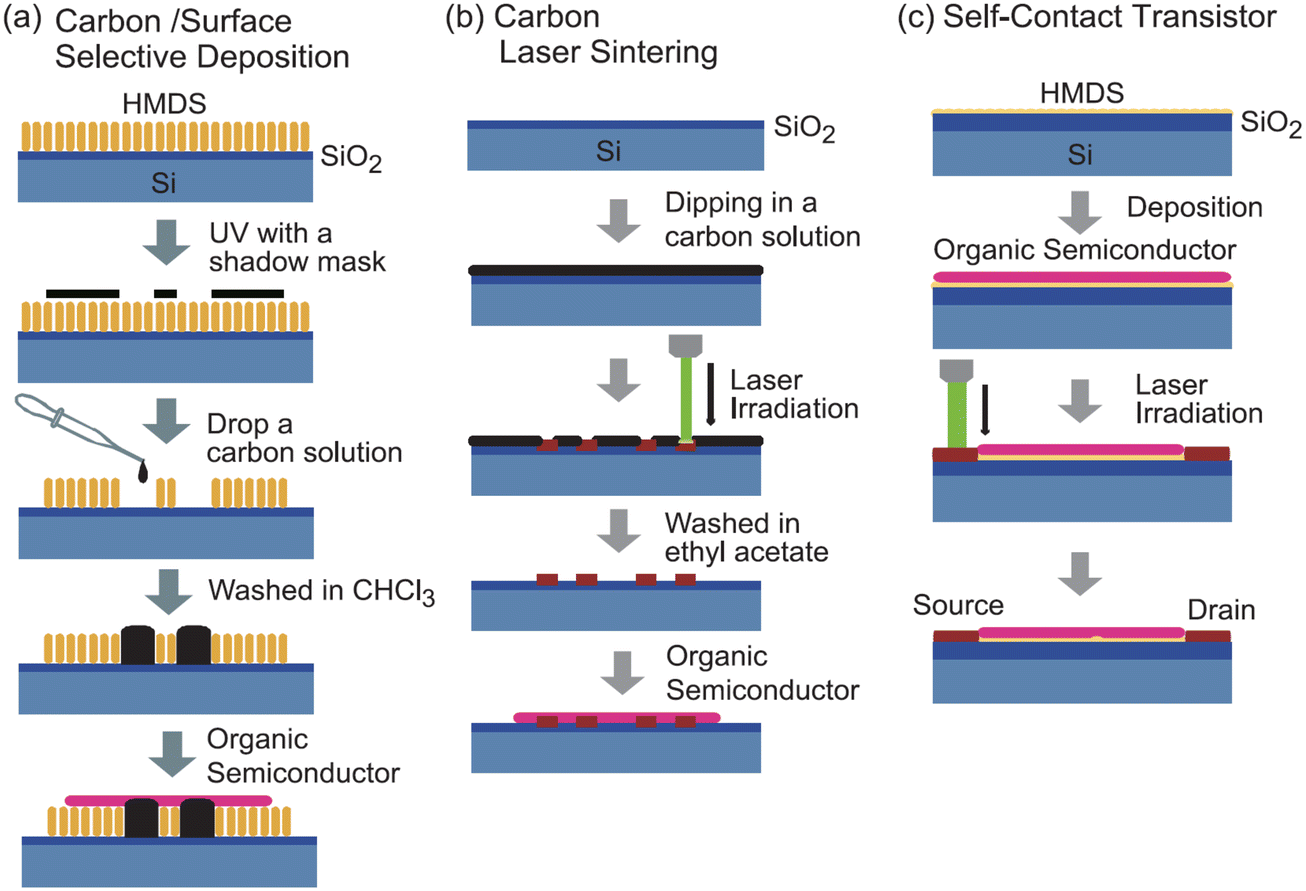 | ||
| Fig. 3 Schematics of (a) solution-processed carbon electrodes, (b) laser-sintered short channel carbon electrodes, and (c) laser-irradiated self-contact transistors. Reprinted with permission from ref. 93. Copyright 2011, The Chemical Society of Japan. | ||
2.3. “Self-contact” transistors using chemically doped organic-conductor electrodes
Many OSCs, regardless of their carrier type, undergo carbonization through heat treatment by laser irradiation.94,95 The carbonized part in the OSC layer has sufficient conductivity for using as transistor electrodes (Fig. 3(b)). Such simple method for creating carbon electrodes by laser irradiation is applicable for various OSCs such as pentacene, oligothiophene, phthalocyanine, and C60.96 This type of transistor is called a “self-contact organic transistor” because the electrode is created directly from the OSC itself (Fig. 3(c)). Unfortunately, the carbon electrode in the self-contact transistors shows a relatively large contact resistance comparable to that of the bottom-contact Au-electrode device, because of the morphological mismatch at the electrode–OSC boundary caused by the laser irradiation. However, the unique concept provides an opportunity to reduce the contact resistance using organic electrodes of CT complexes/salts directly formed by the OSC layer. Since most OSCs are either electron donors or acceptors, they can form highly conducting CT complexes/salts such as (TTF)(TCNQ) with appropriate chemical doping. Thus, the OSC layer can be selectively converted into organic electrodes to form chemical-doped self-contact transistors.97–99Fig. 4 shows an example of a combination of a tetramethyl-TTF (TMTTF) semiconductor and a (TMTTF)(TCNQ) electrode. The TMTTF film on a polystyrene-coated substrate is selectively converted into (TMTTF)(TCNQ) by the vacuum deposition of TCNQ through a metal mask and the subsequent thermal annealing. The resulting self-contact transistors exhibit low contact resistance and good carrier-transport characteristics, similar to standard top-contact Au-electrode devices. In addition to vacuum evaporation, inkjet printing has been applied for doping. The inkjet method allows to use organic electrodes derived from CT salts, which are usually decomposed via vacuum evaporation. Conductive Cu(DMDCNQI)2 electrodes have been fabricated via inkjet printing of a CuI solution on a DMDCNQI semiconductor surface. The resulting transistors show stable n-channel operation and nearly the same performance as those of top-contact Au-electrode devices.99 Furthermore, this doping method has been used to successfully fabricate flexible self-contact transistors composed entirely of organic materials, including electrodes.98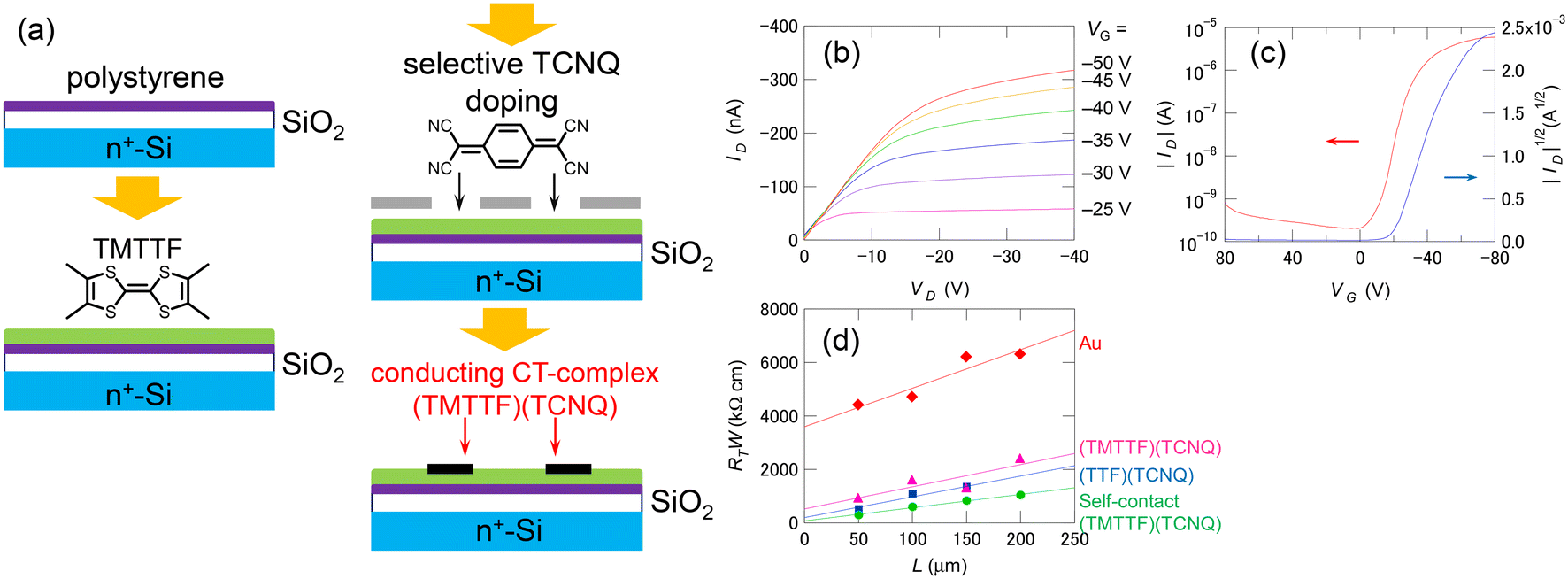 | ||
| Fig. 4 (a) Schematics of the fabrication procedure of the chemical-doped self-contact transistors. (b) Output and (c) transfer characteristics of a self-contact transistor based on TMTTF/(TMTTF)(TCNQ). (d) Channel length dependence of the total resistance RT normalized with respect to the channel width W for the top-contact transistors. Reprinted with permission from ref. 97. Copyright 2013, AIP Publishing. | ||
After chemical doping, the CT complex has a partially filled energy band. In the case of Cu(DMDCNQI)2, the CT complex is an anion-radical salt and this ensures that the electron transport is maintained by the organic acceptor. In Cu(DMDCNQI)2, Cu is Cu4/3+ and DMDCNQI has a −2/3e charge. Therefore, the resulting LUMO band derived from DMDCNQI is 1/3 filled. When the center of the Cu(DMDCNQI)2 band is located at the same position as the original LUMO level of DMDCNQI (4.65 eV), after the band formation with a bandwidth of 0.8 eV and electrons are only 1/3 filled in the entire band, the Fermi level is shifted down by approximately 0.1 eV from the original LUMO level (Fig. 5a). On the other hand, the DA complex forms energy bands from both the donor and acceptor molecules. Upon band formation from both the donor and acceptor molecules, the Fermi level is expected to be located in between the HOMO of the donor and LUMO of the acceptor. Consequently, the Fermi level is located close to the energy level of the semiconductor layer (Fig. 5b).
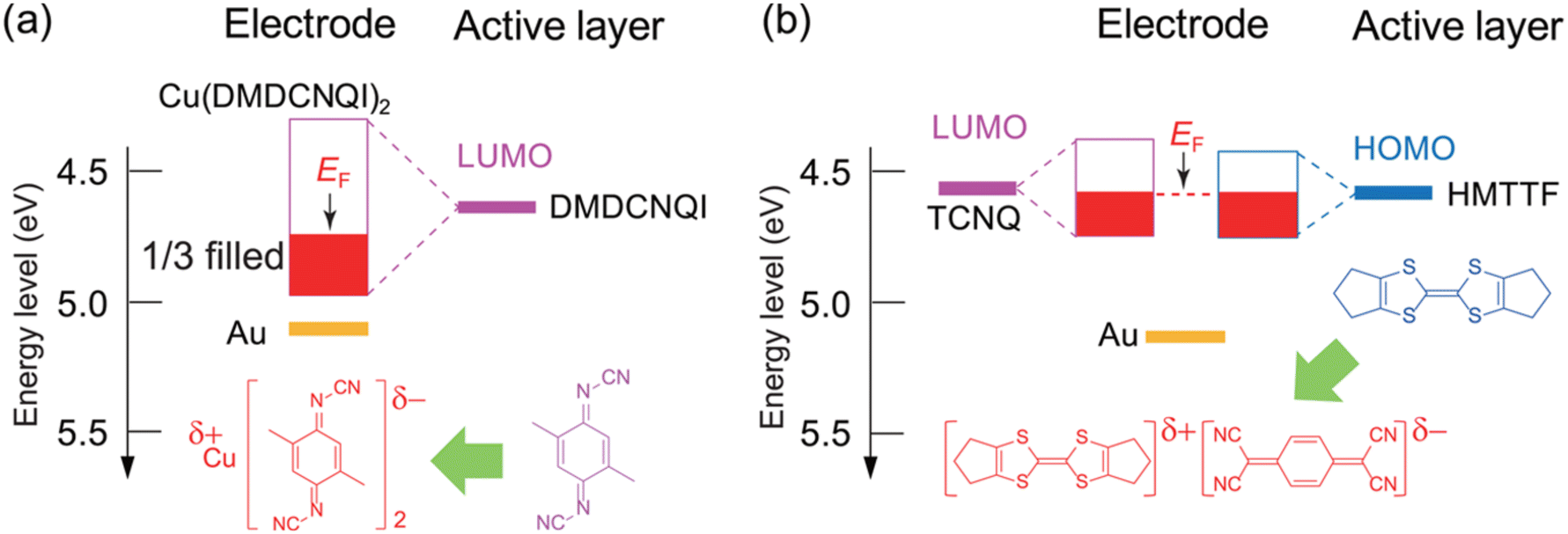 | ||
| Fig. 5 Energy-level alignment between the semiconductors and electrodes in self-contact (a) DMDCNQI/Cu(DMDCNQI)2 and (b) HMTTF/(HMTTF)(TCNQ) transistors. Reprinted with permission from ref. 88. Copyright 2014, American Chemical Society. | ||
The reported self-contact transistors have used redox-active materials such as TTF derivatives and DMDCNQI, which are well-known from studies on organic conductors. However, conventional OSCs of aromatic hydrocarbons such as pentacene and C60 hardly form atmospherically stable and highly conducting organic conductors. Thus, doping techniques have not been widely used in OFETs, in contrast to inorganic silicon, in which chemical doping has become standard. In the case of OSCs that do not form stable CT complexes, the conductivity in the doped electrode area is temporarily improved, but diffusion of dopants occurs, resulting in a decrease in the conductivity of the electrode. CT complexes that are stable as solids or crystals with strong intermolecular forces are necessary for self-contact transistors. Therefore, highly conductive organic conductors with excellent OSCs must be developed to expand the application of the chemical-doped devise fabrication.
3. Organic conductors based on organic semiconductors
The essence of self-contact transistors is that both the active layer and the electrode are comprised from the same OSC materials to reduce the energy level and morphological mismatch at the interface, thereby significantly suppressing the contact resistance. Thus, an important requirement for high-performance self-contact transistors is that OSCs possess both excellent inherent transistor properties and abilities to form highly conducting CT complexes/salts. In this regard, TTF derivatives may become ideal candidates because they can mostly exhibit p-channel behaviour with high mobility and can easily form highly conducting CT complexes/salts.42,90,100–102 However, such TTF-based OSCs are usually difficult to maintain long-term air stability due to their low ionisation potential of about 4.8 eV.103–106 Meanwhile, highly conducting CT complexes/salts have been reported even for non-TTF-type donors, such as perylene107–112 and fluoranthene,113–116 dithiapyrene,117–123 and chalcogen-bridged acenes,124–136 but the transistor properties of these donors are still limited.137–144 This section describes a new class of non-TTF-based organic conductors derived from high-performance OSC materials.3.1. BTBT-based CT salts
Benzothieno[3,2-b][1]benzothiophene (BTBT) is a promising π-electron skeleton for high-performance p-channel OSCs that exhibits high intrinsic mobility and chemical stability.145–163 The excellent hole transport capability of the BTBT materials is attributed to their two-dimensional (2D) crystal/electronic structures including short intermolecular contacts between the sulphur atoms with large HOMO coefficients. The high stability of the BTBT derivatives is associated with the deep HOMO level (EHOMO), ca. 5.65 eV for the unsubstituted BTBT. This contrasts pentacene (EHOMO = 4.85 eV)164 and TTF derivatives (EHOMO = 4.8 eV). Despite the weak electron-donating ability, BTBT can form CT salts with anions by electrochemical oxidation.165 The first reported BTBT-based CT salt is (BTBT)2PF6, which comprises one PF6 anion for every two BTBT molecules (Fig. 6). Thus, the BTBT molecule is partially oxidised with a +0.5e charge, and the salt forms a typical 3/4-filled band structure. In the crystal, the BTBT molecules form π-stacking 1D columns arranged in a windmill manner, in contrast to a herringbone packing seen in most neutral BTBT derivatives. These unique 1D columns are maintained even when PF6 is replaced by other octahedral anions, such as AsF6, SbF6, and TaF6.166 All of these salts exhibit metallic behaviour. In particular, the AsF6 salt exhibits the highest conductivity of 4100 S cm−1, corresponding to a drift mobility of 16 cm2 V−1 s−1. This value is prominently high among the non-TTF-based organic conductors. The high conductivity is attributed to an extremely large transfer integral of approximately 350 meV between the π-stacking BTBT molecules, as revealed by the thermoelectric power and reflection spectra analyses. All these salts undergo a discontinuous resistivity jump during the cooling process, which can be suppressed by physical hydrostatic pressure on the crystal and chemical modifications of the BTBT molecule as described below. Further cooling makes these salts insulating due to the 1D instability. Magnetic susceptibility and spectroscopic experiments suggest that the insulating state is attributed to a 4kF charge-density wave with a slight dimerisation of BTBT molecules.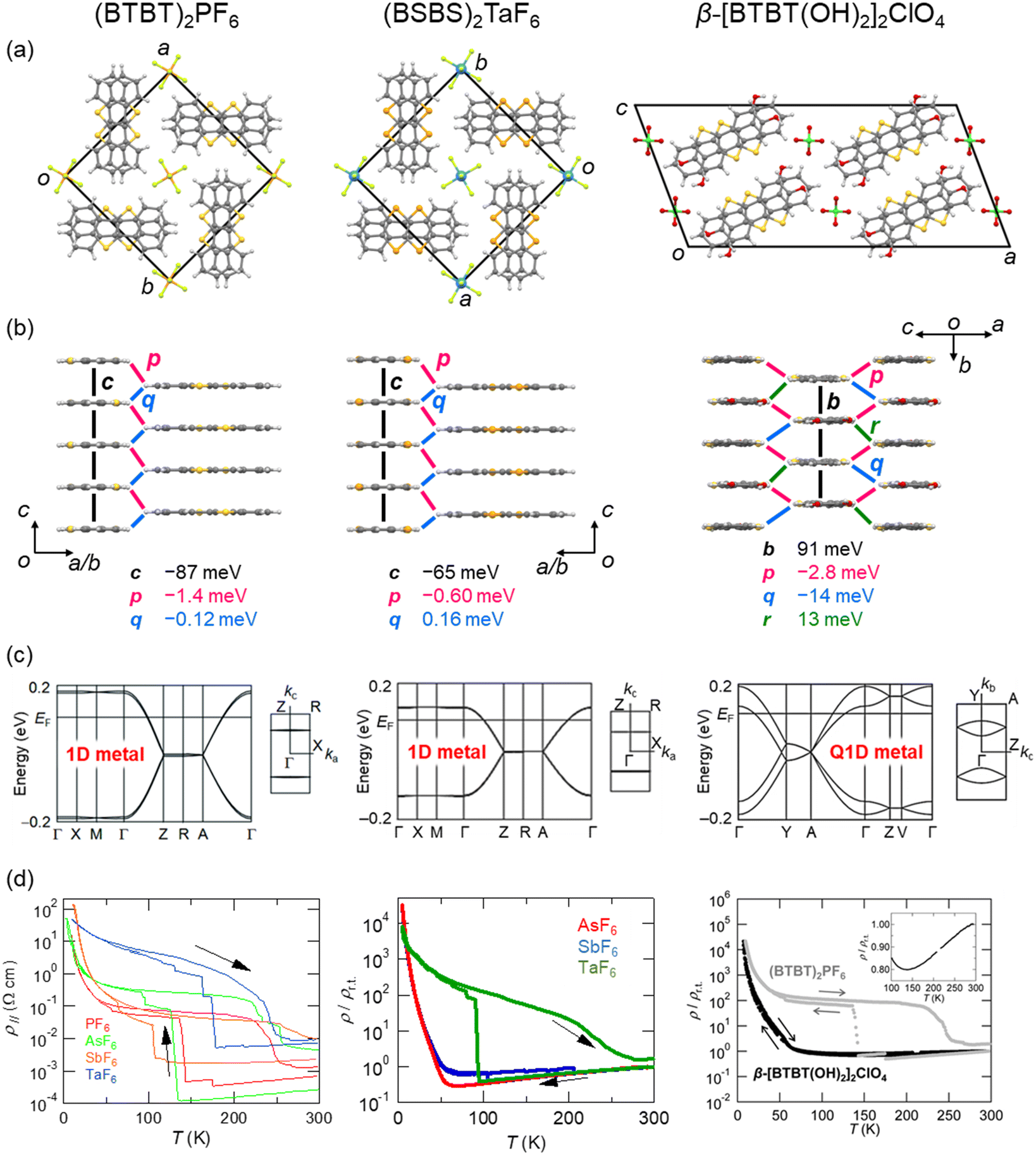 | ||
| Fig. 6 (a and b) Crystal structures of (BTBT)2PF6 (CSD ref code: AFEBIY),165 (BSBS)2TaF6 (CSD ref. code: ZOJREX),167 and β-[BTBT(OH)2]2ClO4 (CSD ref code: XAMRUB),174 respectively. Transfer integrals between the BTBT (BSBS) molecules are shown. Anions are omitted for clarity. (c) Band structures and (d) electrical resistivities of BTBT salts (left, reprinted with permission from ref. 166. Copyright 2016, American Chemical Society), BSBS salts (middle, reprinted with permission from ref. 168. Copyright 2019, American Chemical Society), and BTBT(OH)2 salts (right, reprinted with permission from ref. 174. Copyright 2017, Royal Society of Chemistry), respectively. | ||
Benzoseleno[3,2-b][1]benzoselenophene (BSBS) is a selenium analogue of BTBT, and forms CT salts (BSBS)2X (X = AsF6, SbF6 and TaF6; Fig. 6), which are isostructural to the BTBT salts.167,168 Such heavy chalcogen substitutions have been frequently used in TTF-based organic conductors to enhance the intermolecular interactions, leading to a significant increase of the electrical conductivity and a complete suppression of the metal–insulator transition.169–173 This chemical modification to stabilise the metallic state is also effective in the BTBT system. The discontinuous resistivity jumps seen in the BTBT salts are shifted to lower temperatures or completely suppressed in the BSBS salts. However, the room-temperature conductivities are slightly less than those of the BTBT salts. The metallic state can also be stabilised by controlling the molecular arrangement using hydrogen-bonding interactions. Dihydroxy-substituted BTBT (BTBT(OH)2) involves a catechol substructure with a strong hydrogen-bonding ability, producing a CT salt with a tetrahedral ClO4 anion (Fig. 6), where the donor![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :anion stoichiometry is 2:1.174 The resulting salt, [BTBT(OH)2]2ClO4, forms hydrogen-bond chains through the catechol moiety of BTBT(OH)2 and the ClO4 anion, consequently constructing a sheet-type (so-called β-type) molecular arrangement of BTBT(OH)2. This 2D molecular packing increases the dimensionality of the electronic structure from 1D to quasi-1D (Q1D) and consequently stabilises the metallic state, which is reminiscent of the very early days of TTF-based superconductors.161,162 Note that this molecule shows a quinoid-like bond alternation upon oxidation especially in the central thienothiophene part, as is well-known in oligothiophenes.175–178 These studies demonstrate that functionalised BTBT derivatives are promising electron donors in molecular conductors.
:anion stoichiometry is 2:1.174 The resulting salt, [BTBT(OH)2]2ClO4, forms hydrogen-bond chains through the catechol moiety of BTBT(OH)2 and the ClO4 anion, consequently constructing a sheet-type (so-called β-type) molecular arrangement of BTBT(OH)2. This 2D molecular packing increases the dimensionality of the electronic structure from 1D to quasi-1D (Q1D) and consequently stabilises the metallic state, which is reminiscent of the very early days of TTF-based superconductors.161,162 Note that this molecule shows a quinoid-like bond alternation upon oxidation especially in the central thienothiophene part, as is well-known in oligothiophenes.175–178 These studies demonstrate that functionalised BTBT derivatives are promising electron donors in molecular conductors.
3.2. BTBT-based CT complexes
BTBT molecules form CT complexes when combined with strong organic acceptors. The first report on BTBT-based complexes is in combinations of alkylated BTBT (Cn-BTBT: n = 10) and TCNQ derivatives with a 1:1 composition. The BTBT and TCNQ molecules form an alternating mixed-stack structure.179 Therefore, these complexes do not exhibit metallic conductivity, in contrast to the segregated-stack complexes represented by (TTF)(TCNQ) and the above-mentioned BTBT-based salts. The mixed-stack complexes form a unique layered crystal structure in which the π-conjugated moieties of the BTBT skeleton and TCNQ molecules are arranged in alternating 2D packing, regardless of the number of the fluorine atoms in FmTCNQ (m = 0, 1, 2, and 4) and the alkyl-chain length of the Cn-BTBT (n = 4, 8, and 12; Fig. 7).180 These TCNQ complexes of alkylated BTBTs exhibit basically n-channel semiconducting properties when used as an OFET active layer, as demonstrated in single crystals obtained by recrystallisation, polycrystalline thin films fabricated by vacuum deposition,181 and unidirectional crystalline films prepared by solution coating.182 The electron transport has long-term stability in air, which is ascribed to the sufficiently deep conduction levels of these complexes.183 The dominance of electron transport is explained by the super-exchange mechanism closely related to the orbital symmetry matching, where transfers corresponding to the acceptor-to-acceptor hopping are considerably larger than the donor-to-donor hopping.184–188 Interestingly, these complexes can also be formed by molecular thermal diffusion in bilayers consisting of single-component films189 The pristine bilayers with F4TCNQ stacked on C10-BTBT show p-channel operation as well as single-component C10-BTBT films. In contrast, thermal annealing of the stacked films promotes only n-channel transport, indicating that the CT complexes are formed in the whole film. This carrier polarity conversion can be controlled by adjusting the molar ratio of C10-BTBT and F4TCNQ in the stacked film.
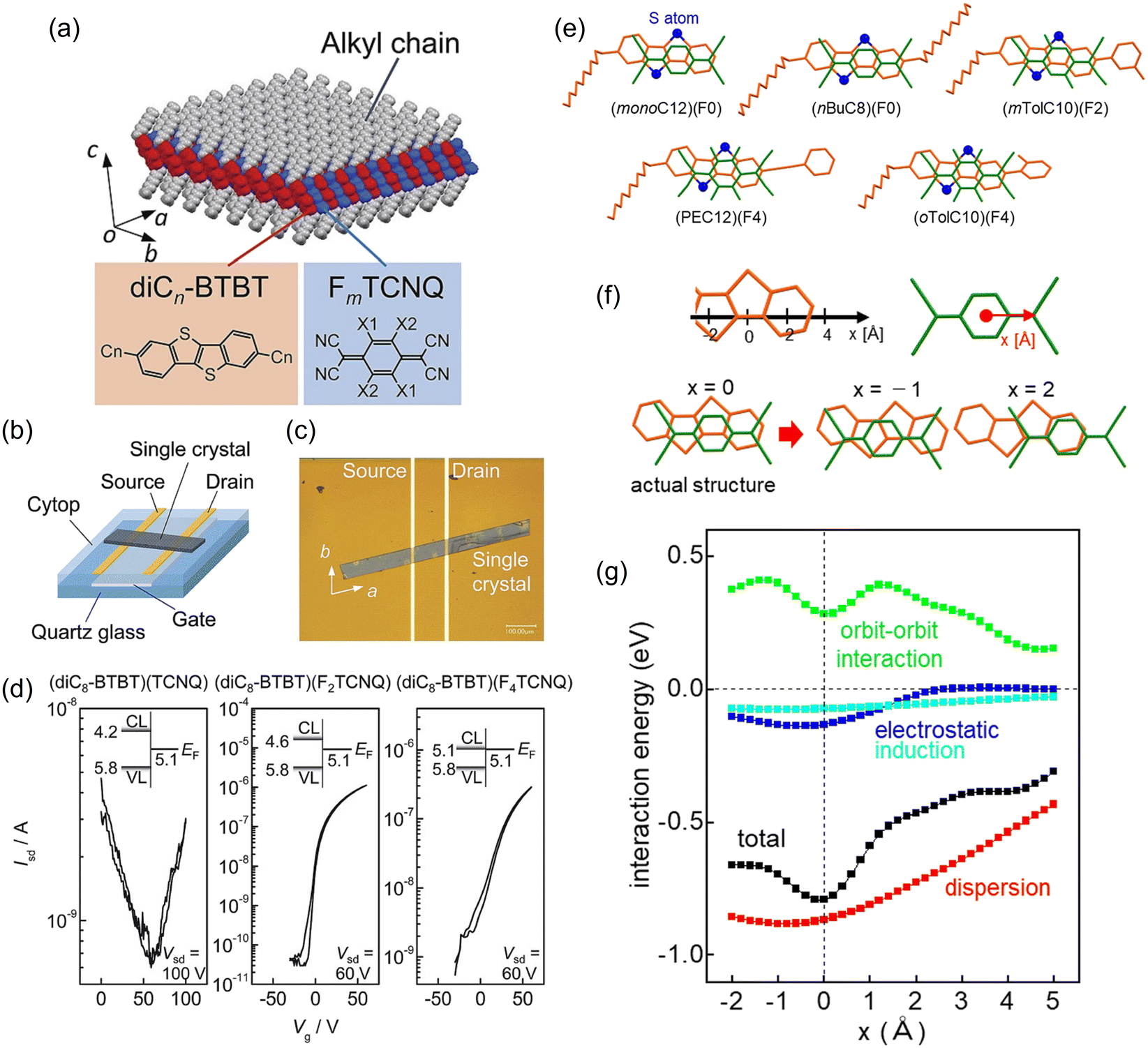 | ||
| Fig. 7 (a) Molecular and crystal structures of (diCn-BTBT)(FmTCNQ) (n = 4, 8, and 12, m = 0, 2, and 4). (b) Schematic and (c) photograph of a single-crystalline OFET fabricated from (diC8-BTBT)(FmTCNQ). (d) Transfer characteristics of (diC8-BTBT)(FmTCNQ). Inset shows the interfacial energy-level alignment between (diC8-BTBT)(FmTCNQ) and the gold source/drain electrode. CL and VL are the conduction and valence levels of (diC8-BTBT)(FmTCNQ), respectively. EF is the Fermi level of the Au source/drain electrode. Reprinted with permission from ref. 180. Copyright 2015, Royal Society of Chemistry. (e) Intermolecular arrangements of five TCNQ complexes BTBT derivatives along the alternating stacking axis. Sulphur atoms are represented by blue-filled circles. (f) Coordinates of BTBT and TCNQ core skeletons for the calculation of the intermolecular interaction energy with a fixed intermolecular distance. Origin of the x axis is set at the actual structure. (g) Profiles of total interaction and respective contributions by dispersion, electrostatic, induction, and short-range orbit-orbit interaction. Reprinted with permission from ref. 199. Copyright 2022, Royal Society of Chemistry. | ||
The unsubstituted BTBT and BSBS can form mixed-stack CT complexes with (fluorinated) TCNQs and a π-extended analogue, tetracyanonaphthoquinodimethane (TNAP).190–196 These complexes have charge transfer degrees of approximately 0.1, irrespective of the acceptor strength, as revealed by the bond length analysis of the acceptor, the CN stretching frequency shifts, and the Raman spectra shifts. Thus, these complexes are classified as neutral complexes. The charge transfer degrees increase in the range of about 0.2 to 0.3 using alkoxy-substituted BTBT derivatives which are slightly stronger donors.197,198 Regardless of the type of BTBT donors, these TCNQ complexes exhibit robust electron transport with excellent ambient and long-term stability. Recently, it has been found that most of the co-crystals of BTBTs and TCNQs have common intermolecular stacking arrangements between the planar donor and acceptor skeletons, irrespective of the type of substituent (Fig. 7).199 The dispersion force is the primary force of attraction between the donor and acceptor and the repulsive short-range (orbit–orbit) interactions play important roles in determining the stacking configuration in the co-crystals. However, it was often claimed that CT interactions could be the main contributors to the attraction in donor–acceptor compounds.
3.3. Extension of dimensionality in non-TTF-type conductors
Solid-state physics in low-dimensional electronic systems has progressed from 1D conductors as typified by (TTF)(TCNQ), and the dimensionality of the electronic structures in organic conductors has been extended by elegant molecular designs. Bis(ethylenedithio)tetrathiafulvalene (BEDT-TTF) is a pioneer TTF to form CT complexes with a rich variety of 2D molecular arrangements depending on the combined anions.200–214 This is attributed to the ethylenedithio group (i.e., 1,4-dithiin ring) on the outside of the BEDT-TTF molecule, which plays two important roles:215–219 1) the sulphur atoms increase the overlap of molecular orbitals in the molecular-short direction. 2) The carbon atoms suppress simple π-stacking by protruding sterically above and below the TTF plane. Such molecular designs have provided a large number of CT salts composed of BEDT-TTF and its analogues in various orientations,5,46,220,221 which have exhibited exotic physical properties such as superconducting transitions above 10 K,222–227 non-equilibrium charge ordering,228–232 metal–Mott insulator transitions,233–238 a zero-band-gap state,239–244 and quantum spin liquids,245–252 further advancing the materials science in organic conductors.Recently, a non-TTF-type 2D CT salt, (BEDT-BDT)PF6 (BEDT-BDT = benzo[1,2-g:4,5-g]bis(thieno[2,3-b][1,4]dithiin)), has been reported (Fig. 8).253 In this salt, the donor comprises a thienoacene π-core like BTBT and ethylenedithio groups similar to BEDT-TTF. This report has demonstrated that introducing substituents with high structural degrees of freedom, such as ethylenedithio groups, is quite effective in extending the dimensionality of non-TTF-type conductors as well as TTF systems. In contrast, the conventional non-TTF-type conductors mostly have a 1D crystal/electronic structure (as seen in perylene, fluoranthene, phthalocyanine, chalcogen-bridged acene, and dithiapyrene systems). The BEDT-BDT salt is formed by a 2D donor arrangement classified as a θ-type (herringbone-like) packing in a 1:1 composition with an octahedral anion PF6. This contrasts to the neutral BEDT-BDT crystal,254 which forms a pitched π-stacking similar to that of rubrene, one of the most high-performance OSCs.255–258 The salt is a genuine Mott insulator with a hole localised in a molecule without any dimerisation in contrast to representative Mott insulators based on BEDT-TTF with a κ-type dimerised molecular packing,259 and exhibits semiconducting behaviour because of the strong electronic correlations. The 2D electronic structure of the salt has been confirmed by tight-binding band calculations and magnetic susceptibility measurements. The 2D crystal/electronic structure can be finely modulated by chemical modifications using other octahedral anions or selenized donors,260,261 while maintaining the isomorphous structure, allowing other novel physical properties to be explored.
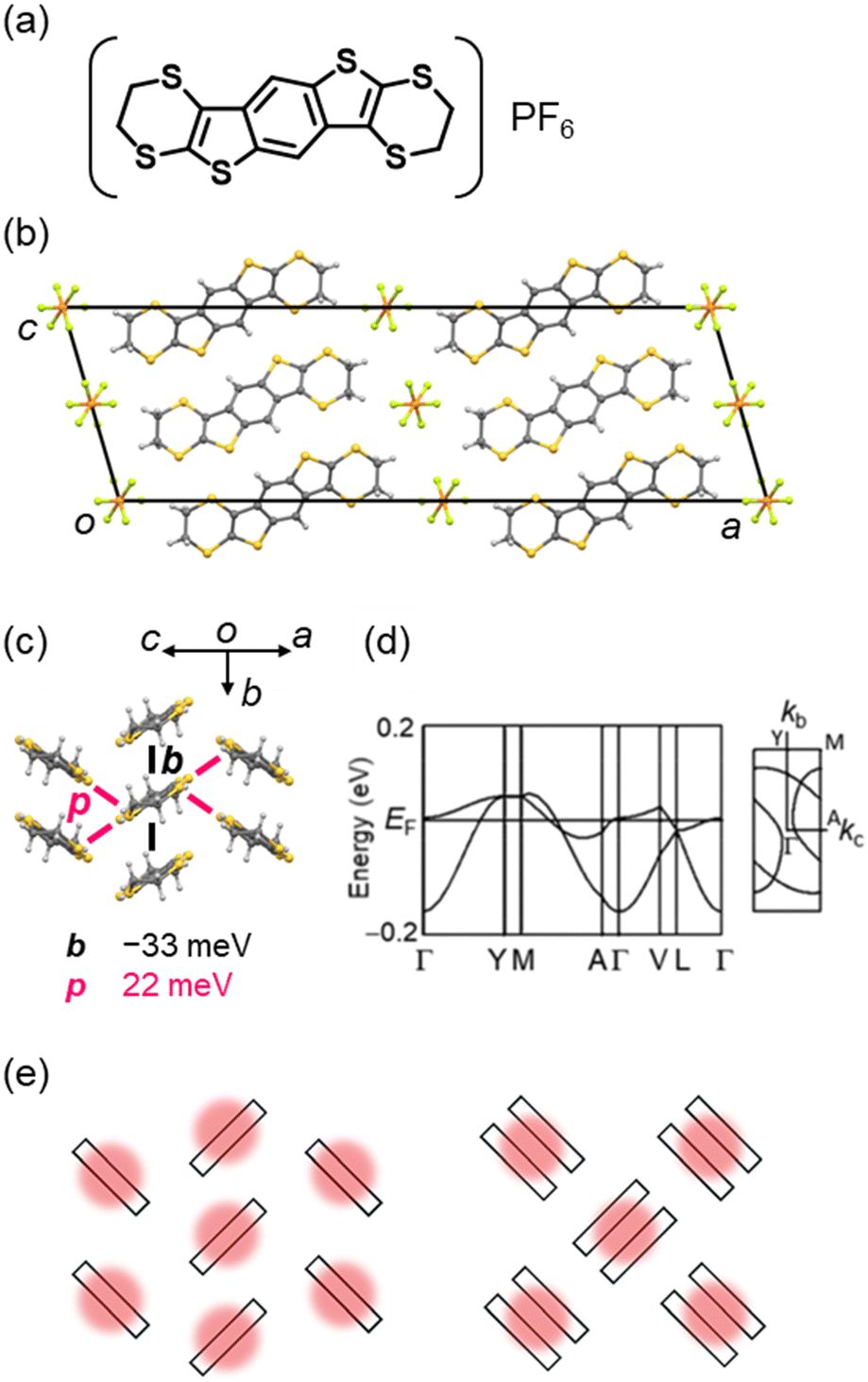 | ||
| Fig. 8 (a) Chemical structure of (BEDT-BDT)PF6. Crystal structures of θ-(BEDT-BDT)PF6 (CSD ref code: FUSFEH),253 viewed along (b) the π-stacking (crystallographic b) direction and (c) the molecular long-axis direction with the intermolecular transfer integrals (PF6 anions are omitted for clarity). (d) Electronic band structures of θ-(BEDT-BDT)PF6. (e) Schematics of the hole distribution in a genuine Mott insulator with a θ-type molecular packing (left) and a representative dimerized Mott insulator with a κ-type molecular packing (right). Reprinted with permission from ref. 253. Copyright 2020, Royal Society of Chemistry. | ||
4. Conclusions
In this highlight, we presented examples of studies using organic conductors as electrodes to suppress the contact resistance (charge injection barrier) at the OSC–electrode interface of organic transistors. Conductive organic materials have similar chemical properties and surface energies in contrast to inorganic metals. Thus, the contact resistance of organic transistors can be significantly reduced by using organic electrodes, in addition to the use of dopants and SAMs as a buffer layer at the OSC–electrode interface. In particular, the transistor fabrication technique using direct chemical doping, which is the standard for silicon semiconductors, is a unique attempt to develop a new fabrication method for organic devices. The development of high-conductivity organic conductors with organic semiconductors is essential for the widespread application of this technique. New semiconductors that achieve not only high mobility but also air stability and redox activity should be developed. Thienoacene-based CT salts, such as BTBT, which have been developed for this purpose, have the potential to create new trends in research on the physical properties of organic conductors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partly supported by the JSPS KAKENHI Grant Numbers 20K15356, 21K14699 and 23K04882, JST CREST Grant Number JPMJCR18J2, and The Mazda Foundation, Izumi Science and Technology Foundation, Research Foundation for the Electrotechnology of Chubu.Notes and references
- D. D. Eley, Nature, 1948, 162, 819–819 CrossRef CAS PubMed.
- H. Akamatu and H. Inokuchi, J. Chem. Phys., 1950, 18, 810–811 CrossRef CAS.
- H. Inokuchi, Bull. Chem. Soc. Jpn., 1951, 24, 222–226 CrossRef CAS.
- H. Akamatsu, H. Inokuchi and Y. Matsunaga, Nature, 1954, 173, 168–169 CrossRef CAS.
- T. Naito, Crystals, 2021, 11, 838 CrossRef CAS.
- T. Naito, Bull. Chem. Soc. Jpn., 2017, 90, 89–136 CrossRef CAS.
- R. Kato, Bull. Chem. Soc. Jpn., 2014, 87, 355–374 CrossRef CAS.
- F. Wudl, G. M. Smith and E. J. Hufnagel, Chem. Commun., 1970, 1453–1454 RSC.
- D. L. Coffen, Tetrahedron Lett., 1970, 2633–2636 CrossRef CAS.
- H. Prinzbach, H. Berger and A. Lütringhaus, Angew. Chem., Int. Ed. Engl., 1965, 4, 435 CrossRef.
- A. Takamizawa and K. Hirai, Chem. Pharm. Bull., 1969, 17, 1931–1936 CrossRef CAS.
- J. Yamada and T. Sugimoto, TTF Chemistry–Fundamentals and Applications of Tetrathiafulvalene, Kodansha & Springer, Tokyo, Japan, 2004 Search PubMed.
- Y. Morita, S. Suzuki, K. Sato and T. Takui, Nat. Chem., 2011, 3, 197–204 CrossRef CAS PubMed.
- Y. Kobayashi, T. Terauchi, S. Sumi and Y. Matsushita, Nat. Mater., 2017, 16, 109–114 CrossRef CAS PubMed.
- S. K. Pal, P. Bag, M. E. Itkis, F. S. Tham and R. C. Haddon, J. Am. Chem. Soc., 2014, 136, 14738–14741 CrossRef CAS PubMed.
- A. Mailman, S. M. Winter, X. Yu, C. M. Robertson, W. Yong, J. S. Tse, R. A. Secco, Z. Liu, P. A. Dube, J. A. K. Howard and R. T. Oakley, J. Am. Chem. Soc., 2012, 134, 9886–9889 CrossRef CAS PubMed.
- J. W. L. Wong, A. Mailman, K. Lekin, S. M. Winter, W. Yong, J. Zhao, S. V. Garimella, J. S. Tse, R. A. Secco, S. Desgreniers, Y. Ohishi, F. Borondics and R. T. Oakley, J. Am. Chem. Soc., 2014, 136, 1070–1081 CrossRef CAS PubMed.
- S. K. Mandal, S. Samanta, M. E. Itkis, D. W. Jensen, R. W. Reed, R. T. Oakley, F. S. Tham, B. Donnadieu and R. C. Haddon, J. Am. Chem. Soc., 2006, 128, 1982–1994 CrossRef CAS PubMed.
- S. K. Pal, M. E. Itkis, F. S. Tham, R. W. Reed, R. T. Oakley and R. C. Haddon, Science, 2005, 309, 281–284 CrossRef CAS PubMed.
- Y. Kobayashi, K. Hirata, S. N. Hood, H. Yang, A. Walsh, Y. Matushita and K. Ishioka, Chem. Sci., 2020, 11, 11699–11704 RSC.
- H. Tanaka, Y. Okano, H. Kobayashi, W. Suzuki and A. Kobayashi, Science, 2001, 291, 285–287 CrossRef CAS PubMed.
- W. Suzuki, E. Fujiwara, A. Kobayashi, Y. Fujishiro, E. Nishibori, M. Tanaka, M. Sakata, H. Fujiwara and H. Kobayashi, J. Am. Chem. Soc., 2003, 125, 1486–1487 CrossRef CAS PubMed.
- T. Tanaka, H. Kobayashi and A. Kobayashi, J. Am. Chem. Soc., 2002, 124, 10002–10003 CrossRef PubMed.
- N. Tenn, N. Bellec, O. Jeannin, L. Piekara-Sady, P. Auban-Senzier, J. Iniguez, E. Canadell and D. Lorcy, J. Am. Chem. Soc., 2009, 131, 16961–16967 CrossRef CAS PubMed.
- H. B. Cui, H. Kobayashi, S. Ishibashi, M. Sasa, F. Iwase, R. Kato and A. Kobayashi, J. Am. Chem. Soc., 2014, 136, 7619–7622 CrossRef CAS PubMed.
- H. Kamo, A. Ueda, T. Isono, K. Takahashi and H. Mori, Tetrahedron Lett., 2012, 53, 4385–4388 CrossRef CAS.
- T. Isono, H. Kamo, A. Ueda, K. Takahashi, A. Nakao, R. Kumai, H. Nakano, K. Kobayashi, Y. Murakami and H. Mori, Nat. Commun., 2013, 4, 1344 CrossRef PubMed.
- A. Ueda, S. Yamada, T. Isono, H. Kamo, A. Nakao, R. Kumai, H. Nakao, Y. Murakami, K. Yamamoto, Y. Nishio and H. Mori, J. Am. Chem. Soc., 2014, 136, 12184–12192 CrossRef CAS PubMed.
- H. Mori, S. Yokomori, S. Dekura and A. Ueda, Chem. Commun., 2022, 58, 5668–5682 RSC.
- A. Ueda, Bull. Chem. Soc. Jpn., 2017, 90, 1181–1188 CrossRef CAS.
- B. Kippelen and J. L. Brédas, Energy Environ. Sci., 2009, 2, 251–261 RSC.
- S. B. Darling and F. You, RSC Adv., 2013, 3, 17633–17648 RSC.
- Y. Cui, Y. Xu, H. Yao, P. Bi, L. Hong, J. Zhang, Y. Zu, T. Zhang, J. Qin, J. Ren, Z. Chen, C. He, X. Hao, Z. Wei and J. Hou, Adv. Mater., 2021, 33, 2102420 CrossRef CAS PubMed.
- C. Xu, Z. Zhao, K. Yang, L. Niu, X. Ma, Z. Zhou, X. Zhang and F. Zhang, J. Mater. Chem. A, 2022, 10, 6291–6329 RSC.
- M. Zhang, L. Zhu, G. Zhou, T. Hao, C. Qiu, Z. Zhao, Q. Hu, B. W. Larson, H. Zhu, Z. Ma, Z. Tang, W. Feng, Y. Zhang, T. P. Russell and F. Liu, Nat. Commun., 2021, 12, 309 CrossRef CAS PubMed.
- L. Zhan, S. Li, X. Xia, Y. Li, X. Lu, L. Zuo, M. Shi and H. Chen, Adv. Mater., 2021, 33, 2007231 CrossRef CAS PubMed.
- F. Huewe, A. Steeger, K. Kostova, L. Burroughs, I. Bauer, P. Strohriegl, V. Dimitrov, S. Woodward and J. Pflaum, Adv. Mater., 2017, 29, 1605682 CrossRef PubMed.
- Q. Zhang, Y. Sun, W. Xu and D. Zhu, Adv. Mater., 2014, 26, 6829–6851 CrossRef CAS PubMed.
- B. Russ, A. Glaudell, J. J. Urban, M. L. Chabinyc and R. A. Segalman, Nat. Rev. Mater., 2016, 1, 16050 CrossRef CAS.
- J. Liu, B. V. D. Zee, R. Alessandri, S. Sami, J. Dong, M. I. Nugraha, A. J. Barker, S. Rousseva, L. Qiu, X. Qiu, N. Klasen, R. C. Chiechi, D. Baran, M. Caironi, T. D. Anthopoulos, G. Portale, R. W. A. Havenith, S. J. Marrink, J. C. Hummelen and L. J. A. Koster, Nat. Commun., 2020, 11, 5694 CrossRef CAS PubMed.
- H. Sirringhaus, Adv. Mater., 2014, 26, 1319–1335 CrossRef CAS PubMed.
- M. Mas-Torrent and C. Rovira, Chem. Rev., 2011, 111, 4833–4856 CrossRef CAS PubMed.
- H. Chen, W. Zhang, M. Li, G. He and X. Guo, Chem. Rev., 2020, 120, 2879–2949 CrossRef CAS PubMed.
- T. Higashino and T. Mori, Phys. Chem. Chem. Phys., 2022, 24, 9770–9806 RSC.
- W. Tang, Y. Huang, L. Han, R. Liu, Y. Su, X. Guo and F. Yan, J. Mater. Chem. C, 2019, 7, 790–808 RSC.
- T. Mori, Bull. Chem. Soc. Jpn., 2016, 89, 973–986 CrossRef CAS.
- C. D. Dimitrakopoulos and P. R. L. Malenfant, Adv. Mater., 2002, 14, 99–117 CrossRef CAS.
- J. Takeya, K. Yamada, K. Hara, K. Shigeto, K. Tsukagoshi, S. Ikehata and Y. Aoyagi, Appl. Phys. Lett., 2006, 88, 112102 CrossRef.
- H. Shimotani, H. Asanuma, J. Takeya and Y. Iwasa, Appl. Phys. Lett., 2006, 89, 203501 CrossRef.
- J. Lee, M. J. Panzer, Y. He, T. P. Lodge and C. D. Frisbie, J. Am. Chem. Soc., 2007, 129, 4532–4533 CrossRef CAS PubMed.
- R. Misra, M. McCarthy and A. F. Hebard, Appl. Phys. Lett., 2007, 90, 052905 CrossRef.
- J. H. Cho, J. Lee, Y. Xia, B. Kim, Y. He, M. J. Renn, T. P. Lodge and C. D. Frisbie, Nat. Mater., 2008, 7, 900–906 CrossRef CAS PubMed.
- H. M. Yamamoto, M. Nakano, M. Suda, Y. Iwasa, M. Kawasaki and R. Kato, Nat. Commun., 2013, 4, 2379 CrossRef PubMed.
- M. Suda and H. M. Yamamoto, Phys. Chem. Chem. Phys., 2018, 20, 1321–1331 RSC.
- H. M. Yamamoto, Bull. Chem. Soc. Jpn., 2021, 94, 2505–2539 CrossRef CAS.
- M. Waldrip, O. D. Jurchescu, D. J. Gundlach and E. G. Bittle, Adv. Funct. Mater., 2020, 30, 1904576 CrossRef CAS.
- C. Liu, G. Li, R. D. Pietro, J. Huang, Y. Y. Noh, X. Liu and T. Minari, Phys. Rev. Appl., 2017, 8, 034020 CrossRef.
- D. Natali and M. Caironi, Adv. Mater., 2012, 24, 1357–1387 CrossRef CAS PubMed.
- S. M. Sze and K. K. Ng, Physics of Semiconductor Devices, John Wiley & Sons, Inc., Hoboken, New Jersey, 3rd edn, 2007 Search PubMed.
- H. Wada, K. Shibata, Y. Bando and T. Mori, J. Mater. Chem., 2008, 18, 4165–4171 RSC.
- H. Ishii, K. Sugiyama, E. Ito and K. Seki, Adv. Mater., 1999, 11, 605–625 CrossRef CAS.
- H. Ishii and K. Seki, IEEE Trans. Electron Devices, 1997, 44, 1295 CrossRef CAS.
- H. Ishii, K. Sugiyama, D. Yoshimura, E. Ito, Y. Ouchi and K. Seki, IEEE J. Sel. Top. Quantum Electron., 1998, 4, 24 CrossRef CAS.
- G. Hill and A. Kahn, Interface electronic properties of organic molecular semiconductors, Organic Light-Emitting Materials and Devices II, Proc. SPIE 3476, 1998 Search PubMed.
- K. Shibata, K. Ishikawa, H. Takezoe, H. Wada and T. Mori, Appl. Phys. Lett., 2008, 92, 023305 CrossRef.
- E. H. Rhoderick and R. H. Williams, Metal-Semiconductor Contacts, Clarendon, Oxford, 2nd edn, 1988 Search PubMed.
- T. Minari, P. Darmawan, C. Liu, Y. Li, Y. Xu and K. Tsukagoshi, Appl. Phys. Lett., 2012, 100, 093303 CrossRef.
- Y. Abe, T. Hasegawa, Y. Takahashi, T. Yamada and Y. Tokura, Appl. Phys. Lett., 2005, 87, 153506 CrossRef.
- J. Soeda, Y. Hirose, M. Yamagishi, A. Nakao, T. Uemura, K. Nakayama, M. Uno, Y. Nakazawa, K. Takimiya and J. Takeya, Adv. Mater., 2011, 23, 3309–3314 CrossRef CAS PubMed.
- A. Nollau, M. Pfeiffer, T. Fritz and K. Leo, J. Appl. Phys., 2000, 87, 4340–4347 CrossRef CAS.
- J. H. Oh, P. Wei and Z. Bao, Appl. Phys. Lett., 2010, 97, 243305 CrossRef.
- B. D. Naab, S. Guo, S. Olthof, E. G. B. Evans, P. Wei, G. L. Millhauser, A. Kahn, S. Barlow, S. R. Marder and Z. Bao, J. Am. Chem. Soc., 2013, 135, 15018–15025 CrossRef CAS PubMed.
- P. Wei, J. H. Oh, G. Dong and Z. Bao, J. Am. Chem. Soc., 2010, 132, 8852–8853 CrossRef CAS PubMed.
- B. D. Naab, S. Himmelberger, Y. Diao, K. Vandewal, P. Wei, B. Lussem, A. Salleo and Z. Bao, Adv. Mater., 2013, 25, 4663–4667 CrossRef CAS PubMed.
- J. T. E. Quinn, J. Zhu, X. Li, J. Wang and Y. Li, J. Mater. Chem. C, 2017, 5, 8654–8681 RSC.
- B. deBoer, A. Hadipour, M. M. Mandoc, T. vanWoudenbergh and P. W. M. Blom, Adv. Mater., 2005, 17, 621–625 CrossRef.
- S. Tatara, Y. Kuzumoto and M. Kitamura, Jpn. J. Appl. Phys., 2016, 55, 03DD02 CrossRef.
- K. Aoshima, S. Arai, K. Fukuhara, T. Yamada and T. Hasegawa, Org. Electron., 2017, 41, 137–142 CrossRef CAS.
- G. Kitahara, K. Aoshima, J. Tsutsumi, H. Minemawari, S. Arai and T. Hasegawa, Org. Electron., 2017, 50, 426–428 CrossRef CAS.
- Y. Yu, M. Kanno, H. Wada, Y. Bando, M. Ashizawa, A. Tanioka and T. Mori, Phys. B, 2010, 405, S378–S380 CrossRef CAS.
- T. Matsushima, Y. Kinoshita and H. Murata, Appl. Phys. Lett., 2007, 91, 253504–253506 CrossRef.
- T. Kadoya, O. Pitayatanakul and T. Mori, Org. Electron., 2015, 21, 106–110 CrossRef CAS.
- K. Shibata, H. Wada, K. Ishikawa, H. Takezoe and T. Mori, Appl. Phys. Lett., 2007, 90, 193509 CrossRef.
- J. Ferraris, D. O. Cowan, V. Walatka and J. H. Perlstein, J. Am. Chem. Soc., 1973, 95, 948–949 CrossRef CAS.
- L. B. Coleman, M. J. Cohen, D. J. Sandman, F. G. Yamagishi, A. F. Garito and A. J. Heeger, Solid State Commun., 1993, 88, 989–995 CrossRef.
- T. E. Phillips, T. J. Kistenmacher, J. P. Ferraris and D. O. Cowan, J. Chem. Soc., Chem. Commun., 1973, 471–472 RSC.
- Y. Takahashi, T. Hasegawa, Y. Abe, Y. Tokura, K. Nishimura and G. Saito, Appl. Phys. Lett., 2005, 86, 063504 CrossRef.
- Y. Takahashi, T. Hasegawa, Y. Abe, Y. Tokura and G. Saito, Appl. Phys. Lett., 2006, 88, 073504 CrossRef.
- D. de Caro, K. Jacob, H. Hahioui, C. Faulmann, L. Valade, T. Kadoya, T. Mori, J. Fraxedas and L. Viau, New J. Chem., 2011, 35, 1315–1319 RSC.
- T. Kadoya, D. de Caro, K. Jacob, C. Faulmann, L. Valade and T. Mori, J. Mater. Chem., 2011, 21, 18421–18424 RSC.
- H. Wada and T. Mori, Appl. Phys. Lett., 2008, 93, 213303 CrossRef.
- H. Wada and T. Mori, Appl. Phys. Lett., 2009, 95, 253307 CrossRef.
- T. Mori, Chem. Lett., 2011, 40, 428–434 CrossRef CAS.
- R. Srinivasan and B. Braren, Chem. Rev., 1989, 89, 1303–1316 CrossRef CAS.
- F. Raimondi, S. Abolhassani, R. Brütsch, F. Geiger, T. Lippert, J. Wambach, J. Wei and A. Wokaun, J. Appl. Phys., 2000, 88, 3659 CrossRef CAS.
- J. Inoue, H. Wada and T. Mori, Jpn. J. Appl. Phys., 2010, 49, 071605 CrossRef.
- S. Tamura, T. Kadoya, T. Kawamoto and T. Mori, Appl. Phys. Lett., 2013, 102, 063305 CrossRef.
- S. Tamura, T. Kadoya and T. Mori, Appl. Phys. Lett., 2014, 105, 023301 CrossRef.
- T. Kadoya, S. Tamura and T. Mori, J. Phys. Chem. C, 2014, 118, 23139–23146 CrossRef CAS.
- C. Rovira, Chem. Rev., 2004, 104, 5289–5317 CrossRef CAS PubMed.
- H. Jiang, X. Yang, Z. Cui, Y. Liu, H. Li, W. Hu and C. Kloc, CrystEngComm, 2014, 16, 5968–5983 RSC.
- R. Pfattner, S. T. Bromley, C. Rovira and M. Mas-Torrent, Adv. Funct. Mater., 2016, 26, 2256–2275 CrossRef CAS.
- M. Kanno, Y. Bando, T. Shirahata, J. Inoue, H. Wada and T. Mori, J. Mater. Chem., 2009, 19, 6548–6555 RSC.
- J. Inoue, M. Kanno, M. Ashizawa, C. Seo, A. Tanioka and T. Mori, Chem. Lett., 2010, 39, 538–540 CrossRef CAS.
- J. Nagakubo, M. Ashizawa, T. Kawamoto, A. Tanioka and T. Mori, Phys. Chem. Chem. Phys., 2011, 13, 14370–14377 RSC.
- T. Higashino, Y. Akiyama, H. Kojima, T. Kawamoto and T. Mori, Crystals, 2012, 2, 1222–1238 CrossRef CAS.
- L. Alcacer and A. H. Maki, J. Phys. Chem., 1974, 78, 215–217 CrossRef CAS.
- H. J. Keller, D. Nöthe, H. Pritzkow, D. Wehe, M. Werner, P. Koch and D. Schweitzer, Mol. Cryst. Liq. Cryst., 1980, 62, 181–199 CrossRef CAS.
- P. Koch, D. Schweitzer, R. H. Harms, H. J. Keller, H. Schäfer, H. W. Helberg, R. Wilckens, H. P. Geserich and W. Ruppel, Mol. Cryst. Liq. Cryst., 1982, 86, 87–101 CrossRef.
- D. Schweitzer, I. Hennig, K. Bender, H. Endres and H. J. Keller, Mol. Cryst. Liq. Cryst., 1985, 120, 213–220 CrossRef CAS.
- V. Gama, M. Almeida, R. T. Henriques, I. C. Santos, A. Domingos, S. Ravy and J. P. Pouget, J. Phys. Chem., 1991, 95, 4263–4267 CrossRef CAS.
- V. Gama, R. Henriques, M. Almeida and L. Alcácer, Synth. Met., 1991, 42, 2553–2556 CrossRef CAS.
- C. Kröhnke, V. Enkelmann and G. Wegner, Angew. Chem., Int. Ed. Engl., 1980, 19, 912–919 CrossRef.
- H. Eichele, M. Schwoerer, C. Kröhnke and G. Wegner, Chem. Phys. Lett., 1981, 77, 311–313 CrossRef CAS.
- V. Enkelmann, B. S. Morra, C. Kröhnke, G. Wegner and J. Heinze, Chem. Phys., 1982, 66, 303–313 CrossRef CAS.
- E. Dormann and G. Sachs, Ber. Bunsen-Ges., 1987, 91, 879–885 CrossRef CAS.
- K. Bechgaard, Mol. Cryst. Liq. Cryst., 1985, 125, 81–89 CrossRef CAS.
- N. Thorup, G. Rindorf, C. S. Jacobsen, K. Bechgaard, I. Johannsen and K. Mortensen, Mol. Cryst. Liq. Cryst., 1985, 120, 349–352 CrossRef CAS.
- K. Nakasuji, H. Kubota, T. Kotani, I. Murata, G. Saito, T. Enoki, K. Imaeda, H. Inokuchi, M. Honda, C. Katayama and J. Tanaka, J. Am. Chem. Soc., 1986, 108, 3460–3466 CrossRef CAS.
- K. Nakasuji, M. Sasaki, T. Kotani, I. Murata, T. Enoki, K. Imaeda, H. Inokuchi, A. Kawamoto and J. Tanaka, J. Am. Chem. Soc., 1987, 109, 6970–6975 CrossRef CAS.
- A. Kawamoto, J. Tanaka, A. Oda, H. Mizumura, I. Murata and K. Nakasuji, Bull. Chem. Soc. Jpn., 1990, 63, 2137–2145 CrossRef CAS.
- Y. Morita, Y. Yakiyama, T. Murata and K. Nakasuji, Solid State Sci., 2008, 10, 1720–1723 CrossRef CAS.
- J. Bendix, K. Bechgaard and J. B. Christensen, RSC Adv., 2021, 11, 14607–14614 RSC.
- Y. Matsunaga, J. Chem. Phys., 1965, 42, 2248–2249 CrossRef CAS.
- E. A. Perez-Albuerne, H. Johnson and D. J. Trevoy, J. Chem. Phys., 1971, 55, 1547–1554 CrossRef CAS.
- L. I. Buravov, G. I. Zvereva, V. F. Kaminskii, L. P. Rosenberg, M. L. Khidekel, R. P. Shibaeva, I. F. Shchegolev and E. B. Yagubskii, J. Chem. Soc., Chem. Commun., 1976, 720–721 RSC.
- L. C. Isett and E. A. Perez-Albuerne, Solid State Commun., 1977, 21, 433–435 CrossRef CAS.
- T. Inabe and Y. Matsunaga, Bull. Chem. Soc. Jpn., 1978, 51, 2813–2816 CrossRef CAS.
- Y. Matsunaga and K. Takayanagi, Bull. Chem. Soc. Jpn., 1980, 53, 2796–2799 CrossRef CAS.
- P. Kathirgamanathan and D. R. Rosseinsky, J. Chem. Soc., Chem. Commun., 1980, 356 RSC.
- H. Tanaka, T. Nogami and H. Mikawa, Chem. Lett., 1982, 11, 727–730 CrossRef.
- T. Nogami, H. Tanaka, S. Ohnishi, Y. Tasaka and H. Mikawa, Bull. Chem. Soc. Jpn., 1984, 57, 22–25 CrossRef CAS.
- K. Takimiya, H. Miyamoto, Y. Aso, T. Otsubo and F. Ogura, Chem. Lett., 1990, 19, 567–570 CrossRef.
- K. Takimiya, Y. Aso, T. Otsubo and F. Ogura, Bull. Chem. Soc. Jpn., 1991, 64, 2091–2102 CrossRef CAS.
- K. Takimiya, A. Ohnishi, Y. Aso, T. Otsubo, F. Ogura, K. Kawabata, K. Tanaka and M. Mizutani, Bull. Chem. Soc. Jpn., 1994, 67, 766–772 CrossRef CAS.
- T. Otsubo, K. Takimiya and Y. Aso, Phosphorus, Sulfur Silicon Relat. Elem., 1998, 136, 447–462 CrossRef.
- T. Ohta, T. Nagano, K. Ochi, Y. Kubozono and A. Fujiwara, Appl. Phys. Lett., 2006, 88, 103506 CrossRef.
- J.-W. Lee, H.-S. Kang, M.-K. Kim, K. Kim, M.-Y. Cho, Y.-W. Kwon, J. Joo, J.-I. Kim and C.-S. Hong, J. Appl. Phys., 2007, 102, 124104 CrossRef.
- H. Jiang, K. K. Zhang, J. Ye, F. Wei, P. Hu, J. Guo, C. Liang, X. Chen, Y. Zhao, L. E. McNeil, W. Hu and C. Kloc, Small, 2013, 9, 990–995 CrossRef CAS PubMed.
- C.-T. Hsieh, C.-Y. Chen, H.-Y. Lin, C.-J. Yang, T.-J. Chen, K.-Y. Wu and C.-L. Wang, J. Phys. Chem. C, 2018, 122, 16242–16248 CrossRef CAS.
- A. L. Briseno, Q. Miao, M.-M. Ling, C. Reese, H. Meng, Z. Bao and F. Wudl, J. Am. Chem. Soc., 2006, 128, 15576–15577 CrossRef CAS PubMed.
- A. L. Briseno, S. C. B. Mannsfeld, X. Lu, Y. Xiong, S. A. Jenekhe, Z. Bao and Y. Xia, Nano Lett., 2007, 7, 668–675 CrossRef CAS PubMed.
- S. Wen, A. Li, J. Song, W. Deng, K. Han and W. A. Goddard, J. Phys. Chem. B, 2009, 113, 8813–8819 CrossRef CAS PubMed.
- L. Zhang, S. M. Fakhouri, F. Liu, J. C. Timmons, N. A. Ran and A. L. Briseno, J. Mater. Chem., 2011, 21, 1329–1337 RSC.
- K. Takimiya, H. Ebata, K. Sakamoto, T. Izawa, T. Otsubo and Y. Kunugi, J. Am. Chem. Soc., 2006, 128, 12604–12605 CrossRef CAS PubMed.
- H. Ebata, T. Izawa, E. Miyazaki, K. Takimiya, M. Ikeda, H. Kuwabara and T. Yui, J. Am. Chem. Soc., 2007, 129, 15732–15733 CrossRef CAS PubMed.
- H. Minemawari, T. Yamada, H. Matsui, J. Tsutsumi, S. Haas, R. Chiba, R. Kumai and T. Hasegawa, Nature, 2011, 475, 364–367 CrossRef CAS PubMed.
- K. Takimiya, S. Shinamura, I. Osaka and E. Miyazaki, Adv. Mater., 2011, 23, 4347–4370 CrossRef CAS PubMed.
- A. Y. Amin, A. Khassanov, K. Reuter, T. Meyer-Friedrichsen and M. Halik, J. Am. Chem. Soc., 2012, 134, 16548–16550 CrossRef CAS PubMed.
- T. Schmaltz, A. Y. Amin, A. Khassanov, T. Meyer-Friedrichsen, H.-G. G. Steinrück, A. Magerl, J. J. Segura, K. Voitchovsky, F. Stellacci and M. Halik, Adv. Mater., 2013, 25, 4511–4514 CrossRef CAS PubMed.
- V. S. Vyas, R. Gutzler, J. Nuss, K. Kern and B. V. Lotsch, CrystEngComm, 2014, 16, 7389–7392 RSC.
- H. Iino, T. Usui and J.-I. Hanna, Nat. Commun., 2015, 6, 6828 CrossRef CAS PubMed.
- G. Schweicher, V. Lemaur, C. Niebel, C. Ruzié, Y. Diao, O. Goto, W. Lee, Y. Kim, J. Arlin, J. Karpinska, A. R. Kennedy, S. R. Parkin, Y. Olivier, S. C. B. B. Mannsfeld, J. Cornil, Y. H. Geerts and Z. Bao, Adv. Mater., 2015, 27, 3066–3072 CrossRef CAS PubMed.
- J.-M. Cho, T. Higashino and T. Mori, Appl. Phys. Lett., 2015, 106, 193303 CrossRef.
- C. Niebel, Y. Kim, C. Ruzié, J. Karpinska, B. Chattopadhyay, G. Schweicher, A. Richard, V. Lemaur, Y. Olivier, J. Cornil, A. R. Kennedy, Y. Diao, W.-Y. Y. Lee, S. Mannsfeld, Z. Bao and Y. H. Geerts, J. Mater. Chem. C, 2015, 3, 674–685 RSC.
- Y. He, M. Sezen, D. Zhang, A. Li, L. Yan, H. Yu, C. He, O. Goto, Y.-L. L. Loo and H. Meng, Adv. Electron. Mater., 2016, 2, 1600179 CrossRef.
- K. He, W. Li, H. Tian, J. Zhang, D. Yan, Y. Geng and F. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 35427–35436 CrossRef CAS PubMed.
- S. Arai, S. Inoue, T. Hamai, R. Kumai and T. Hasegawa, Adv. Mater., 2018, 30, 1707256 CrossRef PubMed.
- T. Higashino, A. Ueda and H. Mori, New J. Chem., 2019, 43, 884–892 RSC.
- T. Kadoya, S. Mano, A. Hori, K. Tahara, K. Sugimoto, K. Kubo, M. Abe, H. Tajima and J. Yamada, Org. Electron., 2020, 78, 105570 CrossRef CAS.
- S. Inoue, K. Nikaido, T. Higashino, S. Arai, M. Tanaka, R. Kumai, S. Tsuzuki, S. Horiuchi, H. Sugiyama, Y. Segawa, K. Takaba, S. Maki-Yonekura, K. Yonekura and T. Hasegawa, Chem. Mater., 2022, 34, 72–83 CrossRef CAS.
- K. Nikaido, S. Inoue, R. Kumai, T. Higashino, S. Matsuoka, S. Arai and T. Hasegawa, Adv. Mater. Interfaces, 2022, 9, 2201789 CrossRef CAS.
- R. Akai, K. Oka, S. Dekura, H. Mori and N. Tohnai, Bull. Chem. Soc. Jpn., 2022, 95, 1178–1182 CrossRef CAS.
- T. Yasuda, T. Doto, K. Fujita and T. Tsutsui, Mol. Cryst. Liq. Cryst., 2006, 444, 219–224 CrossRef CAS.
- T. Kadoya, M. Ashizawa, T. Higashino, T. Kawamoto, S. Kumeta, H. Matsumoto and T. Mori, Phys. Chem. Chem. Phys., 2013, 15, 17818–17822 RSC.
- Y. Kiyota, T. Kadoya, K. Yamamoto, K. Iijima, T. Higashino, T. Kawamoto, K. Takimiya and T. Mori, J. Am. Chem. Soc., 2016, 138, 3920–3925 CrossRef CAS PubMed.
- T. Higashino, T. Kadoya, S. Kumeta, K. Kurata, T. Kawamoto and T. Mori, Eur. J. Inorg. Chem., 2014, 3895–3898 CrossRef CAS.
- T. Kadoya, R. Oki, Y. Kiyota, Y. Koyama, T. Higashino, K. Kubo, T. Mori and J. Yamada, J. Phys. Chem. C, 2019, 123, 5216–5221 CrossRef CAS.
- E. M. Engler and V. V. Patel, J. Am. Chem. Soc., 1974, 96, 7376–7378 CrossRef CAS.
- S. Etemad, Phys. Rev. B, 1976, 13, 2254–2261 CrossRef CAS.
- D. Jérome, A. Mazaud, M. Ribault and K. Bechgaard, J. Phys. Lett., 1980, 41, 95–98 CrossRef.
- R. L. Greene and E. M. Engler, Phys. Rev. Lett., 1980, 45, 1587–1590 CrossRef CAS.
- M. D. Mays, R. D. McCullough, D. O. Cowan, T. O. Poehler, W. A. Bryden and T. J. Kistenmacher, Solid State Commun., 1988, 65, 1089–1092 CrossRef CAS.
- T. Higashino, A. Ueda, J. Yoshida and H. Mori, Chem. Commun., 2017, 53, 3426–3429 RSC.
- J. Cornil, D. Beljonne and J. L. Brédas, J. Chem. Phys., 1995, 103, 842–849 CrossRef CAS.
- D. Beljonne, J. Cornil, R. H. Friend, R. A. J. Janssen and J. L. Brédas, J. Am. Chem. Soc., 1996, 118, 6453–6461 CrossRef CAS.
- T. Okamoto, K. Kudoh, A. Wakamiya and S. Yamaguchi, Chem. – Eur. J., 2007, 13, 548–556 CrossRef CAS PubMed.
- S. S. Zade, N. Zamoshchik and M. Bendikov, Chem. – Eur. J., 2009, 15, 8613–8624 CrossRef CAS PubMed.
- H. Méndez, G. Heimel, A. Opitz, K. Sauer, P. Barkowski, M. Oehzelt, J. Soeda, T. Okamoto, J. Takeya, J. B. Arlin, J. Y. Balandier, Y. Geerts, N. Koch and I. Salzmann, Angew. Chem., Int. Ed., 2013, 52, 7751–7755 CrossRef PubMed.
- J. Tsutsumi, S. Matsuoka, S. Inoue, H. Minemawari, T. Yamada and T. Hasegawa, J. Mater. Chem. C, 2015, 3, 1976–1981 RSC.
- Y. Shibata, J. Tsutsumi, S. Matsuoka, K. Matsubara, Y. Yoshida, M. Chikamatsu and T. Hasegawa, Appl. Phys. Lett., 2015, 106, 143303 CrossRef.
- Y. Shibata, J. Tsutsumi, S. Matsuoka, H. Minemawari, S. Arai, R. Kumai and T. Hasegawa, Adv. Electron. Mater., 2017, 3, 1700097 CrossRef.
- H. Koike, J. Tsutsumi, S. Matsuoka, K. Sato, T. Hasegawa and K. Kanai, Org. Electron., 2016, 39, 184–190 CrossRef CAS.
- L. Zhu, H. Geng, Y. Yi and Z. Wei, Phys. Chem. Chem. Phys., 2017, 19, 4418–4425 RSC.
- K. Iijima, R. Sanada, D. Yoo, R. Sato, T. Kawamoto and T. Mori, ACS Appl. Mater. Interfaces, 2018, 10, 10262–10269 CrossRef CAS PubMed.
- R. Sato, T. Kawamoto and T. Mori, J. Mater. Chem. C, 2019, 7, 567–577 RSC.
- H. Geng, L. Zhu, Y. Yi, D. Zhu and Z. Shuai, Chem. Mater., 2019, 31, 6424–6434 CrossRef CAS.
- Q. Wei, L. Liu, S. Xiong, X. Zhang, W. Deng, X. Zhang and J. Jie, J. Phys. Chem. Lett., 2020, 11, 359–365 CrossRef CAS PubMed.
- S. Takamaru, J. Hanna and H. Iino, Appl. Phys. Express, 2022, 15, 031001 CrossRef.
- R. Sato, M. Dogishi, T. Higashino, T. Kadoya, T. Kawamoto and T. Mori, J. Phys. Chem. C, 2017, 121, 6561–6568 CrossRef CAS.
- N. Castagnetti, A. Girlando, M. Masino, C. Rizzoli and C. Rovira, Cryst. Growth Des., 2017, 17, 6255–6261 CrossRef CAS.
- R. R. Dasari, X. Wang, R. A. Wiscons, H. F. Haneef, A. Ashokan, Y. Zhang, M. S. Fonari, S. Barlow, V. Coropceanu, T. V. Timofeeva, O. D. Jurchescu, J. Brédas, A. J. Matzger and S. R. Marder, Adv. Funct. Mater., 2019, 29, 1904858 CrossRef CAS.
- A. Ashokan, C. Hanson, N. Corbin, J.-L. Brédas and V. Coropceanu, Mater. Chem. Front., 2020, 4, 3623–3631 RSC.
- J. Zhang, J. Jin, H. Xu, Q. Zhang and W. Huang, J. Mater. Chem. C, 2018, 6, 3485–3498 RSC.
- A. A. Dar and S. Rashid, CrystEngComm, 2021, 23, 8007–8026 RSC.
- W. Wang, L. Luo, P. Sheng, J. Zhang and Q. Zhang, Chem. – Eur. J., 2021, 27, 464–490 CrossRef CAS PubMed.
- T. Higashino, M. Dogishi, T. Kadoya, R. Sato, T. Kawamoto and T. Mori, J. Mater. Chem. C, 2016, 4, 5981–5987 RSC.
- L. Fijahi, T. Salzillo, A. Tamayo, M. Bardini, C. Ruzié, C. Quarti, D. Beljonne, S. D'Agostino, Y. H. Geerts and M. Mas-Torrent, J. Mater. Chem. C, 2022, 10, 7319–7328 RSC.
- S. Matsuoka, K. Ogawa, R. Ono, K. Nikaido, S. Inoue, T. Higashino, M. Tanaka, J. Tsutsumi, R. Kondo, R. Kumai, S. Tsuzuki, S. Arai and T. Hasegawa, J. Mater. Chem. C, 2022, 10, 16471–16479 RSC.
- M. Mizuno, A. F. Garito and M. P. Cava, J. Chem. Soc., Chem. Commun., 1978, 18–19 RSC.
- G. Saito, T. Enoki, K. Toriumi and H. Inokuchi, Solid State Commun., 1982, 42, 557–560 CrossRef CAS.
- T. Mori, A. Kobayashi, Y. Sasaki, H. Kobayashi, G. Saito and H. Inokuchi, Chem. Lett., 1982, 11, 1963–1966 CrossRef.
- S. S. P. Parkin, E. M. Engler, R. R. Schumaker, R. Lagier, V. Y. Lee, J. C. Scott and R. L. Greene, Phys. Rev. Lett., 1983, 50, 270–273 CrossRef CAS.
- E. B. Yagubskii, I. F. Shchegolev, V. N. Laukhin, P. A. Kononovich, M. V. Karatsovnik, A. V. Zvarykina and L. I. Buravov, JETP Lett., 1984, 39, 12–16 CAS.
- T. Mori, A. Kobayashi, Y. Sasaki, H. Kobayashi, G. Saito and H. Inokuchi, Chem. Lett., 1984, 13, 957–960 CrossRef.
- T. Mori, A. Kobayashi, Y. Sasaki, H. Kobayashi, G. Saito and H. Inokuchi, Bull. Chem. Soc. Jpn., 1984, 57, 627–633 CrossRef CAS.
- H. Kobayashi, R. Kato, A. Kobayashi, Y. Nishio, K. Kajita and W. Sasaki, Chem. Lett., 1986, 15, 789–792 CrossRef.
- H. Kobayashi, R. Kato, A. Kobayashi, Y. Nishio, K. Kajita and W. Sasaki, Chem. Lett., 1986, 15, 833–836 CrossRef.
- R. Kato, H. Kobayashi, A. Kobayashi, S. Moriyama, Y. Nishio, K. Kajita and W. Sasaki, Chem. Lett., 1987, 16, 507–510 CrossRef.
- A. Kobayashi, R. Kato, H. Kobayashi, S. Moriyama, Y. Nishio, K. Kajita and W. Sasaki, Chem. Lett., 1987, 16, 459–462 CrossRef.
- C. Hotta, J. Phys. Soc. Jpn., 2003, 72, 840–853 CrossRef CAS.
- T. Mori, Bull. Chem. Soc. Jpn., 1998, 71, 2509–2526 CrossRef CAS.
- T. Mori, H. Mori and S. Tanaka, Bull. Chem. Soc. Jpn., 1999, 72, 179–197 CrossRef CAS.
- T. Mori, Bull. Chem. Soc. Jpn., 1999, 72, 2011–2027 CrossRef CAS.
- H. Kobayashi, A. Kobayashi, G. Saito, T. Enoki and H. Inokuchi, J. Am. Chem. Soc., 1983, 105, 297–298 CrossRef CAS.
- H. Kobayashi, T. Mori, R. Kato, A. Kobayashi, Y. Sasaki, G. Saito and H. Inokuchi, Chem. Lett., 1983, 12, 581–584 CrossRef.
- H. Kobayashi, R. Kato, T. Mori, A. Kobayashi, Y. Sasaki, G. Saito and H. Inokuchi, Chem. Lett., 1983, 12, 759–762 CrossRef.
- H. Kobayashi, R. Kato, T. Mori, A. Kobayashi, Y. Sasaki, G. Saito, T. Enoki and H. Inokuchi, Mol. Cryst. Liq. Cryst., 1984, 107, 33–43 CrossRef CAS.
- A. Miyazaki, T. Enoki and G. Saito, Synth. Met., 1995, 70, 793–794 CrossRef CAS.
- H. Mori, J. Phys. Soc. Jpn., 2006, 75, 051003 CrossRef.
- G. Saito and Y. Yoshida, Chem. Rec., 2011, 11, 124–145 CrossRef CAS PubMed.
- H. Urayama, H. Yamochi, G. Saito, K. Nozawa, T. Sugano, M. Kinoshita, S. Sato, K. Oshima, A. Kawamoto and J. Tanaka, Chem. Lett., 1988, 17, 55–58 CrossRef.
- A. M. Kini, U. Geiser, H. H. Wang, K. D. Carlson, J. M. Williams, W. K. Kwok, K. G. Vandervoort, J. E. Thompson and D. L. Stupka, Inorg. Chem., 1990, 29, 2555–2557 CrossRef CAS.
- H. Yamochi, T. Nakamura, T. Komatso, N. Matsukawa, T. Inoue, G. Saito, T. Mori, M. Kusunoki and K. Sakaguchi, Solid State Commun., 1992, 82, 101–105 CrossRef CAS.
- J. A. Schlueter, K. D. Carlson, U. Geiser, H. H. Wang, J. M. Williams, W.-K. Kwok, J. A. Fendrich, U. Welp, P. M. Keane, J. D. Dudek, A. S. Komosa, D. Naumann, T. Roy, J. E. Schirber, W. R. Bayless and B. Dodrill, Phys. C, 1994, 233, 379–386 CrossRef CAS.
- H. Taniguchi, M. Miyashita, K. Uchiyama, K. Satoh, N. Môri, H. Okamoto, K. Miyagawa, K. Kanoda, M. Hedo and Y. Uwatoko, J. Phys. Soc. Jpn., 2003, 72, 468–471 CrossRef CAS.
- T. Kawamoto, T. Mori, A. Nakao, Y. Murakami and J. A. Schlueter, J. Phys. Soc. Jpn., 2012, 81, 023705 CrossRef.
- F. Sawano, I. Terasaki, H. Mori, T. Mori, M. Watanabe, N. Ikeda, Y. Nogami and Y. Noda, Nature, 2005, 437, 522–524 CrossRef CAS PubMed.
- T. Mori, I. Terasaki and H. Mori, J. Mater. Chem., 2007, 17, 4343 RSC.
- F. Kagawa, T. Sato, K. Miyagawa, K. Kanoda, Y. Tokura, K. Kobayashi, R. Kumai and Y. Murakami, Nat. Phys., 2013, 9, 419–422 Search PubMed.
- T. Mori, Phys. Rev. B, 2016, 93, 245104 CrossRef.
- S. Sasaki, K. Hashimoto, R. Kobayashi, K. Itoh, S. Iguchi, Y. Nishio, Y. Ikemoto, T. Moriwaki, N. Yoneyama, M. Watanabe, A. Ueda, H. Mori, K. Kobayashi, R. Kumai, Y. Murakami, J. Müller and T. Sasaki, Science, 2017, 357, 1381–1385 CrossRef CAS PubMed.
- H. Ito, T. Ishiguro, M. Kubota and G. Saito, J. Phys. Soc. Jpn., 1996, 65, 2987–2993 CrossRef CAS.
- K. Kanoda, Hyperfine Interact., 1997, 104, 235–249 CrossRef CAS.
- S. Lefebvre, P. Wzietek, S. Brown, C. Bourbonnais, D. Jérome, C. Mézière, M. Fourmigué and P. Batail, Phys. Rev. Lett., 2000, 85, 5420–5423 CrossRef CAS PubMed.
- H. Mori, M. Kamiya, M. Haemori, H. Suzuki, S. Tanaka, Y. Nishio, K. Kajita and H. Moriyama, J. Am. Chem. Soc., 2002, 124, 1251–1260 CrossRef CAS PubMed.
- T. Kawamoto, K. Kurata and T. Mori, J. Phys. Soc. Jpn., 2018, 87, 083703 CrossRef.
- K. Kajita, T. Ojiro, H. Fujii, Y. Nishio, H. Kobayashi, A. Kobayashi and R. Kato, J. Phys. Soc. Jpn., 1992, 61, 23–26 CrossRef CAS.
- N. Tajima, M. Tamura, Y. Nishio, K. Kajita and Y. Iye, J. Phys. Soc. Jpn., 2000, 69, 543–551 CrossRef CAS.
- S. Katayama, A. Kobayashi and Y. Suzumura, J. Phys. Soc. Jpn., 2006, 75, 054705 CrossRef.
- N. Tajima, S. Sugawara, R. Kato, Y. Nishio and K. Kajita, Phys. Rev. Lett., 2009, 102, 176403 CrossRef PubMed.
- T. Mori, J. Phys. Soc. Jpn., 2013, 82, 034712 CrossRef.
- K. Kajita, Y. Nishio, N. Tajima, Y. Suzumura and A. Kobayashi, J. Phys. Soc. Jpn., 2014, 83, 072002 CrossRef.
- R. Kato, H. Cui, T. Tsumuraya, T. Miyazaki and Y. Suzumura, J. Am. Chem. Soc., 2017, 139, 1770–1773 CrossRef CAS PubMed.
- M. Tamura and R. Kato, J. Phys.: Condens. Matter, 2002, 14, L729–L734 CrossRef CAS.
- Y. Shimizu, K. Miyagawa, K. Kanoda, M. Maesato and G. Saito, Phys. Rev. Lett., 2003, 91, 107001 CrossRef CAS PubMed.
- T. Itou, A. Oyamada, S. Maegawa, M. Tamura and R. Kato, Phys. Rev. B, 2008, 77, 104413 CrossRef.
- T. Isono, H. Kamo, A. Ueda, K. Takahashi, M. Kimata, H. Tajima, S. Tsuchiya, T. Terashima, S. Uji and H. Mori, Phys. Rev. Lett., 2014, 112, 177201 CrossRef PubMed.
- Y. Shimizu, T. Hiramatsu, M. Maesato, A. Otsuka, H. Yamochi, A. Ono, M. Itoh, M. Yoshida, M. Takigawa, Y. Yoshida and G. Saito, Phys. Rev. Lett., 2016, 117, 107203 CrossRef PubMed.
- T. Hiramatsu, Y. Yoshida, G. Saito, A. Otsuka, H. Yamochi, M. Maesato, Y. Shimizu, H. Ito, Y. Nakamura, H. Kishida, M. Watanabe and R. Kumai, Bull. Chem. Soc. Jpn., 2017, 90, 1073–1082 CrossRef CAS.
- M. Shimozawa, K. Hashimoto, A. Ueda, Y. Suzuki, K. Sugii, S. Yamada, Y. Imai, R. Kobayashi, K. Itoh, S. Iguchi, M. Naka, S. Ishihara, H. Mori, T. Sasaki and M. Yamashita, Nat. Commun., 2017, 8, 1821 CrossRef CAS PubMed.
- Y. Zhou, K. Kanoda and T.-K. Ng, Rev. Mod. Phys., 2017, 89, 025003 CrossRef.
- T. Kadoya, S. Sugiura, K. Tahara, T. Higashino, K. Kubo, T. Sasaki, K. Takimiya and J. Yamada, CrystEngComm, 2020, 22, 5949–5953 RSC.
- C. Wang, H. Nakamura, H. Sugino and K. Takimiya, J. Mater. Chem. C, 2018, 6, 3604–3612 RSC.
- V. Podzorov, S. E. Sysoev, E. Loginova, V. M. Pudalov and M. E. Gershenson, Appl. Phys. Lett., 2003, 83, 3504–3506 CrossRef CAS.
- V. Podzorov, E. Menard, A. Borissov, V. Kiryukhin, J. A. Rogers and M. E. Gershenson, Phys. Rev. Lett., 2004, 93, 086602 CrossRef CAS PubMed.
- J. Takeya, M. Yamagishi, Y. Tominari, R. Hirahara, Y. Nakazawa, T. Nishikawa, T. Kawase, T. Shimoda and S. Ogawa, Appl. Phys. Lett., 2007, 90, 102120 CrossRef.
- C. Reese and Z. Bao, Adv. Mater., 2007, 19, 4535–4538 CrossRef CAS.
- T. Kawamoto, K. Kurata, T. Mori and R. Kumai, J. Phys. Soc. Jpn., 2021, 90, 103703 CrossRef.
- T. Kadoya, S. Sugiura, T. Higashino, K. Tahara, K. Kubo, T. Sasaki, K. Takimiya and J. Yamada, Crystals, 2021, 11, 868 CrossRef CAS.
- T. Kadoya, M. Shishido, S. Sugiura, T. Higashino, K. Tahara, K. Kubo, T. Sasaki and J. Yamada, Chem. Lett., 2022, 51, 683–686 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |