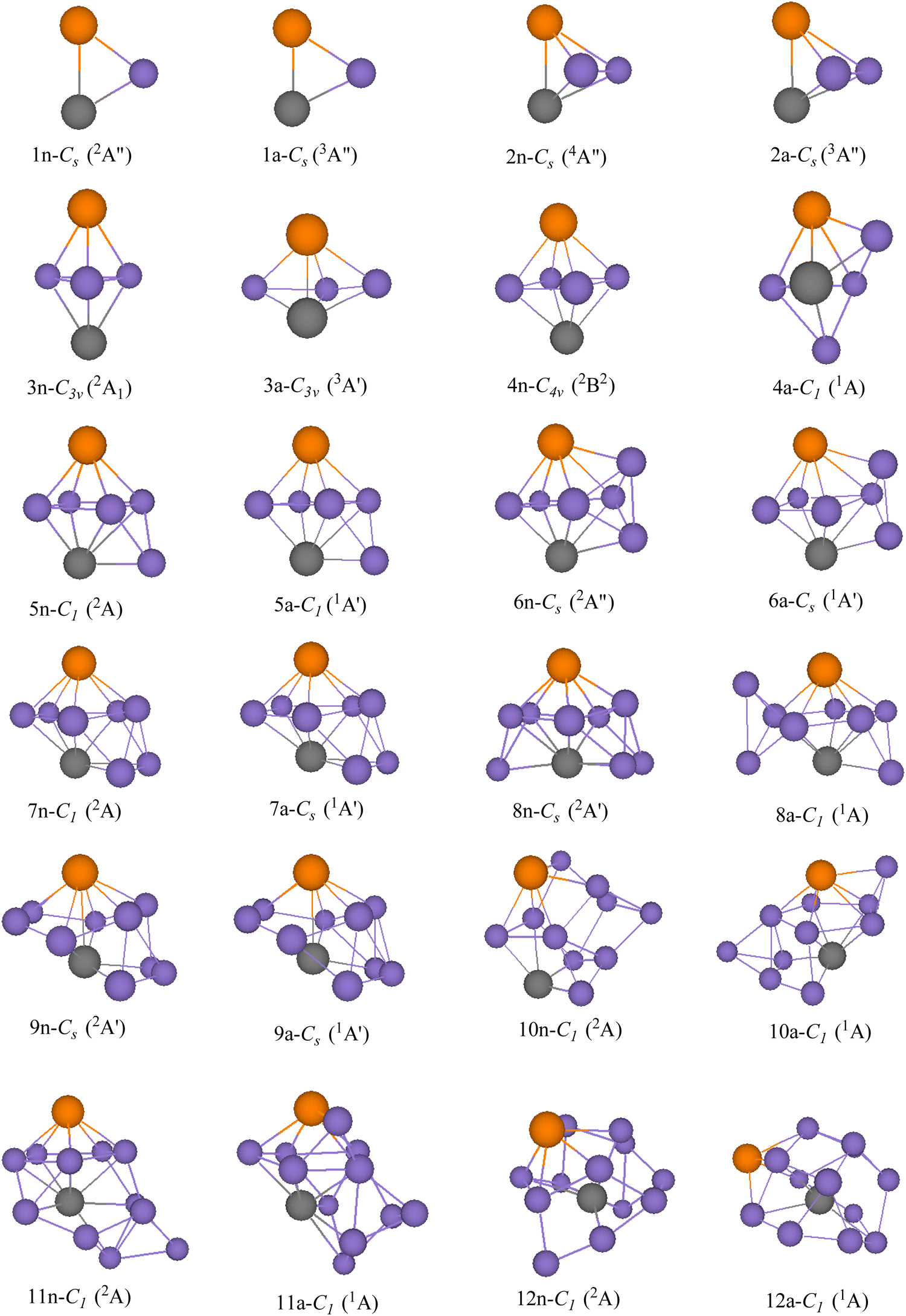Structural evolution and electronic properties of neutral and anionic TiASil (A = Sc, Ti; l ≤ 12): relatively stable TiASi4 as a structural unit†
Received
21st October 2022
, Accepted 24th November 2022
First published on 8th December 2022
Abstract
Simulated photoelectron spectroscopy was conducted to investigate the structural evolution and electronic properties of TiASil (A = Sc, Ti; l ≤ 12) clusters and their anions via the Perdew–Burke–Enzerhof scheme and extensive cluster search using the ABCluster software. The results revealed that the ground-state structures of the TiASil (A = Sc, Ti) clusters generally exhibited similar configurations except for the Ti2Si3, ScTiSi3, and TiScSi10 clusters. Furthermore, the TiASil clusters exhibited an adsorptive evolution pattern, and the TiASi4 unit was considered the basic constituent framework of the structure, excluding several distortions and minor changes. With the increase in the cluster size, the lowest-energy structures varied from the exohedral to the cage structures of the single-metal atom at the center. Regarding the second energy difference data, the neutral TiASi4 clusters exhibited better stability among the neutral and anionic TiASil (A = Sc, Ti; l ≤ 12) clusters. Furthermore, the chemical bonding in the TiASi4 clusters was analyzed by molecular orbitals (MOs), highest occupied MO–lowest unoccupied MO gaps, and adaptive natural density partitioning analyses for the best Ti2Si4 cluster especially, and the results were combined with the natural population analysis data. The hybridization of the spd orbital of the metal atoms, eight localized bonds, and four delocalized bonds may primarily account for the relative stabilities of the neutral, square bipyramidal structure of Ti2Si4. Thus, the TiASi4 clusters may be assembled as the basic units of silicon-based semiconductor clusters of large-size neutral and anionic TiASil.
1. Introduction
Numerous studies have confirmed that the stability of bare silicon clusters can be improved by doping them with metal atoms.1–6 Particularly, transition-metal-doped silicon compounds can be good candidates for developing a framework of assembly materials; they also exhibit optical, electrical, and catalytic properties.7–15 Scandium and titanium are the first two elements in the transition-metal group, and their extranuclear, valence electron structures are represented by 3d14s2 and 3d24s2, respectively. Thus, their largely inactive 3d orbitals can combine with the dangling bonds of bare silicon clusters to form more stable structures than those of the bare clusters and exhibit several unique physicochemical properties.16–22 Therefore, scandium- and titanium-doped silicon clusters have been extensively researched theoretically and experimentally. Concurrently, numerous computational methods, including density functional theory (DFT), have been adopted to explore their structures and properties that can accurately predict target molecules mainly because of the complexities of selecting excited states, the consideration of the spin–orbit coupling, the strong relativistic effects, and the electron correlation.23 The B3LYP functional, B3PW91 methods, CASSCF/CASPT2 methods, Perdew–Burke–Enzerhof (PBE) and mPW2PLYP/aug-cc-pVTZ schemes,24,25 and ccCA-TM, G4, and G4(MP2) methods26,27 were employed in previous study. For some time, researchers have theoretically and experimentally focused on the geometries and properties of single-metal-atom-doped silicon clusters.28,29 For example, Nguyen and coworkers30 investigated the ground-state structures of neutral and anionic ScSi2 and ScSi4–6via the B3LYP functional and CASSCF/CASPT2 methods. The predicted lowest-energy structures were consistent with those predicted in our previous research employing high-level combination methods.26,27 Previous studies employing diverse calculation methods indicated that the anionic ScSi16− cluster exhibited higher stability than its neighbors; moreover, it is predicted to be a magic cluster with better thermodynamics.31–33 Employing DFT and other appropriate methods, the structures and stabilities, as well as physicochemical properties of titanium–silicon clusters, have been theoretically calculated and reported.34–47 For instance, the TiSin cluster system links to the configurations of the encapsulated structures as the cluster size increases. Moreover, numerous studies confirmed that TiSi12 and TiSi16 can exhibit higher relative stabilities than their neighboring clusters,41–46 especially TiSi16 possessed a highly symmetrical stability due to its large HOMO–LUMO gap.47–49 The construction of a building block of a self-assembly nanomaterial is among the main goals of exploring candidates for achieving a stable structure of transition-metal–silicon compounds. Hence, it can be the key to designing and predicting the geometries and electronic structures of multiple metal atoms that are doped into the silicon clusters. The adiabatic electron affinities (AEAs) and vertical detachment energies (VDEs) of Sc2Sin− (n = 2–6) and Sc2Sin (n ≤ 8) clusters have been studied via B3LYP and the composite method of G4(MP2), respectively.50 Employing the generic algorithm coupled with the DFT method, Zhang et al.51 determined the structures and properties of Ti2Sin (n = 3–24) clusters, revealing that the clusters exhibited higher stabilities than the neighboring ones at sizes n = 4, 13, and 21. Furthermore, the PBE and HSE06 functionals revealed that the Ti2Si15 cluster comprised closed electronic shells and might be ideal building blocks of nanomaterials owing to its high stability.52
Although there are several reports on small A2Sil (A = Sc, Ti) neutral clusters, there are only a few systematic reports on their calculation methods, ground-state structures, and electronic properties. Furthermore, identical or different metal atoms that are doped into bare silicon clusters and already applied in the microelectronics industry53 exhibit novel application prospects; for example, LaScSi and Y5Si3 with perfect catalytic stabilities have been applied to ammonia synthesis,13 beyond that MgSc for hydrogen storage materials.54 This calculation is aimed at probing neutral and anionic clusters’ evolution behavior and exploring clusters with higher relative stabilities. Overall, based on TiASil (A = Sc, Ti; l ≤ 12) clusters and their anions, scandium and titanium metal atoms with similar structures were employed as dopants in the bare silicon molecules at low-coordinated sites because both metals exhibit the same electron compositions, with one or two d-electrons.
2. Computational details
Gaussian09 software55 was employed to calculate all the geometries of TiASil (A = Sc, Ti; l ≤ 12) and their anions. The challenge of accurately determining the ground-state structure of clusters is well known. Pure silicon clusters that are doped with multiple transition-metal atoms exhibit several novel geometries, e.g., tubular, pearl-chain style, wheel-like, (V3Si1256 and Cr3Si1257)-stacked naphthalene-like, or dodecahedral structures, compared with those that are doped with single-metal atoms.58 Furthermore, the impossibility of completely determining the other structures on the potential surface is also well known. Fortunately, we employed an unprejudiced global optimization software, namely the ABCluster procedure59,60 with excellent conjectures, to determine the valid isomers of multi-metal–silicon clusters24,25,50 to predict the designs of the unknown structures in this study. The ABCluster procedure uses the “artificial bee colony” arithmetic to proceed efficiently with the global optimization. Thus, one may use the “isomer” components of the ABCluster software combined with Gaussian09 in order to seek out the lowest-energy structure. The “isomer” components will run to generate a geometry with the document of optfile and inp. Through this approach, ≥100 initial neutral or anionic geometries of TiASil (A = Sc, Ti; l ≤ 12) were established and competed. Furthermore, some additional structural designs were included as a supplementary search to reduce the risk of leaking the ground-state structure. First, we assumed that the lowest structures of the bare silicon clusters were replaced or assimilated by one or two metal atoms. Second, we calculated them separately when the ground states of the neutral compounds differed from those of the corresponding anion. Finally, several other reported ground-state structures of metal-atom-doped silicon or boron clusters61 were also designed and calculated; while exploring each structure, the spin multiplicities of 1–6 were assigned to their anionic or neutral species to join the n < 5 species, respectively. The detailed implementation of the entire calculation process is, as follows:
First, the initially unknown structures were optimized via the PBE method coupled with the mixed basis sets (LanL2DZ for the Ti/Sc atoms and 6-31G(d) for the silicon atoms). The following research revealed that the PBE scheme exhibited a competitive performance in predicting the ground-state structures and electronic properties of metal atoms, including transition- and rare-earth-metal-doped silicon clusters.62,63 Next, the PBE/Gen-A (Gen-A: the silicon atoms of the cc-pVTZ basis set,64 Ti/Sc metal atoms for full electronic basis set of def2-TZVP65) level is employed to calculate the low-lying clusters, as well as the harmonic vibrational frequencies. The computational geometries without virtual frequencies were determined as the local minimal site of the entire potential energy surface. Thereafter, the PBE/Gen-B (Gen-B: aug-cc-pVTZ basis set66 for the silicon and Ti/Sc atoms of def2-TZVP) scheme was adopted to calculate the single-point energies since the diffusion property was a considerable step for predicting the thermodynamic energies of the anionic clusters. Moreover, the V2 and Si2 dimers were calculated, and the results correlated excellently with the previous experimental report, further confirming the accuracy of the PBE/Gen-B method.62,63 Regarding the global minimum structures (GMSs) of the Ti2 dimer, the electronic state and bond distance of Ti–Ti were 3Σg and 1.90 Å, respectively, corresponding with 3Σg and 1.94 Å, which had been obtained experimentally.67 It is well known that the PBE/Gen-B method predicted a silicon system that was doped with two 3d metal atoms with high efficiency and low computational cost.
3. Theoretical results and discussion
3.1 GMSs of the neutral and anionic clusters
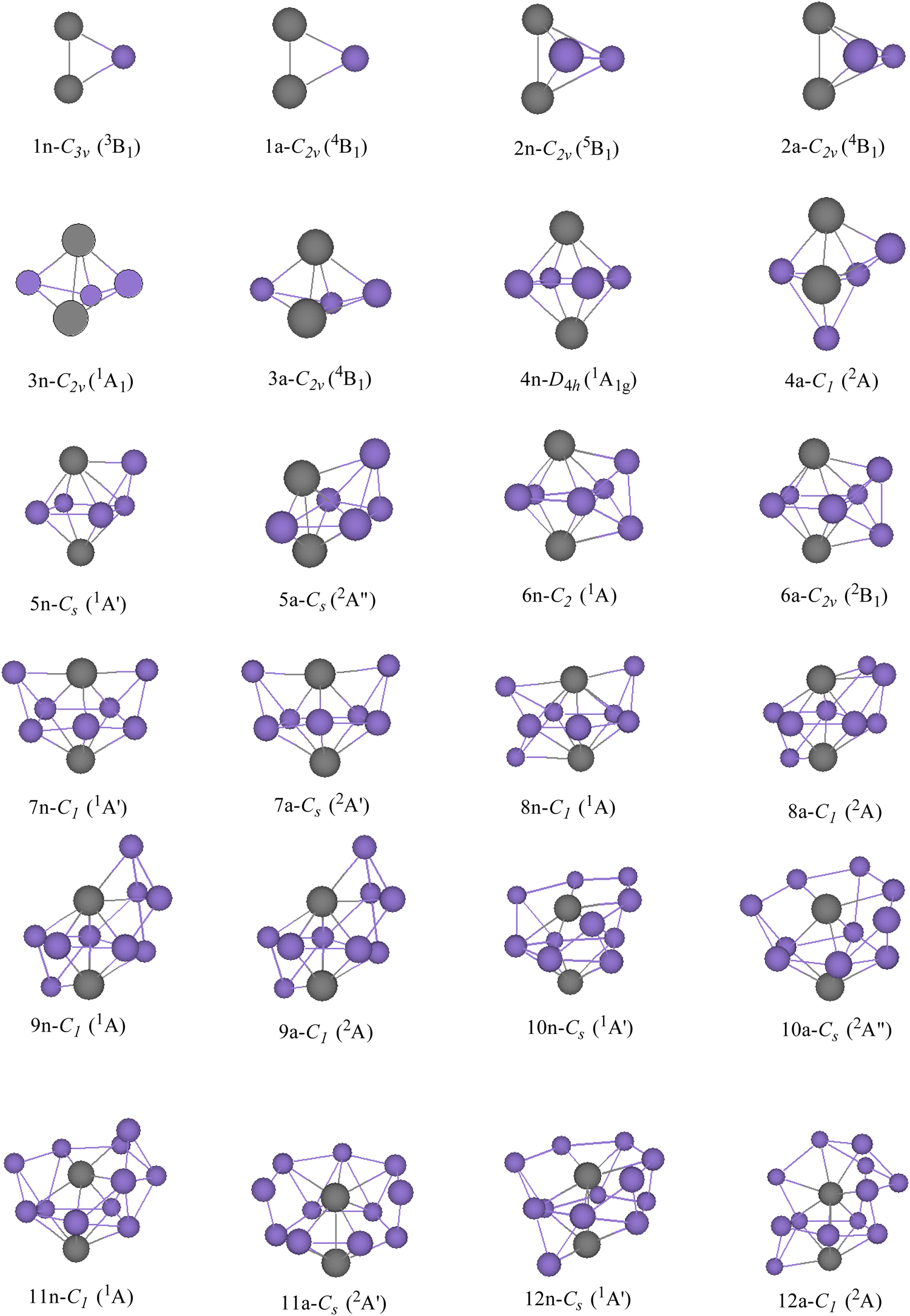
GMSs, as well as the competitive metastable geometries of TiASil (A = Sc, Ti; l ≤ 12) and their anions, were determined by the PBE/Gen-B scheme (Fig. 1 and 2 and Fig. S1–S2, ESI†). GMSs of TiASil (A = Sc, Ti; l < 7), as well as its anionic geometries, exhibited a similar shape except for the neutral Ti2Si3 and ScTiSi3 clusters. GMSs of neutral ScTiSi3 exhibited a C3v symmetry and a 2A1 electronic state, which can be regarded as the swapping of a scandium atom of the neutral or anionic GMS of Sc2Si3 with a titanium atom.50 Notably, the anionic or neutral TiASil (A = Sc, Ti, l < 5) exhibited triplet and quartet electronic states, respectively. The anionic or neutral GMSs of the Ti2Si7 clusters corresponded to the absorption of a titanium atom by the bottom-left position of the GMSs of Ti2Si6. Moreover, two additional silicon atoms were located on either side of one titanium atom. Although the GMSs of TiScSi7 were inconsistent with that of Ti2Si7, the two silicon atoms preferentially combined with the titanium atoms than with the scandium one. Fig. 1 and 2 show that more silicon atoms (the absorbing silicon atoms) were always located next to one titanium atom than to the scandium atoms, starting from the neutral or anionic GMSs of TiASil (A = Sc, Ti; l = 8). This phenomenon might mainly occur because the valence electrons of the titanium atoms are more than those of scandium. The neutral and anionic TiASil (A = Sc,Ti; l > 8) GMSs exhibited similar configurations except for the TiScSi10 cluster. Particularly, the TiASi12 cluster exhibited a cage structure in which a titanium atom was surrounded by many silicon atoms and the geometry of anionic TiScSi12 was viewed by the replacement of the titanium atom by the scandium atom of the GMSs of the corresponding Ti2Si12. TiASil (A = Sc,Ti; l ≤ 12) adopted the absorption-type evolution pattern. Particularly, the GMSs of the TiASil (l ≤ 12) clusters might have gained from the GMSs of the TiASil–1 clusters by absorbing an increasing number of silicon atoms, whether neutral or anionic clusters. Regarding Ti2Sil, if a scandium atom substituted one titanium atom, the GMSs of TiScSil would exhibit occasional deformation. These growing characteristics might become the universal feature of one or more transition-metal atoms that are doped in a silicon cluster.50,62,63 The neutral GMSs of TiASi4 was key to the evolution of the Ti2Sil clusters or TiScSil structures. This is because the basic 1–4–1 construction (1 metal, 4 silicons, 1 metal atom) can be part of a unit despite the presence of some distortions and minor changes. Thus, TiASi4 was assumed as a more appropriate building block for larger-size clusters, even new functional nanomaterials. These findings will facilitate for determination of the GMSs of large-scale clusters of more transition-metal-atom-doped silicon clusters.
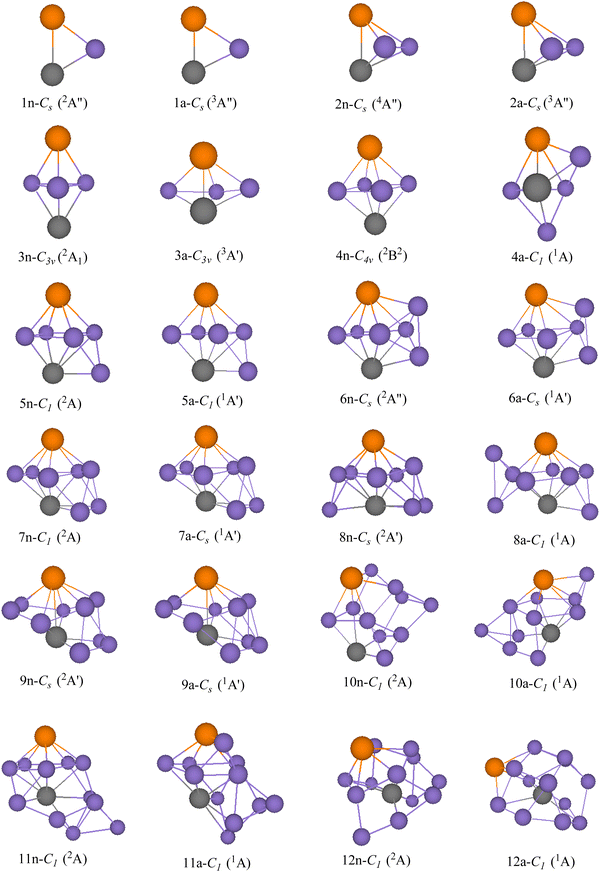 |
| | Fig. 1 GMSs of TiScSil (l ≤ 12) clusters and their anions. The number and purple ball are silicon atoms, n and a stand for neutral and anionic TiScSil, respectively. The orange balls represent scandium; the other gray balls represent titanium. The symmetry and electronic state are given below the structure for each size. | |
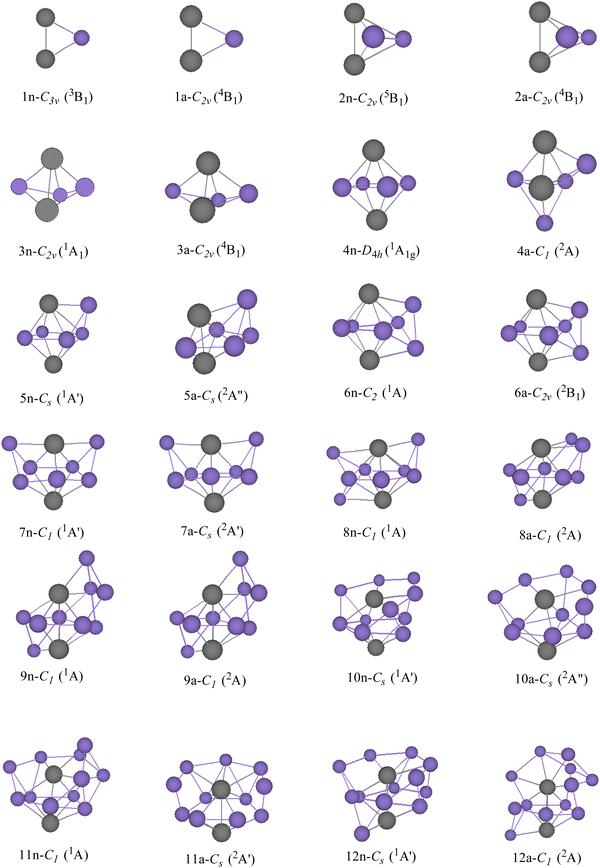 |
| | Fig. 2 GMSs of Ti2Sil (l ≤ 12) clusters and their anions. The number and purple ball are silicon atoms, n and a stand for neutral and anionic Ti2Sil, respectively. The gray balls represent titanium. The symmetry and electronic state are given below the structure for each size. | |
3.2 Simulated photoelectron spectroscopy (PES) of the anionic TiASil anionic (A = Sc, Ti; l ≤ 12) clusters
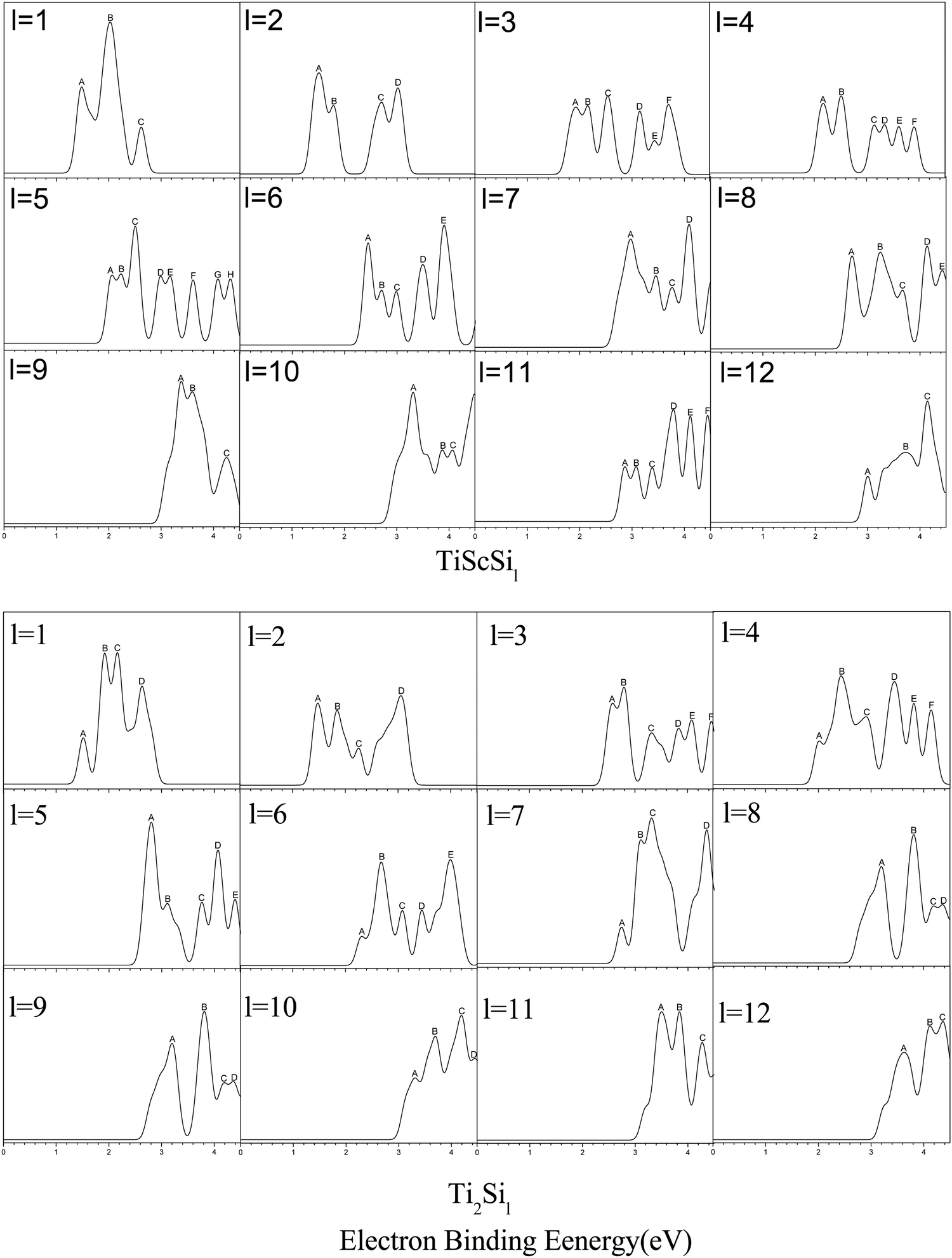
The research on the PES of anionic clusters is highly significant because of the close relationship between the GMSs and low-energy isomers of the clusters and the information and peaks of the corresponding PES diagram.68,69 Namely, PES might be among the most effective methods for verifying the anionic cluster structures, which still lack direct experimental determination methods. Hence, the PES of the anionic TiASil (A = Sc, Ti; l ≤ 12) clusters were simulated via the PBE/Gen-B method (Fig. 3). In the process of producing PES, the parameter of En was referred and defined by the formula of En = EHOMO−n−EHOMO according to the noted Koopmans’ law. The front peak in the position of the HOMO was located in the result of VDE, and additional peaks had the movement to more deep orbitals on the basis of values of En, just as were performed in other reports.50,62,63 The PES of the anionic clusters were calculated and drawn using a Gaussian full-width of 0.2 eV in order to obtain a more appropriate map scale, and within the limits of 4.5 eV because the range showed most of the peaks employing the Multiwfn software.70 The quantities and location of the peaks ranged from l = 1 to 4 for the anionic Ti2Sil and ScTiSil; their theoretical PES exhibited similar results. For instance, Ti2Si3 and ScTiSi3 exhibited six peaks (A–F), which were centered at 2.58, 2.79, 3.32, 3.83, 4.08, and 4.45 eV, and 1.93, 2.15, 2.54, 3.15, 3.43, and 3.70 eV, respectively. Only slight differences were observed in the shoulder and tailing peaks in the PES of Ti2Si1 (B and C peaks), ScTiSi2 (C and D peaks), ScTiSi4 (A and B peaks), and TiSi4 (A, B, and C peaks) clusters. Moreover, the number of peaks for the anionic ScTiSi5 reached its maximum, although three shoulder peaks (A and B, D and E, and G and H) were detected in all eight peaks in the PES drawing. For the TiASi6 (A = Sc, Ti) clusters, PES expressed similarities in their quantities and shapes except for the unresolved peaks of A, B, and C, as well as the additional peaks of D and E at 3.49, 3.9 eV for TiScSi6−, and 3.45 and 4.00 eV for Ti2Si6−, respectively. For the anionic TiScSi11− cluster, six distinct peaks were observed at 2.86, 3.08, 3.39, 3.79, 4.11, and 4.44 eV. The number of peaks ranged from l = 9, 10, and 12 for anionic Ti2Sil and ScTiSil; in the theoretical PES, it was 3 or 4. Particularly, the positions of the peaks were also similar in the TiASi9 (A = Sc, Ti) clusters. Table S1 (ESI†) presents the PES-determined locations of the peaks in the anionic TiASil (A = Sc, Ti; l ≤ 12) clusters. The similarities between the locations and quantities of the peaks of Ti2Sil and TiScSil might be related to their similar construction and valence electron configurations. There are currently no comparable experimental PES data for the anionic clusters. Hopefully, this will motivate the realization of the experimental data of PES of the clusters in the title.
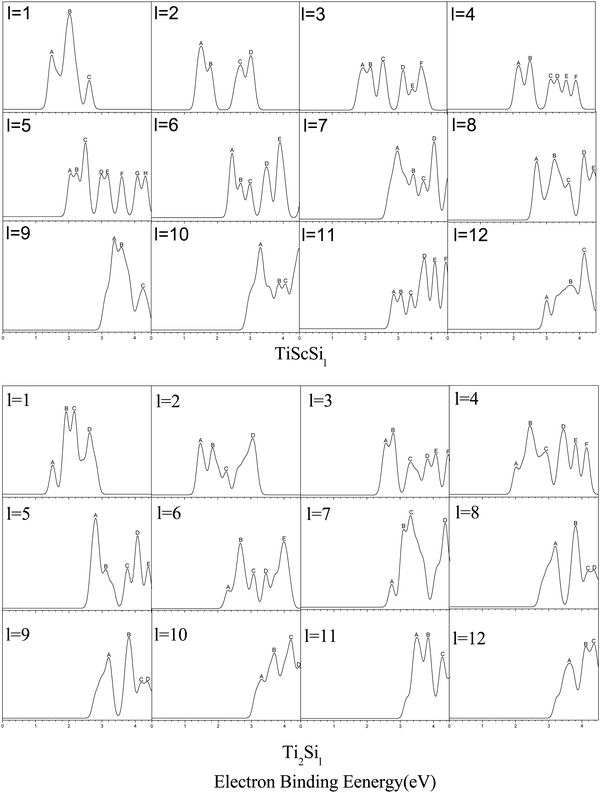 |
| | Fig. 3 Simulated PES spectra of GMSs of anionic TiASil (A = Sc, Ti, l ≤ 12) clusters. | |
3.3 VDEs and AEAs
Although no experimental values were compared with our theoretical values, the PBE/Gen-B//PBE/Gen-A exhibited good performance in predicting two transitional-metal-atom-doped silicon clusters.50,62 Furthermore, Table 1 presents the VDEs and AEAs data of the TiASil (A = Sc, Ti; l ≤ 12) molecules. The table shows that the overall trend of the AEAs and VDEs data increased with the increasing cluster size. There were larger VDEs data for the Ti2Sil clusters than for the TiScSil molecules, except for the Ti2Si4 and Ti2Si9 compounds. Notably, Ti2Si4 exhibited a lower value than its neighbors, indicating that it will gain less energy when the neutral GMSs of the Ti2Si4 compound loses an electron, whether it is VDEs or AEAs.
Table 1 Adiabatic electron affinities (AEAs) and vertical detachment energies (VDEs) for TiASiI (A = Sc, Ti, I ≤ 12) clusters. The values are in eV
| Clusters |
AEAs |
VDEs |
Clusters |
AEAs |
VDEs |
| TiScSi |
1.30 |
1.43 |
Ti2Si |
1.46 |
1.52 |
| TiScSi2 |
1.38 |
1.41 |
Ti2Si2 |
1.41 |
1.44 |
| TiScSi3 |
1.53 |
1.80 |
Ti2Si3 |
1.70 |
2.50 |
| TiScSi4 |
1.75 |
2.10 |
Ti2Si4 |
1.43 |
2.00 |
| TiScSi5 |
2.04 |
2.04 |
Ti2Si5 |
1.90 |
2.67 |
| TiScSi6 |
2.17 |
2.42 |
Ti2Si6 |
2.26 |
2.30 |
| TiScSi7 |
2.58 |
2.72 |
Ti2Si7 |
2.59 |
2.74 |
| TiScSi8 |
2.64 |
2.64 |
Ti2Si8 |
2.61 |
2.79 |
| TiScSi9 |
3.00 |
3.09 |
Ti2Si9 |
2.91 |
2.98 |
| TiScSi10 |
2.81 |
2.96 |
Ti2Si10 |
2.60 |
3.12 |
| TiScSi11 |
2.20 |
2.86 |
Ti2Si11 |
2.74 |
3.19 |
| TiScSi12 |
2.93 |
3.00 |
Ti2Si12 |
2.59 |
3.23 |
3.4 Stability
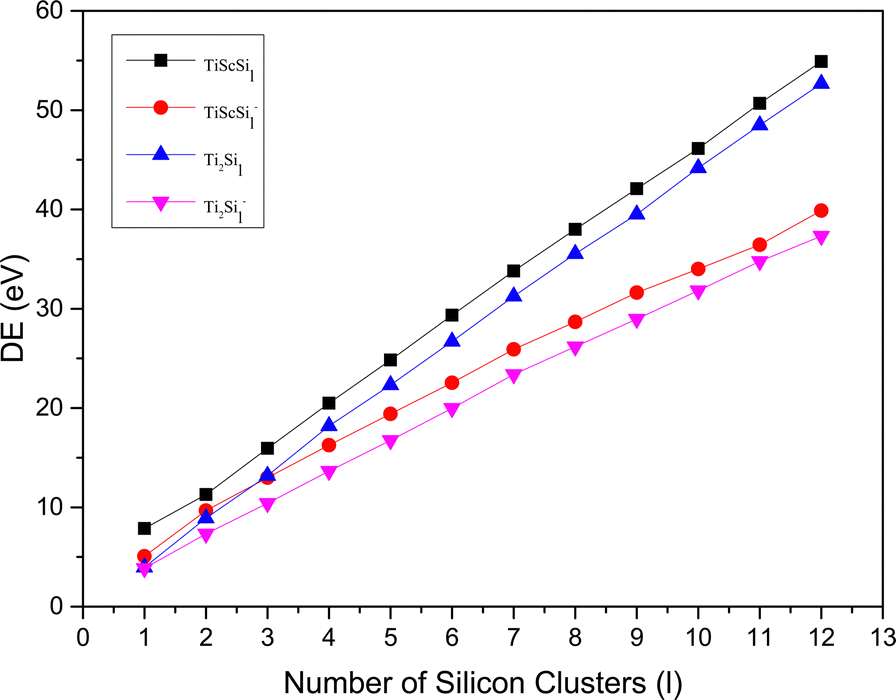
The relative stabilities of the neutral and anionic GMSs of the anionic TiASil (A = Sc, Ti; l ≤ 12) molecules were evaluated through the dissociation energy (DE) and second energy difference (SED) parameters. All the energy data involving DE and SED were processed and plotted (Fig. 4 and 5, respectively). The DE and SED of the neutral anionic clusters were calculated according to the corresponding equations:
| DE (TiASil0/−1) = lE(Si0/−1) + E(Ti) + E(A) − E(TiASil0/−1) |
| SED (TiASil0/−1) = E(TiASil−10/−1) + E(TiASil+10/−1) − 2E(TiASil0/−1) |
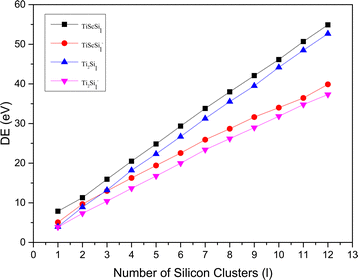 |
| | Fig. 4 DE versus the number of atoms l for TiASil (A = Sc, Ti, l ≤ 12) clusters and their anions. | |
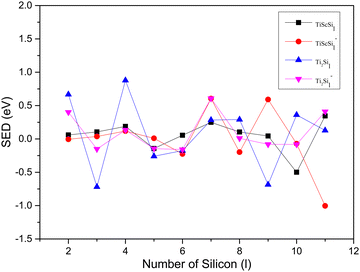 |
| | Fig. 5 SED versus the number of atoms l for TiASil (A = Sc, Ti, l ≤ 12) clusters and their anions. | |
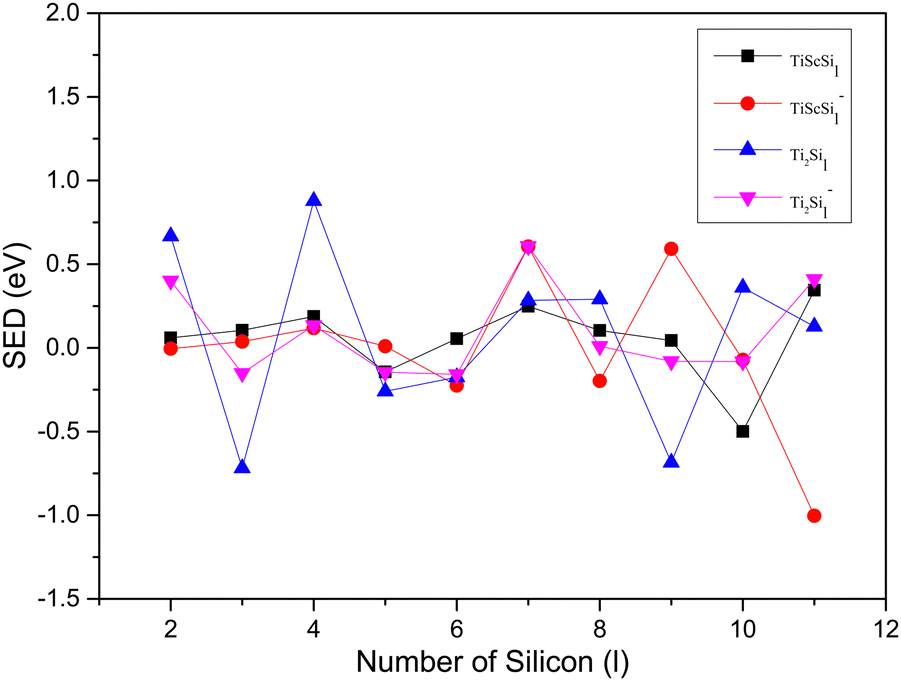
The variation tendencies of the DE curves of the neutral and anionic Ti2Sil clusters were the same for TiScSil. Namely, the energy increased with the increasing cluster size, and the neutral clusters exhibited significantly higher energy values than the anionic clusters. Compared with the Ti2Sil cluster, TiScSil exhibited better relative stability owing to its higher DE value. The results of the energy analysis were similar to those reported for the V2Sin clusters.62 The numerical variation in the SED could reflect the stability of the clusters; thus, the larger the energy value, the better the relative stabilities. Namely, the neutral and anionic TiASi4 and TiASi7 exhibited higher stabilities than their neighboring clusters because of their higher SED data. Evidently, the neutral Ti2Si4 clusters exhibited the best relative stability among the neutral and anionic TiASil clusters (A = Sc, Ti; l ≤ 12).
3.5 Charge transfer
To further elucidate the relationship between the scandium or titanium atoms and the silicon atoms, NPA (natural population analysis) was performed for GMSs via the PBE/Gen-B//PBE/Gen-A method. The data of the electron configuration ([Ar]4s3d4p), the charges on the scandium and titanium atoms, and their dipole moments were determined and listed in Tables 2 and 3. The valence configurations were 4s0.29−1.013d2.82−4.714p0.19−1.62 for the titanium atoms of the neutral and anionic Ti2Sil systems, 4s0.34−0.943d2.95−4.404p0.19−1.51 for the titanium atom, and 4s0.25−1.173d1.54−2.374p0.17−0.64 for the scandium atom in the neutral and anionic TiScSil clusters. The spd hybridization occurred in the 4s, 3d, and 4p orbitals of the transition-metal atoms in the neutral and anionic TiASil systems. The 3d orbitals of the metal atoms were key to their bonding to the silicon clusters. The charges of the titanium atoms ranged from −2.92 (anionic Ti2Si12−) to 0.11 (neutral Ti2Si) a.u. for the Ti2Sil clusters, −0.22 (scandium atoms of anionic ScTiSi10−) to 0.45 (scandium atoms in neutral ScTiSi2), and −2.52 (titanium atoms in anionic ScTiSi12−) to −0.01 (titanium atoms in neutral ScTiSi) a.u. for the TiScSil systems. Overall, compared with the scandium atom, the titanium atoms gained more charges from the silicon atoms, particularly in the anionic TiASil clusters, and this might explain why the titanium atom was wrapped in a cage structure. The dipole moments of the TiASil clusters ranged from 0 to 5.55 D and 1.90 to 7.29 D for the TiScSil and Ti2Sil clusters, respectively. The dipole moments were 0 D and nonpolar for the neutral Ti2Si4 clusters owing to the high symmetry (D4h). If an additional electron is added to the neutral TiASil molecule except for Ti2Si11, TiScSi8, and TiScSi11, it would follow that the extra electron would be assigned to the silicon clusters and the comprising metal atoms.
Table 2 Natural electron configuration and natural charge population (in a.u.) of Ti and Sc atoms and dipole moment (in Debye) for neutral TiScSil (l ≤ 12) clusters and their anions
| Clusters |
Electron configuration |
Charge (Ti/Sc) |
Dipole moment |
| ScTiSi |
Ti:[core]4S(0.89)3d(2.95)4p(0.19)/Sc:[core]4S(0.91)3d(1.54)4p(0.17) |
−0.01/0.39 |
4.10 |
| ScTiSi2 |
Ti:[core]4S(0.56)3d(3.32)4p(0.25)/Sc:[core]4S(0.61)3d(1.75)4p(0.21) |
−0.12/0.45 |
5.53 |
| ScTiSi3 |
Ti:[core]4S(0.48)3d(3.26)4p(0.35/Sc:[core]4S(0.56)3d(1.84)4p(0.30) |
−0.08/0.31 |
1.90 |
| ScTiSi4 |
Ti:[core]4S(0.34)3d(3.37)4p(0.56)/Sc:[core]4S(0.30)3d(2.01)4p(0.41) |
−0.27/0.28 |
2.12 |
| ScTiSi5 |
Ti:[core]4S(0.41)3d(3.52)4p(0.61)/Sc:[core]4S(0.41)3d(2.02)4p(0.39) |
−0.54/0.19 |
3.31 |
| ScTiSi6 |
Ti:[core]4S(0.41)3d(3.50)4p(0.60)/Sc:[core]4S(0.39)3d(2.03)4p(0.44) |
−0.51/0.13 |
2.59 |
| ScTiSi7 |
Ti:[core]4S(0.38)3d(3.77)4p(0.82)/Sc:[core]4S(0.33)3d(1.97)4p(0.47) |
−1.01/0.22 |
3.13 |
| ScTiSi8 |
Ti:[core]4S(0.43)3d(3.75)4p(1.04)/Sc:[core]4S(0.34)3d(2.10)4p(0.46) |
−1.34/0.10 |
3.92 |
| ScTiSi9 |
Ti:[core]4S(0.35)3d(3.31)4p(0.97)/Sc:[core]4S(0.27)3d(2.06)4p(0.53) |
−0.88/0.12 |
3.55 |
| ScTiSi10 |
Ti:[core]4S(0.40)3d(3.71)4p(0.70)/Sc:[core]4S(0.30)3d(2.18)4p(0.45) |
−0.82/0.07 |
5.03 |
| ScTiSi11 |
Ti:[core]4S(0.38)3d(4.02)4p(1.30)/Sc:[core]4S(0.30)3d(2.21)4p(0.49) |
−1.87/0.00 |
4.25 |
| ScTiSi12 |
Ti:[core]4S(0.35)3d(3.91)4p(1.39)/Sc:[core]4S(0.25)3d(2.37)4p(0.52) |
−2.00/−0.13 |
5.11 |
| ScTiSi− |
Ti:[core]4S(0.94)3d(3.05)4p(0.25)/Sc:[core]4S(1.17)3d(1.57)4p(0.24) |
−0.24/0.03 |
3.24 |
| ScTiSi2− |
Ti:[core]4S(0.67)3d(3.46)4p(0.31)/Sc:[core]4S(1.03)3d(1.61)4p(0.28) |
−0.43/0.08 |
3.43 |
| ScTiSi3− |
Ti:[core]4S(0.57)3d(3.35)4p(0.44)/Sc:[core]4S(0.76)3d(1.71)4p(0.35) |
−0.35/0.17 |
3.46 |
| ScTiSi4− |
Ti:[core]4S(0.57)3d(3.39)4p(0.61)/Sc:[core]4S(0.54)3d(2.00)4p(0.36) |
−0.58/0.10 |
5.87 |
| ScTiSi5− |
Ti:[core]4S(0.44)3d(3.58)4p(0.69)/Sc:[core]4S(0.49)3d(2.10)4p(0.47) |
−0.73/−0.06 |
3.12 |
| ScTiSi6− |
Ti:[core]4S(0.41)3d(3.59)4p(0.64)/Sc:[core]4S(0.40)3d(2.08)4p(0.48) |
−0.65/0.03 |
2.52 |
| ScTiSi7− |
Ti:[core]4S(0.38)3d(3.82)4p(0.90)/Sc:[core]4S(0.35)3d(2.07)4p(0.53) |
−1.16/0.04 |
3.40 |
| ScTiSi8− |
Ti:[core]4S(0.39)3d(3.66)4p(0.87)/Sc:[core]4S(0.36)3d(2.24)4p(0.58) |
−0.99/−0.20 |
3.16 |
| ScTiSi9− |
Ti:[core]4S(0.34)3d(3.42)4p(1.00)/Sc:[core]4S(0.28)3d(2.07)4p(0.53) |
−1.01/0.09 |
3.61 |
| ScTiSi10− |
Ti:[core]4S(0.39)3d(3.99)4p(1.05)/Sc:[core]4S(0.32)3d(2.25)4p(0.64) |
−1.51/−0.22 |
2.37 |
| ScTiSi11− |
Ti:[core]4S(0.38)3d(4.14)4p(1.16)/Sc:[core]4S(0.37)3d(2.21)4p(0.62) |
−1.79/−0.22 |
2.74 |
| ScTiSi12− |
Ti:[core]4S(0.34)3d(4.40)4p(1.51)/Sc:[core]4S(0.32)3d(2.20)4p(0.45) |
−2.52/0.05 |
7.29 |
Table 3 Natural electron configuration and natural charge population (in a. u.) of Ti atoms and dipole moment (in Debye) for neutral Ti2Sil (l ≤ 12) clusters and their anions
| Clusters |
Electron configuration |
Charge (Ti) |
Dipole moment |
| Ti2Si |
Ti:[core]4S(0.90)3d(2.82)4p(0.19) |
0.11 |
3.35 |
| Ti2Si2 |
Ti:[core]4S(0.55)3d(3.17)4p(0.24) |
0.07 |
4.99 |
| Ti2Si3 |
Ti:[core]4S(0.44)3d(3.35)4p(0.40) |
−0.18 |
3.97 |
| Ti2Si4 |
Ti:[core]4S(0.34)3d(3.49)4p(0.54) |
−0.35 |
0.00 |
| Ti2Si5 |
Ti1:[core]4S(0.43)3d(3.49)4p(0.65)/Ti2:4S(0.43)3d(3.48)4p(0.56) |
−0.57/−0.47 |
2.50 |
| Ti2Si6 |
Ti:[core]4S(0.41)3d(3.48)4p(0.59) |
−0.49 |
1.85 |
| Ti2Si7 |
Ti1:[core]4S(0.46)3d(3.65)4p(0.82)/Ti2:[core]4S(0.37)3d(3.62)4p(0.60) |
−0.98/−0.59 |
2.21 |
| Ti2Si8 |
Ti1:[core]4S(0.39)3d(3.55)4p(0.81)/Ti2:[core]4S(0.38)3d(3.67)4p(0.73) |
−0.81/−0.80 |
1.42 |
| Ti2Si9 |
Ti1:[core]4S(0.39)3d(3.79)4p(1.04)/Ti2:[core]4S(0.41)3d(3.68)4p(0.79) |
−1.31/−0.89 |
1.40 |
| Ti2Si10 |
Ti1:[core]4S(0.37)3d(3.54)4p(1.15)/Ti2: [core]4S(0.31)3d(3.85)4p(0.71) |
−1.32/−0.86 |
3.07 |
| Ti2Si11 |
Ti1:[core]4S(0.37)3d(3.99)4p(1.40)/Ti2: [core]4S(0.32)3d(3.82)4p(0.68) |
−2.02/−0.80 |
4.00 |
| Ti2Si12 |
Ti1:[core]4S(0.33)3d(3.66)4p(0.75)/Ti2:[core]4S(0.35)3d(3.68)4p(1.32) |
−0.77/−1.67 |
2.11 |
| Ti2Si− |
Ti: [core]4S(1.01)3d(2.96)4p(0.23) |
−0.19 |
3.30 |
| Ti2Si2− |
Ti:[core]4S(0.76)3d(3.19)4p(0.30) |
−0.25 |
3.59 |
| Ti2Si3− |
Ti:[core]4S(0.60)3d(3.24)4p(0.42) |
−0.26 |
3.77 |
| Ti2Si4− |
Ti1:[core]4S(0.57)3d(3.37)4p(0.55)/Ti2:[core]4S(0.53)3d(3.41)4p(0.44) |
−0.50/−0.37 |
5.55 |
| Ti2Si5− |
Ti1:[core]4S(0.46)3d(3.54)4p(0.61)/Ti2:[core]4S(0.42)3d(3.51)4p(0.47) |
−0.62/−0.40 |
4.70 |
| Ti2Si6− |
Ti:[core]4S(0.41)3d(3.60)4p(0.61) |
−0.62 |
1.98 |
| Ti2Si7− |
Ti1:[core]4S(0.43)3d(3.62)4p(0.90)/Ti2:[core]4S(0.39)3d(3.73)4p(0.68) |
−1.03/−0.80 |
3.20 |
| Ti2Si8− |
Ti1:[core]4S(0.39)3d(3.79)4p(0.95)/Ti2:[core]4S(0.40)3d(3.63)4p(0.81) |
−1.20/−0.88 |
1.55 |
| Ti2Si9− |
Ti1:[core]4S(0.38)3d(3.89)4p(1.09)/Ti2:[core]4S(0.40)3d(3.73)4p(0.81) |
−1.46/−0.97 |
2.23 |
| Ti2Si10− |
Ti1:[core]4S(0.36)3d(3.97)4p(1.24)/Ti2:[core]4S(0.34)3d(3.42)4p(0.61) |
−1.82/−0.40 |
3.21 |
| Ti2Si11− |
Ti1:[core]4S(0.29)3d(3.66)4p(0.59)/Ti2:[core]4S(0.34)3d(3.70)4p(1.35) |
−0.75/−1.76 |
4.02 |
| Ti2Si12− |
Ti1:[core]4S(0.38)3d(4.71)4p(1.62)/Ti2:[core]4S(0.37)3d(3.72)4p(0.80) |
−2.92/−0.92 |
3.44 |
3.6 Chemical bond analysis
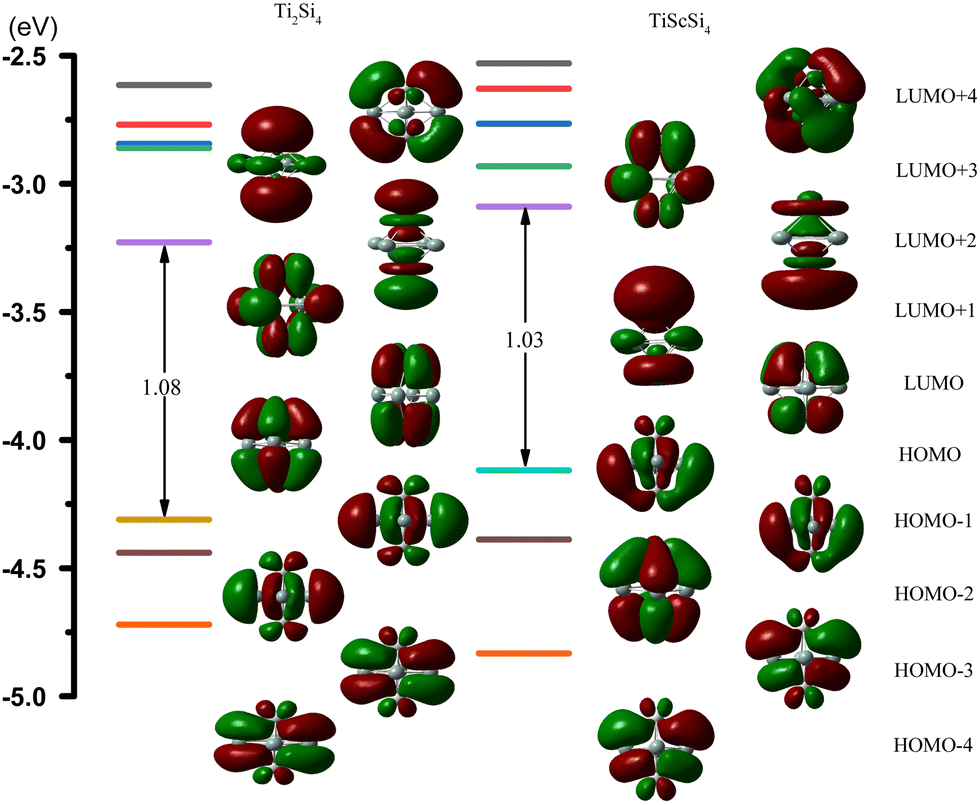
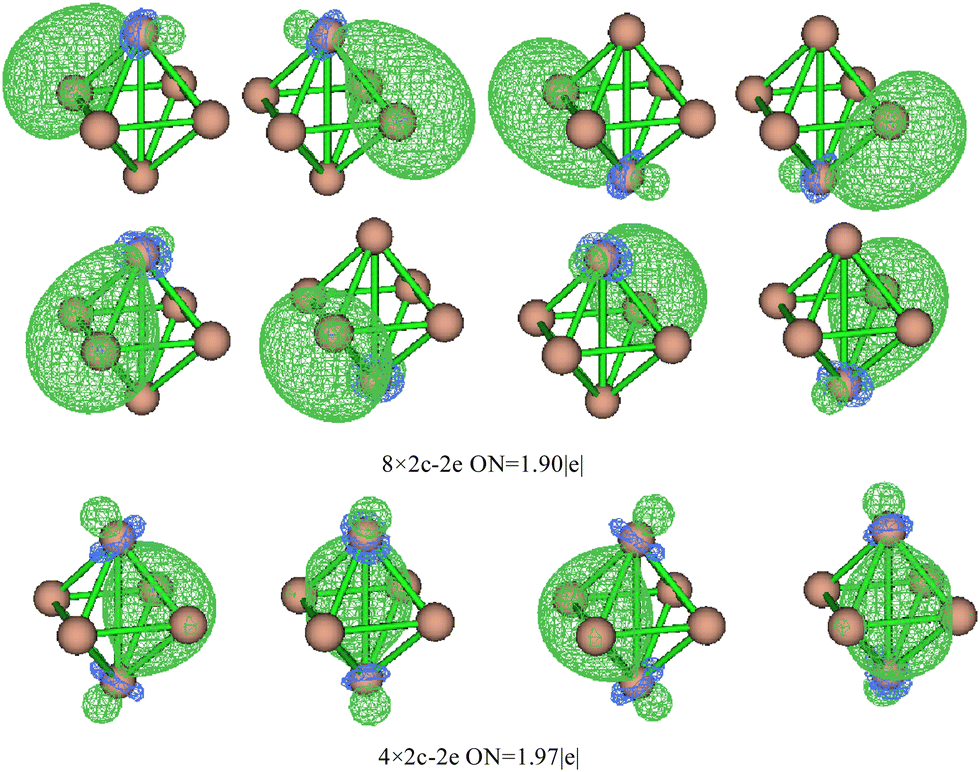
Based on the SED data, the neutral TiASi4 compound exhibited better relative stability. Hence, the highest occupied molecular orbital (HOMO)–lowest unoccupied MO (LUMO) gaps (HLgs) and MOs of the neutral Ti2Si4 and TiScSi4 clusters were drawn to gain further insight into the existing chemical bonds (Fig. 6). The spin multiplicity of neutral TiScSi4 was 2, and only the alpha orbitals were displayed on the chart. The data of HLgs of Ti2Si4 and TiScSi4 were 1.08 and 1.03 eV, respectively, demonstrating that Ti2Si4 was more stable than the neutral TiScSi4 cluster. This result correlates with the values of the SED analysis. For Ti2Si4, HOMO comprised 40.40% of Ti 3dx2y2 and 4.64% of each Si pz of the atomic orbitals (AOs). HOMO−1 comprised 16.39% of the Ti dyz AOs, 7.62% of two silicon AOs, and 24.84% of the other silicon atom py orbitals. HOMO−1 and HOMO−2 were analyzed to be equivalent orbitals because they exhibited the same MO compositions and energies. HOMO−3 comprised 19.18% and 7.17% of each Ti dxz and px AOs, respectively; 23.12% of two silicon pz as HOMO−1 and HOMO−2; and HOMO−3 and HOMO−4 belong to the degenerate orbital. LUMO for Ti2Si4 mainly comprised the pxy AOs (49.28%) of two titanium atoms. For instance, LUMO+1 comprised 33.06% 3dx2y2 AOs of the titanium atom, as well as 1.44% s and 6.35% p AOs of every silicon atom. LUMO+2 in Ti2Si4 comprised s, pz, and dz2 AOs of each titanium atom. Regarding LUMO+3, the AOs comprised the titanium (42.80%) and silicon (3.60%) atoms; regarding LUMO+4, the AOs comprised titanium (33.90%) and silicon (15.76%) atoms. For the neutral TiScSi4 MOs, the HOMO comprised 12.06% and 1.52% of the Ti dyz and Ti py AOs, respectively; 15.60% of the Sc 4dyz, and 24.67% of the two silicon atoms of py AOs. Similar to Ti2Si4, HOMO−1 and HOMO−2 were considered equivalent orbitals, and HOMO−3 and HOMO−4 also reflected the same energy. The LUMO comprised 63.70% of Ti-dxy, 31.22% of Sc-dxy, and 0.89% of the Si-p AOs. Compared with TiScSi4, the HOMO of Ti2Si4 was mainly availed by the metal atoms. Furthermore, the composition of the LUMO almost consisted of metal atoms. Integrating the NPA of TiASi4 revealed that the spd hybridization of the metal atoms was the key to the A–Si and Ti–Si chemical bonds in the neutral TiASi4 clusters. The bonding characteristics of Ti2Si4 were further analyzed by the adaptive natural density partitioning (AdNDP) scheme. The AdNDP tool elucidated the bond localization or delocalization of the Ti–Si bonding of Ti2Si4 according to N-center two electron (Nc–2e) bonds with more or less of N. The graphics (Fig. 7) were visualized using the Multiwfn software.70 The analysis revealed that the Ti2Si4 cluster exhibited eight and six localized and delocalized bonds, respectively. The localized bonds were featured by the eight 2c–2e localized σ-type Ti–Si bonds with 1.90 electrons in each bond, with the components of the bond mainly including the Si-s and p, Ti-s, p, and d AOs. The delocalized orbitals represent the 4c–2e σ-type bonds (ON = 1.97|e|) with the components of bonds mainly including the Si-p and Ti-d AOs. Therefore, the eight and four localized and delocalized bonds might be the key to ensuring the relative stabilities of the neutral square bipyramidal Ti2Si4.
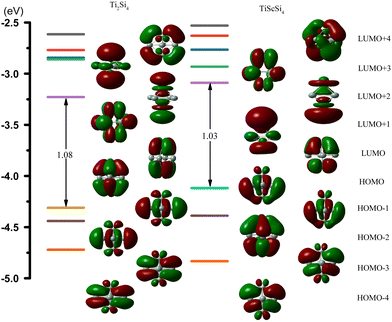 |
| | Fig. 6 Molecular orbitals (MO) and the HOMO–LUMO energy gaps (HLgs) of neutral TiASi4 clusters (A = Sc, Ti). | |
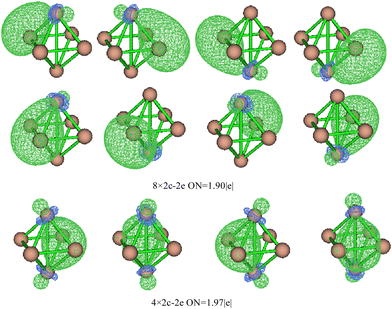 |
| | Fig. 7 Analysis of the AdNDP chemical bonds of neutral Ti2Si4. The balls at the top and bottom are titanium atoms. | |
4. Conclusions
In conclusion, a metal–silicon cluster, TiASil (A = Sc,Ti; l ≤ 12), and its anions were investigated via the PBE method combined with the structural searching procedure of ABCluster. The neutral GMSs of the TiASil clusters reflected an adsorptive evolution pattern and contained a TiASi4 unit (1–4–1) in their interior construction despite some distortions and minor changes. As the cluster size increased, more silicon atoms were preferably combined with the titanium atoms than with the scandium atoms. The simulated PES, VDEs, AEAs, DE, and SED of the TiASil clusters were calculated and analyzed. The neutral TiASi4 clusters exhibited better relative stability among the neutral and anionic TiASil (A = Sc, Ti; l ≤ 12) based on the SED data. Therefore, the chemical bond analysis of the TiASi4 clusters was performed employing their MO, HLgs, and AdNDP data for the best Ti2Si4 cluster especially. Combined with the NPA data, the metal-atom hybridization of the spd orbital, we observed that the eight localized and four delocalized bonds might primarily account for the relative stabilities of the neutral square bipyramidal structure of Ti2Si4. Thus, the Ti2Si4 clusters might be a good candidate for the basic unit of assembling silicon-based semiconductor materials. This study will inspire further experimental verification and application of silicon-based nanosemiconductor materials, availing possible proofs based on the electronic properties of the TiASil clusters.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work has been supported by the Grant (2021LHMS02005 and 2020MS03079) from the Inner Mongolia Natural Science Foundation, by the Inner Mongolia University of Science and Technology Innovation Fund (2019QDL-B46), Inner Mongolia Autonomous Region scientific research project, and the Natural Science Foundation of Shaanxi Province (No. 2021JM-457).
Notes and references
- J. Zhao, Q. Du, S. Zhou and V. Kumar, Chem. Rev., 2020, 120, 9021–9163 CrossRef CAS PubMed.
- J. F. Harrison, Chem. Rev., 2000, 100, 679–716 CrossRef CAS PubMed.
- K. Raghavachari and C. M. Rohifing, J. Chem. Phys., 1988, 89, 2219–2234 CrossRef CAS.
- H. Hiura, T. Miyazaki and T. Kanayama, Phys. Rev. Lett., 2001, 86, 1733–1736 CrossRef CAS PubMed.
- S. N. Khanna, B. K. Rao, P. Jena and S. K. Nayak, Chem. Phys. Lett., 2003, 373, 433–438 CrossRef CAS.
- J. T. Lau, K. Hirsch, Ph Klar, A. Langenberg, F. Lofink, R. Richter, J. Rittmann, M. Vogel, V. Zamudio-Bayer, T. Möller and Bv Issendorff, Phys. Rev. A: At., Mol., Opt. Phys., 2009, 79, 053201 CrossRef.
- M. Ohara, K. Miyajima, A. Pramann, A. Nakajima and K. Kaya, J. Phys. Chem. A, 2002, 106, 3702–3705 CrossRef CAS.
- K. Koyasu, J. Atobe, S. Furuse and A. Nakajima, J. Chem. Phys., 2008, 129, 214301 CrossRef PubMed.
- A. Grubisic, H. Wang, Y. J. Ko and K. H. Bowen, Chem. Phys., 2008, 129, 054302 Search PubMed.
- K. Wang, H. Y. Zhao, L. Miao, Z. Z. Jia, G. J. Yin, X. D. Zhu, R. Moro, B. von Issendorf and L. Ma, J. Phys. Chem. A, 2022, 126, 1329–1335 CrossRef CAS PubMed.
- W. Qin, W. C. Lu, L. Z. Zhao, Q. J. Zang, C. Z. Wang and K. M. Ho, J. Phys.: Condens. Matter, 2009, 21, 455501 CrossRef PubMed.
- H. Yu, L. L. Wang, M. Schaekers, J. L. Everaert, Y. L. Jiang, D. Mocuta, N. Horiguchi, N. Collaert and K. D. Meyer, IEEE Electron Device Lett., 2017, 38, 843–846 CAS.
- J. Wu, Y. Gong, T. Inoshita, D. C. Fredrickson, J. Wang, Y. Lu, M. Kitano and H. Hosono, Adv. Mater., 2017, 29, 1700924 CrossRef PubMed.
- G. F. Zhao, J. M. Sun, Y. Z. Gu and Y. X. Wang, J. Chem. Phys., 2009, 131, 114312 CrossRef PubMed.
- K. Wang, G. J. Yin, Z. Z. Jia, L. Miao, R. Moro, B. von Issendorff and L. Ma, Phys. Chem. Chem. Phys., 2022, 24, 18321–18330 RSC.
- S. J. Lu, Mol. Phys., 2020, 118, e1682209 CrossRef.
- W. Qin, W. C. Lu, L. Z. Zhao, Q. J. Zang, C. Z. Wang and K. M. Ho, J. Phys.: Condens. Matter, 2009, 21, 455501 CrossRef PubMed.
- H. Yu, L. L. Wang, M. Schaekers, J. L. Everaert, Y. L. Jiang, D. Mocuta, N. Horiguchi, N. Collaert and K. D. Meyer, IEEE Electron Device Lett., 2017, 38, 843–846 CAS.
- J. Wu, Y. Gong, T. Inoshita, D. C. Fredrickson, J. Wang, Y. Lu, M. Kitano and H. Hosono, Adv. Mater., 2017, 29, 1700924 CrossRef PubMed.
- H. Mizoguchi, S. W. Park, K. Kishida, M. Kitano, J. Kim, M. Sasase, T. Honda, K. Ikeda, T. Otomo and H. Hosono, J. Am. Chem. Soc., 2019, 141, 3376–3379 CrossRef CAS PubMed.
- J. Li, G. Wang, C. Yao, Y. Mu, J. Wan and M. Han, J. Chem. Phys., 2009, 130, 164514 CrossRef PubMed.
- V. Kumar and Y. Kawazoe, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 073404 CrossRef.
- W. Jiang, N. J. DeYonker, J. J. Determan and A. K. Wilson, J. Phys. Chem. A, 2012, 116, 870–885 CrossRef CAS PubMed.
- C. Dong, L. Han, J. Yang and L. Cheng, Int. J. Quantum Chem., 2019, 119, e25978 CrossRef.
- Y. Liu, J. Yang and L. Cheng, Inorg. Chem., 2018, 57, 12934–12940 CrossRef CAS PubMed.
- J. Lu, J. Yang, Y. Kang and H. Ning, J Mol. Model., 2014, 20, 1–12 CAS.
- J. Lu, J. Yang, Z. Xing and H. Ning, J. Theor. Comput. Chem., 2014, 13, 1450038 CrossRef.
- H. G. Xu, M. M. Wu, Z. G. Zhang, Q. Sun and W. J. Zheng, Chin. Phys. B, 2011, 20, 043102 CrossRef.
- H. G. Xu, Z. G. Zhang, Y. Feng and W. Zheng, Chem. Phys. Lett., 2010, 498, 22–26 CrossRef CAS.
- M. T. Nguyen, Q. T. Tran and V. T. Tran, J. Mol. Model., 2017, 23, 282 CrossRef PubMed.
- J. He, K. Wu, C. Liu and R. Sa, Chem. Phys. Lett., 2009, 483, 30–34 CrossRef CAS.
- J. Wang, Q. M. Ma, R. P. Xu, Y. Liu and Y. C. Li, Phys. Lett. A, 2009, 373, 2869–2875 CrossRef CAS.
- J. U. Reveles and S. N. Khanna, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 035435 CrossRef.
- M. Tomonari and K. Tanaka, Theor. Chem. Acc., 2001, 106, 188 Search PubMed.
- L. J. Guo, X. Liu, G. F. Zhao and Y. H. Luo, J. Chem. Phys., 2007, 126, 234704 CrossRef PubMed.
- C. Deng, L. Zhou, G. Li, H. Chen and Q. Li, J. Cluster Sci., 2012, 23, 975 CrossRef CAS.
- V. Kumar, T. M. Briere and Y. Kawazoe, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 155412 CrossRef.
- M. B. Torres, E. M. Fernández and L. C. Balbás, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 205425 CrossRef.
- L. Guo, G. Zhao, Y. Gu, X. Liu and Z. Zeng, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 195417 CrossRef.
- P. Sen and L. Mitas, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 155404 CrossRef.
- V. Kumar, C. Majumder and Y. Kawazoe, Chem. Phys. Lett., 2002, 363, 319 CrossRef CAS.
- H. Kawamura, V. Kumar and Y. Kawazoe, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 075423 CrossRef.
- M. Tomonari and K. Tanaka, Theor. Chem. Acc., 2001, 106, 188 Search PubMed.
- L. J. Guo, X. Liu, G. F. Zhao and Y. H. Luo, J. Chem. Phys., 2007, 126, 234704 CrossRef PubMed.
- C. Deng, L. Zhou, G. Li, H. Chen and Q. Li, J. Cluster Sci., 2012, 23, 975 CrossRef CAS.
- C. L. Reis and J. M. Pacheco, J. Phys.: Condens. Matter, 2010, 22, 035501 CrossRef CAS PubMed.
- K. Koyasu, M. Akutsu, M. Mitsui and A. Nakajima, J. Am. Chem. Soc., 2005, 127, 4998–4999 CrossRef CAS PubMed.
- M. Ohara, K. Koyasu, A. Nakajima and K. Kaya, Chem. Phys. Lett., 2003, 371, 490 CrossRef CAS.
- H. Kawamura, V. Kumar and Y. Kawazoe, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 193402 CrossRef.
- J. Lu, Q. Lu and X. Li, Theor. Chem. Acc., 2020, 139, 1–9 Search PubMed.
- B. Zhang, J. Wang, Z. Hu and A. Ning, Mater. Res. Express, 2020, 7, 085006 Search PubMed.
- X. Wu, Q. Du, S. Zhou, X. Huang, M. Chen, L. Miao, G. Yin, J. Wang, K. Wang, B. v Issendorff, L. Ma and J. Zhao, Eur. Phys. J. Plus, 2020, 135, 1–15 Search PubMed.
- K. M. Ho, A. A. Shvartsburg, B. Pan, Z. Y. Lu, C. Z. Wang, J. G. Wacker, J. L. Fye and M. F. Jarrold, Nature, 1998, 392, 582–585 CrossRef CAS.
- H. Chen, H. Liang, W. Dai, C. Lu, K. Ding, J. Bin and B. Zhu, Int. J. Hydrogen Energy, 2020, 45, 32260–32268 CrossRef CAS.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- X. Huang, S. J. Lu, X. Q. Liang, Y. Su, L. W. Sai, Z. G. Zhang, J. J. Zhao, H. G. Xu and W. J. Zheng, J. Phys. Chem. C, 2015, 119, 10987–10994 CrossRef CAS.
- B. Yang, H. Xu, X. Xu and W. Zheng, J. Phys. Chem. A, 2018, 51, 9886 CrossRef PubMed.
- S. J. Lu, X. L. Xu, H. G. Xu and W. J. Zheng, J. Chem. Phys., 2018, 148, 244306 CrossRef PubMed.
- J. Zhang and M. Dolg, Phys. Chem. Chem. Phys., 2015, 17, 24173–24181 RSC.
- J. Zhang and M. Dolg, Phys. Chem. Chem. Phys., 2016, 18, 3003–3010 RSC.
- W. L. Li, T. T. Chen, D. H. Xing, X. Chen, J. Li and L. S. Wang, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 6972–6977 Search PubMed.
- J. Lu, Q. Lu and X. Li, Mol. Phys., 2021, 119, e1864042 CrossRef.
- J. Lu, Q. Lu and X. Li, Phys. Chem. Chem. Phys., 2021, 23, 25679–25688 RSC.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456 CrossRef CAS PubMed.
- D. E. Woon Jr. and T. H. Dunning Jr., J. Chem. Phys., 1993, 98, 1358 CrossRef.
- M. Doverstål, B. Lindgren, U. Sassenberg, C. A. Arrington and M. D. Morse, J. Chem. Phys., 1992, 97, 7087–7092 CrossRef.
- W. G. Sun, J. J. Wang, C. Lu, X. X. Xia, X. Y. Kuang and A. Hermann, Inorg. Chem., 2017, 56, 1241–1248 CrossRef CAS PubMed.
- B. L. Chen, W. G. Sun, X. Y. Kuang, C. Lu, X. X. Xia, H. X. Shi and G. Maroulis, Inorg. Chem., 2018, 757, 343–350 CrossRef PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
|
| This journal is © the Owner Societies 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *ab and
Xiao-Jun
Li
c
*ab and
Xiao-Jun
Li
c