
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A perspective on two pathways of photocatalytic water splitting and their practical application systems
Yiwen
Ma
a,
Lihua
Lin
a,
Tsuyoshi
Takata
a,
Takashi
Hisatomi
 a and
Kazunari
Domen
*ab
a and
Kazunari
Domen
*ab
aResearch Initiative for Supra-Materials, Interdisciplinary Cluster for Cutting Edge Research, Shinshu University, Nagano 380-8553, Japan. E-mail: domen@chemsys.t.u-tokyo.ac.jp
bOffice of University Professors, The University of Tokyo, Tokyo 113-86556, Japan
First published on 1st February 2023
Abstract
Photocatalytic water splitting has been widely studied as a means of converting solar energy into hydrogen as an ideal energy carrier in the future. Systems for photocatalytic water splitting can be divided into one-step excitation and two-step excitation processes. The former uses a single photocatalyst while the latter uses a pair of photocatalysts to separately generate hydrogen and oxygen. Significant progress has been made in each type of photocatalytic water splitting system in recent years, although improving the solar-to-hydrogen energy conversion efficiency and constructing practical technologies remain important tasks. This perspective summarizes recent advances in the field of photocatalytic overall water splitting, with a focus on the design of photocatalysts, co-catalysts and reaction systems. The associated challenges and potential approaches to practical solar hydrogen production via photocatalytic water splitting are also presented.
1. Introduction
With a continual increase in global energy consumption and concerns regarding environmental contamination, it is imperative to develop renewable energy resources that are neither dependent on fossil fuels nor emit greenhouse gases.1,2 Solar energy represents an inexhaustible, clean and renewable energy source and has therefore attracted widespread attention.3 However, because the amount of solar energy varies by geographical locations, season and time of day, it is necessary to convert and store this energy in a cost-effective and environmentally friendly manner.4 Hydrogen is a promising energy carrier with a high mass energy density (142 MJ kg−1) and it can serve as an environmentally friendly carbon-free fuel that produces only water as a by-product of combustion.5 In addition, hydrogen is also easily transportable in the forms of liquid hydrogen or via a liquid organic hydrogen carrier.6 Therefore, hydrogen production by solar-driven water splitting, often referred to as the simplest form of artificial photosynthesis, is one of the most important methods of solar energy conversion.There are three main solar-powered water splitting methods: photovoltaic electrolysis,7,8 photoelectrocatalysis7,9–13 and photocatalysis.14–16 Among these, photocatalytic water splitting has attracted considerable interest in recent years because it is one of the most promising technologies for the production of hydrogen energy on a large scale and at low cost.17,18 Many photocatalysts for water photolysis have thus been developed, and the underlying mechanism has been studied in depth over the past five decades. In addition, field testing has demonstrated that photocatalytic water splitting systems can be scaled up without loss of efficiency, which is an important perquisite for practical applications.19 Nevertheless, the generally poor efficiency of this process remains an obstacle to commercialization. Technical and economic analyses have suggested that achieving solar-to-hydrogen (STH) energy conversion efficiencies in the range of 5–10% could make solar-powered hydrogen production via photocatalytic overall water splitting (OWS) economically viable.20,21 Even so, the highest STH value reported to date for an OWS system is only approximately 1%.22 Therefore, the development of high-efficiency photocatalysts and photocatalytic OWS systems is vital to realizing large-scale industrial applications.
The photocatalyst plays a central role in photocatalytic water splitting reactions and must satisfy certain requirements.23 These include a narrow energy bandgap that allows the efficient absorption of solar energy, a suitable band position that provides a sufficient thermodynamic driving force, efficient electron–hole pair separation and transport characteristics (to avoid recombination) and minimal photocorrosion (to maintain stability).24 Photocatalytic OWS systems can be divided into two general categories, these being one- and two-step excitation. In the former, only one photocatalyst participates in the redox reactions of water (that is, the hydrogen evolution reaction and the oxygen evolution reaction).25 In contrast, H2 and O2 are generated over a hydrogen evolution photocatalyst (HEP) and oxygen evolution photocatalyst (OEP), respectively, in a two-step excitation system.26 In these systems, electrons are typically transferred from the OEP to the HEP through electron mediators. Only a few visible-light-driven photocatalysts have been used in one-step excitation systems as a consequence of mismatches in the band alignment of such materials with respect to the water redox potentials.27 Even so, one-step excitation systems have the advantage of being simple, and are being actively studied as an approach to achieving solar hydrogen production on a large scale.28 In the case of two-step excitation systems, also known as Z-scheme processes, the HEP and OEP must simply satisfy the thermodynamic conditions for the corresponding water splitting half-reactions, which greatly extends the range of applicable photocatalysts.29 However, the band alignments of the HEP and OEP and the redox potential of the electron mediator must all be carefully considered in order to realize continuous water splitting reactions. These factors therefore increase the complexity of the design of two-step excitation systems.
Apart from the design of photocatalysts and photocatalytic water splitting systems, it is also important to construct suitable facilities that permit practical high-efficiency water splitting on a large scale. As an example, there has been much progress in research on suspension photocatalysts, and these materials have a wide range of potential applications. Even so, it is difficult to construct and operate large-scale water splitting reactors that require stirring, and it is also challenging to recycle the used photocatalyst. In recent years, immobilization of particulate photocatalysts onto specific substrates has been investigated as an approach to achieving large-scale photocatalytic OWS. Therefore, it will be important to design fixed particulate photocatalytic systems with comparable or even higher performance than suspended systems.22,30
This perspective presents state-of-art of photocatalytic water splitting. Key principles of photocatalysis are first outlined. Specifically, the basics of one-step excitation and Z-scheme OWS are examined. Guidelines are provided for the preparation of efficient photocatalysts and photocatalytic water splitting processes. Subsequently, representative effective approaches to improving photocatalytic performance are introduced. The development of efficient photocatalysts for one-step excitation systems along with cocatalyst loading and surface modification are described in detail. With regard to Z-scheme processes, the choice of redox mediator and the construction of the system also play a vital role and are discussed. Furthermore, the immobilization of particulate photocatalysts on substrates to construct scalable water splitting panels is addressed in detail. Finally, we describe future prospects and challenges associated with realizing large-scale solar water splitting.
2. The basics of photocatalytic overall water splitting
Photocatalytic OWS to produce H2 and O2 is an endergonic reaction with a Gibbs free energy change of ΔG° = 237 kJ mol−1 at 298 K, as shown in eqn (1).31 Thus, an energy input is needed to initiate the reaction, and this energy is obtained from sunlight in the case of solar-driven photocatalytic OWS.32| H2O → H2 + 1/2O2, ΔG° = 237 kJ mol−1 | (1) |
In a one-step excitation system using a single photocatalyst, a semiconductor absorbs photons with energy values greater than or equal to its bandgap energy. As a result, the electrons are excited from the valence band of this material to the conduction band, while holes are generated in the valence band (Fig. 1a).33 These photogenerated electrons and holes are capable of reducing H+ and oxidizing H2O, respectively, if the conduction band minimum (CBM) of the semiconductor is at a more negative potential than the H+/H2 reduction potential (0 V vs. a normal hydrogen electrode (NHE) at pH = 0) and the valence band maximum (VBM) is at a more positive potential than the O2/H2O oxidation potential (+1.23 V vs. NHE at pH = 0).34 Therefore, the theoretical minimum photon energy required for OWS in one-step excitation systems is 1.23 eV.35 Generally, light absorption, the separation and transfer of photogenerated charges, and the surface catalytic reactions of adsorbed species are the three most critical steps determining the performance of a one-step excitation OWS process.36 Obtaining high STH efficiency requires that the bandgap of the photocatalyst is as narrow as possible, such that as much visible light as possible can be absorbed.37 However, a photocatalyst with a narrow bandgap can only provide a weak thermodynamic driving force for the water splitting reaction. The evolution of hydrogen and oxygen on the same photocatalyst also increases the possibility that electrons and holes will recombine in the material and that back reactions will occur on the surface of the catalyst.38 In addition, narrow-bandgap photocatalysts often contain S or N anions and so readily undergo self-oxidation by photoexcited holes, resulting in poor stability against photocorrosion.39 For all these reasons, few photocatalysts have thus far been found to be capable of realizing one-step excitation OWS, especially in the case of visible-light responsive photocatalysts.
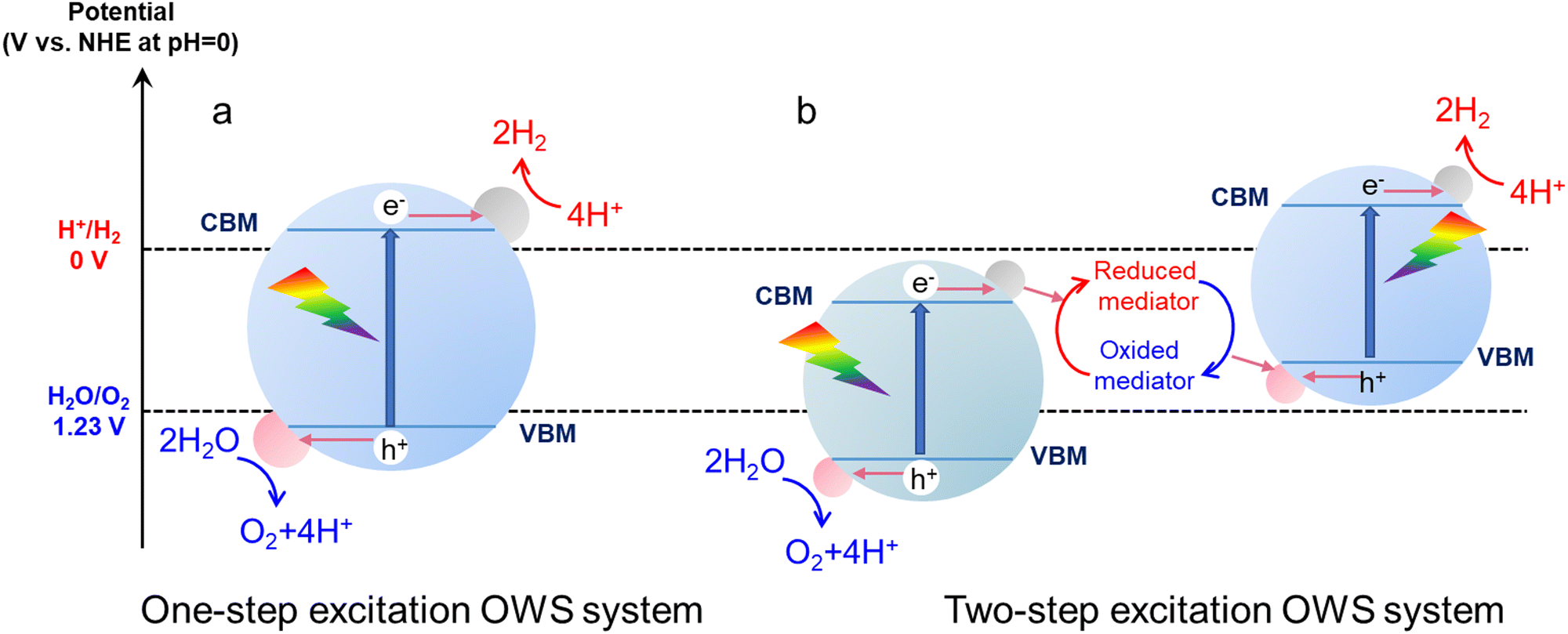 | ||
| Fig. 1 Schematic energy diagrams summarizing (a) one-step excitation and (b) two-step excitation OWS on particulate photocatalysts. | ||
Compared with the challenging requirements associated with one-step excitation, two-step excitation systems allow a wider range of photocatalysts to be considered (Fig. 1b). A typical Z-scheme system includes an HEP and an OEP as well as shuttle redox mediators.40 H+ ions are reduced by electrons in the conduction band of the HEP while mediators in the reduced state are oxidized by holes in the valence band to generate H2 and oxidized mediators, respectively.41 Conversely, the oxidized mediators are transitioned back to their reduced forms by photoexcited electrons on the OEP while H2O molecules are oxidized to O2 molecules by holes.42 Therefore, the OWS process depends on the circulation of redox mediators. A photocatalyst can be used as an HEP in a Z-scheme system if the CBM of the material is more negative than the reduction potential of H+ and the VBM is more positive than the redox potential of the electron mediator.43 The photocatalyst can serve as the OEP if its VBM is more positive than the oxidation potential of H2O and its CBM is more negative than the redox potential of the electron mediator.44 These conditions allow the oxidization of reduced redox species or reduction of oxidized redox species on a thermodynamic basis. Relaxation of these thermodynamic requirements greatly extends the range of photocatalysts applicable to Z-scheme OWS. Some narrow-bandgap photocatalysts used to promote the water splitting half-reaction can be applied in Z-scheme systems and can permit a large portion of the visible light spectrum to be used without reducing the driving force for the water splitting reaction.45 Similar to one-step excitation systems, the extent of light absorption, charge separation and migration along with the occurrence of catalytic surface reactions also affect the performance of Z-scheme water splitting processes. Furthermore, the redox mediators that are employed play important roles in transferring photogenerated charge carriers. Some mediators that have been traditionally used include Fe3+/Fe2+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 46 and I3−/I−47,48 along with complexes such as [Co(bpy)3]3+/[Co(bpy)3]2+ (bpy = 2,2′-bipyridine) and [Co(phen)3]3+/[Co(phen)3]2+ (phen = 1,10-phenanthroline).49 In recent years, Z-scheme OWS systems utilizing physical contact between photocatalyst particles or contact via solid-state electronic conductors have also shown remarkable efficiency.50 The major benefits and limitations of the one- and two-step excitation OWS process are summarized briefly in Table 1.
46 and I3−/I−47,48 along with complexes such as [Co(bpy)3]3+/[Co(bpy)3]2+ (bpy = 2,2′-bipyridine) and [Co(phen)3]3+/[Co(phen)3]2+ (phen = 1,10-phenanthroline).49 In recent years, Z-scheme OWS systems utilizing physical contact between photocatalyst particles or contact via solid-state electronic conductors have also shown remarkable efficiency.50 The major benefits and limitations of the one- and two-step excitation OWS process are summarized briefly in Table 1.
| Advantages | Disadvantages | |
|---|---|---|
| One step excitation OWS system | 1. Simple because of the use of only one type of photocatalyst. | 1. Stringent thermodynamic requirement that the bandgap of the photocatalyst must straddle the redox potentials for water splitting. |
| 2. Easily fixed on a substrate to fabricate water splitting panels for practical application. | 2. Limited number of visible-light-driven photocatalysts known to satisfy the above requirement. | |
| 3. High possibility of backward reactions due to the occurrence of H2 and O2 evolution reactions on the same photocatalyst. | ||
| Two step excitation OWS system | 1. Mild thermodynamic requirement that the bandgap of the HEP and OEP only need to straddle the redox potentials for the corresponding half-reactions. | 1. Complicated because of the use of two types of photocatalysts. |
| 2. Many options for visible-light-driven photocatalysts as HEP and OEP. | 2. Constraints in material selection that the VBM of HEP must more positive than the CBM of OEP. | |
| 3. Spatial separation of the H2 and O2 reactive sites reduces the possibility of back reaction. | 3. Need of a special design to promote charge transfer between HEP and OEP. | |
| 4. High possibility of backward reactions involving redox mediators, if used. | ||
3. One-step excitation overall water splitting based on particle suspension systems
Photocatalysts for OWS systems via one-step excitation have been studied for some time now. Pioneering work in 1972 demonstrated that light energy can be used to decompose water in a stable manner on semiconductors using a TiO2 photoelectrode.51 Since then, many particulate photocatalysts have been shown to promote one-step excitation OWS. Some of these materials exhibit a high apparent quantum yield (AQY) under ultraviolet (UV) light. As an example, Zn-doped Ga2O3 and La-doped NaTaO3 show AQYs of 71% at 254 nm and 56% at 270 nm, respectively.52,53 It should also be noted that there has been remarkable progress in the development of SrTiO3 as a photocatalyst driven by UV light in recent years. Since the first reports regarding the use of NiO-SrTiO3 as a photocatalyst to decompose water vapour in 1980, extensive research has been carried out concerning the modification of SrTiO3.54 These modifications have included cation doping, particle morphology tuning and cocatalyst loading. In 2009, our group demonstrated doping with low-valence cations to introduce oxygen vacancies and decrease the concentration of Ti3+, such as by substituting Na+ for Sr2+ and Ga3+ for Ti4+.55 The defect species responsible for the deactivation of SrTiO3 was Ti3+. Doping of a cation with a valence lower than that of the parent cation introduced oxygen vacancies, which inhibited the formation of Ti3+. Therefore, doping effectively improved the photocatalytic performance of SrTiO3. In subsequent work, SrCl2 was used as a flux to dissolve the Al2O3 dopant and host SrTiO3 particles, thus facilitating Al doping of SrTiO3. This flux also produced SrTiO3 particles with smooth crystal facets. Well-dispersed small particles with a narrow particle size distribution were obtained and these characteristics enhanced charge migration toward surface active sites. On the basis of these modifications, an OWS AQY of 30% was obtained at 360 nm, representing the highest value in this wavelength region reported at that time.56 In addition to doping, modification of the crystal structure and morphology can also affect the performance of this compound. As an example, SrTiO3 grown from TiO2 mesocrystals by topologic epitaxy comprise assembled cubic nanocrystals with a well-defined orientation.57 According to the time-resolved diffuse reflectance spectroscopy and single-particle photoluminescence imaging analysis, photogenerated electrons migrate via trapping-detrapping processes along the nanocube networks inside the mesocrystals and are captured at the larger nanocubes on the external surface, resulting in a prolonged charge lifetime. Therefore, the higher-order structure plays a key role in increasing the charge lifetime by efficient interparticle electron transfer, thereby significantly improving the photocatalytic performance. As a result, SrTiO3 mesocrystals have exhibited a high AQY of 6.7% at 360 nm during the OWS process. Wang's group prepared a SrTiO3/TiO2 heterostructure by hydrothermal crystallization of SrTiO3 on the surface of the TiO2 hollow multi-shelled structures. Owing to the enhanced light absorption and charge separation between SrTiO3 and TiO2, the broccoli-like SrTiO3/TiO2 heterostructure showed fourfold enhancement in the OWS reaction (with AQY of 8.6% at 365 nm) compared with SrTiO3 nanoparticles.58 The facet engineering of SrTiO3 to separate redox active sites has also been investigated (Fig. 2a–c).59 A nanocrystal morphology tailoring strategy was applied to permit the rational design and synthesis of cubic SrTiO3 nanocrystals with morphologies ranging from isotropic facets ({001}, six-faceted) to anisotropic facets ({001} and {110}, 18-faceted) (Fig. 2d–g). Site-selective photodeposition of dual cocatalysts on these specimens resulted in spatial separation of reduction and oxidation sites (Fig. 2h and i). This configuration resulted in a fivefold enhancement of AQY compared with that of six-faceted SrTiO3. Moreover, in 2020, our group achieved OWS with an internal quantum efficiency (IQE) close to unity by applying Al as a dopant to suppress the formation of defects together with a flux treatment to improve the crystallinity of the catalyst.19 In this work, Rh/Cr2O3 and CoOOH cocatalysts were selectively photodeposited on different crystal facets of the semiconductor particles for anisotropic charge transport. This modification separately promoted the hydrogen and oxygen evolution reactions and inhibited the reverse reactions (Fig. 3a–e). A recent study combining dual scale atomic force microscopy (AFM) and imaging with optimized AFM tip sizes found that, within the rather wide pH range of 4 to 6, the {110} and {110} facets of SrTiO3 nanoparticles take on negative and positive surface charges, respectively, indicating that very strong electric fields are likely present within the particles. On this basis, photogenerated electrons and holes will accumulate at {100} and {110} facets, respectively, which is consistent with the observed distribution of cocatalysts on the SrTiO3 surface.60 Overall, these results demonstrate a suitable design for a photocatalyst capable of utilizing photoexcited electrons and holes almost exclusively for the water splitting reaction.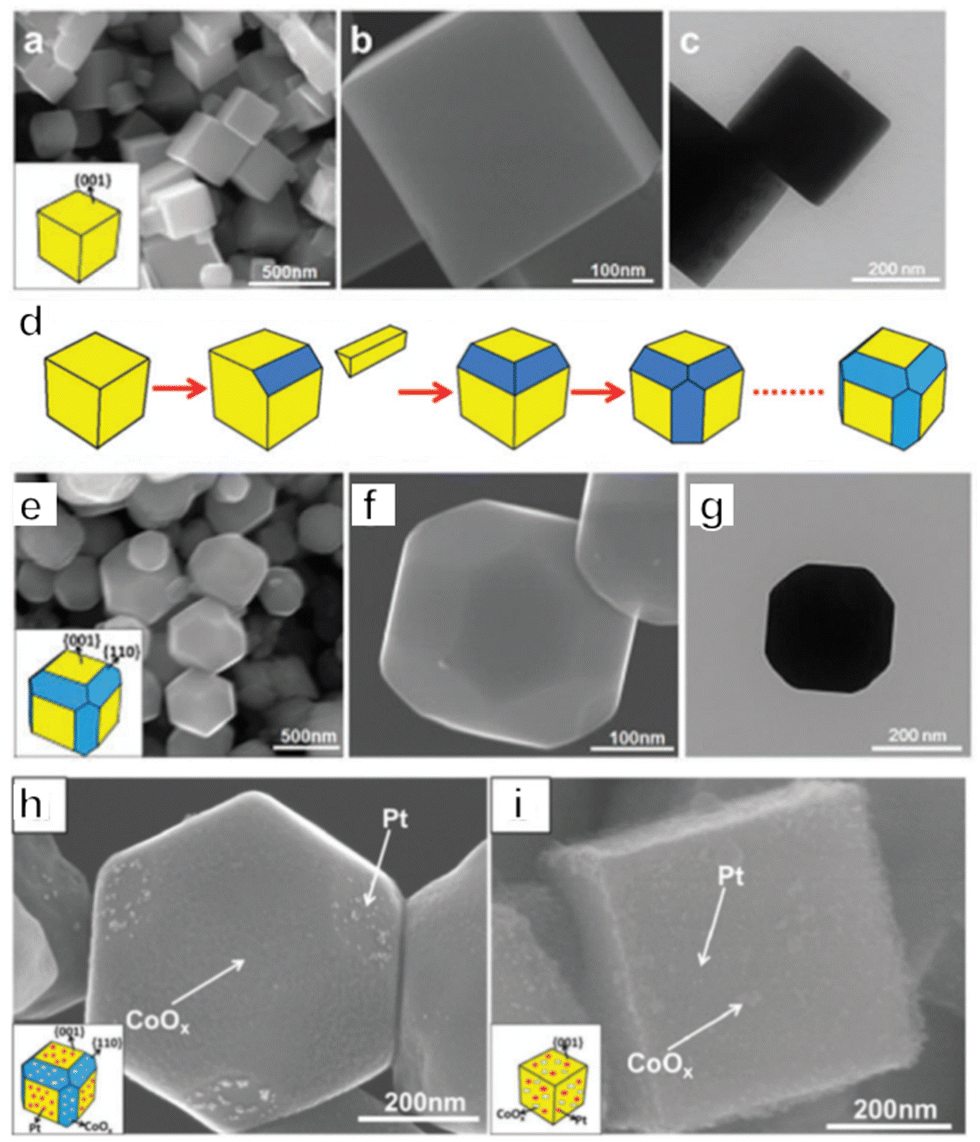 | ||
| Fig. 2 (a–c) Morphology of six-faceted SrTiO3 nanocrystals. (d) Transition of six-faceted to 18-faceted SrTiO3 nanocrystals. (e–g) Morphology of 18-faceted SrTiO3 nanocrystals. (h) SEM images of 18-faceted and (i) six-faceted SrTiO3 nanocrystals with co-catalysts (reprinted with permission from ref. 59 Copyright 2016 The Royal Society of Chemistry). | ||
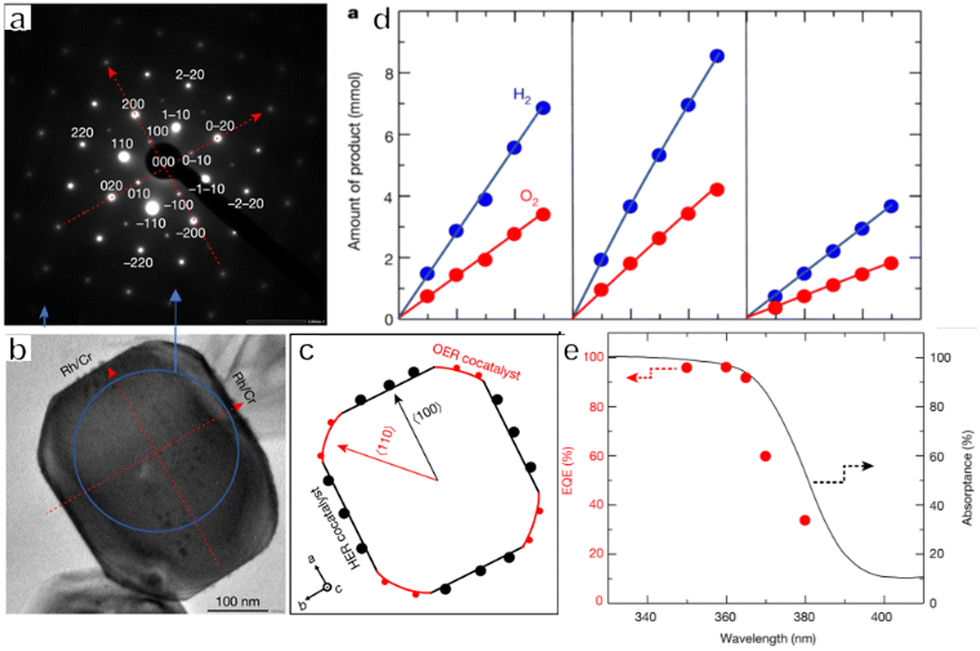 | ||
| Fig. 3 (a) Selected-area electron diffraction pattern and (b) corresponding transmission electron microscopy image of SrTiO3:Al and (c) crystal orientation. (d) Time course of the overall water splitting reaction of SrTiO3:Al loading with Rh/Cr2O3 (left), Rh/Cr2O3/CoOOH (middle) and Rh–Cr oxide (right). (e) UV-Vis diffuse reflectance spectrum of bare SrTiO3:Al (black solid line) and external quantum efficiency (EQE) of Rh/Cr2O3/CoOOH-loaded SrTiO3:Al (red symbols) (reprinted with permission from ref. 19 Copyright 2020 Springer Nature. The Authors, under exclusive license to Springer Nature Ltd). | ||
In addition to metal-containing photocatalysts, low-cost, non-toxic and environmentally friendly metal-free polymers have emerged as a new type of OWS photocatalyst in recent years. Polymeric carbon nitride (PCN) is one such material, and has been intensively studied over the past decade.38,61 PCN prepared by thermal polymerization shows bulk morphology with an amorphous phase, while exhibiting moderate performance in photocatalytic water splitting systems. Many methods, such as co-polymerization, doping and exfoliation, have been applied to modify PCN properties in order to enhance activity.62–65 However, the poor crystallinity of PCN hinders the transfer of charges, thus increasing the probability that photogenerated electrons and holes will recombine, thus lowering the photocatalytic performance of the material. In 2017, Wang's group utilized crystalline polytriazine imide intercalated with Li and Cl ions (PTI/Li+Cl−) as a photocatalyst to achieve OWS under UV light.4 The PTI/Li+Cl− synthesized from a eutectic KCl/LiCl mixture showed improved crystallinity. The structure defects which acted as recombination centres of photogenerated electrons and holes could be decreased as a result of the improved crystallinity. Therefore, the charge carrier lifetime was prolonged and more electrons and holes could participate in water splitting reactions. The bandgap of PTI/Li+Cl− determined by the UV-Vis DRS was around 3.1 eV. Although PTI/Li+Cl− can only respond to UV-light, the bandgap straddled the redox potential for water splitting, which guaranteed that overall water splitting was thermodynamically feasible with a relatively large driving force. After incorporating Pt and Co as dual co-catalysts providing a synergistic effect, an optimized sample showed an AQY of 2.6% at 365 nm during OWS. Subsequently, Wang's group examined the reactive facets of PTI/Li+Cl− on the molecular level using spherical aberration electron microscopy together with theoretical calculations (Fig. 4a–e).66 Assessments of the positions of hydrogen and oxygen evolution cocatalysts loaded using a photodeposition method confirmed that both electrons and holes were primarily transferred to energetically favourable {10![[1 with combining overline]](https://www.rsc.org/images/entities/char_0031_0305.gif) 0} planes. These results also indicated that prismatic {100} planes were more active than basal {0001} planes. On this basis, PTI/Li+Cl− crystals with different aspect ratios were prepared by adjusting the synthesis temperature, and the largest ratio of {1010} to {0001} surface areas (with a value of 5) was obtained at 550 °C. Pt and Co cocatalysts were loaded on the active {1010} facets and an improved photocatalytic OWS performance resulted from the synergistic effect of these cocatalysts, giving an AQY of 8% at 365 nm. It was also found to be possible to further increase the extent of π-conjugation in this material while reducing the number of defects by changing the molten salt species. These modifications promoted charge separation and thus improved the photocatalytic efficiency. As a result, the PTI/Li+Cl− prepared in LiCl/NaCl showed an improved AQY of 12% (λ = 365 nm) during photocatalytic OWS, compared with that prepared in the eutectic LiCl/KCl mixture.67
0} planes. These results also indicated that prismatic {100} planes were more active than basal {0001} planes. On this basis, PTI/Li+Cl− crystals with different aspect ratios were prepared by adjusting the synthesis temperature, and the largest ratio of {1010} to {0001} surface areas (with a value of 5) was obtained at 550 °C. Pt and Co cocatalysts were loaded on the active {1010} facets and an improved photocatalytic OWS performance resulted from the synergistic effect of these cocatalysts, giving an AQY of 8% at 365 nm. It was also found to be possible to further increase the extent of π-conjugation in this material while reducing the number of defects by changing the molten salt species. These modifications promoted charge separation and thus improved the photocatalytic efficiency. As a result, the PTI/Li+Cl− prepared in LiCl/NaCl showed an improved AQY of 12% (λ = 365 nm) during photocatalytic OWS, compared with that prepared in the eutectic LiCl/KCl mixture.67
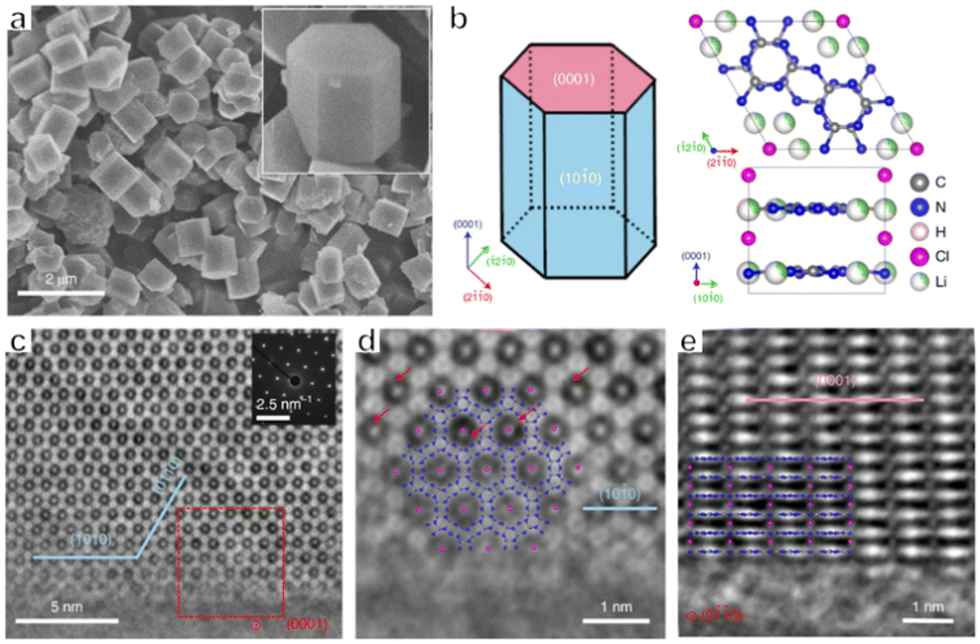 | ||
| Fig. 4 (a) SEM images of PTI/Li+Cl− crystals. (b) Crystal structure of PTI/Li+Cl−. (c) AC-iDPC image of a typical PTI/Li+Cl− crystal aligned along the [0001] direction with corresponding diffraction patterns. (d) Enlarged view of the area in the red box in (c). (e) AC-iDPC image of a typical PTI/Li+Cl− crystal aligned along the [20] direction (reprinted with permission from ref. 66 Copyright 2020 Springer Nature. The Authors, under exclusive license to Springer Nature Ltd). | ||
Photocatalysts intended to promote one-step excitation OWS under visible light have been reported since 2005.68 One of the earliest representative examples of visible-light-driven OWS was based on the GaN:ZnO solid solution system, representing a wurtzite-type semiconductor. The AQY obtained from this system during OWS was approximately 5.1% at 410 nm using a mixed Rh–Cr oxide as the cocatalyst. In addition, hollow GaN:ZnO spheres with a nanostructured morphology were reported to show an AQY of 17.3% at 400 nm.69,70 This value was three times higher than that for the bulk phase, due to the reduced distance required for carrier diffusion to the surface and the increased surface area exposing more active sites. These studies demonstrated that (oxy)nitrides can be used as non-oxide-based photocatalysts for solar energy conversion.
In 2013, the transition metal-based oxynitride photocatalyst ZrO2-modified TaON (ZrO2/TaON), was shown to be active during the OWS reaction under visible light.71 ZrO2/TaON has a bandgap of 2.5 eV and displays an enhanced capacity for water reduction along with an AQY of approximately 0.1% at 420 nm after loading with suitable nanoparticle cocatalysts. However, due to the high defect density of the TaON in this material, the lifetimes of photogenerated electrons and holes were reduced, resulting in poor performance. In subsequent work, amorphous Ta2O5·3.3H2O with a particle size of 15 nm was used as a new precursor to synthesize TaON-based photocatalysts with reduced particle sizes and low defect densities.72 After optimization of the degree of Zr doping and nitridation duration together with loading of Ru/Cr2O3/IrO2 cocatalysts, the ZrO2/TaON photocatalyst decomposed water into hydrogen and oxygen with an AQY of 0.66% at 420 nm. The complex perovskite-type oxynitride LaMgxTa1-xO1+3xN2−3x (x ≥ 1/3) was also found to be active during OWS in response to wavelengths up to 600 nm.73 After fine-tuning of the composition and the application of surface coatings, this material promoted OWS with an AQY of approximately 0.03% at 440 ± 30 nm.
Ta3N5, a nitride semiconductor, is another promising photocatalyst for visible-light-driven OWS due to its simple chemical composition, narrow bandgap of 2.1 eV and band position suitable for both H+ reduction and H2O oxidation.74–76 However, this material was not applied to OWS until 2018, presumably because it contains a high density of defects. Our group synthesized single-crystal nanorods of Ta3N5 on KTaO3via a brief NH3 nitridation. These nanorods were found to be free from grain boundaries and defect states (Fig. 5a–e).77 When decorated with a Rh/Cr2O3 cocatalyst with a core-shell structure, this photocatalyst promoted OWS under visible light with an estimated STH of 0.014% and an AQY of 0.22% at 420 nm (± 25 nm). These findings confirm the importance of obtaining nanostructured single-crystal photocatalysts without structural defects so as to realize effective OWS.
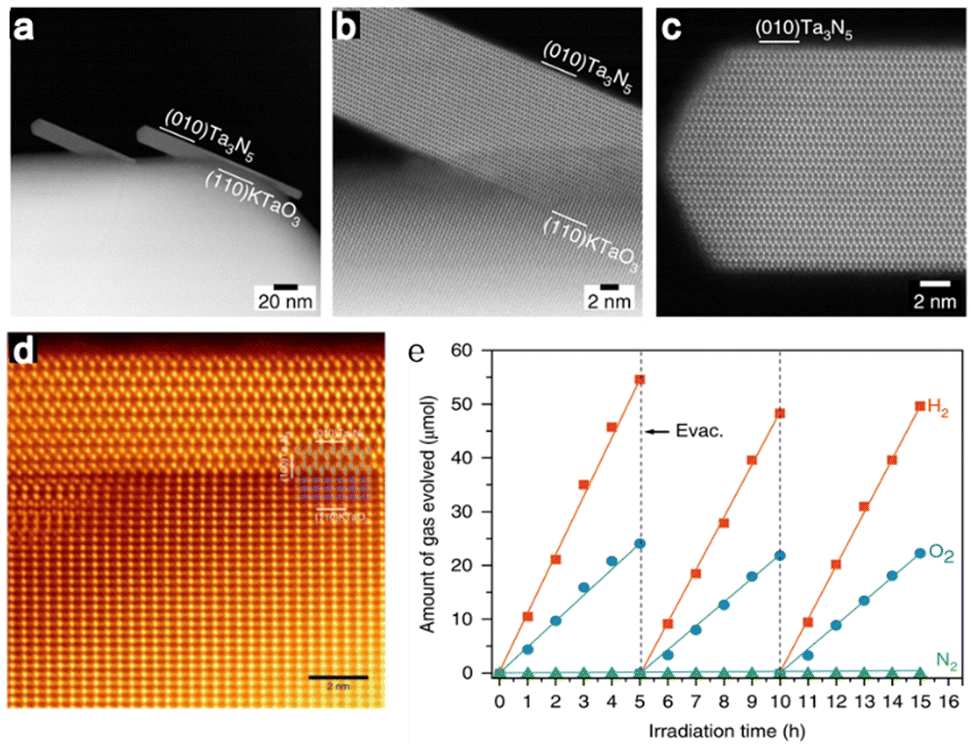 | ||
| Fig. 5 (a–c) ADF-STEM images of Ta3N5/KTaO3. (d) Colourized and magnified ADF-STEM image of Ta3N5 nanorods from the [001] direction. (e) Time courses of OWS on Rh/Cr2O3-modified Ta3N5/KTaO3 under visible light irradiation (reprinted with permission from ref. 77 Copyright 2020 Springer Nature. The Authors, under exclusive license to Springer Nature Ltd). | ||
In addition to (oxy)nitrides, (oxy)sulfides have also attracted attention due to their potential applications to OWS.78–80 Certain metal chalcogenides absorb visible light over a wide range of wavelengths extending as far as the near infrared depending on their composition, owing to their narrow bandgaps.81 Unfortunately, the water oxidation reaction over these materials competes with the oxidation of sulfide ions in the lattice. Therefore, metal sulfides are often prone to photocorrosion and are unstable during the OWS reaction. Compared with sulfides, oxysulfides are more stable, likely because of the hybridization of S 3p and O 2p orbitals in these compounds. Y2Ti2O5S2 (YTOS) is an oxysulfide semiconductor with a layered perovskite-like structure and a bandgap of 1.9 eV. After being modified with IrO2 and Rh/Cr2O3 as cocatalysts for the evolution of O2 and H2, respectively, YTOS exhibited an AQY at 420 nm of 0.36%, with an STH energy conversion efficiency of 7 × 10−3% during the OWS reaction (Fig. 6a–e).82 This relatively poor performance can possibly be attributed to the large particle size of this material, its high defect density, and sluggish surface reactions. A recent study of YTOS using transient diffuse reflectance spectroscopy (TDRS) found that the decay dynamics of the absorption signal in the early stage was associated with the bimolecular recombination of mobile charge carriers.83 In contrast, the late microsecond range was dominated by hole detrapping from exponential tail trap states near the valence band. Based on these data, a theoretical model has been proposed that allows the estimation of various factors that limit efficiency. These include the recombination rate constant and tail state parameters. A diffusion length of 126 nm was estimated from the TDRS results, which is significantly smaller than the typical particle size of YTOS. The calculated internal quantum efficiency was improved substantially, from 7% to 81%, by decreasing the particle size from 10 μm to 500 nm while preserving the crystallinity and density of defects. Therefore, reducing the crystal size is one important approach to improving the performance of this catalyst.
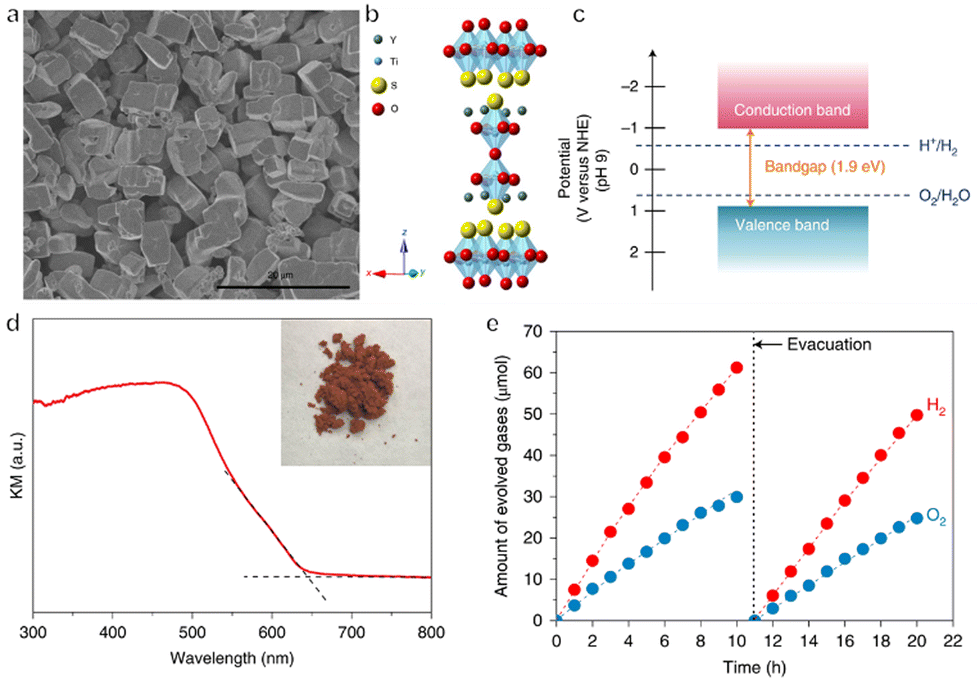 | ||
| Fig. 6 (a) SEM image, (b) crystal structure, (c) band structure and (d) diffuse reflectance spectrum of Y2Ti2O5S2. (e) Time course of overall water splitting on Cr2O3/Rh/IrO2-loaded Y2Ti2O5S2 (reprinted with permission from ref. 82 Copyright 2019 Springer Nature. The Authors, under exclusive license to Springer Nature Ltd). | ||
Some materials have emerged as a new type of visible-light-responsive photocatalysts, different from conventional to inorganic photocatalysts, for a one-step excitation OWS reaction. For example, the most widely studied conjugated polymer of PCN can realize OWS reaction under visible light irradiation after loading with Pt co-catalysts. It was found that the metallic Pt and PtO acted as H2 and O2 evolution co-catalysts, respectively. The activity was significantly enhanced by co-loading of CoOx as an O2 evolution cocatalyst.84 By assembling highly crystalline and ultrathin nanosheets, Zhang's group prepared a three-dimensional porous PCN. The nanosheets presented a 3D interconnected open-framework with an increased specific surface area, which could act as a support to avoid restacking of nanosheets and provide a pathway for the transfer of photogenerated charge carriers. As a result, the 3D PCN nanosheets realized efficient and stable OWS under visible-light irradiation with an AQY of 1.4% at 420 nm.85 In recent years, metal–organic frameworks (MOFs) have been attractive as the photocatalysts in the water splitting reaction due to its semiconductor-like property.86,87 The aluminium-based MOF derived from 2-aminoterephthalic acid H2ATA (Al-ATA MOF) works an O2 evolution photocatalysts, and the benzene ring of ATA2− has been identified as the active site for O2 evolution. By incorporating Ni2+ ions as the H2 evolution site into the Al-ATA MOF by coordination to the amino groups, Huang's group achieved a one-step excitation OWS reaction using the as-obtained Al-ATA-Ni MOF. It was also found the Ni2+ cation coordinated to the amino group of ATA2− not only acted as a H2 evolution site but also enhanced the O2 evolution at the benzene ring bearing the amino group.88 Bu's group applied the MOF of NH2-UiO-66 as a photocatalyst in the OWS reaction. In order to enhance the charge separation, Pt and MoOx were decorated on NH2-UiO-66. Evolution of almost stoichiometric H2 and O2 was observed under visible-light irradiation.89 More recently, Horcajada's group reported a new porous titanium(IV) squarate MOF denoted as IEF-11. This MOF showed a combination of the photo- and electro-activity originating from its Ti-metal nodes and squarate ligands, making IEF-11 an excellent candidate for photocatalysis. The OWS reaction occurred under the irradiation of simulated sunlight or visible-light without noble metals or O2 evolution co-catalysts. A STH of 0.001% was obtained at a reaction temperature of 35 °C under simulated sunlight irradiation.90
4. Two-step excitation overall water splitting based on particle suspension systems
The mismatch between the band position for a photocatalyst and the redox potential of water prevents some photocatalysts from being applied to one-step excitation water splitting. However, using the two-step excitation process that mimics natural photosynthesis, this problem can be addressed to some extent.43 In Z-scheme systems, H2 and O2 are generated on the HEP and OEP, respectively, and holes and electrons in these photocatalysts are quenched by electron mediators to complete the water splitting process.35 HEPs and OEPs only need to satisfy the thermodynamic conditions for the corresponding water splitting half-reaction, which greatly expands the range of available photocatalysts.91 In general, improving the degree of light absorption and charge separation and migration, as well as promoting the surface catalytic reactions of the photocatalyst, are all important, as is also the case for one-step excitation systems. However, there are many additional factors affecting the performance of Z-scheme water splitting processes. As an example, the reactivity and redox potential of the electron shuttle must be carefully considered in order to promote forward electron transfer from the OEP to the HEP.92A Z-scheme model based on inorganic semiconductors was described in 197993 and, in 2001, a suspension system was demonstrated. This system combined Pt-loaded TiO2-anatase as the HEP, TiO2-rutile as the OEP and IO3−/I− as the redox mediator.94 Since this initial report, a variety of narrow-bandgap semiconductors capable of absorbing visible light and applicable to Z-scheme OWS have attracted attention. These have included TaON, BaTaO2N and Rh-doped SrTiO3 as HEPs with WO3, BiVO4 and AgNbO3 as potential OEPs. I, Fe, Co or Mn-based aqueous redox mediators have been widely investigated as components of Z-scheme water splitting systems as a means of promoting electron transfer from OEPs to HEPs. In addition, electron transfer via solid-state electronic conductors and physical contact between photocatalyst particles has also received significant interest in recent years.
The IO3−/I− pair is commonly used as a redox mediator in Z-scheme systems in conjunction with various types of photocatalyst. Li's group synthesized a MgTa2O6−xNy/TaON heterostructure using a one-pot nitridation route.95 The resultant interface had a low defect density that allowed efficient spatial charge separation and effectively suppressed carrier recombination, thereby enhancing the H2 evolution performance. A system based on Pt-loaded MgTa2O6−xNy/TaON as the HEP, PtOx–WO3 as the OEP, and IO3−/I− as the electron mediator demonstrated Z-scheme OWS with an AQY of 6.8% at 420 nm. Subsequently, our group demonstrated a single-crystal particulate BaTaO2N photocatalyst prepared with the assistance of a RbCl flux.96 The stepwise loading of a Pt cocatalyst via impregnation-reduction and subsequent photodeposition generated highly dispersed and uniformly sized Pt active sites on this material. These sites enabled the efficient injection of photogenerated electrons into the Pt cocatalyst to generate H2. When Pt-loaded BaTaO2N was combined with surface-treated WO3 as the OEP, the AQY at 420 nm and the Z-scheme water splitting STH were 4.0% and 0.24%, respectively. This Ta3N5 photocatalyst can also be used as the HEP in Z-scheme water splitting systems following suitable modification. In prior work, one-pot nitridation of Ta2O5 precursors impregnated with Ba(NO3)2 was used to synthesize a Ta3N5/BaTaO2N heterostructure. The Ta3N5 surface in this material was passivated by the BaTaO2N. In addition, electron transfer from the BaTaO2N to the Ta3N5 and hole transfer in the opposite direction were found to be possible. These effects promoted charge separation at the interface of the heterostructure. As a result, Ta3N5/BaTaO2N outperformed Ta3N5, BaTaO2N and physical mixtures of the two when employed as the HEP. OWS was achieved by forming a Z-scheme system with PtOx/WO3 as the OEP and an AQY of 0.1% was obtained at 420 nm.28 In a following work, the same group obtained a heterostructure composed of one-dimensional Ta3N5 nanorods and BaTaO2N nanoparticles by a simple ammonia thermal synthesis. It was demonstrated that intimate contact was formed between Ta3N5/BaTaO2N heterostructure interfaces and therefore photoinduced charge separation was enhanced. In combination with PtOx/WO3 as the OEP and IO3−/I− as the electron mediator, the as-constructed Z-scheme system showed one order of magnitude enhanced solar overall water splitting activity with respect to counterpart systems.97 Recently, a pyrochlore-structural oxynitride Nd2Ta2O5N2 was explored as the HEP which can absorb visible-light up to 620 nm. After modification with a Pt co-catalyst, effective Z-scheme OWS reaction was achieved under visible-light irradiation coupled with the OEP of PtOx/WO3.98 Research has shown that (oxy)sulfides can also be used in Z-scheme systems. Our group established that a series of oxysulfides could be used as HEPs to realize OWS in Z-scheme systems. Sm2Ti2O5S2 (STOS) prepared by a flux method exhibiting higher crystallinity and better photocatalytic activity than that prepared via a traditional solid state reaction. In addition, STOS loaded with a reductive Pt cocatalyst, an oxidative IrO2 cocatalyst, and the modified WO3 were used as the HEP and OEP, respectively.99 The electron mediator I3−/I− was employed to complete the Z-scheme. La5Ti2CuS5O7 and La6Ti2S8O5 were also found to be applicable as HEPs in Z-scheme systems.99 Even though the STH values obtained from such experiments were low (at most 3 × 10−3%), this work demonstrated that narrow-bandgap oxysulfide photocatalysts can be used in visible-light-driven Z-scheme OWS systems. Recently, some metal complex photosensitizers have also been employed in Z-scheme systems. Maeda's group reported a visible-light-driven OWS system based on HCa2Nb3O10 nanosheets sensitized with Ru(II) tris-diimine-type photosensitizers as the HEP combined with a WO3-based OEP and the I3−/I− redox pair.100 An AQY of 2.4% was achieved at 420 nm, which was the highest value reported for a water splitting system utilizing dye sensitizers at that time. There were several reasons for the high AQY of the HCa2Nb3O10-based Z-scheme system. Firstly, the inherent properties of HCa2Nb3O10 nanosheets such as the anisotropic structure, single-crystalline texture, and high surface area were beneficial for dye-sensitized H2 evolution in terms of rapid electron transport through the semiconductor to H2 evolution sites and the adsorption of photosensitizers. Secondly, the Pt co-catalyst was selectively deposited in the interlayer galleries of the HCa2Nb3O10 nanosheet, which prevented the reduction of I3− by confining the H2 evolution cocatalyst to interlayer sites that were inaccessible to anions. Thirdly, modification of HCa2Nb3O10 with Al2O3 can attract I− ions to its surface and prompt the regeneration of photosensitizer. More recently, the same group employed modified Ru dye-sensitized Pt-intercalated HCa2Nb3O10 nanosheets (Ru/Pt/HCa2Nb3O10) with both amorphous Al2O3 and poly(styrenesulfonate) (PSS) as the HEP coupled with a WO3-based photocatalyst as the OEP and the I3−/I− redox shuttle to construct a Z-scheme OWS system. Under the optimized conditions, the AQY at 420 nm was increased to 4.1% and an STH of 0.12% was obtained. Modification of HCa2Nb3O10 nanosheets with PSS can suppress the reduction of I3− but the reaction with I− was also decelerated. Therefore, a significantly enhanced performance was reached by dual modification of the nanosheet photocatalyst with Al2O3 and PSS.101 Similar to one-step excitation OWS, conjugated polymer can also be used in Z-scheme systems. Tang's group constructed a visible-light-driven system by using PCN as the HEP. The highest performance was achieved with the Pt-modified WO3 as the OEP and IO3−/I− as the redox mediator in their work.102
The Fe3+/Fe2+ pair is another commonly used redox mediator. Recently, mixed anion compounds (in particular oxyhalides such as Bi4NbO8Cl and Bi4TaO8Br having single-layer perovskite structures) have been investigated as OEPs due to their efficient O2 evolution performance under visible light when in aqueous solutions containing Fe3+ or Ag+ ions.103,104 When combined with Rh-doped SrTiO3 as the HEP and Fe3+/Fe2+ as an electron mediator, these materials can promote Z-scheme OWS. Theoretical calculations have confirmed that the VBM for Bi4NbO8Cl is composed of highly dispersed O 2p orbitals rather than Cl 3p orbitals as is the case for BiOCl and BiOBr. This band structure likely originates from the relatively strong interactions within and between the Bi–O and Nb–O layers in such materials. The delocalized O 2p orbitals result in a more negative VBM potential than is typical, leading to a narrow bandgap and resistance to oxidative photocorrosion. Unfortunately, the OWS reaction must be carried out at a pH of less than 2.5 because Fe3+ readily precipitates as Fe(OH)3 at higher pH. This requirement makes the Z-scheme system rather unstable and restricts the range of photocatalysts that can be used. For these reasons, the Fe-containing complex [Fe(CN)6]3−/[Fe(CN)6]4−, which can be used under much milder conditions such as a pH of 6–7, has recently been applied as a redox mediator.91,105 The lower redox potential of E° = 0.357 V vs. NHE associated with such systems also favours the storage of solar energy. Li's group reported that BiVO4 and ZrO2-modified TaON could be employed as the OEP and HEP, respectively, to construct a Z-scheme system together with [Fe(CN)6]3−/[Fe(CN)6]4− as the redox mediator (Fig. 7a and b).106 An in situ facet-selective photodeposition was used to apply the dual cocatalysts FeOOH and CoOOH to the {110} facets of the BiVO4. This modification reduced the activation energy barrier to water oxidation and also promoted charge separation and transfer. An Ir cocatalyst was simultaneously deposited on the {010} facets of the BiVO4 and the modified materials demonstrated a superior ability to reduce [Fe(CN)6]3−. In a subsequent Z-scheme OWS reaction, the AQY of the system at 420 nm was 12.3% and the STH was 0.6%. The aforementioned GaN:ZnO solid solution can also be used in the Z-scheme system but the relatively wide bandgap limited the further improvement of its performance. Recently, Liu et al. reported the preparation of GaN:ZnO by heating a mixture of Ga2O3, Zn, and NH4Cl in a sealed evacuated tube. Importantly, the bandgap of GaN:ZnO was reduced from 2.7 to 2.3 eV owing to the high ZnO content. A Z-scheme system was then constructed by using the prepared GaN:ZnO as the OEP and SrTiO3:Rh as the HEP in combination with Fe3+/Fe2+ as the electron mediator. The STH efficiency of 3.7 × 10−2% was achieved with remarkable stability up to 100 h.107 Cooper's group explored a range of conjugated polymer photocatalysts for the hydrogen evolution half-reaction and various metal oxide materials as photocatalysts for oxygen production. It was found that P10 (homopolymer of dibenzo[b,d]thiophene sulfone) and BiVO4 showed the highest activity in the presence of FeCl2 and FeCl3 aqueous solution, respectively, for H2 and O2 half-reactions. Therefore, a Z-scheme system was constructed by using the P10 polymer as the HEP and BiVO4 as the OEP, together with Fe3+/Fe2+ as the redox mediator. Although the STH was very low, this study indicated that linear polymers can be used in the Z-scheme system.108
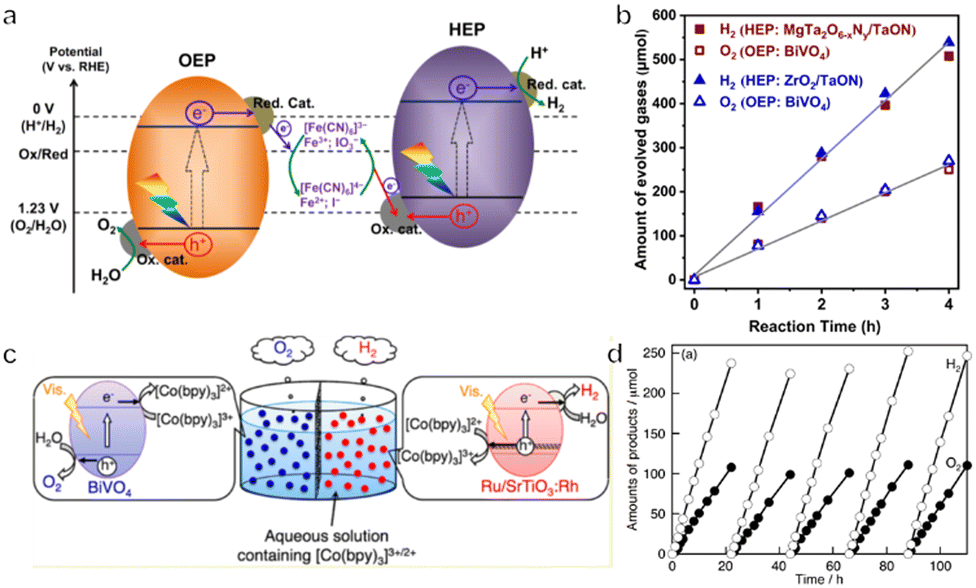 | ||
| Fig. 7 (a) The energy diagram for a two-step photoexcitation system. (b) Time courses of Z-scheme overall water splitting using BiVO4 as the HEP and TaON as the OEP (reprinted with permission from ref. 106 Copyright 2022 Springer Nature). (c) A schematic diagram by using Ru/SrTiO3:Rh, BiVO4 and [Co(bpy)3]2+/3+ as the HEP, OEP and redox mediator, respectively. (d) The time course of overall water splitting using 0.1 g of each photocatalyst under visible light (reprinted with permission from ref. 49 Copyright 2013 American Chemical Society). | ||
Various other metal complexes can also be used as redox mediators, among which cobalt complexes such as [Co(bpy)3]3+/[Co(bpy)3]2+, [Co(phen)3]3+/[Co(phen)3]2+ and [Co(terpy)3]3+/[Co(terpy)3]2+ (terpy = 2,2′:6′,2′′-terpyridine) are commonly employed.49 OWS has been reported to be possible with cobalt complexes as redox mediators in conjunction with Ru/SrTiO3:Rh or (CuGa)1−xZn2xS2 as the HEP and BiVO4 as the OEP (Fig. 7c and d). The redox potential and reactivity of such complexes can also be controlled by changing the central atoms and ligands. Some polyoxometalates with suitable redox potentials and redox reversibility, including [SiW11O39MnIII(H2O)]5−/[SiW11O39MnII(H2O)]6−, are also applicable as the redox couple.109
In early Z-scheme OWS systems, ionic pairs were typically used as redox mediators. In such cases, the mediators were selected by considering the CBM and VBM positions and the reactivity of the HEP and OEP so as to obtain highly efficient OWS. However, ionic redox mediators can absorb some portion of the visible light received by the system or possibly corrode the photocatalyst. In addition, the photocatalyst had to have the capacity to adsorb and desorb the mediator in order to effectively transfer electrons. Finally, the redox mediators would often undergo backward reactions that were thermodynamically more favourable than the water splitting reaction. As a result of all these effects, the use of ionic redox mediators limited the OWS efficiency that could be achieved. To address this issue, solid-state electron mediators such as reduced graphene oxide (RGO) or physical contact are now used to promote interparticle transport of photoexcited charges in Z-scheme systems. Kudo's group conducted extensive research regarding RGO and confirmed that this material can function as an electron mediator (Fig. 8a and b).40,110,111 Specifically, in the case of a Z-scheme system with RGO as the electron mediator, Pt/CuGaS2 as the HEP, and TiO2 as the OEP, the AQY at 380 nm reached 1.3%. It is also possible to construct RGO-based Z-scheme water splitting processes based on polymeric and inorganic photocatalysts. Wang's group linked PCN nanosheets and BiVO4 nanoparticles with RGO via chemical bonding and π–π stacking to form a PCN/RGO/BiVO4 Z-scheme system showing improved photocatalytic efficiency.112 Moreover, Li's group developed a plasmonic-based solid Z-scheme OWS system.113 The Au nanoparticles in this system acted as both charge transfer channels and photoabsorbers due to the surface plasmon resonance effect. Hot electrons in the visible-light-responsive plasmonic Au nanoparticles are thought to have migrated from TiO2 to SrTiO3:Rh through the solid-contact interfaces.
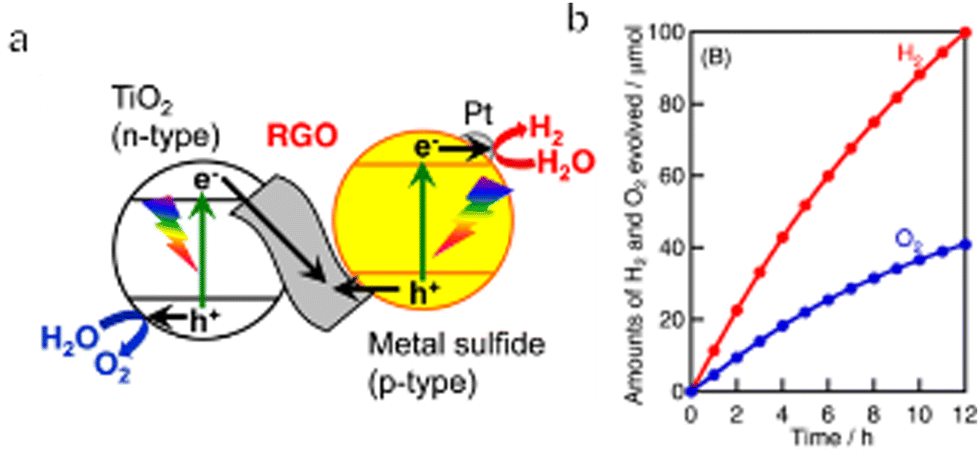 | ||
| Fig. 8 (a) Diagram of a Z-scheme system containing Pt-loaded metal sulfide photocatalyst as the HEP, TiO2 as the OEP and RGO as a solid-state electron mediator. (b) Time course of overall water splitting using Pt-CuGaS2 and RGO-TiO2 under simulated sunlight irradiation (reprinted with permission from ref. 40 Copyright 2014 American Chemical Society). | ||
In addition to redox mediators, some photocatalysts can be made to carry different types of charge by changing the pH of the solution. As a consequence, the HEP and OEP can come into close contact via coulomb interactions to promote charge transfer if they carry opposite charges. Kudo's group reported that particles of Ru/SrTiO3:Rh (as the HEP) and BiVO4 (as the OEP) had opposing charges in an aqueous solution with a pH of 3.5 and so aggregated in response to electrostatic attraction.114 This mechanism permitted photocatalytic water splitting using a Z-scheme suspension under visible light in the absence of an electron mediator. The intimate contact that can be obtained by tuning the pH of the solution is a simple means of realizing charge transfer between the HEP and OEP without an electron mediator. However, the efficiency of such systems may be lower than that obtained using redox shuttles because charge transfer is based solely on physical contact. In addition, acidic conditions may destabilize certain photocatalysts and so reduce their performance. As such, this method is not always applicable and the pH of the reaction solution must be carefully selected based on the photocatalysts being employed. Construction of a heterostructure is another pathway to promote the charge transfer between HEP and OWP without the assistance of an electron mediator. Xu's group used aza-fused microporous polymers (CMP) and C2N ultrathin nanosheets as OEP and HEP, respectively, to construct van der Waals heterostructures. The OWS reaction was successfully achieved by this Z-scheme system irradiated under visible-light.115 More recently, Guo's group reported two-dimensional heterostructures based on PCN nanosheets. Efficient OWS reaction was realized by electrostatic self-assembly of boron-doped and nitrogen-deficient PCN nanosheets obtained by thermal treatment at different temperatures.116
5. Overall water splitting via immobilized particulate systems
To date, one-step or two-step excitation OWS systems have been primarily demonstrated on a lab-scale in the form of particulate suspension systems. However, it is difficult to scale up these processes because maintaining the dispersion of a large quantity of particulate photocatalysts in water requires an additional energy input. Also, recycling the photocatalyst powder from the suspension will greatly increase the cost of the operation. Therefore, it would be preferable to immobilize the photocatalyst powder on a substrate so that the material can be processed into water splitting panels. These panels can then be easily handled and scaled up for practical applications. Even so, it is necessary to design panel structures that allow for the mutual transport of water as the reactant together with the hydrogen and oxygen generated as products. The adhesion of gas bubbles to the surfaces of these panels may also be an issue, as has been observed in trials with planar photoelectrochemical devices.8 In addition, both mass and charge transfer will be limited in these systems, such that the performance of a fixed particle process will generally be lower than that of the corresponding suspended particle system if there is no further optimization. For all these reasons, the construction and modification of immobilized photocatalytic particle systems are critical to maintaining the original performance observed in a suspension system.In the case of one-step excitation OWS systems, accelerating the access of water to the photocatalyst particle layer as well as the release of evolved gases is vital to improving performance. Our group previously attempted to fabricate panels using a Rh2−yCryO3/(Ga1−xZnx)(N1−xOx) photocatalyst in 2014.117 In this work, the photocatalyst was combined with micron-sized hydrophilic silica particles and applied to 5 cm × 5 cm frosted glass plates. The high porosity of the hydrophilic silica layers enabled efficient diffusion of the reactants and products and the resulting panels exhibited comparable OWS activity to that of a suspension of the same photocatalyst. A demonstration reactor based on these panels and having an area of 0.756 m2 was later constructed by fixing a PCN photocatalyst on stainless steel plates.118 Hydrogen evolution proceeded in the presence of a sacrificial agent in these trials and it was confirmed that this panel type photoreactor could be scaled up to a large area without performance losses. As a means of achieving OWS in pure water, which is more challenging, our group constructed a solar-powered photocatalytic water splitting panel based on an Al-doped SrTiO3 photocatalyst immobilized on glass substrates (Fig. 9a–d).30 A panel reactor filled with a 1 mm-deep layer of water was able to release gaseous products smoothly without forced convection. In addition, a flat-panel reactor with a light-receiving area of 1 m2 achieved an STH of 0.4% during water splitting under natural sunlight with an intensity of 65–75 mW cm−2. This work established that easily scalable water splitting panels are a viable approach to performing OWS on a large scale. In 2021, we further improved and expanded this system to produce a 100 m2 array of flat-panel reactors, representing the largest photocatalytic solar hydrogen production unit yet created by far (Fig. 10a–c).119 Purified hydrogen could be automatically recovered from the mixture of hydrogen and oxygen generated as products using commercial polyimide membranes. This system was both durable and safe and exhibited essentially stable operation during 1600 h of continuous illumination under laboratory conditions. Moreover, the system was undamaged following the intentional ignition of the collected hydrogen. A maximum STH energy conversion efficiency of 0.76% was observed. Although this performance is below the range of 5% to 10% required for economically viable solar hydrogen production, safe, large-scale photocatalytic water splitting, gas collection and separation were shown to be feasible.
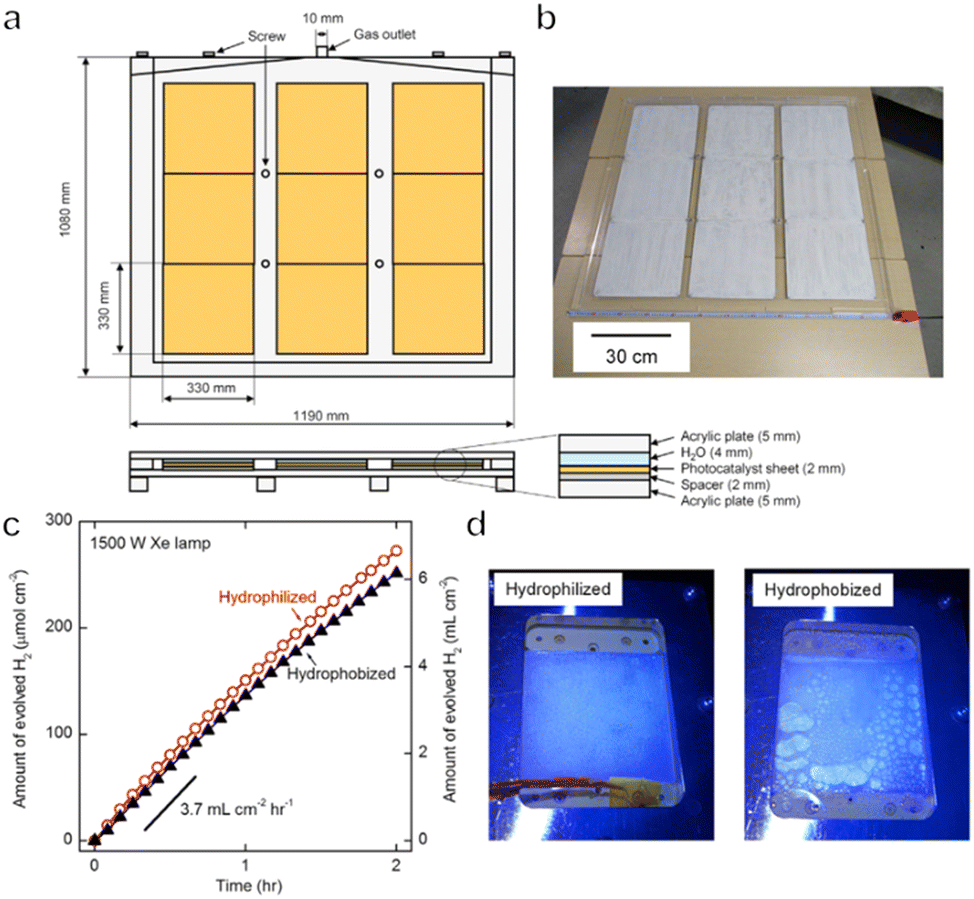 | ||
| Fig. 9 (a) Schematics and (b) photograph of a 1 × 1 m SrTiO3:Al water-splitting panel. (c) Time courses of water splitting over 5 × 5 cm SrTiO3:Al. (d) Photographs acquired with illumination through hydrophilized and hydrophobized windows (reprinted with permission from ref. 30 Copyright 2017 Elsevier Inc). | ||
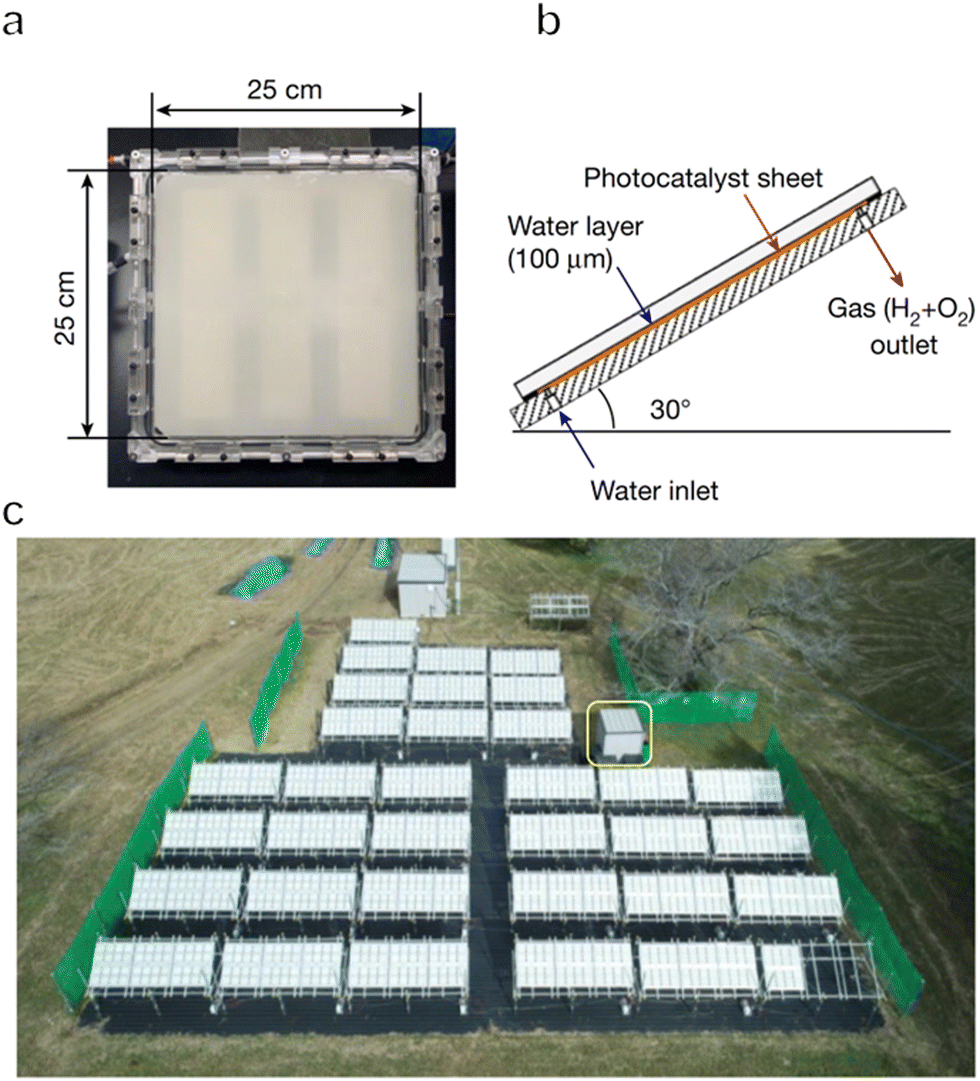 | ||
| Fig. 10 (a) Photograph and (b) diagram of a panel reactor unit (625 cm2). (c) Overhead view of a 100 m2 solar hydrogen production system consisting of 1600 panel reactor units and hut housing gas separation facility, indicated by a yellow box (reprinted with permission from ref. 109 Copyright 2021 Springer Nature. The Authors, under exclusive license to Springer Nature Ltd). | ||
Immobilized two-step excitation systems also have significant potential owing to the applicability of narrow-bandgap photocatalysts to such processes. In this type of system, the HEP and OEP particles are fixed on a conductive layer by various means. Intimate contact between the photocatalysts and the conductive material is the key to facilitating effective charge transfer between the OEP and HEP. Early research used photocatalyst sheets based on SrTiO3 co-doped with La and Rh (SrTiO3:La,Rh) as the HEP, and Mo-doped BiVO4 (BiVO4:Mo) as the OEP embedded in an Au layer for OWS. The as-constructed system showed an AQY of 5.9% under 418 nm monochromatic light, with an STH of 0.2%.50 In subsequent works, charge transfer between HEP and OEP was enhanced by annealing the sheets in air. This was likely due to the formation of an Ohmic contact by annealing metal/semiconductor composites at certain temperatures according to previous studies.120,121 The reduced contact resistance between the semiconductors and the metal layer prompts the charge transfer. In addition, the backward reactions were suppressed by surface modification with Cr2O3 and the STH was improved to 1.1% while the AQY exceeded 30% at 419 nm (Fig. 11a–c).22 This work demonstrated that Z-scheme particulate systems can also be processed into photocatalyst sheets by introducing a suitable conductive layer. To permit large-scale applications, the expensive Au-based conductive layer could be replaced with other less costly conductors. In particular, attention has shifted to carbon materials. Carbon is an earth-abundant and relatively inert conductive material with a reported work function of 5.0 eV, which is close to the values for Au (5.1 eV) and Rh (5.0 eV). Thus, carbon could be used as a suitable conductive layer material for photocatalytic sheets. In addition, carbon is less active than Au with regard to the oxygen reduction reaction and so could inhibit the reverse reaction to a certain extent. As an example, a SrTiO3:La,Rh/C/BiVO4:Mo sheet showed an STH of 1.2% at 331 K and 10 kPa and retained 80% of this efficiency at 91 kPa (Fig. 12a–d).122 In addition to SrTiO3, some narrow-bandgap photocatalysts can also be used as HEPs for Z-scheme sheet systems. In particular, the oxynitride photocatalyst LaMg1/3Ta2/3O2N has been successfully applied in water splitting sheets. RGO was introduced to this system as a solid electron mediator to further enhance charge transfer between the photocatalytic particles above the Au conductive layer. The water splitting activity over this material was 3.5 times greater than that for a RhCrOx/LaMg1/3Ta2/3O2N/Au/BiVO4:Mo sheet system following the deposition of RGO.123 Similarly, the oxysulfide photocatalyst La5Ti2Cu0.9Ag0.1O7S5 (LTCA) can be used as the HEP in combination with BiVO4 as the OEP. After surface modification, a Cr2O3/Rh/Ga-LTCA/Au/BiVO4 sheet exhibited an AQY of 4.9% at 420 nm and an STH of 0.11%.124 Water splitting sheets have additionally been prepared from the solid solution selenide (ZnSe)0.5(CuGa2.5Se4.25)0.5, acting as the HEP, which absorbs at long wavelengths (up to 680–760 nm).125 A Pt/(ZnSe)0.5(CGSe)0.5/Au/BiVO4:Mo sheet showed obvious OWS activity under visible light. The performance of this system was improved by a factor of 3.2 by forming a p–n junction with CdS.126 Interestingly, both the photocorrosion of CdS and the reverse reaction were suppressed by introducing a thin TiO2 coating (Fig. 13a and b). As a result, an AQY of 1.5% at 420 nm was achieved using the resulting Pt-supported TiO2/CdS-(ZnSe)0.5(CuGa2.5Se4.25)0.5/Au/BiVO4:Mo sheet-type Z-scheme OWS system.
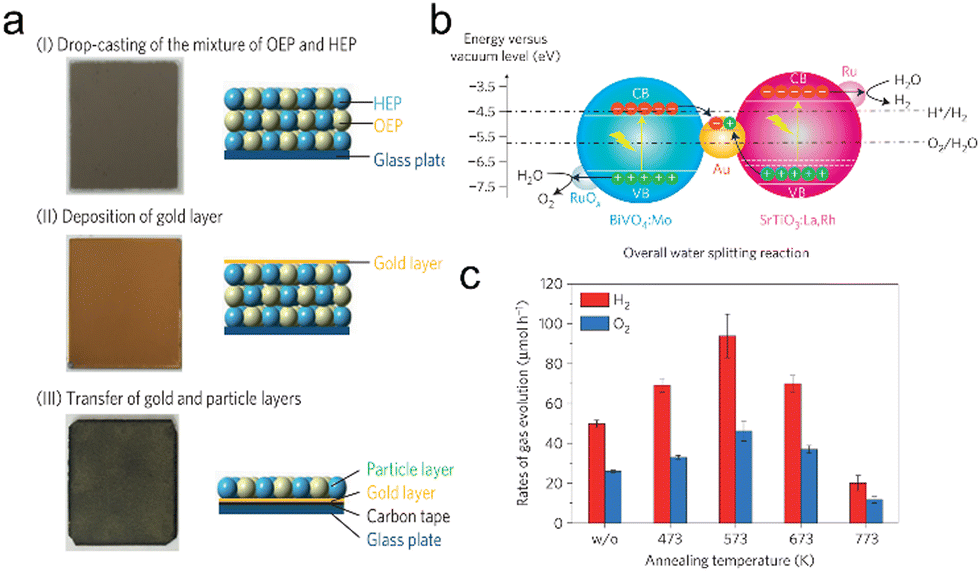 | ||
| Fig. 11 (a) Preparation process and (b) band structure of the SrTiO3:La,Rh/Au/BiVO4:Mo photocatalyst sheet. (c) Overall water splitting performance of SrTiO3:La,Rh/Au/BiVO4:Mo photocatalyst sheets prepared using different annealing temperatures (reprinted with permission from ref. 22 Copyright 2016 Springer Nature. The Authors, under exclusive license to Springer Nature Ltd). | ||
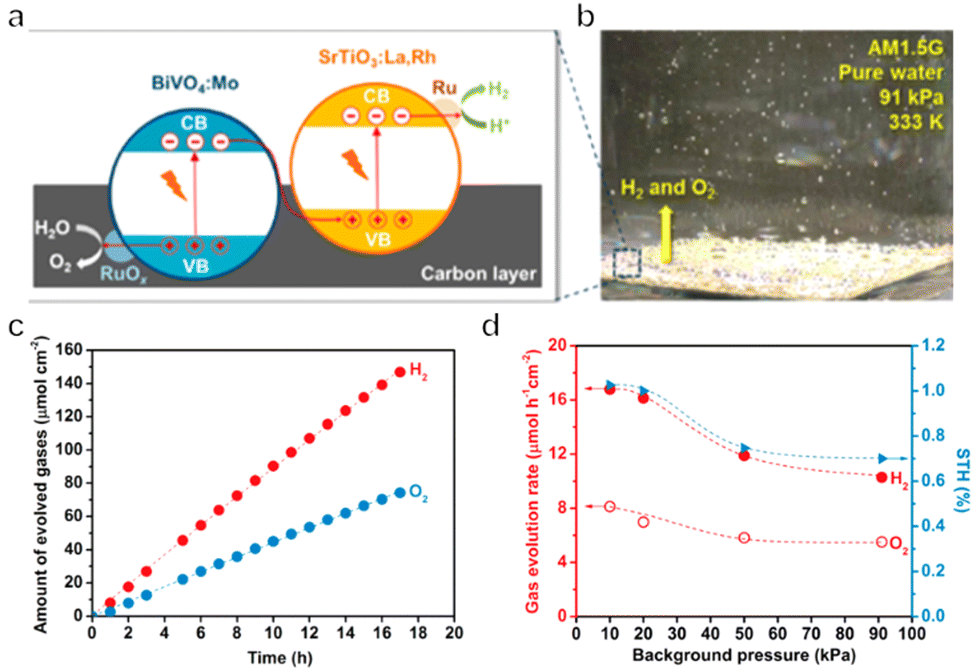 | ||
| Fig. 12 (a) Diagram of aSrTiO3:La,Rh/C/BiVO4:Mo photocatalyst sheet. (b) Photograph of water splitting under AM 1.5G simulated sunlight at 333 K and 91 kPa pressure. (c) Time course of gas evolution during the overall water splitting on a Ru-modified SrTiO3:La,Rh/C/BiVO4:Mo sheet. (d) Effect of background pressure on the photocatalytic activity of a Ru-loaded SrTiO3:La,Rh/C/BiVO4:Mo sheet (reprinted with permission from ref. 122 Copyright 2017 American Chemical Society). | ||
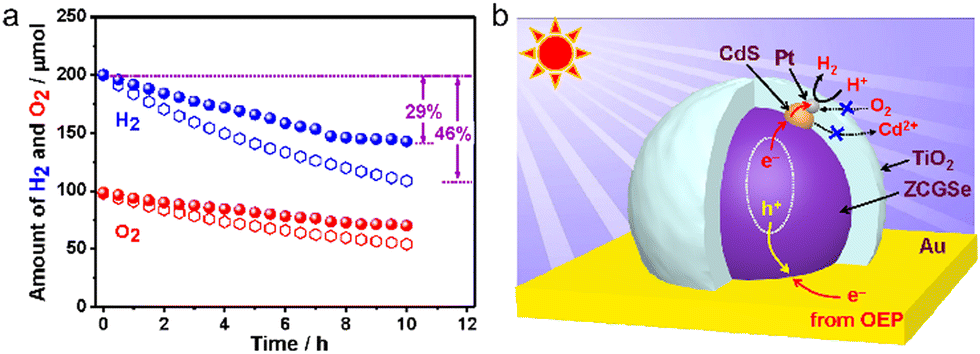 | ||
| Fig. 13 (a) Comparison of the progress of reverse reactions over optimized Pt/TiO2/CdS-ZCGSe/Au/BiVO4:Mo (spheres) and Pt/CdS-ZCGSe/Au/BiVO4:Mo (open hexagons) under dark conditions. (b) Proposed photocatalytic mechanism over HEP Pt/TiO2/CdS-ZCGSe (reprinted with permission from ref. 126 Copyright 2021 American Chemical Society). | ||
To date, one-step OWS panels have been largely based on UV-light-driven photocatalysts, and few visible-light-driven systems have been demonstrated. Therefore, more research work in this field is required along with the development of new photocatalysts. The stability of visible-light-response photocatalysts is an important factor related to the design of Z-scheme water splitting sheets. At the moment, the majority of two-step water splitting sheets are prepared by evaporation or sputtering methods that typically require the application of high vacuum and heating over relatively long periods of time. In contrast to UV-light-driven photocatalysts, visible-light-responsive materials typically contain nitrogen, sulphur or vanadium, and so may be unstable under harsh processing conditions. In addition, cocatalysts and coating layers may be damaged during the various preparation steps. Therefore, developing new methods involving milder conditions will be necessary in order to fabricate visible-light-driven Z-scheme sheets without performance degradation and with reduced cost. The deposition of cocatalysts and coating layers after sheet preparation could also avoid damage, but the loading methods will need to be carefully optimized.
6. Conclusions and perspectives
This perspective summarized the guiding principles for photocatalysts and systems intended to achieve OWS via one- or two-step photoexcitation. The progress in this field was also surveyed. Although Al-doped SrTiO3 has exhibited high efficiency under UV light, the majority of photocatalysts still show relatively low performance, especially visible-light-driven materials. This poor performance can be ascribed to the high recombination rate for photogenerated charges, the sluggish surface reactions and the significant back reactions on these catalysts. Mismatched band alignments with respect to the water redox potential and insufficient thermodynamic driving forces are additional obstacles associated with narrow-bandgap photocatalysts. Various Z-scheme systems partly mitigate these problems, but more complex processes are required. In addition, while some HEPs capable of absorbing at long wavelengths have been developed, few narrow-bandgap OEPs are available for use in Z-scheme water splitting systems. Immobilizing photocatalyst particles on substrates to produce water splitting panels is a potential approach to practical industrial scale OWS, as opposed to suspension-based systems. UV-light-driven water splitting panels based on one-step excitation have been shown to evolve H2 and O2 on a large scale and in a stable manner over prolonged periods. In contrast, visible-light-driven OWS water splitting panels have not yet been reported. Highly efficient two-step excitation water splitting panel systems have been demonstrated based on either Au or carbon conducting layers as alternatives to the use of ionic couples. Even so, further increases in the performance of these sheet systems and size scale up while reducing costs remains difficult.To date, the best STH energy conversion efficiencies achieved by one-step Al-doped SrTiO3 and Z-scheme SrTiO3:La,Rh/Au/BiVO4:Mo water splitting systems are in the order of approximately 0.7 and 1.0%, respectively. These values therefore remain below the minimum requirement of 5% proposed by the United States Department of Energy for practical large-scale applications. As such, both photocatalysts and water splitting systems must be improved. One possible means of increasing the STH is to employ photocatalysts absorbing at longer wavelengths. Some narrow-bandgap photocatalysts, such as (oxy)nitrides, (oxy)sulfides and conjugated polymers, could be candidates after further optimization. Some of the most widely studied oxide materials could also be utilized as visible-light-driven photocatalysts with suitable modification. More specifically, the preparation of highly crystalline photocatalysts with appropriate particle sizes will be indispensable for increasing the longevity of electrons and holes migrating to reactive surfaces, as demonstrated by trials using inorganic and polymeric photocatalysts. The selective deposition of cocatalysts on different facets of the main catalyst to achieve spatial separation of H2 and O2 evolution is an efficient means of suppressing charge recombination and enhancing the water redox reaction. Moreover, applying passivation layers to the cocatalyst and/or photocatalyst surfaces can inhibit back reactions. The key characteristics of the materials for the development of highly efficient photocatalyst for OWS reaction are summarized in Table 2. In the case of Z-scheme OWS systems, the selection of appropriate electron mediators is vital to achieving highly efficient charge transfer between the HEP and OEP. Ionic redox shuttles are generally less expensive and provide better contact with the HEP and OEP, but issues related to competing reactions and instability remain to be solved, such as by exploring new ionic couples. Solid-state electron mediators are an alternative option that could address challenges related to ionic redox shuttles, particularly low-cost carbon-based mediators. Further enhancing the electron transfer efficiency and the development of new fabrication methods could permit the application of solid-state electron mediators providing superior performance on a large scale. It is worth noting that back reactions may occur on the HEP and OEP but also on the electron mediators in Z-scheme systems. Therefore, techniques for inhibiting these reactions on electron mediators should be examined in the future. Additional effort should be dedicated to the study of visible-light-driven water splitting panels based on one- or two-step excitation systems. In particular, there is still much room to optimize the system design and further reduce construction costs, such as by exploring new substrates and catalyst fixation methods. The separation and storage of the gaseous H2 and O2 generated by such processes as well as safety and transportation costs are also critical aspects of photocatalytic water splitting systems. Additional resources must be devoted to achieve progress in these aspects. The design of efficient photocatalysts, scalable systems and innovative technologies for the construction and operation of large-scale photocatalyst chemical plants is expected to eventually lead to a sustainable society based on hydrogen energy in the future.
| Characteristic | Description |
|---|---|
| Bandgap | Photocatalysts should possess a narrow bandgap to harvest sunlight because more than 50% of the solar energy distributes in the visible-light region. |
| Band alignment | The band edge potentials of photocatalysts must satisfy the thermodynamic conditions for one- and two-step excitation OWS reactions. Ideally, the photocatalysts should have more negative CBM and more positive VBM potentials to possess stronger driving forces for reduction and oxidization reactions, respectively. |
| Crystallinity | Photocatalysts should have high crystallinity to reduce the density of defects acting as recombination centres and prolong the lifetime of photogenerated charge carriers. |
| Particle size | Photocatalysts should have smaller particle sizes to reduce the migration distance of electrons and holes to the surface before the recombination. |
| Facet | Photocatalysts should expose nonequivalent specific facets to promote the spatial separation of electrons and holes and of reduction and oxidization sites. |
| Surface | Photocatalysts should have surface properties suitable for providing surface active sites and loading cocatalysts. |
Author contributions
Y. M. Wrote the manuscript. L. L. Revised the manuscript. T. T. and T. H. Reviewed this work. K. D. Supervised the writing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the artificial hoto-synthesis Project (ARPChem) of the New Energy and Industrial Technology Development Organization (NEDO).References
- L. Lin, T. Hisatomi, S. Chen, T. Takata and K. Domen, Trends Chem., 2020, 2, 813–824 CrossRef CAS.
- Z. Wang, C. Li and K. Domen, Chem. Soc. Rev., 2019, 48, 2109–2125 RSC.
- S. Chen, T. Takata and K. Domen, Nat. Rev. Mater., 2017, 2, 17050 CrossRef CAS.
- L. Lin, C. Wang, W. Ren, H. Ou, Y. Zhang and X. Wang, Chem. Sci., 2017, 8, 5506–5511 RSC.
- L. Li, X. Mu, W. Liu, Z. Mi and C.-J. Li, J. Am. Chem. Soc., 2015, 137, 7576–7579 CrossRef CAS PubMed.
- C. Wulf and P. Zapp, Int. J. Hydrogen Energy, 2018, 43, 11884–11895 CrossRef CAS.
- J. H. Kim, D. Hansora, P. Sharma, J.-W. Jang and J. S. Lee, Chem. Soc. Rev., 2019, 48, 1908–1971 RSC.
- I. Y. Ahmet, Y. Ma, J.-W. Jang, T. Henschel, B. Stannowski, T. Lopes, A. Vilanova, A. Mendes, F. F. Abdi and R. van de Krol, Sustain Energ Fuels, 2019, 3, 2366–2379 RSC.
- J. H. Kim, J.-W. Jang, Y. H. Jo, F. F. Abdi, Y. H. Lee, R. van de Krol and J. S. Lee, Nat. Commun., 2016, 7, 13380 CrossRef CAS PubMed.
- L. Pan, J. H. Kim, M. T. Mayer, M.-K. Son, A. Ummadisingu, J. S. Lee, A. Hagfeldt, J. Luo and M. Grätzel, Nat. Catal., 2018, 1, 412–420 CrossRef CAS.
- M. B. Costa, M. A. de Araújo, M. V. de L. Tinoco, J. F. de Brito and L. H. Mascaro, J. Energy Chem., 2022, 73, 88–113 CrossRef CAS.
- S. Chen, T. Liu, Z. Zheng, M. Ishaq, G. Liang, P. Fan, T. Chen and J. Tang, J. Energy Chem., 2022, 67, 508–523 CrossRef CAS.
- S. S. Kalanur and H. Seo, J. Energy Chem., 2022, 68, 612–623 CrossRef CAS.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- L. Lin, H. Ou, Y. Zhang and X. Wang, ACS Catal., 2016, 6, 3921–3931 CrossRef CAS.
- Y.-Y. Wang, Y.-X. Chen, T. Barakat, Y.-J. Zeng, J. Liu, S. Siffert and B.-L. Su, J. Energy Chem., 2022, 66, 529–559 CrossRef CAS.
- L. Lin, W. Ren, C. Wang, A. M. Asiri, J. Zhang and X. Wang, Appl. Catal. B: Environ., 2018, 231, 234–241 CrossRef CAS.
- L. Lin, Z. Yu and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 6164–6175 CrossRef CAS PubMed.
- T. Takata, J. Jiang, Y. Sakata, M. Nakabayashi, N. Shibata, V. Nandal, K. Seki, T. Hisatomi and K. Domen, Nature, 2020, 581, 411–414 CrossRef CAS PubMed.
- Q. Wang and K. Domen, Chem. Rev., 2019, 120, 919–985 CrossRef PubMed.
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520–7535 RSC.
- Q. Wang, T. Hisatomi, Q. Jia, H. Tokudome, M. Zhong, C. Wang, Z. Pan, T. Takata, M. Nakabayashi, N. Shibata, Y. Li, I. D. Sharp, A. Kudo, T. Yamada and K. Domen, Nat. Mater., 2016, 15, 611–615 CrossRef CAS PubMed.
- Y. Moriya, T. Takata and K. Domen, Coord. Chem. Rev., 2013, 257, 1957–1969 CrossRef CAS.
- T. Hisatomi and K. Domen, Nat. Catal., 2019, 2, 387–399 CrossRef CAS.
- Y. Zheng, L. Lin, B. Wang and X. Wang, Angew. Chem., Int. Ed., 2015, 54, 12868–12884 CrossRef CAS PubMed.
- H. Wang, L. Zhang, Z. Chen, J. Hu, S. Li, Z. Wang, J. Liu and X. Wang, Chem. Soc. Rev., 2014, 43, 5234–5244 RSC.
- K. Takanabe, T. Uzawa, X. Wang, K. Maeda, M. Katayama, J. Kubota, A. Kudo and K. Domen, Dalton Trans., 2009, 10055–10062 RSC.
- Y. Qi, S. Chen, M. Li, Q. Ding, Z. Li, J. Cui, B. Dong, F. Zhang and C. Li, Chem. Sci., 2017, 8, 437–443 RSC.
- K. Maeda, ACS Catal., 2013, 3, 1486–1503 CrossRef CAS.
- Y. Goto, T. Hisatomi, Q. Wang, T. Higashi, K. Ishikiriyama, T. Maeda, Y. Sakata, S. Okunaka, H. Tokudome, M. Katayama, S. Akiyama, H. Nishiyama, Y. Inoue, T. Takewaki, T. Setoyama, T. Minegishi, T. Takata, T. Yamada and K. Domen, Joule, 2018, 2, 509–520 CrossRef CAS.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- J. H. Kim and J. S. Lee, Adv. Mater., 2019, 31, 1806938 CrossRef PubMed.
- Z. Zhang and J. T. Yates, Chem. Rev., 2012, 112, 5520–5551 CrossRef CAS PubMed.
- Y. Wang, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 68–89 CrossRef CAS PubMed.
- S. Chen, Y. Qi, C. Li, K. Domen and F. Zhang, Joule, 2018, 2, 2260–2288 CrossRef CAS.
- G. Zhang, Z.-A. Lan and X. Wang, Chem. Sci., 2017, 8, 5261–5274 RSC.
- F. K. Kessler, Y. Zheng, D. Schwarz, C. Merschjann, W. Schnick, X. Wang and M. J. Bojdys, Nat. Rev. Mater., 2017, 2, 17030 CrossRef CAS.
- D. Zheng, C. Pang and X. Wang, Chem. Commun., 2015, 51, 17467–17470 RSC.
- F. Jiang, G. Gunawan, T. Harada, Y. Kuang, T. Minegishi, K. Domen and S. Ikeda, J. Am. Chem. Soc., 2015, 137, 13691–13697 CrossRef CAS PubMed.
- K. Iwashina, A. Iwase, Y. H. Ng, R. Amal and A. Kudo, J. Am. Chem. Soc., 2015, 137, 604–607 CrossRef CAS PubMed.
- Y. Kageshima, Y. Gomyo, H. Matsuoka, H. Inuzuka, H. Suzuki, R. Abe, K. Teshima, K. Domen and H. Nishikiori, ACS Catal., 2021, 11, 8004–8014 CrossRef CAS.
- Q. Yuan, D. Liu, N. Zhang, W. Ye, H. Ju, L. Shi, R. Long, J. Zhu and Y. Xiong, Angew. Chem., Int. Ed., 2017, 56, 4206–4210 CrossRef CAS PubMed.
- K. Hideki, S. Yasuyoshi, I. Akihide and K. Akihiko, Bull. Chem. Soc. Jpn., 2007, 80, 2457–2464 CrossRef.
- Q. Wang, T. Hisatomi, S. S. K. Ma, Y. Li and K. Domen, Chem. Mater., 2014, 26, 4144–4150 CrossRef CAS.
- Z. Song, T. Hisatomi, S. Chen, Q. Wang, G. Ma, S. Li, X. Zhu, S. Sun and K. Domen, ChemSusChem, 2019, 12, 1906–1910 CrossRef CAS PubMed.
- K. Sayama, R. Yoshida, H. Kusama, K. Okabe, Y. Abe and H. Arakawa, Chem. Phys. Lett., 1997, 277, 387–391 CrossRef CAS.
- M. Higashi, R. Abe, T. Takata and K. Domen, Chem. Mater., 2009, 21, 1543–1549 CrossRef CAS.
- K. Sayama, K. Mukasa, R. Abe, Y. Abe and H. Arakawa, Chem. Commun., 2001, 2416–2417 RSC.
- Y. Sasaki, H. Kato and A. Kudo, J. Am. Chem. Soc., 2013, 135, 5441–5449 CrossRef CAS PubMed.
- Q. Wang, Y. Li, T. Hisatomi, M. Nakabayashi, N. Shibata, J. Kubota and K. Domen, J. Catal., 2015, 328, 308–315 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- Y. Sakata, T. Hayashi, R. Yasunaga, N. Yanaga and H. Imamura, Chem. Commun., 2015, 51, 12935–12938 RSC.
- H. Kato, K. Asakura and A. Kudo, J. Am. Chem. Soc., 2003, 125, 3082–3089 CrossRef CAS PubMed.
- K. Domen, S. Naito, M. Soma, T. Onishi and K. Tamaru, J. Chem. Soc., Chem. Commun., 1980, 543–544 RSC.
- T. Takata and K. Domen, J. Phys. Chem. C, 2009, 113, 19386–19388 CrossRef CAS.
- Y. Ham, T. Hisatomi, Y. Goto, Y. Moriya, Y. Sakata, A. Yamakata, J. Kubota and K. Domen, J. Mater. Chem. A, 2016, 4, 3027–3033 RSC.
- P. Zhang, T. Ochi, M. Fujitsuka, Y. Kobori, T. Majima and T. Tachikawa, Angew. Chem., Int. Ed., 2017, 56, 5299–5303 CrossRef CAS PubMed.
- Y. Wei, J. Wang, R. Yu, J. Wan and D. Wang, Angew. Chem., Int. Ed., 2019, 58, 1422–1426 CrossRef CAS PubMed.
- L. Mu, Y. Zhao, A. Li, S. Wang, Z. Wang, J. Yang, Y. Wang, T. Liu, R. Chen, J. Zhu, F. Fan, R. Li and C. Li, Energy Environ. Sci., 2016, 9, 2463–2469 RSC.
- S. Su, I. Siretanu, D. van den Ende, B. Mei, G. Mul and F. Mugele, Adv. Mater., 2021, 33, 2106229 CrossRef CAS PubMed.
- D.-D. Zheng, X.-N. Cao and X.-C. Wang, Angew. Chem., Int. Ed., 2016, 55, 11512–11516 CrossRef CAS PubMed.
- J. Zhang, G. Zhang, X. Chen, S. Lin, L. Möhlmann, G. Dołęga, G. Lipner, M. Antonietti, S. Blechert and X. Wang, Angew. Chem., Int. Ed., 2012, 51, 3183–3187 CrossRef CAS PubMed.
- Y. Zheng, Z. Yu, F. Lin, F. Guo, K. Alamry, L. Taib, A. Asiri and X. Wang, Molecules, 2017, 22, 572 CrossRef PubMed.
- S. Yang, Y. Gong, J. Zhang, L. Zhan, L. Ma, Z. Fang, R. Vajtai, X. Wang and P. M. Ajayan, Adv. Mater., 2013, 25, 2452–2456 CrossRef CAS PubMed.
- J. Zhang, Y. Chen and X. Wang, Energy Environ. Sci., 2015, 8, 3092–3108 RSC.
- L. Lin, Z. Lin, J. Zhang, X. Cai, W. Lin, Z. Yu and X. Wang, Nat. Catal., 2020, 3, 649–655 CrossRef CAS.
- M. Liu, C. Wei, H. Zhuzhang, J. Zhou, Z. Pan, W. Lin, Z. Yu, G. Zhang and X. Wang, Angew. Chem., Int. Ed., 2022, 61, e202113389 CAS.
- K. Maeda, T. Takata, M. Hara, N. Saito, Y. Inoue, H. Kobayashi and K. Domen, J. Am. Chem. Soc., 2005, 127, 8286–8287 CrossRef CAS PubMed.
- Y. Li, L. Zhu, Y. Yang, H. Song, Z. Lou, Y. Guo and Z. Ye, Small, 2015, 11, 871–876 CrossRef CAS PubMed.
- K. Maeda, K. Teramura, D. Lu, T. Takata, N. Saito, Y. Inoue and K. Domen, Nature, 2006, 440, 295 CrossRef CAS PubMed.
- K. Maeda, D. Lu and K. Domen, Chem. – Eur. J., 2013, 19, 4986–4991 CrossRef CAS PubMed.
- J. Xiao, S. Nishimae, J. J. M. Vequizo, M. Nakabayashi, T. Hisatomi, H. Li, L. Lin, N. Shibata, A. Yamakata, Y. Inoue and K. Domen, Angew. Chem., Int. Ed., 2022, 61, e202116573 CAS.
- C. Pan, T. Takata, M. Nakabayashi, T. Matsumoto, N. Shibata, Y. Ikuhara and K. Domen, Angew. Chem., 2015, 127, 2998–3002 CrossRef.
- G. Hitoki, A. Ishikawa, T. Takata, J. N. Kondo, M. Hara and K. Domen, Chem. Lett., 2002, 736–737 CrossRef CAS.
- W.-J. Chun, A. Ishikawa, H. Fujisawa, T. Takata, J. N. Kondo, M. Hara, M. Kawai, Y. Matsumoto and K. Domen, J. Phys. Chem. B, 2003, 107, 1798–1803 CrossRef CAS.
- M. Zhong, T. Hisatomi, Y. Sasaki, S. Suzuki, K. Teshima, M. Nakabayashi, N. Shibata, H. Nishiyama, M. Katayama, T. Yamada and K. Domen, Angew. Chem., Int. Ed., 2017, 56, 4739–4743 CrossRef CAS PubMed.
- Z. Wang, Y. Inoue, T. Hisatomi, R. Ishikawa, Q. Wang, T. Takata, S. Chen, N. Shibata, Y. Ikuhara and K. Domen, Nat. Catal., 2018, 1, 756–763 CrossRef CAS.
- A. Ishikawa, T. Takata, J. N. Kondo, M. Hara, H. Kobayashi and K. Domen, J. Am. Chem. Soc., 2002, 124, 13547–13553 CrossRef CAS PubMed.
- A. Ishikawa, Y. Yamada, T. Takata, J. N. Kondo, M. Hara, H. Kobayashi and K. Domen, Chem. Mater., 2003, 15, 4442–4446 CrossRef CAS.
- F. Zhang, K. Maeda, T. Takata and K. Domen, Chem. Commun., 2010, 46, 7313–7315 RSC.
- M. Yashima, K. Ogisu and K. Domen, Acta Crystallogr., Sect. B, 2008, 64, 291–298 CrossRef CAS PubMed.
- Q. Wang, M. Nakabayashi, T. Hisatomi, S. Sun, S. Akiyama, Z. Wang, Z. Pan, X. Xiao, T. Watanabe, T. Yamada, N. Shibata, T. Takata and K. Domen, Nat. Mater., 2019, 18, 827–832 CrossRef CAS PubMed.
- V. Nandal, R. Shoji, H. Matsuzaki, A. Furube, L. Lin, T. Hisatomi, M. Kaneko, K. Yamashita, K. Domen and K. Seki, Nat. Commun., 2021, 12, 7055 CrossRef CAS PubMed.
- G. Zhang, Z.-A. Lan, L. Lin, S. Lin and X. Wang, Chem. Sci., 2016, 7, 3062–3066 RSC.
- X. Chen, R. Shi, Q. Chen, Z. Zhang, W. Jiang, Y. Zhu and T. Zhang, Nano Energy, 2019, 59, 644–650 CrossRef CAS.
- H. L. Nguyen, Adv. Mater., 2022, 34, 2200465 CrossRef CAS PubMed.
- S. Navalón, A. Dhakshinamoorthy, M. Álvaro, B. Ferrer and H. García, Chem. Rev., 2022, 123, 445–490 CrossRef PubMed.
- Y. An, Y. Liu, P. An, J. Dong, B. Xu, Y. Dai, X. Qin, X. Zhang, M.-H. Whangbo and B. Huang, Angew. Chem., Int. Ed., 2017, 56, 3036–3040 CrossRef CAS PubMed.
- J. Zhang, T. Bai, H. Huang, M.-H. Yu, X. Fan, Z. Chang and X.-H. Bu, Adv. Mater., 2020, 32, 2004747 CrossRef CAS PubMed.
- P. Salcedo-Abraira, A. A. Babaryk, E. Montero-Lanzuela, O. R. Contreras-Almengor, M. Cabrero-Antonino, E. S. Grape, T. Willhammar, S. Navalón, E. Elkäim, H. García and P. Horcajada, Adv. Mater., 2021, 33, 2106627 CrossRef CAS PubMed.
- Y. Qi, Y. Zhao, Y. Gao, D. Li, Z. Li, F. Zhang and C. Li, Joule, 2018, 2, 2393–2402 CrossRef CAS.
- S. Yoshino, A. Iwase, Y. Yamaguchi, T. M. Suzuki, T. Morikawa and A. Kudo, J. Am. Chem. Soc., 2022, 144, 2323–2332 CrossRef CAS PubMed.
- A. J. Bard, J. Photochem., 1979, 10, 59–75 CrossRef CAS.
- R. Abe, K. Sayama, K. Domen and H. Arakawa, Chem. Phys. Lett., 2001, 344, 339–344 CrossRef CAS.
- S. Chen, Y. Qi, T. Hisatomi, Q. Ding, T. Asai, Z. Li, S. S. K. Ma, F. Zhang, K. Domen and C. Li, Angew. Chem., Int. Ed., 2015, 54, 8498–8501 CrossRef CAS PubMed.
- Z. Wang, Y. Luo, T. Hisatomi, J. J. M. Vequizo, S. Suzuki, S. Chen, M. Nakabayashi, L. Lin, Z. Pan, N. Kariya, A. Yamakata, N. Shibata, T. Takata, K. Teshima and K. Domen, Nat. Commun., 2021, 12, 1005 CrossRef CAS PubMed.
- B. Dong, J. Cui, Y. Gao, Y. Qi, F. Zhang and C. Li, Adv. Mater., 2019, 31, 1808185 CrossRef PubMed.
- H. Zou, Y. Qi, S. Du, L. Liu, X. Xin, Y. Bao, S. Wang, Z. Feng and F. Zhang, Chem. Commun., 2022, 58, 10719–10722 RSC.
- G. Ma, S. Chen, Y. Kuang, S. Akiyama, T. Hisatomi, M. Nakabayashi, N. Shibata, M. Katayama, T. Minegishi and K. Domen, J. Phys. Chem. Lett., 2016, 7, 3892–3896 CrossRef CAS PubMed.
- T. Oshima, S. Nishioka, Y. Kikuchi, S. Hirai, K.-I. Yanagisawa, M. Eguchi, Y. Miseki, T. Yokoi, T. Yui, K. Kimoto, K. Sayama, O. Ishitani, T. E. Mallouk and K. Maeda, J. Am. Chem. Soc., 2020, 142, 8412–8420 CrossRef CAS PubMed.
- S. Nishioka, K. Hojo, L. Xiao, T. Gao, Y. Miseki, S. Yasuda, T. Yokoi, K. Sayama, T. E. Mallouk and K. Maeda, Sci. Adv., 2022, 8, eadc9115 CrossRef CAS PubMed.
- D. J. Martin, P. J. T. Reardon, S. J. A. Moniz and J. Tang, J. Am. Chem. Soc., 2014, 136, 12568–12571 CrossRef CAS PubMed.
- H. Fujito, H. Kunioku, D. Kato, H. Suzuki, M. Higashi, H. Kageyama and R. Abe, J. Am. Chem. Soc., 2016, 138, 2082–2085 CrossRef CAS PubMed.
- X. Tao, Y. Zhao, L. Mu, S. Wang, R. Li and C. Li, Adv. Energy Mater., 2018, 8, 1701392 CrossRef.
- T. Shirakawa, M. Higashi, O. Tomita and R. Abe, Sustain Energ Fuels, 2017, 1, 1065–1073 RSC.
- Y. Qi, J. Zhang, Y. Kong, Y. Zhao, S. Chen, D. Li, W. Liu, Y. Chen, T. Xie, J. Cui, C. Li, K. Domen and F. Zhang, Nat. Commun., 2022, 13, 484 CrossRef CAS PubMed.
- K. Liu, B. Zhang, J. Zhang, W. Lin, J. Wang, Y. Xu, Y. Xiang, T. Hisatomi, K. Domen and G. Ma, ACS Catal., 2022, 12, 14637–14646 CrossRef CAS.
- Y. Bai, K. Nakagawa, A. J. Cowan, C. M. Aitchison, Y. Yamaguchi, M. A. Zwijnenburg, A. Kudo, R. S. Sprick and A. I. Cooper, J. Mater. Chem. A, 2020, 8, 16283–16290 RSC.
- K. Tsuji, O. Tomita, M. Higashi and R. Abe, ChemSusChem, 2016, 9, 2201–2208 CrossRef CAS PubMed.
- A. Iwase, Y. H. Ng, Y. Ishiguro, A. Kudo and R. Amal, J. Am. Chem. Soc., 2011, 133, 11054–11057 CrossRef CAS PubMed.
- A. Iwase, S. Yoshino, T. Takayama, Y. H. Ng, R. Amal and A. Kudo, J. Am. Chem. Soc., 2016, 138, 10260–10264 CrossRef CAS PubMed.
- Z. Pan, G. Zhang and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 7102–7106 CrossRef CAS PubMed.
- S. Wang, Y. Gao, Y. Qi, A. Li, F. Fan and C. Li, J. Catal., 2017, 354, 250–257 CrossRef CAS.
- Y. Sasaki, H. Nemoto, K. Saito and A. Kudo, J. Phys. Chem. C, 2009, 113, 17536–17542 CrossRef CAS.
- L. Wang, X. Zheng, L. Chen, Y. Xiong and H. Xu, Angew. Chem., Int. Ed., 2018, 57, 3454–3458 CrossRef CAS PubMed.
- D. Zhao, Y. Wang, C.-L. Dong, Y.-C. Huang, J. Chen, F. Xue, S. Shen and L. Guo, Nat. Energy, 2021, 6, 388–397 CrossRef CAS.
- A. Xiong, G. Ma, K. Maeda, T. Takata, T. Hisatomi, T. Setoyama, J. Kubota and K. Domen, Catal. Sci. Technol., 2014, 4, 325–328 RSC.
- M. Schröder, K. Kailasam, J. Borgmeyer, M. Neumann, A. Thomas, R. Schomäcker and M. Schwarze, Energy Technol., 2015, 3, 1014–1017 CrossRef.
- H. Nishiyama, T. Yamada, M. Nakabayashi, Y. Maehara, M. Yamaguchi, Y. Kuromiya, Y. Nagatsuma, H. Tokudome, S. Akiyama, T. Watanabe, R. Narushima, S. Okunaka, N. Shibata, T. Takata, T. Hisatomi and K. Domen, Nature, 2021, 598, 304–307 CrossRef CAS PubMed.
- B. P. Luther, S. E. Mohney, T. N. Jackson, M. A. Khan, Q. Chen and J. W. Yang, Appl. Phys. Lett., 1997, 70, 57–59 CrossRef CAS.
- C.-Y. Hsu, W.-H. Lan and Y. S. Wu, Appl. Phys. Lett., 2003, 83, 2447–2449 CrossRef CAS.
- Q. Wang, T. Hisatomi, Y. Suzuki, Z. Pan, J. Seo, M. Katayama, T. Minegishi, H. Nishiyama, T. Takata, K. Seki, A. Kudo, T. Yamada and K. Domen, J. Am. Chem. Soc., 2017, 139, 1675–1683 CrossRef CAS PubMed.
- Z. Pan, T. Hisatomi, Q. Wang, S. Chen, A. Iwase, M. Nakabayashi, N. Shibata, T. Takata, M. Katayama, T. Minegishi, A. Kudo and K. Domen, Adv. Funct. Mater., 2016, 26, 7011–7019 CrossRef CAS.
- S. Sun, T. Hisatomi, Q. Wang, S. Chen, G. Ma, J. Liu, S. Nandy, T. Minegishi, M. Katayama and K. Domen, ACS Catal., 2018, 1690–1696 CrossRef CAS.
- S. Chen, G. Ma, Q. Wang, S. Sun, T. Hisatomi, T. Higashi, Z. Wang, M. Nakabayashi, N. Shibata, Z. Pan, T. Hayashi, T. Minegishi, T. Takata and K. Domen, J. Mater. Chem. A, 2019, 7, 7415–7422 RSC.
- S. Chen, J. J. M. Vequizo, Z. Pan, T. Hisatomi, M. Nakabayashi, L. Lin, Z. Wang, K. Kato, A. Yamakata, N. Shibata, T. Takata, T. Yamada and K. Domen, J. Am. Chem. Soc., 2021, 143, 10633–10641 CrossRef CAS PubMed.
| This journal is © the Owner Societies 2023 |