
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Covalently linked pyrene antennas for optically dense yet aggregation-resistant light-harvesting systems†
Lubna
Salah
 a,
Saad
Makhseed
a,
Basma
Ghazal
b,
Ahmed
Abdel Nazeer
c,
Marc K.
Etherington
d,
Carlito S.
Ponseca Jr.
e,
Chunyong
Li
f,
Andrew P.
Monkman
f,
Andrew
Danos
*f and
Ali
Shuaib
*g
a,
Saad
Makhseed
a,
Basma
Ghazal
b,
Ahmed
Abdel Nazeer
c,
Marc K.
Etherington
d,
Carlito S.
Ponseca Jr.
e,
Chunyong
Li
f,
Andrew P.
Monkman
f,
Andrew
Danos
*f and
Ali
Shuaib
*g
aDepartment of Chemistry, Faculty of Science, Kuwait University, P. O. Box 5969, Safat 13060, Kuwait
bOrganometallic and Organometalloid Chemistry Department, National Research Centre, Giza, Egypt
cOrganometallic and Organometalloid Department, National Research Centre, Dokki, Cairo, 12622, Egypt
dDepartment of Mathematics, Physics & Electrical Engineering, Northumbria University, Ellison Place, Newcastle upon Tyne, NE1 8ST, UK
eMathematics and Natural Science Department, Gulf University for Science and Technology, Kuwait
fDepartment of Physics, Durham University, South Road, Durham, DH1 3LE, UK. E-mail: andrew.danos@durham.ac.uk
gBiomedical Engineering Unit, Department of Physiology, Faculty of Medicine, Kuwait University, P. O. Box 24923, Safat 13110, Kuwait. E-mail: ali.shuaib@ku.edu.kw
First published on 8th September 2023
Abstract
In this study we present a novel energy transfer material inspired by natural light-harvesting antenna arrays, zinc(II) phthalocyanine-pyrene (ZnPcPy). The ZnPcPy system facilitates energy transfer from 16 covalently linked pyrene (Py) donor chromophores to the emissive central zinc(II) phthalocyanine (ZnPc) core. Nearly 98% energy transfer efficiency is determined from the changes in emission decay rates between free MePy to covalently linked Py, supported by comparisons of photoluminescence quantum yields using different excitation wavelengths. A comparative analysis of ZnPcPy and an equivalent mixture of ZnPc and MePy demonstrates the superior light-harvesting performance of the covalently linked system, with energy transfer rates 9705 times higher in the covalently bound system. This covalent strategy allows for very high loadings of absorbing Py chromophores to be achieved while also avoiding exciton quenching that would otherwise arise, with the same strategy widely applicable to other pairs of Főrster resonance energy transfer (FRET) chromophores.
Introduction
Molecular light-harvesting is arguably the most important physicochemical process supporting life on Earth.1,2 With rare exceptions (e.g., geothermal and nuclear energy),3 almost all our energy is derived directly or indirectly from the absorption of sunlight.4 While inorganic light-harvesting technologies such as silicon photovoltaics have advanced significantly during the past 50 years, its sophistication and scale is still miniscule compared to the equivalent evolved molecular process: photosynthesis.5,6 In photosynthesis, the light-harvesting complex features many light-absorbing pigments, which transfer absorbed energy to the central reaction center to drive the endothermic fixation of atmospheric carbon dioxide.7 Mirroring this approach, the well-developed field of photocatalysis also uses light absorption to activate a wide range of otherwise inaccessible chemistry.8–10Improving the performance and feasibility of photocatalysts often requires increasing their absorption cross-section,11 allowing higher reaction turnover with smaller amounts of catalyst or more complete absorption of a fixed quantity of illumination.12 However, it is often challenging to engineer altered absorption bands without also impacting the photocatalytic reaction pathway.13 Once again taking inspiration from photosynthesis,14 it is instead possible to develop separate absorbing pigments which activate a non-absorbing reaction centre by Főrster resonance energy transfer (FRET).15 In this way the separate parts of the overall photocatalysis system can be better specialized to their specific roles. Similar specialisation and energy transfer between components is also employed to achieve high performance in hyperfluorescent OLEDs16–20 and in triplet–triplet annihilation upconversion systems.21,22
While the photosynthesis light-harvesting complex is exquisitely optimsed by millions of years of evolution, equivalent synthetic light-harvesting platforms are not as advanced.23–25 Typically, the FRET acceptor and the FRET donor are simply mixed together, either in solution or immobilized in (or along) a polymer host.26,27 Often this approach puts limitations on the types and concentrations of light-absorbers to be used though, as aggregation of these can lead to unwanted non-radiative decay that competes with emission and/or FRET.28–30 For example, small aromatic hydrocarbons such as perylene or pyrene (Py) possess strong absorption bands, making them appealing for use as molecular absorbers. However, their flat and electron-rich structure also makes them highly susceptible to aggregation, with many studies reporting chemical modifications or other strategies designed to prevent this.31,32
In previous work we have successfully synthesised ZnPcPy.33 Here we consider the optical and energy transfer properties of this material, taking inspiration from the photosynthetic antenna array. In that biological system, aggregation is avoided with the absorbing dyes covalently fixed to the FRET acceptor, and unable to diffuse freely to form aggregate states.34 Similarly here, without modification of the Py absorbing unit, ZnPcPy exhibits strong Py absorption with efficient zinc(II)-phthalocyanine (ZnPc) emission in solution, significantly outperforming two-component mixtures at equivalent concentrations, and able to access chromophore concentrations that would be otherwise detrimental to the unbound Py unit.
Results & discussion
Synthesis
The synthesis of zinc(II) phthalocyanine-pyrene (ZnPcPy) was carried out using alkyne-azide click chemistry, as previously reported in a published paper33 and depicted in ESI,† Scheme S1. The structure of ZnPcPy was confirmed, as reported in the previous published study,33 by several spectroscopic techniques including nuclear magnetic resonance (NMR), Fourier transform infrared (FT-IR), and MALDI-TOF, as well as CHN elemental analysis.33 The molecular structures of zinc(II) phthalocyanine (ZnPc), methyl-pyrene (MePy), and ZnPcPy are illustrated in Fig. 1. | ||
| Fig. 1 Structures of ZnPc, MePy, and ZnPcPy. The specific conformation of ZnPcPy shown is for illustrative purposes only. | ||
Optical properties
We first investigated the steady-state absorption and emission properties of ZnPcPy, its separate analogous components (ZnPc and MePy), and equivalent mixtures of those components in dilute THF solution. Fig. 2(A) shows the typical B-band for ZnPc at 345 nm and the Q-band at 667 nm. The main Q-band redshifted to 687 nm in ZnPcPy, possibly due to the extended conjugation system associated with the appended Py units35 (either through-bond or through-space), or by effective solvation of the ZnPc core by the surrounding Py units. The B-band of ZnPcPy was not resolvable underneath the strong absorption bands of the covalently bound Py units, which retained the same spectral shape as those of the unbound MePy.36 Considering that each ZnPcPy molecule contains 16 bound Py subunits, it is surprising that the absorbance intensity of the 2 μM ZnPcPy solution does not match more closely that of an equivalent mixture of 2 μM ZnPc and 32 μM MePy. This could indicate some transfer of oscillator strength when Py is incorporated into the ZnPc electronic system, although the extinction coefficient remains very high overall: 4.76 × 105 M−1 cm−1 at 345 nm, compared to 4.28 × 104 M−1 cm−1 for MePy.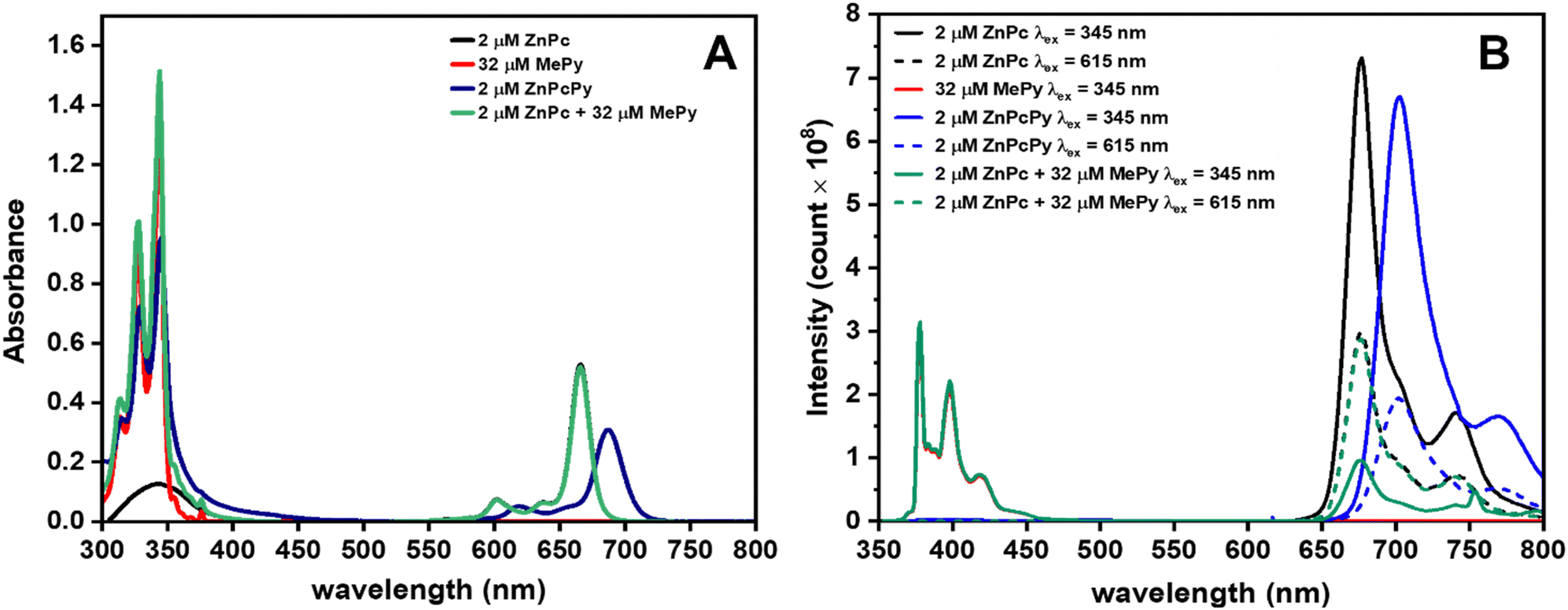 | ||
| Fig. 2 The absorption and emission properties for different compounds in THF. (A) Absorption spectra were acquired for 2 μM ZnPc, 32 μM MePy, 2 μM ZnPcPy, and a mixture containing 2 μM ZnPc and 32 μM MePy. (B) Emission spectra were recorded for the same samples, excited at either 345 or 615 nm. | ||
Comparing the overlapping positions of the MePy emission (Fig. 2(B), 350–400 nm, and Fig. S5, ESI†) with the ZnPc B-band absorption indicates the potential for FRET between the two subcomponents in the joined ZnPcPy compound. Indeed, qualitative evidence for this FRET is evident in the emission spectra of ZnPcPy, which when excited in the Py absorption band (345 nm) emits nearly exclusively from the Pc core (negligible Py emission, Fig. S3, ESI†). The identity of this ZnPc emission is confirmed by comparing with the emission spectra from both ZnPcPy and ZnPc when these are excited directly into their Q-band (∼615 nm), yielding emission only at ∼700 nm. In strong contrast, when a mixture of ZnPc and MePy is excited at 345 nm, simultaneous emission from both separate materials is observed. This immediately demonstrates the superior FRET rate in ZnPcPy, which is able to outcompete and quench the radiative emission of MePy.
To further develop upon the qualitative insights from the steady-state spectra and intensities, the photoluminescence quantum yields (PLQYs) and emission decay lifetimes of the solutions were also measured. As summarised in Table 1, for MePy excited at 345 nm we observe a PLQY of 6.5% and an emission decay lifetime of 17.3 ns. For ZnPc, the excited-state behaviour is very similar regardless of whether excitation occurs at 345 nm or 615 nm, with PLQYs of 25/25% and lifetimes of 3.6/3.5 ns, respectively. This indicates fast and efficient internal conversion from the higher excited states in ZnPc (which can be excited using 345 nm) down to the lowest singlet state, with no significantly active loss pathways interrupting transfer to S1.
| Parameter | 2 μM ZnPc | 2 μM MePy | 2 μM ZnPcPy | Mixture of 2 μM ZnPc + 32 μM MePy |
|---|---|---|---|---|
| λ abs (nm) | 345, 667 | 310, 330, 345 | 314, 330, 345, 687 | 314, 327, 344, 667 |
| λ F (nm) | 677 | 369, 376 | 696 | 377, 398, 678 |
ε (M−1 cm−1) (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ε) 345 nm/667 or 687 nm ε) 345 nm/667 or 687 nm |
63500 (4.80)/264000 (5.42) |
43438 (4.64)/— |
476000 (5.68)/154500 (5.19) |
42781 (4.63)/260000 (5.41) |
| τ (ns) | — | 17.3 | 0.3 | 17.2 |
| λ em = 400 nm | ||||
| λ ex = 345 nm | ||||
| τ (ns) | 3.6/3.5 | —/— | 3.2/3.1 | 3.7/3.6 |
| λ em = 700 nm | ||||
| λ ex = 345/615 nm | ||||
| PLQY | —/— | 0.065/— | 0.0002/— | 0.053/— |
| λ em = 400 nm | ||||
| λ ex = 345/615 nm | ||||
| PLQY | 0.25/0.25 | —/— | 0.16/0.18 | 0.043/0.24 |
| λ em = 700 nm | ||||
| λ ex = 345/615 nm |
In ZnPcPy the optical properties are also strongly independent of the excitation wavelength. The PLQYs derived using 345 nm or 615 nm excitation are 16 and 18%, respectively, with only a vanishingly small contribution from the ∼400 nm Py band (Fig. S3, ESI†). The lifetime of the ∼700 nm emission band is also very similar to that of ZnPc and approximately invariant with the choice of excitation wavelength (3.2 and 3.1 ns at 345 and 615 nm excitation, respectively, decays and fits shown in Fig. S8–S11, ESI†). As in ZnPc, this invariance of both the emission lifetime and PLQY also indicates a very fast and efficient transfer of excitation from higher excited states-including the Py groups-down to the ZnPc-centred S1 state by both internal conversion and intramolecular FRET. In the presence of a hypothetical competing loss pathways, we would not expect that exciting the Py groups (345 nm excitation) would be able to give the same overall PLQY as exciting the ZnPc core directly (with 615 nm, which avoids any losses as excitons transfer from Py to ZnPc). Indeed, by comparing the lifetimes of the 400 nm Py emission in MePy (17.3 ns) and in ZnPcPy (0.3 ns), we can attribute the increased decay rate to FRET and estimate an energy transfer rate of 3.3 × 109 s−1. This rate of excitation transfer is significantly faster than the radiative and nonradiative decay rates for MePy (3.8 × 106 and 5.4 × 107 s−1, respectively), which explains how the ZnPcPy compound can avoid losses associated with the low PLQY of MePy emission, even when this group is predominately excited (with 345 nm). Based on the ratio of decay rates in MePy, this corresponds to a FRET efficiency of ∼98%, which is also consistent with the unchanged PLQYs in ZnPcPy regardless of the excitation wavelength.
Compared with ZnPcPy, the energy transfer properties of an equivalent concentration mixture of ZnPc and MePy are considerably inferior. When excited using 345 nm light, the PLQY of both the MePy and ZnPc emission bands were very low (5.3 and 4.3% respectively), indicating some emission directly from the low-efficiency MePy and poor FRET transfer to the ZnPc. On the contrary, when the same ZnPc in the mixture was excited directly using 615 nm, the PLQY was largely unaffected by the presence of MePy (24%). Additionally, the emission lifetime of MePy hardly changed in the presence of ZnPc, decreasing from 17.3 to 17.2 ns, which corresponds to a FRET rate of only 3.4 × 105 s−1 and a FRET efficiency of 0.6%. Considering again the radiative and non-radiative decay rates of MePy (3.8 × 106 and 5.4 × 107 s−1, respectively), it is clear that nearly all photons absorbed by untethered MePy are wasted. To achieve the same FRET rate as ZnPcPy, the mixture would require a concentration of ZnPc 9705 times higher; i.e. 32 μM of MePy with 17 mM of ZnPc. This extreme ratio of ZnPc would itself defeat the design strategy and purpose of a molecular antenna array, in which many absorbers are intended to funnel energy to a less concentrated emission or reaction centre.
Transient absorption spectra and kinetics were also investigated to gain insights into the internal energy transfer in ZnPcPy upon excitation at 345 nm and 615 nm (Fig. 3). The results demonstrate that the excited-state absorption spectra of ZnPcPy remain unchanged when excited at either of these wavelengths, suggesting that no additional decay pathways compete with the FRET process from Py to ZnPc groups. If alternative decay pathways were present, one would anticipate detecting an additional band in the excited-state absorption spectrum corresponding to a quenching state when excited at 345 nm but absent when excited at 615 nm. Furthermore, there is no indication of excited state absorptions or ground state bleaches from the Py chromophore (MePy measured separately, Fig. S7, ESI†), indicating FRET on a more rapid timescale than the measurement. We note that due to different ground state absorption intensities and pump beam photon fluxes, we do not attribute any meaning to the different intensities of the transient absorption spectra using the different excitation wavelengths.
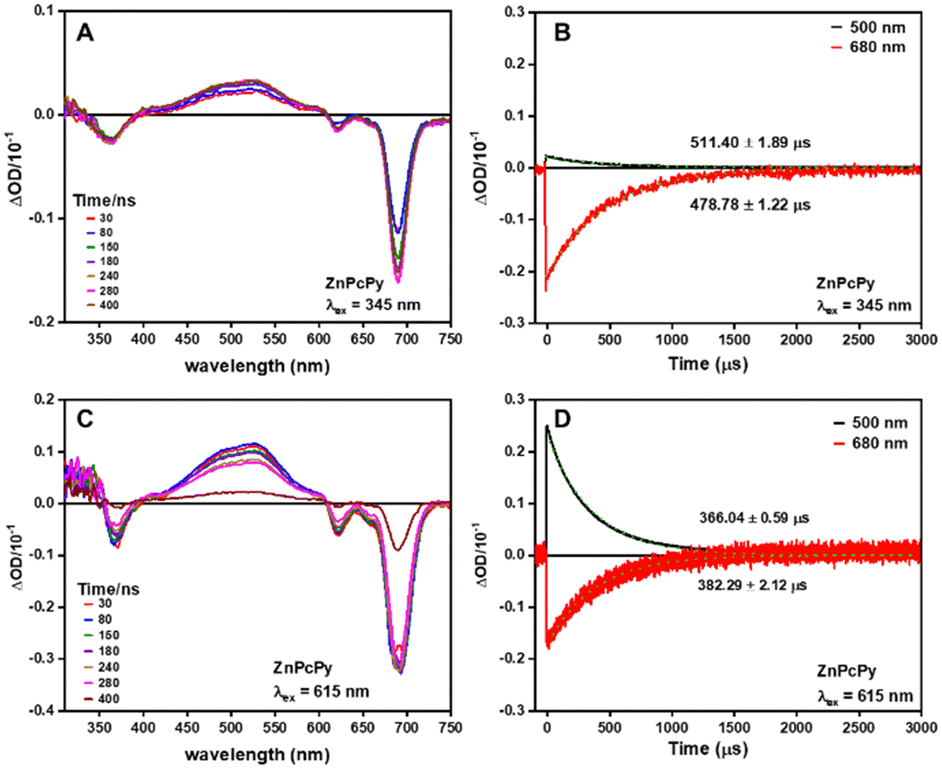 | ||
| Fig. 3 Transient absorption spectra (left) and decay traces (right) of ZnPcPy in THF when excited at (A), (B) 345 nm and (C), (D) 615 nm, respectively. | ||
The enhanced FRET efficiency observed in ZnPcPy can be primarily attributed to the covalent attachment of Py groups to the ZnPc core, which ensures consistent close proximity between chromophores. Given the strong dependence of FRET rate on distance, this short distance between chromophores can overcome the apparently small FRET spectral overlap. In contrast, the simple mixture of ZnPc and MePy experiences larger and more dynamic inter-chromophore distances controlled by molecular diffusion, leading to a lower FRET rate. Even with identical molar concentration of chromophores in both ZnPcPy and the simple mixture, the importance of structural rigidity and fixed distances in preventing stacking and aggregation should not be underestimated. The covalent tethering of Py groups to the ZnPc core additionally creates a stable and well-defined spatial arrangement, reducing the accessibility of conformations that lead to intramolecular Py aggregates and maintaining consistent chromophore distances. This covalently linked configuration also decreases the probability of Py aggregates forming between molecules, as the concentrations of other ZnPcPy molecules is 16 times lower than the equivalent concentration of Py molecules in the mixture, with correspondingly 16 times lower rate of aggregate-forming molecular collisions.
Conclusions
In conclusion, we have shown that covalently linked pyrene antennas offer a promising approach for developing efficient and aggregation-resistant light-harvesting systems. Inspired by nature's photosynthetic antenna arrays, ZnPcPy exhibits exceptional energy transfer efficiency that greatly surpasses simple mixtures. Covalent bonds between the chromophores ensure consistent distances necessary for energy transfer, while the rigidity and geometric constraints help to prevent aggregate formation. While pyrene itself is not optimized for solar energy absorption, this strategy can be widely applied to other pairs of FRET-enabled chromophores to target different absorption ranges, even when the FRET overlap is small. Overall, covalently linked FRET systems showcase the power of nature-inspired design principles for engineering advanced light-harvesting systems, paving the way for further innovation and applications in the areas of photocatalysis, emissive materials for OLEDs, and solar energy management in the form of upconversion.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Kuwait Foundation for the Advancement of Science (KFAS) for their financial support (grant number P115-14-SC05). The authors thank the Kuwait University for financial support and the RSPU facility no. (GS 01/01, GS 03/01, GS 01/08, GS 02/13, GS 01/05, GS 01/03, and GS 02/01). Additionally, we extend our thanks to the Chemistry Department at Kuwait University for providing access to their analytical facility, specifically the Matrix-Assisted Laser Desorption/Ionization (MALDI).References
- G. Schlau-Cohen, Interface Focus, 2015, 5, 20140088 CrossRef CAS PubMed.
- H. Shang, M. Li and X. Pan, Plants, 2023, 12, 1173 CrossRef CAS PubMed.
- V. M. Friebe, Hacking Photosynthesis, PhD thesis, Research and graduation internal, Vrije UniversiteitAmsterdam, 2017 Search PubMed.
- M. A. Moran and W. L. Miller, Nat. Rev. Microbiol., 2007, 5, 792–800 CrossRef CAS PubMed.
- O. Inganäs and V. Sundström, Ambio, 2016, 45, 15–23 CrossRef PubMed.
- B. C. Tashie-Lewis and S. G. Nnabuife, Chem. Eng. J. Adv., 2021, 8, 100172 CrossRef CAS.
- M. Ballottari, J. Girardon, L. Dall’Osto and R. Bassi, Biochim. Biophys. Acta, Bioenerg., 2012, 1817, 143–157 CrossRef CAS PubMed.
- T. J. Blackburn, S. M. Tyler and J. E. Pemberton, Anal. Chem., 2022, 94, 515–558 CrossRef CAS PubMed.
- L. L. Tinker, N. D. McDaniel and S. Bernhard, J. Mater. Chem., 2009, 19, 3328–3337 RSC.
- V. Jain, R. K. Kashyap and P. P. Pillai, Adv. Opt. Mater., 2022, 10, 2200463 CrossRef CAS.
- Z. Zafar, S. Yi, J. Li, C. Li, Y. Zhu, A. Zada, W. Yao, Z. Liu and X. Yue, Energy Environ. Mater., 2022, 5, 68–114 CrossRef CAS.
- J. Ge, Y. Zhang and S.-J. Park, Materials, 2019, 12, 1916 CrossRef CAS PubMed.
- Q. Van Le, V.-H. Nguyen, T. D. Nguyen, A. Sharma, G. Rahman and D. L. T. Nguyen, Chem. Eng. Sci., 2021, 237, 116547 CrossRef.
- M. R. Wasielewski, Acc. Chem. Res., 2009, 42, 1910–1921 CrossRef CAS PubMed.
- G. A. Jones and D. S. Bradshaw, Front. Phys., 2019, 7, 100 CrossRef.
- K. Stavrou, S. Suresh, D. Hall, A. Danos, N. Kukhta, A. Slawin, S. Warriner, D. Beljonne, Y. Olivier and A. Monkman, Adv. Opt. Mater., 2022, 10(17), 2200688 CrossRef CAS.
- D. Hall, K. Stavrou, E. Duda, A. Danos, S. Bagnich, S. Warriner, A. M. Slawin, D. Beljonne, A. Köhler and A. Monkman, Mater. Horiz., 2022, 9, 1068–1080 RSC.
- K. Stavrou, A. Danos, T. Hama, T. Hatakeyama and A. Monkman, ACS Appl. Mater. Interfaces, 2021, 13, 8643–8655 CrossRef CAS PubMed.
- N. Haase, A. Danos, C. Pflumm, P. Stachelek, W. Brütting and A. P. Monkman, Mater. Horiz., 2021, 8, 1805–1815 RSC.
- L. Salah, M. K. Etherington, A. Shuaib, A. Danos, A. A. Nazeer, B. Ghazal, A. Prlj, A. T. Turley, A. Mallick and P. R. McGonigal, J. Mater. Chem. C, 2021, 9, 189–198 RSC.
- A. Danos, R. W. MacQueen, Y. Y. Cheng, M. Dvořák, T. A. Darwish, D. R. McCamey and T. W. Schmidt, J. Phys. Chem. Lett., 2015, 6, 3061–3066 CrossRef CAS PubMed.
- R. W. MacQueen, Y. Y. Cheng, A. N. Danos, K. Lips and T. W. Schmidt, RSC Adv., 2014, 4, 52749–52756 RSC.
- B. Ghazal, D. E. Nevonen, L. Salah, A. Shuaib, V. N. Nemykin and S. Makhseed, Dyes Pigm., 2023, 111361 CrossRef CAS.
- A. Pinnola, J. Exp. Bot., 2019, 70, 5527–5535 CrossRef CAS PubMed.
- X. Li, L. E. Sinks, B. Rybtchinski and M. R. Wasielewski, J. Am. Chem. Soc., 2004, 126, 10810–10811 CrossRef CAS PubMed.
- N. J. Davis, R. W. MacQueen, D. A. Roberts, A. Danos, S. Dehn, S. Perrier and T. W. Schmidt, J. Mater. Chem. C, 2016, 4, 8270–8275 RSC.
- B. Zhang, G. Lyu, E. A. Kelly and R. C. Evans, Adv. Sci., 2022, 2201160 CrossRef PubMed.
- A. Husain, A. Ganesan, L. Salah, P. Kubát, B. Ghazal and S. Makhseed, ChemPlusChem, 2022, 87, e202200275 CrossRef CAS PubMed.
- V. Sundström and T. Gillbro, J. Chem. Phys., 1985, 83, 2733–2743 CrossRef.
- J. S. Huff, P. H. Davis, A. Christy, D. L. Kellis, N. Kandadai, Z. S. Toa, G. D. Scholes, B. Yurke, W. B. Knowlton and R. D. Pensack, J. Phys. Chem. Lett., 2019, 10, 2386–2392 CrossRef CAS PubMed.
- E. Aksoy, A. Danos, C. Varlikli and A. P. Monkman, Dyes Pigm., 2020, 183, 108707 CrossRef CAS.
- E. Aksoy, A. Danos, C. Li, A. P. Monkman and C. Varlikli, J. Phys. Chem. C, 2021, 125, 13041–13049 CrossRef CAS.
- A. Husain, A. Ganesan, M. Sebastian and S. Makhseed, Dyes Pigm., 2021, 184, 108787 CrossRef CAS.
- L. Bar Eyal, R. Ranjbar Choubeh, E. Cohen, I. Eisenberg, C. Tamburu, M. Dorogi, R. Ünnep, M.-S. Appavou, R. Nevo and U. Raviv, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 9481–9486 CrossRef PubMed.
- J. Ohshita, Y. Hatanaka, S. Matsui, Y. Ooyama, Y. Harima and Y. Kunugi, Appl. Organomet. Chem., 2010, 24, 540–544 CrossRef CAS.
- G. B. Ray, I. Chakraborty and S. P. Moulik, J. Colloid Interface Sci., 2006, 294, 248–254 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp02586a |
| This journal is © the Owner Societies 2023 |