
Metamorphic oxygen-evolving molecular Ru and Ir catalysts
Nataliia
Vereshchuk
 ab,
Marcos
Gil-Sepulcre
a,
Abolfazl
Ghaderian
ab,
Jan
Holub
ad,
Carolina
Gimbert-Suriñach
ac and
Antoni
Llobet
*ac
ab,
Marcos
Gil-Sepulcre
a,
Abolfazl
Ghaderian
ab,
Jan
Holub
ad,
Carolina
Gimbert-Suriñach
ac and
Antoni
Llobet
*ac
aInstitute of Chemical Research of Catalonia (ICIQ), Barcelona Institute of Science and Technology (BIST), Avda. Països Catalans 16, 43007 Tarragona, Spain. E-mail: allobet@iciq.cat
bDepartament de Química Física i Inorgànica, Universitat Rovira i Virgili, Marcel·lí Domingo s/n, 43007 Tarragona, Spain
cDepartament de Química, Universitat Autònoma de Barcelona, Cerdanyola del Vallès, 08193 Barcelona, Spain
dDepartment of Inorganic Chemistry, University of Chemistry and Technology, Prague, CZ-16628 Prague, Czech Republic
First published on 2nd December 2022
Abstract
Today sustainable and clean energy conversion strategies are based on sunlight and the use of water as a source of protons and electrons, in a similar manner as it happens in Photosystem II. To achieve this, the charge separation state induced by light has to be capable of oxidising water by 4 protons and 4 electrons and generating molecular oxygen. This oxidation occurs by the intermediacy of a catalyst capable of finding low-energy pathways via proton-coupled electron transfer steps. The high energy involved in the thermodynamics of water oxidation reaction, coupled with its mechanistic complexity, is responsible for the difficulty of discovering efficient and oxidatively robust molecules capable of achieving such a challenging task. A significant number of Ru coordination complexes have been identified as water oxidation catalysts (WOCs) and are among the best understood from a mechanistic perspective. In this review, we describe the catalytic performance of these complexes and focus our attention on the factors that influence their performance during catalysis, especially in cases where a detailed mechanistic investigation has been carried out. The collective information extracted from all the catalysts studied allows one to identify the key features that govern the complex chemistry associated with the catalytic water oxidation reaction. This includes the stability of trans-O–Ru–O groups, the change in coordination number from CN6 to CN7 at Ru high oxidation states, the ligand flexibility, the capacity to undergo intramolecular proton transfer, the bond strain, the axial ligand substitution, and supramolecular effects. Overall, combining all this information generates a coherent view of this complex chemistry.
 Nataliia Vereshchuk | Nataliia Vereshchuk obtained her bachelor's and master's degree in Chemical Engineering from the National Technical University of Ukraine. In 2021 earned her PhD degree from the Institute of the Chemical Research of Catalonia (Spain) under the supervision of Prof. Antoni Llobet. She is currently a postdoctoral fellow in Prof. Katherine Marica's team at Dartmouth College (Hanover, NH), where she works on developing Multifunctional Materials for Chemical Sensing based on MOFs and COFs. |
 Marcos Gil-Sepulcre | Marcos Gil-Sepulcre obtained his PhD from the Autonomous University of Barcelona (UAB) in 2018. The same year he joined Prof. Antoni Llobet's team at the Institute of Chemical Research of Catalonia (ICIQ), where he worked on the development of molecular-based catalysts and hybrid materials applied to water oxidation. He is currently Marie Skłodowska–Curie postdoctoral fellow in Prof. Serena DeBeer's group at Max Planck Institute for Chemical Energy Conversion (MPI CEC), where he works on spectroscopy applied to CO2 reduction catalysts. |
 Abolfazl Ghaderian | Abolfazl Ghaderian defended his PhD in Chemistry with Honor (Cum laude) in Ruthenium complexes as molecular water oxidation catalysts at ICIQ in 2020. His research internship was done at Prof. Ivana Ivanovic's lab at the Friedrich Alexander University Erlangen-Nürnberg, in Germany within the framework an international collaborative project in 2019. During his studies, he won several prizes e.g. the Gold prize in “Explaining Their Research in a Video” of the Chemistry – A European Journal video competition for explaining “Water Oxidation with Ru-bda” highlighted by ChemistryViews website. |
 Jan Holub | Jan Holub defended his PhD in 2016 under the tutelage of Prof. J.-M. Lehn at the Institut de Science et d’Ingénierie Supramoléculaires, Strasbourg, France studying metallosupramolecular self-assembly. He then joined the group of Prof. A. Llobet at Institut Català d’Investigació Química, Tarragona, Spain where he studied the use of molecular complexes for electrocatalytic activation of various small molecules. After a short period at the University of Cambridge, Cambridge, United Kingdom with Prof. G. Bernardes, he obtained a junior position at the University of Chemistry and Technology, Prague, Czech Republic focusing on guided metalo-driven self-assembly for preparation of large functional architectures. |
 Carolina Gimbert-Suriñach | Carolina Gimbert obtained her PhD at UAB under the supervision of Prof. A. Vallribera. After one year as assistant professor at the same university, she moved to UNSW to undertake postdoctoral research with Prof. S. B. Colbran. Then she worked at ICIQ in Prof. A. Llobet group as postdoctoral fellow and research group coordinator. After a short stay at UB as Serra Húnter professor, she started as Ramón y Cajal fellow and co-leader of CatSyNanoMat group at UAB. Her scientific interests are in the field of photocatalysis as well as organic and hybrid materials with application to artificial photosynthesis. |
 Antoni Llobet | Antoni Llobet received his PhD from the Autonomous University of Barcelona (UAB) in 1985. Then he was a Postdoctoral Fellow at the University of North Carolina with T. J. Meyer and at Texas A&M University with D. T. Sawyer and A. E. Martell. He is currently a Professor of Chemistry at UAB and Group Leader at ICIQ. |
1. Introduction
1.1 General context
Global warming is responsible for the climate change situations1 that are already leading to natural catastrophes placing billions of people at risk of heatwaves, water shortages, and a range of other problems. In addition, recent mathematical models predict the collapse of the Atlantic circulation2 and crossing the point of no return for the conventional earth system trajectories in the Anthropocene,3 putting additional dramatism into the immediate future of planet earth and us as humankind. This global warming is mainly due to the vast amounts of CO2 generated from the combustion of fossil fuels. The recent IEA report4 stated that the total energy supply in 2018 was 14 Gtoes (140 Ecals), with 85% of this supply coming from burning fossil fuels, thus directly causing a continuous rise of the CO2 concentration in the atmosphere, further worsening the global warming and the increasingly repeating extreme climate episodes suffered recently.There is thus an urgent need to shift from fossil to green and sustainable energy resources. Among several options, water splitting with sunlight (hν-WS) stands out as one of the most promising strategies in short to medium term.5 As indicated in eqn (1), water splitting consists of the transformation of water into H2, a clean fuel, and O2 as a side product. This reaction is thermodynamically uphill by 113.5 kcal mol−1,6 with the energy input ideally covered by omnipresent sunlight (hν). The H2 generated in this way is termed “solar hydrogen”.
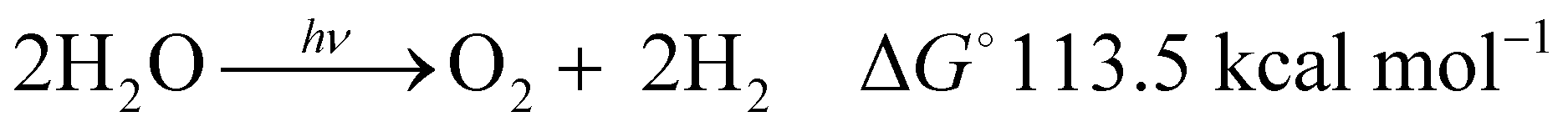 | (1) |
The WS reaction can be written as the sum of two redox half-reactions that involve the oxidation of water and the reduction of protons as indicated in eqn (2) and (3):
| 2H2O → O2 + 4H+ + 4e− E° = 1.23 V vs. NHE at pH = 0 | (2) |
| 2H+ + 2e− → H2 E° = 0.00 V vs. NHE at pH = 0 | (3) |
The bias-free overall water splitting with sunlight can be achieved via photocatalysis or by using a photoelectrochemical cell (PEC) using solid-state (photo)anodes and (photo)cathodes. For efficient functioning of PECs, the water oxidation and proton reduction reactions should occur at the lowest possible overpotentials. Therefore, identifying appropriate catalysts is an imperative and crucial part of any serious study. This report will focus our discussion on the water oxidation catalyst.
Nature's solar fuels generation strategy also involves the oxidation of water to dioxygen using the so-called CaMn4O5 cubane cluster as a catalyst, where the Mn centres have an octahedral type of coordination, and the Ca centre is seven coordinated. The Mn and Ca atoms are coordinated with oxo bridging groups with additional coordination by nitrogen and oxygen atoms from imidazole and carboxylate amino acid residues and by aqua ligands, as shown in Fig. 1. This CaMn4O5 catalyst is situated in photosystem II (PSII) in the chloroplast and is commonly known as the oxygen-evolving complex (OEC).7,8 There are several hypothetical mechanisms proposed associated with photodamage to PSII depending on whether light energy is initially absorbed by the photosynthetic pigment, the Mn cluster, or both.9,10 Further, the formation of singlet oxygen by the interaction of molecular oxygen with the excited triplet state of chlorophyll has been identified as one of the main causes of the oxidative stress that causes the degradation,9,11 requiring the PSII to reassemble every 20 to 40 minutes.12 Assuming that the OEC-PSII turnover is at ms time scale, it makes roughly 1.2–2.4 million turnovers (TONs) before it is replaced.
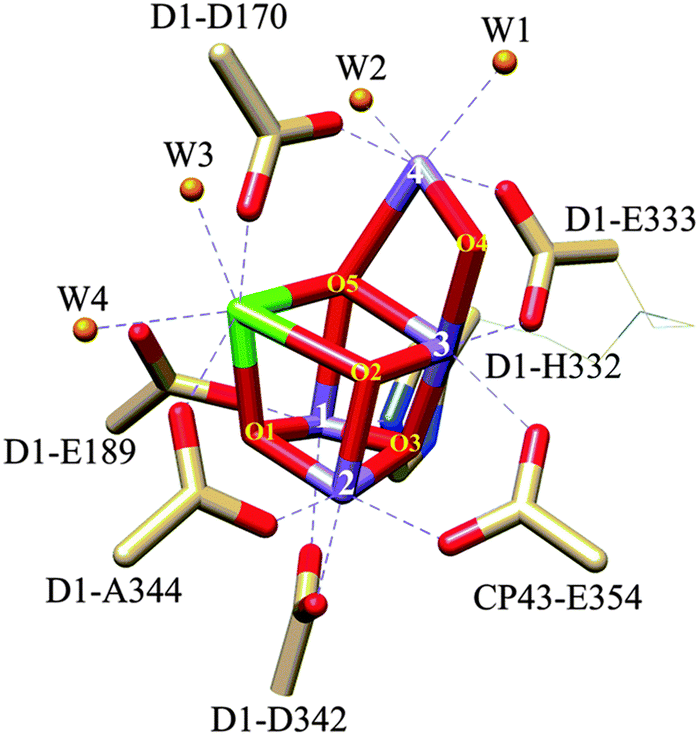 | ||
| Fig. 1 Drawing of the crystal structure of the OEC-PSII. The Mn4O5Ca cubane type of structure and the dangling Mn(4) are shown with capped sticks. The bonds for the additional ligands completing the octahedral type of coordination of the Mn and Ca metal centres are depicted with dashed lines. The histidine and carboxylates residues, represented with capped sticks, are indicated in the drawing, whereas the aqua ligands are represented with spheres and are labelled as W1–W4. Please note that W in this drawing does not stand for the tungsten metal.13 Colour code: Mn, purple; Ca, green; O, red; O from aqua ligands, orange; N, blues; C, light brown. Reproduced from ref. 85 with permission from the PCCP Owner Societies. | ||
1.2 The development of molecular WOCs
The development of artificial water oxidation catalysts (WOCs) has been mainly carried out via solid-state oxides and molecular complexes. Whereas the first WOC based on metal oxides dates as far back as 1903,14 the first properly characterised molecular WOC is the so-called blue dimer cis,cis-{[RuIII(H2O)(bpy)2]2(μ-O)}4+, 1, (bpy is 2,2′-bipyridine; see Fig. 2 for a drawing of this complex) which was reported only in 1982.15,16 All the ligands and complexes discussed in the present work are labelled and displayed in Charts 1 and 2 respectively. The development of WOCs is a complex task because the catalyst has to manage the removal of four electrons from two water molecules, which need to be stored on the complex, concurrently remove four protons and finally form an O–O bond. Nevertheless, for both solid-state and molecular catalysts, we have witnessed spectacular development since the turn of the present century.17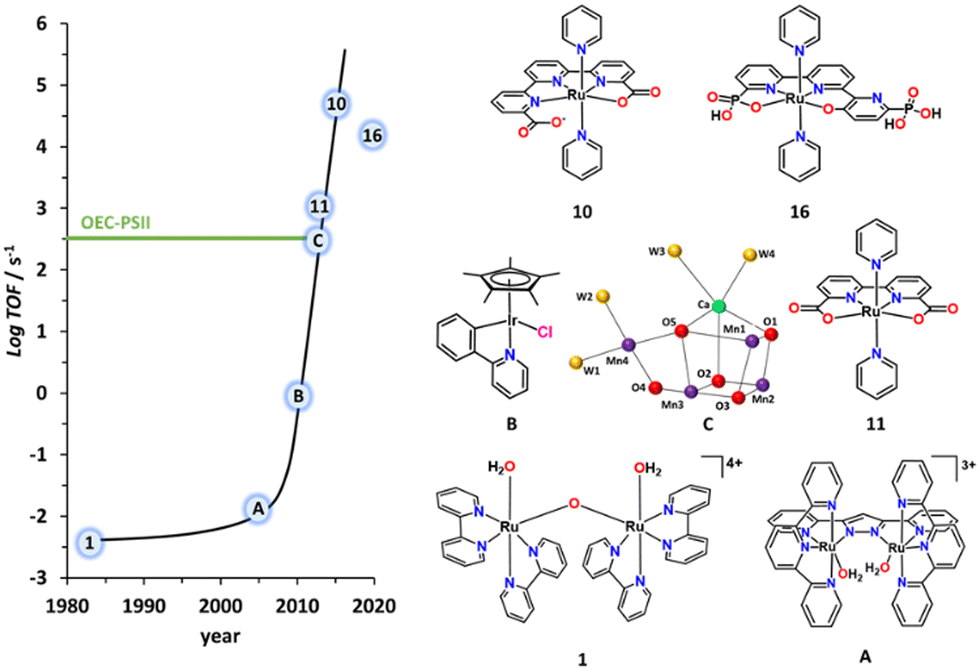 | ||
| Fig. 2 Graph representing the TOF of the indicated molecular catalysts vs the year they were published. The numerical labels on the catalyst structure are keyed in Chart 2. A is {[RuII(H2O)(trpy)]2(μ-bp)}3+ (bp = 3,5-bis(2-pyridyl)pyrazolate),17B is [IrCl(Cp*)(phpy)] where (phpy = 2-(pyridin-2-yl)benzen-1-ide)17 and C represents a simplified ball and stick drawing of the OEC-PSII shown in Fig. 1. | ||
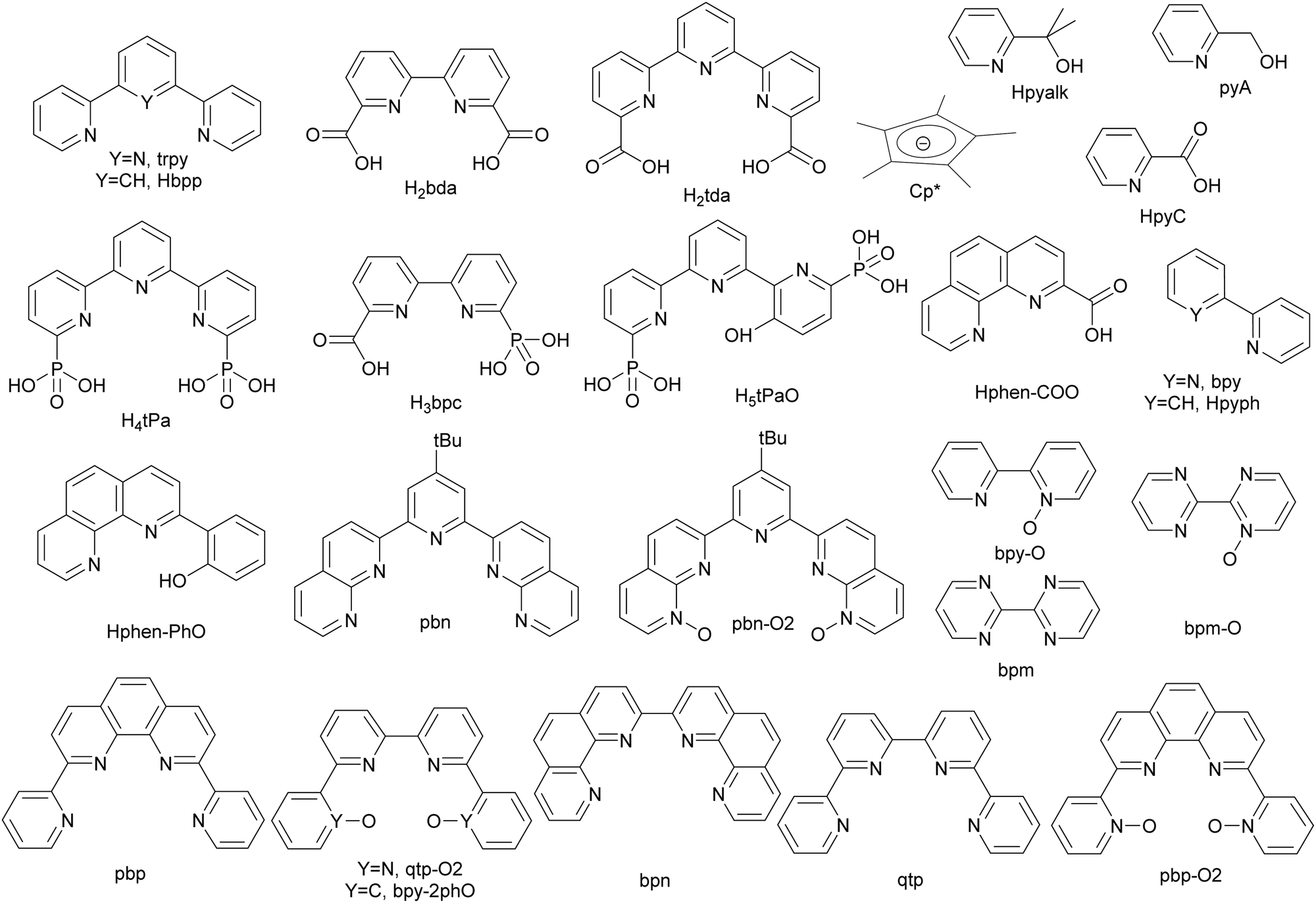 | ||
| Chart 1 Ligands discussed in this report. | ||
 | ||
| Chart 2 Complexes discussed in this report. | ||
In particular, for molecular WOCs, this development can be grasped by examining the turnover frequencies achieved by the reported catalysts over time, as displayed in Fig. 2. This molecular WOC development has been accomplished thanks to the molecular toolkit that provides a large number of transition metal complexes with different geometries and oxidation states, together with an infinite number of organic ligands that can coordinate the metal centre. The judicious combination of all these elements has resulted in powerful and robust molecular WOCs.18–21
Although a number of different transition metal complexes have been reported as potential WOCs,15 the development has been mainly led by Ru complexes22 for reasons that will be discussed herein. Today, Ru catalysts are highly efficient and rugged, giving more than a million TONs with no sign of deactivation23 and with TOFs in the range of 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s−1 when driven electrochemically.24,25 This is about two orders of magnitude faster than the operation of the OEC at PSII, although the latter is driven by light, which is the ultimate goal of building PEC devices.
000 s−1 when driven electrochemically.24,25 This is about two orders of magnitude faster than the operation of the OEC at PSII, although the latter is driven by light, which is the ultimate goal of building PEC devices.
The achievement of robust molecular WOCs has been made thanks to the information extracted from the electrochemical and spectroscopic characterisation of reaction intermediates and complemented with theoretical studies, mainly based on DFT calculations. This has provided a complete picture of the different factors that govern the performance of Ru-WOCs, including fine-tuning their working potentials and critical activation energies.21,26 In addition, a valuable piece of information that needs to be addressed in developing robust and efficient WOCs is the identification of the potential deactivation pathways that can occur in parallel or associated with the generated reactive intermediates during the catalysis. Understanding the occurrence of deactivation reactions allows for designing WOCs while minimising or even suppressing these processes. However, this is a very infrequent investigation due to the difficulty involved in analysing spent or transformed catalysts due to the multiple potential products that can be generated under turnover conditions. In the present review, we focus on describing water oxidation catalysts that evolve into new species, describing both catalytic and deactivated species and the central pathways that lead to them or towards the formation of the corresponding oxide.
1.3 Molecular water oxidation catalysts: requirements and pitfalls
Transition metal complexes capable of behaving as water oxidation catalysts have to be able to store four holes in addition to generating O–O bonds. The former implies necessarily that the metal center will have to cycle through different oxidation states unless the holes can be stored in the auxiliary ligands. Except for a couple of examples reported recently27–29 where the electron transfer (ET) occurs only at the ligands, the main role of the metal center consists of achieving higher oxidation states (storing holes) at relatively low potentials and generating a sufficiently reactive M–O group where the O–O bond formation can occur. A strategy to achieve reactive oxidation states at relatively low potentials consists of the use of a Ru(II)–OH2 group that can undergo two consecutive proton-coupled electron transfer (PCET) processes and reach the reactive Ru(V)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O species as is the case for instance of [RuII(trpy)(bpy)(H2O)]2+, 2 (see Scheme 1).30 This way, two holes have been stored at the metal center, ready to accept two electrons, for instance, from an incoming water molecule and generate a Ru(III)–OOH group. However, if the complex contains two aqua ligands such as in cis-[RuII(bpy)2(H2O)2]2+, 3, and in the mentioned blue dimer 1, then four-hole storage can be achieved at reasonably low redox potentials generating cis-[RuVI(bpy)2(O)2]2+ and cis,cis-{[RuV(O)(bpy)2]2(μ-O)2}4+, respectively. The PCET strategy described here is also used in nature by several oxidative metalloproteins31 as well as by the OEC-PSII. There, the tetranuclear Mn complex stores four holes, achieving a reactive Mn–O group responsible for O–O bond formation as described in the Kok cycle.32–34 An additional strategy to influence the redox potentials of the reactive species is via the electronic nature of the ligands bonded to the metal center. In general, electron donors will decrease redox potentials, whereas electron withdrawers will increase them.26,35 Especially drastic effect is exerted by replacing neutral ligands with anionic ones. For example, for [RuCl(trpy)(bpy)]+, 4, the potential of the Ru(III/II) couple appears at 0.90 V vs. NHE. All redox potentials discussed here are vs. NHE unless otherwise mentioned. In sharp contrast, replacing the trpy ligand with bis-pyridylphenylate (bpp−) in [RuIICl(bpp)(bpy)], 5, shifts the potential cathodically by 800 mV down to 0.10 V.36
O species as is the case for instance of [RuII(trpy)(bpy)(H2O)]2+, 2 (see Scheme 1).30 This way, two holes have been stored at the metal center, ready to accept two electrons, for instance, from an incoming water molecule and generate a Ru(III)–OOH group. However, if the complex contains two aqua ligands such as in cis-[RuII(bpy)2(H2O)2]2+, 3, and in the mentioned blue dimer 1, then four-hole storage can be achieved at reasonably low redox potentials generating cis-[RuVI(bpy)2(O)2]2+ and cis,cis-{[RuV(O)(bpy)2]2(μ-O)2}4+, respectively. The PCET strategy described here is also used in nature by several oxidative metalloproteins31 as well as by the OEC-PSII. There, the tetranuclear Mn complex stores four holes, achieving a reactive Mn–O group responsible for O–O bond formation as described in the Kok cycle.32–34 An additional strategy to influence the redox potentials of the reactive species is via the electronic nature of the ligands bonded to the metal center. In general, electron donors will decrease redox potentials, whereas electron withdrawers will increase them.26,35 Especially drastic effect is exerted by replacing neutral ligands with anionic ones. For example, for [RuCl(trpy)(bpy)]+, 4, the potential of the Ru(III/II) couple appears at 0.90 V vs. NHE. All redox potentials discussed here are vs. NHE unless otherwise mentioned. In sharp contrast, replacing the trpy ligand with bis-pyridylphenylate (bpp−) in [RuIICl(bpp)(bpy)], 5, shifts the potential cathodically by 800 mV down to 0.10 V.36
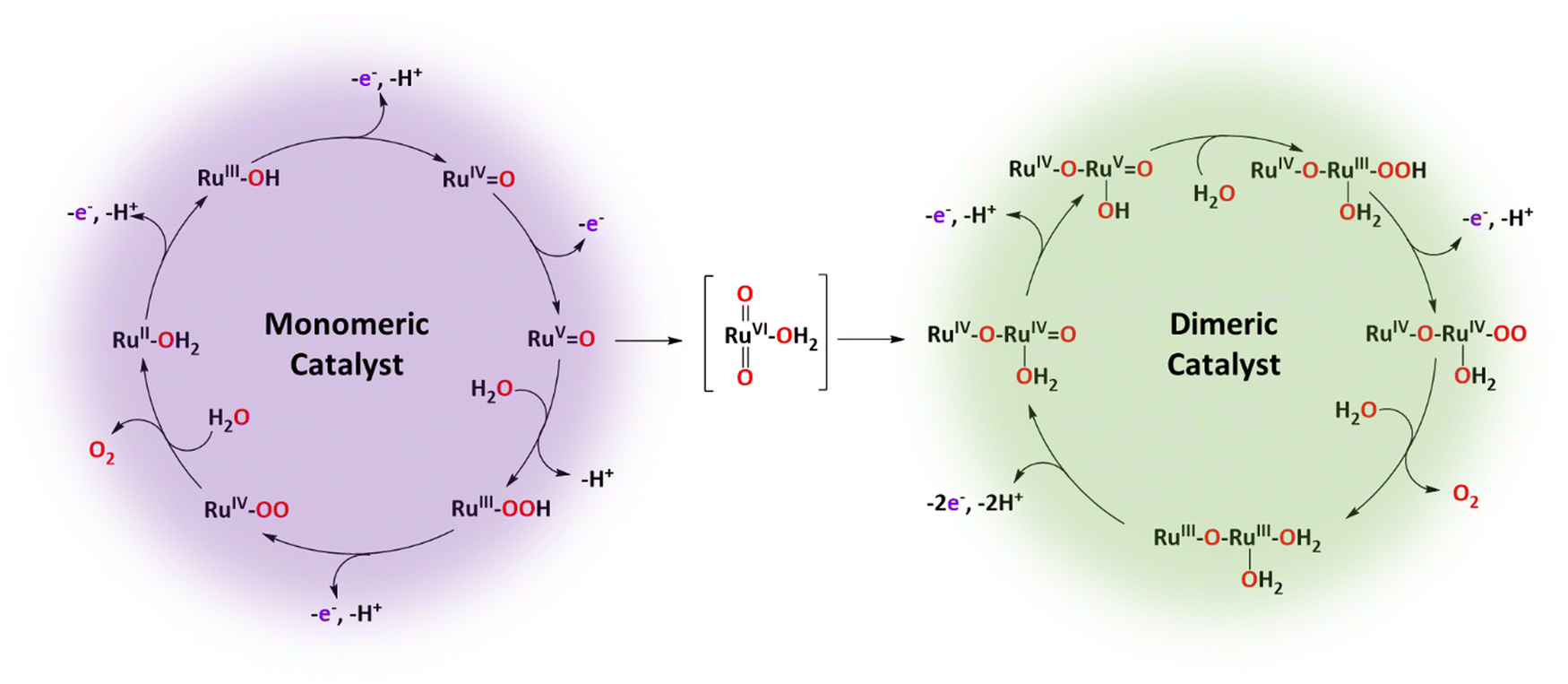 | ||
| Scheme 1 Catalytic water oxidation cycles followed by complex [RuII(trpy)(bpy)(H2O)]2+, 2, based on its monomeric (left) and oxo-bridged dimeric (right) species. In the center is shown the trans-[(trpy)RuVI(O)2(H2O)]2+, 7, the complex that acts as a gate between the two catalytic cycles. The trpy and bpy auxiliary ligands that complete the octahedral coordination sphere of the Ru centers are not shown. | ||
For transition metal complexes, access to different oxidation states can significantly change the electronic nature of the metal center. Thus, the preferred geometry, coordination number, and type of basicity (soft/hard) depend on the oxidation state. For instance, Ru(II) is considered a soft acid and thus prefers N-donor type ligands to O-donor ones. However, Ru(III) and Ru(IV) are considered hard acids and thus prefer ligands that bond via O-atom donors.37 Indeed, there is rich chemistry for Ru(III) and Ru(IV) ions containing oxo ligands that can act as terminal or bridging ligands giving rise to dimers and/or oligomers. For Ru(III) a paradigm is the blue dimer cis,cis-{[RuIII(H2O)(bpy)2]2(μ-O)}4+, 1, mentioned earlier.38,39 The electronic nature of the metal center at the different oxidation states where the species need to go through the catalytic cycle will pose a significant challenge to the stability of the M–L bond of the auxiliary ligands that complement the coordination sphere of the Ru–OH2/Ru–O groups. The M–L bond breaking can drive the catalytic cycle towards ligand loss and the formation of other species that might not be catalytically active. The latter will obviously be responsible for derailing the catalytic cycle towards side pathways that lead to inactive species.
1.4 Compilation of ligand effects extracted from the behaviour of model catalysts
The rich chemistry of Ru complexes, which can display different oxidation states, coordination numbers and geometries, has fostered the discovery of a large number of reactions that are catalysed by them.40,41 This rich chemistry is, in turn, responsible for a variety of pathways followed by Ru complexes during the water oxidation catalytic reaction. The chosen paths are not generally ascribed to a single factor but rather to a combination of them, as described below. The following section is divided into six subsections where a particular aspect of the complex reactivity is highlighted. This allows introducing, step by step, the key factors that govern the complex chemistry associated with the catalytic water oxidation reaction offering a coherent view. Section 2.1 focuses on the effects produced by the relative stability of the high oxidation states of Ru and, in particular, the tendency to stabilise trans-O–Ru–O groups. Section 2.2 underlines the need to avoid easily oxidisable ligands as well as the geometrical consequences of using tridentate facial ligands such as Cp*, in this case, for Ir complexes. Section 2.3 overviews Ru complexes based on FAME (Flexible, Adaptative, Multidentate, Equatorial) ligands, their capacity to change from CN6 to CN7 at high oxidation states and the beneficial effects of intramolecular proton transfer. A particular case will be described where the steric effects radically change the whole mechanistic behaviour and finally leads to partial ligand decoordination and oxidation. Section 2.4 describes the chemistry related to Ru complexes that contain a phenolato moiety bonded to the Ru center and its evolution via multiple steps to a carboxylate as the water oxidation reaction proceeds. Section 2.5 describes a related ligand oxidation effect but now with complexes that undergo N-oxide formation favoured by the release of bond strain. It is also shown how depending on the rigidity of the ligands, other pathways, such as axial ligand substitution, can compete with N-oxide formation. And finally, Section 2.6 displays how the presence of additional supramolecular effects such as CH–π, π–π, and anion–π interactions can change the reactivity from the established one in the absence of them.2. Description of the reported major results for evolving WOCs
The following describes the key properties of a few Ru complexes reported in the literature that have been tested as water oxidation catalysts that evolve to a different species upon turnover conditions. In particular, we will focus on examples where the newly formed species behave as water oxidation catalysts. In some cases, they perform even better than their initial starting complex. The combination of examples described below will provide a global view of the different factors that influence molecular water oxidation catalyst performance and thus will be helpful for the design of new, improved catalysts.2.1 Is one site really enough? or the coordination preferences of different oxidation states of Ru
A number of mononuclear monoaqua Ru-based complexes have been reported in the literature as WOCs.42–44 Most of them are proposed to carry out the WO reaction in a relatively similar manner following the roller coaster water nucleophilic attack (WNA) mechanism.45 A paradigmatic example is [RuII(trpy)(bpy)(H2O)]2+, 2, whose proposed reaction mechanism is described in Scheme 1, left.46Initially, the Ru(II)–OH2 complex (auxiliary ligands are omitted for clarity) reaches the Ru(IV)O species via two consecutive PCET processes. Then an additional ET generates the highly reactive species Ru(V)O that is proposed to undergo a water nucleophilic attack with a solvent water molecule, to form the Ru(III)–OOH peroxide species that further suffers an additional PCET process to form Ru(IV)–OO. This subsequently releases dioxygen and coordinates an aqua ligand that regenerates the initial complex, consequently closing the catalytic cycle.
Interestingly, under turnover conditions at Eapp = 1.60 V, the initial mononuclear complex is slowly but progressively transformed into an oxo-bridged dinuclear complex losing one of the initial bpy ligands. This transformation occurs via a multi-step process where initially, Ru(V) mononuclear complex [(trpy)(bpy)RuVO]3+, loses a bpy ligand according to eqn (4), generating the Ru(V) species trans-[(trpy)RuV(O)2(H2O)]+. At the applied potential, the latter is oxidised to its Ru(VI) species 7, as indicated in eqn (5). Finally, 7 and 2 react to form the dinuclear complex 6 (eqn (6)) that originates the second water oxidation catalytic cycle shown on the right-hand side of Scheme 1.
| [(trpy)(bpy)RuVO]3+ + 2H2O → trans-[(trpy)RuV(O)2(H2O)]+ + bpyH+ + H+ | (4) |
| trans-[(trpy)RuV(O)2(H2O)]+–1e− → trans-[(trpy)RuVI(O)2(H2O)]2+, 7 | (5) |
| trans-[(trpy)RuVI(O)2(H2O)]2+, 7 + [RuII(trpy)(bpy)(H2O)]2+, 2 → trans-[(trpy)(bpy)RuIV–O–RuIV(O)(H2O)(trpy)]4+, 6 + H2O | (6) |
Once formed, complex trans-[(trpy)RuVI(O)2(H2O)]2+, 7, constitutes the gate that bridges the mononuclear with the dinuclear catalytic cycles, as indicated in Scheme 1. Complex 6, responsible for the dinuclear catalytic pathway, is an excellent catalyst that gives more than 14000 cycles with no sign of decomposition.46
The driving force for the bpy loss is the strong trans effect that the Ru(V)O group exerts over the Ru–N bpy bond, weakening it. Furthermore, the trans-Ru(O)2 entity is also known to generate very stable complexes47,48 and thus is an additional driving force for the bpy loss. Examples of Ru complexes undergoing similar bpy losses have been reported recently.49 In this line, it is essential to highlight here that the mononuclear Ru complex cis-[RuII(bpy)2(H2O)2]2+, 3, acts as WOC but is progressively transformed into trans-Ru(O)2 species again due to the significant stability of the trans O–Ru–O groups.48 Unfortunately, the latter does not have the thermodynamic driving force to be able to act as a WOC and thus represents a non-productive deactivation pathway.
Related examples have been reported for complexes proposed to undergo partial decoordination of bpy and other polypyridyl-based ligands and the formation of the corresponding N-oxide50 that will be described in more detail in the next section.50–55
2.2 Non oxidatively robust WOCs, the benzyl and picolyl case
The thermodynamics of water oxidation requires an E° = 1.23 V (or a ΔG° = 123 kcal mol−1). It is important to compare the thermodynamics of water oxidation versus that of organic substrate oxidation. For instance, the oxidation of ethylbenzene to 1-phenylethanol and hydrogen carried out by the water, similarly to the water-splitting reaction described in eqn (1), requires a ΔG° = 22.3 kcal mol−1 as indicated in the equation below.| PhCH2Me + H2O → PhCH(OH)Me + H2 | (7) |
Eqn (7) clearly illustrates that organic oxidation is a much more favoured reaction from a thermodynamic perspective than water oxidation by more than 90 kcal mol−1. This implies a large amount of energy; thus, it is imperative to use oxidatively robust ligands (with significant kinetic barriers to oxidation). Otherwise, the ligands attached to the metal center will be irreversibly oxidised, sometimes all the way to CO2. A large number of examples have been described in the literature showing that when transition metal complexes that contain easily oxidisable groups such as picolyl (CH2-py) or sulphur groups are tested as WOCs, they invariably suffer irreversible deactivation processes.56–60
The case of [RuII(bda)(pyA)(pic)], 8a, (bda is [2,2′-bipyridine]-6,6′-dicarboxylato; pic is 4-methylpyridine; pyA is 2-pyridinemethanol; see Chart 1 for ligand drawings) is another interesting example that illustrates the oxidation fragility of the picolyl group. Under an applied potential of 1.5 V 8a, which contains a 2-pyridinemethanol group, is readily and quantitatively oxidised to the corresponding picolinic acid (pyC, Chart 1) complex, [RuIII(bda)(pyC)(pic)], 8b, as illustrated in Scheme 2. Interestingly, the new ligand generated in situ becomes an oxidatively robust ligand.61
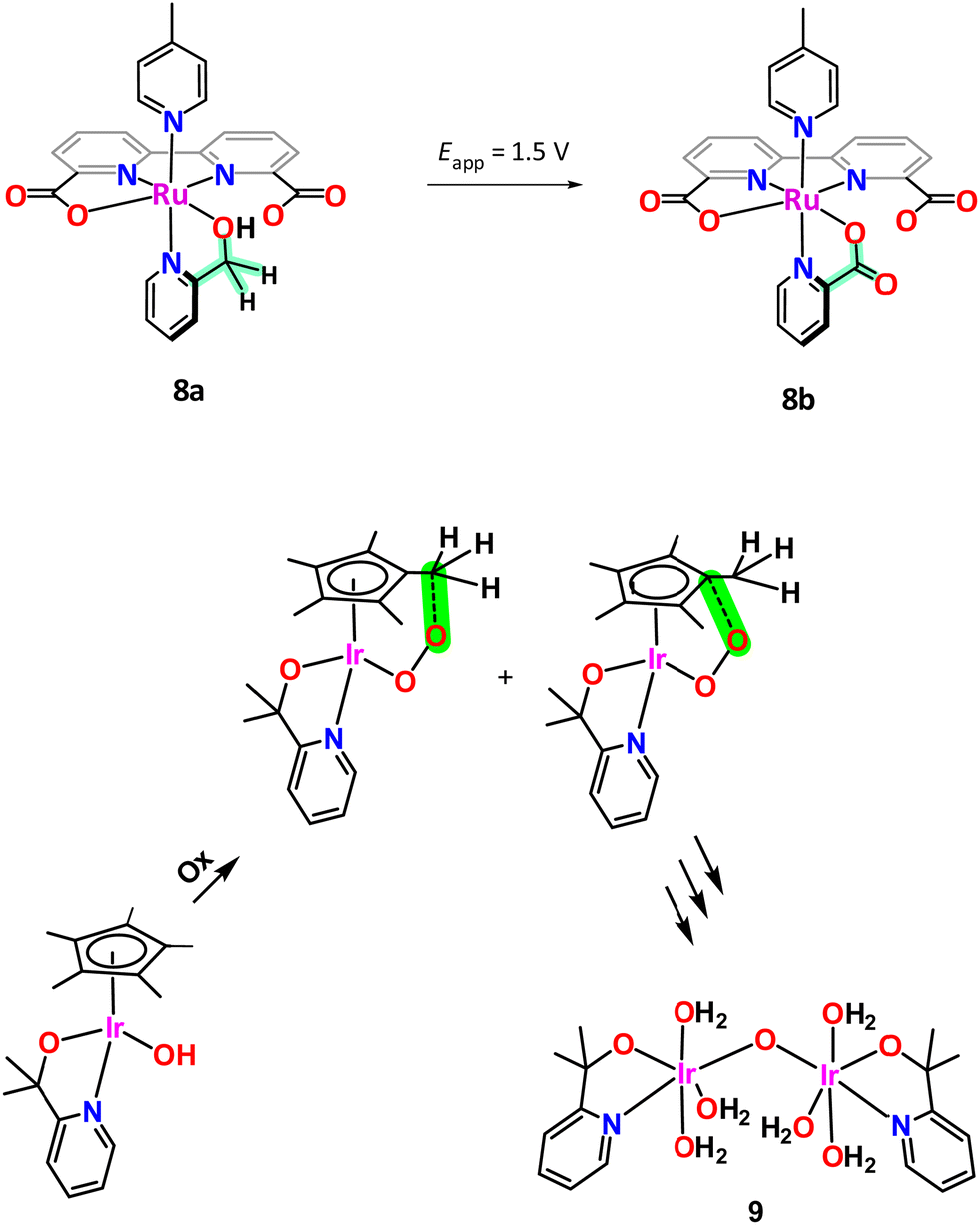 | ||
| Scheme 2 In situ ligand oxidations. Top, the CH2 of the picolyl site (highlighted in green) of [RuII(bda)(pyA)(pic)], 8a, is oxidised to the corresponding acid at an applied potential of 1.5 V to form [RuIII(bda)(pyC)(pic)], 8b. Bottom, the [IrIII(Cp*)(pyalk)(OH)] complex is oxidized to the active species {[IrIV(pyalk)(H2O)3]2(μ-O)}4+, 9, losing the Cp* ligand. | ||
An interesting case of ligand transformation has been described for mononuclear Ir complexes containing the Cp* ligand that acts in a tridentate facial manner in [IrIII(Cp*)(L)(OH)] (HL = 2-phenylpyridine (Hpyph) or 2-(2′-pyridyl)-2-propanolate (Hpyalk))62 and [IrIII(Cp*)(L)(OH)]+ (L = 2,2′-bipyridine (bpy))63 that were initially reported by Crabtree et al.64 as water oxidation catalysts. Later, Macchioni et al.,65–67 showed that the Cp* ligand in the IrIII-pyalk complex underwent oxidative transformation all the way to HCOOH and MeCOOH, which finally led to complete Cp* ligand loss. This transformation is highly favoured because the potentially reactive high oxidation state species [IrV–OOH] and/or [IrV–O] have the perfect geometry to interact intramolecularly with the C–Me moiety of the Cp* ligand as indicated in the bottom of Scheme 2. Further analysis of the active species in the homogeneous phase for the complex [IrIII(Cp*)(pyalk)(OH)] led to the proposal of a bis-oxo Ir dinuclear complex, {[IrIV(pyalk)(H2O)2]2(μ-O)2}2+, as the catalytically active species.68 However, after additional careful analysis, including a DFT combined with an array of spectroscopic techniques, favours a single oxo bridge dinuclear Ir complex, {[IrIV(pyalk)(H2O)3]2(μ-O)}4+, 9, as the active species as shown in Scheme 2, bottom.69
2.3 Steric effects at high oxidation states with FAME ligands and the entropic factor
The best WOCs today are Ru complexes containing FAME ligands, defined as Flexible, Adaptative, Multidentate auxiliary ligands that occupy the Equatorial position of the Ru coordination geometry. The best examples of Ru complexes with FAME ligands are the [Ru(tda)(py)2], 1021 (see Fig. 3), and [Ru(bda)(py)2], 11,70,71 and their related family of complexes.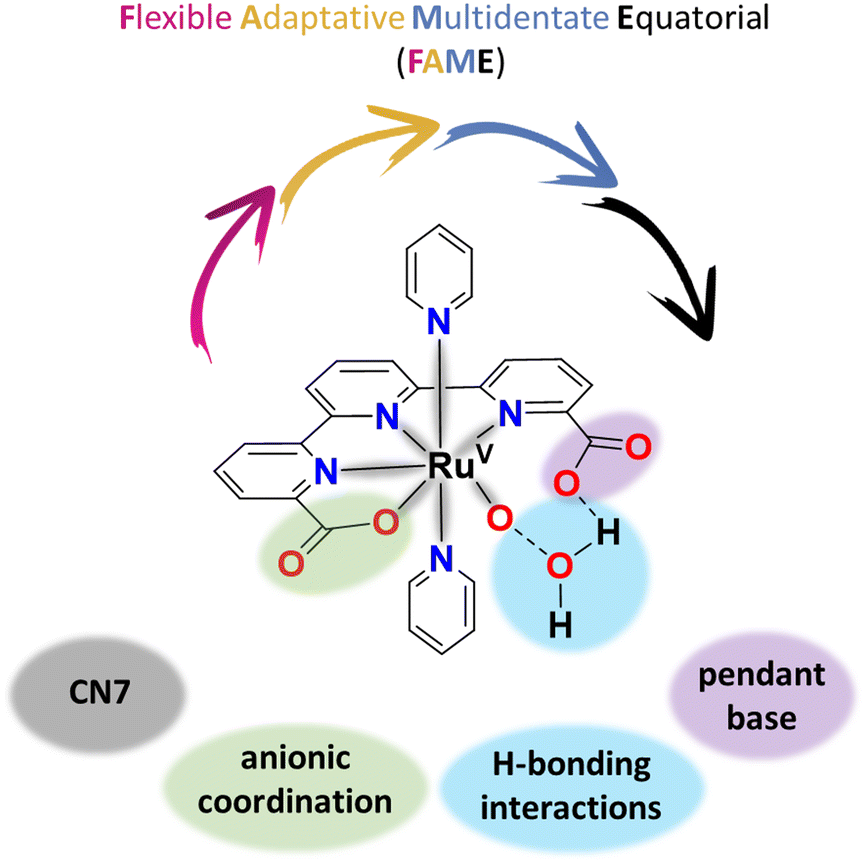 | ||
| Fig. 3 Drawing of the [RuV(O)(tda)(py)2] complex, containing the tda2− FAME ligand, interacting with an exogenous solvent H2O molecule via H-bonding before the O–O bond formation step. | ||
For the particular case of the tda2− pentadentate FAME ligand, it adapts to the coordination demands of the Ru metal center at different oxidation states both from a geometrical and an electronic perspective. From a geometrical perspective, this involves the rearrangement of the first coordination sphere of the Ru complexes from a six-coordinated (CN6) metal with pseudo-octahedral geometry at oxidation states II and III to a potential seven-coordinated (CN7) pentagonal bipyramidal type of geometry at oxidation states IV and V. From an electronic perspective, the pentagonal bipyramidal geometry brings additional accessibility to high oxidation states by the coordination of one more Lewis's base atom donor (carboxylate). In addition, FAME ligands can affect the kinetics of water oxidation catalysis, primarily through second coordination sphere effects. In this context, for complexes where the O–O bond formation is the rate-determining step, and the operating mechanism is WNA. The intramolecular proton transfer is the critical phenomenon that drastically reduces the free activation energies.
On the other hand, for the complexes such as the [RuII(bda)(MeO-isoq)2], 12, that follow an interaction of two M–O units (I2M) mechanism, supramolecular π–π stacking interaction via the axial ligands is the crucial element that provides a low energy pathway. The positive π–π stacking interaction significantly reduces the entropic cost of bringing two molecules sufficiently close to form O–O bond as graphically depicted in the calculated transition state shown in Fig. 4 for the proposed catalytic cycle of 12.71
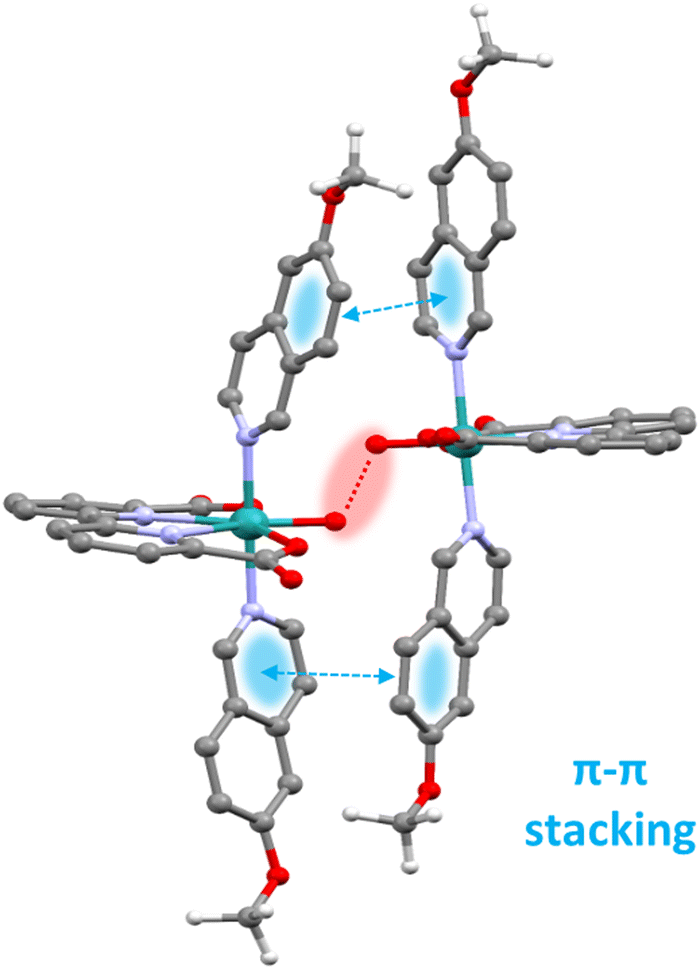 | ||
| Fig. 4 Drawing of the transition state structure calculated for the [RuII(bda)(MeO-isoq)2], 12, catalyst at the O–O bond formation (red highlight). The blue highlight shows the supramolecular π–π stacking attractive bonding interaction among the aromatic groups of the MeO-isoq axial ligands. | ||
Recently the preparation and performance of a Ru-tda analogue, [RuII(HtPa)(py)2]−, 13, where the carboxylate has been substituted by phosphonate groups, have been reported.22 The structural consequences of changing a carboxylate group by phosphonate can be graphically observed by comparing the structures of the Ru(IV) diamagnetic seven coordinated catalyst precursors [RuIV(tda-κ-N3O2)(py)2]2+, 14, and [RuIV(tPa-κ-N3O2)(py)2], 15, displayed in Fig. 5.
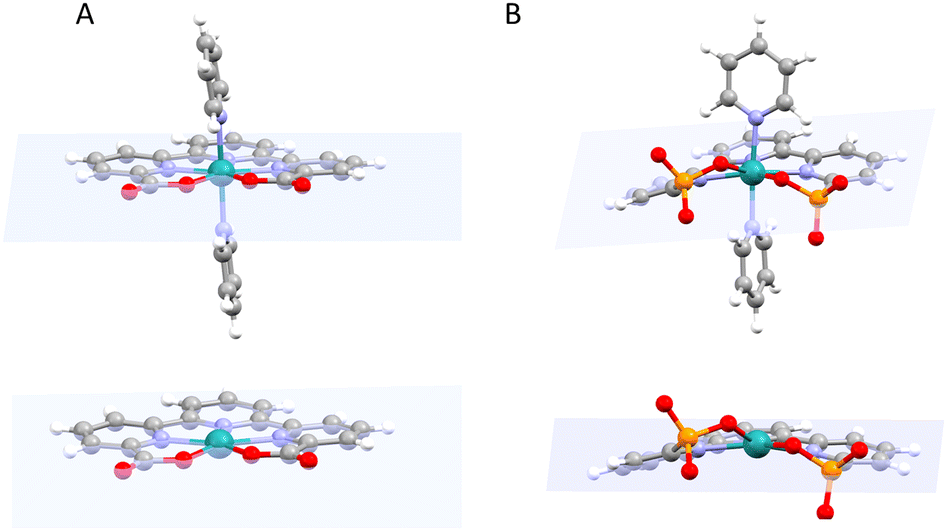 | ||
| Fig. 5 Top, structures of the Ru(IV) diamagnetic seven coordinated catalyst precursors [RuIV(tda-κ-N3O2)(py)2]2+14 (A), (X-ray structure) and [RuIV(tPa-κ-N3O2)(py)2], 15 (B), (DFT calculated structure). The best plane containing the three pyridyl rings of trpy moiety is depicted in light blue. Bottom, a different view omits the axial ligands. | ||
While the X-ray structure of 14 shows all the coordinating atoms of the tda2− ligand nearly on the equatorial plane. In contrast, the coordinating oxygen atoms of the phosphonate complex 15, are significantly off the equatorial plane. Such distortion breaks the planarity of the trpy moiety of the tPa4− ligand diminishing the aromatic π delocalisation. This difference in geometry/planarity between 14 and 15 results in weaker Ru–O bonds for the phosphonic complex and is caused mainly by the different local geometric parameters of the phosphonic acid group (P, pseudo Td) as compared to the carboxylate (C, pseudo C2v). From an electronic perspective, the phosphonate group acts as a stronger Sigma donor than the carboxylate, leading in general to lower redox potentials and higher pKa.26
During turnover conditions, the initial Ru–tPa complex 13 evolves to a new complex [RuIII(tPaO-κ-N2OPOC)(py)2]2−, 16, that contains a pyridyloxy group bonded to the metal center following the steps indicated in Scheme 3. In detail, complex 13 undergoes two consecutive PCETs to generate [RuIV(tPa-κ-N3O2)(py)2], 15. At oxidation state IV in basic solution, 15, undergoes OH− substitution to form either [RuIV(OH)(tPa-κ-N2O)(py)2]− or [RuIV(O)(HtPa-κ-N2O)(py)2]−, depending on the pH. The generation of the RuIV–OH (or RuIVO) occurs simultaneously with the breaking of both the Ru–N and Ru–O bonds of one of the pyridyl-phosphonato arms of the tPa4− ligand. The decoordination is driven by the steric congestion of the phosphonate group that precludes the formation of CN7 species such as [RuIV(O)(HtPa-κ-N3O)(py)2]−. Further oxidation leads to the formation of highly reactive [RuV(O)(tPa-κ-N2O)(py)2]− species that contains a dangling pyridyl-phosphonato arm with a low energy rotation barrier at the C–C bond of non-bonded external pyridyl group of the trpy moiety. This generates the perfect space disposition to undergo intramolecular oxygen atom transfer to the C-atom in the para position with regard to the phosphonate group of the non-bonded pyridyl ring. The intramolecular O-atom insertion into the C–H bond then generates the catalytically active complex [RuIII(tPaO-κ-N2OPOC)(py)2]2−, 16.
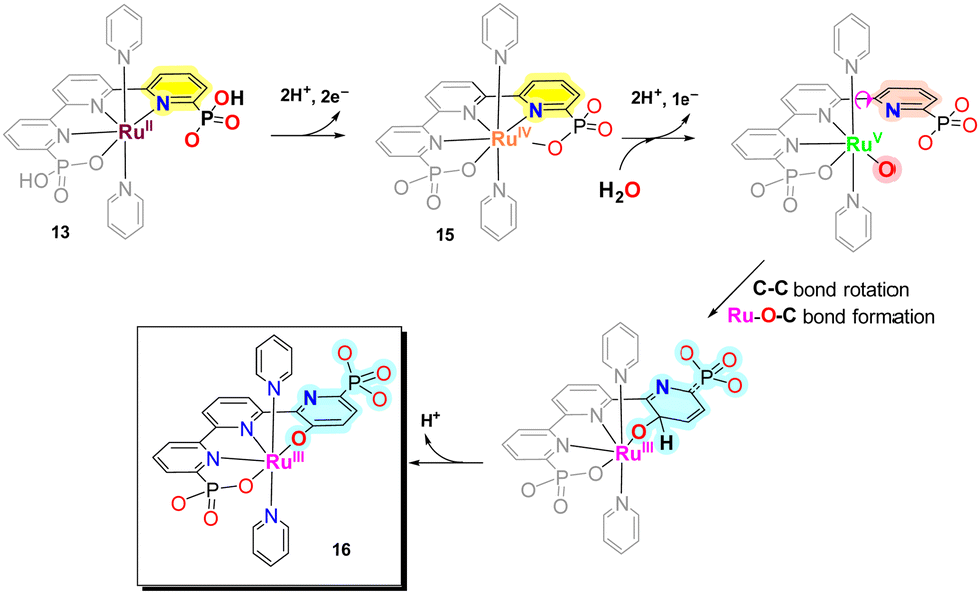 | ||
| Scheme 3 Formation of [RuIII(tPaO-κ-N2OPOC)(py)2]2−, 16, from [RuII(H2tPa)(py)2], 13, after a suit of reactions involving oxidations (top) and oxygen atom insertion into an aromatic CH bond, highlighted in cyan (bottom). | ||
Based on electrochemical evidence and DFT calculations, complex 16 is proposed to catalyse water oxidation via the WNA mechanism depicted in Fig. 6. This mechanism involves two-electron oxidation of 16 to form the RuVO species that is responsible for the formation of the O–O bond and generates the RuIII–OOH intermediate. This O–O bond formation is the rate-determining step (rds) of the catalytic cycle, and the structure of the calculated TS is shown in Fig. 6 in the center of the catalytic cycle.
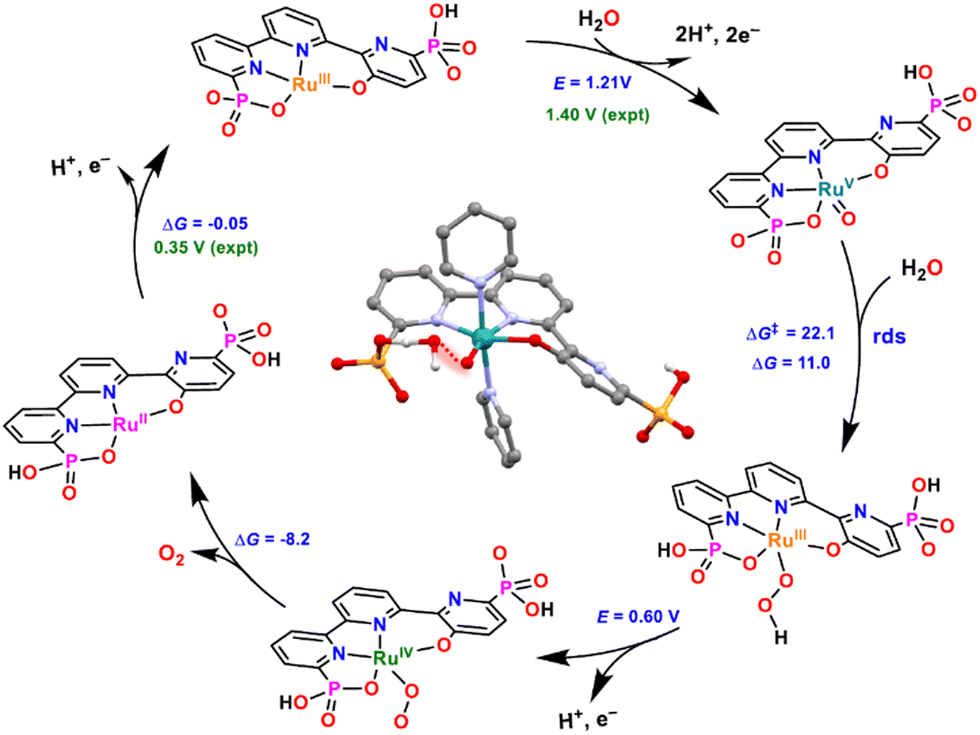 | ||
| Fig. 6 Calculated catalytic cycle for catalyst [RuIII(tPaO-κ-N2OPOC)(py)2]2−, 16, at pH 7. Redox potentials in V (experimental in green and calculated in blue) and ΔG and ΔG‡ in kcal mol−1. Axial pyridyl ligands are omitted for clarity. Inside, optimised calculated TS structure for the O–O bond formation step (highlighted in red). Colour code: Ru, cyan; P, orange; N, blue; O, red; C, black; H, white. They were reproduced from ref. 23 with permission from the American Chemical Society. | ||
It is interesting to point out the presence of a hydrogen bonding between the incoming water molecule and the adjacent phosphonate that provides access to an intramolecular proton transfer. Overall, this produces a very low energy pathway for the rds, resulting in the fastest water oxidation catalysts reported to date at pH 7 with a TOFmax of 16000 s−1 at an overpotential of 530 mV. The latter has an overpotential of 75 mV lower than the one reported earlier for 10.25
2.4 In situ phenolate transformation into carboxylate
The above-described phenolate complex 16 is a powerful WOC. However, during turnover, in the long run, it further evolves into a new catalyst where the pyridinoxo species is transformed into a carboxylate [RuII(bpc)(py)2]−, 17 (see Chart 1 for a drawing of the H3bpc ligand).72–74 The phenolate to carboxylate transformation involves breaking a C–C bond between two pyridyl units of the tPaO3− ligand. It has been proposed to occur by several pathways outlined in Scheme 4 that have support in laboratory-performed oxidations as well as in biological transformations of pyridine derivatives.75–78 | ||
| Scheme 4 Potential reaction pathways for the formation of [Ru(bpc)(py)2]−, 17, from [RuIII(tPaO-κ-N2OPOC)(py)2]2−, 16. See text for details. | ||
The first path involves the oxidation of the pyridinoxo group concomitant with aromatic ring opening as shown on the top of Scheme 4, followed by the formation of pyrrole that finally leads to the phosphonate carboxylate bpc ligand formation. The second potential mechanism is shown in the center of Scheme 4. It involves an oxygen insertion into the pyridyl group forming an ortho-pyridyl-catecholate group that, after successive transformations, leads to the Ru-bpc complex. A third option involves an aromatic ring opening after the aromatic hydroxylation leading to an amide derivative, which can be subsequently hydrolysed into the carboxylic group. Multiple oxidations during the process are also possible. Finally, it further evolves to a carboxylic acid of the final Ru-bpc complex 17, as indicated at the bottom of Scheme 4.
A related example where a phenolate group is transformed into a carboxylate has also been described for [RuII(trpy)(phen-PhO)]+, 18, that upon oxidation generates the active catalyst [RuII(trpy)(phen-PhCOO)]+, 19.73,79 Another in situ phenolate-carboxylate transformation worth mentioning is the one suffered by the complex [RuIII(bpy-2phO)(pic)2]+, 20, where the phenolate groups are progressively transformed into a mono phenoxo-catecholate 20′, whose crystal structure has been solved, and that involves necessarily a bimolecular catalyst–catalyst interaction. Further phenoxo-catecholate transformation leads now to the symmetric complex, 20′′, that is then further oxidised all the way to the bpy-biscarboxylate ligand, as shown in Scheme 5.73,79 It is interesting to realise here that the final complex reached after all these ligand-based oxidative transformations of 20, is the complex [RuIII(bda)(pic)2]+11′, that is one of the first complexes described with FAME ligands.80
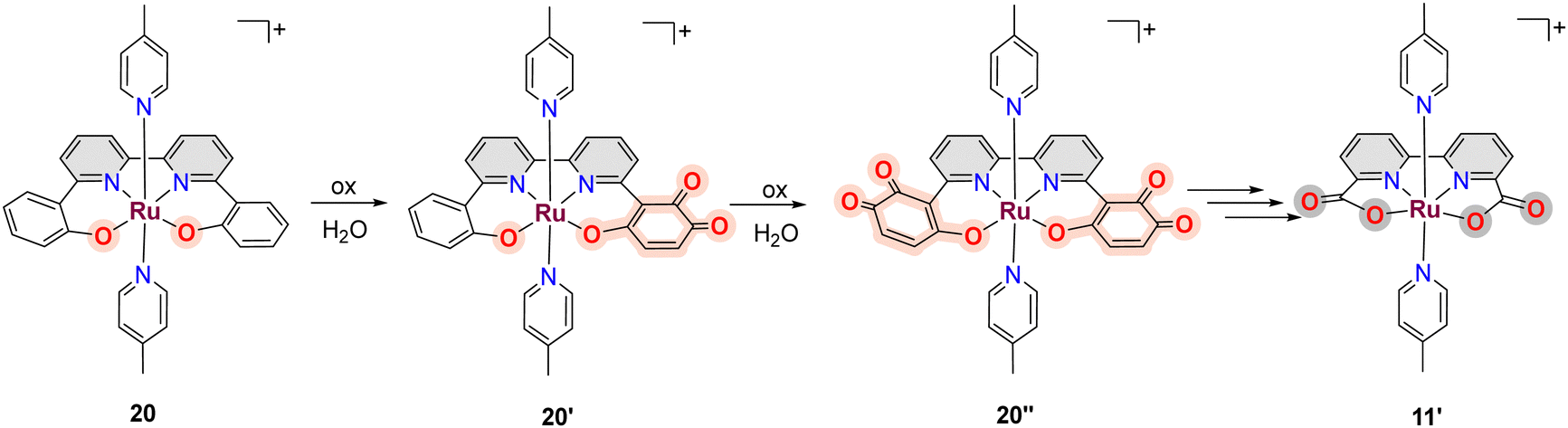 | ||
| Scheme 5 Formation of [RuIII(bda)(pic)2]+, 11′, from [RuIII(bpy-2phO)(py)2]+, 20, after a suite of oxidation reactions involving the initial phenolate groups of the bpy-2phO ligand. An X-ray crystal structure was obtained for 20′ supporting this oxidation pathway. | ||
2.5 N-Oxide formation: the release of bond strain
A recent report has shown the Ru complex, [RuII(pbn)(pic)2]2+, 21, containing the penta-N-dentate ligand 2,2′-(4-(tert-butyl)pyridine-2,6-diyl)bis(1,8-naphthyridine) (pbn), undergoing N-oxide formation via an intramolecular O-atom transfer from a Ru–O bond in a similar manner as has been described in the previous section, but where the O-transfer occurs at the N-atom of a naphthyridyl group forming the pbn-O2 ligand (see Chart 1 for a drawing of the ligands).52 In this manner, after a second N-oxide formation, the initial pentadentate N5 ligand changes its coordination mode from a κ-N5 to κ-N3O2. Interestingly, a DFT analysis53 of the reaction mechanism for this complex suggests the possibility that the O–O bond formation occurs in an intramolecular manner between the Ru–O group and the unbonded N–O of the pbn-O2 ligand. Similarly, a couple of reports have shown that the mono-aqua Ru complexes such as [RuII(trpy)(bpy)(H2O)]2+, 2,52 and [RuII(trpy)(bpm)(H2O)]2+, 22,55 can also undergo an intramolecular O-atom transfer to form the corresponding bpy-N-oxide (bpy-O) and bpyrimidine N-oxide (bpm-O) ligands. In this case, the driving force is the weakening of the Ru–N bond trans to the Ru–O group and the highly favoured formation of the trans-O–Ru–O entity.Further, two more complexes based on planar tetra-N-dentate ligands 2,2′:6′,2′′:6′′,2′′′-quaterpyridine (qtp) [RuII(qtp)(pic)2]2+, 23,54 and 2,9-di(pyridin-2-yl)-1,10-phenanthroline (pbp) [RuII(pbp)(py)2]2+, 24,51 have also been described to undergo intramolecular N-oxide formation generating their corresponding bis oxide analogues qtp-O2 and pbp-O2 respectively as shown in Scheme 6. After these reactions, the new ligands coordinate the metal center in a κ-N2O2 fashion. Here the N-oxide formation occurs because the geometry of the NO2-qtp and NO2-bpp release the bond strain imposed by the N4 ligand, as can be observed in Fig. 7. For complex, [RuII(qtp-O2)(py)2], 25, the crystal structure shows that the Ru–O bond is slightly tilted over the equatorial plane to accommodate the new N2O2-ligand geometry adequately.
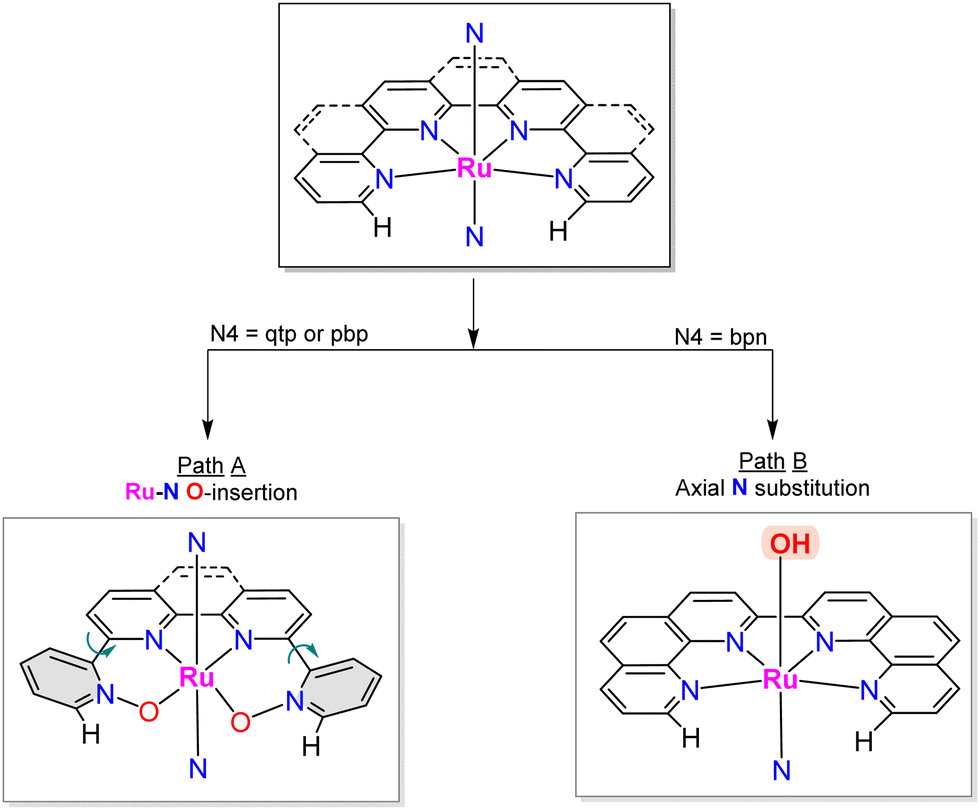 | ||
| Scheme 6 Different pathways followed by Ru complexes with equatorial tetra-N dentate (N4) ligands (top) depending on the degree of rotation of their peripheral pyridyl groups. The N on the axial coordination site of Ru represents either a pyridine or a picoline ligand. | ||
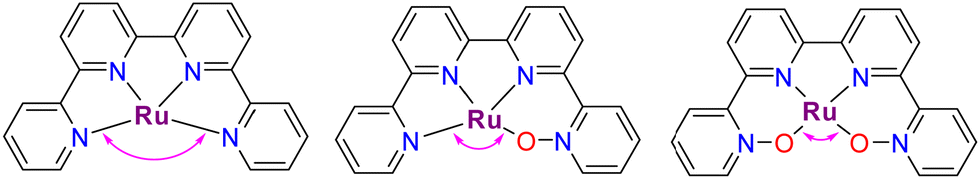 | ||
| Fig. 7 Drawing of the equatorial structure showing qualitatively the external coordination angle (in fuchsia) involved in the coordination modes κ-N4, κ-N3O, and κ-N2O2, assuming that all coordination atoms and Ru are situated at the same plane. | ||
Interestingly, a related complex [RuII(bpn)(pic)2]2+, 26,81 containing the 2,2′-bis(1,10-phenanthroline) (bpn) ligand, does not undergo N–O formation because the rigidity of the phenanthroline groups doesn’t allow the needed rotation to facilitate the intramolecular O-transfer. Instead, the complex undergoes axial ligand substitution forming the complex [RuII(bpn)(pic)(H2O)]2+, 27, where the active Ru-aqua group occupies the axial position. This interplay between rotation and substitution depending on ligands is graphically summarised in Scheme 6. Unfortunately, this complex, upon oxidation, undergoes dimerisation forming an oxo bridge between the two metal centers, {[RuIII(bpn)(pic)]2(m-O)}4+, 28, that blocks the activity of the complex as WOC.
2.6 Ru-bda evolving on graphitic materials: the role of the surface
Recently, it has been shown that the reaction of [RuII(bda)(DMSO)2] with the bridging ligand 4,4′-bpy generates a range of linear oligomeric complexes of the general formula, {[RuII(bda-κ-N2O2)(m-4,4′-bpy)]n(m-4,4′-bpy)}, with “n” up to 10 units.82 The space-filling DFT calculated structure for n = 10, {[RuII(bda-κ-N2O2)(m-4,4′-bpy)]10(4,4′-bpy)}, 29, is displayed in Fig. 8. Complex 29 has been shown to possess a huge affinity towards graphitic surfaces such as graphite or multi-walled carbon nanotubes (CNTs) thanks to the multiple (up to 40) CH–π interactions that occur between the H-atoms of the bda2− ligands and the π-system of the graphitic material.83,84 Anchored on CNTs, complex 29 behaves as a molecular electrocatalytic anode but nevertheless progressively evolves to a new complex where the bda2− ligands change their coordination mode from bda-κ-N2O2 to bda-κ-NO, generating the new molecular hybrid material {[RuII(bda-κ-NO)(H2O)2(4,4′-bpy)]10(m-4,4′-bpy)}, 30, anchored on CNT. As the coordination mode of the bda2− changes from bda-κ-N2O2 to bda-κ-NO, the metal center reacts with two water molecules to complete the octahedral coordination. This transformation is depicted in Fig. 9 for the dimeric analogue {[RuII(bda-κ-N2O2)(py)]2(m-4,4′-bpy)}, 29′, that generates {[RuII(H2O)2(bda-κ-NO)(py)]2(m-4,4′-bpy)} 30′, where the interaction of the bda2− ligand with graphitic surfaces favouring a partial decoordination via additional π–π and anion–π interactions is shown. Evidence for this partial decoordination of the pyridyl-carboxylate arm of the bda2− ligands and concomitant water coordination include EPR spectroscopy and electrochemistry, as well as further corroboration by DFT calculations. Both 29 and 30 are powerful water oxidation catalysts, and their proposed catalytic cycles are illustrated in Scheme 7.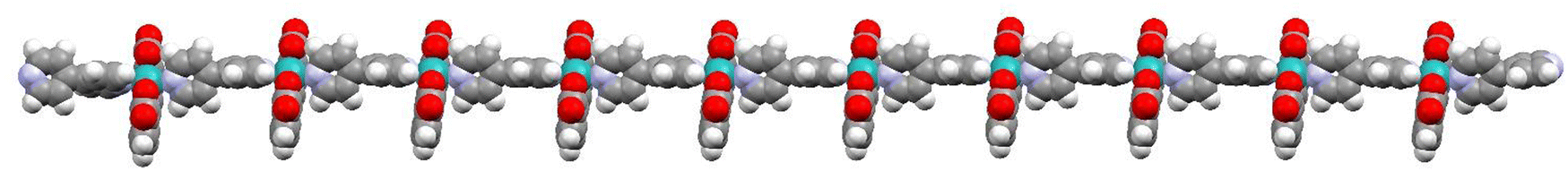 | ||
| Fig. 8 Space filling structure of oligomer 29 in the gas phase based on the PM6-D3H4 method. Colour code: Ru, light blue; O, red; N, blue; C, grey; H, white. In this model, the distance between the N-atoms in the extremes of the oligomer (non-bonded to Ru) turns out to be 11.96 nm. It is reproduced from ref. 82 with permission from American Chemical Sociaty. | ||
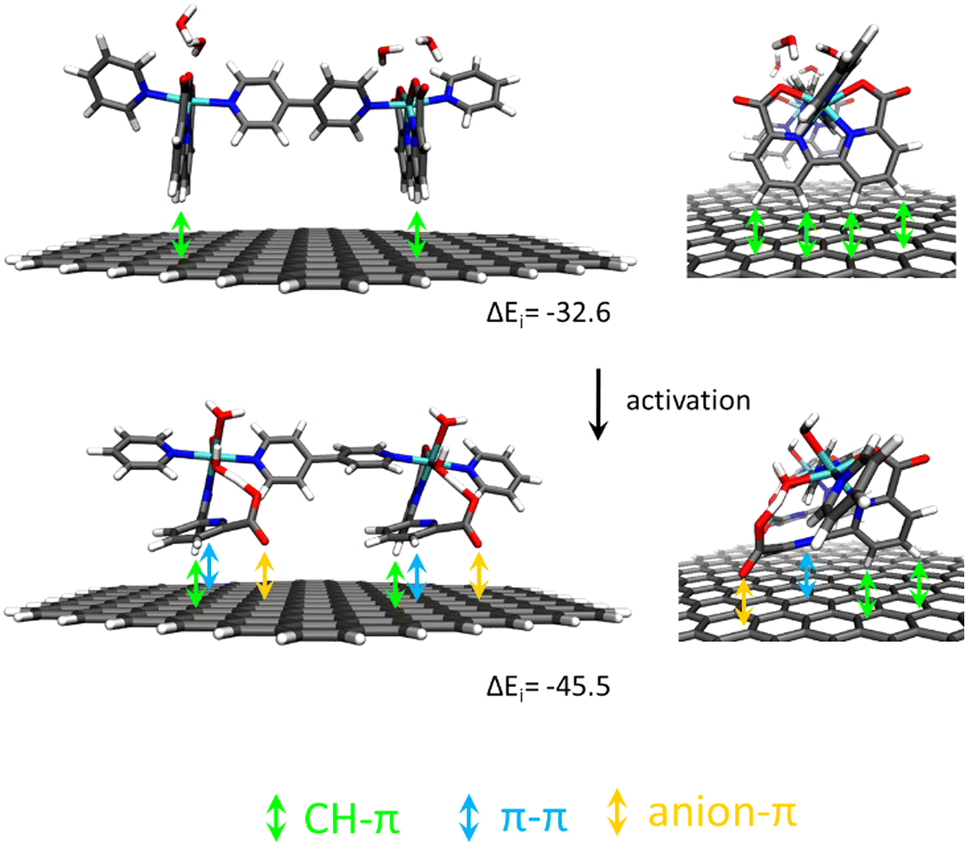 | ||
| Fig. 9 Schematic representation of non-covalent interactions in a dimer model of 29′@graphite (top) and after its transformation into 30′@graphite (bottom) with semi-empirically calculated interaction energies per Ru center ΔEi in kJ mol−1. Reproduced from ref. 82 with permission from the American Chemical Society. | ||
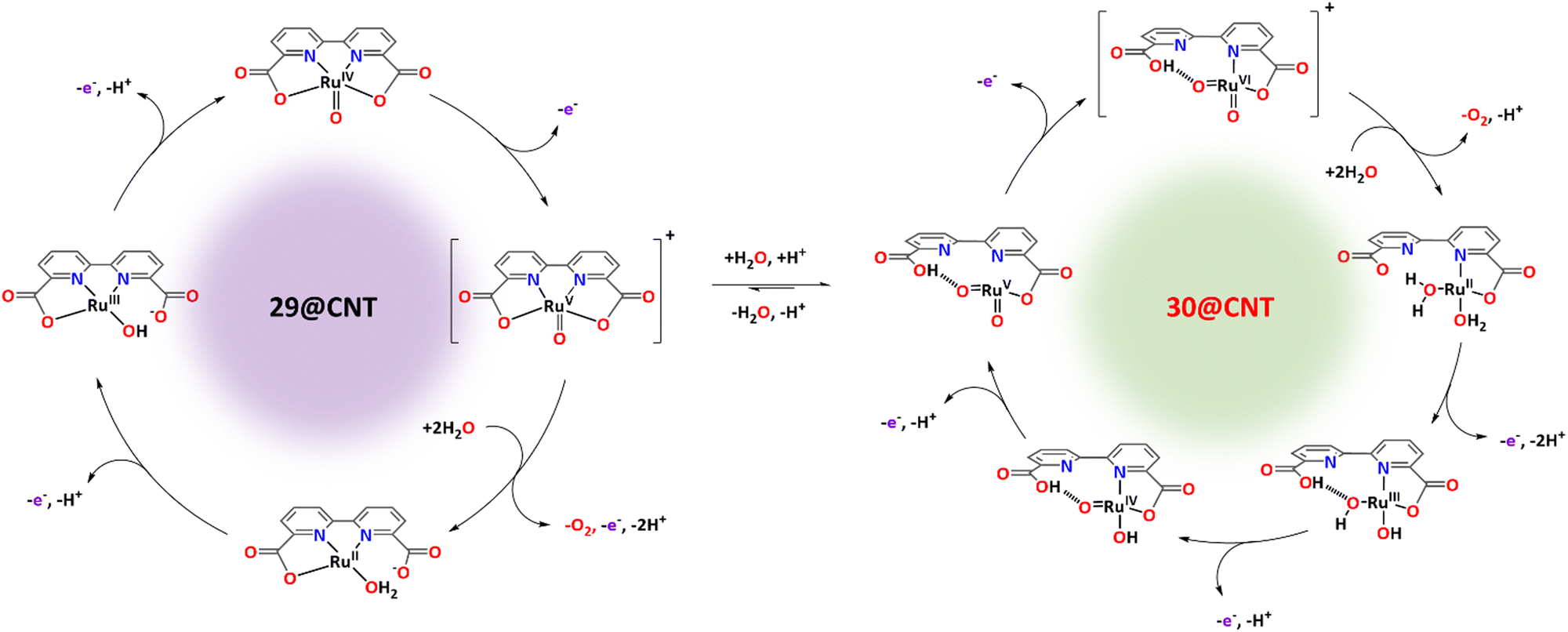 | ||
| Scheme 7 Proposed water oxidation catalytic cycles undergone by representative monomer of {[RuII(bda-κ-N2O2)(4,4′-bpy)]10(4,4′-bpy)}, 29, and {[RuII(bda-κ-NO)(H2O)2(4,4′-bpy)]10(4,4′-bpy)}, 30, and their interconversion promoted by graphitic surfaces. See text for additional details. Axial pyridyl ligands are not shown. | ||
The hybrid material 30@CNT achieves current densities in the range of 200 to 260 mA cm−2 at pH 7 under the applied potential of 1.45 V, using minimal amounts of mass (0.1 nmol cm−2 of Ru at the GC electrode and 16.7 nmol cm−2 at the CNTs) with TOFs and TONs in the range of 3.7 × 104 s−1 and of 9.4 × 104, respectively. The high activity of the complex is associated with the ligand flexibility and the second coordination sphere effects exerted by the decoordinated arm of the bda ligand that provides the necessary stabilisation by H-bonding for a low energy catalytic cycle. On the other hand, the high stability of the Ru water oxidation catalyst is associated with molecular catalyst–surface interactions, especially during turnover conditions. The oligomeric nature of the molecule provides multiple sites of the anchorage at the surface that strongly enhances the overall stability of the hybrid catalyst at the CNT anodic surface via dynamic bonding.
3. Conclusions and future prospects
Water oxidation catalysis is one of the key reactions that need to be understood and mastered in order to be able to build efficient PECs for the generation of solar fuels. Molecular species stand out as superior for the design of water oxidation catalysts. In comparison to metal oxides, molecular species offer many choices from the molecular toolkit, such as tailoring the first and second coordination sphere effects. Moreover, discrete molecular complexes with different nuclearities can be obtained using different bridging ligands. These can be considered analogues of the highly reactive site-isolated metal species discovered recently in the oxide field.85 Today, the best molecular water oxidation catalysts have TOFmax two orders of magnitude higher than nature's OEC-PSII,86 and about five orders of magnitude higher than the best metal oxides.87–89 Nevertheless, the judicious choice of ligands and metal centers allowing building superior WOCs is by no means an easy task due to several reasons. Firstly, there is a large variety of ligands and metal centers with different oxidation states from which to choose. Secondly, the transition metal complex must endure a number of stringent conditions in order to carry out the water oxidation reaction in a catalytic manner.The main complexities that need to be fully comprehended for designing molecular WOCs have been described in the previous section and will be further discussed below. All the catalysts presented in this review have been based mainly on Ru complexes because they constitute the most prominent family of molecular WOCs where a detailed analysis has been done, especially regarding the integrity of the catalyst during and after catalysis. An additional feature of the Ru complexes that allow the emergence of such a catalyst-evolution analysis is their substitution inertness at both Ru(II) and Ru(III) oxidation states90,91 that minimises the potential substitution of the initial auxiliary ligands by solvent aqua ligands as would be the case of their Fe counterparts.
Water oxidation thermodynamics is endothermic by 113.5 kcal mol−1, as indicated in eqn (1). For related organic oxidations, such as PhCH2OH, this involves only 22.3 kcal mol−1 (eqn (7)). Therefore, the only option for a catalyst to survive a suicidal bimolecular interaction is to use ligands that undergo only kinetically very slow ligand oxidation. In this respect, using benzyl or picolyl groups, as shown for complex 8a, is unsuitable due to its rapid oxidation. This illustrates the utmost importance of the rational approach to designing robust oxidation-resistant ligands for efficient and long-lasting molecular WOCs catalysts.
Water oxidation catalysis involves the removal of 4e− from two water molecules, which implies that a catalyst capable of carrying out this reaction needs to be able to store four holes. Thus, the auxiliary ligands bonded to the transition metal center will need to be stable over a range of different oxidation states, as exemplified with the [RuII(trpy)(bpy)(H2O)]2+, 2, a complex that needs to reach Ru(V) oxidation state (See Scheme 1, left). At this oxidation state, the RuO group exerts a strong trans effect on the bonded bpy ligand provoking its decoordination, followed by a concomitant formation of a trans ORuO group, which is a thermodynamic sink in this type of Ru chemistry, and thus constitutes an additional driving force for ligands loss.
An elegant solution for the ligands to comply with the electronic and geometrical demands of the metal center at different oxidation states is using the tda2− ligand (Chart 1) within the framework of the so-called FAME ligands. In complex [Ru(tda)(py)2], 10, the pentadentate tda2− ligand binds to Ru(II) in a tetradentate manner. However, when the metal center reaches oxidation states IV and V it can become pentadentate, thus helping to decrease the energy needed to access such a high oxidation state and increasing the Ru coordination number from 6 to 7, which additionally involves a geometrical change in the first coordination sphere of the Ru center that evolves from the typical octahedral to a pentagonal bipyramid one. Further, the flexibility of the FAME ligands also allows for a second coordination sphere effect that provides the right spatial disposition for an intramolecular proton transfer. The latter strongly decreases the energy activation where it is most appreciated, at the rds.
The steric factor can also play a crucial role, as exemplified in the case of [RuII(HtPa)(py)2]−, 13, which is similar to Ru-tda 10, but where the trpy biscarboxylate ligands tda2− have been substituted by the trpy bisphosphonate ligand tPa4−. The increased steric hindrance between the phosphonato group and the Ru–O groups, as compared to the case of the carboxylate in the Ru-tda case (P Tdvs. C C2v), is the crucial parameter that drives the partial decoordination of the tPa4− ligand followed by O-atom insertion into the CH bond of the pyridyl moiety. The latter is further entropically favoured by the right spatial disposition of the two groups occurring in an intramolecular manner. This entropic factor is also the driving force for a number of ligand transformations in molecular water oxidation catalysts, as is the case for complexes [RuII(qtp)(pic)2]2+, 23, and [RuII(pbp)(py)2]2+, 24, that suffer the intramolecular oxidation of their pyridyl groups to the corresponding N-oxides.
Finally, weak supramolecular interactions have also been shown to play a critical role, as is the example of [RuII(bda)(MeO-isoq)2], 12. In this case, the MeO-isoq axial ligand that contains a π-extended system strongly decreases the energy of activation for the formation of a dinuclear complex that leads to the O–O bond formation in the case of the I2M mechanisms. Another exceptional example that constitutes an additional turn of the screw of the importance of supramolecular interaction is the formation of {[RuII(bda-κ-NO)(H2O)2(μ-4,4′-bpy)]10(4,4′-bpy)}, 30, from {[RuII(bda-κ-N2O2)(μ-4,4′-bpy)]10(4,4′-bpy)}, 29, at the surface of multi-walled carbon nanotubes. The supramolecular catalyst-surface π–π bonding interactions irreversibly favour the partial decoordination of one of the arms of the bda2− ligand concomitant with solvent coordination and the formation of the Ru(OH2)2 group. Interestingly, the latter phenomenon has never been observed in homogeneous phase for the Ru-bda complex 11.
The evolution of the oxygen-evolving catalysts described here outlines the richness associated with molecular Ru complexes in combination with the complexity associated with the 4H+/4e− water oxidation reaction to dioxygen. The richness of the Ru chemistry is due to both a large number of reactions that the complex can follow at relatively close energies and to the different geometries and coordination numbers that can be accessed by the metal center influenced by the large number of ligands that can be used. Under such a scenario, relatively small effects such as steric encumbrance or weak π–π interaction can have a crucial impact that dramatically changes the catalyst performance. Finally, entropy is one of the most critical factors that need to be considered, as is exemplified by various intramolecular reactions that drive the evolution of the catalysts. Further, the entropic factor is also crucial for the decrease of activation energies at the O–O bond formation WNA rate-determining step via an intramolecular proton transfer.
The present performance of WOCs could already be used for niche technological applications. However, replacing Ru with more earth–abundant transition metals such as Mn, Fe, or Cu would be highly desirable for a massive application such as covering the world's energy demand. For this purpose, all the knowledge extracted with the work described here with Ru complexes should be transferred to the first-row transition metal complexes but pay special attention to their intrinsic electronic properties. Especially challenging are substitution kinetics mentioned earlier in the presence of water. Nevertheless, an incipient body of complexes is emerging that needs to be largely increased and their performance highly improved to develop potential technological applications.27,92–95
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from Ministerio de Ciencia e Innovación, FEDER and AGAUR through grants PID2019-111617RB-I00, SO-CEX2019-000925-S, RYC2019-027423-I and 2017-SGR-1631 is gratefully acknowledged.Notes and references
- IPCC, 2021: Climate Change 2021: The Physical Science Basis ecosystems, 2021.
- N. Boers, Nat. Clim. Change, 2021, 11, 680–688 CrossRef.
- W. Steffen, J. Rockström, K. Richardson, T. M. Lenton, C. Folke, D. Liverman, C. P. Summerhayes, A. D. Barnosky, S. E. Cornell, M. Crucifix, J. F. Donges, I. Fetzer, S. J. Lade, M. Scheffer, R. Winkelmann and H. J. Schellnhuber, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 8252–8259 CrossRef CAS.
- International Energy Agency (2021), World Energy Outlook 2021.
- SUNRISE initiative: https://sunriseaction.com/sunrise-initiative.
- M. Kondo, H. Tatewaki and S. Masaoka, Chem. Soc. Rev., 2021, 50, 6790–6831 RSC.
- L. v Kulik, B. Epel, W. Lubitz and J. Messinger, J. Am. Chem. Soc., 2007, 129, 13421–13435 CrossRef CAS.
- W. Lubitz, E. J. Reijerse and J. Messinger, Energy Environ. Sci., 2008, 1, 15–31 RSC.
- A. Krieger-Liszkay, C. Fufezan and A. Trebst, Photosynth. Res., 2008, 98, 551–564 CrossRef CAS.
- A. Zavafer, M. H. Cheah, W. Hillier, W. S. Chow and S. Takahashi, Sci. Rep., 2015, 5, 16363 CrossRef CAS PubMed.
- F. Bashir, A. U. Rehman, M. Szabó and I. Vass, Photosynth. Res., 2021, 149, 93–105 CrossRef CAS.
- D. J. Vinyard, S. L. Badshah, M. R. Riggio, D. Kaur, A. R. Fanguy and M. R. Gunner, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 18917–18922 CrossRef CAS.
- R. Pokhrel and G. W. Brudvig, Phys. Chem. Chem. Phys., 2014, 16, 11812–11821 RSC.
- A. Coehn and M. Gläser, Z. Anorg. Chem., 1902, 33, 9–24 CrossRef CAS.
- S. W. Gersten, G. J. Samuels and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 4029–4030 CrossRef CAS.
- J. A. Gilbert, D. S. Eggleston, W. R. Murphy, D. A. Geselowitz, S. W. Gersten, D. J. Hodgson and T. J. Meyer, J. Am. Chem. Soc., 1985, 107, 3855–3864 CrossRef CAS.
- R. Matheu, P. Garrido-Barros, M. Gil-Sepulcre, M. Z. Ertem, X. Sala, C. Gimbert-Suriñach and A. Llobet, Nat. Rev. Chem., 2019, 3, 331–341 CrossRef CAS.
- M. D. Kärkäs, O. Verho, E. V. Johnston and B. Åkermark, Chem. Rev., 2014, 114, 11863–12001 CrossRef.
- M. D. Kärkäs and B. Åkermark, Chem. Rec., 2016, 16, 940–963 CrossRef.
- D. W. Shaffer, Y. Xie and J. J. Concepcion, Chem. Soc. Rev., 2017, 46, 6170–6193 RSC.
- R. Matheu, M. Z. Ertem, C. Gimbert-Suriñach, X. Sala and A. Llobet, Chem. Rev., 2019, 119, 3453–3471 CrossRef CAS PubMed.
- Y. Pushkar, Y. Pineda-Galvan, A. K. Ravari, T. Otroshchenko and D. A. Hartzler, J. Am. Chem. Soc., 2018, 140, 13538–13541 CrossRef CAS PubMed.
- J. Creus, R. Matheu, I. Peñafiel, D. Moonshiram, P. Blondeau, J. Benet-Buchholz, J. García-Antón, X. Sala, C. Godard and A. Llobet, Angew. Chem., Int. Ed., 2016, 55, 15382–15386 CrossRef CAS.
- N. Vereshchuk, R. Matheu, J. Benet-Buchholz, M. Pipelier, J. Lebreton, D. Dubreuil, A. Tessier, C. Gimbert-Suriñach, M. Z. Ertem, A. Llobet, M. Z. Ertem and A. Llobet, J. Am. Chem. Soc., 2020, 142, 5068–5077 CrossRef CAS PubMed.
- R. Matheu, M. Z. Ertem, J. Benet-Buchholz, E. Coronado, V. S. Batista, X. Sala and A. Llobet, J. Am. Chem. Soc., 2015, 137, 10786–10795 CrossRef CAS.
- R. Matheu, M. Z. Ertem, C. Gimbert-Suriñach, J. Benet-Buchholz, X. Sala and A. Llobet, ACS Catal., 2017, 7, 6525–6532 CrossRef CAS.
- M. Gil-Sepulcre, P. Garrido-Barros, J. Oldengott, I. Funes-Ardoiz, R. Bofill, X. Sala, J. Benet-Buchholz and A. Llobet, Angew. Chem., Int. Ed., 2021, 60, 18639–18644 CrossRef CAS.
- P. Garrido-Barros, D. Moonshiram, M. Gil-Sepulcre, P. Pelosin, C. Gimbert-Suriñach, J. Benet-Buchholz and A. Llobet, J. Am. Chem. Soc., 2020, 142, 17434–17446 CrossRef CAS PubMed.
- H.-Y. Du, S.-C. Chen, X.-J. Su, L. Jiao and M.-T. Zhang, J. Am. Chem. Soc., 2018, 140, 1557–1565 CrossRef CAS.
- B. A. Moyer, M. S. Thompson and T. J. Meyer, J. Am. Chem. Soc., 1980, 102, 2310–2312 CrossRef CAS.
- F. P. Guengerich, ACS Catal., 2018, 8, 10964–10976 CrossRef CAS PubMed.
- J. Kern, R. Chatterjee, I. D. Young, F. D. Fuller, L. Lassalle, M. Ibrahim, S. Gul, T. Fransson, A. S. Brewster, R. Alonso-Mori, R. Hussein, M. Zhang, L. Douthit, C. de Lichtenberg, M. H. Cheah, D. Shevela, J. Wersig, I. Seuffert, D. Sokaras, E. Pastor, C. Weninger, T. Kroll, R. G. Sierra, P. Aller, A. Butryn, A. M. Orville, M. Liang, A. Batyuk, J. E. Koglin, S. Carbajo, S. Boutet, N. W. Moriarty, J. M. Holton, H. Dobbek, P. D. Adams, U. Bergmann, N. K. Sauter, A. Zouni, J. Messinger, J. Yano and V. K. Yachandra, Nature, 2018, 563, 421–425 CrossRef CAS.
- H. Dau and M. Haumann, Biochim. Biophys. Acta, Bioenerg., 2007, 1767, 472–483 CrossRef CAS PubMed.
- W. Ames, D. A. Pantazis, V. Krewald, N. Cox, J. Messinger, W. Lubitz and F. Neese, J. Am. Chem. Soc., 2011, 133, 19743–19757 CrossRef CAS.
- A. Llobet, Inorg. Chim. Acta, 1994, 221, 125–131 CrossRef CAS.
- T. Ono, S. Qu, C. Gimbert-Surinach, M. A. Johnson, D. J. Marell, J. Benet-Buchholz, C. J. Cramer and A. Llobet, ACS Catal., 2017, 7, 5932–5940 CrossRef CAS.
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533–3539 CrossRef CAS.
- F. Liu, J. J. Concepcion, J. W. Jurss, T. Cardolaccia, J. L. Templeton and T. J. Meyer, Inorg. Chem., 2008, 47, 1727–1752 CrossRef CAS.
- I. López, S. Maji, J. Benet-Buchholz and A. Llobet, Inorg. Chem., 2015, 54, 658–666 CrossRef.
- C. Gunanathan and D. Milstein, Chem. Rev., 2014, 114, 12024–12087 CrossRef CAS.
- F. Nahra and C. S. J. Cazin, Chem. Soc. Rev., 2021, 50, 3094–3142 RSC.
- J. J. Concepcion, J. W. Jurss, J. L. Templeton and T. J. Meyer, J. Am. Chem. Soc., 2008, 130, 16462–16463 CrossRef CAS.
- S. Neudeck, S. Maji, I. Lo, F. Meyer and A. Llobet, J. Am. Chem. Soc., 2013, 2–5 Search PubMed.
- L. Vigara, M. Z. Ertem, N. Planas, F. Bozoglian, N. Leidel, H. Dau, M. Haumann, L. Gagliardi, C. J. Cramer and A. Llobet, Chem. Sci., 2012, 3, 2576–2586 RSC.
- S. Romain, L. Vigara and A. Llobet, Acc. Chem. Res., 2009, 42, 1944–1953 CrossRef CAS PubMed.
- I. López, M. Z. Ertem, S. Maji, J. Benet-Buchholz, A. Keidel, U. Kuhlmann, P. Hildebrandt, C. J. Cramer, V. S. Batista and A. Llobet, Angew. Chem., Int. Ed., 2014, 53, 205–209 CrossRef.
- B. A. Moyer and T. J. Meyer, Inorg. Chem., 1981, 20, 436–444 CrossRef CAS.
- X. Sala, M. Z. Ertem, L. Vigara, T. K. Todorova, W. Chen, R. C. Rocha, F. Aquilante, C. J. Cramer, L. Gagliardi and A. Llobet, Angew. Chem., Int. Ed., 2010, 49, 7745–7747 CrossRef CAS PubMed.
- J. B. C. Mack, K. L. Walker, S. G. Robinson, R. N. Zare, M. S. Sigman, R. M. Waymouth and J. du Bois, J. Am. Chem. Soc., 2019, 141, 972–980 CrossRef CAS.
- A. K. Ravari, G. Zhu, R. Ezhov, Y. Pineda-Galvan, A. Page, W. Weinschenk, L. Yan and Y. Pushkar, J. Am. Chem. Soc., 2020, 142, 884–893 CrossRef CAS PubMed.
- Y. Pineda-Galvan, A. K. Ravari, S. Shmakov, L. Lifshits, N. Kaveevivitchai, R. Thummel and Y. Pushkar, J. Catal., 2019, 375, 1–7 CrossRef CAS.
- D. Moonshiram, Y. Pineda-Galvan, D. Erdman, M. Palenik, R. Zong, R. Thummel and Y. Pushkar, J. Am. Chem. Soc., 2016, 138, 15605–15616 CrossRef CAS PubMed.
- M. Z. Ertem and J. J. Concepcion, Inorg. Chem., 2020, 59, 5966–5974 CrossRef CAS.
- Y. Liu, S. M. Ng, S. M. Yiu, W. W. Y. Lam, X. G. Wei, K. C. Lau and T. C. Lau, Angew. Chem., Int. Ed., 2014, 53, 14468–14471 CrossRef CAS PubMed.
- Y. Wang, Z. Rinkevicius and M. S. G. Ahlquist, Chem. Commun., 2017, 53, 5622–5624 RSC.
- R. B. Baar and F. C. Anson, J. Electroanal. Chem. Interfacial Electrochem., 1985, 187, 265–282 CrossRef CAS.
- B. Radaram, J. A. Ivie, W. M. Singh, R. M. Grudzien, J. H. Reibenspies, C. E. Webster and X. Zhao, Inorg. Chem., 2011, 50, 10564–10571 CrossRef CAS PubMed.
- J. Yang, J. An, L. Tong, B. Long, T. Fan and L. Duan, Inorg. Chem., 2019, 58, 3137–3144 CrossRef CAS PubMed.
- A. C. Sander, A. Schober, S. Dechert and F. Meyer, Eur. J. Inorg. Chem., 2015, 4348–4353 CrossRef CAS.
- J. Zhou, W. Xi and J. K. Hurst, Inorg. Chem., 1990, 29, 160–167 CrossRef CAS.
- A. Ghaderian, J. Holub, J. Benet-Buchholz, A. Llobet and C. Gimbert-Suriñach, Inorg. Chem., 2020, 59, 4443–4452 CrossRef CAS PubMed.
- N. D. Schley, J. D. Blakemore, N. K. Subbaiyan, C. D. Incarvito, F. D’Souza, R. H. Crabtree and G. W. Brudvig, J. Am. Chem. Soc., 2011, 133, 10473–10481 CrossRef CAS PubMed.
- J. D. Blakemore, N. D. Schley, D. Balcells, J. F. Hull, G. W. Olack, C. D. Incarvito, O. Eisenstein, G. W. Brudvig and R. H. Crabtree, J. Am. Chem. Soc., 2010, 132, 16017–16029 CrossRef CAS.
- T. K. Michaelos, D. Y. Shopov, S. B. Sinha, L. S. Sharninghausen, K. J. Fisher, H. M. C. Lant, R. H. Crabtree and G. W. Brudvig, Acc. Chem. Res., 2017, 50, 952–959 CrossRef CAS PubMed.
- A. Savini, P. Belanzoni, G. Bellachioma, C. Zuccaccia, D. Zuccaccia and A. Macchioni, Green Chem., 2011, 13, 3360–3374 RSC.
- C. Zuccaccia, G. Bellachioma, S. Bolaño, L. Rocchigiani, A. Savini and A. Macchioni, Eur. J. Inorg. Chem., 2012, 1462–1468 CrossRef CAS.
- D. B. Grotjahn, D. B. Brown, J. K. Martin, D. C. Marelius, M.-C. Abadjian, H. N. Tran, G. Kalyuzhny, K. S. Vecchio, Z. G. Specht, S. A. Cortes-Llamas, V. Miranda-Soto, C. van Niekerk, C. E. Moore and A. L. Rheingold, J. Am. Chem. Soc., 2011, 133, 19024–19027 CrossRef CAS PubMed.
- S. W. Sheehan, J. M. Thomsen, U. Hintermair, R. H. Crabtree, G. W. Brudvig and C. A. Schmuttenmaer, Nat. Commun., 2015, 6, 6469 CrossRef CAS PubMed.
- K. R. Yang, A. J. Matula, G. Kwon, J. Hong, S. W. Sheehan, J. M. Thomsen, G. W. Brudvig, R. H. Crabtree, D. M. Tiede, L. X. Chen and V. S. Batista, J. Am. Chem. Soc., 2016, 138, 5511–5514 CrossRef CAS.
- L. Duan, F. Bozoglian, S. Mandal, B. Stewart, T. Privalov, A. Llobet and L. Sun, Nat. Chem., 2012, 4, 418–423 CrossRef CAS PubMed.
- C. J. Richmond, R. Matheu, A. Poater, L. Falivene, J. Benet-Buchholz, X. Sala, L. Cavallo and A. Llobet, Chem. – Eur. J., 2014, 20, 17282–17286 CrossRef CAS.
- N. Vereshchuk, J. Holub, M. Gil-Sepulcre, J. Benet-Buchholz and A. Llobet, ACS Catal., 2021, 11, 5240–5247 CrossRef CAS.
- Y. Liu, G. Chen, S.-M. Yiu, C.-Y. Wong and T.-C. Lau, ChemCatChem, 2018, 10, 501–504 CrossRef CAS.
- D. W. Shaffer, Y. Xie, D. J. Szalda and J. J. Concepcion, J. Am. Chem. Soc., 2017, 139, 15347–15355 CrossRef CAS PubMed.
- F. Khasaeva, N. Vasilyuk, P. Terentyev, M. Troshina and A. T. Lebedev, Environ. Chem. Lett., 2011, 9, 439–445 CrossRef CAS.
- G. B. Daware and P. R. Gogate, Ultrason. Sonochem., 2020, 67, 105120 CrossRef CAS.
- Z. Xu, H. Liu, J. Niu, Y. Zhou, C. Wang and Y. Wang, J. Hazard. Mater., 2017, 327, 144–152 CrossRef CAS PubMed.
- S. Takenaka, R. Nomura, A. Minegishi and K. Yoshida, BMC Microbiol., 2013, 13, 62 CrossRef CAS.
- H. N. Kagalwala, L. Tong, R. Zong, L. Kohler, M. S. G. G. Ahlquist, T. Fan, K. J. Gagnon and R. P. Thummel, ACS Catal., 2017, 7, 2607–2615 CrossRef CAS.
- L. Duan, A. Fischer, Y. Xu and L. Sun, J. Am. Chem. Soc., 2009, 131, 10397–10399 CrossRef CAS.
- A. Ghaderian, A. Franke, M. Gil-Sepulcre, J. Benet-Buchholz, A. Llobet, I. Ivanović-Burmazović and C. Gimbert-Suriñach, Dalton Trans., 2020, 49, 17375–17387 RSC.
- M. Gil-Sepulcre, J. O. Lindner, D. Schindler, L. Velasco, D. Moonshiram, O. Rüdiger, S. DeBeer, V. Stepanenko, E. Solano, F. Würthner and A. Llobet, J. Am. Chem. Soc., 2021, 143, 11651–11661 CrossRef CAS PubMed.
- D. Schindler, M. Gil-Sepulcre, J. O. Lindner, V. Stepanenko, D. Moonshiram, A. Llobet and F. Würthner, Adv. Energy Mater., 2020, 2002329 CrossRef CAS.
- M. A. Hoque, M. Gil-Sepulcre, A. de Aguirre, J. A. A. W. Elemans, D. Moonshiram, R. Matheu, Y. Shi, J. Benet-Buchholz, X. Sala, M. Malfois, E. Solano, J. Lim, A. Garzón-Manjón, C. Scheu, M. Lanza, F. Maseras, C. Gimbert-Suriñach and A. Llobet, Nat. Chem., 2020, 12, 1060–1066 CrossRef CAS PubMed.
- C. Copéret, Acc. Chem. Res., 2019, 52, 1697–1708 CrossRef.
- K. André, H. Michael and D. Holger, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 16035–16040 CrossRef.
- N. Toshima and T. Yonezawa, New J. Chem., 1998, 22, 1179–1201 RSC.
- S. Cao, F. (Feng) Tao, Y. Tang, Y. Li and J. Yu, Chem. Soc. Rev., 2016, 45, 4747–4765 RSC.
- C. Roy, B. Sebok, S. B. Scott, E. M. Fiordaliso, J. E. Sørensen, A. Bodin, D. B. Trimarco, C. D. Damsgaard, P. C. K. Vesborg, O. Hansen, I. E. L. Stephens, J. Kibsgaard and I. Chorkendorff, Nat. Catal., 2018, 1, 820–829 CrossRef CAS.
- L. Helm and A. E. Merbach, Chem. Rev., 2005, 105, 1923–1960 CrossRef CAS.
- I. Rapaport, L. Helm, A. E. Merbach, P. Bernhard and A. Ludi, Inorg. Chem., 1988, 27, 873–879 CrossRef CAS.
- P. Pelosin, M. Gil-Sepulcre, P. Garrido-Barros, D. Moonshiram, J. Benet-Buchholz, C. Gimbert-Suriñach and A. Llobet, iScience, 2020, 23, 101378 CrossRef CAS.
- M. Gil-Sepulcre and A. Llobet, Nat. Catal., 2022, 5, 79–82 CrossRef CAS.
- C. Panda, J. Debgupta, D. Díaz Díaz, K. K. Singh, S. sen Gupta and B. B. Dhar, J. Am. Chem. Soc., 2014, 136, 12273–12282 CrossRef CAS PubMed.
- S. Pattanayak, D. R. Chowdhury, B. Garai, K. K. Singh, A. Paul, B. B. Dhar and S. sen Gupta, Chem. – Eur. J., 2017, 23, 3414–3424 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |