
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organometallic chemistry confined within a porphyrin-like framework
Michał J.
Białek
 a,
Karolina
Hurej
a,
Hiroyuki
Furuta
*b and
Lechosław
Latos-Grażyński
*a
a,
Karolina
Hurej
a,
Hiroyuki
Furuta
*b and
Lechosław
Latos-Grażyński
*a
aDepartment of Chemistry, University of Wrocław, 14 F. Joliot-Curie, 50383 Wrocław, Poland. E-mail: lechoslaw.latos-grazynski@chem.uni.wroc.pl
bDepartment of Chemistry and Biochemistry, Graduate School of Engineering, Kyushu University, 744 Moto-oka, Nishi-ku, Fukuoka 819-0395, Japan. E-mail: furuta.hiroyuki.165@m.kyushu-u.ac.jp
First published on 28th February 2023
Abstract
The world of modified porphyrins changed forever when an N-confused porphyrin (NCP), a porphyrin isomer, was first published in 1994. The replacement of one inner nitrogen with a carbon atom revolutionised the chemistry that one is able to perform within the coordination cavity. One could explore new pathways in the organometallic chemistry of porphyrins by forcing a carbon fragment from the ring or an inner substituent to sit close to an inserted metal ion. Since the NCP discovery, a series of modifications became available to tune the coordination properties of the cavity, introducing a fascinating realm of carbaporphyrins. The review surveys all possible carbatetraphyrins(1.1.1.1) and their spectacular coordination and organometallic chemistry.
 Michał J. Białek | Michał Białek was born in 1987. He graduated from the Wrocław University of Science and Technology in 2011. His PhD studies under the supervision of Lechosław Latos-Grażyński at the University of Wrocław focused on the organometallic chemistry and aromaticity of carbaporphyrinoids with an azulene motif. After completing PhD in 2017, he joined prof. Rafal Klajn's group at the Weizmann Institute of Science, Israel, a year later as a postdoc. His current research at the University of Wrocław covers a range of topics from the organometallic chemistry of modified porphyrinoids to chemistry under confinement within host–guest systems or macrocycles. |
 Karolina Hurej | Karolina Hurej was born in 1988 in Lubin, Poland. In 2017, she completed her PhD under the supervision of Lechosław Latos-Grażyński. Her investigations were dedicated to carbaporphyrinoids and their rhodium and ruthenium complexes. In 2018, Karolina won the “Mobilność Plus” scholarship from the Polish Minister of Science and Higher Education and joined the group of Dr Paweł Dydio at the Institute of Supramolecular Chemistry and Engineering, France. Currently, she works at the University of Wrocław. Her research interests include C–H activation within the carbaporphyrinoid frameworks and in simple organic molecules using porphyrin-based catalysts. |
 Hiroyuki Furuta (left) and Lechosław Latos-Grażyński (right) | Hiroyuki Furuta (left) received his BSc (1980), MSc (1982), and PhD (1986) degrees from Kyoto University. After postdoctoral research at the Mitsubishi-Kasei Institute of Life Sciences, Tokyo, and at the University of Texas, Austin, he started his independent academic career in 1992 as an Associate Professor at Oita University, where the “N-confused porphyrin” was found. In 1997, he moved to Kyoto University as an Associate Professor. In 2002, he was named a Full Professor in Chemistry at Kyushu University. In 2022, he retired from Kyushu University and became professor emeritus. His current research focuses on the synthesis of N-confused porphyrinoids with novel functionality. Lechosław Latos-Grażyński (right) received his PhD in 1978 from the University of Wrocław for the work supervised by Bogusława Jeżowska-Trzebiatowska. After postdoctoral research on highly oxidized iron porphyrins with Alan L. Balch and Gerd N. La Mar at the University of California, Davis (1979–1981), he returned to Wrocław, where he achieved professorship in 1992 and was nominated as the Head of the Organic Chemistry Division in 2002. Lechosław Latos-Grażyński is interested in the chemistry of porphyrins and N-confused porphyrins and their analogues, including various carbaporphyrinoids. His current interests include the synthesis of new porphyrinoids, their coordination and organometallic chemistry, and NMR spectroscopy of paramagnetic systems. |
Introduction
Porphyrinoids, defined as porphyrin-inspired macrocycles, have undergone systematic evolution extending the original concepts.1–10 The new research directions mainly originate from seminal porphyrin isomer11–15 and heteroporphyrin4,16–20 studies. Porphyrinoids may be seen as a unique platform for coordination/organometallic chemistry, which creates a specific environment allowing for the observation of unusual oxidation and electronic states of bound metal cations. Consequently, they have created an opportunity to address fundamental issues investigated in such seemingly distant fields as the aromaticity of porphyrinoids, including those which adopt a Möbius topology,21–24 organometallic copper(II) compounds,25,26 the contraction of benzene to cyclopentadiene,27,28 the inclusion of a d-electron subunit into a π-electron conjugation,29,30 catalysis,31–33 dioxygen activation,34 or serving as a π-coordination platform to accommodate the Ru4(CO)9 cluster(s).35Since its discovery by Rothemund,36meso-tetraarylporphyrin S1-1 has become a ubiquitous building block used in biomimetic chemistry to investigate metalloporphyrin-catalysed reactions or as working elements of molecular devices. The progress in the field has been confirmed by the development of efficient synthetic strategies for preparing meso-tetraarylporphyrins introduced by Adler and Longo37 and subsequently by Lindsey38 in a version adjusted for milder conditions. Remarkably, the synthesis of meso-tetraaryl-N-confused porphyrin12,13 (2-aza-21-carbaporphyrin, inverted porphyrin) S1-2 – a constitutional isomer of S1-1, formed in the Lindsey-type condensation of pyrrole and arylaldehyde – triggered the interest in the original class of macrocycles, 21-carbaporphyrins, to be later extended to a broader group of carbaporphyrinoids.
This specific subclass of core-modified porphyrins, i.e., 21-carbaporphyrins, can be formally constructed after a confinement of a carbocyclic moiety within the porphyrin S1-1 scaffold. A perfect match between the metal cation radii and the crevice diameter, so significant for the coordination chemistry of porphyrins, remains conserved in 21-carbaporphyrin. This provides a crucial incentive to explore unique macrocyclic organometallic chemistry. Today, replacing a porphyrin's single pyrrole ring with cyclopentadiene or N-confused pyrrole rings, as shown in Scheme 1, seems to be a natural strategy for creating porphyrin-type frames containing a paradigmatic [CNNN] coordination core. The several advances in the broader field of carbaporphyrinoids are comprehensively accounted for in a series of recent review articles.1,39–43
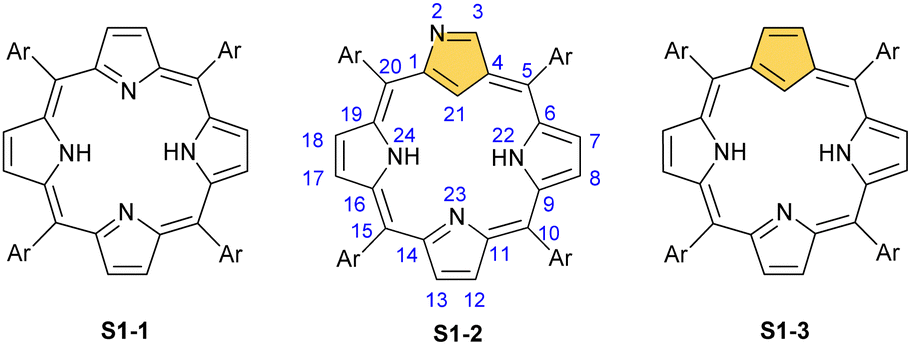 | ||
| Scheme 1 meso-Tetraarylporphyrin S1-1 and its carba analogues: N-confused porphyrin S1-2 and meso-tetraaryl-21-carbaporphyrin S1-3. | ||
For the purpose of this review, we would like to remain focused on a basic porphyrin scaffold – tetraphyrin(1.1.1.1) with four five-membered rings closed in a macrocycle with meso-bridges. The apparent structural resemblance of N-confused porphyrin S1-2 and 21-carbaporphyrin S1-3 frames to those coined “the working horses” of metalloporphyrin chemistry – meso-tetraarylporphyrin or β-octaethylporphyrin – provides the well-founded expectation for similar roles of S1-2 and S1-3 in organometallic chemistry. The conformation and strain within the macrocycle remain unaltered. However, by forcing a carbon fragment into close contact with the metal centre, organometallic reactions occur that would otherwise not progress if there was no confinement of these two species. Consequently, because of macrocyclic stabilisation, one is also able to witness and detect unstable transient forms solving mechanistic riddles. Eventually, the formation of organometallic complexes with suitably labile carbon–metal bond(s) can create an avenue to practical applications for catalysis, in a way the pincer ligands are famously used for.44–47 Finally, seemingly slight scaffold modifications are powerful means for determining new structure–property relationships in the spectral properties and reactivity of modified macrocycles.
The start of the review seems to be predictable because of the authors’ involvement in the originating field. The enlightening modification, namely N-confusion, introduced a carbon atom into the cavity and started a broad family of carbaporphyrins. The next step in creating an organometallic environment imprinted in porphyrin was introducing a carbocyclic unit. Thus, the presentation is split into a few complementary parts (Scheme 2). The first section covers the chemistry of N-confused porphyrin (S2-2); the second one addresses the properties of further modifications, including X-confused (S2-3) and neo-confused porphyrins (S2-4). The third part is focused on azuliporphyrin (S2-5) and benziporphyrin (S2-6). The fourth one presents true carbaporphyrins (S2-7). The fifth section introduces ‘higher complexity’ systems containing more than one carbon atom in the inner core. Finally, the last one looks at selected examples closely related to the previous sections.
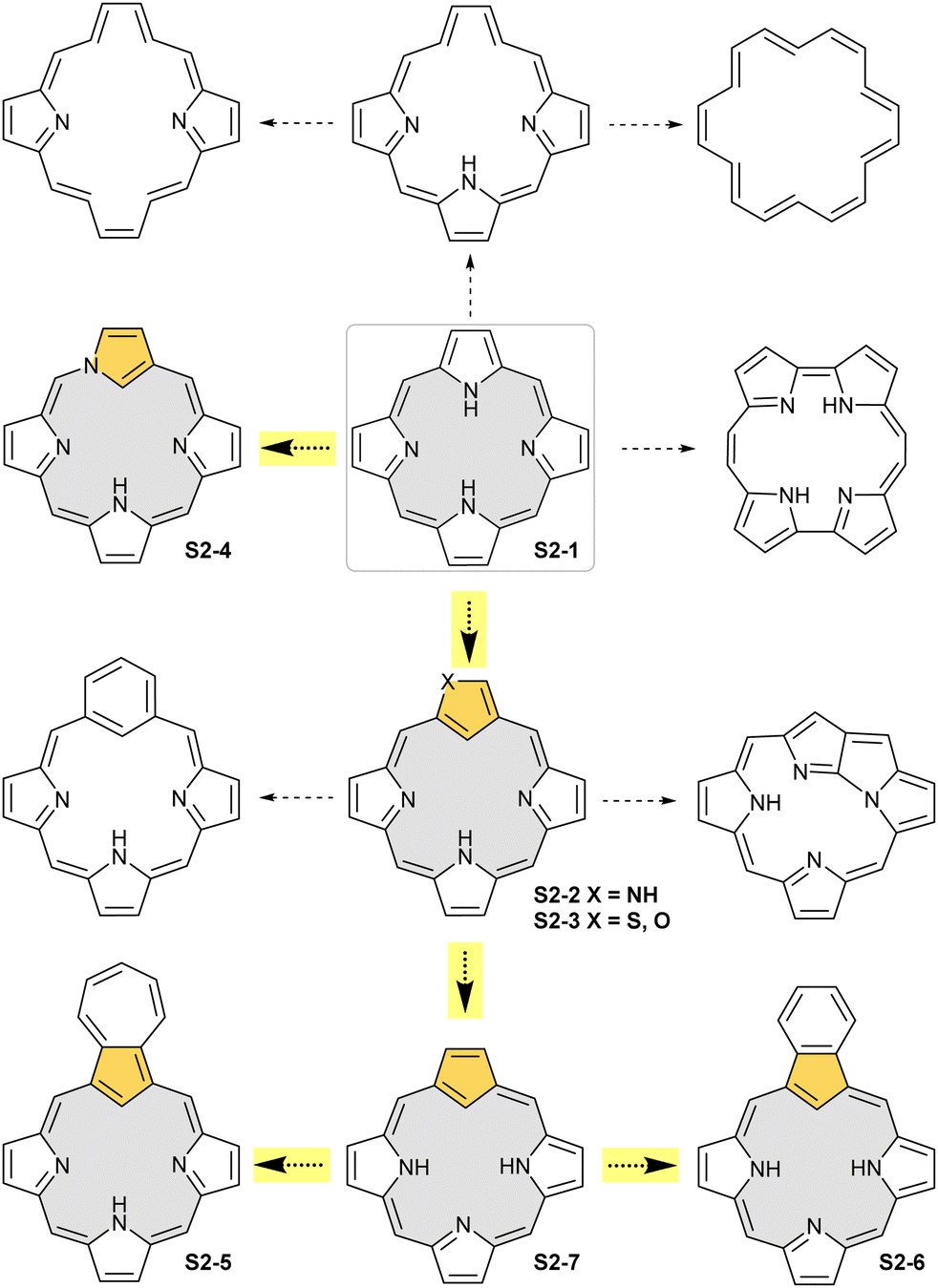 | ||
| Scheme 2 Basic structural motifs leading from regular porphyrin (S2-1) to carbaporphyrin (S2-7) and isomeric porphyrins (S2-2 and S2-4), while preserving the regular porphyrin framework. The inner parts of the described macrocyclic frameworks are grey-coloured to underline the leading motif together with an orange-coloured crucial five-membered ring that donates an inner carbon atom for coordination. Highlighted arrows indicate general pathways followed in this review. The remaining structures underline the great richness of the modified and isomeric porphyrinoid family, including other carbaporphyrinoids, not covered by this review. | ||
NCP – an origin of carbaporphyrinoids
The story of carbaporphyrins started with discovering meso-tetraaryl-2-aza-21-carbaporphyrin (N-confused porphyrin, inverted porphyrin) as a side-product of Lindsey-type condensation of pyrrole and arylaldehyde that typically leads to regular meso-tetraarylporphyrin. Our two groups isolated and determined the structure of this molecule independently as early as 1994.12,13 However, the preliminary suggestions addressing the potential existence and the structure of this peculiar porphyrin isomer were reported by Aronoff and Calvin in 194348 and Pauling in 1944 in his laboratory notes.15Originally, N-confused porphyrin was identified as an additional macrocyclic product of meso-tetraarylporphyrin synthesis carried out under classical Lindsey conditions.12,13 The first studies reported relatively low yields (4% Latos-Grażyński and 5–7% Furuta). The first preparative breakthrough required the synthesis of predesigned dipyrromethanes containing a pyrrole ring attached at a β-position (N-confused pyrrole), and the procedure afforded a 25% yield.49 The absolute game-changer resulted from the optimised Lindsey-type condensation of regular pyrrole and arylaldehyde. Lindsey et al. upgraded the process, eventually achieving the impressive ca. 40% yield of S1-2. Here, the skillful selection of an acidic catalyst, namely methanesulfonic acid, was crucial.50 This paved the way for extensive multidirectional studies of NCP – the constitutional porphyrin isomer with a carbon atom in the coordination core and the pyrrolic nitrogen atom at its perimeter. Finally, an appropriate design and the [3+1] reaction of protected N-confused tripyrrane and 2,5-bis(hydroxymethyl)pyrrole resulted in the formation of the N-confused isomer of porphine (S3-2).51
N-confusion could not go unnoticed in spectroscopic characterisation studies.12,52 The macrocycle containing an N-confused pyrrolic moiety is still aromatic, thus exhibiting a strong Soret band at 440 nm in an electronic spectrum and the overall red-brown colour of the chloroform solution. These features are typical as long as the experiment is carried out in a solvent with low polarity. In polar solvents such as DMF, the solution becomes green, Q-bands shift, and the Soret gets broader and slightly less intense. The differences are much more apparent in 1H NMR spectroscopy. In chloroform-d, strong macrocyclic aromaticity shifts upfield the inner proton signals and is a consequence of a sharp peak at ca. −5 ppm from an internal C(21)–H. In contrast, the solution in DMF-d7 reveals a different pattern, with a C(21)–H signal at 0.76 ppm and an increasing peak at 13.54 ppm. This alone allowed for a conclusion that two tautomers of NCP exist (S1-2 three inner protons – 3H and S3-1 (Scheme 3) two inner protons – 2H) and that one is able to control the formation of a particular tautomer by picking the right solvent52,53 and, as further studies show, substituents.53,54 Indeed, the signal at 13.54 ppm comes from the NH proton located outside the cavity. The conjugation pathway in S3-1 is formally disturbed; thus, the macrocyclic aromaticity is somewhat diminished.
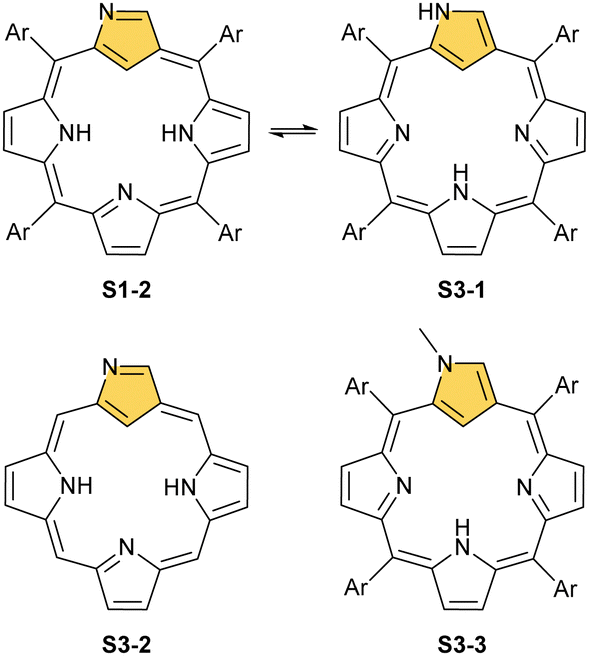 | ||
| Scheme 3 N-confused porphyrin tautomers (S1-2 – 3H and S3-1 – 2H) and other derivatives. | ||
The described aromaticity control within a preserved porphyrin scaffold of NCP shows a clear difference with the behaviour of the regular porphyrin. The tautomeric structural reorganisation of N-confused porphyrin S1-2 implies substantial coordination flexibility.
Reactivity of N-confused pyrrole – fundamental routes
Typically, such reactions are facilitated by the presence of a base. The reactivity of dihaloderivatives, including 1,2-bis(bromomethyl)benzene as a representative example, was tested as a logical next step in such studies.58 As expected, a reaction in an inert atmosphere provides, aside from an N(2)-alkylated product, a dimer S4-2 in which two NCP units are bridged through N(2) positions using an o-xylene moiety. Significantly, the presence of dioxygen triggers an alternative route. An annulation of a transient N(2)-substituted derivative S4-1 (Scheme 4) involves the reaction of an adjacent bromomethyl site with a C(3) atom. This affords, after oxidation, pyrrolo[1,2-b]isoquinoline-embedded macrocycle S4-3.
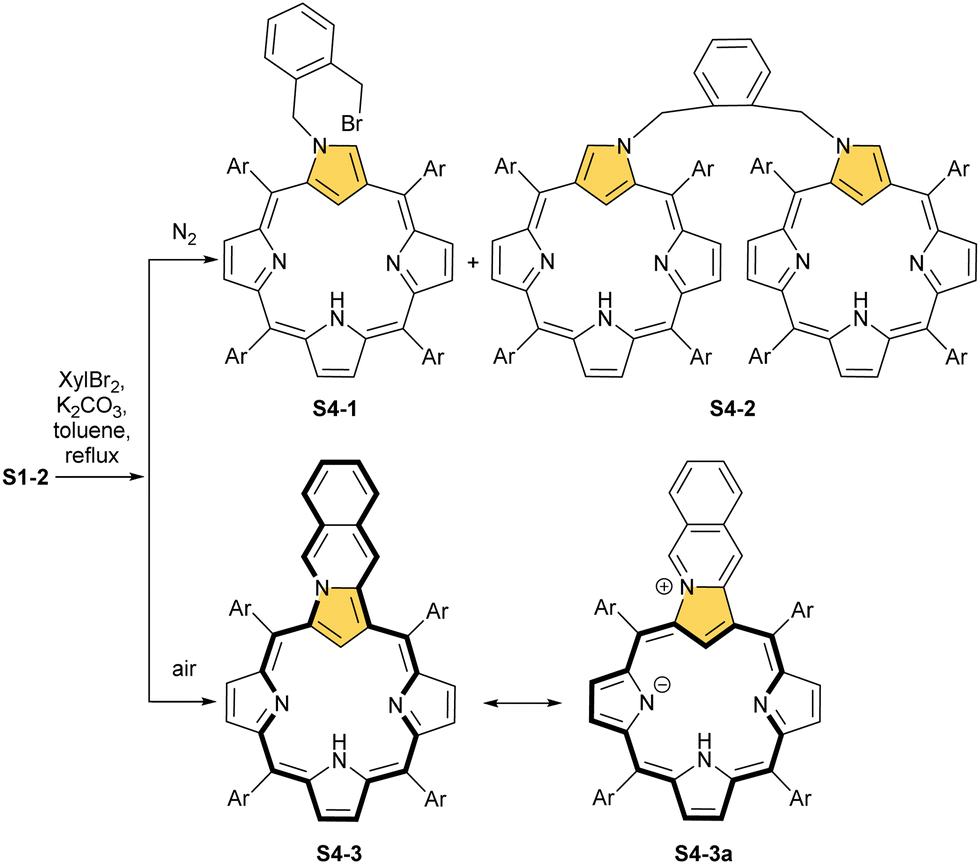 | ||
| Scheme 4 N(2)-Substituted derivatives of N-confused porphyrin S1-2. | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond forms. Accordingly, the molecular pattern related to S1-2 was enforced, which in the properties is reminiscent of a 3-oxo N-confused porphyrin (lactam) derivative.61
C double bond forms. Accordingly, the molecular pattern related to S1-2 was enforced, which in the properties is reminiscent of a 3-oxo N-confused porphyrin (lactam) derivative.61
 | ||
| Scheme 5 C(3)-Substituted derivatives of N-confused porphyrin S1-2. | ||
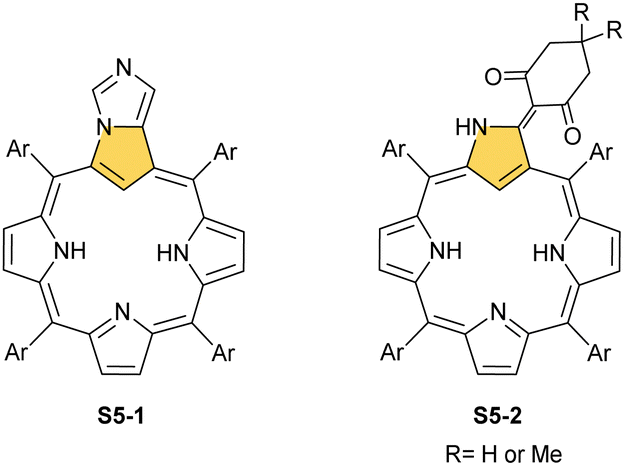
A different porphyrin skeleton, yet with inner 3H, can be warranted by the location of bulky substituents at the C(3) position. A phosphonate group of S6-1 (Scheme 6) (introduced in a reaction of NCP with trialkyl phosphite) is attached through a P–C(3) linkage.62 Phosphonylation of NCP occurs regioselectively. Initially, the reduction at the meso C(5) position was detected in this substitution process. Eventually, the reduced intermediary species was oxidised with 2,3-dichloro-5,6-dicyano-p-benzoquinone (DDQ) to form the final derivative S6-1.
 | ||
| Scheme 6 Selected results of C(3) reactivity in NCP. | ||
A spectacular C(3)-centred reactivity of N-confused porphyrin can be illustrated by the formation of covalently linked dimers represented here by S6-2.63S6-2 is readily produced by acid-catalysed dimerization of N-confused porphyrin S1-2 carried out in a toluene/dichloromethane mixture under reflux. In S6-2, two NCP subunits are linked by the C(3)–C(3′) bond of 1.468(3) Å length (Fig. 1).
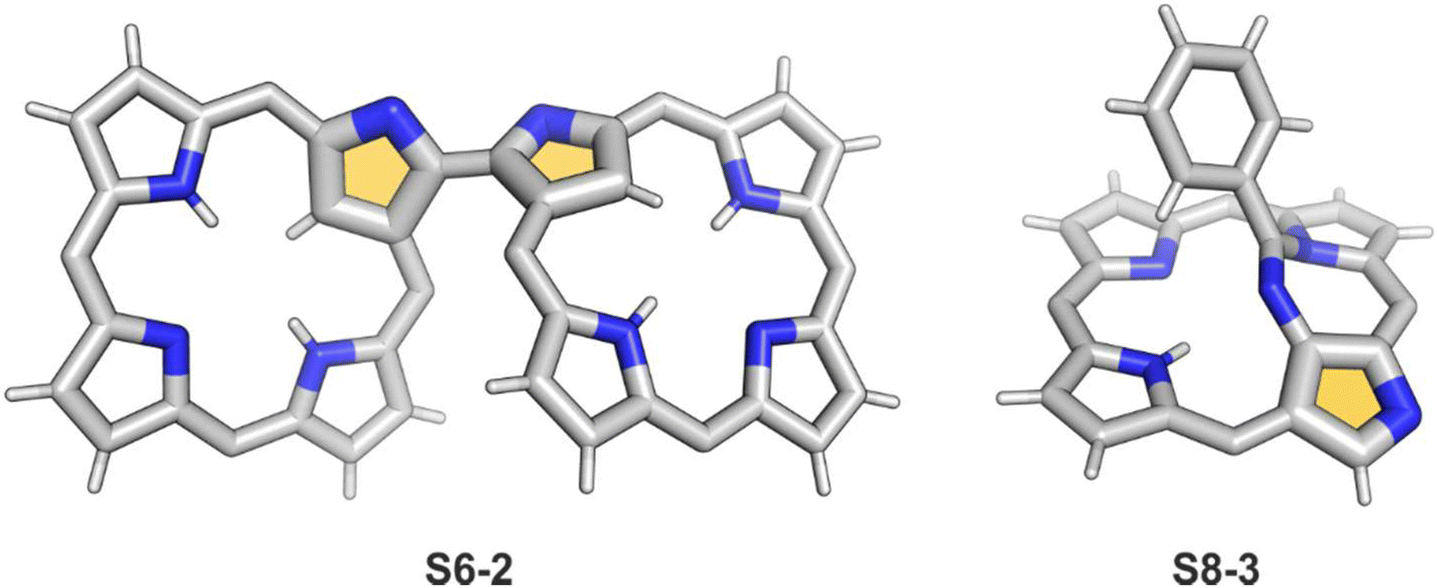 | ||
| Fig. 1 Products of C(3)/C(21) reactivity in NCP. meso-Substituents not shown. | ||
 | ||
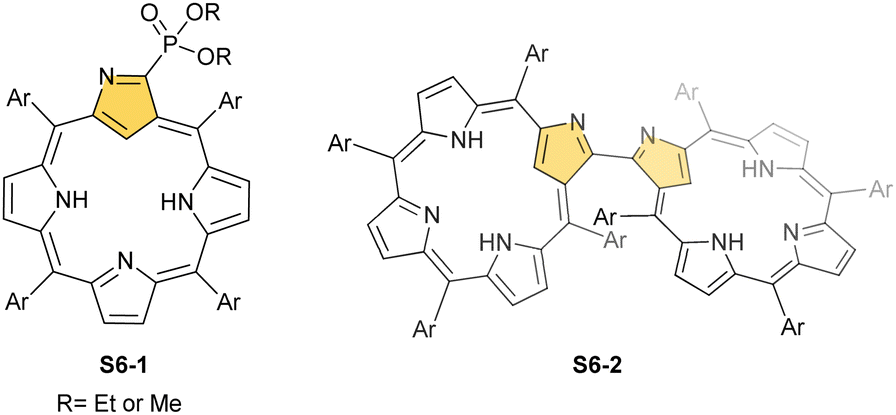
| Scheme 7 Phosphorylation of N-confused porphyrin S1-2 using potassium diphenylphosphide. | ||
Another C(21) functionalisation example is regioselective nitration leading to S8-1 (Scheme 8). The process occurs solely at the C(21) position when using HNO2 or diluted HNO3 acids.65 The reduction of the introduced NO2 group generates the respective amino derivative S8-2 – a suitable starting point for further transformations.66 The addition of arylaldehyde to S8-2 results in intramolecular annulation, which delivers two isomeric products (S8-3 and S8-4). In fact, in the oxidative step, the carbon atom of the imine moiety forms a single bond with one of two available internal NH atoms (N(22)H or N(24)H), completing the closure of novel seven-membered rings. Interestingly, S8-3, with the C(21)/N(24) link (Fig. 1), forms in higher yields. Upon heating, the isomers interconvert, yet the original preference under thermodynamic equilibrium is preserved. A reaction of N-confused porphyrin and paraformaldehyde (at elevated temperatures and under basic conditions) provides another illustrative example of intramolecular ring fusion.67 The initial incorporation of the formyl group at the C(21) position is followed by a bridge formation reaching the adjacent N(22) or N(24) nitrogen atoms. Consequently, this step leads to fusion, making the six-membered rings in S9-1 (Scheme 9) and S9-2 with a clear preference toward a C(21)/N(24) bond of S9-1.
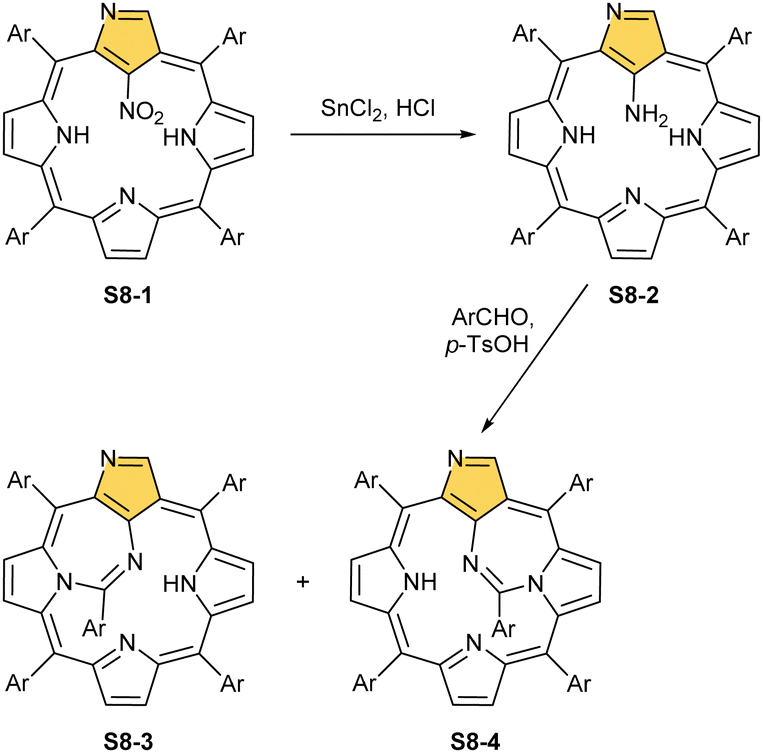 | ||
| Scheme 8 Reactivity of 21-nitro N-confused porphyrin S8-1. | ||
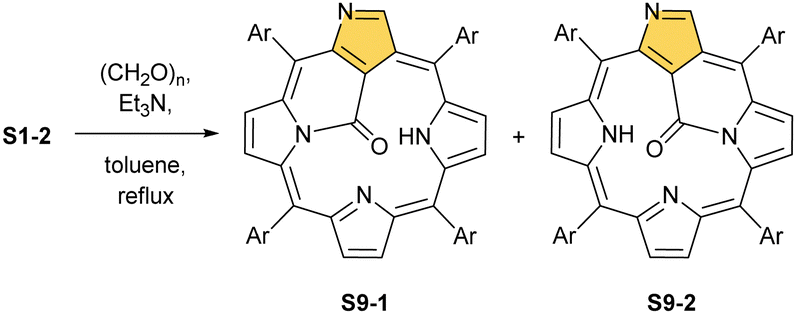 | ||
| Scheme 9 Reactivity of N-confused porphyrin with paraformaldehyde. | ||
It is significant to emphasise that the N-confused porphyrin's conformationally flexible nature creates a route for a peculiar intramolecular fusion concept.68,69 Thus, the regioselective bromination of S1-2 with NBS affords 21-bromo N-confused porphyrin S10-1 (Scheme 10).70 In a pyridine solution, S10-1 readily converts into N-fused porphyrin, NFP-Br (S10-3), a new type of porphyrinoid with a fused pyrrole ring in the macrocyclic core.71 Presumably, the presence of a bulky bromide substituent at the centre of NCP-Br S10-1 accelerates the flip of the N-confused ring. The geometry of transient species S10-2 imposes an adjacency of C(3) and N(22) atoms. Their proximity facilitates the oxidative ring fusion resulting in the formation of the C(3)–N(22) bond. The formation of NFP-Br creates a novel coordination platform, allowing for an unprecedented architecture of metal binding.72 The macrocyclic framework of N-fused porphyrin is markedly rearranged. After the N-confused pyrrole ring flip, the originally internal C(21) atom resides outside the coordination cavity.
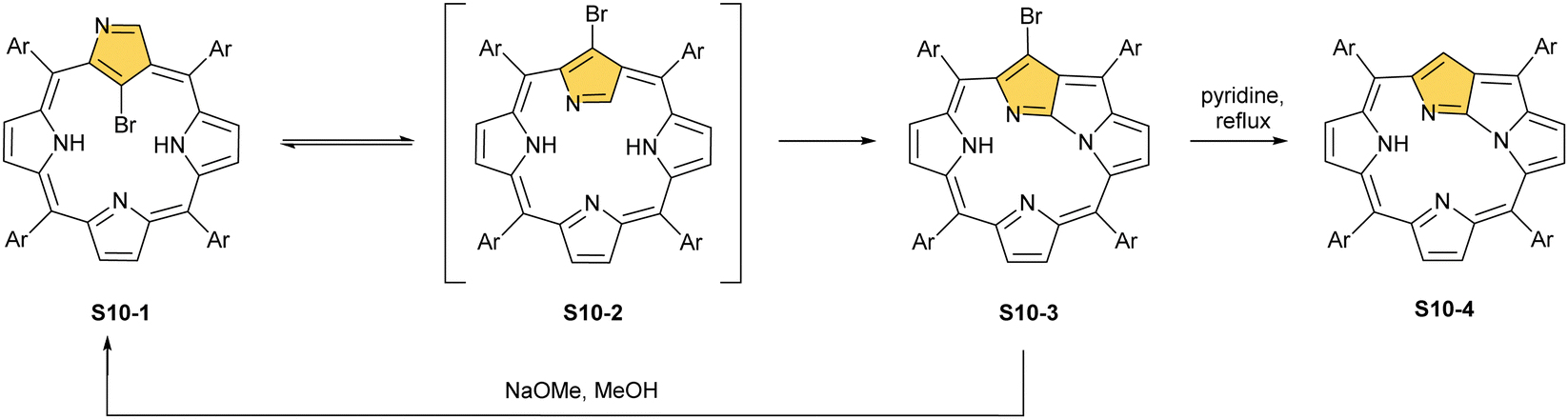 | ||
| Scheme 10 Reactivity of 21-bromo N-confused porphyrin S10-1. | ||
Furthermore, debromination of S10-3via reflux in pyridine afforded a fundamental frame of NFP S10-4. The addition of sodium methoxide to S10-3 results in the recovery of the NCP-Br frame, albeit after the cleavage of the N(22)–C(3) bond, the substitution by the methoxy group occurs at the C(3) position. Such a rearrangement can be induced by various nucleophiles, including thiols,73 which can be subsequently removed from the macrocycle.
Interestingly, S10-3 can be considered as a convenient starting point to generate suitably C(21)-substituted NCP due to the significant exposure of now the outer C(21) atom and an accessible NFP–NCP conversion.74 Thus, a facile Br(21) substitution with an alkynyl moiety produced an appropriate NFP alkynyl derivative S11-1 (Scheme 11). The subsequent conversion triggered by methoxide creates 21-alkynyl-substituted NCP S11-2. The further internal fusion of the triple bond with the adjacent NH provides etheno-bridged N-confused porphyrin S11-3 and, in terms of electronic structure, the endocyclic extension of the porphyrin π-system.74,75 In contrast to previously mentioned fusions, this one occurs regioselectively with the N(22) atom (S11-3), probably due to kinetic reasons. The exposed C(21) position of NFP S10-4 may also be functionalised with another NFP unit in an oxidative coupling reaction forming a covalently linked dimer S12-1.76 The NFP-NCP conversion triggered by methoxide allows for a stereoselective transformation of S12-1 (Scheme 12) into the original structure of S12-2, in which the subunits are C(21)-covalently linked (Fig. 2). The C(21)–C(21)′ bond distance is similar to the C(3)–C(3)′ bond length determined for S6-2, i.e., 1.480(7) Å, despite possible steric strains.
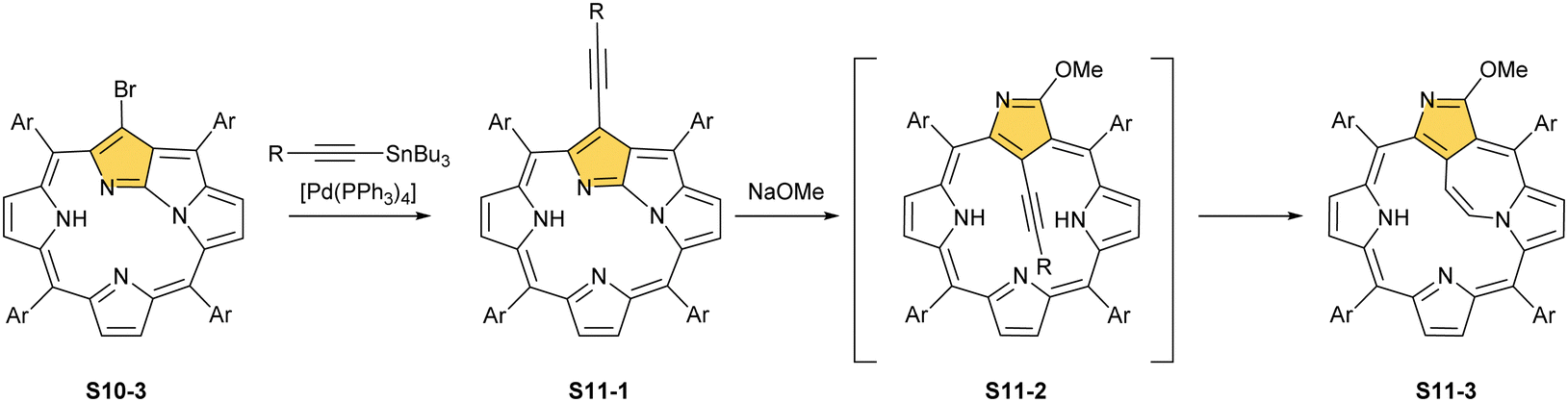 | ||
| Scheme 11 Reactivity of 21-bromo N-fused porphyrin S10-3. | ||
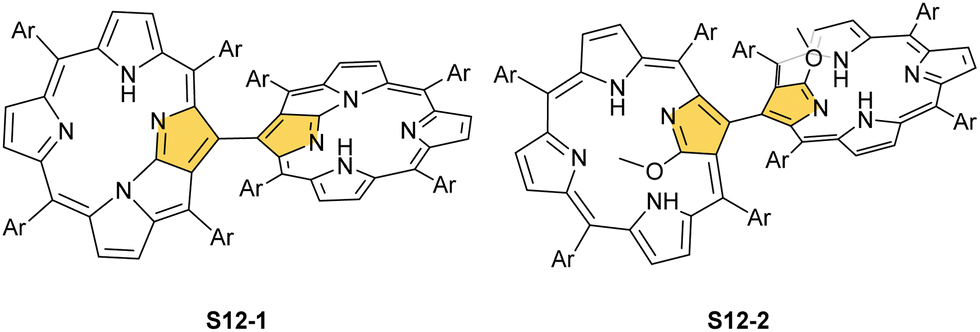 | ||
| Scheme 12 C(21)–C(21′) dimers. | ||
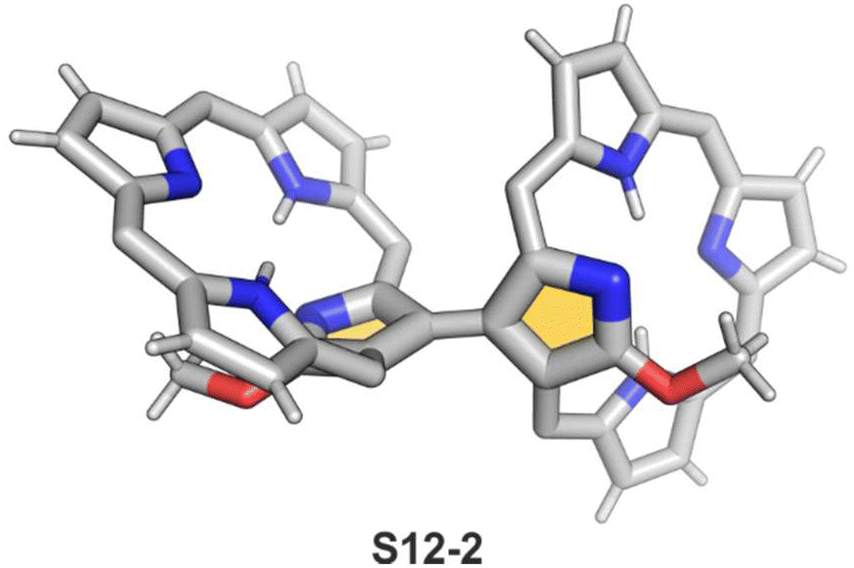 | ||
| Fig. 2 C(21)–C(21′) NCP dimer. meso-Substituents not shown. | ||
Organometallic chemistry
Such induction of metal–carbon interactions provides a unique opportunity to analyse them and translate the results into more reactive and dynamic (e.g., catalytic) systems. S1-2 enabled extensive studies on organometallic chemistry in macrocyclic surroundings.
One can notice that the coordination crevices of NCP can be adjusted in several ways via specific internal or peripheral modifications. Alternatively, a response to axial coordination to the transition metal ion can be probed. Fundamentally, the preferences for an oxidation state and/or tendency to form a direct M–C bond vary considerably. Thus, the higher oxidation states prefer the ligand structure related to tautomer S1-2. The lower oxidation state revealed the preference for the ligand structure visualised already for tautomer S3-1. The developments in the organometallic chemistry of N-confused porphyrin created a firm background for further, more general explorations of the organometallic chemistry of carbaporphyrinoids. In the following paragraphs, we will describe the highlights of organometallic chemistry in N-confused porphyrin surroundings.
In general, three fundamentally different modes of coordination can be identified for the inner core C(21) atom of the N-confused pyrrole ring defined by the C(21) donor geometry (Scheme 13):
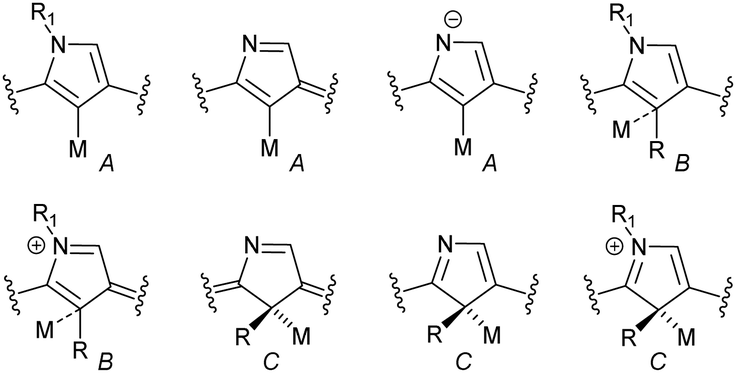 | ||
| Scheme 13 Different coordination modes of N-confused porphyrin. | ||
• σ-bonding of trigonal (sp2) carbanion (A),
• side-on coordination involving the trigonal (sp2) unit (B),
• σ-bonding of tetrahedral (sp3) carbanion (C).
Zinc(II), when inserted using zinc(II) acetate, coordinates to three internal nitrogen atoms and the external axial acetate ligand.77 The side-on η1 coordination of zinc(II) to the C(21) atom was also documented. NCP accommodates the structure of the S1-2 tautomer; thus, the zinc(II) charge is compensated by the equatorial ligand. The availability of the outer N(2) donor centre resulted in the formation of a tetranuclear zinc(II) NCP dimer S14-1 (Scheme 14). Two additional zinc(II) cations are involved in the construction of the bridging unit (Fig. 3). The reaction of S14-1 with Et4NOH leads to a more compact dimer, S14-2 – the natural conjunction of a zinc(II) preferred coordination number and [CH,NNN] core coordination. Finally, the saturation with pyridine breaks the dimer S14-2 affording a monomer S14-3. The Zn⋯C distance from the X-ray structure in S14-3 is 2.414(10) Å.
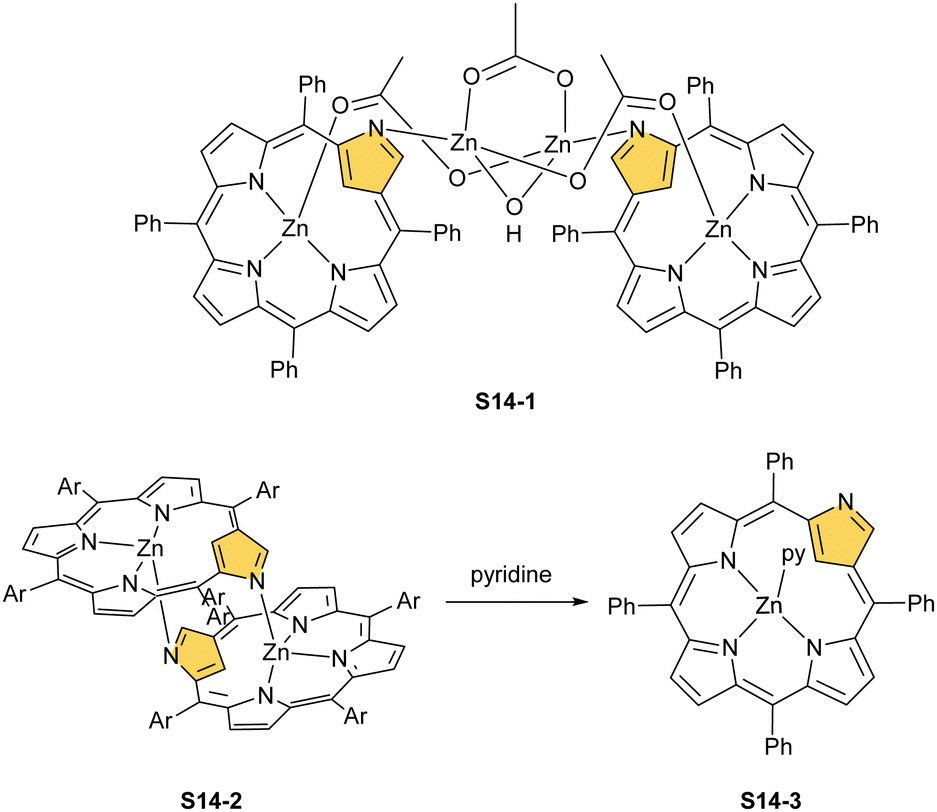 | ||
| Scheme 14 Zinc(II) complexes of N-confused porphyrin derivatives. | ||
 | ||
| Fig. 3 Oligoporphyrinic assemblies induced by zinc(II) coordination. meso-Substituents not shown. | ||
The structure of S14-2 was also confirmed by X-ray crystallography, showing Z(cis)-symmetry with vectors from C(3) to N(2) pointing in the same direction (Fig. 3).78 The similar behaviour of cadmium(II) and mercury(II) N-confused porphyrins was detected, although [Hg(NCP)]2S15-3 is somewhat unstable. The Zn⋯C distance in the dimer S14-2 increases to 2.534(8) Å. Interestingly, after mixing Zn and Cd dimers together, the heterodimer formation was observed during equilibration due to N(2) de- and recoordination. Inserting mercury(II) into N(2)-methyl NCP, which adopts an S3-1-like cavity with one inner NH, resulted in a monomeric complex S15-1 (Scheme 15) supplemented with chloride for the charge balance.79 The same coordination mode can be found in the ZnII and CdII complexes of phosphonylated N-confused porphyrin (S6-1)62 or the zinc(II) complexes of the C(3)–C(3)′ dimer (S6-2).80 Complex S15-1 exhibits the Hg⋯C(21) distance of 2.711(6) Å and, together with similar derivatives, demonstrate the agostic interaction, which has significant consequences in communication between closely located interacting atoms. In the case of S15-1 and related species, the scalar coupling of the internal H(21) and 199Hg is reflected in an appropriately split 1H NMR pattern. Depending on NCP substituents, e.g., mono-5-phenyl substitution, coordination trimers S15-4 can be formed as sole products (Fig. 3).81,82
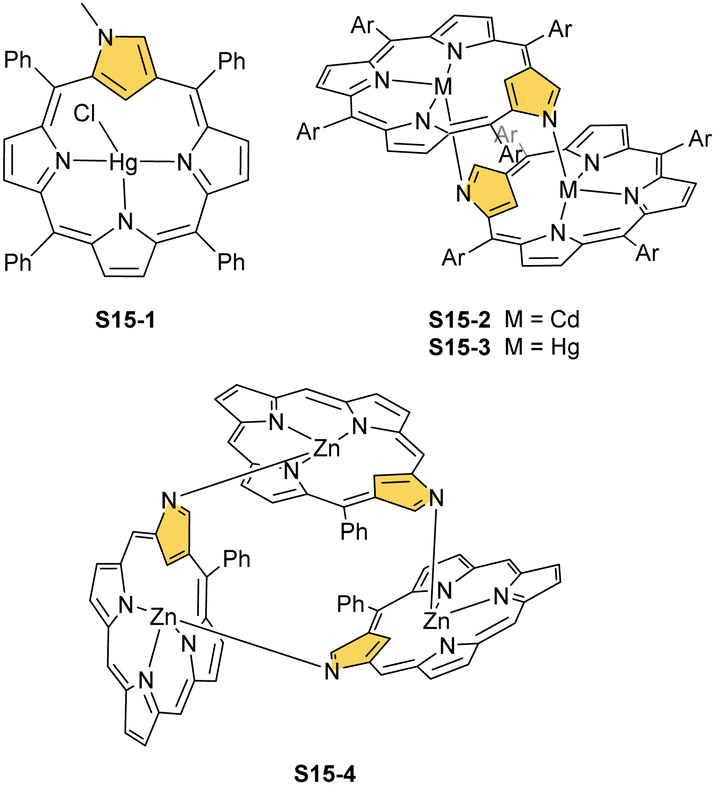 | ||
| Scheme 15 Complexes of N-confused porphyrin derivatives with mercury(II), cadmium(II), and zinc(II). | ||
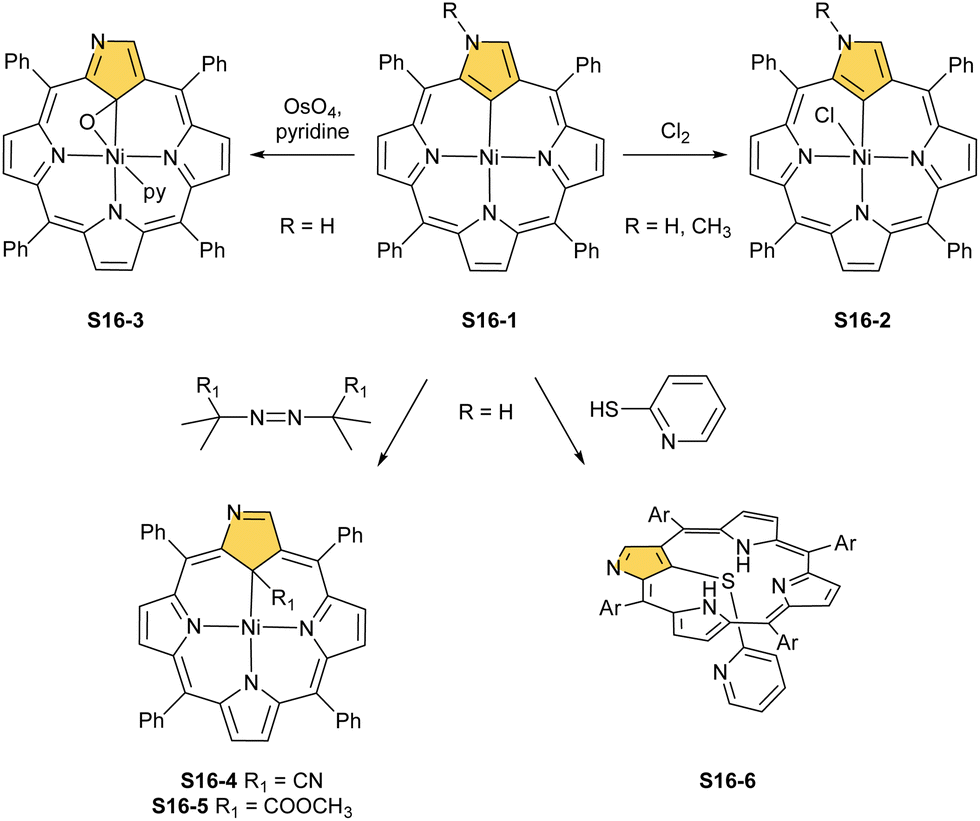 | ||
| Scheme 16 Reactivity of nickel(II) N-confused porphyrin S16-1. | ||
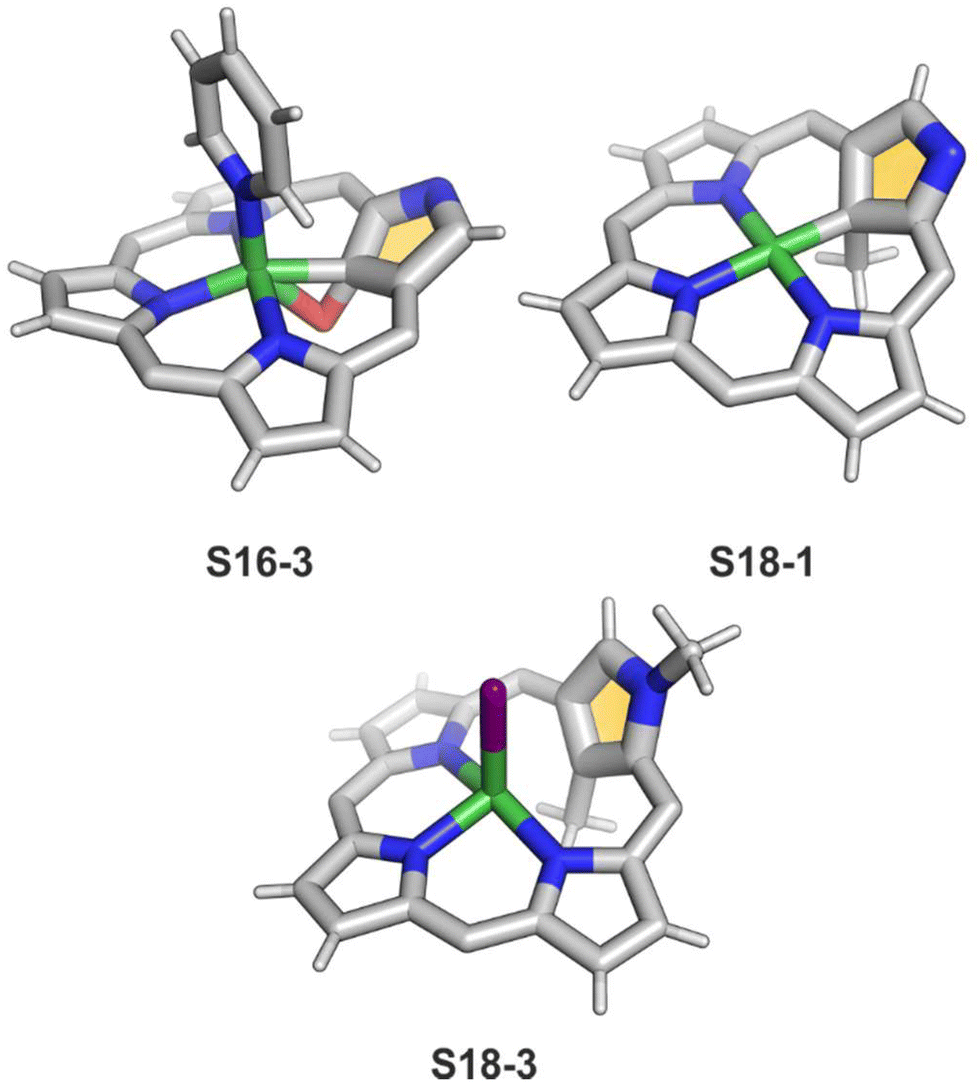
A different oxidation product can be obtained in a reaction of S16-1 and OsO4.84 During the reaction, an oxygen atom is transferred onto the C(21) atom (S16-3). As confirmed by the X-ray structure (Fig. 4), organometallic bonding is still in place being indicated by a short C(21)–Ni distance of 2.069(4) Å. In the course of this process, the one-electron oxidation of nickel(II) takes place, and the charge is balanced by O(21) coordination in its phenolic state (C(21)–O(21) distance equals 1.377(5) Å). The nickel(III) coordination sphere is supplemented by pyridine used in the reaction. A very interesting transformation is enabled by the: (I) initial internal C–H activation by a nickel(II) ion, (II) adjustable coordination properties of the NCP macrocycle, i.e., the possibility of nickel(III) stabilisation, (III) close proximity to the metal centre and the carbon fragment. The same molecular motif is obtained after nickel(II) is inserted into 3-ethoxy NCP and then exposed to air.85 As the primarily formed nickel(II) 3-ethoxy NCP complex S17-1 (Scheme 17) is unstable, transformation leads to a 3-ethoxy complex S17-2. The oxidation of the nickel(II) centre with [bis(trifluoroacetoxy)iodo]benzene (PIFA) conducted in the presence of ethanol leads to the incorporation of an additional ethoxy substituent at the C(21) position. This complex S17-3 bears additional chloride at the nickel(III) centre, which also interacts with the inner ethoxy group as indicated by a Ni–O(21) contact of 2.331(3) Å. The Ni–C(21) distance is 2.065(3) Å. Both inner oxide (in S17-2) and ethoxide (in S17-3) can be removed in a reversible reduction using NaBH4, leading to the starting nickel(II) complex S17-1. This study points to differences in reactivity induced just by adding a 3-ethoxy substituent. In other studies, when nickel(II) NCP (S16-1) is exposed to azo radical initiators, like 2,2′-azobis(2-methylpropionitrile) (AIBN) and dimethyl 2,2′-azobis(2-methylpropionate), the isolated product contains the new C(21) substituent originating evidently from the azo-source.86 In the case of AIBN, it is a CN group (S16-4) and, for the second one, an ester moiety (S16-5).
 | ||
| Fig. 4 Coordination motifs in nickel(II) NCP complexes. meso-Substituents not shown. | ||
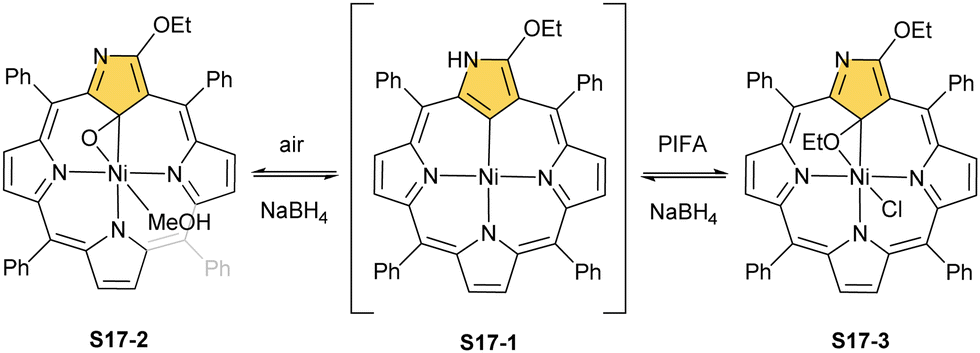 | ||
| Scheme 17 Reactivity of nickel(II) 3-ethoxy N-confused porphyrin S17-1. | ||
As the final complex is diamagnetic, square-planar nickel(II) is expected. Thus, a C(21)–Ni bond must remain intact for the charge balance. The transformation involves a nickel(III) N-confused porphyrin intermediate, which eventually coordinates the generated radical. Then, an R1 substituent is transferred onto C(21) with the cleavage of either the C–CN or the C–COOMe bond and reductive elimination of a remaining carbon fragment from the metal centre. After demetallation, free 21-substituted porphyrins can be obtained. The coordination sphere flexibility of nickel has its limitation. After the reaction of S16-1 and 2-mercaptopyridine, the C(21)–S bond is formed, but the complex is demetallated at the same time (S16-6), in contrast to cobalt(II) and ruthenium(II) chemistry (see below).33 Substitution at the activated C(21) position will be a frequent motif reported throughout this review.
Another example of S16-1 reactivity is regioselective methylation which occurs upon mixing with methyl iodide.87 Firstly, a methyl group is attached at the C(21) position forming diamagnetic nickel(II) 21-methyl N-confused porphyrin S18-1 (Scheme 18). The reversible concerted addition of HX converts diamagnetic S18-1 into paramagnetic S18-2. In contrast, it was proposed that nickel(II) β-alkylated NCP S19-1 (Scheme 19) undergoes the protonation of the internal, coordinated C(21) atom after adding TFA affording aromatic S19-2.88 Similarly, reversible C-protonation is observed in the case of N-methylated NCP.89
The methylation of S16-1 at the C(21) position may also be followed by the reaction at the peripheral N(2) atom to afford the paramagnetic dimethylated derivative nickel(II) 2,21-dimethyl NCP (S18-3). The reaction route does not include the alternative path, namely the peripheral methylation to form diamagnetic nickel(II) 2-Me NCP like S19-1. However, the treatment of S19-1 with methyl iodide results in the methylation of the C(21) position.
 | ||
| Scheme 18 Regioselective alkylation of nickel(II) N-confused porphyrin S16-1. | ||
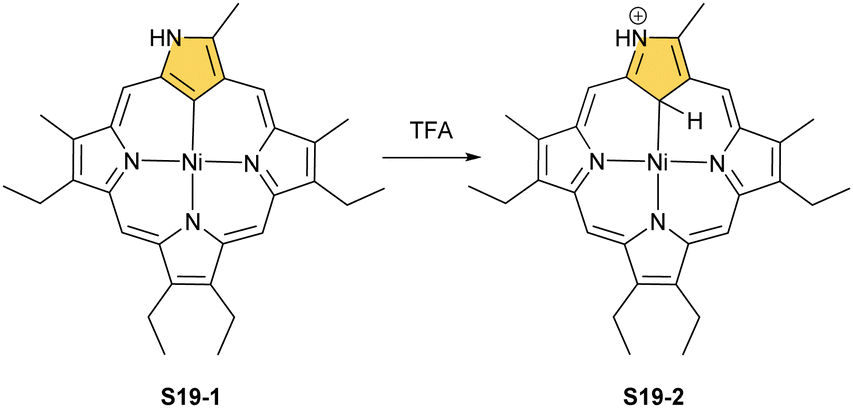 | ||
| Scheme 19 Protonation of nickel(II) β-alkylated N-confused porphyrin S19-1. | ||
In contrast to methylation with methyl iodide, perfluoroalkyl iodides react selectively at the C(21) position of S16-1.90 The molecular structures of S18-1 and S18-3 were determined by X-ray crystallography (Fig. 4). The Ni–C(21) bond length in S18-1 (2.005(6) Å) indicates direct bonding to the clearly sp3-hybridised carbon atom. The structure of S18-3, with iodide attached to nickel(II), gives a longer distance (2.406(9) Å) corresponding to the strong Ni⋯C(21) interaction enforced by macrocyclic constraints, leaving C(sp2) substituted with the methyl group.
An application of substrates with more halide atoms attached can promote the formation of covalently linked dimers. Such a reaction was reported with dihalomethanes as the precursors of bridging units. The representative treatment of S16-1 with dihalomethanes in the presence of a base afforded preferentially the dimer S18-4.91 Overwhelming majority of the product (even above 90%) acquired the N(2)–C(21)′ CH2 linkage (S18-4). The only other dimeric product identified is S18-5 with the N(2)–CH2–N(2)′ bridge.
Coordination with nickel(II) may induce the formation of a strapped NCP dimer complex S20-1 (Scheme 20) when starting from 3,7,23,27-tetrabromo[36]octaphyrin bearing two unsubstituted meso positions.92 It was suggested that the initial NiII complexation occurs in two NNNC pseudo-cavities (Ni–C: 1.885(4) Å; Fig. 5). It imposes proximity between positions 5,27 and 7,25, leading to a fusion reaction and bromides leaving positions 7 and 27. This step is followed by debromination at the perimeter. The whole molecule exhibits weak paratropic features due to the domination of the 36π-electron ring current.
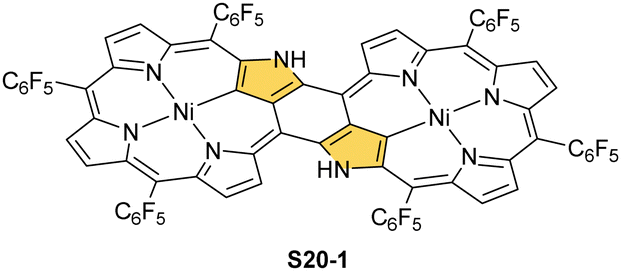 | ||
| Scheme 20 Nickel(II) NCP dimer S20-1. | ||
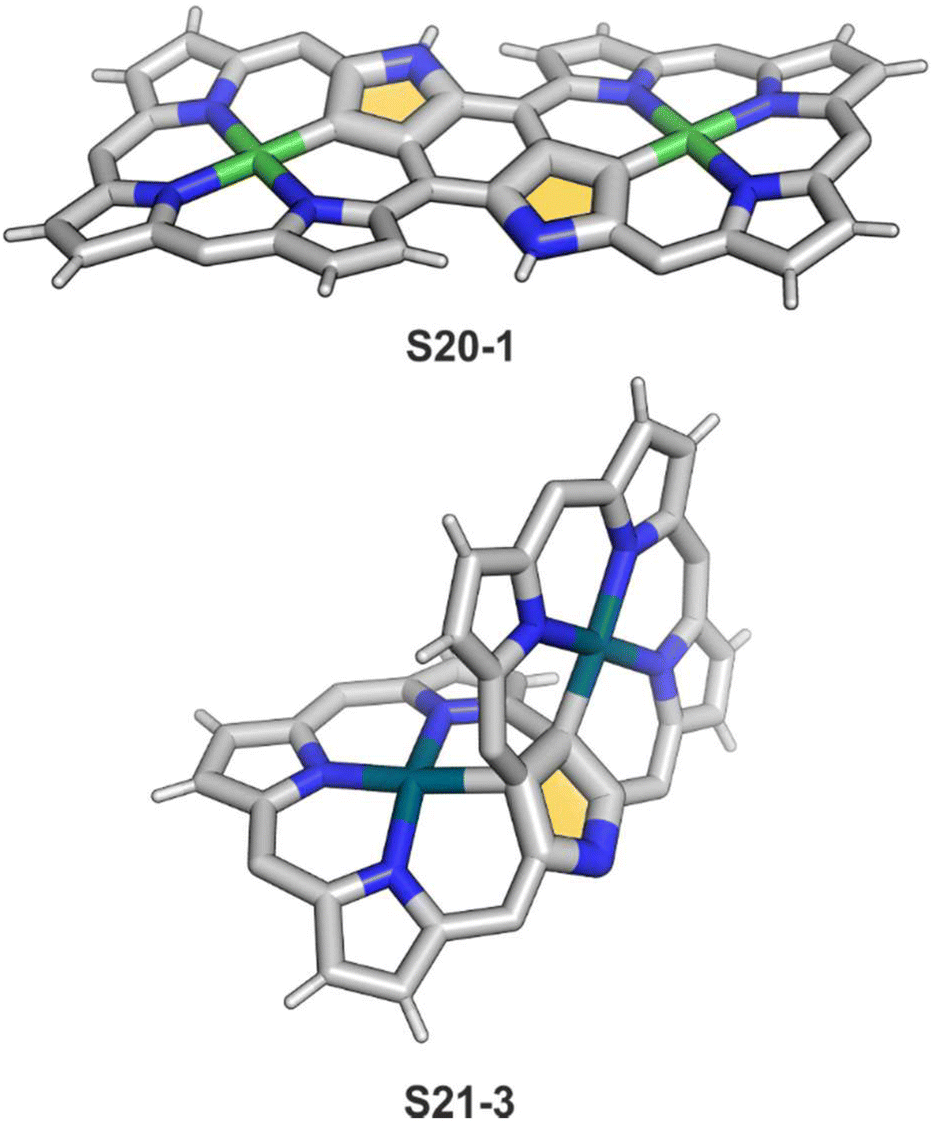 | ||
| Fig. 5 Bis-N-confused porphyrin skeletons formed after expanded porphyrin coordination. meso-Substituents not shown. | ||
Two other metals accompanying nickel in the 10th group, i.e., palladium and platinum, efficiently react with NCP. A simple insertion of palladium(II) into NCP using palladium(II) acetate in boiling chloroform affords a regular square-planar complex S21-1 (Scheme 21),93 analogous to the nickel(II) complex S16-1.
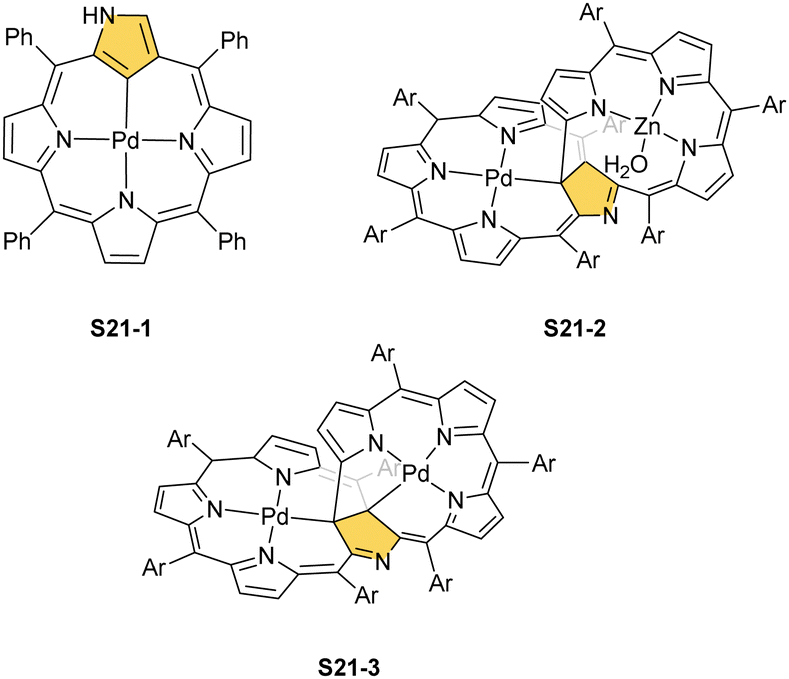 | ||
| Scheme 21 Palladium(II) complexes of N-confused porphyrin derivatives. | ||
Platinum(II) NCP S22-1 (Scheme 22) is formed efficiently in a reaction of [PtCl2(MeCN)2] and NCP with perfluorinated meso-phenyls (–C6F5) in boiling 1,2-dichlorobenzene.94 This complex undergoes swift regioselective methylation on the activated C(21) atom (S22-2) like the nickel analogue does. Interestingly, a C(3),C(21)-disubstituted complex of palladium(II) can be obtained in an unexpected transformation using [32]heptaphyrin and palladium acetate at room temperature.95 Initially, in the course of the insertion, palladium(II) is accommodated into a [CNNN] surrounding. The structure of formed intermediary species promoted the profound heptaphyrin bond rearrangement affording a peculiar architecture S21-2 with the readily identified moiety based on the skeleton of 3,21-disubstitued NCP. A more prolonged reaction with a 20 eq. excess of Pd(OAc)2 leads to interesting dipalladium(II) species S21-3 (Fig. 5).
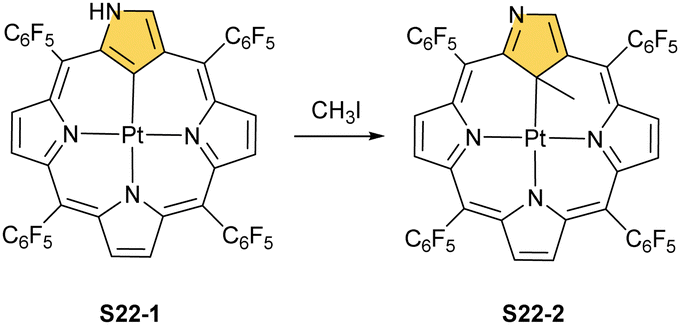 | ||
| Scheme 22 Regioselective methylation of platinum(II) N-confused porphyrin S22-1. | ||
In fact, the outcome of the regular palladium(II) or platinum(II) insertion depends enormously on the choice of conditions. As already mentioned, the insertion of palladium(II) under mild conditions afforded regular palladium(II) NCP S21-1. Two additional products, S23-1 (Scheme 23) and S23-2, were identified once boiling toluene was used as a solvent.93 As one can see, the presence of outer N(2) facilitates the formation of isomeric Pd2L2 dimers, in which each palladium(II) ion coordinates two core nitrogen atoms of one NCP subunit and the outer N(2) atom from the second subunit (Fig. 6). This geometry brings palladium(II) close to a proximal meso-aryl substituent. The proximity of palladium(II) and the ortho-CH fragment seems to be instrumental in activating the Cortho–H bond and eventually forming the palladium(II)–Cortho bond.
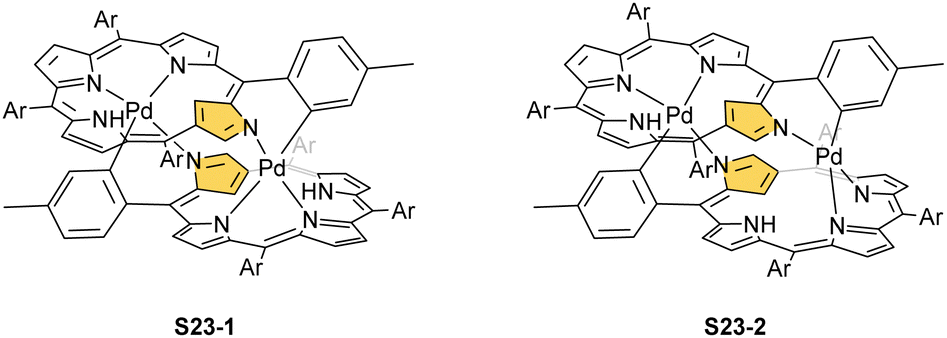 | ||
| Scheme 23 meso-Aryl-coordinated palladium(II) NCP-based complexes. | ||
 | ||
| Fig. 6 meso-Aryl-coordinated NCP-based complexes. Other meso-substituents not shown. | ||
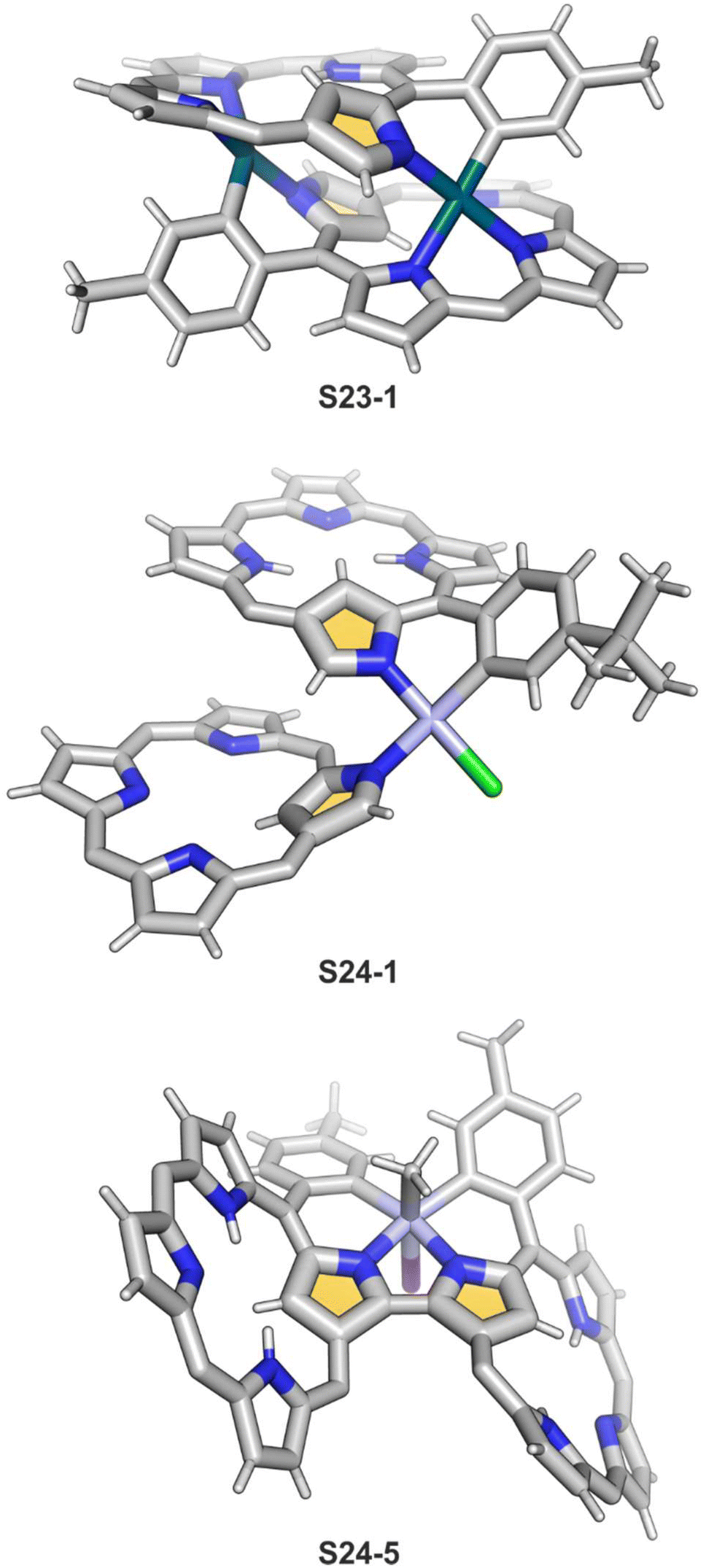
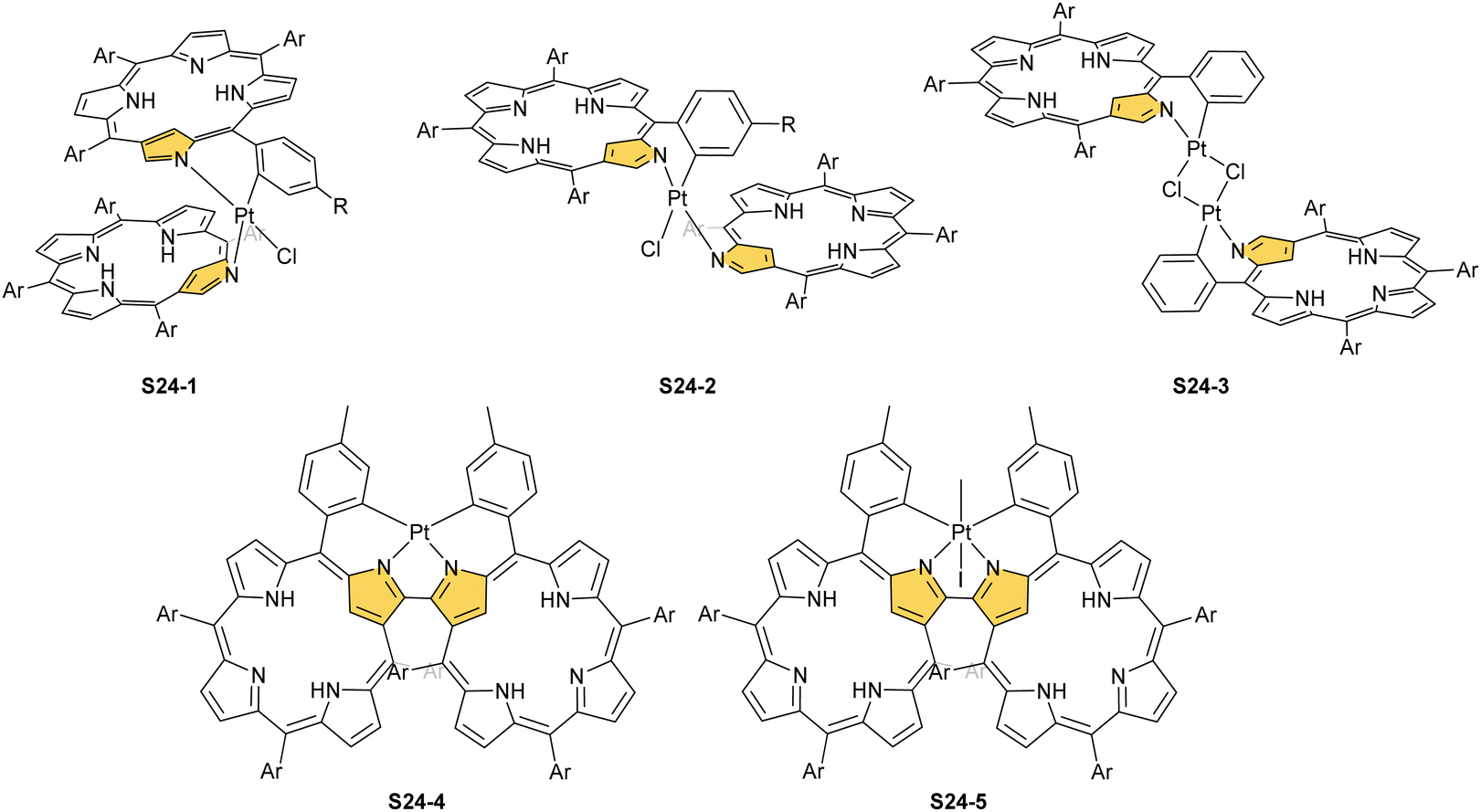
The insertion of platinum(II) (0.5 eq. PtCl2) into NCP, carried out in boiling toluene, affords the diporphyrinic species [PtCl(NCP)2] S24-1 (Scheme 24) and S24-2.96 Two NCP subunits are solely linked via perimeter N(2), N(2)′, and Cortho donors coordinated to a single platinum(II) cation. The fourth coordination site is occupied by chloride. The cis vs. trans location of Cortho and Cl is responsible for forming two isomers, with S24-1 (cis) existing as a major product (Fig. 6). In the specific structures S24-1 and S24-2, both the cavities of NCP subunits remain unoccupied, potentially leaving some room for the accommodation of other metal cations to form composite trimetallic species. The reaction outcome changes quite drastically when 1 eq. of platinum(II) chloride is used. The regular platinum(II) NCP and the diporphyrinic species [PtCl(NCP)2] S24-1 and S24-2 can be detected but as minor side-products. The major product [PtCl(NCP)]2 forms as a dimeric and symmetric structure S24-3 in which two chloride ligands bridge the subunits.
 | ||
| Scheme 24 meso-Aryl-coordinated platinum(II)/(IV) NCP-based complexes. | ||
The preorganised environment of a covalently linked NCP C(3)–C(3)′ dimer S6-1 enforces the specific external coordination to a platinum ion.97 Two proximal N(2) and N(2)′ atoms and two adjacent Cortho positions participate in platinum(II) coordination, providing an external CCNN environment (S24-4). The NCP cores remain available for further coordination. When treated with methyl iodide, the platinum(II) centre of S24-4 undergoes oxidative addition yielding the platinum(IV) complex with equatorial (PtIV–Cortho) and axial (PtIV–CH3) organometallic bonding (S24-5; Fig. 6).
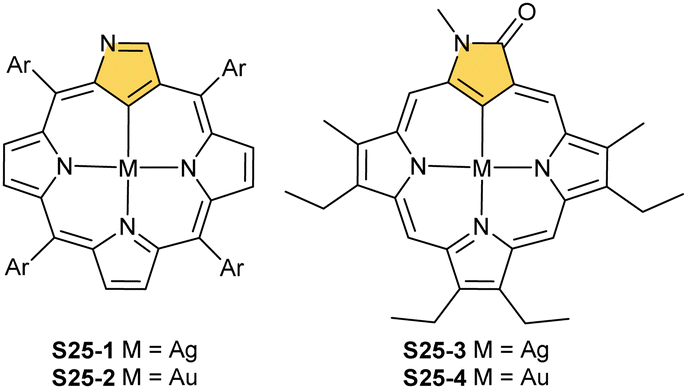 | ||
| Scheme 25 Silver(III) and gold(III) complexes of N-confused porphyrin and its derivative. | ||
Thus, the dissociation of inner core protons of tautomer S1-2 (two (N)H and one (C)H) generates a trianionic, aromatic macrocyclic ligand allowing the initial insertion of silver(I) to be followed by two-electron oxidation with an excess of silver(I). The metal centre can be removed through acidification.99 Interestingly, when the complex is substituted with an ethoxy group at the C(3) position, demetallation is accompanied by the halogenation of the C(21) atom coming from the HX molecule.
The treatment of NCP with [AuCl(S(CH3)2)] in refluxing toluene yields small amounts of gold(III) NCP S25-2, with the source adopting the same roles (insertion and oxidising reagent).100 Remarkably, N-fused porphyrin S10-4 is mainly produced while using regular NCP. Before complexation, C(21) bromination must be performed to obtain S25-2 efficiently. The reaction of activated 21-Br NCP (S10-3) and [AuCl(S(CH3)2)] provides desired S25-2. The organometallic contact of 2.016(12) Å clearly points to direct bonding.
One can form a bimetallic species by using imidazole-fused NCP S5-1.59 In the first step, the silver(III) imidazole-fused NCP is formed. Subsequently, the methylation with methyl iodide yields cationic species S26-1 (Scheme 26), in which the methyl group is attached to the outer nitrogen atom. S26-1 reacted with Ag2O acquires the initial N-heterocyclic carbene functionality. The following metallation with [AuCl(S(CH3)2)] affords a AgIII/AuI hybrid S26-2 (Fig. 7). The insertions of silver and gold to N(2)-methyl (β-substituted) N-confused porphyrin were probed as well.89 The inner core of N(2)-methyl N-confused porphyrin S3-3 contains only two dissociable hydrogen atoms (NH and C(21)H); the frame of tautomer S3-1 and, as predictable, tends to promote the stabilisation of metal(II) cations. Consequently, the silver(III) or gold(III) insertions have been accompanied by oxygenation at position C(3) to form the lactam units of S25-3 and S25-4.
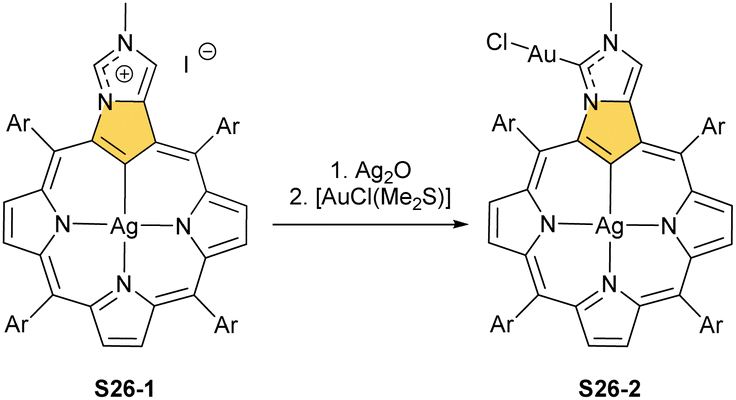 | ||
| Scheme 26 Gold-carbene coordination at the periphery of silver(III) imidazole-fused NCP S26-1. | ||
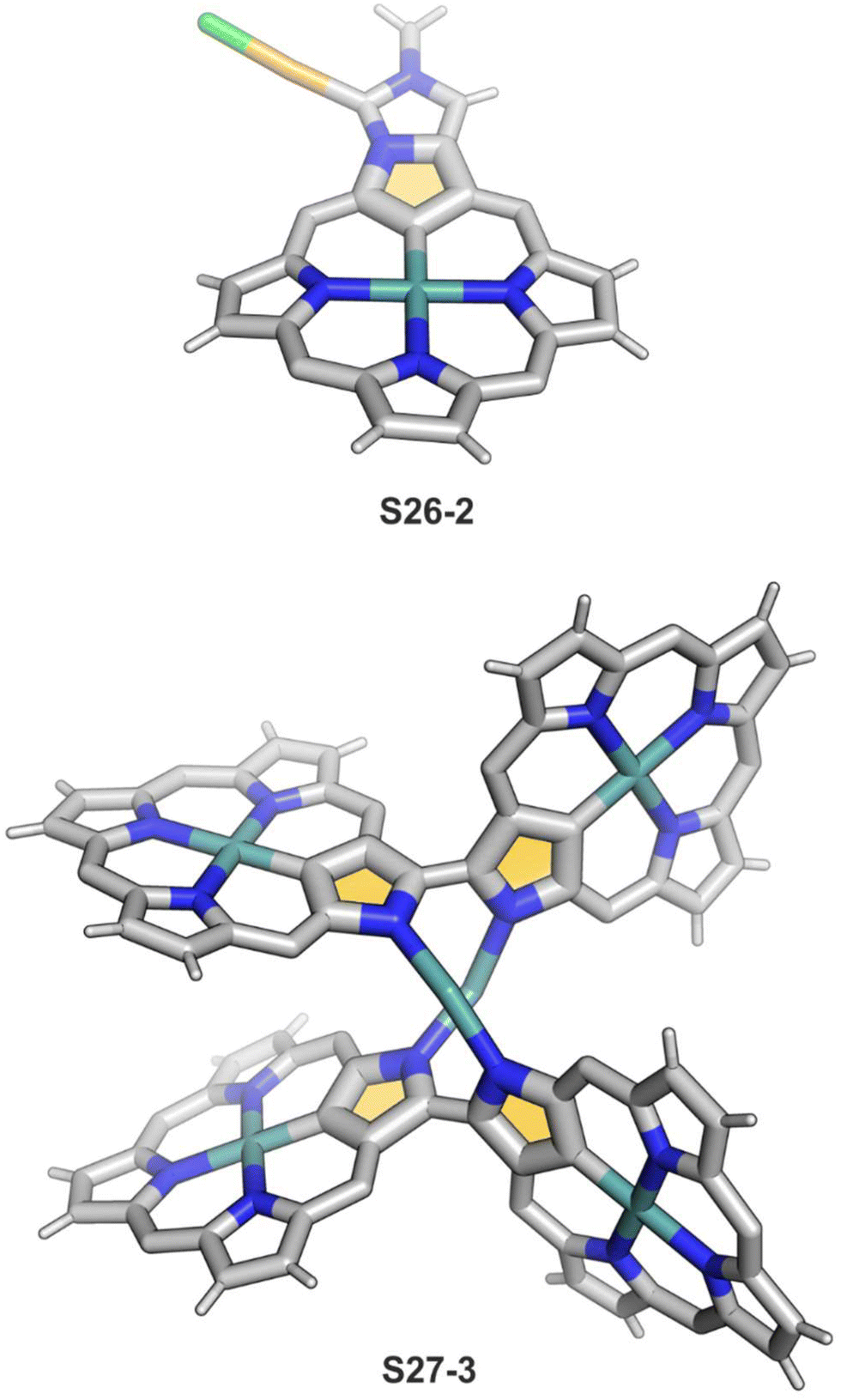 | ||
| Fig. 7 Specific silver(III) NCP coordination motifs. meso-Substituents not shown. | ||
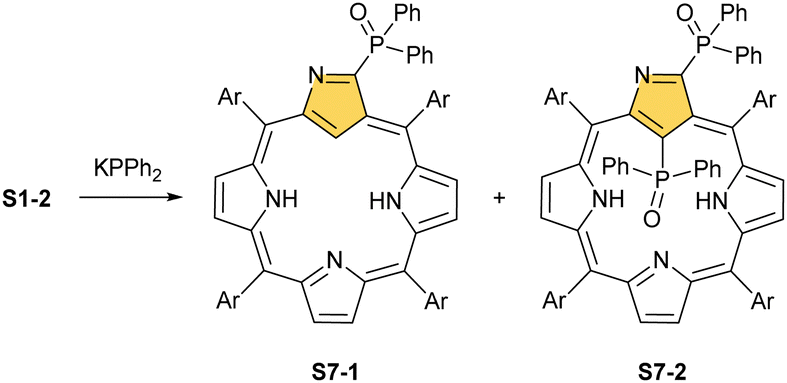
When silver(III) NCP S25-1 reacts with KPPh2, it gives C(21) substitution regioselectively with diphenylphosphine (S27-1, Scheme 27).64 At the same time, the macrocycle gets demetallated, probably due to the difficulties of a new form in stabilising AgIII. The substituent can be oxidised to a diphenylphosphoryl form. The interesting fact is that the C(21)–P bond is cleaved after the reinsertion of silver [from Ag(CF3COO)], meaning one returns to starting S25-1. Another example is a reaction with dimethylamine.101 After C(21) substitution with –N(CH3)2, the silver ion leaves the molecule. It was also shown that the outer N(2)C(3) bond in the silver(III) complex might act as an electrophilic site, and alcohol addition to the C(3) position is possible.102 Silver(I) trifluoroacetate-catalysed transformation yields a methoxy substituent after a reaction with ethanol and, per analogy, with different alcohols.
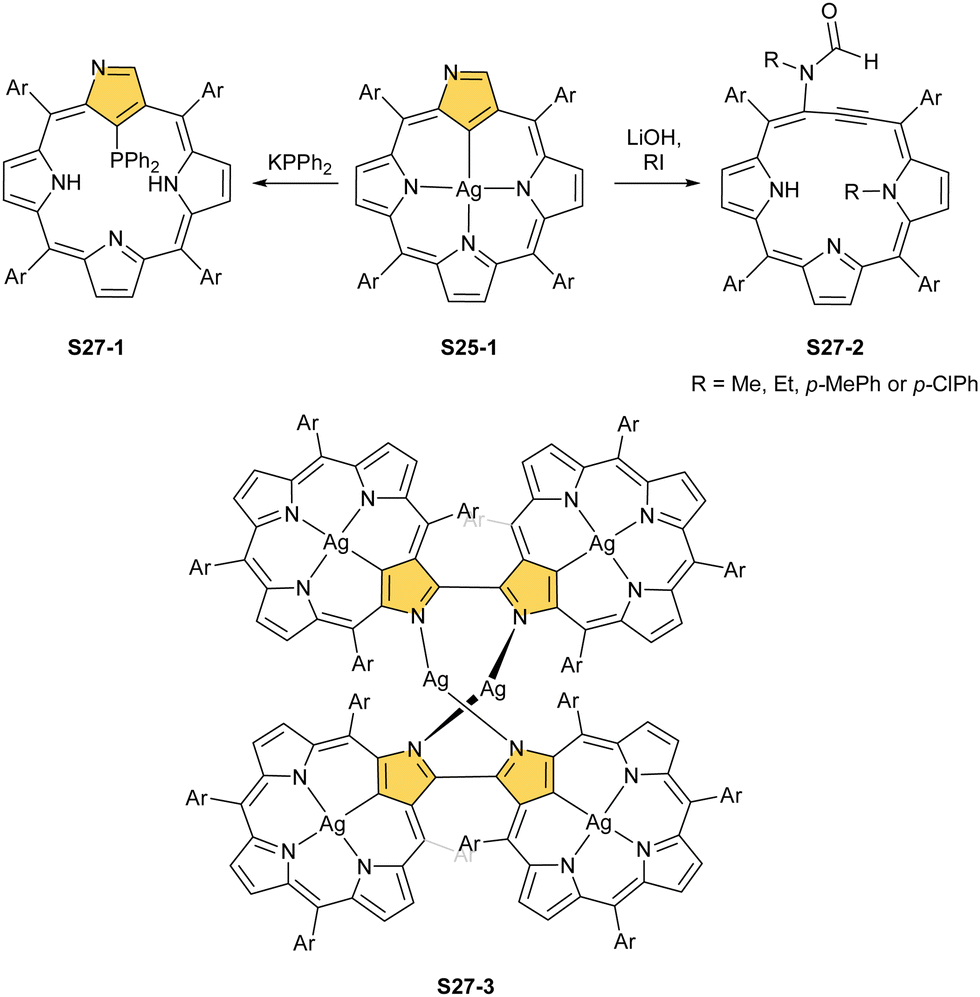 | ||
| Scheme 27 Reactivity of silver(III) N-confused porphyrin S25-1. | ||
An interesting transformation is induced when S25-1 reacts with methyl iodide and lithium hydroxide in THF.103 N(2) and N(22) get methylated, which is associated with the silver(III) centre removal. More importantly, the bond between the C(3) and C(4) atoms breaks, leaving the C(3) atom oxidised in a formyl functionality held merely by the N(2)–C(3) bond. The formed S27-2 remains cyclised with C(21) and C(4) atoms forming a triple bond. The so-called porphyrin remains aromatic with a −3.89 ppm chemical shift for the N(22) methyl group. Nickel(II) NCP and the free ligand did not exhibit the same type of reactivity.
Finally, the insertion of silver into C(3),C(3)′-dimer S6-2 using Ag(CF3SO3) results in unprecedented species.104 Besides the regular silver(III) insertion (two cavities of S6-2 occupied by the metal ions), the outer N(2) atom coordination is instrumental and shown in “the dimer of dimers” ensemble S27-3. Two perimetral silver(I) cations coordinate linearly two N(2) nitrogen atoms from the different dimeric subunits (Fig. 7). The cationic charge of S27-3 is balanced by two nonbonding counterions forming the [AgIII4AgI2L2] complex eventually.
The third metal completing the discussed coinage group, i.e., copper, is known for forming organometallic compounds adopting three fundamental oxidation states, i.e., copper(I), copper(II), and copper(III). Thus, its organometallic chemistry in the NCP environment varies significantly once related to the heavier counterparts – silver and gold.
The reaction of copper(II) acetate and NCP under mild conditions (inert atmosphere, THF, 298 K) results in the formation of paramagnetic square-planar copper(II) N-confused porphyrin S28-1 (Scheme 28) belonging to the small group of organocopper(II) species.105 The stabilisation of the copper(II) centre implies that the ligand frame acquires the geometry of tautomer S3-1 and N(2) nitrogen is protonated. Accordingly, 2-Me NCP S3-3, having already the appropriately prearranged molecular structure, readily coordinates to copper(II) to form S28-4. Protonation (HCl) or methylation (CH3I) of S28-1 occurs at the initially activated C(21) centre and is followed by the axial coordination of accompanying anions. Thus, in the case of S28-4, already N(2)-methylated, the inner core methylation at C(21) was detected, eventually generating copper(II) 2,21-dimethyl NCP S28-5. The reactive organometallic copper(II) NCP complexes (S28-1 and S28-4) gain additional stabilisation once perfluorophenyl substituents are located at meso positions.106 Such a complex (S28-1) was shown to undergo a swift and reversible one-electron metal-centred oxidation with DDQ to form diamagnetic square-planar copper(III) (S28-2).25 This is facilitated by switching to the 3H tautomer of NCP. On the other hand, the alternative insertion procedure (Cu(OAc)2, NCP, reflux in toluene, O2) provides harsh conditions that result in controlled degradation.
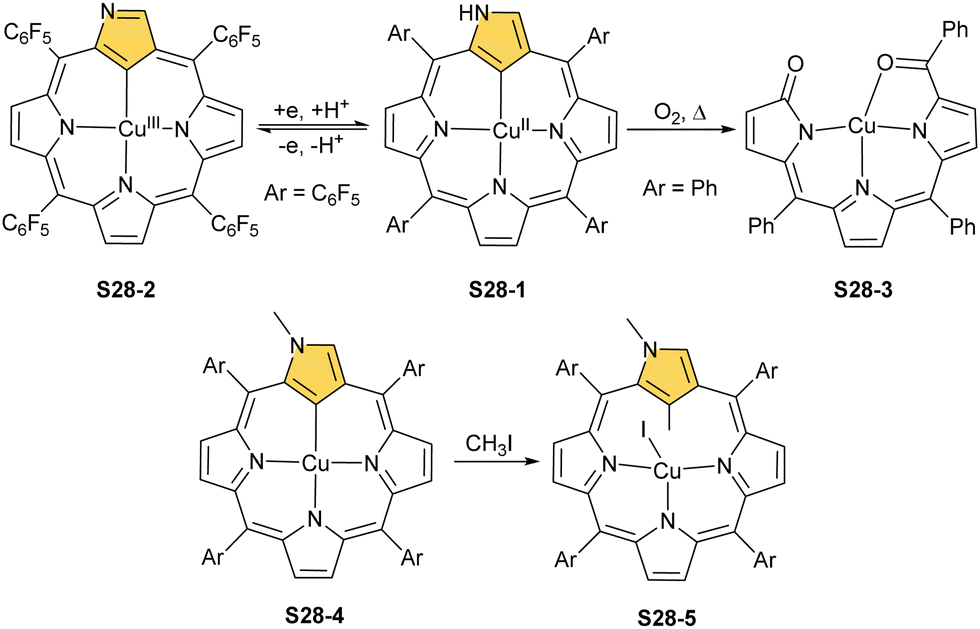 | ||
| Scheme 28 Reactivity of copper(II) N-confused porphyrin S28-1. | ||
Oxygenolysis of S28-1 includes cleavage of C–C bonds adjacent to the N-confused pyrrolic ring and eventually, this subunit linked to one meso phenyl is extruded from the framework.107 The remaining tripyrrinone-like fragment of S28-3 incorporates C(6) and C(20) carbon atoms of starting NCP, albeit transformed into the carbonyl units. In fact, the degradation proceeds regioselectively and introduces a larger structural transformation than the photooxidation of a free macrocycle.108,109 The tripyrrinone derivative conserves the coordination to copper(II), completing the fourth coordination site with the terminal C(20)–carbonyl oxygen atom. Oxygenation of copper(II) N(2)-methyl NCP S28-4 under milder conditions (reflux in a mixture of chloroform and acetonitrile, O2) results in an insertion of an oxygen atom into the Cu–C(21) bond affording copper(II) oxo-NCP (or 21-hydroxy NCP) S29-1 (Scheme 29).110
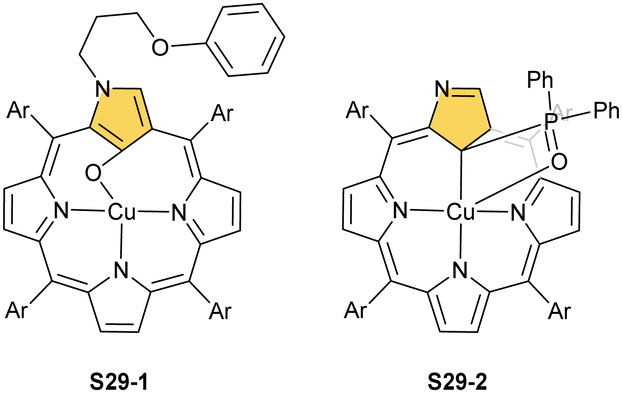 | ||
| Scheme 29 Copper(II) complexes of N-confused porphyrin derivatives. | ||
The CuII–C(21) distance increases to 2.530(3) Å, consistent with the side-on η1-type interaction. On the other hand, the regular CuII–O bond has been encountered (Cu–O(21) 1.900(2) Å). The O(21)–C(21) distance is relatively short (1.307(4) Å) but longer than the typical bond length of the regular carbonyl group. One can conclude that, in S29-1, the ligand preserves features of 21-hydroxy NCP. Finally, there is an example of copper(II) insertion into C(21)-substituted NCP. A diphenylphosphoryl functionalised porphyrin obtained in a reaction of [AgIII(NCP)], KPPh2 was used.111 The insertion occurs under mild conditions and, in contrast to silver, a metal complex with C(21) substitution is formed (S29-2). The C(21) atom gets rehybridised to sp3 and the CuII–C(21) contact is in a bonding range (2.154(3) Å; Fig. 8). The phosphoryl oxygen atom is located at the centre of porphyrin and forms an axial bond yielding a square pyramid coordination mode.
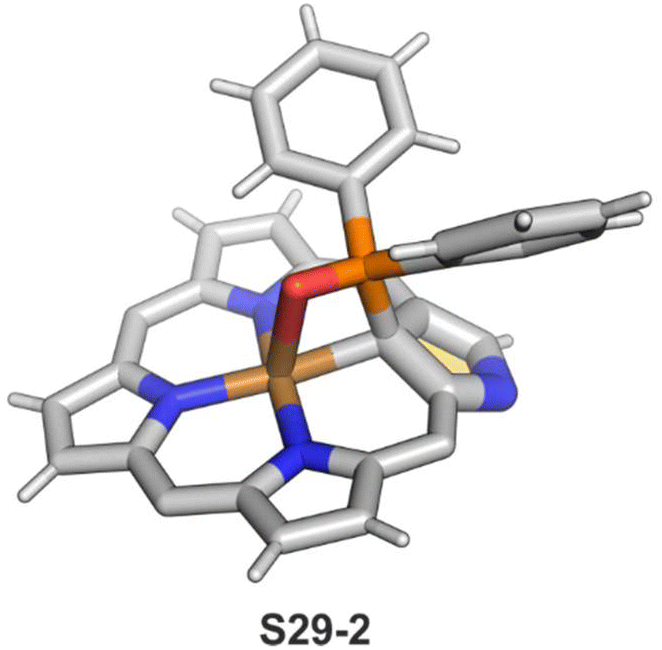 | ||
| Fig. 8 Copper(II) organometallic complex with C(21)-substituted NCP. meso-Substituents not shown. | ||
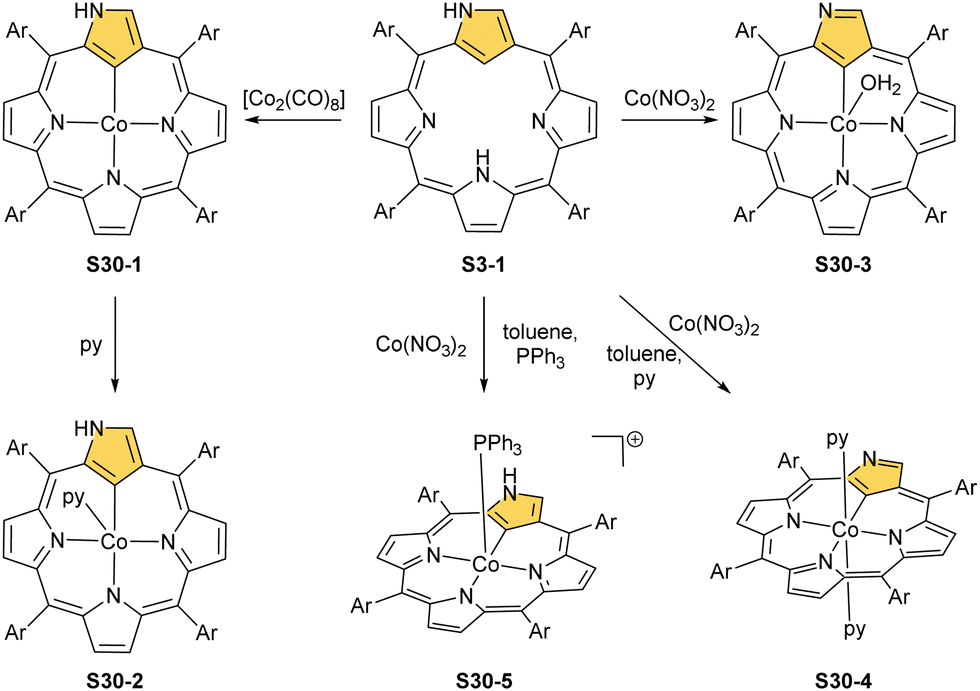 | ||
| Scheme 30 Cobalt(II) insertion into N-confused porphyrin S3-1. | ||
The cobalt(II) NCP S30-1 reacts with nitric oxide, which effectively binds as the fifth axial ligand.112 In this process, carried out in dichloromethane solution, a peculiar transformation occurs. The complex activates two solvent molecules and forms a C(21) 2-chlorovinyl substitution (S31-1, Scheme 31).
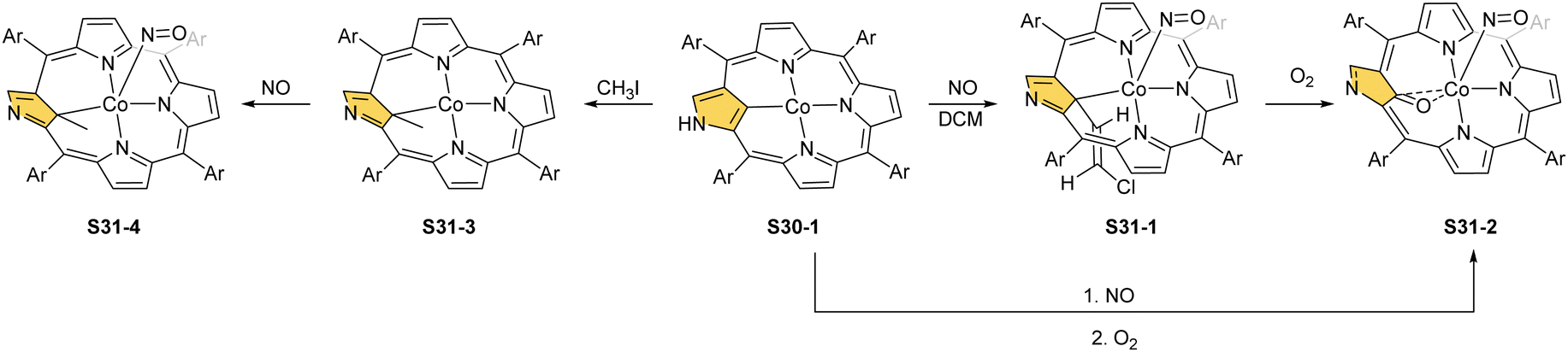 | ||
| Scheme 31 Reactivity of cobalt(II) N-confused porphyrin S30-1. | ||
The organometallic bond is preserved as illustrated by the Co–C(21) distance (2.046(5) Å); thus, C(21) becomes tetragonal (Fig. 9), and the electronic state of the centre is {Co(NO)}8. Prolonged NO bubbling through solutions of S30-1 and S31-1 results in oxygen atom insertion into the Co–C(21) bond of S30-1 associated, however, for S31-1 with the extrusion of a 2-chlorovinyl substituent. It was suggested that the incorporated oxygen atom came from NO contamination. The formed species (S31-2) can be characterised as a {Co(NO)}7 electronic state. The C(21)–O(21) distance suggests a double bond (1.301(9) Å) η2-interaction with the metal centre (Fig. 9). This is in line with Co–O(21) and Co–C(21) distances (2.051(5) and 2.175(8) Å, respectively). Treatment of S30-1 with methyl iodide produces a C(21)-methylated complex S31-3, which reacts in the next step with NO, forming a structural analogue of S31-1, i.e., S31-4 (Fig. 9).116 This {Co(NO)}8 complex can be then reduced to a stable {Co(NO)}9 anion using [Co(Cp*)2].
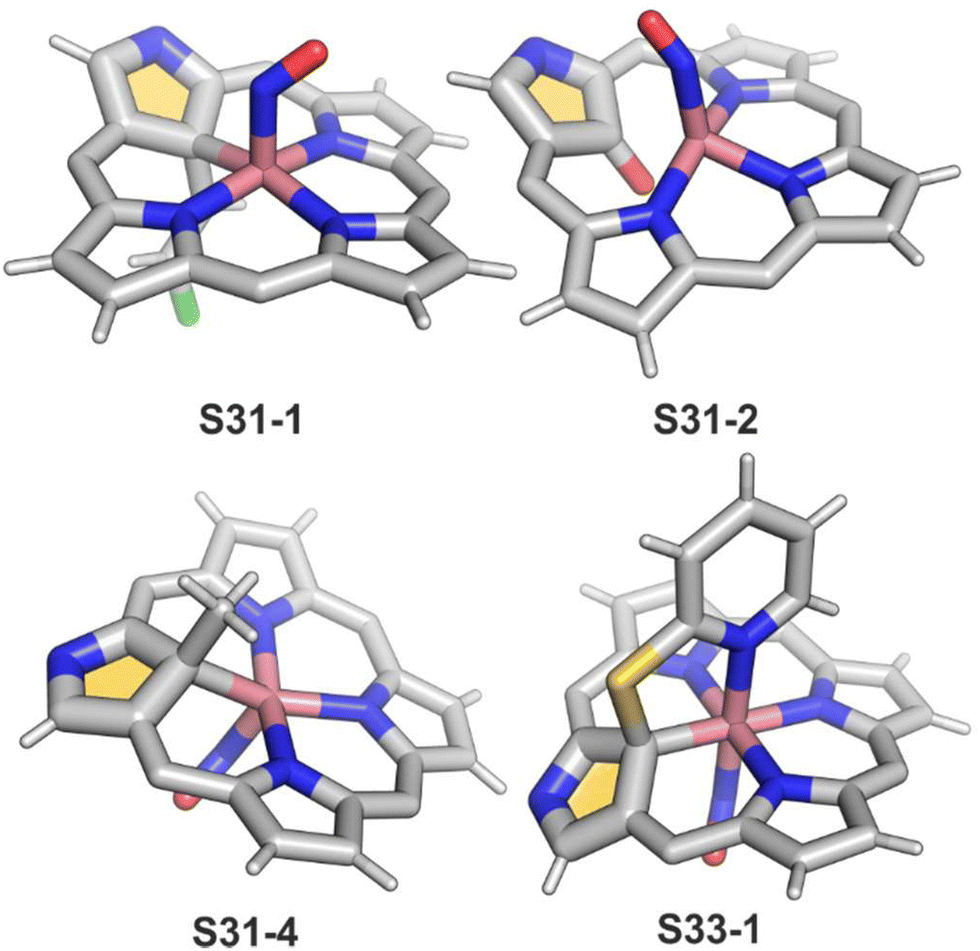 | ||
| Fig. 9 Cobalt-coordinated NCP derivatives. meso-Substituents not shown. | ||
The insertion of cobalt(II) into N(2)-substituted N-confused porphyrin S32-1 (Scheme 32) using Co(SCN)2 in a mixture of acetonitrile/toluene gives quite unexpected results, however, evidently controlled by the nature N(2) substitution.117 The p-xylyl substitution connected through one of the aliphatic carbon atoms leads to a product with a cobalt(II) centre coordinating to three nitrogen atoms and supplemented with SCN− for the charge balance. Thus, the inner C(21) atom links the benzyl moiety derived from toluene (S32-3). Switching N(2) substitution to methyl, benzyl, or ethoxycarbonylmethyl leads to C(21) cyanation (S32-2), assuming the cleavage of the former SCN− anion assisted by a cobalt cation.
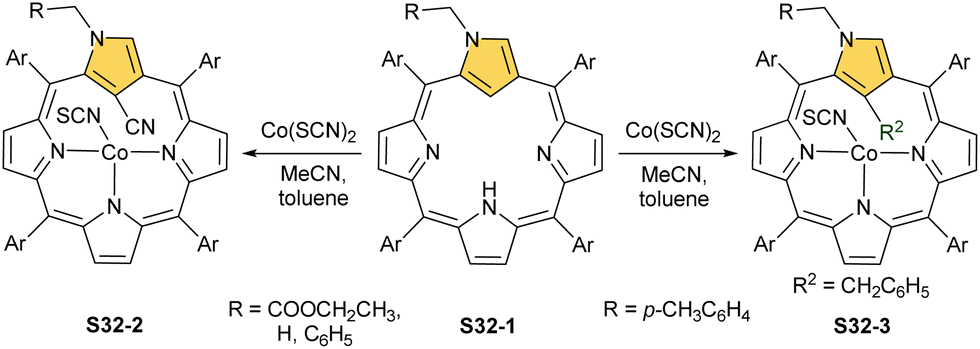 | ||
| Scheme 32 Cobalt(II) insertion into N(2)-substituted N-confused porphyrin S32-1. | ||
The Co–C(21) distances of S32-2 and S32-3 type species are in a range of 2.52–2.56 Å suggesting the effective side-on Co⋯C(21) interactions. Different charge distributions over C(21) caused by the N(2) substituents explain the identified differences in reactivity. When S32-1 containing the ethoxycarbonylmethyl N(2) substituent is used but with another cobalt(II) source, namely CoCl2, one can still obtain a C(21)-benzylated CoII complex via toluene activation, the structural analogue of S32-3 but with axially coordinated chloride.118,119 These results clearly outline that moderately subtle molecular changes in N-confused porphyrin substitution and/or of reaction conditions may result in remarkably different outcomes pointing at the richness of chemistry conceivable to be induced. These results also suggest that, at first, the C(21)–H bond is activated by adjacent cobalt(II) firmly located via coordination with three nitrogen donors. Then, a vulnerable inner C(21) atom undergoes substitution. The order of actions may also be reversed. The already mentioned reaction of a nickel(II) regular complex of NCP with mercaptopyridine leads to the C(21) substitution and demetallation (S16-6).33
Prearranged macrocycle S16-6 can be remetallated with cobalt(II) (reflux of S16-6 and Co(NO3)2 in THF) to the transient cobalt(II) complex. The subsequent one-electron oxidation produces six-coordinate cobalt(III) 21-substituted N-confused porphyrin S33-1 (Scheme 33) with a direct organometallic bond to an sp3-hybridised C(21) atom (Co–C(21): 2.022(4) Å, Fig. 9). The axial coordination is supplemented with a nitrogen atom from attached pyridine accompanied by the trans-located nitrite-N anion as the sixth ligand. Alternatively, S33-1 can be directly formed starting from cobalt(II) insertion into NCP in the presence of mercaptopyridine. Mechanistically, the process naturally involves activating the C(21)H unit, which precedes the formation of S33-1.
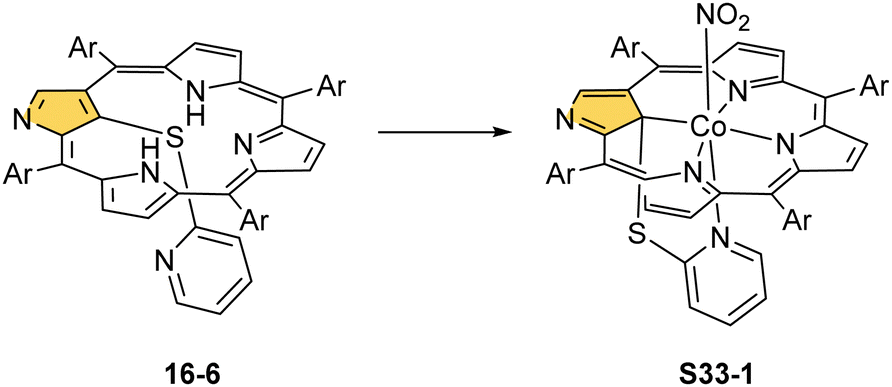 | ||
| Scheme 33 Preparation of cobalt(III) 21-substituted N-confused porphyrin S33-1. | ||
In contrast to cobalt, the heavier elements of group 9 of the periodic table, i.e., rhodium and iridium, demonstrated a preference for metal(I) and metal(III) oxidation states in NCP environments.
A product formed by reacting NCP and [RhCl(CO)2]2 (under relatively mild conditions of boiling dichloromethane) constitutes a bimetallic species (S34-2). None of the rhodium(I) cations managed to activate the internal C(21)–H bond; thus, one RhI centre is bound through N(22) and N(23) atoms and its coordination sphere is supplemented with two carbonyl groups (Fig. 10).120 The other centre is attached through the external N(2) donor and the coordination is completed by two carbonyl groups and one chloride to balance the rhodium(I) charge. By using only 0.5 eq. of the dirhodium source, selective outer-only coordination can be achieved (S34-1, Scheme 34).121
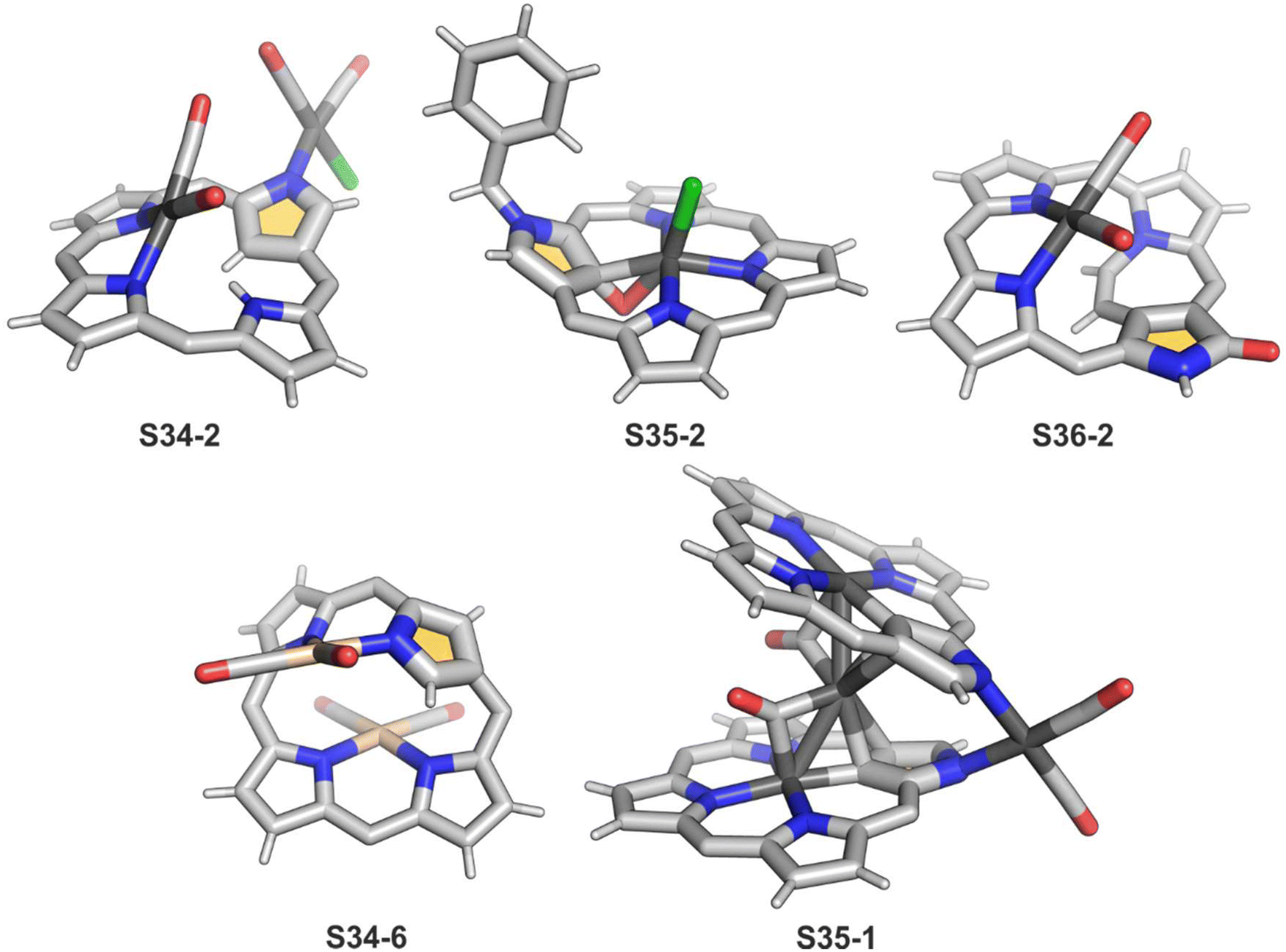 | ||
| Fig. 10 Rhodium and iridium coordination motifs. meso-Substituents of NCP not shown. | ||
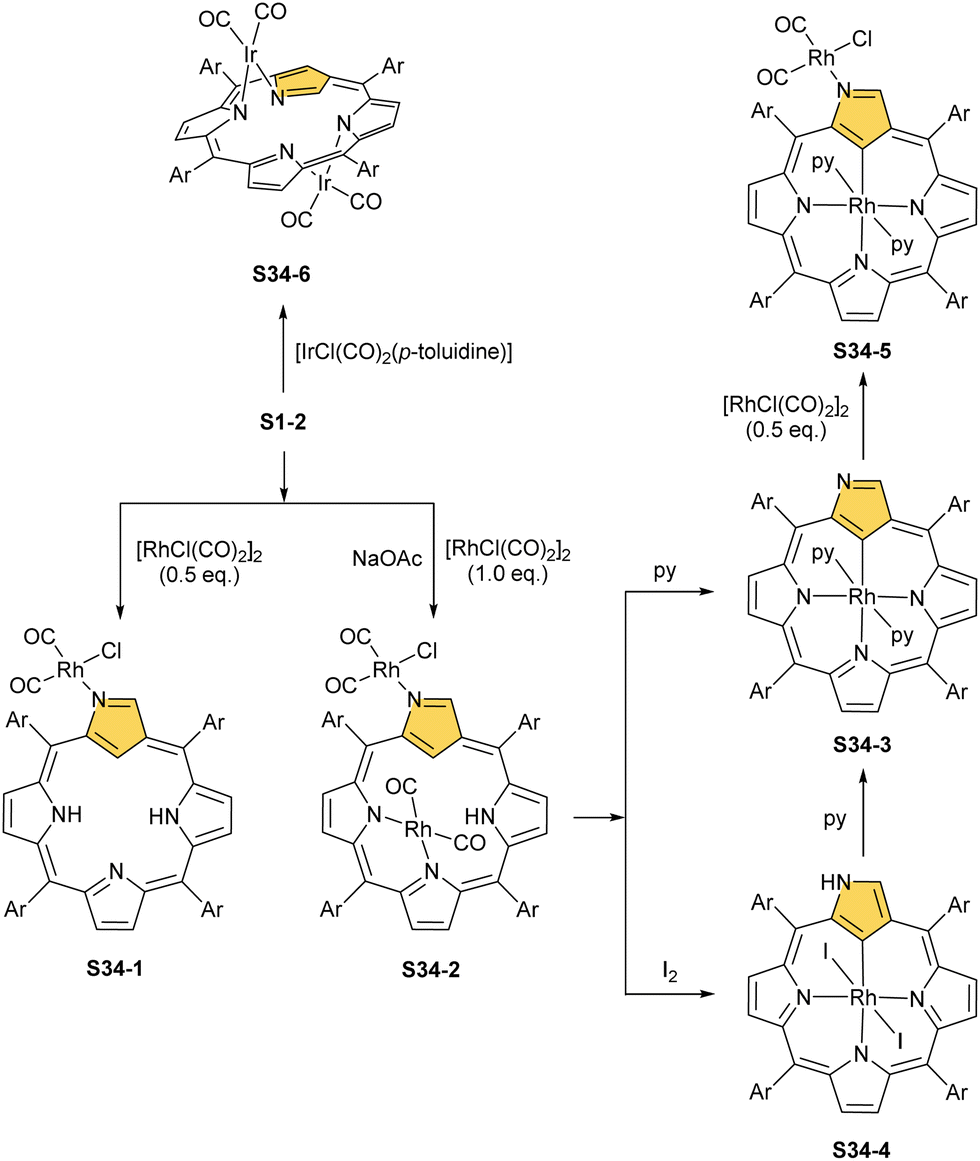 | ||
| Scheme 34 Iridium and rhodium N-confused porphyrin complexes. | ||
In a similar trial, a reaction of 2 eq. of [IrCl(CO)2(p-toluidine)] with NCP (in boiling toluene) gives two Ir(CO)2 fragments coordinated to two nitrogen atoms each (S34-6).122 In this case, a complex reminiscent of a dirhodium(I) complex with regular porphyrin120 is obtained. Such a coordination mode means one ion coordinates to N(22) and N(23) and the other one to N(24) and N(2) (Fig. 10). Surprisingly, the inverted ring flips to facilitate coordination, but no N-fusion is observed. The structure is confirmed by X-ray crystallography and through the observation of chemical shifts for protons H(21) and H(3) in 1H NMR spectra. As the macrocycle preserves aromaticity, H(21) relocates from −5.01 ppm to 10.11 ppm. The complex is stable and the conversion to a regular form with Ir–C(21) is not observed.
However, for rhodium, one is able to obtain a regular complex with an organometallic bond when the metal centre gets oxidised.121,123 Pyridine enables the oxidation of S34-2 to form monorhodium(III) species S34-3. The outer RhI ion is removed and the macrocycle acts as a trianionic ligand stabilising rhodium(III) accompanied by two axial pyridines. It is possible to transform to the bimetallic species when an additional 0.5 eq. of the dirhodium(I) source is added to isolated S34-3. It forms a mixed rhodium(III)/(I) system S34-5. A situation looks different when iodine is added to the solution of S34-2 at room temperature. The resulting complex becomes paramagnetic and possesses two axial iodide ligands, which suggests a rhodium(IV) centre (S34-4), while the porphyrin acts as a dianionic species. The reaction with pyridine initiates the reduction of the metal centre to rhodium(III) and yields diamagnetic S34-3 as a consequence.
Another fascinating construction (S35-1, Scheme 35) is formed when [RhCl(CO)2]2 reacts with NCP in boiling toluene.124 An identified [Rh4(NCP)2(CO)4] complex is a dimer bridged through Rh coordination; thus, there are three coordination motifs (Fig. 10): (I) the Rh1 ion binds to both N(2) and N(2)′ atoms introducing the stable N-confused porphyrin motif, (II) Rh2 and Rh4 cations inserted into C(21)NNN cores form the C(21)–Rh bonds, and (III) Rh3 with Rh2 and Rh4 (the Rh2⋯Rh3 distance is ca. 2.71 Å) afford an Rh3 cluster. The rest of the coordination sphere is filled up by carbonyl groups but also the η1 interaction of Rh3 with C(21) and C(21)′ atoms (2.262(5) and 2.214(5) Å). Only one isomer has been identified for S35-1. The total oxidation state of the multimetallic species must be fully stabilised by two trianionic macrocycles, as there are no counterions in the crystal lattice. Thus, a total of 6+ charge has to be distributed over four rhodium centres to yield a diamagnetic species.
A rhodium insertion in a boiling mixture of dichloromethane and acetonitrile into an N(2)-substituted NCP and without protection from oxygen results in a C(21) atom oxygenation in the final rhodium(III) complex (S35-2; Fig. 10).110 The Rh–C and Rh–O distances indicate direct η2 bonding (Rh–C(21) 2.170(3) and Rh–O(21) 2.043(2) Å) with an internal carbonyl (C(21)–O(21) 1.330(4) Å), similarly to an oxygenated complex with cobalt (S31-2).
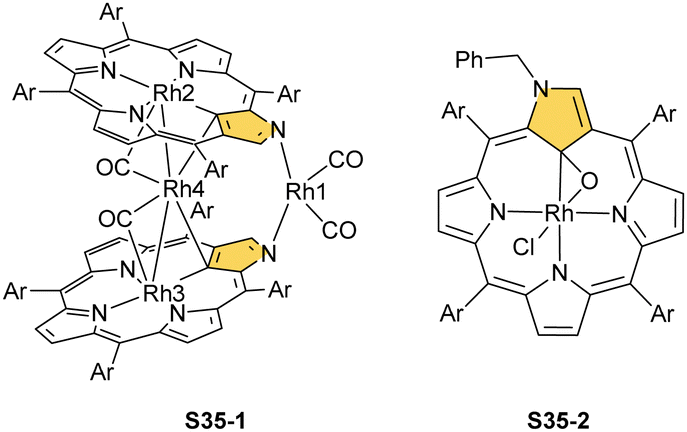 | ||
| Scheme 35 Rhodium complexes of N-confused porphyrin derivatives. | ||
Eventually, the internally locked system S11-3 was tested as a potential non-trivial molecular platform to accommodate rhodium(n).74 A reaction of S11-3 with [RhCl(CO)2]2 yields a dirhodium(I) complex S36-1 (Scheme 36), an analogue of S34-2. The double bond of the bridging ethene is preserved. The internally incorporated Rh(CO)2 moiety, linked to two pyrrolic nitrogen atoms, is located at the opposite porphyrinic side with respect to the ethene bridge. Interestingly, after treatment with silica gel, S36-1 transforms to S36-2via demethylation, forming a lactam moiety. This is accompanied by decoordination of an N(2)-attached centre (Fig. 10).
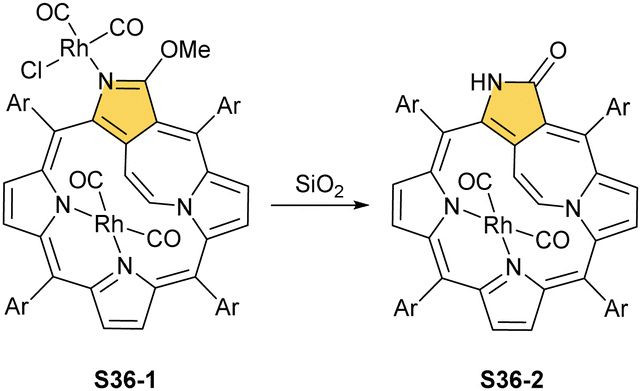 | ||
| Scheme 36 Rhodium(I) complexes of N-confused porphyrin derivatives. | ||
Iron(II,III) NCP and their mono-, dimethylated, or C(21)O derivatives were characterised by 1H and 2H NMR spectra, revealing the typical isotropic shift patterns of paramagnetic iron porphyrins in given oxidation and spin states.125–127 The high-spin bromoiron(II) NCP S37-1 (Scheme 37) displays a distinctively large isotropic shift of the inner H(21) atom (812 ppm, 298 K), which indicates that an FeII⋯(C(21)–H) agostic interaction also reflected in its molecular structure (Fig. 11).125 In the case of C(21)-methylated NCP derivatives, the characteristic C–Me resonances occur in a unique window (420–520 ppm) for iron(III) C(21)-methylated NCP, which remains in contrast to relatively small values found for iron(II) C(21)-methylated NCP (50–80 ppm).127
 | ||
| Scheme 37 Reactivity of iron(II) N-confused porphyrin S37-1. | ||
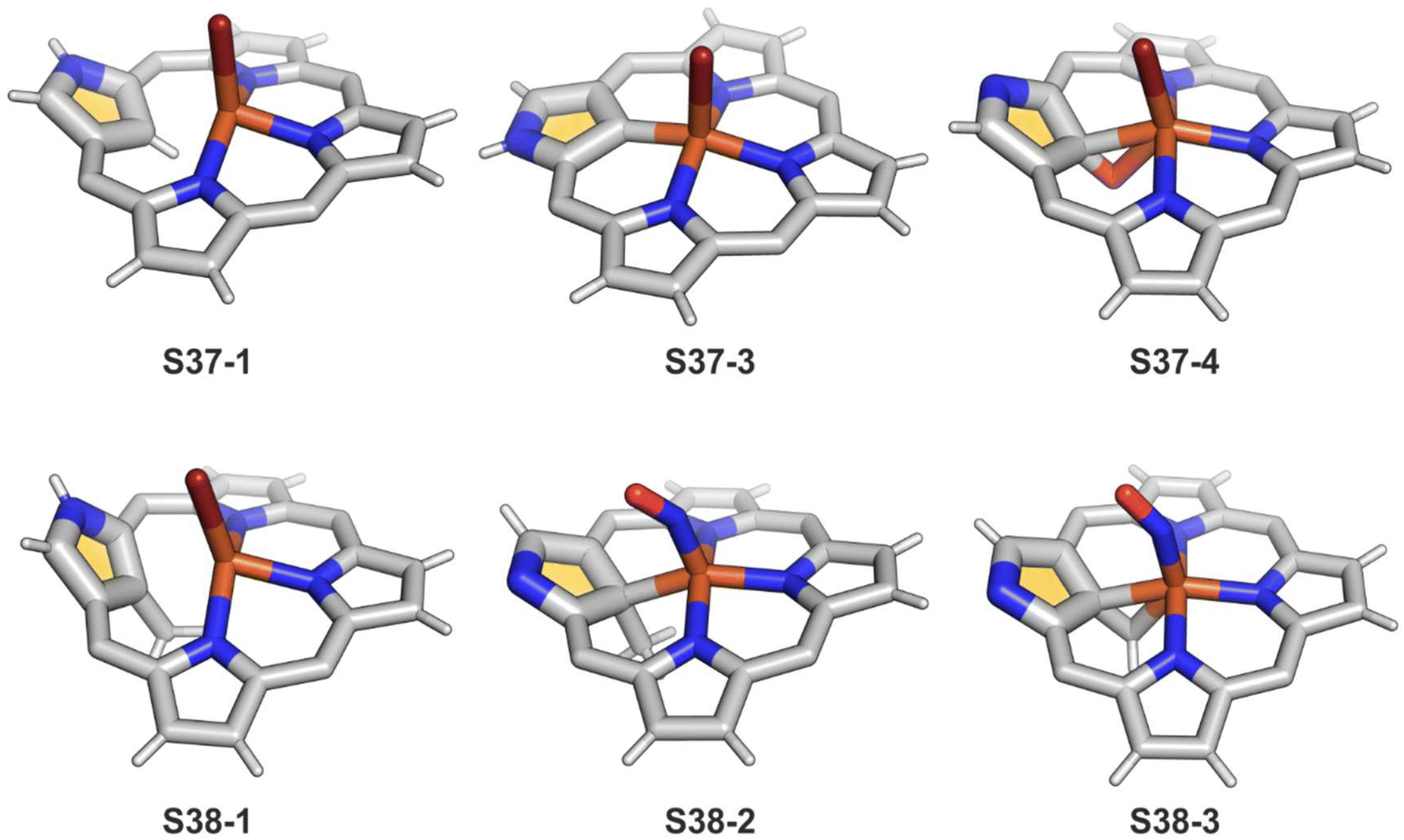 | ||
| Fig. 11 Iron coordination motifs for C(21)-substituted and regular NCP. meso-Substituents not shown. | ||
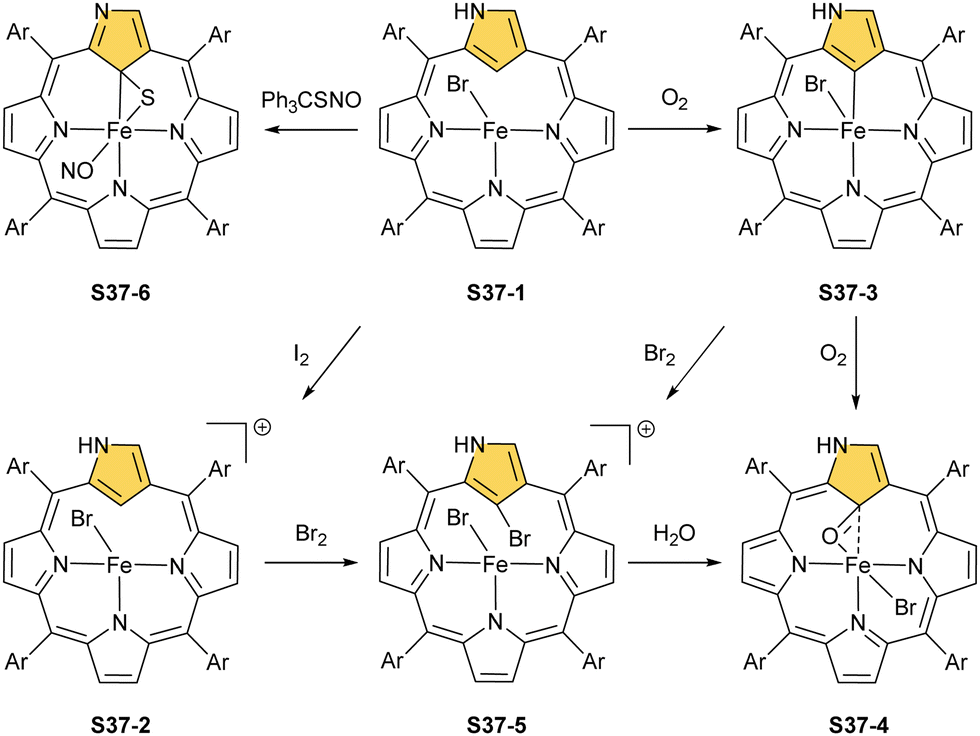
Aside from important spectroscopic findings, organometallic chemistry was also intensively explored. Oxygen-sensitive high-spin iron(II) NCP S37-1 is formed in a reaction of NCP with FeBr2 heated in a mixture of MeCN/THF.128 Iron(II) coordinates three inner nitrogen atoms and one axial bromide. The coordinated NCP is in its S3-1 tautomeric form, having protonated N(2). The framework constraints result in the mentioned enforced equatorial agostic interactions with C(21)–H as confirmed by a close Fe⋯C(21) distance of 2.361(10) Å. A replacement of a bromide anion with a thiolate ligand in a reaction of S37-1 and sodium 4-methylbenzenethiolate produces a structurally related compound with a comparable Fe⋯C(21) distance of 2.398(3) Å.
The treatment of S37-1 with iodine causes the one-electron oxidation of iron(II) to produce iron(III) NCP S37-2. Iron(II) NCP S37-1 exposed to dioxygen quickly transforms, at first, into five-coordinate intermediate-spin iron(III) NCP S37-3, creating a direct Fe–C(21) σ-bond (Fig. 11).126S37-2 demonstrates sensitivity toward dioxygen as well. The further exposure of S37-3 results in an oxygen atom insertion into the Fe–C(21) bond, forming high-spin iron(III) C(21)-oxygenated NCP S37-4, similarly as presented already for copper, cobalt, or rhodium NCPs. The bond distances (Fe–C(21) 2.199(7), Fe–O(21) 2.041(5), and C(21)–O21 1.299(7) Å) point at the iron(III) η2 interaction with the internal carbonyl group (Fig. 11). The addition of Br2 to the solution containing S37-2 or S37-3 results in bromination at C(21), yielding S37-5 in accord with typical N-confused porphyrin reactivity. The subsequent reaction with water finally results in forming the mentioned C(21)O fragment (S37-4). The demetallation of S37-4 with strong acid affords 21-hydroxy NCP.129 The X-ray structure of the isolated free base is disordered over the inversion centre, but it points at a C(21)–OH rather than a C(21)O distance (1.497(5) Å).
Incorporating a sulphur atom into Fe–C(21) was also documented. It is achieved in the reaction of S37-1 with S-nitrosotriphenylmethanethiol giving S37-6.130 An NO molecule dissociates from nitrosothiol and is captured as an axial ligand replacing bromide. The transformation also involves a C–S bond cleavage followed by the incorporation of a liberated sulphur atom into the Fe–C(21) bond. S37-6 is diamagnetic and acquires the {Fe(NO)}6 electronic structure. The C(21) atom acquires the tetrahedral geometry and forms a stable organometallic bond as the Fe–C(21) distance equals 2.051(6) Å. The C(21)–S(21) bond length equals 1.763(5) Å being close to a single bond. The Fe–S(21) distance (2.2432(17) Å) confirms the binding of the thiolate-like sulphur atom to the iron centre. Thus, species S37-6 incorporates the ferrathiacyclopropane motif into its structure. Similar chemistry was observed for S38-1 (Scheme 38) (Fig. 11) – the C(21)-methylated analogue of S37-1.131
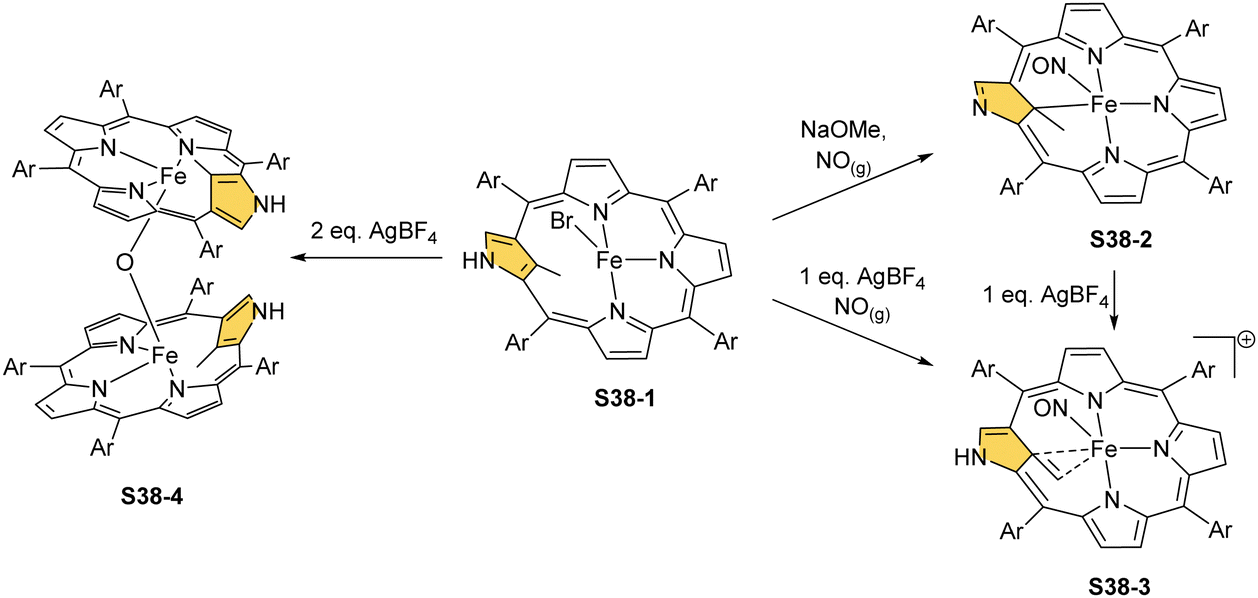 | ||
| Scheme 38 Reactivity of iron(II) 21-methyl N-confused porphyrin S38-1. | ||
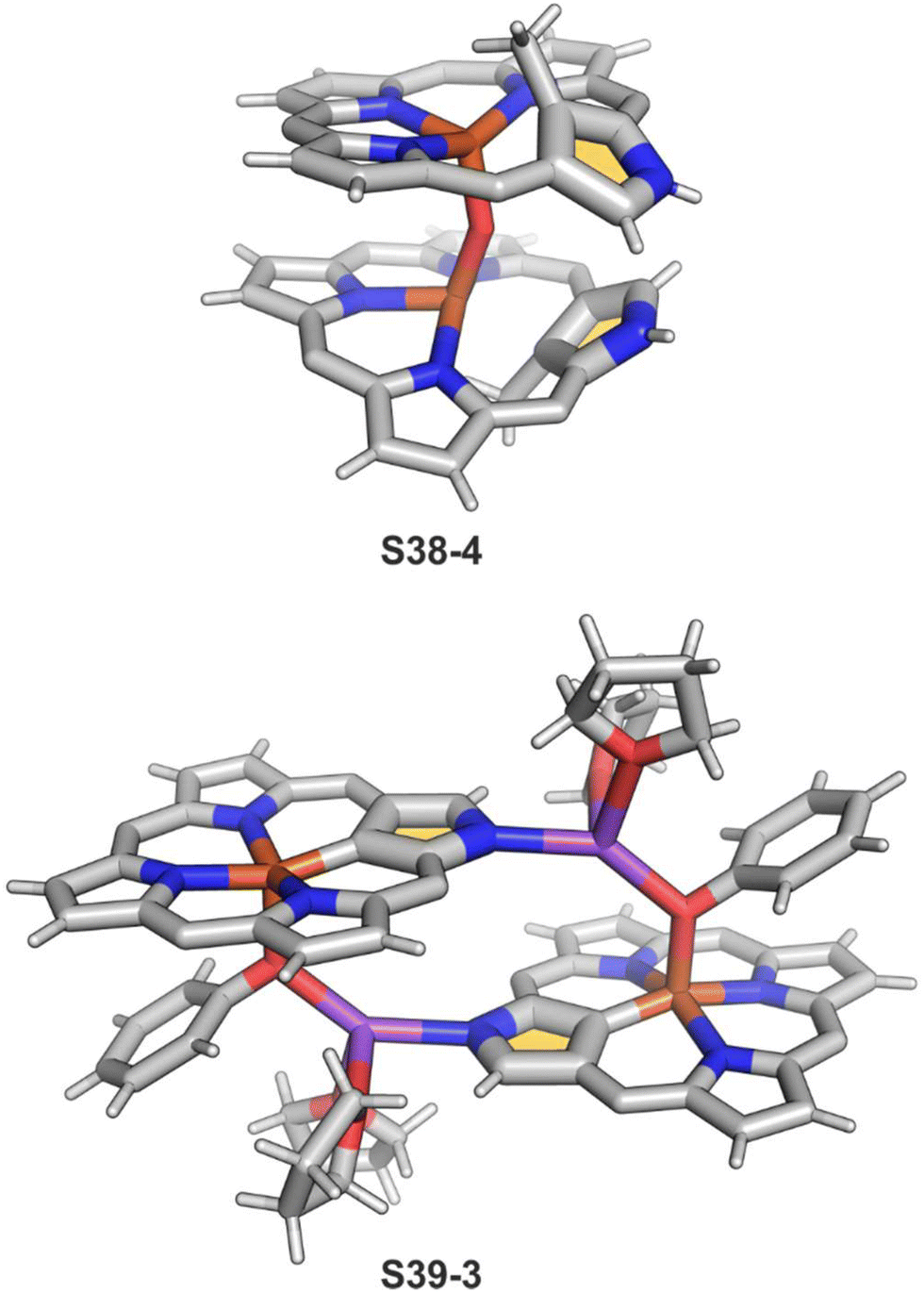
The replacement of bromide with NO triggers the subsequent rearrangement leading to the formation of S38-2 with a characteristic direct Fe–C(21) σ-bond (2.072(6) Å) and a {Fe(NO)}7 configuration (Fig. 11). After the gentle oxidation of S38-2 with AgBF4, activation of the inner methyl occurs and a double bond between CH2 and C(21) forms with a length equal to 1.439(4) Å (S38-3; Fig. 11). The short contacts with the metal centre suggest that the formed alkene unit participates in an η2 type π-bonding (distances of Fe–C(21) and Fe–CH2 are equal to 2.103(3) and 1.974(3) Å). The reaction of iron(II) 21-Me NCP S38-1 with two equivalents of AgBF4 under anaerobic conditions yields an air-sensitive product which, after exposure to air, transforms into a μ-oxy-bridged iron(III) dimeric complex [(FeIIINCP)2O](BF4)2S38-4 (Fig. 12). Two antiferromagnetically coupled high-spin iron(III) centres are bridged by the oxygen dianion. However, there is no direct organometallic σ-bonding as the distances for C(21)⋯Fe contacts are 2.345(8) and 2.319(7) Å in both subunits.
 | ||
| Fig. 12 Bridged dimeric iron NCP complexes. meso-Substituents not shown. | ||
Over the years, other synthetic routes were successfully probed to construct the molecular system containing two iron N-confused porphyrin subunits. Thus, one can abstract the H(21) atom and axial bromide from bromoiron(II) NCP S37-1 with sodium benzeneselenate.132 This opens an opportunity to form a plain head-to-tail type [(FeIINCP)2] dimer S39-1 (Scheme 39). Here, the outer N(2) and N(2)′ nitrogen atoms act as the donors creating the typical iron(II)–N bonds with the iron centres form twin subunits in a similar fashion as previously encountered for the zinc(II) [(ZnIINCP)2] analogue S14-2. The C(3)N(2) and C(3)′N(2)′ vectors in S39-1 point in opposite directions, which remains in contrast to the mutual arrangement of zinc(II) NCP or cadmium(II) NCP subunits in the respective dimers.78
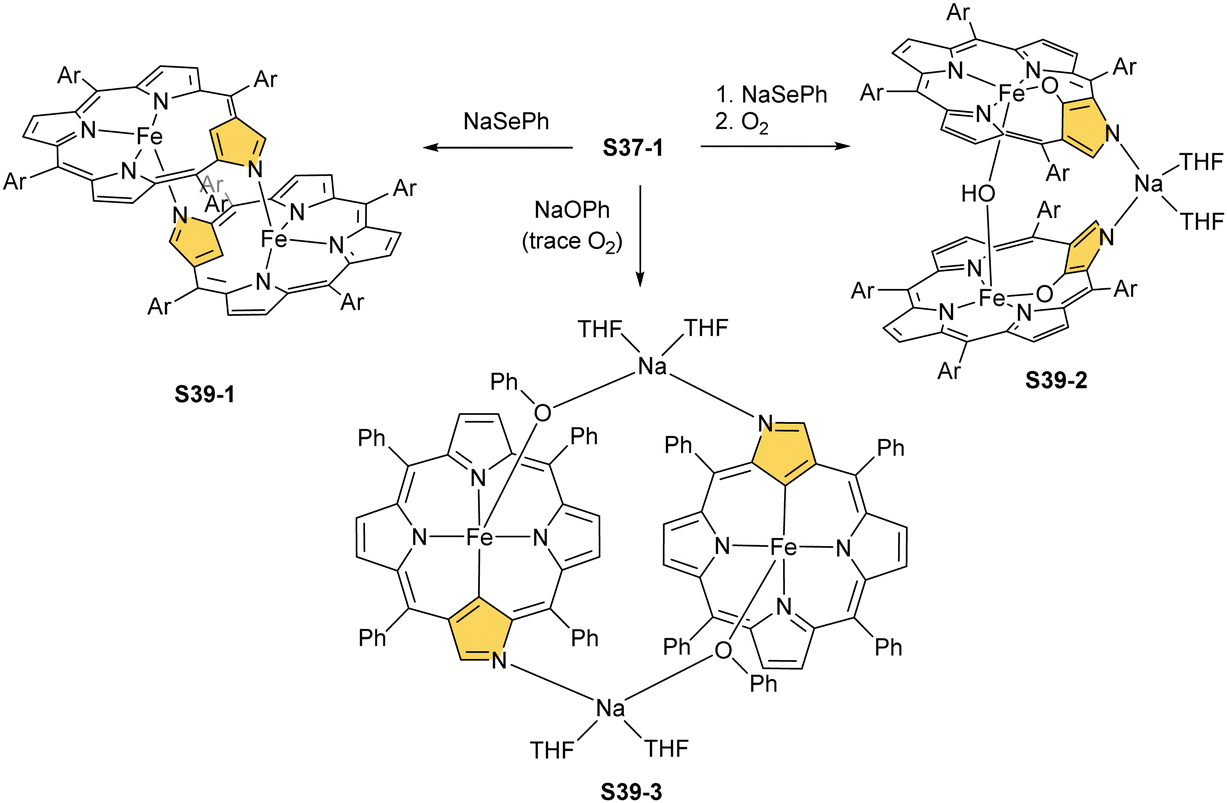 | ||
| Scheme 39 Reactivity of iron(II) N-confused porphyrin S37-1 with NaSePh and NaOPh. | ||
S39-1 is highly reactive towards dioxygen as its exposition to air results in the oxidation of iron(II) to iron(III) as well as in the oxygenation of C(21) positions. It eventually generates a diiron(III) species (S39-2). The fundamental dimeric arrangement was conserved, though the μ-hydroxy bridge connecting two iron(III) ions is incorporated. The additional linkage is identified in the structure of S39-2 as a Na(THF)2+ cation that coordinates N(2) and N(2)′ atoms. The bond pattern of the oxygenated N-confused pyrrolic fragment of S39-2 (Fe–O(21) 1.957(4) Å, Fe–C(21) 2.297(6) Å, and C(21)–O(21) 1.336(6) Å) seems to reflect a framework of 21-hydroxy NCP. The reactivity of bromoiron(II) NCP S37-1 was also tested in the presence of sodium phenolate under strictly anaerobic conditions.133 A replacement of bromide with phenoxide is initially detected. Introducing trace amounts of dioxygen into the reaction mixture is sufficient to carry iron(II) to iron(III) one-electron oxidation. It is still insufficient for feasible oxygen incorporation into the Fe–C(21) bond or the formation of μ-oxy (μ-hydroxy) bridges. The formed complex S39-3 captures each iron(III) centre in a square pyramid mode with a direct Fe–C(21) bond (1.950(5) Å; Fig. 12). Two FeIIINCP subunits are symmetrically bridged with phenolate coordinated to the iron(III) centre as well as a sodium cation interacting with the phenolate oxygen atom and an N(2) atom of the second subunit.
Substantial progress in ruthenium N-confused porphyrin chemistry has been recently noted. The formation of regular organometallic ruthenium(II) N-confused porphyrin S40-1 (Scheme 40) was initially described. Insertion is performed in boiling chlorobenzene using triruthenium dodecacarbonyl [Ru3(CO)12] as a metal source.33S40-1 contains an axially coordinated carbonyl ligand and pyridine after its subsequent addition. The Ru–C(21) distance of 2.015(5) Å clearly shows direct bonding. The inner carbon is activated, as demonstrated in a reaction of S40-1 with 2-mercaptopyridine. The reaction involves the formation of the covalent C(21)–S bond and trigonal to tetrahedral C(21) rehybridisation accompanied by pyridine side-arm coordination generating S40-2. The Ru–C(21) distance (2.118(3) Å) is still in a bonding range. The oxidation of S40-2 with m-chloroperoxybenzoic acid yields the sulphone derivative S40-3, resembling the analogous cobalt N-confused porphyrin chemistry.134 Also, 2-amino- and 2-hydroxypyridine react with S40-1 generating coordinating side-arms.134 Interestingly, in contrast to a mercapto derivative, they form a C(21)–N bond through a nitrogen atom from pyridine. The oxygen or nitrogen donors derived from the original 2-amine or 2-hydroxy substituents of pyridine coordinate to the ruthenium(II) centre in the typical side-arm geometry. In the case of the oxygen atom in S40-4, the C–O bond length is 1.264(4) Å, suggesting a double bond and dearomatization of a pyridine ring. A similar rearrangement has been detected in the case of S40-5 in which the structural parameters of the side arm reflect the frame of pyridin-2-imine (CNH bond: 1.305(3) Å). The characteristic of S40-5, the upfield relocated resonance of the NH hydrogen atom, has been unambiguously localised in the 1H NMR spectrum of S40-5 at 0.16 ppm.
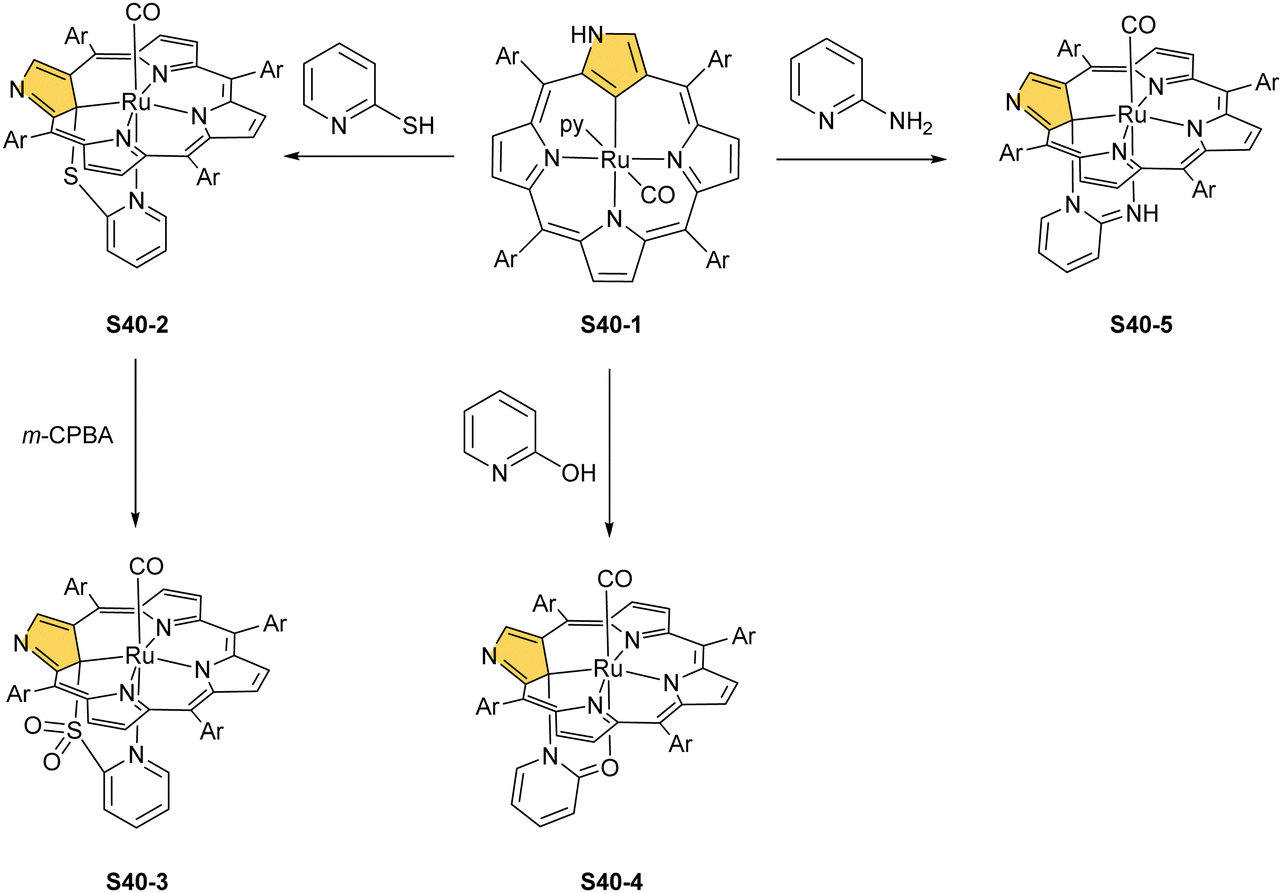 | ||
| Scheme 40 Reactivity of ruthenium(II) N-confused porphyrin S40-1. | ||
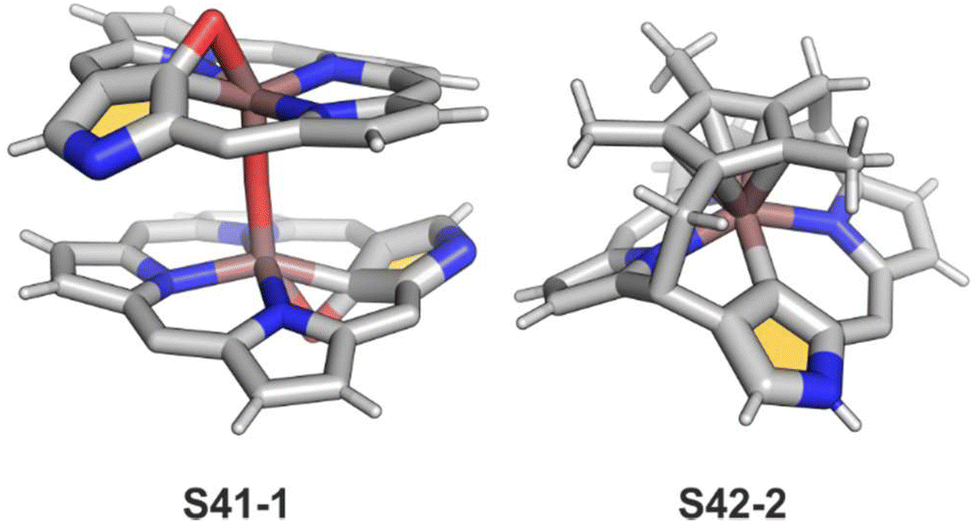
Oxidation of ruthenium(II) porphyrin S40-1 using (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) carried out under an argon atmosphere (in refluxing o-dichlorobenzene) affords two diamagnetic, isomeric ruthenium(IV) N-confused porphyrin μ-oxy-bridged complexes S41-1 (Scheme 41) and S41-2. They differ solely by alignments of the N(2)C(3) and N(2)′C(3)′ vectors.135 The C(21) environments in S41-1 and S41-2, as determined by X-ray crystallography (Fig. 13), are only slightly tetrahedrally distorted. The bond distances (e.g., in S41-1 Ru–C(21) 2.121(7) Å, Ru–O 1.988(5) Å, C(21)–O 1.371(7) Å, C(4)–C(21) 1.462(7) Å, and C(1)–C(21) 1.461(8) Å) point a borderline situation between a direct bonding and the η2 interaction between the CO moiety and the ruthenium(IV) centre resembling the relevant structural features of the respective iron(III) 21-oxo N-confused porphyrin S37-4 or {Co(NO)}7 21-oxo NCP S31-2.
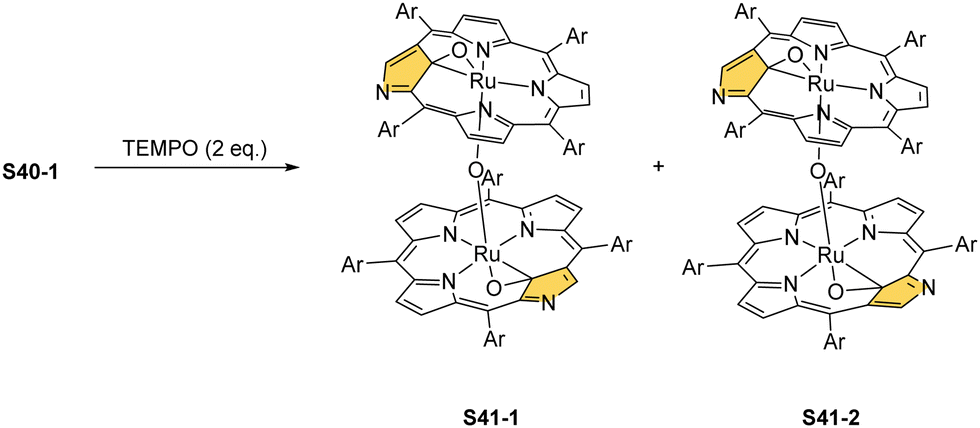 | ||
| Scheme 41 Oxidation of ruthenium(II) N-confused porphyrin S40-1. | ||
 | ||
| Fig. 13 Ruthenium coordination motifs. Regular meso-substituents not shown. | ||
Complexes of a quite distinct molecular architecture are formed once [RuCp*(CH3CN)3]PF6 is used as a ruthenium source (Cp* – pentamethylcyclopentadiene).136 Mixing it with NCP and refluxing the dichloroethane solution result in N-confused porphyrin incorporating the [RuCp*] π-coordinated meso-phenyl ring S42-1 (Scheme 42). The process is regioselective as the meso-phenyl ring adjacent to N(2) is solely involved in π-coordination. The outer nitrogen donor seems instrumental in providing regioselectivity, presumably stabilising the intermediate form which directs the final step, i.e., π-coordination. The protection of meso-aryls with bulky substituents (3,5-di-tert-butylphenyl rings) redirects the reaction to build the transient ruthenium(IV) sitting-atop structure. The ruthenium(IV) coordination forced the adjacency of Cp* methyl groups and two trans meso carbon atoms [C(5) and C(15)] of the NCP skeleton. This imposed vicinity induced a peculiar reaction in which two methyl groups of Cp* formed two regular covalent C–C bonds with proximal C(5) and C(15) meso carbon atoms. Eventually, the process results in sitting atop ruthenocenophane S42-2, described alternatively as ruthenium(IV) N-confused calix[4]phyrin strapped with a cyclopentadienyl ligand (Fig. 13). Ruthenium(IV) coordinates all inner donor atoms, including C(21) (the Ru–C(21) distance: 2.065(4) Å).
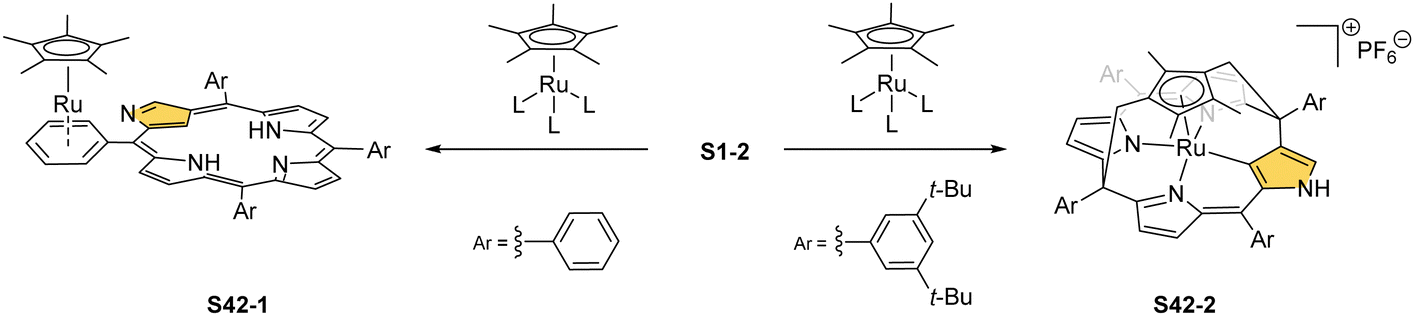 | ||
| Scheme 42 N-confused porphyrins containing [RuCp*] motifs. | ||
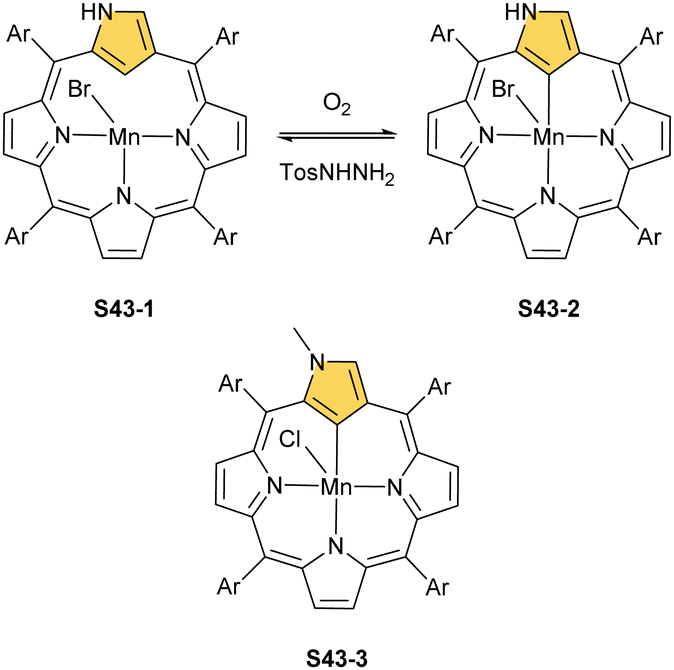 | ||
| Scheme 43 Manganese(II)/(III) complexes of N-confused porphyrin and 2-methyl N-confused porphyrin. | ||
Manganese(II) NCP may also acquire a dimeric form in the absence of additional ligands contending for axial positions.138 Thus, dimer S44-1 (Scheme 44) is directly formed as a product of a reaction of NCP and [Mn2(CO)10] in toluene. Similar to other dimers, the fourth coordination site of the first [Mn(NCP)] building block is occupied by the N(2)′ atom of the second one imposing the head-to-tail arrangement of subunits. Subsequently, dimer S44-1 can be cleaved when a strong ligand, for example, pyridine, competes for the axial position to eventually form monomeric monopyridine species S44-2. The species S44-1, exposed to dioxygen, undergoes one-electron oxidation to produce S44-3. Manganese(II) to manganese(III) oxidation is accompanied by the formation of the regular [CNNN] donor set, similarly as described for S43-2. Organometallic manganese(III) N-confused porphyrin S44-3 incorporates an equatorial trianionic ligand, which conserves features of the NCP tautomer S1-2. Two axial pyridine ligands complete the coordination sphere of manganese(III). The manganese(III) 2-methyl NCP (S43-3) is obtained directly in a reaction of 2-methyl NCP and manganese(II) acetate, followed by ligand exchange with sodium chloride.139 A prolonged heating of dimeric S44-1 with [Mn2(CO)10] leads to the ligand-centred reduction of a single subunit involving hydrogenation at C(10) and C(20) meso positions affording a heterogenous dimanganese(II) ensemble S44-4. It is noteworthy that the opposite directionalities (trans vs. cis) of N(2)C(3) vectors have been identified for S44-1 and S44-4 in the solid-state structures.
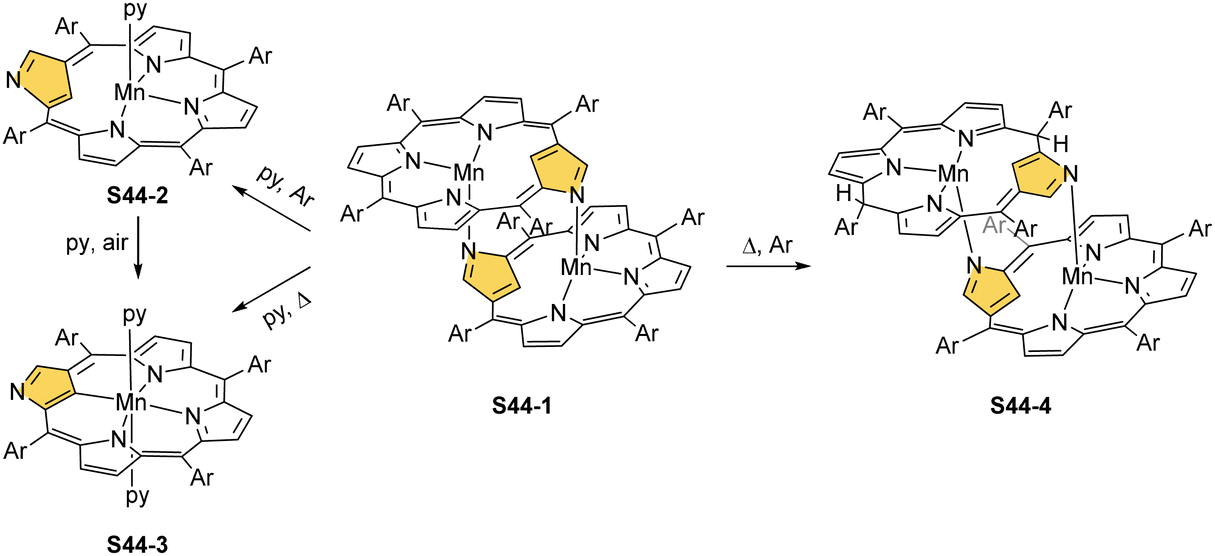 | ||
| Scheme 44 Reactivity of the manganese(II) NCP dimer S44-1. | ||
In a reaction of a manganese(I) source, [MnBr(CO)5], and 21-substituted NCP (in the presence of K2CO3), the manganese(I) N-fused porphyrin S45-1 (Scheme 45) is synthesised instead of the expected regular manganese NCP.72 NCP with electron-withdrawing C(21)-substituents like NO2 and Cl produces mostly not NFP but a manganese(II) dimeric complex S44-1 with C(21)-substituents detached.
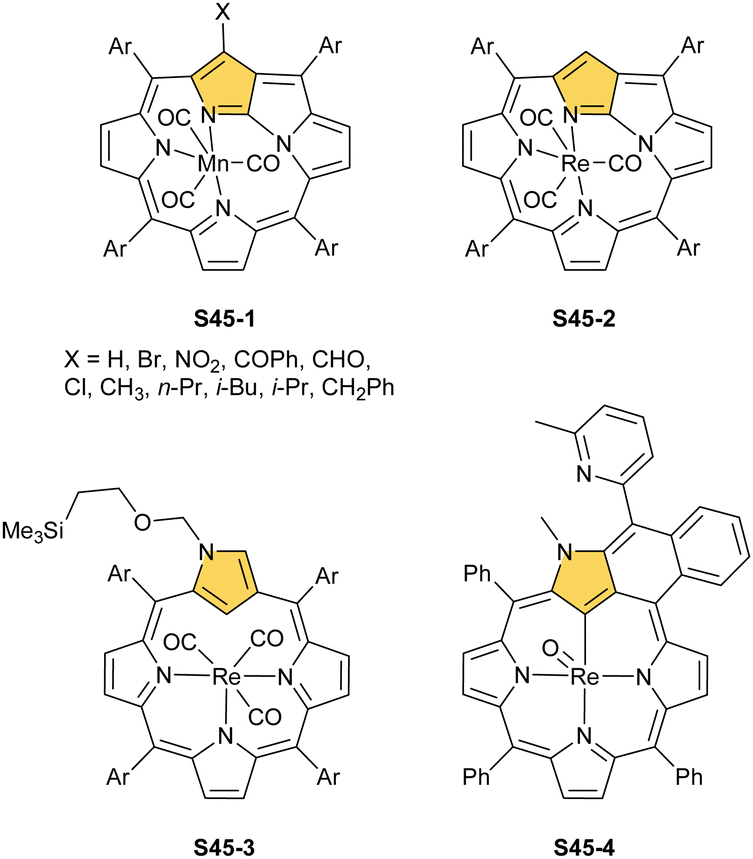 | ||
| Scheme 45 Manganese(I) and rhenium(I)/(V) complexes of N-confused porphyrin derivatives. | ||
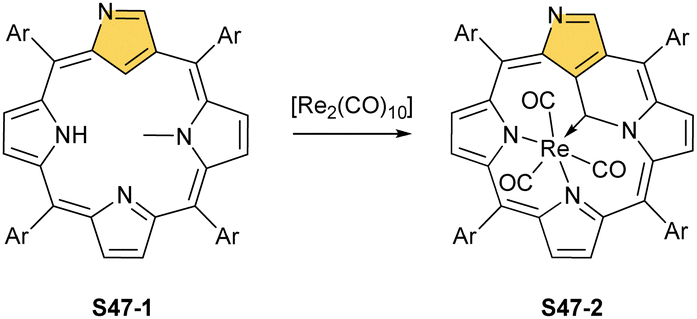
Similar reactivity, as reported for manganese(I) NCP, has been documented for manganese's heavier counterpart – rhenium. An attempt to insert rhenium into NCP using [Re2(CO)10] results in the formation of the rhenium tricarbonyl complex bearing N-fused ligand S45-2.140 Naturally, the protection of the N(2) position with a substituent prevents the N-fusion affording rhenium(I) tricarbonyl 2-substituted NCP S45-3.141,142 The formed S45-3 contains the octahedral rhenium(I) ion facially bound to three inner nitrogen atoms. Its coordination sphere is supplemented with three carbonyl ligands located on the opposite face. The C(21)–H fragment remained intact. When a suitable substituent is involved, N(2) protection can be readily removed to obtain rhenium(I) tricarbonyl NCP, which is prone to transform into rhenium(I) tricarbonyl NFP S45-2.
Finally, the insertion of rhenium into 2-methyl NCP using [ReBr(CO)5] in the presence of 2,6-dimethylpyridine (2,6-lutidine) creates oxorhenium(V) peripherally π-extended N-confused porphyrin S45-4.143 The regular [CNNN] surrounding of oxorhenium(V) can be noticed (the Re–C(21) bond length equals 2.050(5) Å). The same product can be synthesised when rhenium(I) tricarbonyl 2-methyl NCP S46-1 (Scheme 46) (Fig. 14) reacts with [ReBr(CO)]5 and 2,6-lutidine. The presence of 2,6-lutidine is essential because a fusion at the NCP perimeter occurs concomitantly to the rhenium(V) insertion. One of the methyl groups from 2,6-lutidine forms a C–C bond with the ortho carbon atom of meso-phenyl adjacent to C(3). Simultaneously, the same carbon atom forms the double bond with proximal C(3). The oxidation and oxygenation of initial rhenium(I) to oxorhenium(V) were confirmed by X-ray crystallography.
 | ||
| Scheme 46 Oxygenation of a rhenium(I) NCP complex S46-1. | ||
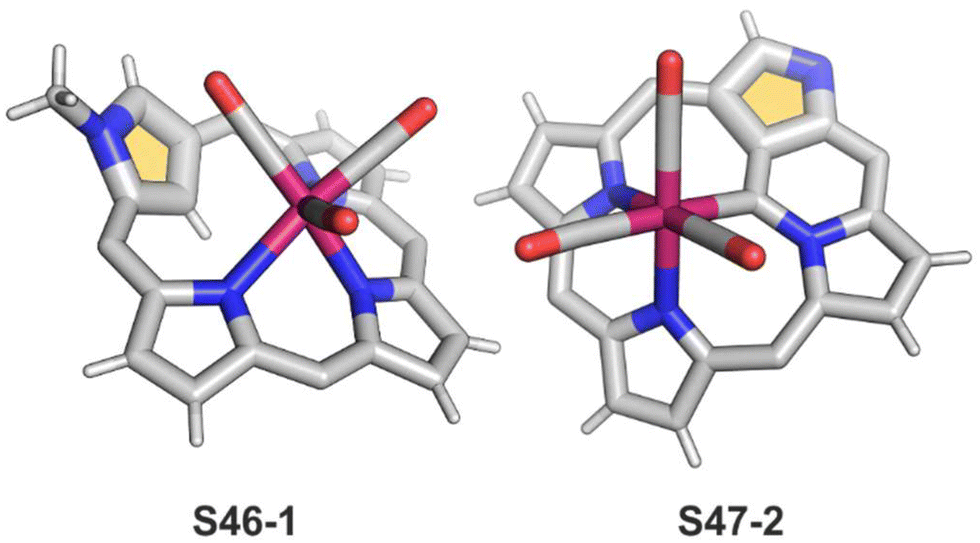 | ||
| Fig. 14 Rhenium(I) NCP complexes. meso-Substituents not shown. | ||
The interesting oxygenation of S46-1 occurs with suitable oxygen atom donors (t-BuOOH and TEMPO).144 The tricarbonyl ruthenium(I) N(2)-methylated NCP complex acquires two or three oxygen atoms during oxygenation and transforms into S46-2 and S46-3, respectively. Aside from oxorhenium centre formation, the attachment of the oxygen atom at C(3) affords a lactam unit (the lactam function is confirmed by the CO bond length: 1.256(6) Å). Finally, the incorporation of the third oxygen atom, which eventually links rhenium(V) and C(21) atoms, finalises the process, as evident in the structure of S46-3. The Re–C(21) distance increases to 2.245(9) Å from 2.070(4) Å for S46-2. The C(21)–O(21) distance equal to 1.415(11) Å reflects the single bond character. Interestingly, interconversion between S46-2 and S46-3 can occur when one uses PPh3 as a reductant and an oxygen atom scavenger and pyridine N-oxide as an oxidant.
Eventually, a different type of reactivity is induced when inner core methylated derivatives are explored.145 The reaction of N(22)-methyl NCP S47-1 (Scheme 47) and [Re2(CO)10] produces a single carbon bridge between the originally methyl-substituted site and an adjacent C(21) atom (aside from accompanied but predictable N-fusion). The bridge is then coordinated to the metal centre (S47-2). The bridging carbon atom acts as a carbene ligand, coordinated to the tricarbonyl rhenium(I) unit, also bound to the other two available inner nitrogen donors (Fig. 14). The Re–C bond length equals 2.140(5) Å. Its carbene character is confirmed by the characteristic 13C NMR signal located at 196.4 ppm. The alternative insertion of rhenium(I) into 21-methyl NCP leads to a mixture of internally bridged rhenium(I) NCP-derivatives with a clear preference for the C(21)–N(24) fused one.
 | ||
| Scheme 47 Rhenium(I) insertion into 22-methyl N-confused porphyrin S47-1. | ||
In the unique for the field studies, it was demonstrated that erbium(III) and ytterbium(III) ions could form stable complexes with NCP.146,147 Reactions of erbium(III) or ytterbium(III) amides (Ln(N(SiMe3)2)3·x[LiCl(THF)3]) with N-confused porphyrin in bis(methoxyethyl) ether are finalised by the isolation of the respective N-confused porphyrin complexes, S48-1 (Scheme 48) and S48-2. Their stability is assured by the facial coordination of anionic tripodal cobaltocene decorated by three P-phosphonate arms. The stabilising interaction involves the coordination of three phosphoryl oxygen atoms located on a single face of a coordination polyhedron. The opposite face embraces three inner pyrrolic nitrogen atoms of NCP. In the case of S48-1, the η2-agostic interaction with the inner C(21)–H can be noticed with a Er⋯C(21) distance of 2.595(6) Å (Fig. 15).
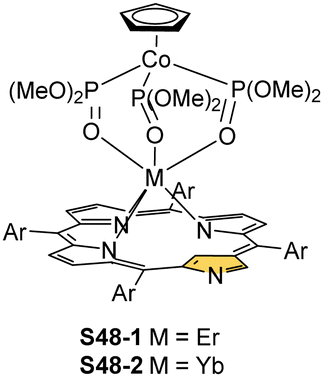 | ||
| Scheme 48 The unique erbium(III) and ytterbium(III) complexes of NCP derivatives. | ||
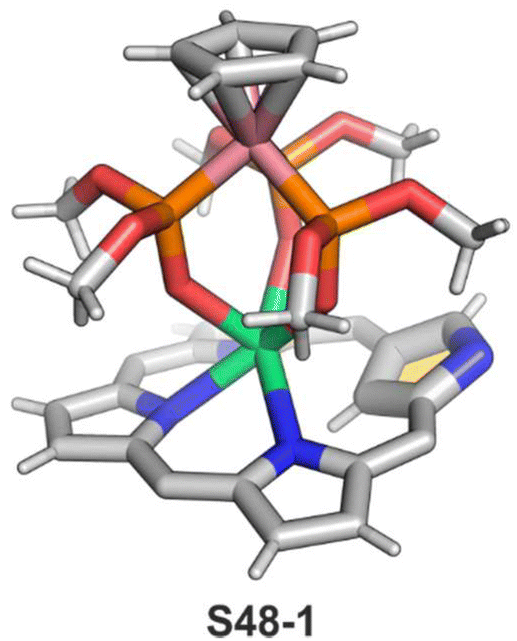 | ||
| Fig. 15 Erbium double-decker NCP complex. meso-Substituents not shown. | ||
N-Confused porphyrin was quite extensively probed as an appropriate macrocyclic setting for the coordination of main group metal ions and metalloids known for their small ionic radii, exemplified here by boron(III), phosphorus(V), and silicon(IV).
Thus, insertions of boron(III) or oxophosphorus(V) prompt the inversion of the N-confused pyrrole ring, followed by N-fusion, which creates the contracted three nitrogen [NNN] coordinating surrounding of N-fused porphyrin. Thus, the σ-phenyl boron(III) N-fused porphyrin S49-1 (Scheme 49) cation and oxophosphorus(V) dihydro-N-fused porphyrin S49-2 were identified.148,149 Evidently, the apparent mismatch between the core of N-confused porphyrin and boron(III) or phosphorus(V) radii was instrumental for N-confused porphyrin to N-fused porphyrin conversions. Altogether, seemingly distant coordination centres boron(III) and phosphorus(V) and transition metal cations: rhenium(I) and manganese(I) enforce the identical NCP to the NFP rearrangement, fine-tuning the size of the macrocyclic core.
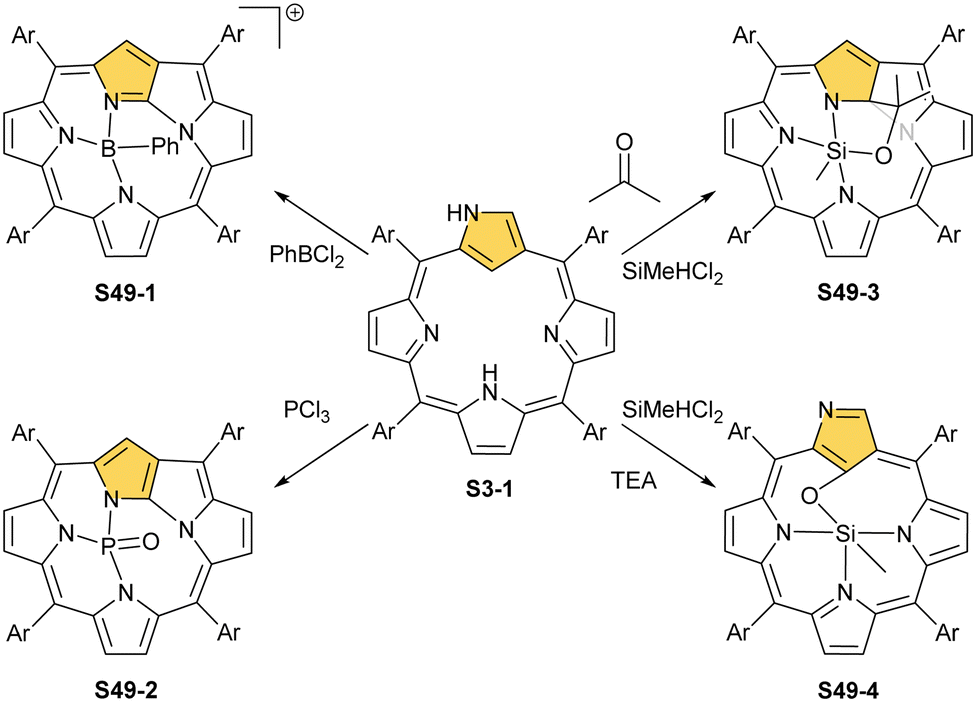 | ||
| Scheme 49 Reactivity of N-confused porphyrin S3-1 with boron(III), phosphorus(V), and silicon(IV) ions. | ||
The similar values of the cationic radii of phosphorus(V) (rcat = 0.38 Å) and silicon(IV) (rcat = 0.40 Å) are consistent with the fact that the resembling types of NCP molecular rearrangements were discovered in the course of silicon(IV) insertions. Thus, the reaction of N-confused porphyrin and dichloromethylsilane in the presence of triethylamine and aldehydes of ketones results in the formation of the methylsilicon(IV) complex of a NFP derivative (S49-3). The novel macrocycle is substituted at the inner C(9) position with a hydroxyalkyl moiety derived from introduced aldehyde or ketone.150 Interestingly, the fundamental NCP frame is preserved when the silicon(IV) insertion is carried out without even traces of aldehydes or ketones in the reaction mixture. Under these conditions, the methylsilicon(IV) 21-hydroxy NCP (S49-4) is solely isolated.
The other subgroup forms from regular complexes with NCP after C(21)H activation. The examples here are complexes with MoII and two axial piperidines (S50-1, Scheme 50),151 SnIV with two axial chlorides (S50-2),152,153 and SbV with two anions as well (S50-3).154,155 Note that both SnIV and SbV have the same coordination environment and both complexes are neutral. This, once again, points to the exceptional flexibility of NCP in the stabilisation of different oxidation states.
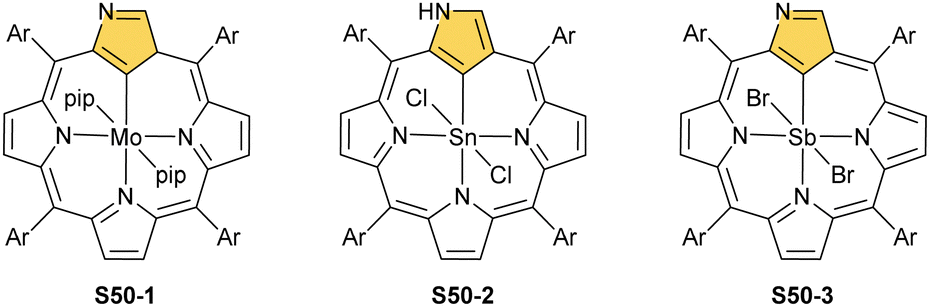 | ||
| Scheme 50 Molybdenum(II), tin(IV), and antimony(V) complexes of N-confused porphyrin. | ||
At present, the lithium N-confused porphyrin S51-1 (Scheme 51) completes the set of main group N-confused porphyrins. A reaction of NCP or N(2)-methyl NCP with lithium bis(trimethylsilyl)amide results in the incorporation of lithium(I) in the carbaporphyrinic crevice (S51-1).156 Lithium(I) coordinates with three inner nitrogen atoms. The LiI⋯C(21)–H interaction is reflected by the Li⋯C(21) distance which is equal to 2.363(6) Å. It increases to 2.482(4) Å after the axial coordination of the THF molecule (S51-2).
In summary, the vast abundance of organometallic motifs has been provided by N-confused porphyrin and spans from direct σ-bonds and π-coordination to weaker agostic interactions. Nonetheless, most of them are enforced and stabilised by macrocyclic confinement of the metal ion in the proximity of the carbon fragment.
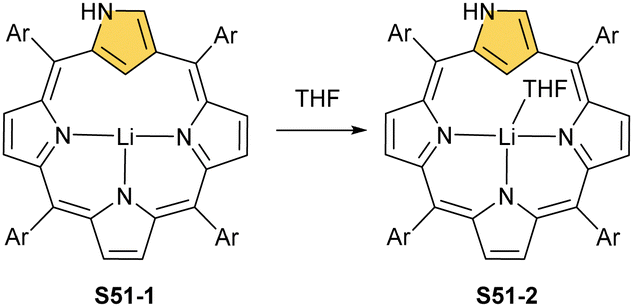 | ||
| Scheme 51 Coordination motifs of a lithium(I) N-confused porphyrin. | ||
X-confused and other “peculiar” 21-carbaporphyrins
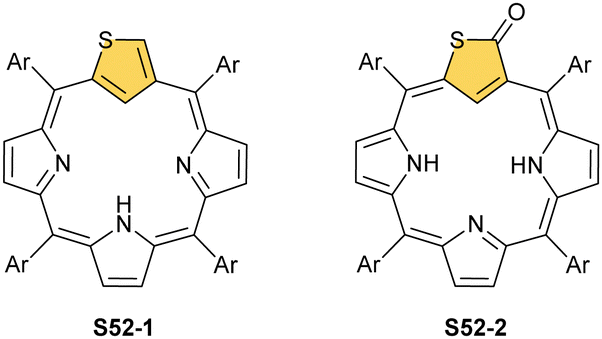
The N-confusion concept opened the route to the rich chemistry of N-confused porphyrin S1-2 and subsequently to N-confused expanded porphyrins and heteroporphyrins.3,42,157 The continuation of the story brings O-confusion and S-confusion concepts, affording 2-hetero-21-carbaporphyrins eventually.158–160 In a macrocyclic environment, a heteroatom can function as a donor toward various metal ions, offering insight into the coordination properties of furan, thiophene, selenophene, or tellurophene.4,161 The heteroatom confusion (X-confusion) concept was also successfully applied in constructing a nontrivial macrocyclic environment for organometallic chemistry.The syntheses of 2-hetero-21-carbaporphyrins are more complex when compared to straightforward condensation affording S1-2. The appropriately prearranged building block including an X-confused heterocycle (furan or thiophene substituted at 2- and 4-positions) must be prepared for the final condensation. Thus, S-confused thiaporphyrin (2-thia-21-carbaporphyrin) S52-1 (Scheme 52) is formed in the condensation of 2,4-bis(phenylhydroxymethyl)thiophene, pyrrole, and arylaldehyde catalysed with BF3·Et2O.158 A prolonged oxidation using DDQ in excess results in C(3)-oxygenation and aromatic 2-thia-3-oxo-21-carbaporphyrin S52-2. Thus, the specific reactivity at the C(3) position has been detected. A direct comparison of N-confused porphyrin S1-2 and S-confused thiaporphyrin S52-1 concludes that the thia-derivative demonstrates the less malleable character of a coordination crevice. Only two dissociable hydrogen atoms are located in the cavity of S52-1, whereas the tautomeric equilibrium of S1-2 allowed for switching between two or three such hydrogen atoms (see above). The oxygenation of S52-1 provides the macrocycle S52-2 with a coordination core prone to act as a trianionic ligand. The borderline macrocyclic aromaticity was detected for S52-1, whereas the evident aromatic behaviour was established for S52-2 as documented by the appropriate chemical shift of the inner H(21) atom (S52-1 4.76 ppm and S52-2 −5.31 ppm). Compared to the S-confusion procedure, the synthesis of O-confused oxaporphyrin remained a demanding task because of the peculiar furan activity. An acid-catalysed condensation of 2,4-bis(phenylhydroxymethyl)furan, pyrrole, and arylaldehyde readily yields the strongly aromatic macrocycle S53-1.159S53-1 (Scheme 53) is structurally related to the hypothetical 3-dihydro-2-oxa-21-carbaporphyrin and contains an appended pyrrole ring linked to the tetrahedral C(3) atom.
 | ||
| Scheme 52 S-confused thiaporphyrin S52-1 and 3-oxo S-confused thiaporphyrin S52-2. | ||
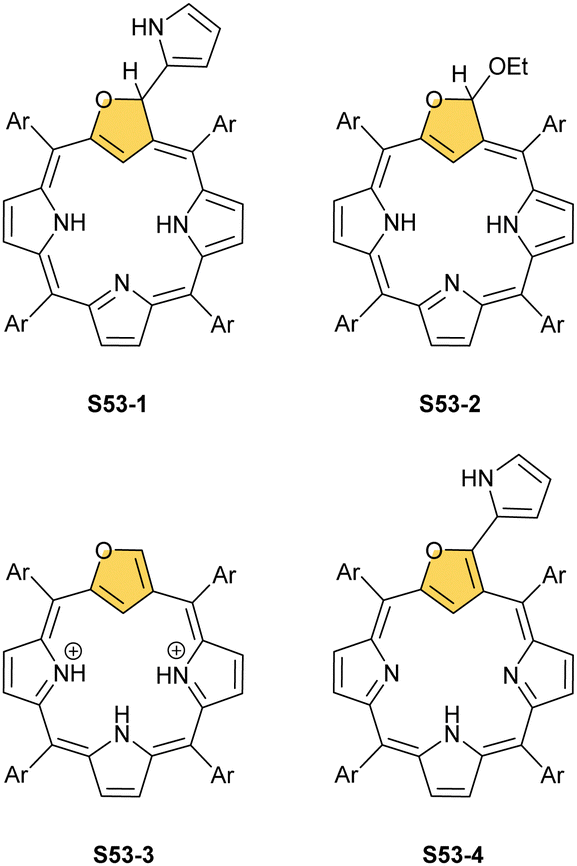 | ||
| Scheme 53 O-confused oxaporphyrinoids. | ||
Eventually, the condensation was purposely directed toward the formation of the 2-oxa-21-carbaporphyrin derivative S53-2 without an appended pyrrole. However, the 3-ethoxy substitutent was accommodated and turned out to be a “good” leaving group.160 The careful titration of S53-2 with trifluoroacetic acid (TFA) produced the true O-confused oxaporphyrin solely stabilised in its diprotonated form S53-3. The process is reversible, and S53-2 can be rebuilt from S53-3 simply by adding sodium ethoxide. An oxidised version of S53-1, that is, S53-4, structurally resembles the 3-(2′-pyrrolyl) N-confused porphyrin derivative, formed in the reaction of S1-2 with pyrrole162 and was identified in the form of metal complexes.163
Modifications at the C(3) position are reflected by pronounced changes in overall macrocyclic aromaticity. Thus, the borderline macrocyclic aromaticity was detected for S53-3 and S53-4 but the clear aromatic features were established for S53-1 and S53-2. Two other original strategies to construct carbaporphyrinoids containing the [CNNN] core were explored, generating S54-1 (Scheme 54) and neo-confused porphyrin S55-1 (Scheme 55). S54-1 forms when N-substituted pyrazole-3,5-dialdehyde is used in the typical condensation with tripyrrane.164 As both outer positions (2 and 3) are now occupied with nitrogen atoms, the perimetral substitution cannot occur, yet the unprotected meso position is vulnerable to oxygenations (S54-3).165,166
 | ||
| Scheme 54 Pyrazole-containing NCP analogues. | ||
 | ||
| Scheme 55 Neo-confused porphyrin S55-1 and its benzo-fused derivative S55-2. | ||
The borderline aromaticity of S54-1 is consistent with the H(21) NMR resonance located at 5.34 ppm. When an outer nitrogen atom is substituted by phenyl rather than methyl, the synthesis yields phlorin, which incorporates a tetrahedral C(5)H2meso bridge.
A prominent example of a 21-carbaporphyrin group is called neo-confused porphyrin and originally was synthesised as its benzo-fused derivative S55-2167–169 and subsequently, as intact neo-confused porphyrin S55-1.169 Neo-confused porphyrin S55-1, similarly to N-confused porphyrin S1-2, is classified as a constitutional isomer of regular porphyrin which preserves its structural frame.41 The neo-confused pyrrole ring is incorporated into a porphyrin scaffold at N(1) and C(3) positions. This particular connectivity allows for full π-conjugation and marked macrocyclic aromaticity as expressed in the chemical shifts of inner H(21) atoms 1.32 ppm (S55-1) and −0.74 ppm (S55-2).167
Organometallic chemistry
The coordination properties of X-confused heteroporphyrins were tested, albeit the scope of their investigation was significantly smaller compared to the extensive studies on N-confused porphyrin.Inserting cadmium(II) and zinc(II) into S-confused thiaporphyrin S52-1 results in the formation of the respective S56-1 (Scheme 56) and S56-2 complexes.170 The macrocycle acts as a monoanionic ligand. Three nitrogen atoms and the C(21)H fragment of the S-confused thiophene are located at equatorial positions. The metal charge is compensated by the apical chloride coordination. The distinctive 1H NMR resonances of the inner core H(21) atom, associated with the S-confused thiophene ring, are located at 1.71 and 1.86 ppm for S56-2 and S56-1, respectively. The peculiar proximity of the S-confused thiophene fragment to the cadmium(II) ion induces direct scalar coupling between the spin-active nucleus of cadmium (111/113Cd) and the adjacent 1H nucleus (JCdH = 10.0 Hz). The Cd⋯C(21)H interaction is clearly reflected by significant changes of C(21) chemical shifts: S56-1 92.9, S56-2 88.2 ppm vs. 123.7 ppm for S52-1. The molecular structure of S56-2 confirmed the side-on cadmium⋯thiophene interaction with the close Cd⋯C(21) contact of 2.615(7) Å (Fig. 16).
 | ||
| Scheme 56 Zinc(II) and cadmium(II) complexes of S-confused thiaporphyrin. | ||
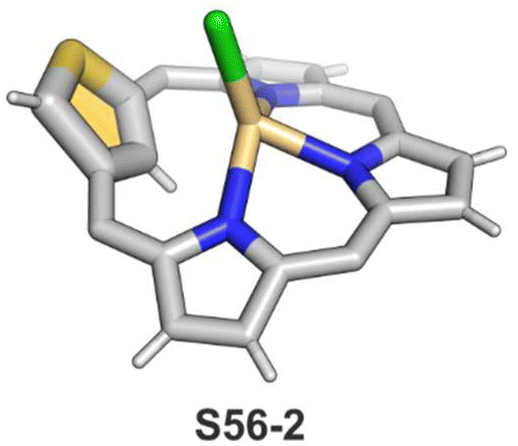 | ||
| Fig. 16 Agostic interactions in cadmium(II) SCP. meso-Substituents not shown. | ||
O-Confused oxaporphyrin and its derivatives (S53-1, S53-2, and S53-3) applied as macrocyclic ligands toward selected metal ions (Zn/Cd/Ni/Cu2+ and Cu/Ag3+) demonstrate the peculiar flexibility of its molecular and electronic structures firmly providing, however, the equatorial [CNNN] surrounding. It controls the subtle interplay between their structural flexibility, perimeter substitution, coordination, and aromaticity. The macrocyclic tuning is easily triggered by the addition/elimination of a nucleophile located at the C(3) position. The tetrahedral–trigonal rearrangements originating at the C(3) atom spread out their consequences to the whole macrocyclic scaffold.
The reaction of S53-1 with nickel(II) or palladium(II) chloride produces weakly aromatic S57-1 (Scheme 57) and S57-2, respectively.159 The structurally prearranged macrocycle S53-4 affords S57-1 as well. The molecular structure of S57-1 shows a direct metal–carbon bond with a length of 1.892(4) Å. In contrast to nickel(II) or palladium(II), the insertion of zinc(II) or cadmium(II) into S53-4 affords the side-on species S57-3 and S57-4 resembling the geometry of the thia analogue S56-2.163 Thus, macrocycle S53-4 acts as a monoanionic ligand. Compensation of the metal(II) charge requires axial chloride coordination. Coordination cores encountered for O-confused oxaporphyrins like S53-1 are well-matched for stabilising higher oxidation states of metal ions. The dissociation of three inner protons (two NH and one C(21)H) leads to a trianionic equatorial surrounding. Thus, the reaction between silver(I) acetate and S53-1 in acetonitrile in the presence of ethanol results in S58-1.159 The tetrahedral geometry around the C(3) atom, required to preserve the aromaticity, is clearly demonstrated. The addition of TFA to S58-1 (Scheme 58) yielded strongly aromatic S58-2via the elimination of the ethoxy group.
 | ||
| Scheme 57 Complexes of the O-confused oxaporphyrin derivative S53-1. | ||
 | ||
| Scheme 58 Protonation of the silver(III) O-confused oxaporphyrin derivative S58-1. | ||
A reaction between the 3-ethoxy-substituted O-confused oxaporphyrin and silver(I) acetate produces the respective AgIII complex S59-1.160 An addition of TFA to S59-1 (Scheme 59) generates weakly aromatic silver(III) “true” O-confused oxaporphyrin S59-2 formed by the elimination of the ethoxy group. The subsequent intramolecular oxidation of S59-2, accompanied by water C(3)-addition and two-electron-reduced silver(I) extrusion, affords carbaporpholactone S59-3, the aromatic 21-carbaporphyrinoid. S59-3 acts as a trianionic ligand capable of again accommodating silver(III) (S59-4; the Ag–C(21) bond of 2.013(5) Å).
 | ||
| Scheme 59 Reactivity of the silver(III) O-confused oxaporphyrin derivative S59-1. | ||
Sliver(III) carbaporpholactone S59-4 can be treated with potassium diphenylphosphide yielding the mixture of two derivatives with diphenylphosphanyl S60-1 (Scheme 60) or diphenylphosphoryl S60-2 moieties attached at the C(21) position.26 The coordination of the highly oxidised silver(III) and C(21)H activation are prerequisite factors for phosphorylation, as S59-3 is inactive toward potassium diphenylphosphide under similar conditions. The subsequent oxidation with DDQ produces exclusively S60-2, and the insertion of silver(I) into S60-2 recovers S59-4.
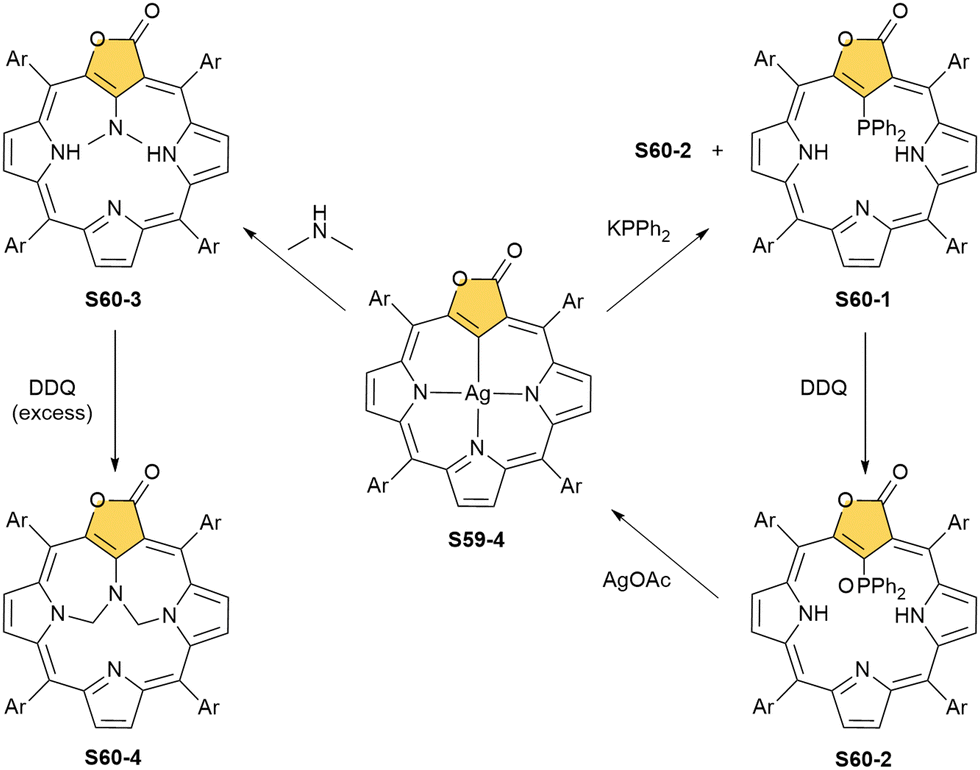 | ||
| Scheme 60 Reactivity of silver(III) 21-carbaporpholactone S59-4. | ||
The reaction of dimethylamine and S59-4 reveals analogous regioselectivity of substitution.101 The process results in the formation of S60-3. Stepwise oxidation of the dimethylamine derivative leads finally to the internally bridged carbaporpholactone S60-4, which incorporates the [5.7.5.7.5] pentacyclic ring into its macrocyclic scaffold.
O-confused oxaporphyrins with an appended pyrrole ring (S53-1 and S53-4) and carbaporpholactone S59-4 create unique macrocyclic surroundings to form organocopper(II) or alternatively organocopper(III) compounds. The reaction of copper(II) acetate and S53-1 under mild conditions results in the quantitative formation of the organocopper(III) compound S61-1 (Scheme 61), which preserves the scaffold of maternal S53-1.171 A straightforward insertion of copper(II) into S53-4 quantitatively produces paramagnetic organocopper(II) species S61-2, which conserves the geometry of starting S53-4. The transformation of S61-1 in the course of the reaction with dioxygen, which includes the oxidation of macrocycle and the reduction of copper(III) to copper(II), generates S61-2 as well. The one-electron oxidation of S61-2 forms organocopper(III) species S61-3 resembling the analogous silver(III) molecule S58-2.
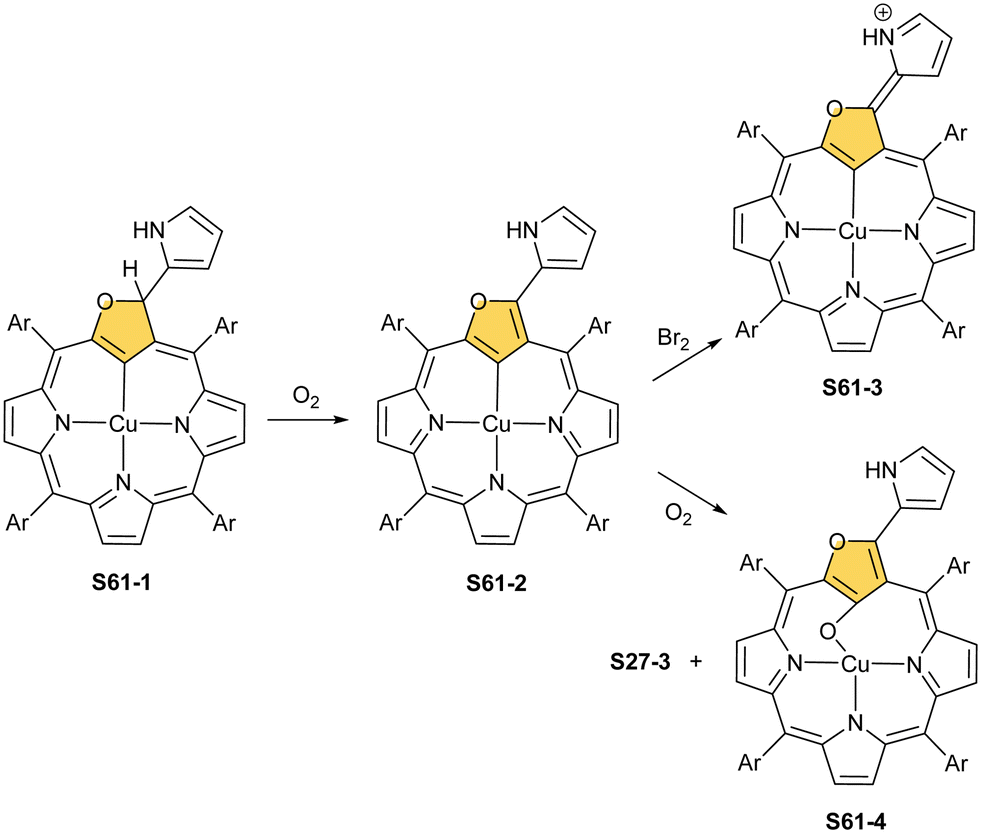 | ||
| Scheme 61 Reactivity of copper(III) O-confused oxaporphyrin derivative S61-1. | ||
The electron paramagnetic resonance spectroscopic features of S61-2 correspond to a copper(II) oxidation state. The crystallographic analysis of S61-2 revealed the practically planar structure and confirmed the formation of a direct CuII–C bond (1.939(4) Å) in which C(21) acts similarly to an sp2 σ-carbanion. The degradation of S61-2 in a reaction with dioxygen yields known from NCP chemistry copper(II) tripyrrinone S27-3. This process is accompanied by regioselective oxygenation at the inner C(21) position to form complex S61-4.
The reaction of copper(II) acetate with S60-2 under mild conditions affords S62-1 (Scheme 62).26 The EPR spectrum recorded for S62-1 reveals typical features associated with the copper(II) electronic state resembling copper(II) N-confused porphyrin and copper(II) 21-carbaporpholactone derivatives. The coordination environment of copper(II) forms a square pyramid with the equatorial positions occupied by the cavity donors [CNNN] and an oxygen atom located apically. The Cu–C(21) bond length equals 2.232(2) Å. The furanone is sharply tipped out of the [CNNN] plane, allowing the coordination to the copper(II) ion through the sp3-hybridised C(21) atom.
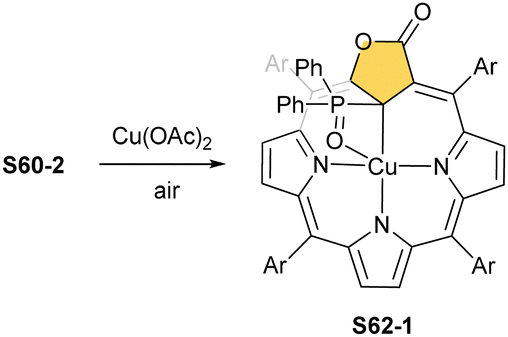 | ||
| Scheme 62 Synthesis of copper(II) 21-carbaporpholactone derivative S62-1. | ||
The coordination chemistry of pyrazole containing 21-carbaporphyrin S54-1, neo-confused porphyrin S55-1, and benzo-fused S55-2 was explored in the scope limited solely to nickel(II) and palladium(II).
Treatment of S54-1 with either nickel(II) or palladium(II) leads to regular 21-carbaporphyrin-like complexes with an organometallic C(21)–MII bond. Insertion into a phlorin variant ends up with an appropriate 21-carbaporphyrin complex S63-1 (Scheme 63) as well.164
 | ||
| Scheme 63 Nickel(II) and palladium(II) complexes of the pyrazole-containing NCP analogue, neo-confused porphyrin, and its benzo-fused derivative. | ||
A similar organometallic potential was demonstrated for neo-confused porphyrins, like S55-1 and S55-2 producing typical for 21-carbaporphyrins planar S63-2 and S63-3 complexes.167,172
Azuliporphyrin and benzocarbaporphyrin
At this stage, it is essential to recall that the leitmotif macrocycles of this review have to preserve the fundamental skeleton of porphyrin or heteroporphyrin, clearly incorporating a 21-carbaporphyrin-like [CNNN] coordination core. Accordingly, they encompass X-confused (S2-1, S52-1, and S53-3) or neo-confused (S63-2) five-membered rings. The presented data analysis clearly reveals that this class can be readily extended by applying the fusion concept. Namely, the fusion of the five-, six-, or seven-membered rings to the already transformed five-membered ring conserves the major porphyrinic core, markedly modifying the overall properties of the resulting carbaporphyrin. In this manner, a collection of 21-carbaporphyrinoids have been impressively enriched, as shown in Scheme 64, demonstrating the relationship between N-confused porphyrin S1-2 and its fused derivatives (S4-3 and S5-1). A similar fusion-type relationship was described above for neo-confused porphyrin S55-1 and benzo-fused neo-confused porphyrin S55-2.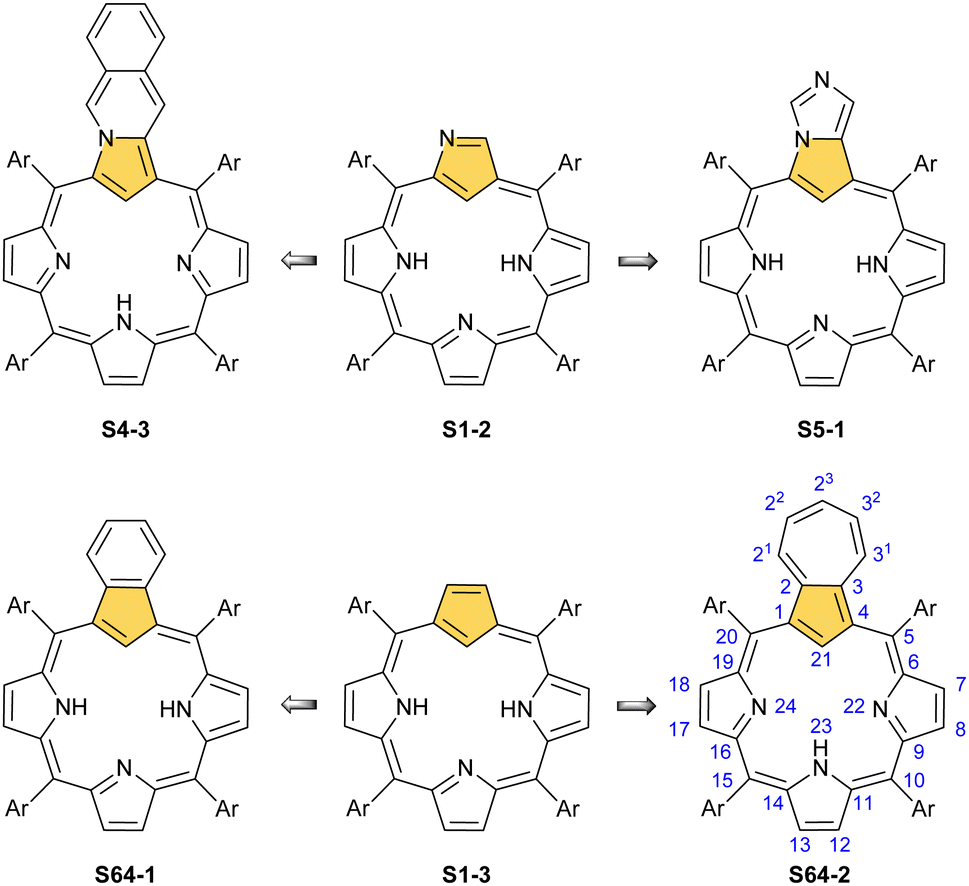 | ||
| Scheme 64 N-confused porphyrin S1-2 with its fused derivatives, and 21-carbaporphyrin S1-3 with its fused derivatives. | ||
A fusion approach, illustrated above, performs perfectly well when applied to a regular meso-tetraaryl-21-carbaporphyrin S1-3. Namely, the fusions of six- or seven-membered rings assemble benzocarbaporphyrin S64-1 or azuliporphyrin S64-2, respectively.
Retrospectively, β-alkylated benzocarbaporphyrin S65-1 (Scheme 65) belongs to the group of macrocycles synthesised just after N-confused porphyrin, which played a founding role in launching a carbaporphyrinoid field. Serendipitously, in their effort to form azuliporphyrin S65-2, Berlin et al. identified β-alkylated benzocarbaporphyrin S65-1.173 It occurs that the applied reaction conditions induced a peculiar transformation of the initially formed azuliporphyrin S65-2 into benzocarbaporphyrin S65-1 as firmly proven in the course further investigations by Lash.174 This discovery led to optimising the synthetic pathway leading to S65-1. Under base-catalysed conditions, azuliporphyrin S65-2 reacts with tert-butyl hydroperoxide yielding benzocarbaporphyrin S65-1 and its formyl-substituted derivative.
 | ||
| Scheme 65 β-Alkylated benzocarbaporphyrin S65-1 and β-alkylated azuliporphyrin S65-2. | ||
Subsequently, the “3+1” methodology, by which porphyrinoid systems are constructed from a tripyrrane and a cyclic dialdehyde, i.e., azulene-1,3-dicarbaldehyde, was successfully applied to generate β-alkylated azuliporphyrin S65-1.174 An alternative [3+1] strategy using the azulene analogue of the tripyrranes was developed to form β-substituted azuliporphyrins.175–177 The related [3+1] methodology procedure affords 10,15-diaaryl-23-thia and 23-selenaazuliporphyrins (S66-5, R = H).178 Finally, meso-tetraaryl-23-thiaazuliporphyrin forms in a reaction of plain azulene, arylaldehyde, and thiatripyrrane (S66-5, R = Ar).179
Benzocarbaporphyrin S66-2 and its 23-heteroanalogue S66-3 are directly synthesised starting from carbatripyrrin S66-1, as shown in Scheme 66.175,180 Diheterocarbaporphyrins S66-4 can be obtained following a similar synthetic approach.180,181
 | ||
| Scheme 66 Synthesis of 23-hetero-, 22,23-dihetero- and regular benzocarbaporphyrins from carbatriphyrin S66-1. Respective 23-heteroazuliporphyrin S66-5. | ||
Eventually, a one-pot Rothemund-type synthesis of meso-tetraarylazuliporphyrin S64-2 was developed. Thus, azulene, pyrrole, and arylaldehyde were shown to react under typical Lindsey conditions with a boron trifluoride etherate catalyst to form meso-tetraarylazuliporphyrin S64-2 up to 20% yield.182,183 Treatment of S64-2 with tert-butyl hydroperoxide results (similarly as for S65-2) in the contraction to a fused benzene ring yielding meso-tetraarylbenzocarbaporphyrin S64-1 and its 21- or 22-formyl-substituted derivatives.182,183 The contraction of the seven-membered ring and Rothemund-like condensation are feasible because of the specific electronic structure of azulene. Azulene is more nucleophilic on its five-membered ring – leading to its ability to undergo a macrocyclic condensation reaction similarly to pyrrole. On the other hand, the seven-membered ring is more electrophilic, thus, prone to nucleophile attacks. It leads to a ring contraction and the formation of benzocarbaporphyrin and its derivatives with a formyl group (formed from the extruded carbon atom) located on a fused benzo-ring.
These relationships between S64-1/S64-2 and S65-1/S65-2 as well as the intrinsic properties of azuliporphyrin itself are important for encountered coordination abilities in both types of carbaporphyrinoids. Generally, benzocarbaporphyrins S64-1/S65-1 demonstrate the spectroscopic and structural features of aromatic porphyrinoids, as confirmed with a H(21) chemical shift of −5.3 ppm for a meso-substituted derivative.182 In contrast to S64-1/S65-1, azuliporphyrins S64-2/S65-2 provide a case of borderline aromaticity,182,184,185 with a formally disrupted π-delocalization pathway. The resonance contributors of S64-2/S65-2 with charge separation favouring the positive charge accumulation over the seven-membered ring are responsible for the residual aromatic features. This is apparent after the protonation of S64-2. The inner H(21) shifts from 3.3 to −0.5 ppm.182
Analysing the potential coordination properties, one can notice that S64-1/S65-1 and S64-2/S65-2 complementarily resemble tautomers of N-confused porphyrins – S1-2 and S3-1. Therefore, benzocarbaporphyrins S64-1/S65-1 can act as trianionic macrocyclic ligands (C(21)H, N(22)H, and N(24)H dissociable protons), whereas azuliporphyrins S64-2/S65-2 acquire the dianionic form (C(21)H and N(23)H dissociable protons). An adjustment of the anionic charge of the core delivers a substantial driving force for the contraction of the azulene fragment to suitably stabilise specific coordination motifs.179,186 The total anionic charge of a macrocyclic crevice can be rationally modified via replacing one or more pyrrole rings with a heterocycle, e.g. thiophene, incorporating a heteroatom instead of an NH (see Scheme 66).179,180,187,188
Tropone-fused carbaporphyrin S67-1 (Scheme 67) was generated in the typical procedure originally aimed to form 23-methoxyazuliporphyrin.189 The process involves a classical [3+1] condensation of methoxyazulitripyrrane and pyrrole dialdehyde. Presumably, the spontaneous demethylation affords the transient 23-hydroxyazuliporphyrin, which transforms into isomeric tropone-fused carbaporphyrin S67-1. The molecule preserves the structural frame of azuliporphyrin but acts as a trianionic aromatic ligand resembling benzocarbaporphyrin, including its aromatic π-delocalization pathway.
 | ||
| Scheme 67 Tropone-fused carbaporphyrin S67-1 and 22-methyl-benzocarbaporphyrin S67-2. | ||
It is essential to note that the nature of the coordination cavity can be modified by appropriate inner core substitution. The cavity of azuliporphyrin was significantly altered when 2-substituted azulene was used for condensation. 21-Alkylazuliporphyrin lacks removable H(21) in the cavity, suffering as well from the imposed increased crowdedness.177
In the case of S64-1/S65-1, the inner C(21) position was modified in a reaction with FeCl3 in methanol.190 The conditions lead to the dimethoxy substitution of the inner carbon atom. The direct inner core alkylation was also explored as the controlling means. Alkyl iodide mixed with S65-1 in a base presence results in preferential N(22) alkylation yielding 22-methyl-benzocarbaporphyrin S67-2.191
The coordination environments of mentioned benzocarba- and azuliporphyrinoids make them natural platforms for performing fascinating organometallic transformations.
Coordination of fused 21-carbaporphyrins
An occurrence of a C(21) atom in the inner cavity of benzocarbaporphyrins S65-1 and S64-1 or azuliporphyrins S65-2 and S64-2 is crucial for the arrangement of suitable [CNNN] surroundings.Consistent with their prearranged cores, the coordination of “classical” coordination probes, i.e., metal(II) cations, is favoured by azuliporphyrins, whereas the coordination of metal(III) cations seems to be preferred by benzocarbaporphyrins.
Thus, benzocarbaporphyrin treated with silver(I) acetate efficiently forms silver(III) benzocarbaporphyrin S68-1 (Scheme 68),192 compared to unsuccessful trials with trifluoroacetate salt.193 Silver(III) benzocarbaporphyrin S68-1 reproduces the essential structural features of analogous silver(III) N-confused porphyrin S25-1, including the formation of a direct Ag–C(21) bond. A similar procedure affords gold(III) benzocarbaporphyrin S68-2. Gold(I) iodide acts poorly as a gold source; thus, gold(III) acetate is preferred. Interestingly, a meso-substituted derivative S64-1 occurred to be superior when compared to β-substituted derivatives due to the side reactivity of S65-1, which was not observed in the course of silver(III) insertions. The same organometallic silver(III) environment can be achieved using a benzo homologue of S65-1, i.e., naphtho[2,3-b]carbaporphyrin.194 A respective complex could also be obtained for another 3H macrocycle yielding silver(III) tropone-fused carbaporphyrin S68-3.189
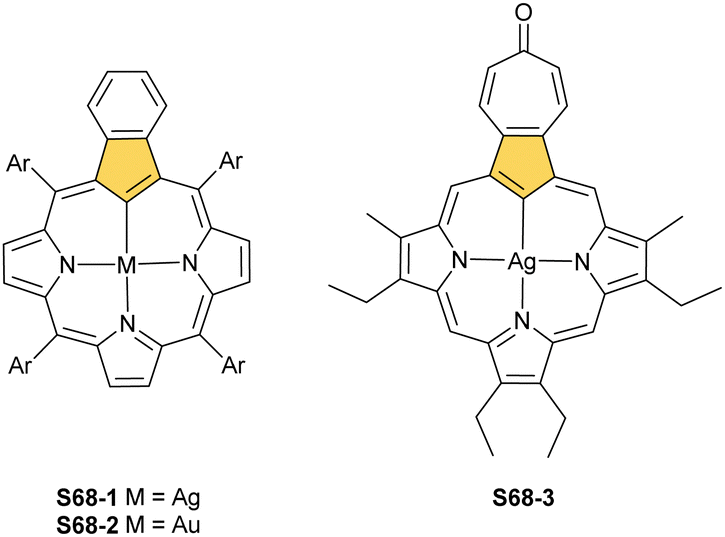 | ||
| Scheme 68 Silver(III) and gold(III) complexes of benzocarbaporphyrin and silver(III) tropone-fused carbaporphyrin. | ||
In attempts to insert silver(III) ions into azuliporphyrin S65-2, the profound transformation of the original carbaporphyrinoid scaffold was discovered (Scheme 69). Thus, silver(III) benzocarbaporphyrin S69-1 and its formyl-substituted derivatives were unambiguously identified in this process.186 Presumably, initial coordination of a silver(III) ion within S65-2 induces contraction of the seven-membered ring, forming eventually a benzocarbaporphyrin framework which could naturally stabilise a tricationic centre. It was speculated that the silver(III) species mediates the activation and attack of an active oxygen species on the azulene ring. Several insertion trials into S65-2 using various gold sources did not give any insertion or insertion and contraction products.
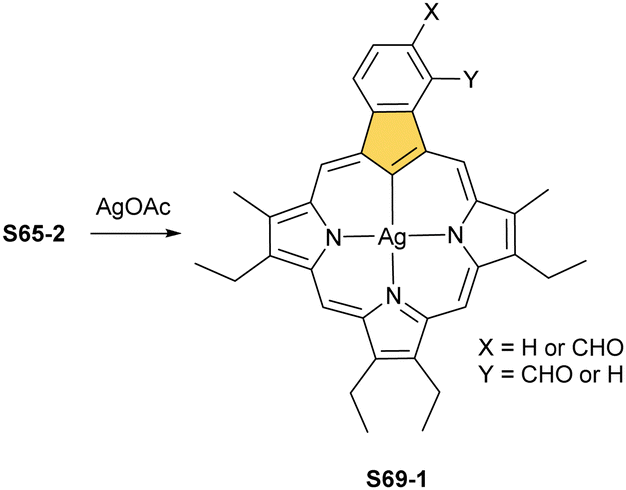 | ||
| Scheme 69 Silver(III) benzocarbaporphyrin S69-1 and its formyl-substituted derivatives. | ||
A similar dioxygen activation, as reported for silver species, could be expected for its lighter counterpart – copper. In this case, however, the activation occurs during the treatment of azuliporphyrin S64-2 with copper(II) acetate.195 The product isolated in the reaction contains an oxygen atom inserted between the metal centre and the C(21) atom (S70-2).
The electronic structure of S64-2/S65-2 strongly disfavours the C(sp3) configuration of the inner C(21) atom in contrast to those of S64-1/S65-1 and N-confused porphyrin S1-2. The oxygenated ligand trapped in S70-2 acquires a structure related to 21-hydroxyazuliporphyrin as suggested by the close to phenolic-like C(21)–O(21) bond distance of 1.302(3) Å. The incorporated oxygen bridge pushes the C(21) atom away from the metal centre (C(21)⋯Cu distance is 2.474(2) Å) and the azulene fragment is significantly canted with respect to the porphyrin mean [18]annulene plane. Considering an affinity of the metal(II) cation to azuliporphyrin, it is justified to assume that the formation of a regular complex of CuII with an organometallic bond precedes the formation of S70-2. Such a hypothesis was tested and indeed, under strictly anaerobic conditions, without an excess of the copper(II) source, appropriate species S70-1 (Scheme 70) is obtained.196 This elusive, very reactive form, trapped by EPR spectroscopy, transforms into S70-2 as soon as exposed to dioxygen, reflecting the incorporation of an oxygen atom into the extremely sensitive CuII–C(21) bond. The acidic demetallation of S70-2 afforded 21-hydroxyazuliporphyrin S70-3, but in solution, its oxo tautomer S70-4 (identified in the solid state – C(21)O 1.268(3) Å) prevails.197 Subsequently, S70-4 was used as a dianionic macrocyclic ligand for other metal ions, for example, nickel(II), palladium(II), and platinum(II).197 As discussed above, regular azuliporphyrins form typical [CNNN] organometallic complexes with various metal(II) cations. Thus, a reaction with metal(II) acetate or chloride (in the case of platinum) in DMF provides the stable complexes of respective ions [e.g., S70-5 for palladium(II)].198 The MII–C(21) distances of 1.896(3) Å for nickel(II) and 1.980(3) Å for palladium(II) clearly indicate the successful deprotonation and formation of an organometallic bond.
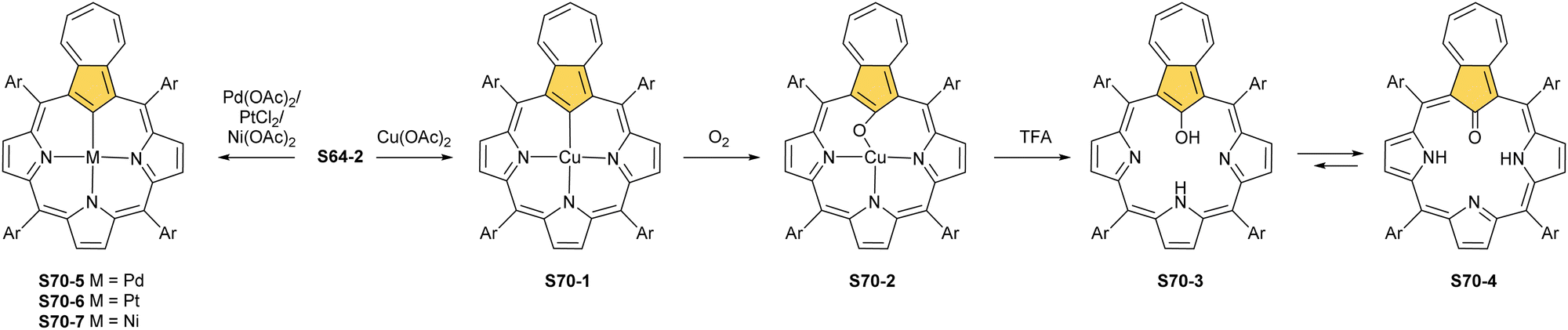 | ||
| Scheme 70 Synthesis of azuliporphyrin complexes with various metal(II) cations. | ||
23-Heterobenzocarbaporphyrin S66-3 possesses the same number of dissociable inner protons as regular azuliporphyrin S64-2/S65-2 and is expected to preferentially accommodate metal(II) cations. The nickel(II) and palladium(II) reactivities are similar, provided that the reaction with the former is conducted under anaerobic conditions.199 The heating of the obtained complex S71-1 (Scheme 71) under aerobic conditions leads to the oxygenation of the inner C(21) atom and demetallation of the macrocycle to give a 23-hetero-21-oxobenzocarbaporphyrin S71-2. 23-Tellurabenzocarbaporphyrin also forms a stable palladium(II) complex, in contrast to the stability of the macrocycle itself.180 In it, the tellurophene ring is reactive towards dioxygen forming a derivative with a hydroxyl group attached (forming an internal Te–OH motif) resembling 21-telluraporphyrin reactivity.200
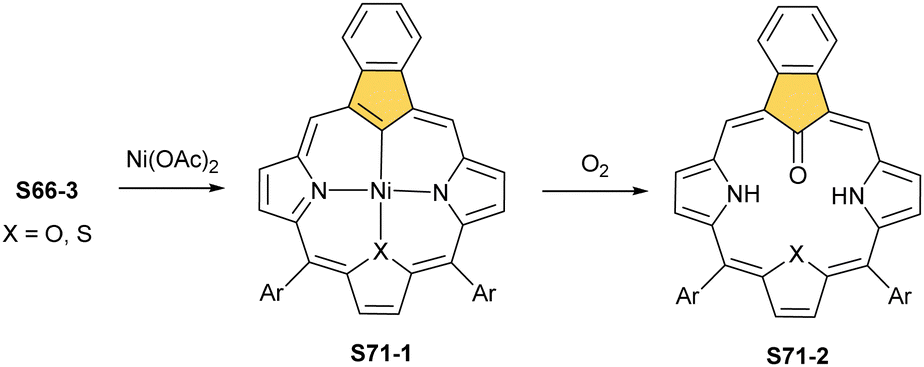 | ||
| Scheme 71 Synthesis of nickel(II) 23-heterobenzocarbaporphyrin and its reactivity with dioxygen. | ||
Interestingly, palladium(II) 23-thiabenzocarbaporphyrin S72-2 can also be obtained when 23-thiaazuliporphyrin with one inner proton [H(21)] is used in the insertion reaction.179 The primarily obtained PdII complex (S72-1) (Scheme 72) acquires square-planar geometry and is cationic because of the charge imbalance. This enforces the participation of resonance contributors with charge accumulated over the seven-membered ring, rendering it prone to nucleophilic attacks and, consequently, contraction.
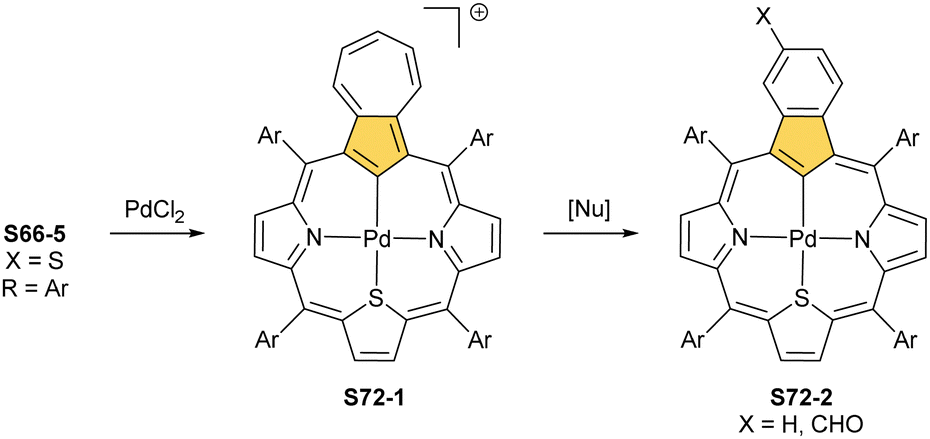 | ||
| Scheme 72 Synthesis of palladium(II) 23-heteroazuliporphyrin and its nucleophile-induced contraction. | ||
An interesting transformation occurs during palladium(II) insertion into N(22)-methylated benzocarbaporphyrin S67-2. When heated briefly with palladium(II) acetate in acetonitrile, the ligand produces an expected insertion product (S73-1) (Scheme 73), yet as a minor fraction.201 The major product was identified as palladium(II) 21-methylbenzocarbaporphyrin S73-2 (Fig. 17), which forms from initially generated S73-1. Thus, N(22)-methyl migrates to the inner C(21) atom and creates the C(sp3) donor centre eventually, as reflected by the C(21) characteristic chemical shift of 45.3 ppm in 13C NMR spectra. A similar transformation occurs for 23-methylbenzocarbaporphyrin, yet the process is significantly slower, suggesting the increased number of methyl relocation steps which are required for the passage to S73-2.191
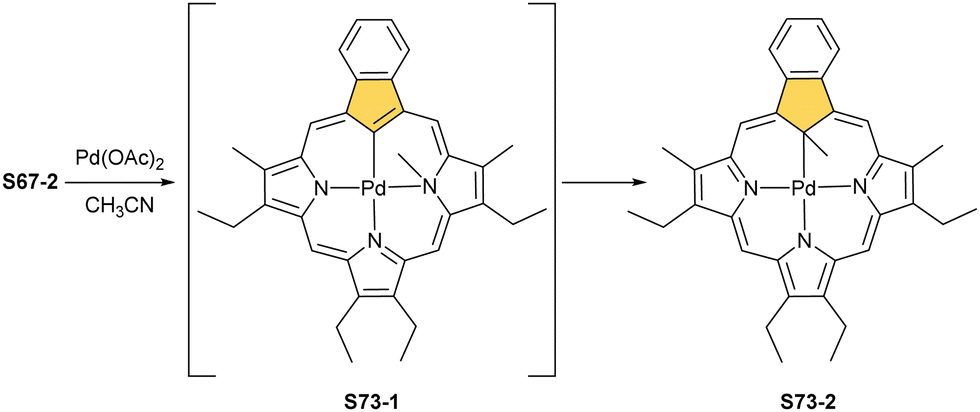 | ||
| Scheme 73 Methyl group migration after palladium insertion into methylated benzocarbaporphyrin S67-2. | ||
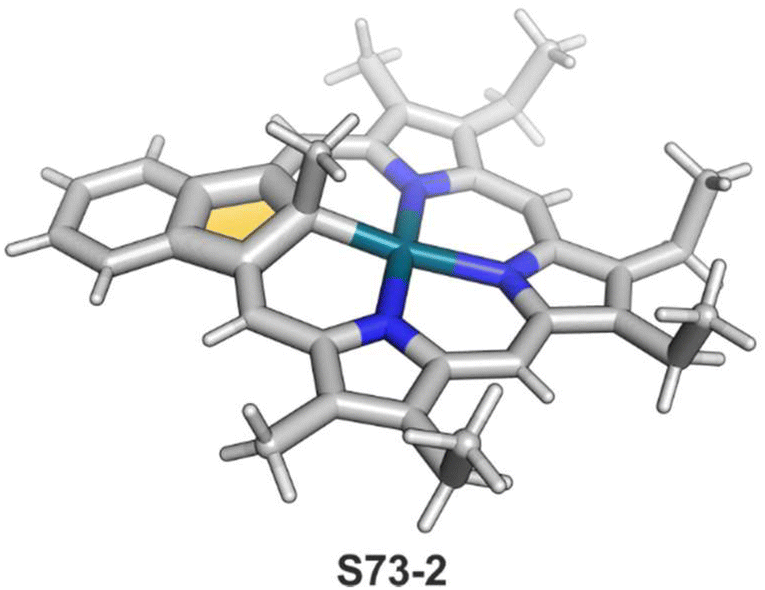 | ||
| Fig. 17 Palladium(II) C(21)-methylated benzocarbaporphyrin. | ||
21-Methylazuliporphyrin S74-1 (Scheme 74) was treated with palladium(II) acetate affording solely the product of C-demethylation S74-2, synthesised previously in regular insertion into azuliporphyrin S65-2.177 The inner core charge of transient palladium(II) 21-methylazuliporphyrin is not sufficient to stabilise divalent metal ions and the C(21) atom could not adopt the necessary C(sp3) hybridisation.
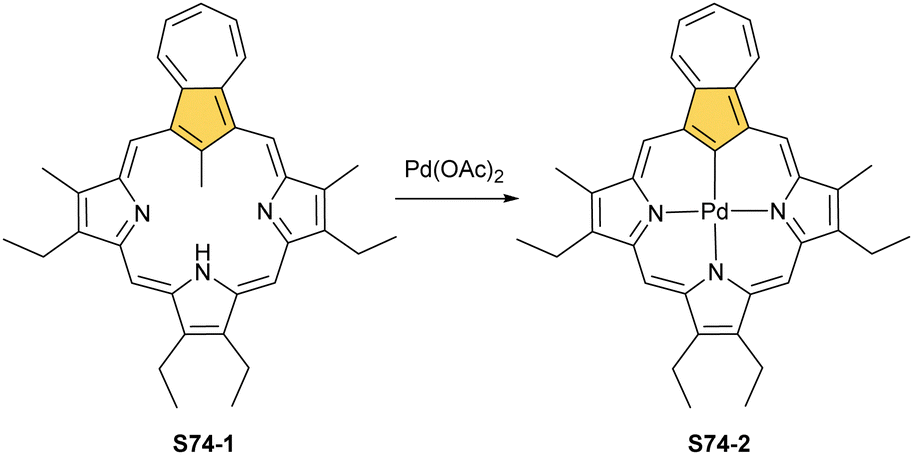 | ||
| Scheme 74 Palladium(II) insertion into 21-methylazuliporphyrin. | ||
As shown for several metal(II) cations, the [CNNN] coordination cavity of azuliporphyrins facilitates the generation of M–C σ-bonding. Significantly, it was encountered that the azulene moiety in the metal(II) azuliporphyrin provides a specific, suitable π-surface which reveals a distinct affinity to accommodate ruthenium(0) clusters, including Ru4(CO)9. Azulene incorporated into the azuliporphyrin S64-2 scaffold constitutes an independent organometallic centre.35 Two conceptually different organometallic motifs are merged in a unique three-dimensional architecture. It was documented that these particular coordination abilities of azulene introduced into a carbaporphyrinoid frame are preserved in other suitable systems producing a series of bimetallic complexes [M(S64-2)(Ru4(CO)9)], in which M = Ni, Pd, Pt, Ru, or Co.35,196
A reaction between [Ru3(CO)12] and azuliporphyrin S64-2 leads primarily to the insertion of a ruthenium(II) ion into the [CNNN] cavity to yield ruthenium(II) azuliporphyrin S75-1 (Scheme 75), which involves the formation of a direct Ru–C(21) bond. The reaction does not stop there and, in the presence of an excess of [Ru3(CO)12], hybrid S75-2 forms (Fig. 18). The formed Ru4(CO)9 cluster possesses a tetrahedral ruthenium scaffold and coordinates to both the 5-membered and 7-memberd rings of the azulene moiety through the three basal ruthenium atoms (Ru1, Ru2, and Ru3).
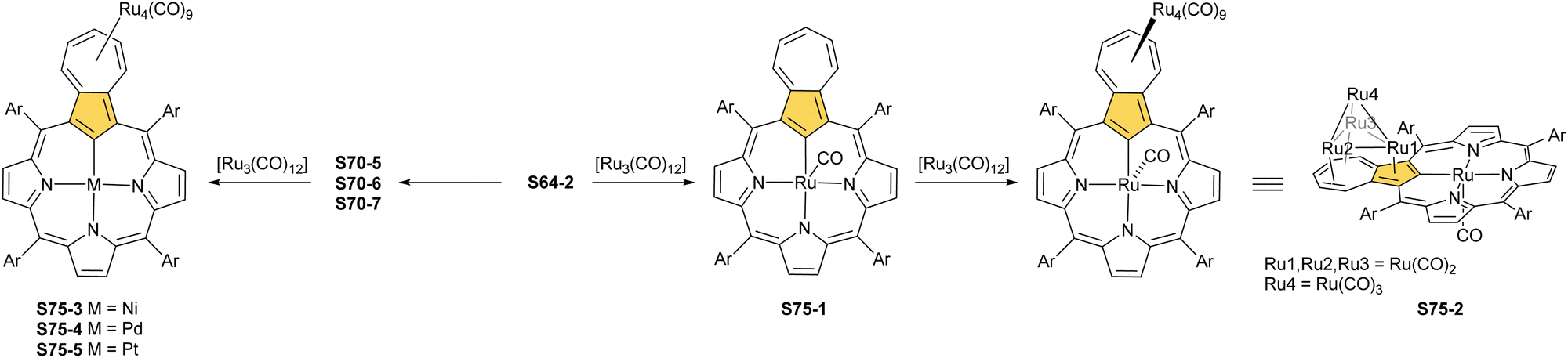 | ||
| Scheme 75 Reactivity of azuliporphyrin S64-2 and its metal(II) complexes with [Ru3(CO)12]. | ||
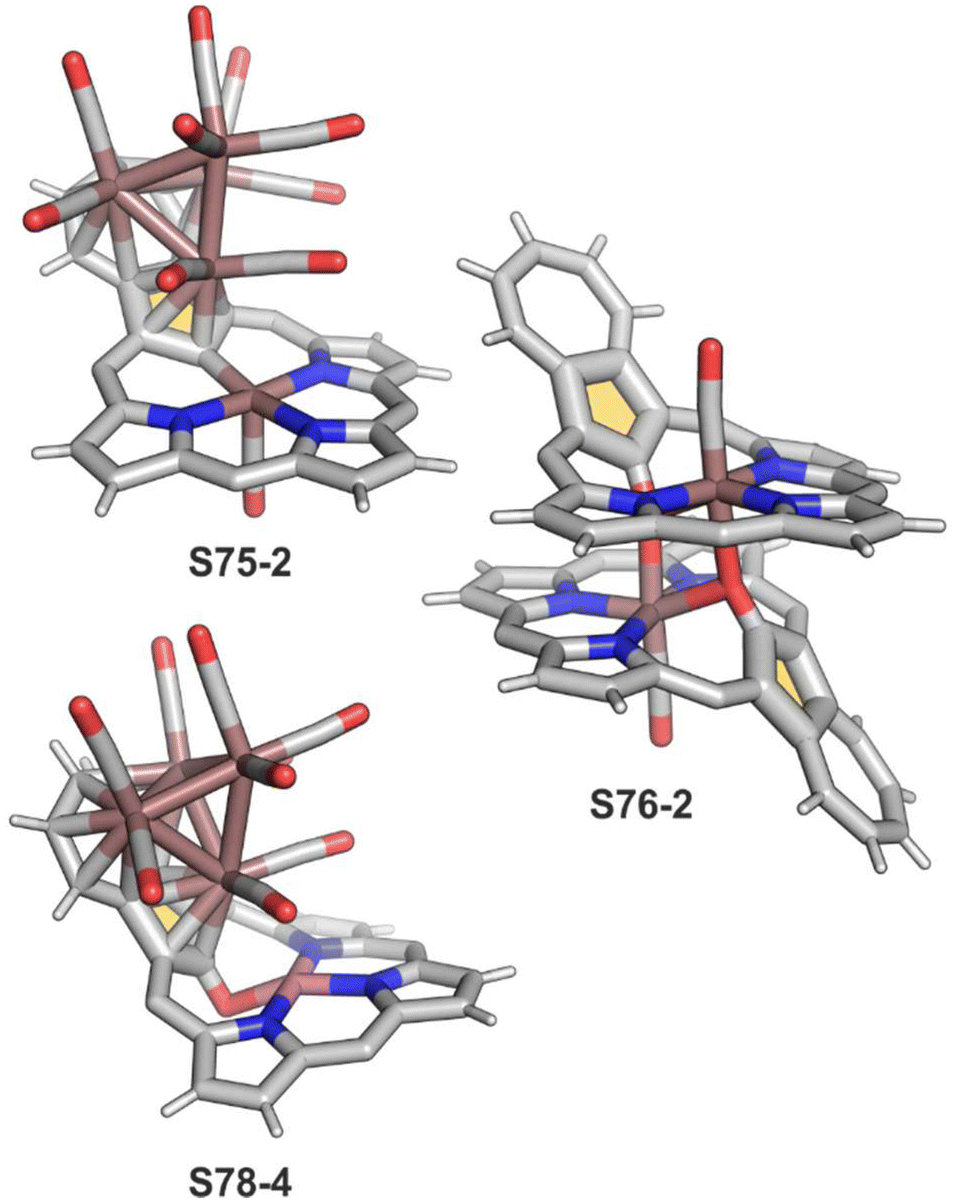 | ||
| Fig. 18 Azulene- and cavity-ruthenium-coordinated azuliporphyrins. meso-Substituents not shown. | ||
The coordination can be correlated with significantly upfield-shifted proton and carbon signals (H(23) shifts for 4.80 ppm). The molecular structure shows a significant bending of the azulene moiety to efficiently accommodate the cluster, while the RuII–C(21) distance remains in a regular binding range (2.017(6) Å for S75-1 with sixth, axial N,N′-dimethylaminopyridine ligand and 2.039(9) Å for S75-2). Besides intriguing organometallic chemistry, S75-2 demonstrates the special geometric features enabling the forced proximity of metal ions: the ruthenium(II) centre located in the [CNNN] cavity and the ruthenium(0) atom coordinating the five-membered ring (Ru1). Their contact is much shorter than the van der Waals radius sum (the RuII⋯Ru0 distance in S75-2 is 3.1596(18) Å vs. 2.9–2.7 Å within the Ru4 cluster), indicating possible intermetallic interactions. If at least one axial position of a coordination centre is unoccupied, then the other hybrids may potentially form. Accordingly, a series of bimetallic complexes S75-3, S75-4, S75-5, and S78-2 were synthesised, taking as substrates regular nickel(II), palladium(II), platinum(II), and cobalt(II) azuliporphyrins.
Regular ruthenium(II) azuliporphyrin S75-1 demonstrates an additional reactivity as well. The Ru–C(21) bond is quite stable, yet in time, in the presence of dioxygen, especially in a heated solution, S75-1 transforms into S76-1 (Scheme 76) at the oxygenated C(21) position.202 The formed complex exists in equilibrium with its dimer S76-2 in solution (Fig. 18), as long as no potential axial ligand (e.g., pyridine) is present. The C(21)⋯Ru distance increases to 2.523(13) Å in a complex with a sixth axial n-butanol ligand and the length of a seemingly single C–O bond is 1.314(16) Å.
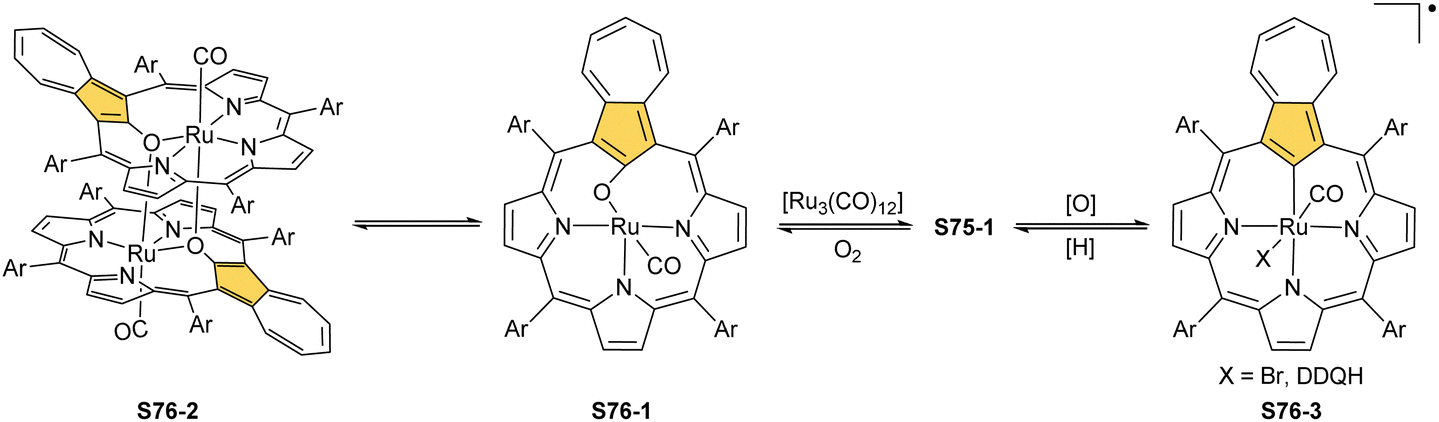 | ||
| Scheme 76 Reactivity of ruthenium(II) azuliporphyrin S75-1. | ||
The oxygen bridge of S76-1 or S76-2 can be removed in reduction with [Ru3(CO)12] to recover S75-1. On the other hand, S75-1 can get one-electron oxidised to an organoradical S76-3 with spin density noticeably distributed over the azulene's seven-membered ring.
Ruthenium(II) 23-thiaazuliporphyrin S77-1 (Scheme 77) can neither form a cluster nor a radical since the sixth coordination site is occupied with a chloride ligand.179 It can be obtained from a reaction with a range of ruthenium sources, including [RuCl2X]2, in which X is an additional π-ligand (cyclooctadiene (cod) or p-cymene). This implies the formation of the axial carbonyl ligand, present in the final product, during the insertion reaction. Yet, an attempt to synthesise the complex from different ruthenium compounds resulted, at times, in a second product, an alternative one-electron oxidised possibility – a ruthenium(III) complex with two axial chlorides (S77-2).
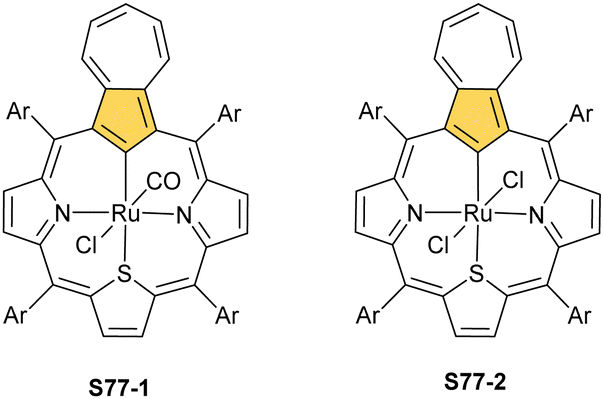 | ||
| Scheme 77 Ruthenium(II) 23-thiaazuliporphyrin S77-1 and ruthenium(III) 23-thiaazuliporphyrin S77-2. | ||
Regular cobalt(II) azuliporphyrin S78-1 (Scheme 78) forms smoothly in a reaction of azuliporphyrin S64-2 and cobalt(II) salts.196 The complete insertion occurs, inferring the activation of the C(21)–H bond and the formation of a new one, CoII–C(21), with a length of 1.930(2) Å. Treatment of S78-1 with triruthenium dodecacarbonyl [Ru3(CO)12] yields the paramagnetic Co–Ru4 hybrid S78-2. Interestingly, this low-spin cobalt(II) complex can coordinate a dioxygen molecule (S78-3), which intramolecularly oxygenates the organometallic Co–C(21) bond, forming S78-4.
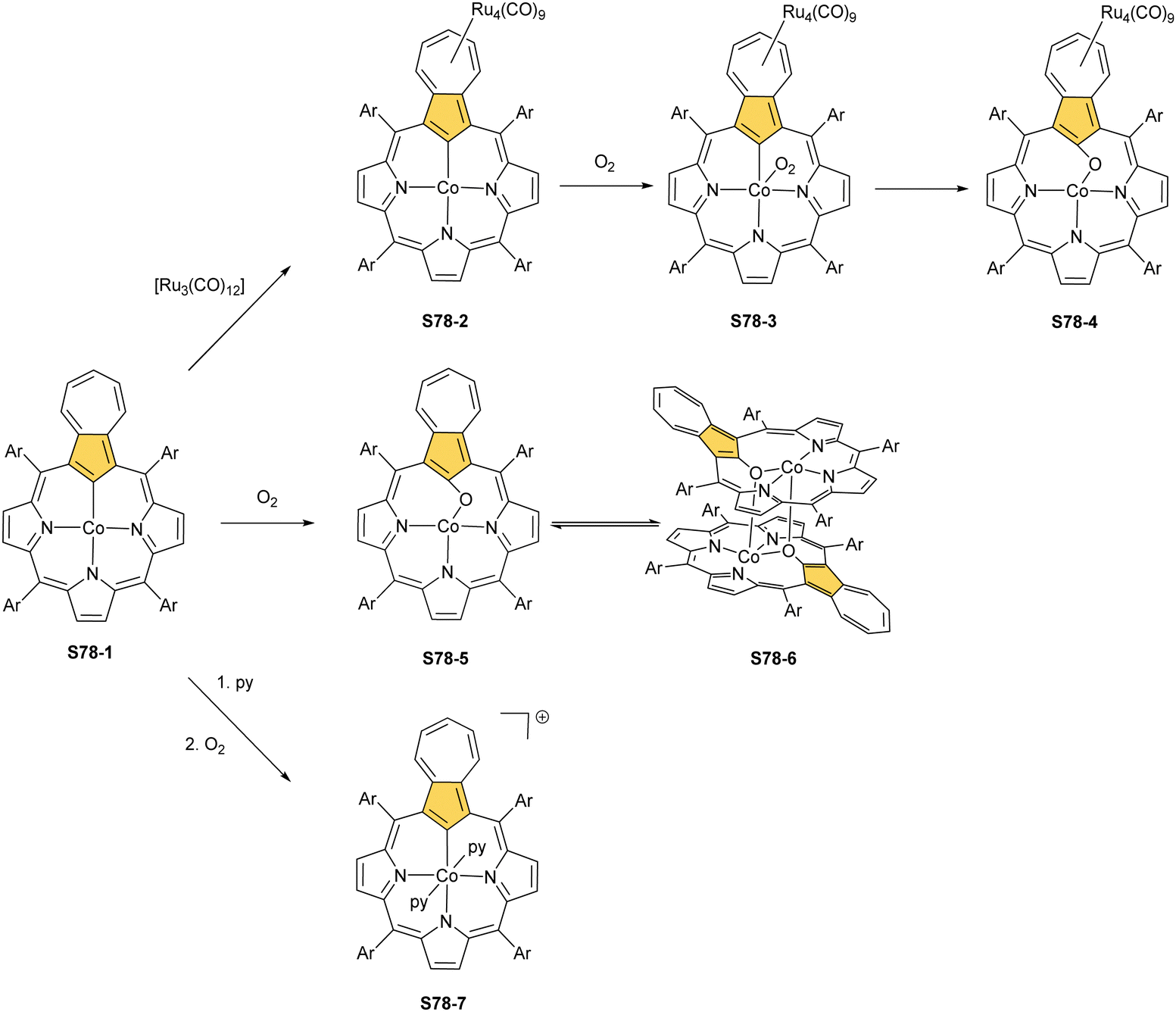 | ||
| Scheme 78 Reactivity of cobalt(II) azuliporphyrin S78-1. | ||
Due to the incorporation of the oxygen bridge in S78-4, the azulene moiety is even more deflected from the porphyrin plane, which forces even closer contact between metal centres, compared to S78-2, with a CoII⋯Ru0 distance of 2.9799(12) Å (Fig. 18). Meanwhile, the Co⋯C(21) distance measures 2.364(8) Å. The C(21)–O bond length is 1.288(8) Å, situating it on a border between the single and double bonds. Perhaps, it is one of the reasons for the relatively close distance of the metal centre to the C(21) atom. The low-spin cobalt(II) species S78-4 coordinates a dioxygen molecule but without any further reactivity observed. S78-1 gets oxygenated even faster (probably a cluster in S78-2 performs a sterically protective role) to form S78-5, which can dimerise (S78-6), similarly to the respective ruthenium(II) complex (S76-1 into S76-2). The C(21)–O distance is 1.303(4) Å and the CoII⋯C(21) separation equals 2.730(4) Å, far greater than in S78-1, but it is also significantly more distant than in a hybrid complex S78-4 (2.363(7) Å). Monomeric S78-5 and, in particular, dimeric S78-6 reveal spectroscopic features that clearly indicate the high-spin cobalt(II) ground electronic state.
This observation provides proof of a significant alteration of the overall electronic structure of Co–Ru hybrids in comparison to regular cobalt(II) azuliporphyrin. Dimer S78-6 is readily cleaved into monomeric subunits by pyridine addition which competes for the axial position. The addition of pyridine to S78-1 under strictly anaerobic conditions followed by exposure to dioxygen results eventually in one-electron cobalt(II) to cobalt(III) oxidation and diamagnetic organocobalt(III) S78-7 is formed. Analogously to a cationic PdII complex S72-1, S78-7 demonstrates increased aromaticity due to its cationic nature.
A similar complex forms in the insertion of the heavier congener of cobalt–rhodium. At first, a reaction of benzocarbaporphyrin with [RhCl(CO)2]2 yields a rhodium(I) complex S79-1 (Scheme 79).203 This asymmetric species binds the Rh(CO)2 moiety through N(22) and N(23) in the typical side-on fashion. It does not have a direct Rh–C(21) bond and the relevant distance equals 3.1775(12) Å. When S79-1 is heated in pyridine, oxidation to rhodium(III) occurs with parallel incorporation of rhodium(III) into the [CNNN] crevice of S79-2. This naturally narrows the Rh–C(21) distance to 1.988(2) Å. The analogous iridium(III) benzocarbaporphyrin S79-3 was produced via the treatment of S65-1 with an appropriate iridium(I) source. The same reactivity was also observed for naphthocarbaporphyrin.204 Internally methylated benzocarbaporphyrin was tested in the search for the organometallic environment of rhodium(III).191
 | ||
| Scheme 79 Rhodium(I)/(III) and iridium(III) complexes of β-alkylated benzocarbaporphyrin. | ||
When 23-methylbenzocarbaporphyrin S80-1 (Scheme 80) is heated in toluene with the rhodium(I) source, a methyl group migration occurs, resembling the palladium(II) 22-methylbenzocarbaporphyrin S73-1 behaviour. Finally, the methyl group lands at the C(21) position, yielding transient rhodium(III) 21-methylbenzocarbaporphyrin perfectly prearranged for the next step in this peculiar process. Thus, the rhodium(III) in the structurally forced adjacency to C(21)–Me activates one of the C–H methyl bonds and forms a rhodacyclopropane motif of S80-2. The methylene bridge enforces a rehybridisation of the C(21) atom. The coordination of the methylene group does not replace but rather supplements the C(21) atom coordination. Both Rh–C(21) and Rh–CMe distances are similar, being slightly longer for the in-plane bond to C(21) (2.1329(11) Å vs. 1.9932(11) Å to CMe). This specific C–H activation leads to a neutral complex stabilising rhodium(III) in the crevice.
 | ||
| Scheme 80 Rhodium insertion into 23-methylbenzocarbaporphyrin S80-1, yielding rhodium(III) 21-methylbenzocarbaporphyrin with a rhodacyclopropane motif S80-2. | ||
Rhodium insertion into S62-2 can bring another yet related product during prolonged heating with xylene isomers.205 Regardless of the xylene isomer, the insertion is accompanied by activation of xylene methyl groups, followed by axial coordination, yielding S81-1 (Scheme 81 and Fig. 19). This allows for RhIII stabilisation within azuliporphyrin with two starting inner protons. The distance from RhIII to C(21) (1.966(3) Å) is similar to the one from the methylene group in the axial position (2.073(4) Å). Interestingly, a similar procedure with toluene failed to produce an analogue of S81-1 (presumably because of a too-low boiling point of toluene when compared to xylene).
 | ||
| Scheme 81 Rhodium and iridium insertion into β-alkylated benzocarbaporphyrin in xylene isomers or acetone/toluene mixture. | ||
 | ||
| Fig. 19 Axial and equatorial organometallic rhodium(III) complexes of azuliporphyrin. | ||
Yet, activating a methyl group in acetone is possible when the initial insertion is carried out in acetonitrile and the formed rhodium azuliporphyrin is directly transferred into a mixture of acetone and toluene to afford S81-2, especially under basic conditions. The treatment of azuliporphyrin S65-2 with [IrCl(COD)]2 in xylenes leads to solvent activation as well. The solely isolated iridium(III) azuliporphyrin S81-3 incorporates the axially coordinated methylbenzoyl ligand presumably formed by the oxygenation of the originally linked σ-methylbenzyl unit. Here, again, both organometallic bonds are of similar distance (Ir–C(21): 1.985(7) Å and Ir–C(O): 1.981(7) Å). A similar insertion trial conducted in toluene did not provide any metallated derivative.
Fundamental 21-carbaporphyrin
The exploration of carbaporphyrin chemistry, reported in the previous sections, brought us to the most fundamental motif one could imagine to be explored in organometallic chemistry encompassed in a porphyrinic framework, namely 21-carbaporphyrin (S1-3). Here, one of the inner nitrogen atoms is simply replaced with a carbon atom.21-Carbaporphyrin, sometimes called true carbaporphyrin, was first reported as a β-substituted derivative. In fact, the publication occurred soon after N-confused porphyrin, but till present, this subfield remains to be extensively investigated. This state may be related to demanding synthetic pathways which afford S82-1/S82-2/S82-3 (Scheme 82), or S1-3. Thus, the first 21-carbaporphyrin S82-1/S82-2 was reported in 1996 by Berlin.206 The reaction of triformylcyclopentadiene with tripyrrane in the presence of HBr/glacial acetic acid as the catalyst gives 2-formyl-21-carbaporphyrins S82-1 and S82-2 in 7.8% and 6.5% yields, respectively. The synthesis and characterisation of S82-1 were refined in 1997 by Lash and Hayes.207
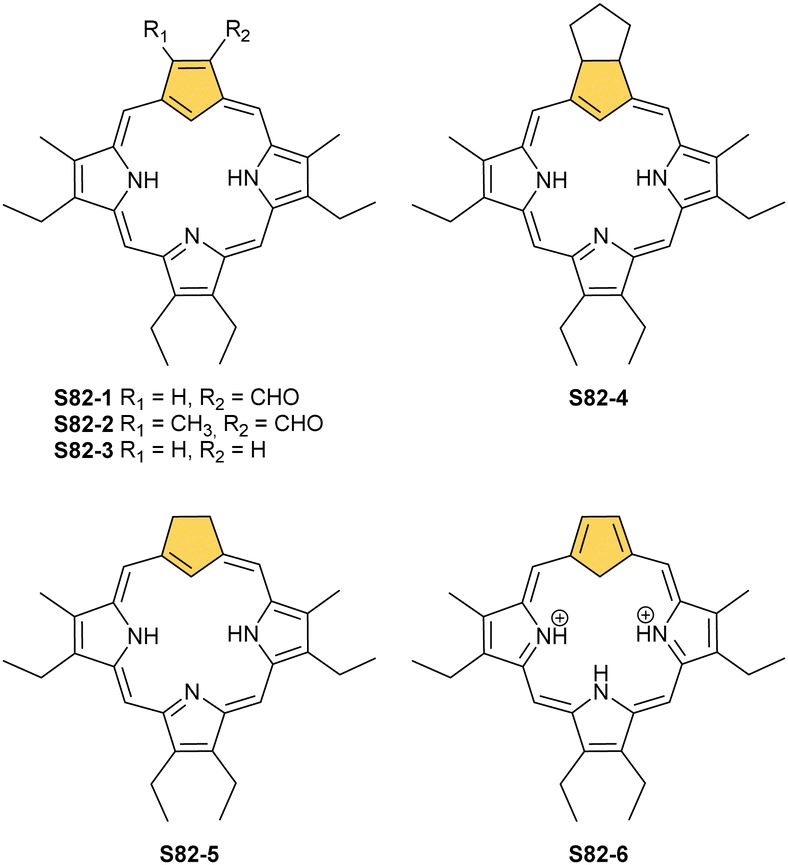 | ||
| Scheme 82 β-Alkylated 21-carbaporphyrinoids. | ||
Macrocycle S82-1 (and other similar derivatives) is fully aromatic, as clearly indicated by the −7.23 ppm chemical shift of the inner H(21) atom. The preliminary metallations with zinc(II) and nickel(II) were described as unsuccessful.206 Another molecule reported in a path to 21-carbaporphyrins is 21-carbachlorin isolated initially as the derivative, which incorporates an additional fused five-membered ring S82-4.208 A version with a non-substituted five-membered ring (S82-5) was also reached after the suitable carbocycle substrate redesign.209S82-5 is formed in the [3+1] MacDonald condensation using 3-ethoxymethylenecyclopentene-1-carbaldehyde and tripyrrane catalysed by TFA. The subsequent oxidation with aqueous ferric chloride solution results in S82-5 with an 11–16% yield. The subsequent careful oxidation of S82-5 with one equivalent of DDQ affords β-alkylated-21-carbaporphyrin S82-3 with a remarkable outcome (70%). The facile diprotonation of S82-5 produces a dication S82-6, which accommodates the additional protons at the single available pyrrolic nitrogen atom and – noticeably – at the rehybridised tetrahedral C(21) atom. The dication preserves the strong diatropic character of maternal S82-3, as reflected by the 1H chemical shift of C(21)H2 (−8.27 ppm). A 21-chlorinated derivative of S82-5 has also been obtained.210 In this case, the subsequent oxidation with DDQ results in dechlorinated S82-3 and its derivatives in which R1 and/or R2 get substituted with chlorine atoms (2-chloro- or 2,3-dichloro-21-carbaporphyrins). In general, β-alkylated 21-carbaporphyrins S82-1/S82-2/S82-3 bear some resemblance to widely used octaethylporphyrin.
A significant synthetic effort was also devoted to creating meso-substituted 21-carbaporphyrins, which resemble, at least in their general scaffolds, extensively explored meso-tetraarylporphyrins. Chronologically, this area has been initialised by the synthesis of 10,15-dimesityl-21-carba-23-thiaporphyrin S83-1 (Scheme 83), and its reduced congener S83-2.211 The synthesis of carbachlorin S83-2 involves the one-pot reaction of the mixture of the carba analogues of tripyrrane with 2,5-bis(mesitylhydroxymethyl)thiophene catalysed by BF3·Et2O, eventually followed by oxidation with p-chloranil (tetrachloro-1,4-benzoquinone). The subsequent careful oxidation of S83-2 with only one equivalent of DDQ at room temperature gives S83-1 with a 25% yield. The intriguing minor side-product S83-3, which incorporates a chloranil-derived fragment linked to the C(21) and N(22) atoms, was isolated as well. The macrocycles are aromatic, as reflected by the H(21) chemical shifts of −5.16 and −4.79 ppm for S83-2 and S83-1, respectively.
 | ||
| Scheme 83 21-Carba-23-thiaporphyrin derivatives. | ||
In separate studies, another oxidation product was recognised, namely the C(21)–C(21)′ bridged dimer S83-4.212 Bis(carbathiaporphyrin) S83-4 was obtained with 46% yield by oxidation of S83-1 with DDQ. The two-electron reduction of S83-4 with zinc dust results in splitting the C(21)–C(21′) bond, recovering the monomer S83-1. The reactivity of the incorporated cyclopentadiene ring has been noted. For instance, the outer rim [C(2), C(3)] fragment reacts with hydrochloride or hydrobromide, yielding doubly halogenated 21-carbachlorins. Similarly, oxygenation products were identified, which form during the protonation of a molecule with ca. 10 eq. of TFA.
The rational synthesis of meso-tetraaryl-21-carbaporphyrin S1-3 was eventually reported.213 As cyclopentadiene tends to be unstable and reactive, a tripyrrane derivative used in condensation contains a 1,3-ferrocenyl unit S84-1 (Scheme 84). Thus, incorporating a cyclopentadiene moiety into the meso-tetraarylporphyrin framework results in S1-3 with a suggested transient formation of ferrocenylporphyrins like S84-2. The molecular design of S1-3 preserves all essential virtues of the original tetrapyrrolic architecture of meso-tetraarylporphyrin, including the perfect match between the ionic radii of an inserted metal and the size of the macrocyclic [CNNN] core, as well as steric protection provided by thoughtfully chosen meso-aryl substituents. The molecule is aromatic, similar to β-substituted analogues S82-1/S82-2/S82-3. The characteristic H(21) chemical shift of aromatic S1-3 equals −5.86 ppm and changes to −6.26 ppm after the formation of dication. Diprotonation of S1-3 is accompanied by C(21)-centred protonation and the formation of a methylenic C(21)H2 unit.
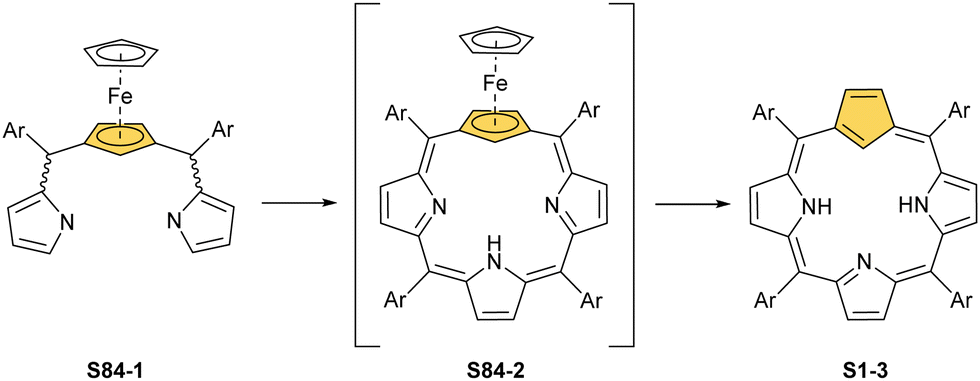 | ||
| Scheme 84 Synthesis of meso-tetraaryl-21-carbaporphyrin. | ||
Organometallic chemistry of 21-carbaporphyrins
Palladium(II) seems to be a metal cation of choice to initially test the coordination properties of carbaporphyrinoids, including 21-carbaporphyrins.
The feasible rearrangements have been well illustrated, focusing on inner core methylated β-substituted 21-methyl-21-carbaporphyrin S85-1 (Scheme 85) and 22-methyl-21-carbachlorin S86-1 (Scheme 86). The insertion of palladium(II) into S85-1 yields neutral S85-2.209,214 The coordination enforced rehybridisation of the C(21) atom from a trigonal one to a tetrahedral one. The insertion of a palladium(II) ion into S86-1 initially produces palladium(II) N(22)-methylated 21-carbachlorin S86-2. The insertion requires the inner C(21)–H bond activation. The subsequent stepwise migration of the methyl group, accompanied by oxidation, affords palladium(II) C(21)-methylated carbaporphyrin S86-3.209
 | ||
| Scheme 85 Palladium insertion into β-substituted 21-methyl-21-carbaporphyrin S85-1. | ||
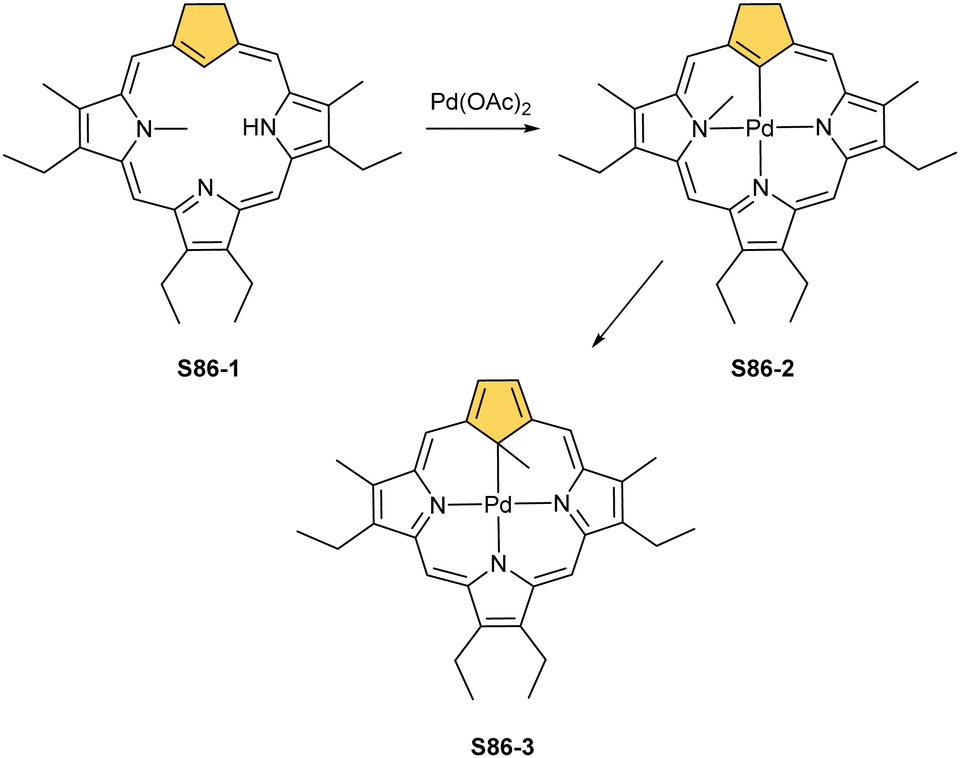 | ||
| Scheme 86 Methyl group migration after palladium insertion into N(22)-methylated 21-carbachlorin S86-1. | ||
21-Carba-23-thiaporphyrin S83-1 and its counterpart S83-2 are predestined to act as macrocyclic dianionic ligands because of a sulphur atom.211 Both carbaporphyrinoids form palladium(II) complexes S87-1 (Scheme 87) and S87-2. However, in these cases, in contrast to S86-3 and S85-2, the organometallic bond is formed between the palladium(II) and sp2-hybridised inner carbon centres.
 | ||
| Scheme 87 Palladium(II) complexes of 21-carba-23-thiaporphyrin and 21-carba-23-thiachlorin. | ||
The typical palladium(II) insertion is effective for S1-3 as well, forming complex S88-2 (Scheme 88).213 The coordination of palladium(II) stabilises the macrocyclic scaffold of the tautomer with sp3-hybridised C(21), prone to act as a dianionic ligand. Silver(III), gold(III), and nickel(II) complexes of β-substituted carbaporphyrins and their C(21)- or N(22)-methylated derivatives were also reported.214
 | ||
| Scheme 88 Synthesis of palladium(II) 21-carbaporphyrin. | ||
A reaction of S89-1 (Scheme 89) with [RhCl(CO)2]2 brings, at first, the side-on rhodium(I) complex S89-2 with two inner adjacent nitrogen atoms solely engaged in coordination.204S89-2 can be heated in pyridine solution and be efficiently transformed into regular rhodium(III) 21-carbaporphyrin S89-3, acquiring the classical [CNNN] binding surroundings.
 | ||
| Scheme 89 Synthesis of the rhodium(I) and rhodium(III) complexes of the β-substituted 21-carbaporphyrin derivative. | ||
Somewhat analogously to already described rearrangements of azuliporphyrin S64-2/S65-2 into benzocarbaporphyrin S64-1/S65-1, meso-tetraaryl-m-benziporphyrin or the inner carbon-alkylated derivative undergo an extrusion of one of the m-phenylene outer carbon atoms, when coordinated with rhodium(III) or gold(III) ions.28,215,216 In the first step, the reaction with [RhCl(CO)2]2 affords nonaromatic rhodium(III) m-benziporphyrin S90-1 (Scheme 90) with a typical equatorial [CNNN] surrounding of central metal ion to be accompanied by axially coordinated chloride and carbonyl ligands.28 This species, when exposed to dichloromethane on a silica gel column under oxidative conditions, transforms into aromatic rhodium(III) m-benziporphyrin S90-3, which incorporates the rhodacyclopropane motif substituted with a formyl group. S90-1 converts into S90-3 during prolonged reflux in dichloromethane through an initial formation of S90-2 containing a 2-chlorovinyl moiety generated from the solvent. S90-2 and S90-3, when mixed with basic alumina, are immediately transformed into a mixture of rhodium(III) 21-carbaporphyrins S90-4, S90-5, and S90-6. The C(sp3) centre in S90-4 binds to the RhIII ion (Rh–C(21), 2.116(7) Å) and, simultaneously, a carbon atom from a CHCHO substituent (Rh–C, 2.060(9) Å) forming a rhodacyclopropane motif (Fig. 20).
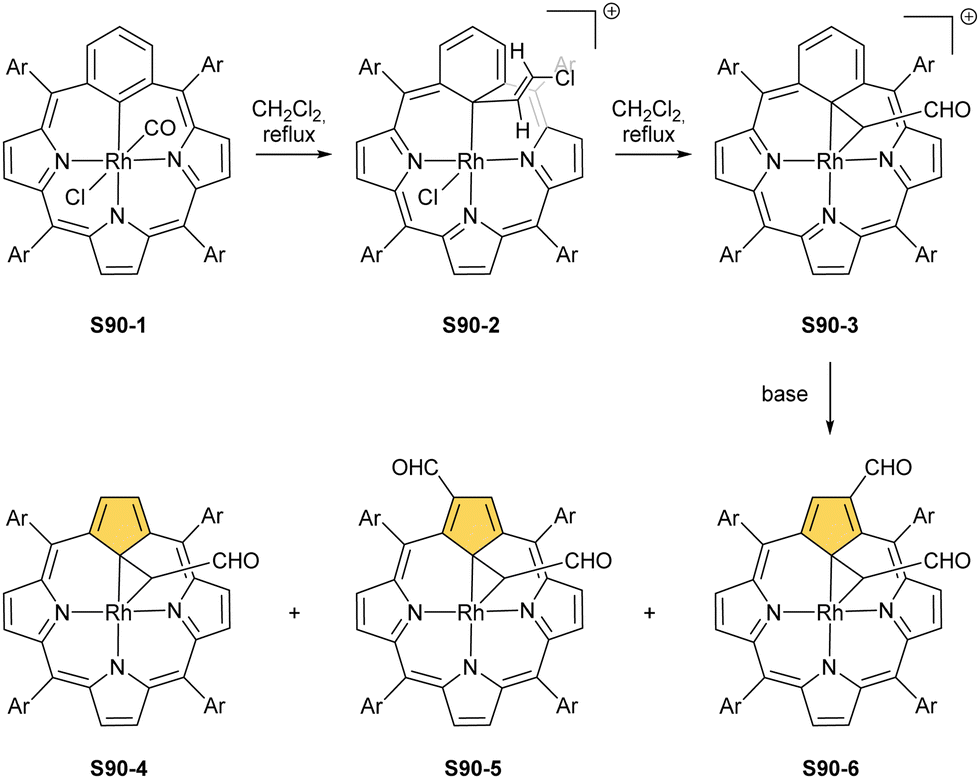 | ||
| Scheme 90 Reactivity of rhodium(III) meta-benziporphyrin S90-1. | ||
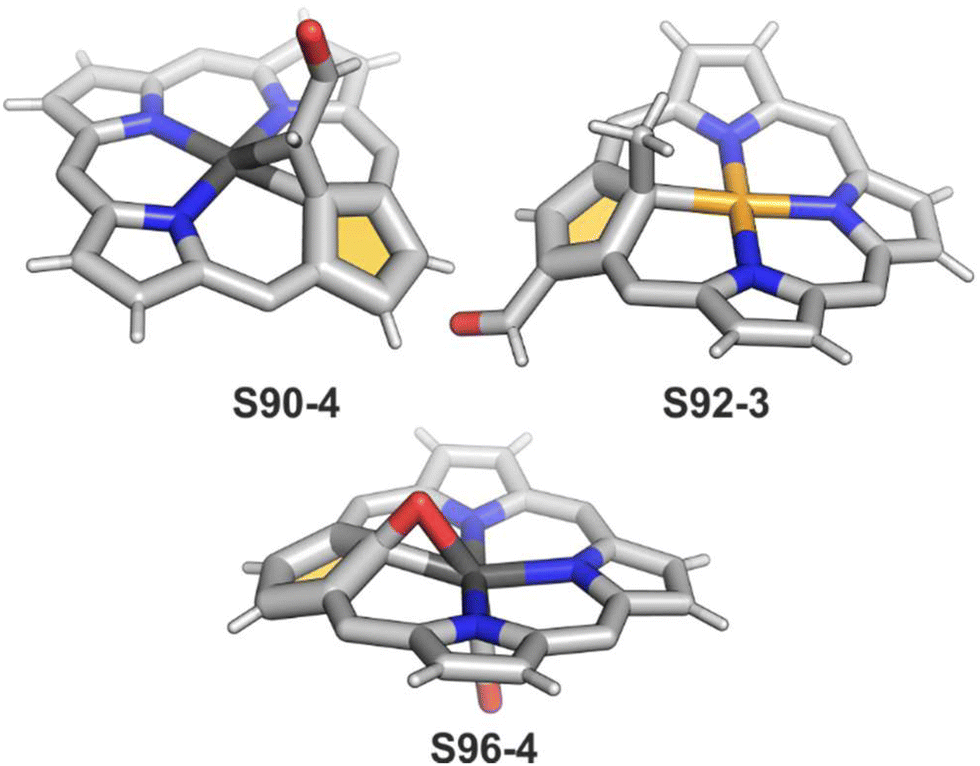 | ||
| Fig. 20 Contraction-derived carbaporphyrin complexes. meso-Substituents not shown. | ||
Consequently, the rhodium(III) and gold(III)-triggered transformations of a m-phenylene ring embedded in 22-alkyl-m-benziporphyrins were explored.215,216 When S91-1 (Scheme 91) is mixed with basic alumina or treated with selected basic reagents in solution (triethylamine, 2,4,6-trimethylpyridine), it quickly converts into a mixture of rhodium(III) 21-(μ-ethylidene)-21-carbaporphyrins S91-2 and two diastereomers of rhodium(III) 2-formyl-21-(μ-ethylidene)-21-carbaporphyrin.
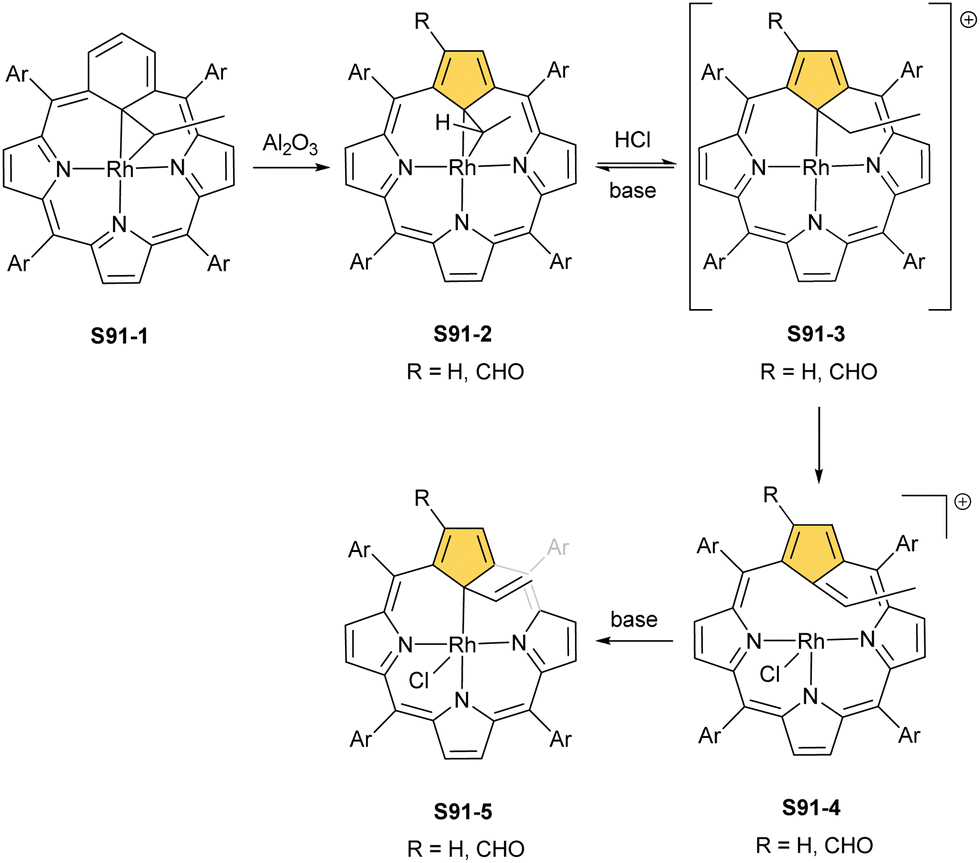 | ||
| Scheme 91 Contraction of rhodium(III) 22-(μ-ethylidene)-m-benziporphyrin S91-1. | ||
An exposure of S91-2 to hydrogen chloride potentially results in the formation of rhodium(III) 21-ethylcarbaporphyrin S91-3. The thermodynamically reversible protonation is centred at the μ-ethylidene substituent in S91-2. The protonation/deprotonation is convoluted with the cleavage/reformation of rhodacyclopropane. Subsequent two-electron oxidation of cationic S91-3 yields S91-4, which formally incorporates the 6-methylfulvene motif into the 21-carbaporphyrin frame. The side-on interaction between rhodium(III) and the C(21)C(25) double bond of fulvene was suggested. The situation changes after adding a nitrogen base (e.g., Et3N) to the solution of S91-4 as it activates isomerisation to rhodium(III) 21-vinylcarbaporphyrin S91-5.
Treatment of 22-methyl-m-benziporphyrin with sodium tetrachloroaurate(III) in dichloromethane results in quantitative transformation into gold(III) 22-methyl-m-benziporphyrin S92-1 (Scheme 92). This species is chemically unstable and readily converts into a mixture of gold(III) 21-methyl-21-carbaporphyrins S92-2 and S92-3.215 In striking contrast, the insertion of gold(III) to regular m-benziporphyrin, carried out under similar conditions, produced solely unreactive gold(III) m-benziporphyrin. Thus, the inner alkylation of m-phenylene is a prerequisite for initiating the contraction.
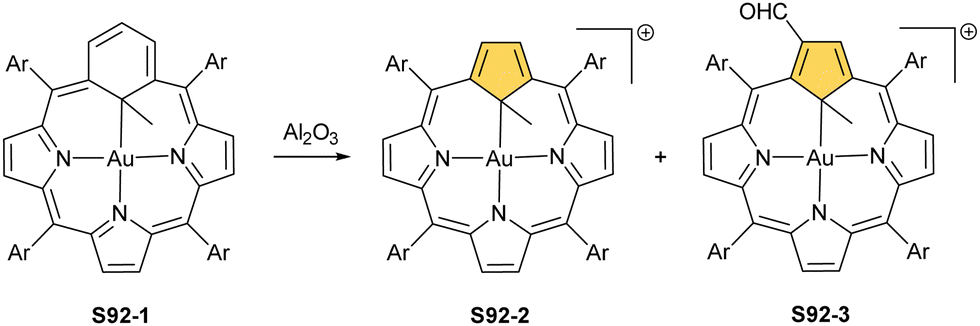 | ||
| Scheme 92 Contraction of gold(III) 22-methyl-m-benziporphyrin S92-1. | ||
As gold(III) coordination preferences differ from rhodium(III), the methyl group is not involved in direct binding with the gold(III) centre in S92-1 or products of contraction (S92-2 and S92-3). The feature is clearly indicated by the Au⋯Cmethyl distance of 2.868(13) Å as compared to that of 2.073(11) Å for Au–C(21) determined for gold(III) 2-formyl-21-methyl-carbaporphyrins S92-3 (Fig. 20). The presence of the formyl group at the perimeter of S92-3 allows for its peculiar transformation. In the presence of Et3N, the 2-formyl group in S92-3 is substituted by the hydroxyl one. It forms cationic gold(III) 2-hydroxy-21-methyl-21-carbaporphyrin S93-2, eventually affording S93-1 (Scheme 93) under basic conditions. S93-1 is only weakly aromatic, as shown by the 1H NMR pattern. With the addition of acid, the equilibrium shifts towards S93-2, which regains the marked macrocyclic aromaticity.
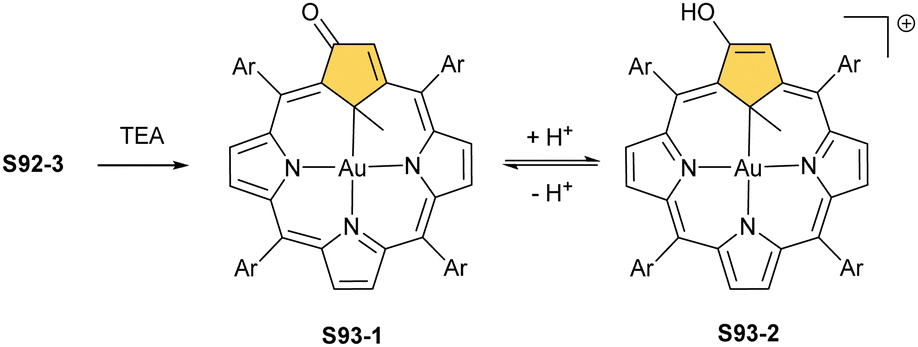 | ||
| Scheme 93 Reactivity of gold(III) 2-formyl-21-methyl-21-carbaporphyrin S92-3. | ||
It is possible to induce the contraction of a differently introduced benzene ring, namely the one in para-benziporphyrin, through gold coordination.217 An extrusion of the external carbon atom has been suggested in the case of m-benziporphyrin contraction. In the case of isomeric p-benziporphyrin, an alternative route of contraction has been encountered. Namely, the inner carbon atom of p-phenylene, which directly interacts with a coordinating metal ion, is, in due course, extruded.217,218 The gold(III)-stimulated contraction of p-phenylene to cyclopentadiene can be encountered in a reaction of p-benziporphyrin with sodium tetrachloroaurate(III) and potassium carbonate in dichloromethane.217 The macrocycle in the gold(III) complex S94-1 (Scheme 94) after contraction acts as trianionic ligand. The [CNNN] equatorial surrounding was detected for S92-3 as well, albeit the enforced by C-methylation geometry around C(21) is tetrahedral. It is noteworthy that S94-1 experiences the reversible protonation centred at the inner C(21) atom with the formation of a less aromatic cationic derivative S94-2.
 | ||
| Scheme 94 Protonation of gold(III) 21-carbaporphyrin S94-1. | ||
Treatment of p-benziporphyrin with palladium(II) chloride in acetonitrile yields S95-1 (Scheme 95). In the presence of potassium carbonate, this species transforms into palladium(II) 21-carbaporphyrin S88-2 and palladium(II) 21-formyl-21-carbaporphyrin S95-2.27 The molecular structure of S95-2 shows the conventional square-planar coordination environment of the palladium(II) centre with the relatively short Pd⋯C(formyl) distance (2.531(2) Å). The Pd–C(21) distance of 2.084(2) Å clearly reflects the σ-bonding interaction.
 | ||
| Scheme 95 Contraction of palladium(II) p-benziporphyrin S95-1. | ||
It is also important to note that the fused benzoanologue of meso-tetraaryl-p-benziporphyrin, i.e., meso-tetraaryl-1,4-naphthiporphyrin, when exposed to the coordination of palladium(II) or gold(III), undergoes the contraction of the innner phenylene ring to appropriate palladium(II) or gold(III) benzocarbaporphyrin complexes.217,218 It additionally reconfirms contraction centred at the inner carbon atom forced to interact with metal centres, as opposed to contractions in m-benziporphyrins.
Eventually, rhodium(III) coordination was reported to induce the contraction of p-benziporphyrin.219 Once again, similar to palladium(II) or gold(III) chemistry, rhodium(III) p-benziporphyrin S96-1 (Scheme 96), when heated under basic conditions, contracts smoothly to rhodium(III) 21-methylene-21-carbaporphyrin as the major aromatic product (S96-2). The molecule incorporates a rhodacyclopropane motif also identified in the products of rhodium(III) 22-alkylated m-benziporphyrin contractions.
 | ||
| Scheme 96 Contraction of rhodium(III) p-benziporphyrin S96-1. | ||
The bound methylene bridge can be reversibly protonated, affording the 21-methylated derivative S96-3. S96-2 undergoes oxygenation when acidified with gaseous HCl and transferred onto basic alumina. The oxygenation leads to rhodium(III) 21-oxo-21-carbaporphyrin S96-4, with the metal ion interacting with both atoms from the C(21)–O(21) fragment. Thus, S96-4 gains an oxygen bridge between C(21) and Rh atoms without breaking the organometallic bond (Fig. 20). The respective distances are 2.099(2) Å [Rh–C(21)], 2.035(2) Å [Rh–O(21)], and 1.343(6) Å [C(21)–O(21)]. Additionally, the methylene bridge gets oxidised as well, forming an axially coordinated carbonyl group. Also, the rhodium(III) centre in S96-4 coordinates to a double bond acquiring a mixed σ/π organometallic coordination (S96-5). It occurs when the centre is deprived of the carbonyl group in a reaction with methanolate and then exposed to an alkene, e.g., 1-pentene.
To summarise, we showed the richness of organometallic interactions between the metal centre and the inner carbon atom of monocarbaporphyrins. A selection of more common motifs is presented in Table 1.
| *Italics for the distance to substituent’s carbon. **The closest contact [in both cases it is C(21)]. |
|---|
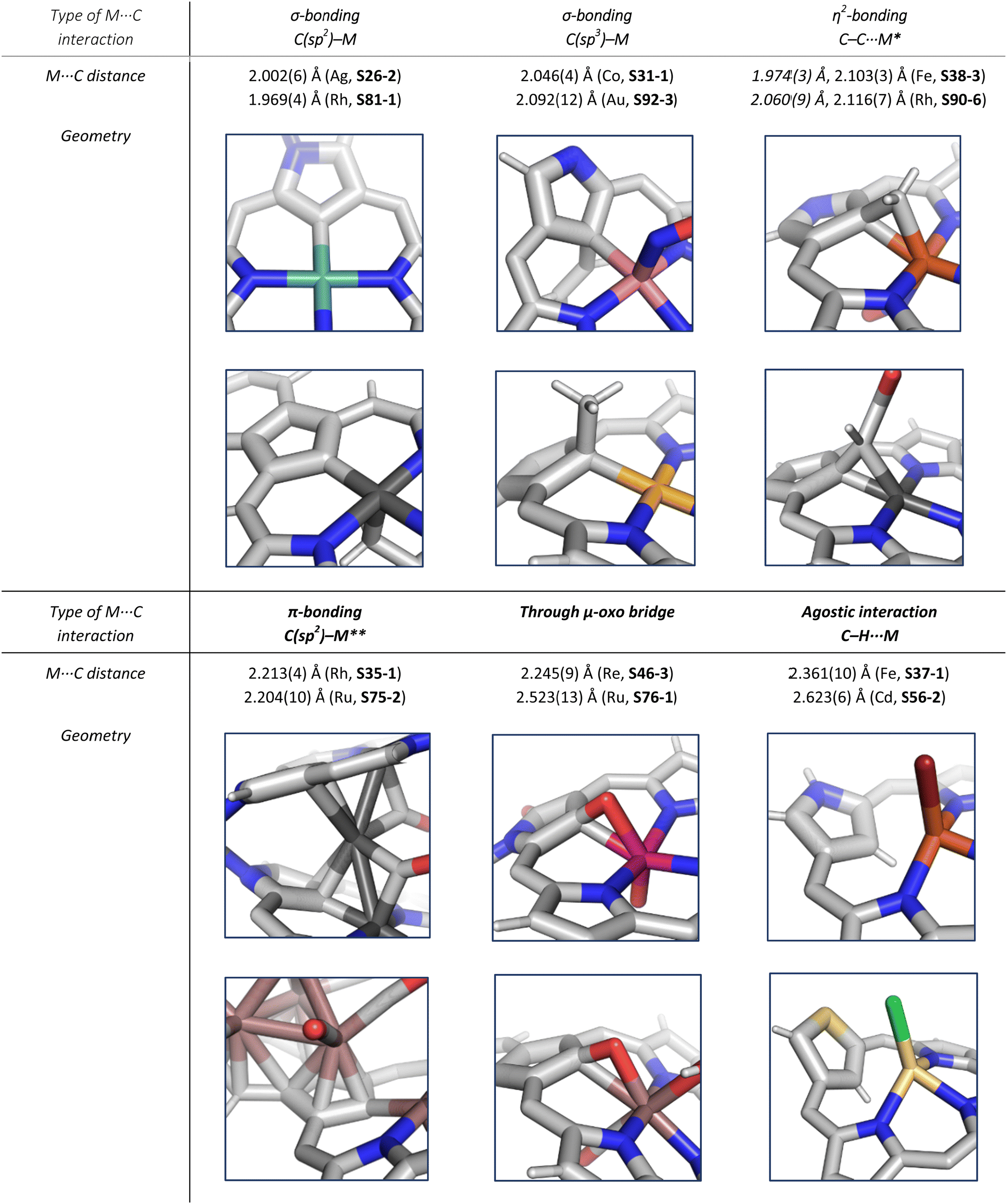
|
Many-carbon tetraphyrin and related systems
In previous sections, an overview of various monocarbaporphyrins, completed with meso-tetraaryl-21-carbaporphyrin, was presented. In particular, the utility as platforms for macrocyclic organometallic chemistry was addressed. Over recent years, a dynamic development of monocarbaporphyrinoid organic chemistry has been seen that resulted in the availability of an impressive portfolio of ligands, mainly with a [CNNN] donor set. Consequently, a promising direction of expansion is expected to present di-, tri-, and tetracarbaporphyrins.This way, one can establish a playground to explore the novel aspects of the chemistry of fundamental nature. This will include specific metal binding modes and intermediaries between the regular bond and the van der Waals interaction. The distinctive macrocyclic environment creates a long-sought chance to trap unique organometallic intermediates, typically postulated in the catalytic process, shedding some light on their real nature. The basic strategies to construct multicarbaporphyrins are directly derived from monocarbaporphyrin chemistry taking advantage of well-elaborated syntheses, which initially allowed for the incorporation of a single carbocyclic building block.
N-confusion
The N-confusion adopted to more than one pyrrolic ring seemed to be the strategy to be explored while aiming for multicarbaporphyrins. The probabilities of double or triple N-confusion in a simple condensation seem unlikely for statistical reasons.Construction of the [CCNN] frame was preceded by forming a predesigned building block, i.e., N-confused dipyrromethane and further condensation with an aldehyde. Such an approach yields cis-doubly N-confused porphyrin S97-1.220 Additionally, as the reaction is conducted in chloroform stabilised with ethanol, an outer position on one of the inverted rings becomes substituted with an ethoxy group. In total, the macrocycle has three inner dissociable protons in the cavity. Macrocycle S97-1 (Scheme 97) is only borderline aromatic as the delocalisation pathway has a formal disruption. It results in a relatively small upfield relocation of the inner H(21) and H(22) atoms, with chemical shifts of 3.52 and 3.22 ppm, respectively.
 | ||
| Scheme 97 cis-Doubly N-confused porphyrin S97-1 and benzene-fused doubly N-confused porphyrin S97-2. | ||
The specific core modification of cis-doubly N-confused porphyrin (the isomeric frame as compared to S97-1, please notice the alternative location of outer nitrogen atoms) was presented recently.221 It took the shape of a methine bridge between the inner carbon atoms (S97-2). The complete macrocycle possesses a shrunk cavity with a motif similar to the already presented in the chemistry of NCP, which led to N-heterocyclic carbene embedded in an NCP framework (S47-2).145 The bridge also imposes a different π-delocalization pathway, and that the molecule is more aromatic than the unbridged congener, as the chemical shift of the inner bridge proton equals −1.85 ppm.
Another possible variation is introducing N-confused pyrrole rings in the mutual translocation. Thus, trans-doubly N-confused porphyrin S98-2 was obtained in a self-condensation of N-confused dipyrromethene carbinol.222 In this reaction, N-confused N-fused porphyrin S98-1 (Scheme 98) forms at first and later transforms into S98-2 under basic conditions in an alcohol environment. The process is accompanied by ethoxylation of the outer carbon atom. The formed S98-2 is strongly aromatic in contrast to its cis counterpart, S97-1. The inner CH protons [H(21) and H(23)] resonate at −4.34 and −4.36 ppm, respectively. An attempt to open a semi-fused macrocycle with thiophenol instead of alcohol results in more complex chemistry. It affords a substitution of the outer N-confused pyrrole carbon atoms of both rings, generating S98-3.223 It is reduced when compared to S98-2. Its electronic structure varies significantly from the aromatic counterpart, as the π-delocalization pathway is interrupted, so the inner CH protons resonate at 8.29 ppm. It can be reversibly oxidised to an aromatic form S98-4 (the diagnostic CH signal relocates to −4.05 ppm).
 | ||
| Scheme 98 Reactivity of N-confused N-fused porphyrin S98-1. | ||
Another approach was applied to obtain trans-doubly N-confused porphyrin with two oxo substituents on outer carbon atoms.224S99-1 (Scheme 99) can be efficiently obtained in condensation of N-methylated building units, N-confused tripyrrane and N-confused dicarbinol (condensation [3+1]), followed by oxidation in which lactam functionalities are formed. The molecule possesses the 16π-electron delocalisation pathway, leading to its antiaromaticity. This is depicted with a chemical shift of inner protons equal to 12.98 ppm. After two-electron reduction with NaBH4, one gets an aromatic macrocycle (S99-2; the contributor with separated charges is shown), with inner protons at −1.88 ppm. By using DDQ instead of chloranil for oxidation, the tetraoxygenated product S99-3 is formed instead of S99-1.225
 | ||
| Scheme 99 trans-Doubly N-confused porphyrin derivatives. | ||
Incorporation of five-membered rings – cyclopentadiene and fused derivatives
Simple building blocks, including those incorporating azulene, indene, or cyclopentadiene, were used as the rewarding units to construct cis- or trans-dicarbaporphyrins. A first example is β-substituted cis-diazuliporphyrins S100-1 (Scheme 100) which also accounts for the first case of mesoionic porphyrinoid.226 A diazulene precursor is condensed with dipyrromethene. The compound has been isolated in the solid state as monoprotonated hydrochloride or hydrobromide salts. In solution, the isolated species also exists in a cationic form, and the inner CH atoms resonate at −0.05 ppm (bromide as a counterion).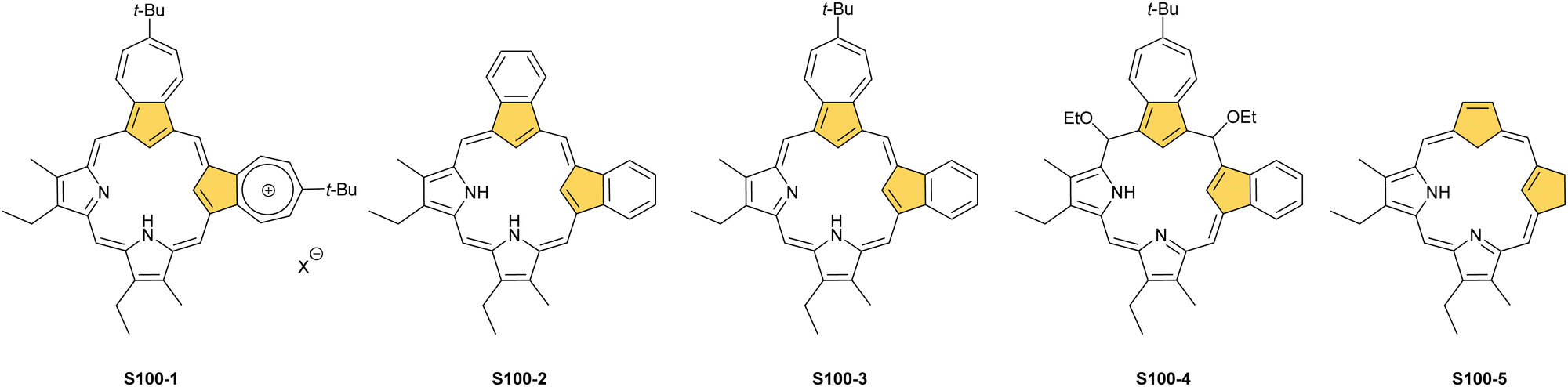 | ||
| Scheme 100 β-Substituted cis-dicarbaporphyrins. | ||
Similarly, cis-dibenzocarbaporphyrin S100-2 is obtained from a diindene precursor and dipyrromethane.227 The porphyrinoid is fully aromatic (the H(21), H(22) chemical shift locates at −6.24 ppm) and contains four inner protons. Protons from nitrogen atoms can be removed using 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) to form macrocyclic dianions. The alternative stepwise protonation is centred at the inner carbon atoms, which requires their trigonal to tetrahedral rehybridisation.
Eventually, the azuli-benzocarba hybrid S100-3 was produced, preceded by azulene and indene coupling to form a building unit for [2+2] condensation.228,229 Its aromaticity is hampered by azulene. Still, the inner H(21) and H(22) atoms demonstrate the upfield relocation (chemical shifts equal 1.6 and 3.5 ppm). The protonation of S100-3 promotes their further upfield repositioning, similar to azuliporphyrins. Structurally, the cavity of S100-3 contains three removable protons, i.e., the intermediate situation between cis-diazuli S100-1, which could stabilise dications in its neutral state, and cis-dibenzocarba S100-2 analogues. S100-3 reacts with silver(I) acetate in ethanol, yet the expected silver(III) coordination does not occur and instead two ethoxy groups are added at meso positions flanking azulene to form nonaromatic S100-4.229
cis-Dicarbaporphyrin, which incorporates two five-membered carbocycles, seemed to be logically targeted as the next step. The procedure resembles that applied for S100-1 and S100-2 starting, however, using dicyclopentadienomethane.230 Peculiar dicarbachlorin S100-5, which contains an internal methylene group, was isolated (Fig. 21). Still, S100-5 is fully aromatic (H(21) and H(22) shifts are −6.68 and −4.03 ppm) and, despite one deprotonated nitrogen atom, the cavity possesses four protons. This way, all kinds of environments occur by varying the carbon-providing units.
 | ||
| Fig. 21 Dicarbaporphyrinoids with C(sp2) and C(sp3) in the cavity. | ||
The translocation of the respective carbocyclic rings afforded a separate subclass of dicarbaporphyrins. The elaborate procedures usually involve a condensation of activated azulene or indene fragments with two units containing pyrrole or a heterocycle. Due to less directional synthesis, the yields are sometimes lower than for the respective cis analogues. The first dicarbaporphyrinoid with two opposite benzocarba moieties (S101-1, Scheme 101) was reported as early as 1999.231 The properties of S101-1 resemble those reported for cis-dibenzocarbaporphyrin S100-2. The inner protons resonate at −5.7 ppm, reflecting strong diatropicity. The macrocycle is somewhat less stable in solution than 22,24-dioxa232 and the cis analogues.
 | ||
| Scheme 101 β-Substituted trans-dicarbaporphyrins. | ||
Azulene-based tripyrrane condensed with an indene derivative gives mixed azulene-indene porphyrinoid S101-2.233 The macrocycle is aromatic as its cis counterpart S100-3 with inner H(21) and H(23) signals detected below 2 ppm.
Diazuliporphyrinoids can be formed in a reaction of azulene and heterocycle-based dicarbinols.234–236 The condensation leads to the formation of diazuliporphyrinogen, e.g., S102-1 (Scheme 102 and Fig. 22). S102-1 is naturally nonaromatic and more flexible than the target porphyrin. The oxidation of S102-1 with controlled amounts of DDQ yields mixtures of the porphyrin analogue S102-2 (Fig. 22), its radical cation S102-3, and dication S102-4. These three species constitute a peculiar multielectron redox system. The neutral form S102-2 is not aromatic as indicated with a H(21), H(23) chemical shift of 7.02 ppm. On the other hand, dication S102-4 reveals a significant degree of diatropicity (the H(21), H(23) signal shifts to 1.48 ppm) evidently related to the accumulation of the positive charge over the seven-membered ring. Similar reactivity was observed for dioxa-,235 diselena-, and ditelluraderivatives.236
 | ||
| Scheme 102 Reactivity of diazuliporphyrinogen S102-1. | ||
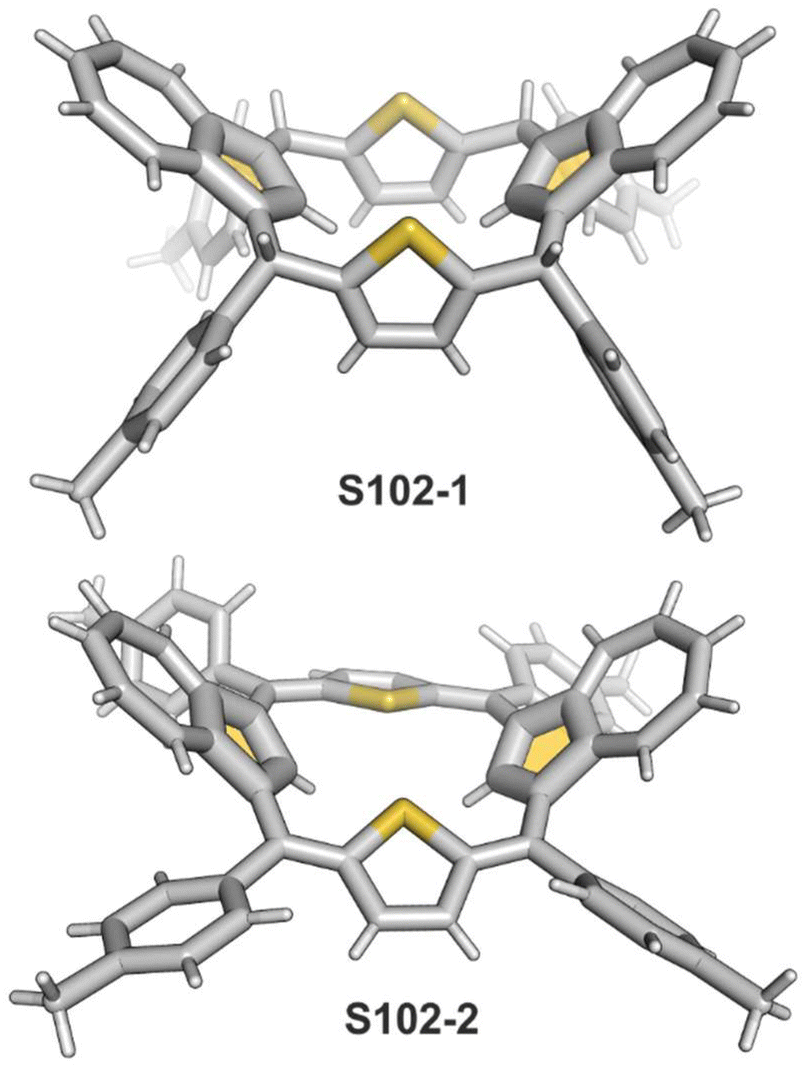 | ||
| Fig. 22 Diazuliporphyrinoids. meso-Substituents preserved to show changes in conformation after oxidation. | ||
Finally, a molecule bridging the features of porphyrinoids and the final, presented in this section group of potential ligands – macrocyclic polycarbenes – was described.237 Macrocycle S103-1 (Scheme 103) is obtained in a click-reaction between alkyne-disubstituted carbazole and a diazide derivative, followed by the N-methylation of two formed triazole rings with trimethyloxonium tetrafluoroborate. The final dication (S103-1) is colourless, indicating carbazole-centred aromaticity rather than the presence of any macrocyclic ring current as typical for porphyrins.
 | ||
| Scheme 103 Carbazole-triazolylidene porphyrin S103-1. | ||
Tri- and tetracarbamacrocycles
Macrocycles preserving some structural features of the regular porphyrin that contain more than two inner carbon atoms are really scarce. Over the years, quatyrin,238 a porphyrin with four inner carbon atoms, has been a target structure and remains to be synthesised. Still, there are a few examples of tri- or tetracarbamacrocyles that can potentially serve as multiorganometallic macrocyclic ligands.A reaction of thiophene-based biscarbinol with arylaldehyde and azulene catalysed by BF3·Et2O yields azuliporphyrinogen-type isomeric structures exemplified by S104-1 (Scheme 104), containing sulphur and three carbon atoms in the core.239 The subsequent oxidation with DDQ produces a rare organic tetracation S104-2 with four C(sp2) meso bridges. The positive charge is preferentially located at the meso carbon atoms. Significantly, the macrocyclic π-delocalization is absent, and the macrocycle is nonplanar. S104-2 is quite susceptible to nucleophilic attacks centred at meso positions.
 | ||
| Scheme 104 Triazuliporphyrinogens S104-1 and S104-2. | ||
A simple condensation of azulene with arylaldehyde (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio) affords meso-tetraaryl-tetraazuliporphyrinogen S105-1 (Scheme 105) as a mixture of stereoisomers,240,241 similar as with paraformaldehyde (which produces the unsubstituted derivative – calix[4]azulene).240 Oxidation of S105-1 leads to a tetracation S105-2, resembling S104-2, with no indication of macrocyclic aromaticity (inner protons resonate at 11.34 ppm).
:1 molar ratio) affords meso-tetraaryl-tetraazuliporphyrinogen S105-1 (Scheme 105) as a mixture of stereoisomers,240,241 similar as with paraformaldehyde (which produces the unsubstituted derivative – calix[4]azulene).240 Oxidation of S105-1 leads to a tetracation S105-2, resembling S104-2, with no indication of macrocyclic aromaticity (inner protons resonate at 11.34 ppm).
 | ||
| Scheme 105 Tetraazuliporphyrinogen S105-1 and oxidised S105-2. | ||
Another interesting macrocycle, described soon after NCP, can be generated by the flash vacuum thermolysis of an appropriately designed pyridine derivative (e.g., 3-diazo-4-hydroxypyridine).242 It transforms into 3-carbonyl-3H-pyrrole that tetramerizes forming S106-1 (Scheme 106). The macrocycle could be viewed as the tetra(neo-confused) porphyrin, but due to carbonyl moieties at meso positions, there is no macrocyclic aromaticity present (inner protons resonate at 8.78 ppm). The synthesis of the poorer soluble tetrabenzo homologue of S106-1 was also described.
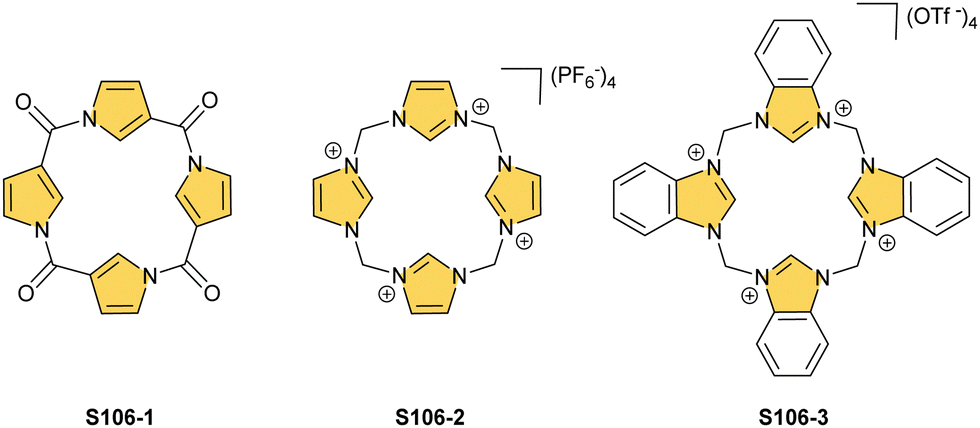 | ||
| Scheme 106 Tetra(neo-confused) porphyrin-like macrocycles. | ||
An original group of macrocycles containing a [CCCC] set of donors that reveal the high coordination potential have been presented in the literature. Our attention will be focused on a subset with the evidently porphyrin-like framework. Their electronic structure is principally different from that classified as typical for carbaporphyrinoids. They have a molecular structure containing four N-heterocyclic carbene (NHC) donors. However, the high structural similarity of molecular skeletons to so far described porphyrinoids and porphyrinogens in particular, straightforward and efficient syntheses, and the essential flexibility of frameworks bring a promise of interesting organometallic chemistry. The synthesis involves bridging two bisimidazoylmethanes to obtain the final tetracationic species S106-2.243,244 Benzohomologue S106-3 was obtained following a similar procedure.245 Naturally, the fundamental framework has various modifications, including heteroatoms like boron at meso positions to decrease the overall charge and tune the molecular properties.246–249 These macrocycles also have exciting organometallic chemistry, e.g., with actinides,250 iron,247 or chromium.251 The all-carba ligands (S106-2 and S106-3) allow following the organometallic reactivity in more polar solvents than that in the case of neutral porphyrin complexes. Additionally, anion exchange can be applied to tune the solubility of complexes in different organic media.243,244
Organometallic chemistry of di-, tri-, and tetracarbaporphyrinoids
Although several examples of di-, tri-, and tetracarbaporphyrinoids have been described in a previous section, only a handful of coordination examples exist in the literature, which remains in contrast to monocarba analogues. It indicates that the field is still in its infancy and that novel, exciting possibilities to stabilise extraordinary organometallic motifs still await exploration.Starting from cis-doubly N-confused porphyrin S97-1, in contrast to NCP, it stabilises MIII complexes with both silver and copper.220 Silver(III) complex S107-1 (Scheme 107) forms after stirring the ligand with silver(I) acetate, while the stable copper(III) square-planar coordination (S107-2) is provided after stirring with copper(II) acetate. The lengths of organometallic bonds in both molecules are within standard ranges, i.e., 1.987(8) and 2.011(8) Å for AgIII–C and 1.939(3) and 1.934(4) Å for CuIII–C. The treatment of S97-1 with palladium(II) acetate in refluxing toluene afforded palladium(II) dicarbaporphyrin S107-3.252 Its formation is clearly associated with toluene activation as the p-tolyl or m-tolyl moiety has been attached in S107-3 to one of the inner carbon atoms (Fig. 23). Such C-substitution has been detected with benzene in a mixture with toluene or m-xylene as well. It is noteworthy that the new C(21)–C(aryl) bond is formed directly to the phenyl ring and not the potentially more reactive methyl group. The C–Pd distances vary significantly between the coordination of tetrahedrally distorted C(21) (2.202(9) Å) and the regular C(sp2) (1.971(15) Å) inner carbon atom.
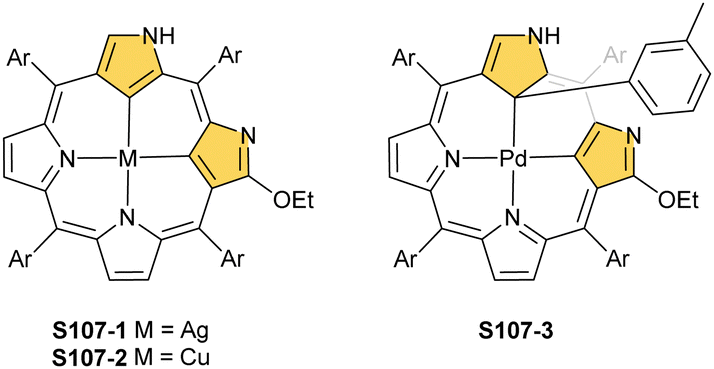 | ||
| Scheme 107 cis-Doubly N-confused porphyrin complexes. | ||
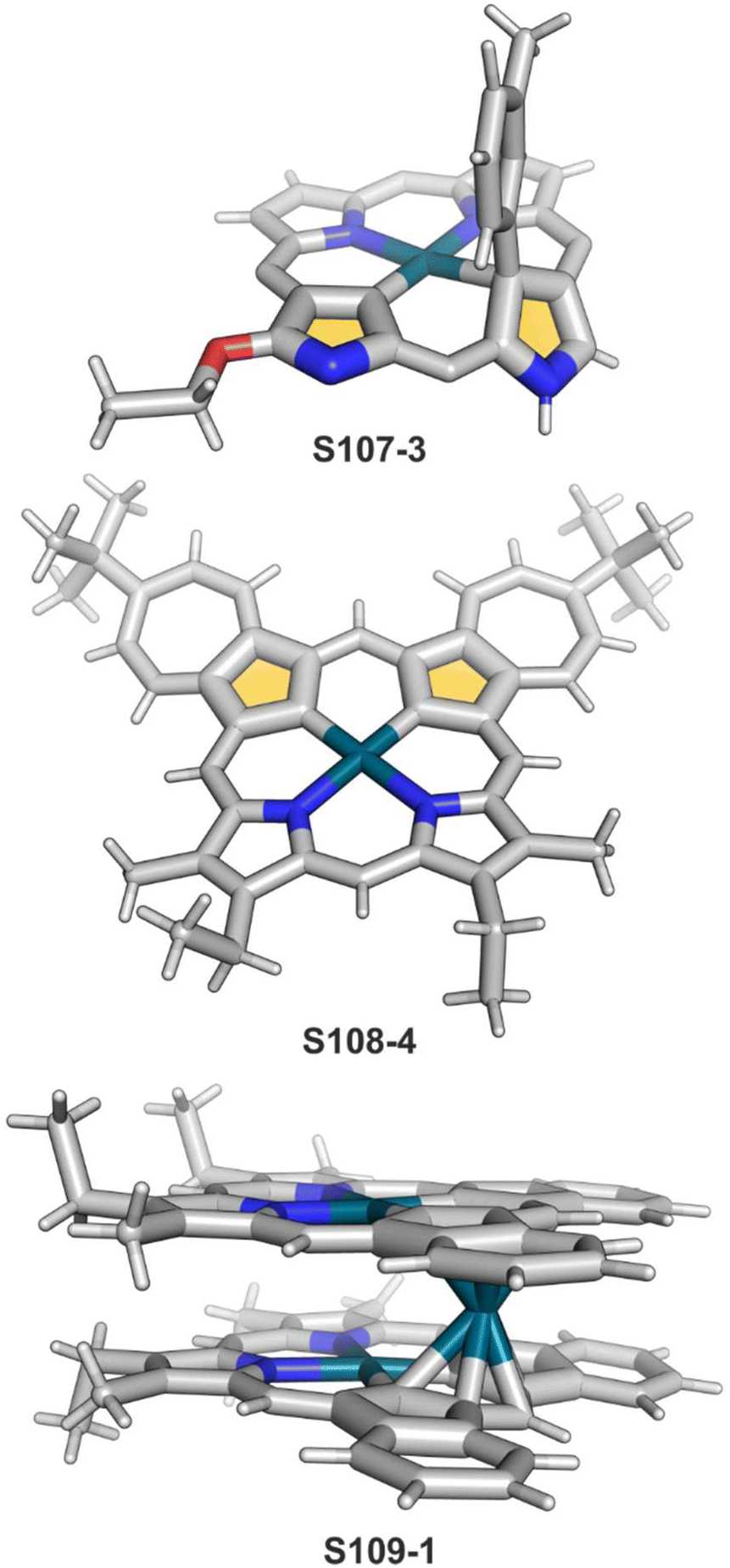 | ||
| Fig. 23 Palladium coordination motifs for dicarbaporphyrins. meso-Substituents in S107-3 not shown. | ||
trans-Doubly N-confused porphyrin S98-3 containing four inner dissociable protons may potentially act as a tetraanionic ligand but behaves very similarly to the cis isomer S97-1. Namely, a reaction with copper(II) acetate results in copper(III) insertion with one inverted pyrrole unit accommodating an outer proton on the outer nitrogen atom.222 This points to a degree of flexibility of such ligands resembling NCP. A similar outcome of silver(III) or copper(III) of coordination (S108-1 and S108-2, Scheme 108) was observed in a reaction with isophlorin S98-3 and its aromatic counterpart S98-4.223,253 A reaction of S98-3 with [AuCl(S(CH3)2)] yields an analogous gold(III) complex (S108-3), yet to conduct gold(III) insertion, preceding bromination of inner CH positions is necessary.253
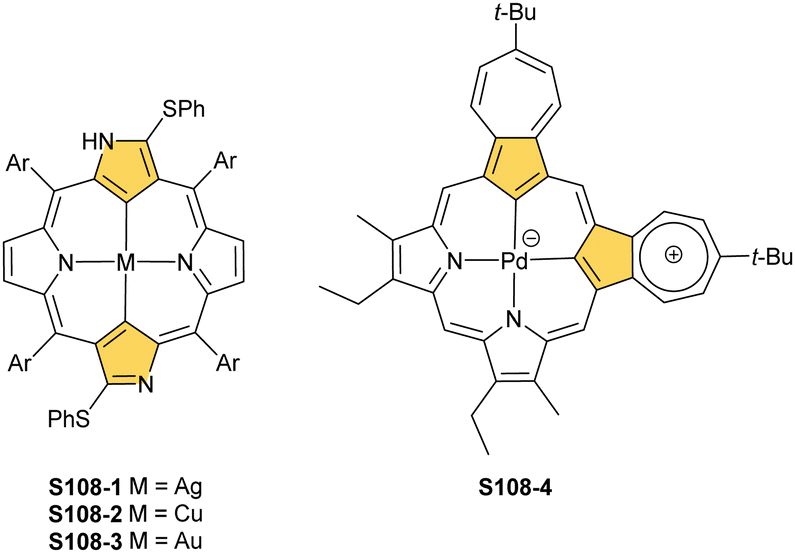 | ||
| Scheme 108 Trans-Doubly N-confused porphyrin complexes and the palladium(II) cis-diazuliporphyrin derivative. | ||
cis-Diazuliporphyrin S100-1, after deprotonation, is dianionic. Thus, it is a suitable platform to accommodate a palladium(II) ion. After a reaction of diazuliporphyrin bromide S100-1 with a palladium source, the neutral complex S108-4 is formed,226 which continues to have a mesoionic electronic structure as the free ligand has (Fig. 23).
The dibenzocarba analogue (S100-2) reacts with palladium(II) acetate under similar conditions, yet the product is strikingly different.227 The formed species acquires the dianionic nature as four inner protons are removed after complete palladium(II) coordination. Eventually, the process affords a stable [Pd3L2] dimer (S109-1) (Scheme 109) with two inserted palladium(II) cations and one palladium(IV) playing the bridging role (Fig. 23). The palladium(IV) centre links two subunits via effective π-coordination interactions. The inner PdII–C(21) distances are typical (ca. 2.0 Å), while the closest contacts with palladium(IV) forming the η5 motif with each unit are in a range of 2.223(9) to 2.515(8) Å.
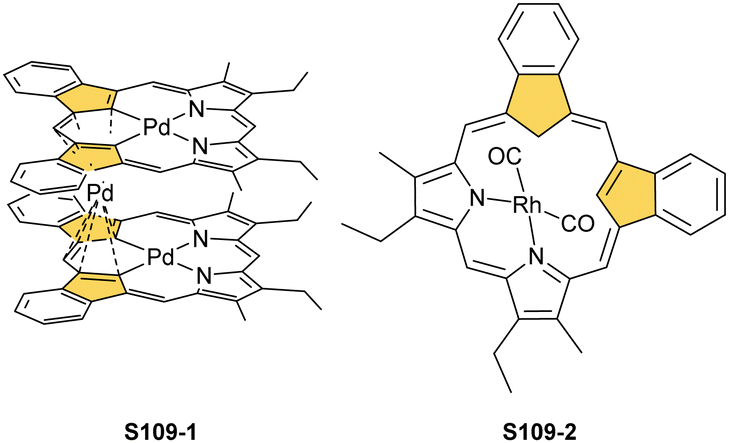 | ||
| Scheme 109 cis-Dibenzocarbaporphyrin complexes. | ||
Coordination to a rhodium(I) ion requires the rehybridisation of one of the inner carbon atoms to provide the environment with one deprotonated inner nitrogen atom.204 This is how two coordinated nitrogen atoms secure the neutrality of the formed complex S109-2. Two uncoordinated C(21) and C(22) carbon atoms are confirmed to possess different sp2 and sp3 hybridisations.
The mentioned porphyrin–carbene hybrid (S103-1) gets activated by deprotonation with lithium bis(trimethylsilyl)amide to form a dilithium complex S110-1 (Scheme 110).237 It can be transmetallated with scandium(III) using a trichloride source. The formed complex S110-2 possesses apically attached chloride and a THF molecule at the same macrocyclic side. To probe any possible macrocyclic ring currents, the ligands on the scandium(III) centre have been replaced with a cyclopentadienyl anion (from LiCp), but no upfield shift of the Cp resonances was detected in the 1H NMR spectrum.
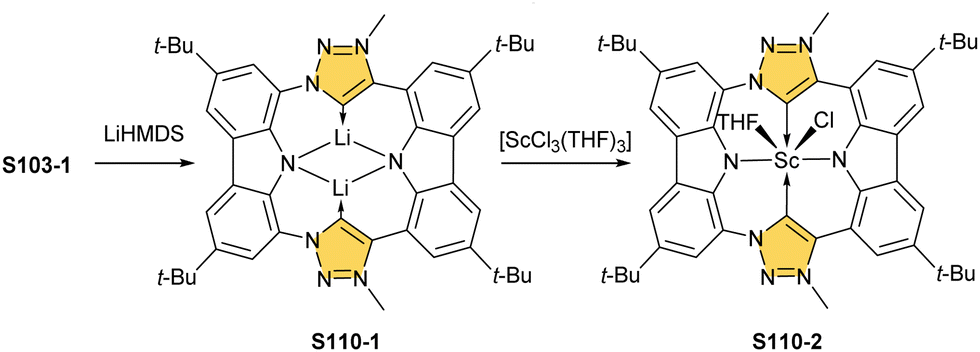 | ||
| Scheme 110 Dilithium carbene complex of carbazole-triazolylidene porphyrin S110-1 and its transmetallation. | ||
Let us remain with nonaromatic trans-dicarbaporphyrinoids in which the only known coordination of ruthenium was encountered for azuliporphyrinogens, including dithiadiazuliporphyrinogen S102-1.254
Reactions of S102-1 with [Ru3(CO)12] result in the formation of ruthenium π-bound clusters, similarly, as reported for azuliporphyrin S64-2. In this case, though, the cavity persists in being unoccupied. The complex is formed purely due to azulene π-coordination. As there are two azulene moieties, both participate, and each accommodates identical Ru4(CO)9 units forming S111-1 (Scheme 111). Otherwise, a transient cluster Ru2(CO)5 may be attached, providing that there is not enough of the ruthenium(0) source accessible or the reaction time is too short. Interestingly, the number of products is determined by the conformation of parental dithiaporphyrinogen S102-1. The one having all four meso substituents at the same side of the macrocyclic frame yields three isomeric complexes varying by clusters attached to different sides of azulene surfaces (S111-2 to S111-4) heavily influencing the overall molecular conformation (Fig. 24). The tri- (S104-1) and tetraazuliporphyrinogen (S105-1) also provide S111-2-like complexes with one (S112-1, Scheme 112) or two (S112-2) azulene fragments remaining unbound.
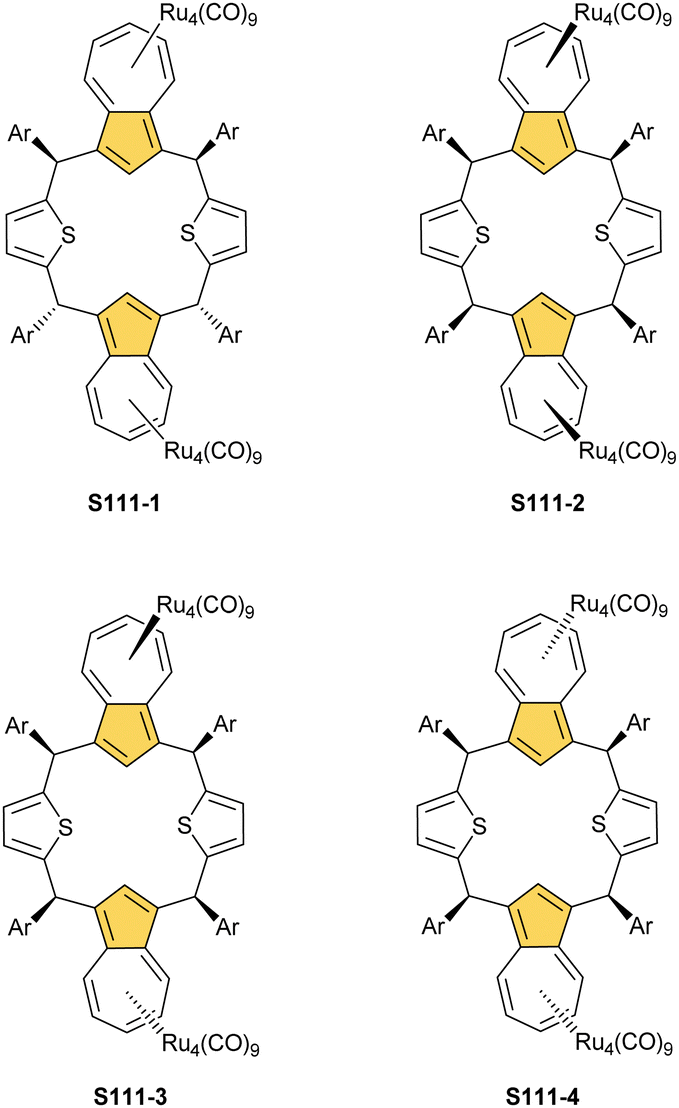 | ||
| Scheme 111 Diazuliporphyrinogens with azulene rings coordinated with tetraruthenium clusters. | ||
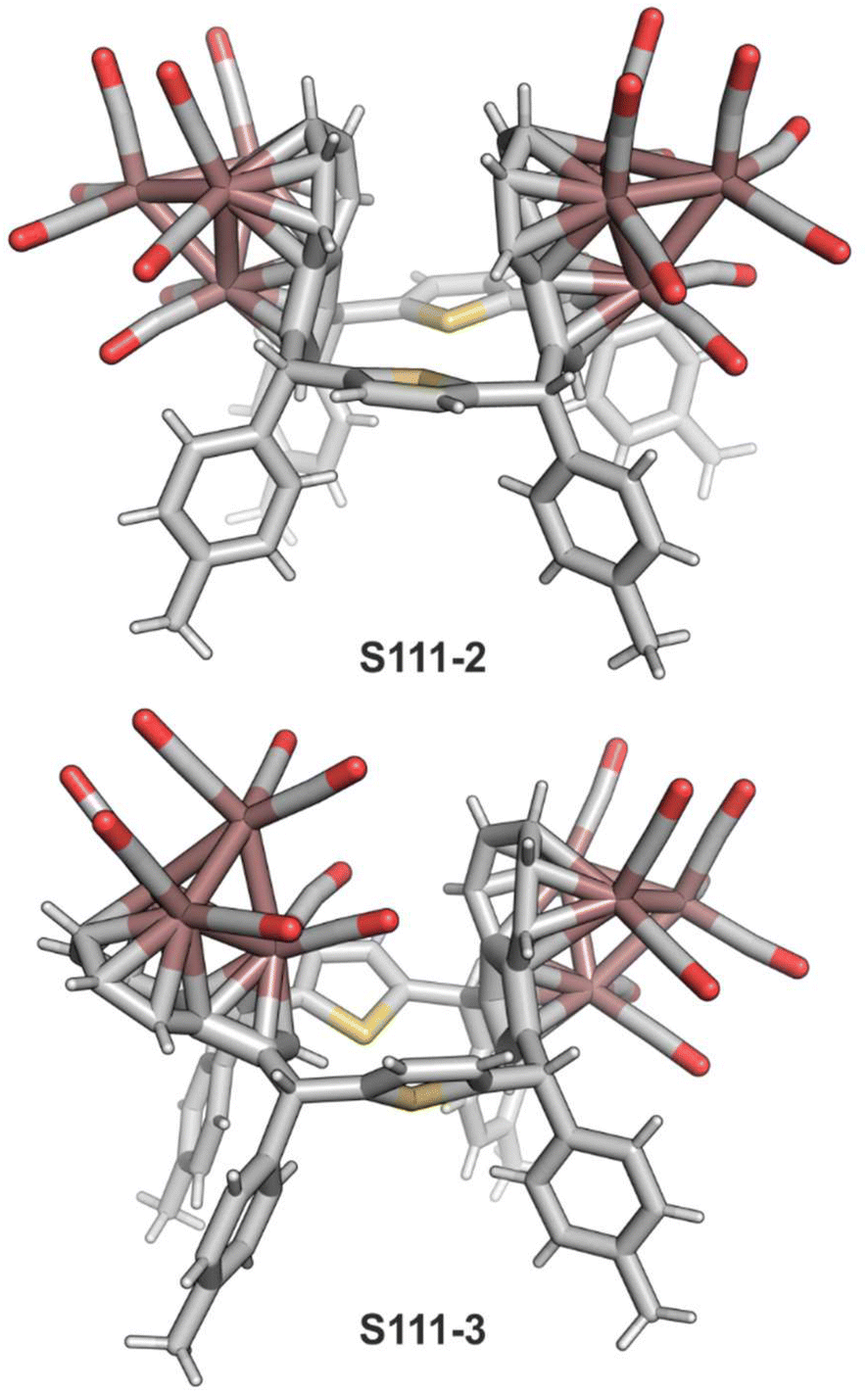 | ||
| Fig. 24 Diazuliporphyrinogens π-coordinated with ruthenium clusters. meso-Substituents preserved. | ||
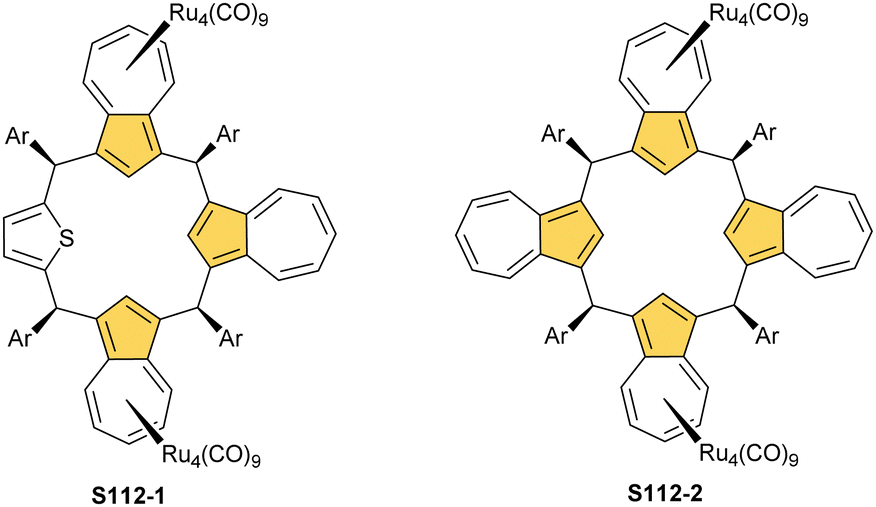 | ||
| Scheme 112 Triazuliporphyrinogen and tetraazuliporphyrinogen with ruthenium clusters. | ||
N-Heterocyclic carbene macrocycles S106-2 and S106-3 with porphyrinogen topology present quite rich organometallic chemistry taking advantage of the unique [CCCC] core and the peculiar conformational flexibility.
Starting from S106-2, this species reacts easily with Ag2O in acetonitrile to form [Ag4L2]4+S113-1 (Scheme 113).255 Four silver(I) ions, which prefer a linear coordination mode, form a Ag4 square motif in which each cation is bound with NHC from the top and bottom rings. To reach such an arrangement, a conformationally flexible molecule S106-2 has oriented all four five-membered rings perpendicularly to the N8 mean plane. The average Ag–C distance is ca. 2.09 Å, whereas the Ag⋯Ag contacts are ca. 3.21 Å. The benzimidazole-containing derivative S106-3 was also probed.245S114-1 (Scheme 114) acquired the identical conformation to S113-1 with respective average distances being ca. 2.11 Å for Ag–C and around 3.00 Å for Ag⋯Ag. Silver(I) species S113-1 may also serve as a nontrivial substrate for transmetallation. Accordingly, a reaction of S113-1 with [AuCl(S(CH3)2)] results in a replacement of AgI for AuI to create S113-2 with the preserved dimolecular frame of S113-1.255 Here, metalophylic interactions are also prominent as the Au⋯Au contact equals 3.1482(5) Å and the Au–C bond lengths are around 2.03 Å (Fig. 25).
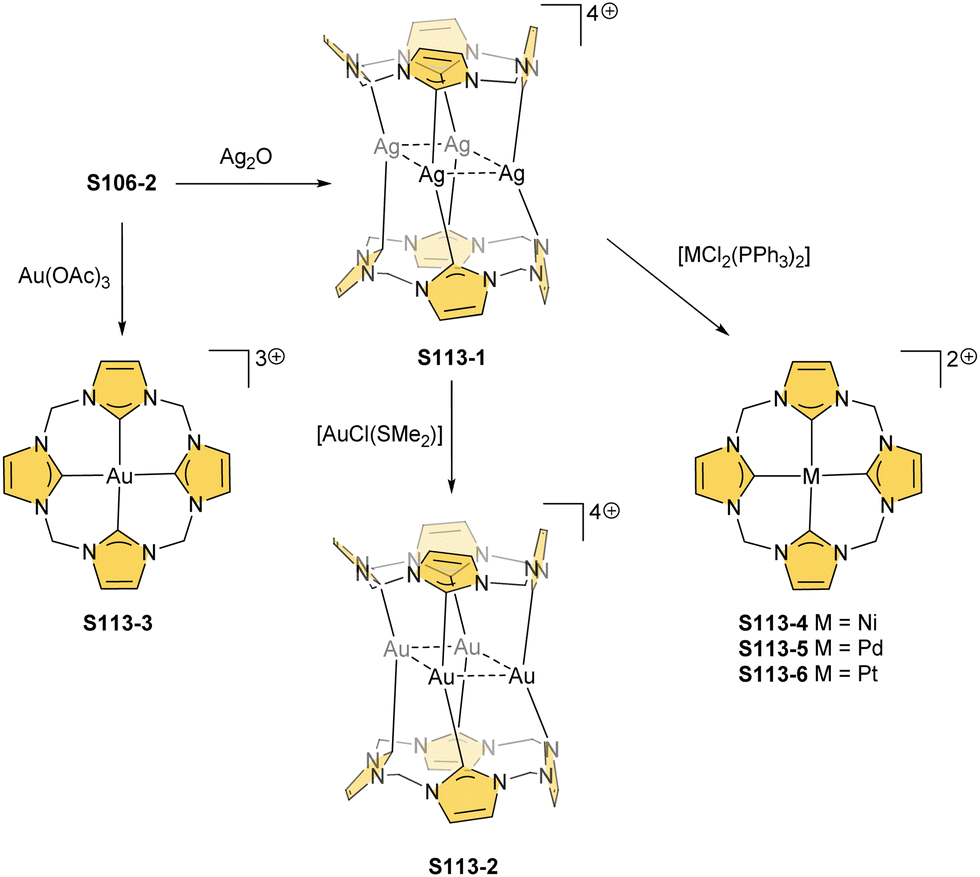 | ||
| Scheme 113 Carbene silver and gold complexes with calix[4]imidazolium. | ||
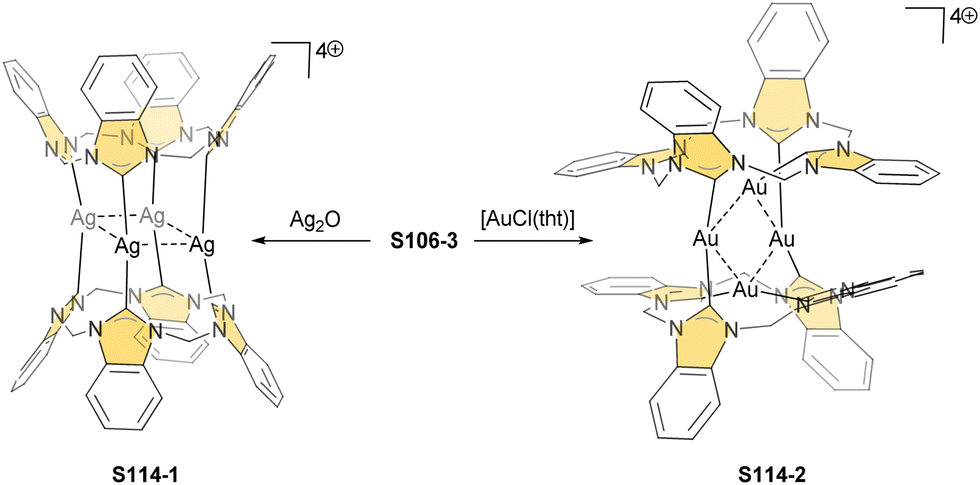 | ||
| Scheme 114 Coordination of benzo-fused calix[4]imidazolium with silver and gold salts. | ||
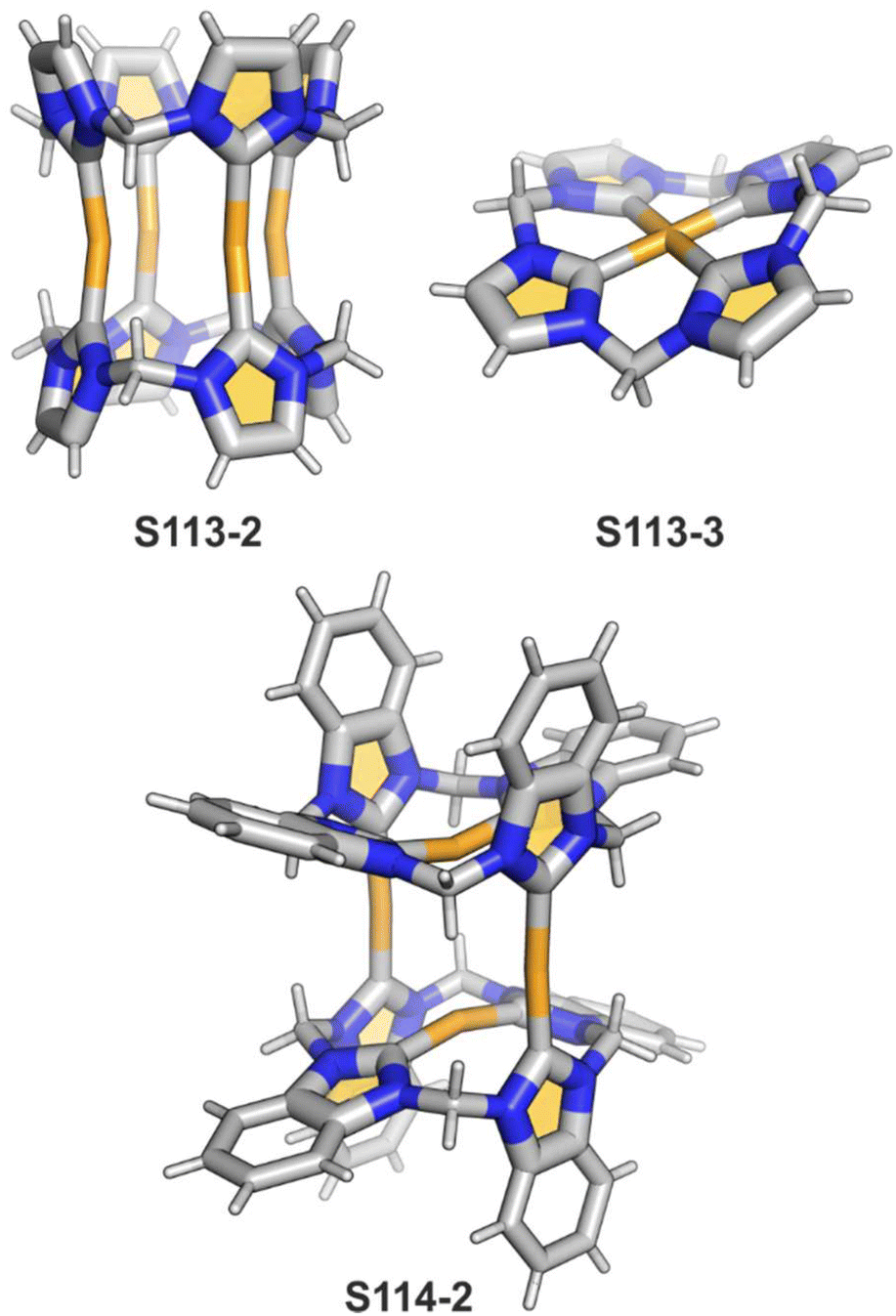 | ||
| Fig. 25 Carbene gold complexes. | ||
Transmetallation of S114-1 does not provide a related and expected gold(I) product. Interestingly, the direct treatment of S106-3 with chloro(tetrahydrothiophene)gold(I) generates [Au4L2]4+S114-2 with the same stoichiometry as in S113-2, although structurally significantly different (Fig. 25).245 The X-ray-determined molecular structure of S114-2 clearly shows two gold(I) cation ions coordinating to two trans ‘in-plane’ benzimidazoles (Au–C: 2.03 Å), while two other gold(I) cations link rings perpendicular to the mean plane (Au–C: 2.02 Å) affording the dimer. Eventually, S106-2 was also directly reacted with gold(III) acetate and provided a mononuclear, regular complex with four Au–C bonds in a planar arrangement (S113-3; Fig. 25).245 The bonds are, on average, 1.99 Å long, a bit shorter than determined in the respective gold(I) complex S113-2.
An attempt of transmetallation of S113-1 with [MCl2(PPh3)2], in which M = Ni, Pd, Pt, was elaborated as an effective route to generate respective mononuclear complexes S113-4, S113-5, and S113-6.255 As typical for low-spin nickel(II) porphyrins, the incorporation of nickel(II) into S106-2 leads to the most deformed structure of S113-4. At the same time, the larger congeners S113-5 and S113-6 remain relatively planar. The bond length range for Ni–C is between 1.851(3) and 1.886(3) Å, while for PdII and PtII, it is slightly below 2.00 Å.
The direct metallation of S106-2 with iron(II) occurs in a reaction with [Fe(N{Si(CH3)3}2)2].244 It opens a route to interesting coordination chemistry on its own.256–258 The formed tetracarbene dicationic complex S115-1 (Scheme 115) demonstrates the features of the diamagnetic low-spin iron(II) electronic state (the Fe–C distances of ca. 1.91 Å). The iron(II) centre coordinates axially two acetonitrile ligands apart from four equatorial carbon donors. The molecules can be exchanged for DMSO and, interestingly, also for two carbonyl ligands forming S115-2, yet the distances from the centre to the inner carbon atoms remain unaltered (Fig. 26). A reaction of S115-1 with in situ formed nitric oxide results in pentacoordinate S115-3 and slightly longer Fe–C bonds (ca. 1.95 Å; Fig. 26). Despite the {Fe(NO)}7 electronic structure, a diamagnetic-like 1H NMR spectrum is observed together with a characteristic EPR signal. S115-1 can be efficiently oxidised to the iron(III) complex S115-4 by mixing it with thianthrenyl hexafluorophosphate.259 This involves only slight variations in the coordination environment with Fe–C distances close to 1.95 Å.
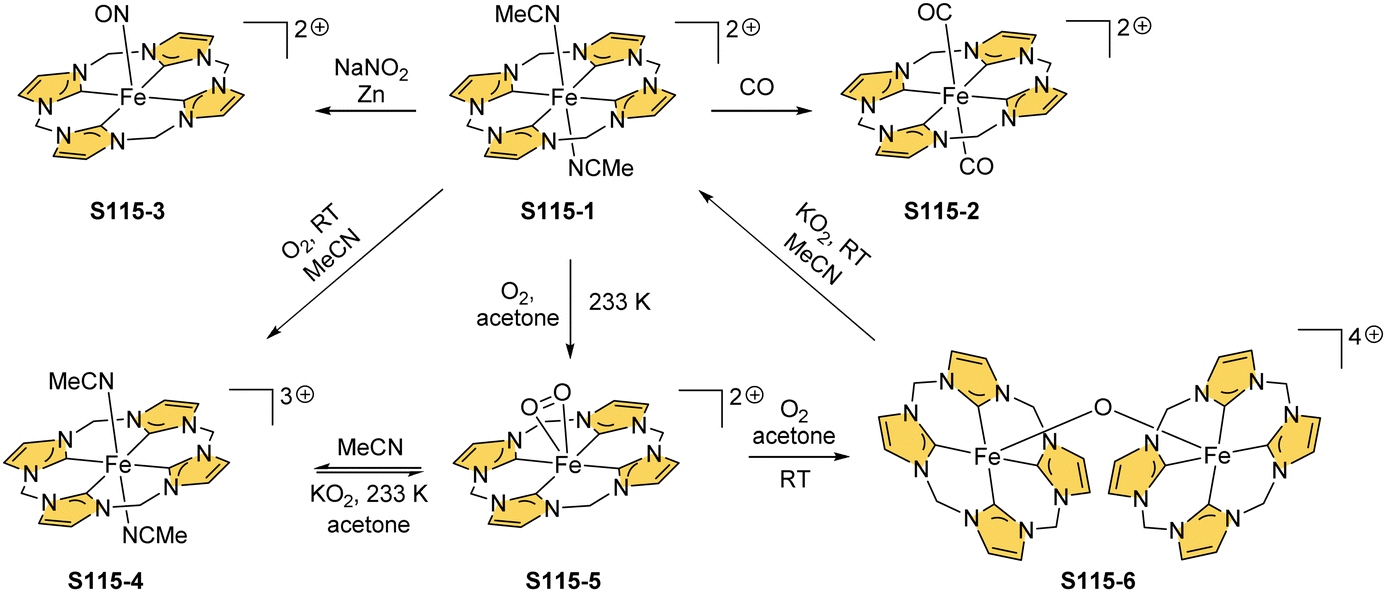 | ||
| Scheme 115 Reactivity of iron(II) calix[4]imidazolium S115-1. | ||
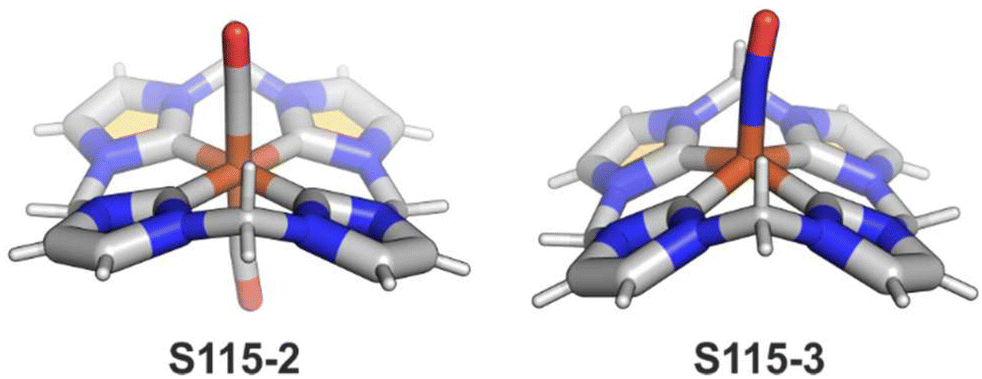 | ||
| Fig. 26 Iron tetracarbene complexes with calix[4]imidazolium. | ||
The oxidation of S115-1 also occurs in acetonitrile exposed to dioxygen.258 When a solvent is switched to acetone, and the reaction occurs at low temperature, a diamagnetic form with activated dioxygen (S115-5) can be detected and even isolated. This species can be alternatively generated by treating S115-4 with KO2. By elevating the temperature to ambient conditions, S115-5 transforms into the μ-oxydimer S115-6 (Fig. 27). The Fe–C bonds are, on average, 1.95 Å long, as in S115-4, while the Fe–O distance is 1.7322(7) Å. The dimer, in turn, can be split and reduced back to S115-1 with a range of agents, including KO2 in acetonitrile. Further iron carbene oxidative reactivity was explored, yet with ligands beyond porphyrin-like geometry.260,261
 | ||
| Fig. 27 μ-Oxy(peroxy)-bridged carbene complexes. | ||
Another dioxygen-activating centre can be successfully introduced into S106-2 in a reaction with CoCl2 yielding S116-1 (Scheme 116).262 In the solid state, the cobalt tetracarbene complex S116-1 acquires a single axial acetonitrile ligand. The CoII–C distances are in a range of 1.889(2) to 1.901(2) Å. After exposure to air, the solution S116-1 undergoes a colour change, forming a diamagnetic compound. The X-ray-determined molecular structure reflects the formation of the μ-peroxy dicobalt(III) species S116-3 (Fig. 27). It was confirmed that the transformation proceeds through a reactive superoxy intermediate S116-2. Finally, after mixing S116-3 with acetic acid, the mononuclear CoIII complex S116-4 is recovered.
 | ||
| Scheme 116 Reactivity of cobalt(II) calix[4]imidazolium S116-1. | ||
The insertion of copper(II) opens a route to interesting redox chemistry as well. S106-2 treated with copper(II) acetate in DMSO affords the copper(III) complex S117-1 (Scheme 117) structurally analogous to the gold(III) derivative S113-3.263 Alternatively, the reaction of S106-2 with copper(I) acetate leads to a dimeric counterpart of the respective dimeric sliver(I) S113-1 or gold(I) S113-2 species with four copper(I) cations bridging two macrocyclic units (S117-2). Interestingly, S106-2 mixed with copper(II) acetate and a base (tetrabutylammonium acetate) yields a completely different product S117-4 in high yields. The reaction is shown to proceed through a copper(I) species (S117-3). Both macrocycles in S117-4 have one of the inner positions oxygenated (Fig. 28); thus, three, not four, copper(I) cations provide stabilising C–Cu–C links. S117-4 is also significantly more resistant to oxygenation. The demetallation of S117-4 with acid produces the oxygenated macrocycle S117-5. Surprisingly, the oxygen atom originates from the acetate anion rather than water or dioxygen, as proven by 18O labelling.
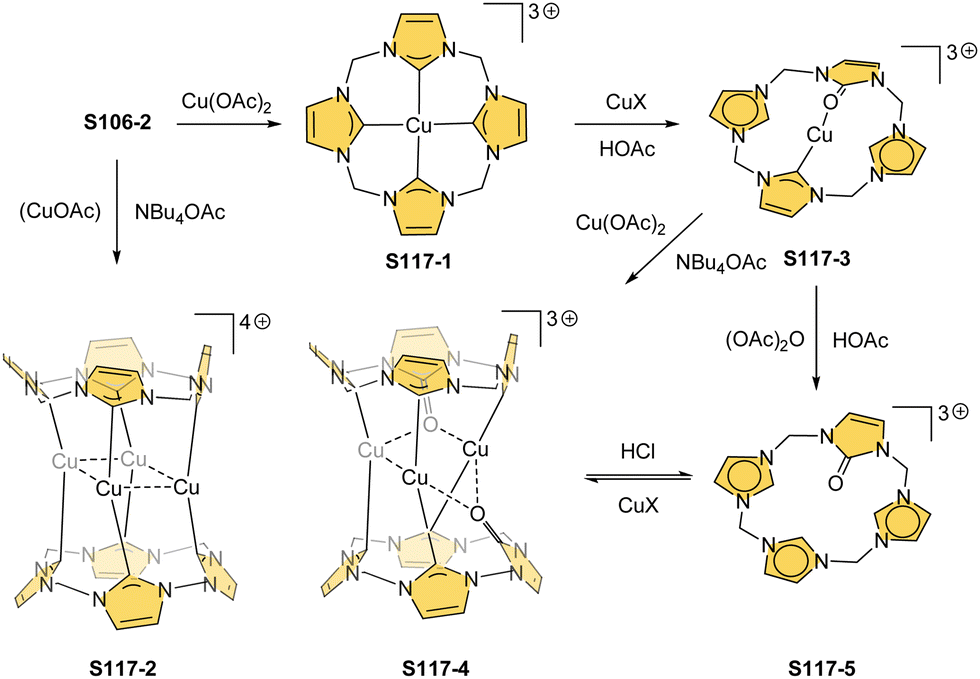 | ||
| Scheme 117 Reactivity of calix[4]imidazolium with copper(I)/(II) ions. | ||
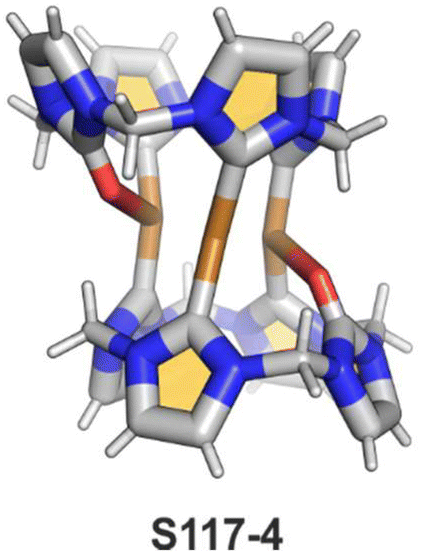 | ||
| Fig. 28 Copper carbene complex with inner-C position oxygenated. | ||
For the sake of completeness, it remains to mention that the meso-dimethylamino-substituted derivatives of the platinum(II) tetracarbene dicationic complex S118-1 (Scheme 118) were also reported.264 This species forms using a template approach.
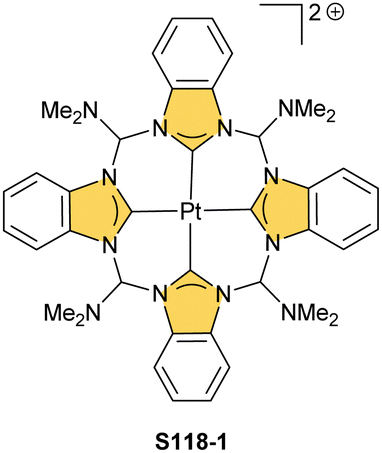 | ||
| Scheme 118 Platinium(II) tetracarbene dicationic complex S118-1. | ||
Beyond carbaporphyrin(1.1.1.1)
In the last section, selected macrocycles, which are arbitrarily chosen as being representative of the further developments in the carbaporphyrinoid field, will be described. Formally, they do not meet all outlined initially criteria. Namely, the requirement to contain four five-membered rings and/or four meso-carbon bridges is not accomplished. However, their structural similarity to the already presented molecules and specific properties secured their place in the review.N-confused calix[4]phyrins
The first subgroup contains N-confused porphyrinoids with one or two meso bridges possessing hybridisation C(sp3), a clear reference to the nonaromatic macrocycles presented above.Calix[4]phyrin with one C(sp3) bridge substituted with two methyl groups is obtained when condensation leading to NCP gets enriched with acetone.265 This produces S119-1 (Scheme 119) with the oxygenated C(3) atom in 3% aside from also formed NCP. The macrocycle acquires a 2H cavity and a lactam moiety at the confused pyrrole ring. Expectedly, S119-1 proved to be a suitable ligand for nickel(II) (S119-2) and, significantly, copper(II) (S119-3) forming a direct and stable organometallic bond (1.905(4) and 2.007(4) Å, respectively). In further explorations,266 the chlorozinc(II) centre was also formed in a reaction of S119-1 with Zn(CH3COO)2 followed by the saturation of the mixture with NaCl (S119-4). Here, the Zn⋯C(21) distance increases to 2.67(1) Å from 2.41–2.53 Å for zinc(II) complexes with NCP (S14-2 and S14-3).
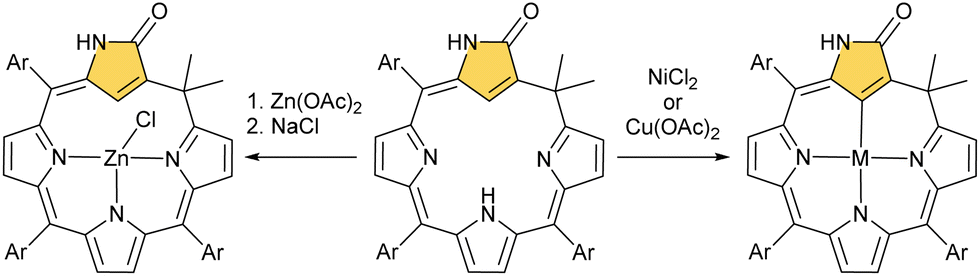 | ||
| Scheme 119 Complexes of the N-confused calix[4]phyrin derivative S119-1. | ||
Calix[4]phyrin complexes have more intensive infrared absorption than the respective NCP derivatives, which was also noted in the study of macrocycles with two C(sp3) meso bridges and one or two N-confused rings.267 The macrocycles are formed in a condensation of substituted N-confused dipyrromethene. Both macrocycles, S120-1 (Scheme 120) and S120-2, possess a 2H cavity that was matched with the platinum(II) centre in a reaction with [PtCl2(PhCN)2]. Interestingly, only S120-4 (with two N-confused pyrroles) shows a significant degree of nonplanarity (Fig. 29). Yet both, S120-3 and S120-4, stabilise organometallic bonds with an average distance of 2.00 Å. Another, not fully oxidised example – porphodimethene S120-5 possesses two adjacent meso bridges C(sp3)-hybridised.268 This ligand stabilises CuIII and NiII metal centres.
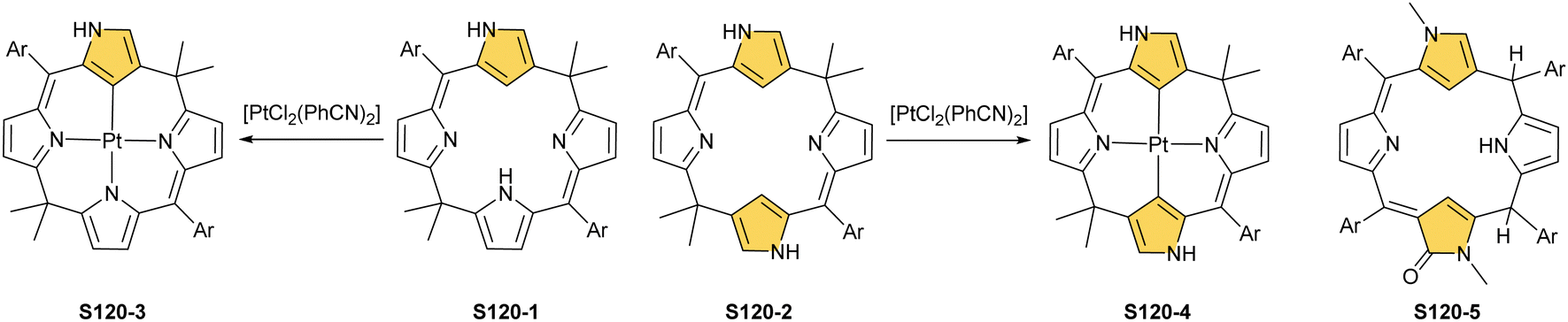 | ||
| Scheme 120 Complexes of N-confused calix[4]phyrin derivatives S120-1 and S120-2 and porphodimethene S120-5. | ||
 | ||
| Fig. 29 Platinum(II) dicarbacalix[4]phyrin. meso-Aryls not shown. | ||
There are also known singly and doubly N-confused macrocycles with all four meso bridges sp3-hybridised called calix[4]pyrroles, yet their main application so far was sensing and anion recognition.269–271
Carbacorroles
Another subgroup contains carbaporphyrinoids lacking one meso bridge, namely carbacorroles and N-confused corroles.Attempts to synthesise 21-silaporphyrin resulted in the serendipitous discovery of a transformation of 21-silaphlorin into nonaromatic isocarbacorrole S121-1.272 Similar trials to form 21-phosphaporphyrin led to the formation of the P-confused analogue.273
Insertion of silver or copper ions into isocarbacorrole S121-1 (Scheme 121) gives two structurally related organometallic complexes of “true” carbacorrole (S121-2 (Fig. 30) and S121-3) in which the metal(III) ions are bound by three pyrrolic nitrogen atoms and a tetrahedrally hybridised inner carbon atom of the cyclopentadiene ring with aryl substitution. The Ag–C(sp3) (2.046(5) Å) bond length is similar to that in silver(III) carbaporphyrinoids in which a trigonal carbon atom coordinates to the metal ion.
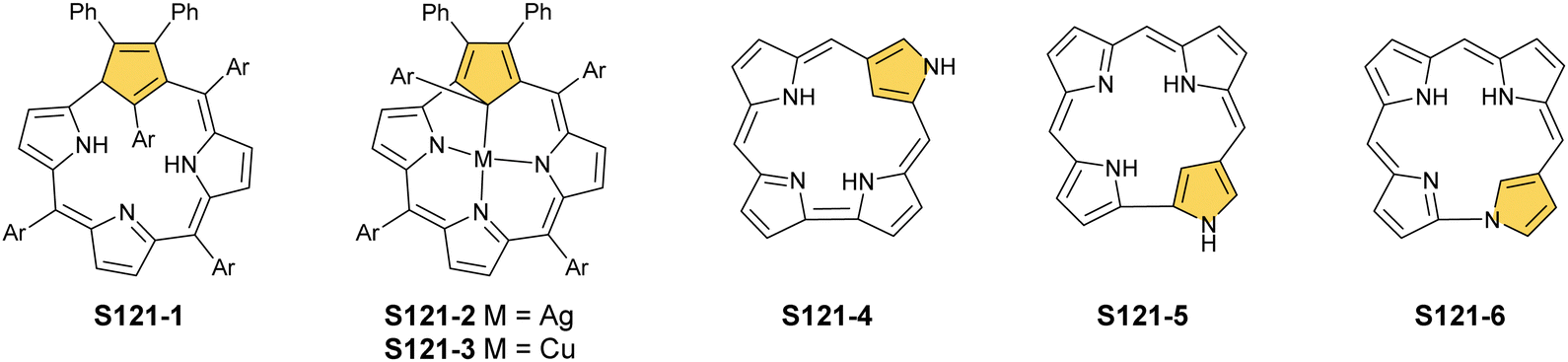 | ||
| Scheme 121 Carbacorroles. | ||
 | ||
| Fig. 30 Selected carbacorrole frameworks. meso-Substituents not shown. | ||
The appropriately designed substrates incorporating one ‘N-confused pyrrolic ring’ allowed for synthesising two types of modified carbabilanes (a noncyclic precursor of carbaporphyrin missing one meso-carbon link).274,275 After oxidation of carbabilane, a Cα–Cα′ pyrrole–pyrrole bond forms closing two macrocycles – N-confused corrole S121-4 and S121-5. Alternatively, the N–Cα pyrrole–pyrrole linkage yields the neo-confused version, i.e., norrole S121-6 (Fig. 30). Their 1H NMR spectra indicate a significant degree of the diatropicity of these contracted carbaporphyrinoids with the inner CH signal located at −0.91, 1.84, and 1.21 ppm for S121-4, S121-5, and S121-6, respectively. It is despite a seemingly interrupted π-delocalization pathway in the typically shown resonance contributors. Also, the oxanorrole derivative has been recently synthesised.276
S121-4 and S121-5 accommodate copper(III), which seems to be a natural central cation for the trianionic cavity.275 The insertion proceeds from copper(II) acetate and gives stable S122-1 (Scheme 122) and S123-1 (Scheme 123).
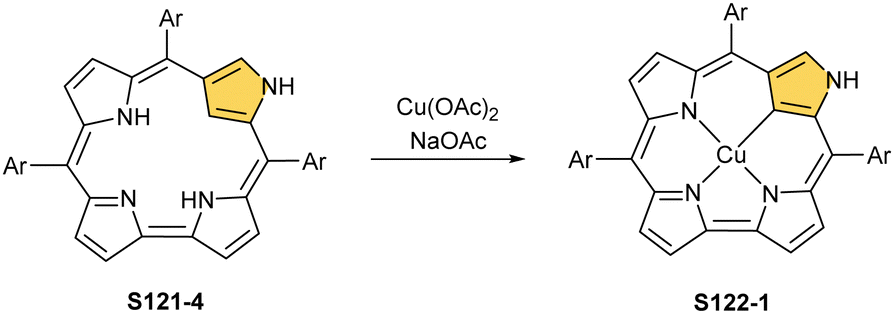 | ||
| Scheme 122 Synthesis of copper(III) N-confused corrole S122-1 from S121-4. | ||
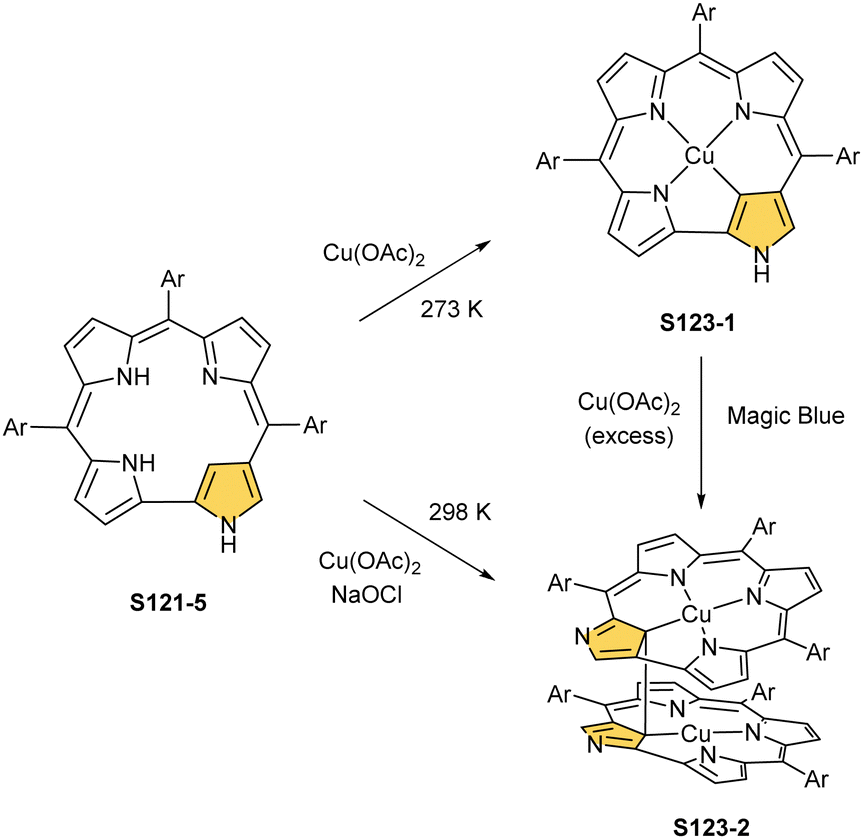 | ||
| Scheme 123 Synthesis of copper(III) N-confused corrole S123-1 and its dimer S1123-2 from S121-5. | ||
However, in contrast to S122-1, complex S123-1 exhibits further reactivity. Under an excess of copper(II) salt or after adding the magic blue oxidant, it undergoes oxidative dimerization forming a C–C bond between inner coordinated carbon atoms yielding S123-2 (Fig. 31). The C–C bond length equals 1.552(7) Å, reflecting a single bond between two C(sp3) centres.
 | ||
| Fig. 31 Carbacorrole inner-C dimers. meso-Substituents not shown. | ||
Similar to S121-6, benzonorrole S124-1 (Scheme 124) can be obtained, first, as a copper(III) complex and then through demetallation.277 Interestingly, S124-1 in a free form can dimerise upon further oxidation, forming trans (S124-2; Fig. 31) and cis (S124-3) isomers with directly linked inner carbon atoms derived from each subunit. Solid-state structures indicated that the linkage is short enough (<1.36 Å) to be described as a double bond. The confirmation comes from the test reduction of S124-3 with the hydrazine derivative that gives S124-4 (Fig. 31). Here, the C–C bond length increases to 1.476(3) Å and the process can be reversed via reoxidation.
 | ||
| Scheme 124 Reactivity of benzonorrole S124-1 with DDQ. | ||
Carbabilane with an indole moiety at each end of the acyclic molecule affords, after macrocyclisation, porphyrinoid S125-1 (Scheme 125) linked through a direct N–N bond, among other products.278 As shown in the scheme, the macrocycle is stabilised through the coordination of a copper(III) centre, which is inserted during macrocyclisation. Copper(III) removal with zinc and acid leads directly to starting carbabilane. An effective π-conjugation through the N–N linkage can be inferred, among others, from the 1H NMR spectra of S125-1.
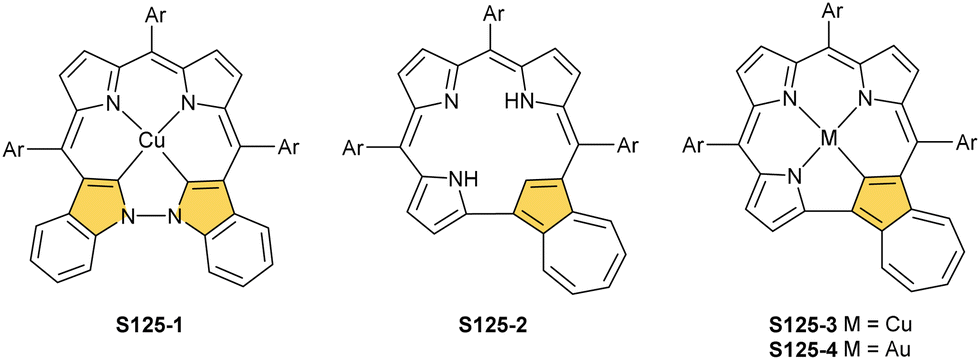 | ||
| Scheme 125 Diazadicarbacorrole S125-1 and carbacorrole, containing azulene. | ||
Finally, copper(III) was a central ion of choice to demonstrate the coordination abilities of carbacorrole, with one pyrrole replaced with an azulene fragment as seen in S125-2.279 Interestingly, this carbaporphyrinoid is formed from simple substrates and the predesigned building blocks were not required. Azulene in S125-2 forms a direct bond to one of pyrrole rings. The aromaticity level resembles that of regular azuliporphyrin S64-2 (the inner H(21) chemical shift in S125-2 equals 3.19 ppm). Three inner protons can be replaced easily with MIII cations; thus, CuIIIS125-3 and AuIIIS125-4 complexes were isolated. The CuIII–C(21) organometallic bond stabilised within the framework of S125-3 is 1.886(10) Å long.
Annulene-containing porphyrins
Vacataporphyrin S126-1 (Scheme 126) represents a next macrocyclic frame to be mentioned, i.e., a porphyrin deprived one inner nitrogen atom, which pushes it further on an axis from porphyrin to annulenes.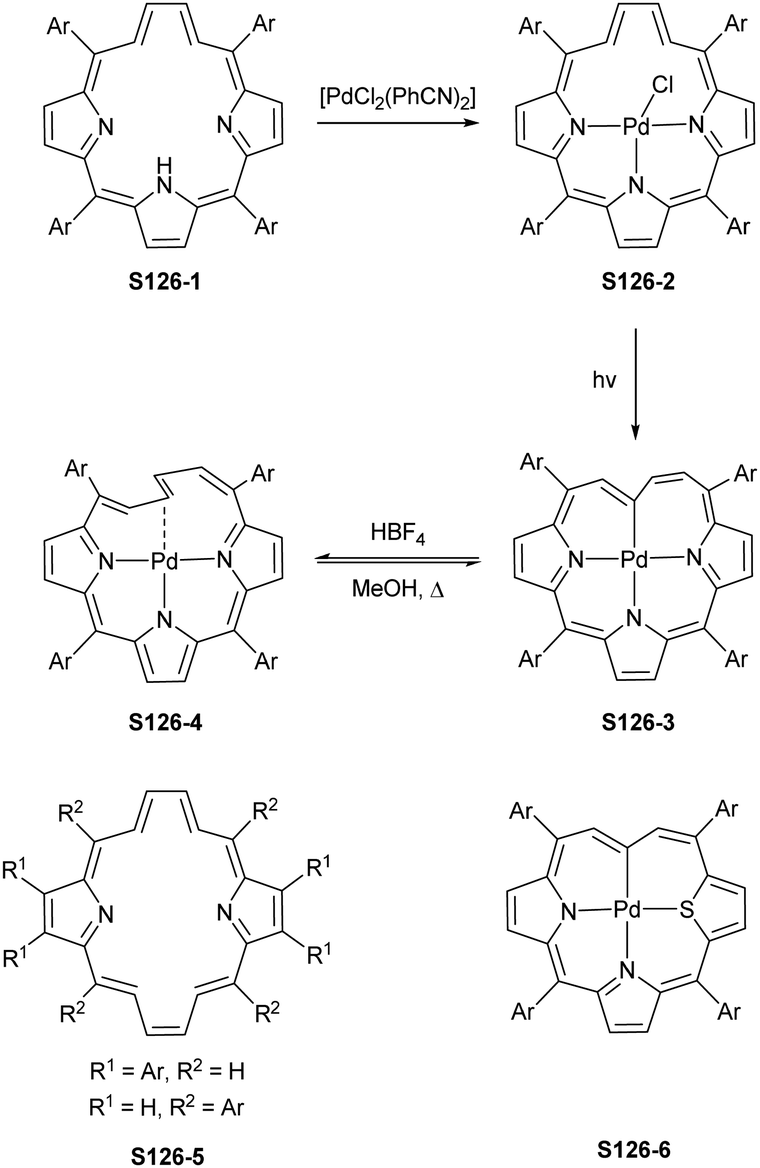 | ||
| Scheme 126 Vacata- (S126-1), divacata- (S126-5), and allyliporphyrin (S126-6) frameworks. | ||
meso-Tetraarylvacataporphyrin S126-1 was initially generated from 21-telluraporphyrin through acidolytic removal of the tellurium atom.280 The macrocycle remains aromatic with the two inner CH protons from a remained diene fragment resonating at −2.50 ppm. S126-1 can be coordinated with palladium(II), among others.281
A reaction with [PdCl2(PhCN)2] yields S126-2 with no direct C–Pd bonding (Fig. 32), yet the forced proximity facilitates further transformations. After exposing the complex to light, the transformation occurs in which one of the carbon atoms gets coordinated, forming S126-3. The latter complex is still aromatic, as confirmed with the 1H NMR spectra, but the spectrum changes after either protonation or methylation with methyl iodide in the presence of a halide acceptor (AgBF4), indicating features of paratropicity. The σ-coordinated carbon atom accepts a proton (or methyl) forming S126-4, but because palladium(II) prefers a tetracoordinate environment, it strongly interacts with the closest double bond (η2-coordination), altering the conformation of a whole fragment and switching the topology of π orbitals from the Hückel aromatic to the Möbius antiaromatic 18π-electron system.
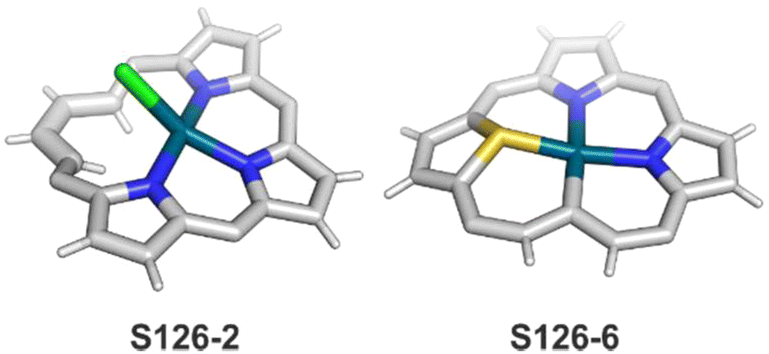 | ||
| Fig. 32 Palladium(II) vacata- and allyliporphyrin complexes. meso-Substituents not shown. | ||
A logical extension of a vacata-like annulene–porphyrin hybrid concept comes with divacataporphyrin S126-5. A half porphyrin–half annulene tetraphenyl-β-substituted derivative was elegantly synthesized by Lash and co-workers in a McMurry coupling reaction.282 Alternatively, an extrusion of two tellurium atoms from 21,23-ditelluraporphyrin yields meso-tetraaryl-21,23-divacataporphyrin.283
Allyliporphyrin constitutes another approach towards annulenes, in which one removes outer β-carbon atoms from a cyclopentadiene unit of theoretical carba(hetero)porphyrin.284 The macrocycle forms in the course of a multistep reaction completed with condensation of an appropriate thiophene-allyl precursor with dipyrromethene. Similar to vacataporphyrins, this dynamic system also shows aromatic features and, due to the availability of two inner protons, forms the palladium(II) complex S126-6 (Fig. 32) with direct Pd–C σ-bonding.
Metallocenoporphyrins
The final group of porphyrinoids to be discussed are classified as metalloceneporphyrins, in which the annulene model of aromaticity is not natural at all. Ferrocene can be introduced into the porphyrinic framework through 1,3-positions from the same ring, replacing one of the pyrrolic rings. Then, the oxidation and work-up processes yield 21-carbaporphyrin (true carbaporphyrin) S1-3.213An alternative incorporation mode of ferrocene was explored as well. Namely, the metallocene moiety can be also built-in through one carbon atom from each cyclopentadienyl ring (1,1′-binding mode) using appropriate precursors.29 Consequently, a stable three-dimensional ferrocenothiaporphyrin S127-1 (Scheme 127) is constructed (Fig. 33). Despite being highly nonplanar, the organometallic system seems to be fully conjugated. The 1H NMR spectrum of S127-1 shows antiaromatic features, with inner protons from the ferrocenyl unit located at 11.30 ppm. This peculiar metallamacrocycle can be reversibly reduced with two electrons providing S127-3 – an aromatic congener. The later molecule exhibits the inner proton signal at 1.73 ppm as expected.
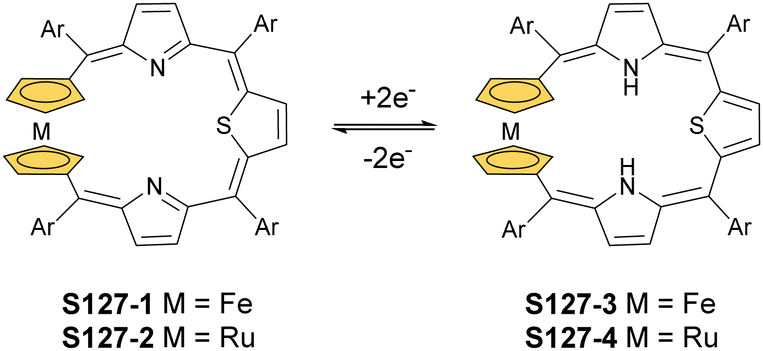 | ||
| Scheme 127 Metallocenothiaporphyrins. | ||
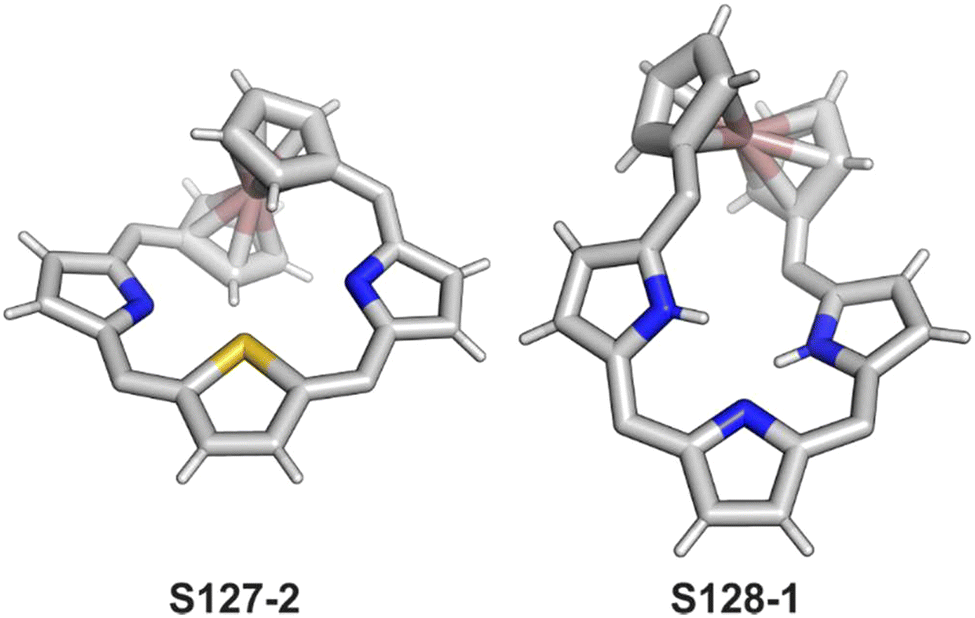 | ||
| Fig. 33 Metallocenoporphyrinoids. meso-Substituents not shown. | ||
Similar molecular qualities are observed for ruthenocenothiaporphyrin S127-2 and its reduced form S127-4.30 The two Cp rings in S127-2 adopt an anticlinal eclipsed conformation. The most interesting electronic properties were detected for all-aza ruthenocenoporphyrin S128-1 (Scheme 128) as it acquires a conformation completely different from S127-1 or S127-2. The monoprotonated cation of S128-1 adopts a peculiar conformation (Fig. 33), with the Cp rings almost eclipsed. It results in the macrocyclic diatropicity of S128-1 and its protonated forms. It means that after changing the conformation from that of S127-2 to that of S128-1, the antiaromaticity switches to aromaticity without changing the number of accessible π-electrons. Therefore, it has been confirmed that the macrocyclic π conjugation transmitted across a d-electron metallocene is conformationally controlled.
 | ||
| Scheme 128 Ruthenocenoporphyrin S128-1. | ||
Conclusions
The well-thought-out decision has been made to explore the “family ties” of carbaporphyrinoids starting from Vogel's original idea, namely, to approach carbaporphyrins from an annulene chemist's point of view.285–287 According to our expectations, such an overview brings an appropriate perspective to the present achievements in the field. In longer distances, the promising perspectives can be founded presumably achievable via well-planned synthesis of intriguing structures still preserving the principal porphyrinic skeleton. Emanuel Vogel pioneered a very stimulating concept in his enlightening and stimulating overview, introducing the idea of novel porphyrin isomers. Namely, he approached a porphyrin frame from an “annulene-chemist perspective”. Thus, an all-carbon forefather of a porphyrin skeleton may be constructed by incorporating two CH2 and two CHCH bridges to the maternal [18]annulene 1, resulting in peculiar aromatic tetracyclopentadienic hydrocarbon 4 (Scheme 129). To date, such hydrocarbon, later named quatyrin,238 and its oxidised derivative didehydroquatyrin remain to be synthesised, albeit the importance of 4 to both annulene and porphyrin fields has been highlighted.288 Significantly, the relevant molecular pattern has been identified in the tetraazuliporphyrin tetracation S105-2.241
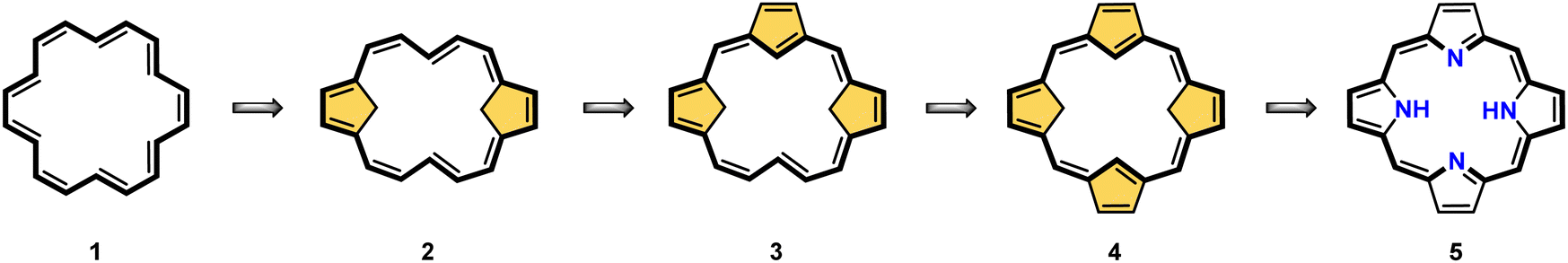 | ||
| Scheme 129 Visualisation of bridging patterns affording [18]annulene 1 transformation into quatyrin 4 and subsequently into porphyrin 5. | ||
Eventually, one can notice that Vogel's concept can be readily adapted to visualise many carbaporphyrins, which preserve the fundamental geometric features of regular porphyrin (Schemes 130–132). Accordingly, quatyrin 4 can be recognised as a fundamental molecular framework to be merely modified by “nitriding” [incorporating, e.g., one (6 or 9), two (7 or 11), three (8 or 12) or four (5) nitrogen atoms in the location of choice instead of carbon atom(s)] (Scheme 130). In the limits of this review, one can present all feasible and equally intriguing structures resulting from the very conceptually rewarding visualisations. The most intriguing or the most promising have been arbitrarily selected, presenting the construction of regular carbaporphyrins (Scheme 130), N-confused porphyrins (Scheme 131, the whole set explored by using the DFT approach289,290), neo-confused porphyrins (Scheme 132, the motif theoretically studied291), and meso-aza carbaporphyrins (Scheme 132).
 | ||
| Scheme 130 Inner core stepwise “nitriding” of quatyrin (an inner core described as: [set of donor atoms], hybridisations: C′ tetrahedral, C trigonal, N trigonal). | ||
 | ||
| Scheme 131 Inner core/perimeter four nitrogen atom “nitriding” of quatyrin to generate N-confused and di-, tri-, and tetra-N-confused porphyrins (an inner core described as: [set donor atoms], hybridisations: C′ tetrahedral, C trigonal, and N trigonal). | ||
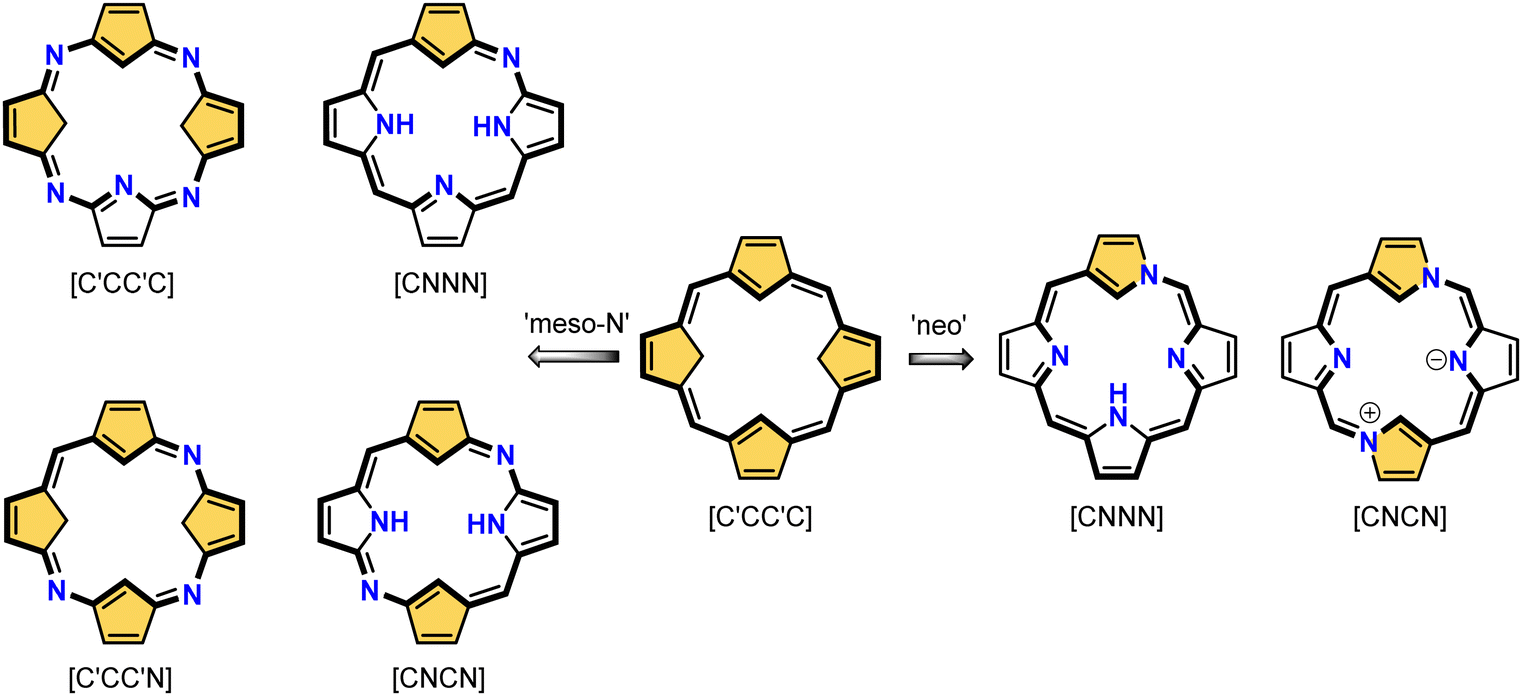 | ||
| Scheme 132 Convoluted inner core/meso- or α-C perimeter four nitrogen atom “nitriding” of quatyrin to generate neo-confused and di-neo-confused porphyrins or meso-aza-carbaporphyrins (an inner core described as: [set donor atoms], hybridisations: C′ tetrahedral, C trigonal, and N trigonal). | ||
Finally, regular porphyrin 5 and quatyrin 4 can be used as two opposite starting points to create prototypes of carbaporphyrinoids via N-confusion or nitriding concepts, respectively (Scheme 133).
 | ||
| Scheme 133 Complementary strategies: inner core/perimeter four nitrogen atom nitriding of quatyrin (blue arrows) and stepwise N-confusion of regular porphyrin (blue arrow nitriding and purple arrow N-confusion). | ||
This all shows a plentiful set of ideas for forming porphyrins containing a carbon atom within the cavity exposed to interactions with a metal ion. As we presented, experimental explorations started in the 1990s are far from being completed. They should bring many new and original results and deepen our knowledge of organometallic chemistry under macrocyclic confinement and the further application of these fascinating systems in the years to come.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI Grant Number JP20H00406 and NCN Grant Numbers 2020/39/D/ST4/00021 and 2019/35/D/ST4/00361. Publication of this article was supported by the Excellence Initiative – Research University program for the University of Wrocław.Notes and references
- T. D. Lash, Chem. Rev., 2017, 117, 2313–2446 CrossRef CAS PubMed.
- M. Toganoh and H. Furuta, Chem. Commun., 2012, 48, 937–954 RSC.
- T. Tanaka and A. Osuka, Chem. Rev., 2017, 117, 2584–2640 CrossRef CAS PubMed.
- T. Chatterjee, V. S. Shetti, R. Sharma and M. Ravikanth, Chem. Rev., 2017, 117, 3254–3328 CrossRef CAS PubMed.
- L. Cuesta and J. L. Sessler, Chem. Soc. Rev., 2009, 38, 2716–2729 RSC.
- G. Anguera and D. Sánchez-García, Chem. Rev., 2017, 117, 2481–2516 CrossRef CAS PubMed.
- S. Shimizu, Chem. Rev., 2017, 117, 2730–2784 CrossRef CAS PubMed.
- Y. Matano, Chem. Rev., 2017, 117, 3138–3191 CrossRef CAS PubMed.
- R. Orłowski, D. Gryko and D. T. Gryko, Chem. Rev., 2017, 117, 3102–3137 CrossRef PubMed.
- C. Brückner, J. Akhigbe and L. P. Samankumara, Handbook of Porphyrin Science, World Scientific Publishing Company, 2012, vol. 31, pp. 1–275 Search PubMed.
- E. Vogel, M. Köcher, H. Schmickler and J. Lex, Angew. Chem., Int. Ed. Engl., 1986, 25, 257–259 CrossRef.
- P. J. Chmielewski, L. Latos-Grażyński, K. Rachlewicz and T. Głowiak, Angew. Chem., Int. Ed. Engl., 1994, 33, 779–781 CrossRef.
- H. Furuta, T. Asano and T. Ogawa, J. Am. Chem. Soc., 1994, 116, 767–768 CrossRef CAS.
- D. Dolphin, R. H. Felton, D. C. Borg and J. Fajer, J. Am. Chem. Soc., 1970, 92, 743–745 CrossRef CAS.
- M. O. Senge, Angew. Chem., Int. Ed., 2011, 50, 4272–4277 CrossRef CAS PubMed.
- M. J. Broadhurst, R. Grigg and A. W. Johnson, J. Chem. Soc. C, 1971, 3681–3690 RSC.
- A. Ulman and J. Manassen, J. Am. Chem. Soc., 1975, 97, 6540–6544 CrossRef CAS PubMed.
- L. Latos-Grażyński, J. Lisowski, M. M. Olmstead and A. L. Balch, J. Am. Chem. Soc., 1987, 109, 4428–4429 CrossRef.
- W. Haas, B. Knipp, M. Sicken, J. Lex and E. Vogel, Angew. Chem., Int. Ed. Engl., 1988, 27, 409–411 CrossRef.
- E. Vogel, P. Röhrig, M. Sicken, B. Knipp, A. Herrmann, M. Pohl, H. Schmickler and J. Lex, Angew. Chem., Int. Ed. Engl., 1989, 28, 1651–1655 CrossRef.
- M. Stępień, L. Latos-Grażyński, N. Sprutta, P. Chwalisz and L. Szterenberg, Angew. Chem., Int. Ed., 2007, 46, 7869–7873 CrossRef PubMed.
- A. Osuka and S. Saito, Chem. Commun., 2011, 47, 4330–4339 RSC.
- M. Stępień, N. Sprutta and L. Latos-Grażyński, Angew. Chem., Int. Ed., 2011, 50, 4288–4340 CrossRef PubMed.
- M. Pawlicki and L. Latos-Grażyński, Chem. – Asian J., 2015, 10, 1438–1451 CrossRef CAS PubMed.
- H. Maeda, Y. Ishikawa, T. Matsuda, A. Osuka and H. Furuta, J. Am. Chem. Soc., 2003, 125, 11822–11823 CrossRef CAS PubMed.
- N. Grzegorzek, M. Pawlicki, L. Szterenberg and L. Latos-Grażyński, J. Am. Chem. Soc., 2009, 131, 7224–7225 CrossRef CAS PubMed.
- B. Szyszko, L. Latos-Grażyński and L. Szterenberg, Angew. Chem., Int. Ed., 2011, 50, 6587–6591 CrossRef CAS PubMed.
- K. Hurej, M. Pawlicki, L. Szterenberg and L. Latos-Grażyński, Angew. Chem., Int. Ed., 2016, 55, 1427–1431 CrossRef CAS PubMed.
- I. Simkowa, L. Latos-Grażyński and M. Stępień, Angew. Chem., Int. Ed., 2010, 49, 7665–7669 CrossRef CAS PubMed.
- I. Grocka, L. Latos-Grażyński and M. Stępień, Angew. Chem., Int. Ed., 2013, 52, 1044–1048 CrossRef CAS PubMed.
- J. B. dela Cruz, M. Ruamps, S. Arco and C.-H. Hung, Dalton Trans., 2019, 48, 7527–7531 RSC.
- J. B. dela Cruz and C.-H. Hung, Catal. Sci. Technol., 2021, 11, 2144–2154 RSC.
- T. Miyazaki, T. Yamamoto, S. Mashita, Y. Deguchi, K. Fukuyama, M. Ishida, S. Mori and H. Furuta, Eur. J. Inorg. Chem., 2018, 203–207 CrossRef CAS.
- K. Rachlewicz, L. Latos-Grażyński and E. Vogel, Inorg. Chem., 2000, 39, 3247–3251 CrossRef CAS PubMed.
- M. J. Białek and L. Latos-Grażyński, Chem. Commun., 2014, 50, 9270–9272 RSC.
- P. Rothemund, J. Am. Chem. Soc., 1936, 58, 625–627 CrossRef CAS.
- A. D. Adler, F. R. Longo, J. D. Finarelli, J. Goldmacher, J. Assour and L. Korsakoff, J. Org. Chem., 1967, 32, 476 CrossRef CAS.
- J. S. Lindsey, I. C. Schreiman, H. C. Hsu, P. C. Kearney and A. M. Marguerettaz, J. Org. Chem., 1987, 52, 827–836 CrossRef CAS.
- M. Pawlicki and L. Latos-Grażyński, Handbook of Porphyrin Science, World Scientific Publishing, Singapore, 2010, vol. 2, pp. 103–192 Search PubMed.
- A. Berlicka and L. Latos-Grażyński, J. Porphyrins Phthalocyanines, 2020, 24, 1–20 CrossRef CAS.
- P. Pushpanandan and M. Ravikanth, Top. Curr. Chem., 2021, 379, 26 CrossRef CAS PubMed.
- M. Toganoh and H. Furuta, Chem. Rev., 2022, 122, 8313–8437 CrossRef CAS PubMed.
- T. D. Lash, Acc. Chem. Res., 2016, 49, 471–482 CrossRef CAS PubMed.
- K. Hurej and L. Latos-Grażyński, in Metal-Ligand Co-operativity: Catalysis and the Pincer-Metal Platform, ed. G. van Koten, K. Kirchner and M.-E. Moret, Springer International Publishing, Cham, 2021, pp. 181–225 Search PubMed.
- J. Zhang, G. Leitus, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2006, 45, 1113–1115 CrossRef CAS PubMed.
- B. Guo, E. Otten and J. G. de Vries, in Metal-Ligand Co-operativity: Catalysis and the Pincer-Metal Platform, ed. G. van Koten, K. Kirchner and M.-E. Moret, Springer International Publishing, Cham, 2021, pp. 321–377 Search PubMed.
- H.-T. Zhang and M.-T. Zhang, in Metal-Ligand Co-operativity: Catalysis and the Pincer-Metal Platform, ed. G. van Koten, K. Kirchner and M.-E. Moret, Springer International Publishing, Cham, 2021, pp. 379–449 Search PubMed.
- S. Aronoff and M. Calvin, J. Org. Chem., 1943, 08, 205–223 CrossRef CAS.
- B. Y. Liu, C. Brückner and D. Dolphin, Chem. Commun., 1996, 2141–2142 RSC.
- G. R. Geier, D. M. Haynes and J. S. Lindsey, Org. Lett., 1999, 1, 1455–1458 CrossRef CAS PubMed.
- T. Morimoto, S. Taniguchi, A. Osuka and H. Furuta, Eur. J. Org. Chem., 2005, 3887–3890 CrossRef CAS.
- H. Furuta, T. Ishizuka, A. Osuka, H. Dejima, H. Nakagawa and Y. Ishikawa, J. Am. Chem. Soc., 2001, 123, 6207–6208 CrossRef CAS PubMed.
- T. Ishizuka, R. Sakashita, O. Iwanaga, T. Morimoto, S. Mori, M. Ishida, M. Toganoh, K. Takegoshi, A. Osuka and H. Furuta, J. Phys. Chem. A, 2020, 124, 5756–5769 CrossRef CAS PubMed.
- R. Sakashita, Y. Oka, H. Akimaru, P. E. Kesavan, M. Ishida, M. Toganoh, T. Ishizuka, S. Mori and H. Furuta, J. Org. Chem., 2017, 82, 8686–8696 CrossRef CAS PubMed.
- P. J. Chmielewski and L. Latos-Grażyński, J. Chem. Soc., Perkin Trans. 2, 1995, 503–509 RSC.
- W. Qu, T. Ding, A. Cetin, J. D. Harvey, M. J. Taschner and C. J. Ziegler, J. Org. Chem., 2006, 71, 811–814 CrossRef CAS PubMed.
- M. Toganoh, T. Yamamoto, T. Hihara, H. Akimaru and H. Furuta, Org. Biomol. Chem., 2012, 10, 4367–4374 RSC.
- P. J. Chmielewski, Org. Lett., 2005, 7, 1789–1792 CrossRef CAS PubMed.
- D. Ren, S. Koniarz, X. Li and P. J. Chmielewski, Chem. Commun., 2020, 56, 4836–4839 RSC.
- X. Li, B. Liu, P. Yi, R. Yi, X. Yu and P. J. Chmielewski, J. Org. Chem., 2011, 76, 2345–2349 CrossRef CAS PubMed.
- I. Schmidt and P. J. Chmielewski, Tetrahedron Lett., 2001, 42, 6389–6392 CrossRef CAS.
- B. Liu, X. Li, X. Xu, M. Stępień and P. J. Chmielewski, J. Org. Chem., 2013, 78, 1354–1364 CrossRef CAS PubMed.
- P. J. Chmielewski, Angew. Chem., Int. Ed., 2004, 43, 5655–5658 CrossRef CAS PubMed.
- N. Grzegorzek, L. Latos-Grażyński and L. Szterenberg, Org. Biomol. Chem., 2012, 10, 8064–8075 RSC.
- Y. Ishikawa, I. Yoshida, K. Akaiwa, E. Koguchi, T. Sasaki and H. Furuta, Chem. Lett., 1997, 453–454 CrossRef CAS.
- N. Kashiwagi, T. Akeda, T. Morimoto, T. Ishizuka and H. Furuta, Org. Lett., 2007, 9, 1733–1736 CrossRef CAS PubMed.
- B. Liu, X. Li, J. Maciołek, M. Stępień and P. J. Chmielewski, J. Org. Chem., 2014, 79, 3129–3139 CrossRef CAS PubMed.
- L. Szteremberg and L. Latos-Grażyński, J. Porphyrins Phthalocyanines, 2001, 05, 474–480 CrossRef.
- M. Toganoh and H. Furuta, J. Phys. Chem. A, 2009, 113, 13953–13963 CrossRef CAS PubMed.
- H. Furuta, T. Ishizuka, A. Osuka and T. Ogawa, J. Am. Chem. Soc., 1999, 121, 2945–2946 CrossRef CAS.
- M. Toganoh and H. Furuta, Chem. Lett., 2019, 48, 615–622 CrossRef CAS.
- S. Ikeda, M. Toganoh and H. Furuta, Inorg. Chem., 2011, 50, 6029–6043 CrossRef CAS PubMed.
- S. Touden, Y. Ikawa, R. Sakashita, M. Toganoh, S. Mori and H. Furuta, Tetrahedron Lett., 2012, 53, 6071–6074 CrossRef CAS.
- M. Toganoh, T. Kimura and H. Furuta, Chem. – Eur. J., 2008, 14, 10585–10594 CrossRef CAS PubMed.
- M. Toganoh, T. Kimura and H. Furuta, Chem. Commun., 2008, 102–104 RSC.
- T. Ishizuka, A. Osuka and H. Furuta, Angew. Chem., Int. Ed., 2004, 43, 5077–5081 CrossRef CAS PubMed.
- H. Furuta, T. Ishizuka and A. Osuka, J. Am. Chem. Soc., 2002, 124, 5622–5623 CrossRef CAS PubMed.
- H. Furuta, T. Morimoto and A. Osuka, Inorg. Chem., 2004, 43, 1618–1624 CrossRef CAS PubMed.
- Y.-C. Chen, J.-Y. Tung, T.-K. Liu, W.-J. Tsai, H.-Y. Lin, Y.-C. Chang and J.-H. Chen, Dalton Trans., 2018, 47, 14774–14784 RSC.
- M. Siczek and P. J. Chmielewski, Angew. Chem., Int. Ed., 2007, 46, 7432–7436 CrossRef CAS PubMed.
- M. Toganoh, H. Ogawa, T. Morimoto and H. Furuta, Supramol. Chem., 2009, 21, 324–330 CrossRef CAS.
- T. Morimoto, H. Uno and H. Furuta, Angew. Chem., Int. Ed., 2007, 46, 3672–3675 CrossRef CAS PubMed.
- P. J. Chmielewski and L. Latos-Grażyński, Inorg. Chem., 1997, 36, 840–845 CrossRef CAS.
- Z. Xiao, B. O. Patrick and D. Dolphin, Inorg. Chem., 2003, 42, 8125–8127 CrossRef CAS PubMed.
- H. He, Z. Ye, D. Shimizu, I. Sumra, Y. Zhang, Z. Liang, Y. Zeng, L. Xu, A. Osuka, Z. Ke and H. W. Jiang, Chem. – Eur. J., 2022, 28, e202103272 CAS.
- H.-W. Jiang, Q.-Y. Chen, J.-C. Xiao and Y.-C. Gu, Chem. Commun., 2009, 3732–3734 RSC.
- P. J. Chmielewski, L. Latos-Grażyński and T. Głowiak, J. Am. Chem. Soc., 1996, 118, 5690–5701 CrossRef CAS.
- T. D. Lash, D. T. Richter and C. M. Shiner, J. Org. Chem., 1999, 64, 7973–7982 CrossRef CAS.
- T. D. Lash and A. L. Von Ruden, J. Org. Chem., 2008, 73, 9417–9425 CrossRef CAS PubMed.
- H.-W. Jiang, Q.-Y. Chen, J.-C. Xiao and Y.-C. Gu, Chem. Commun., 2008, 5435–5437 RSC.
- I. Schmidt and P. J. Chmielewski, Chem. Commun., 2002, 92–93 RSC.
- A. Nakai, J. Kim, T. Tanaka, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2021, 60, 26540–26544 CrossRef CAS PubMed.
- H. Furuta, N. Kubo, H. Maeda, T. Ishizuka, A. Osuka, H. Nanami and T. Ogawa, Inorg. Chem., 2000, 39, 5424–5425 CrossRef CAS PubMed.
- D.-H. Won, M. Toganoh, H. Uno and H. Furuta, Dalton Trans., 2009, 6151–6158 RSC.
- T. Yoneda, S. Saito, H. Yorimitsu and A. Osuka, Angew. Chem., Int. Ed., 2011, 50, 3475–3478 CrossRef CAS PubMed.
- H. Furuta, K. Youfu, H. Maeda and A. Osuka, Angew. Chem., Int. Ed., 2003, 42, 2186–2188 CrossRef CAS PubMed.
- P. J. Chmielewski, B. Durlej, M. Siczek and L. Szterenberg, Angew. Chem., Int. Ed., 2009, 48, 8736–8739 CrossRef CAS PubMed.
- H. Furuta, T. Ogawa, Y. Uwatoko and K. Araki, Inorg. Chem., 1999, 38, 2676–2682 CrossRef CAS.
- I. Sumra, M. Irfan, H. He, Y. Zhang, F. Hao, J. Lin, A. Osuka, Z. Zeng and H.-W. Jiang, Eur. J. Org. Chem., 2021, 4440–4443 CrossRef CAS.
- M. Toganoh, T. Niino and H. Furuta, Chem. Commun., 2008, 4070–4072 RSC.
- N. Grzegorzek, M. Pawlicki and L. Latos-Grażyński, J. Org. Chem., 2009, 74, 8547–8553 CrossRef CAS PubMed.
- H.-W. Jiang, F. Hao, Q.-Y. Chen, J.-C. Xiao, S.-B. Liu and Y.-C. Gu, J. Org. Chem., 2010, 75, 3511–3514 CrossRef CAS PubMed.
- F. Hao, T. Zhang, D. Yu, X. Yang, H.-W. Jiang, J.-C. Xiao and Q.-Y. Chen, Org. Lett., 2022, 24, 1716–1721 CrossRef CAS PubMed.
- P. J. Chmielewski, Angew. Chem., Int. Ed., 2005, 44, 6417–6420 CrossRef CAS PubMed.
- P. J. Chmielewski, L. Latos-Grażyński and I. Schmidt, Inorg. Chem., 2000, 39, 5475–5482 CrossRef CAS PubMed.
- H. Maeda, A. Osuka, Y. Ishikawa, I. Aritome, Y. Hisaeda and H. Furuta, Org. Lett., 2003, 5, 1293–1296 CrossRef CAS PubMed.
- H. Furuta, H. Maeda and A. Osuka, Org. Lett., 2002, 4, 181–184 CrossRef CAS PubMed.
- J. Wojaczyński, M. Popiel, L. Szterenberg and L. Latos-Grażyński, J. Org. Chem., 2011, 76, 9956–9961 CrossRef PubMed.
- J. Wojaczyński and L. Latos-Grażyński, Chem. – Eur. J., 2010, 16, 2679–2682 CrossRef PubMed.
- W.-C. Lin, D.-Z. Hsaio, W.-P. Chang, J.-H. Chen, S.-S. Wang and J.-Y. Tung, Polyhedron, 2012, 42, 243–248 CrossRef CAS.
- N. Grzegorzek, E. Nojman, L. Szterenberg and L. Latos-Grażyński, Inorg. Chem., 2013, 52, 2599–2606 CrossRef CAS PubMed.
- C.-H. Hung, C.-H. Peng, Y.-L. Shen, S.-L. Wang, C.-H. Chuang and H. M. Lee, Eur. J. Inorg. Chem., 2008, 1196–1199 CrossRef CAS.
- I. Zilbermann, E. Maimon, R. Ydgar, A. I. Shames, E. Korin, L. Soifer and A. Bettelheim, Inorg. Chem. Commun., 2004, 7, 1238–1241 CrossRef CAS.
- K. B. Fields, J. T. Engle, S. Sripothongnak, C. Kim, X. P. Zhang and C. J. Ziegler, Chem. Commun., 2011, 47, 749–751 RSC.
- J. D. Harvey and C. J. Ziegler, Chem. Commun., 2004, 1666–1667 RSC.
- C.-H. Chuang, W.-F. Liaw and C.-H. Hung, Angew. Chem., Int. Ed., 2016, 55, 5190–5194 CrossRef CAS PubMed.
- Y.-C. Chen, J.-Y. Tung, T.-K. Liu, W.-J. Tsai, C.-Y. Lin, H.-Y. Lin, S.-S. Li, J.-H. Chen and Y.-C. Chang, Polyhedron, 2019, 159, 233–238 CrossRef CAS.
- Y.-C. Wang, J.-H. Chen, S.-S. Wang and J.-Y. Tung, Inorg. Chem., 2013, 52, 10711–10713 CrossRef CAS PubMed.
- Y.-Y. Lai, Y.-C. Chang, J.-H. Chen, S.-S. Wang and J.-Y. Tung, Dalton Trans., 2016, 45, 4854–4862 RSC.
- A. Srinivasan, H. Furuta and A. Osuka, Chem. Commun., 2001, 1666–1667 RSC.
- A. Srinivasan, M. Toganoh, T. Niino, A. Osuka and H. Furuta, Inorg. Chem., 2008, 47, 11305–11313 CrossRef CAS PubMed.
- M. Toganoh, J. Konagawa and H. Furuta, Inorg. Chem., 2006, 45, 3852–3854 CrossRef CAS PubMed.
- T. Niino, M. Toganoh, B. Andrioletti and H. Furuta, Chem. Commun., 2006, 4335–4337 RSC.
- M. Toganoh, T. Niino, H. Maeda, B. Andrioletti and H. Furuta, Inorg. Chem., 2006, 45, 10428–10430 CrossRef CAS PubMed.
- K. Rachlewicz, S.-L. Wang, C.-H. Peng, C.-H. Hung and L. Latos-Grażyński, Inorg. Chem., 2003, 42, 7348–7350 CrossRef CAS PubMed.
- K. Rachlewicz, S.-L. Wang, J.-L. Ko, C.-H. Hung and L. Latos-Grażyński, J. Am. Chem. Soc., 2004, 126, 4420–4431 CrossRef CAS PubMed.
- K. Rachlewicz, D. Gorzelańczyk and L. Latos-Grażyński, Inorg. Chem., 2006, 45, 9742–9747 CrossRef CAS PubMed.
- W.-C. Chen and C.-H. Hung, Inorg. Chem., 2001, 40, 5070–5071 CrossRef CAS PubMed.
- C.-H. Hung, S.-L. Wang, J.-L. Ko, C.-H. Peng, C.-H. Hu and M.-T. Lee, Org. Lett., 2004, 6, 1393–1396 CrossRef CAS PubMed.
- C.-H. Hung, W.-M. Ching, G.-F. Chang, C.-H. Chuang, H.-W. Chu and W.-Z. Lee, Inorg. Chem., 2007, 46, 10941–10943 CrossRef CAS PubMed.
- W.-M. Ching and C.-H. Hung, Chem. Commun., 2012, 48, 4989–4991 RSC.
- C.-H. Hung, W.-C. Chen, G.-H. Lee and S.-M. Peng, Chem. Commun., 2002, 1516–1517 RSC.
- C.-H. Hung, C.-H. Chang, W.-M. Ching and C.-H. Chuang, Chem. Commun., 2006, 1866–1868 RSC.
- T. Miyazaki, K. Fukuyama, S. Mashita, Y. Deguchi, T. Yamamoto, M. Ishida, S. Mori and H. Furuta, ChemPlusChem, 2019, 84, 603–607 CrossRef CAS PubMed.
- O. Iwanaga, K. Fukuyama, S. Mori, J. T. Song, T. Ishihara, T. Miyazaki, M. Ishida and H. Furuta, RSC Adv., 2021, 11, 24575–24579 RSC.
- T. Yamamoto, K. Mitsuno, S. Mori, S. Itoyama, Y. Shiota, K. Yoshizawa, M. Ishida and H. Furuta, Chem. – Eur. J., 2018, 24, 6742–6746 CrossRef CAS PubMed.
- D. S. Bohle, W.-C. Chen and C.-H. Hung, Inorg. Chem., 2002, 41, 3334–3336 CrossRef CAS PubMed.
- J. D. Harvey and C. J. Ziegler, Chem. Commun., 2002, 1942–1943 RSC.
- S.-H. Peng, M. H. R. Mahmood, H.-B. Zou, S.-B. Yang and H.-Y. Liu, J. Mol. Catal. A: Chem., 2014, 395, 180–185 CrossRef CAS.
- M. Toganoh, T. Ishizuka and H. Furuta, Chem. Commun., 2004, 2464–2465 RSC.
- M. Toganoh, S. Ikeda and H. Furuta, Inorg. Chem., 2007, 46, 10003–10015 CrossRef CAS PubMed.
- M. Toganoh and H. Furuta, Chem. Lett., 2005, 34, 1034–1035 CrossRef CAS.
- T. Yamamoto, M. Toganoh, S. Mori, H. Uno and H. Furuta, Chem. Sci., 2012, 3, 3241–3248 RSC.
- T. Yamamoto, M. Toganoh and H. Furuta, Dalton Trans., 2012, 41, 9154–9157 RSC.
- M. Toganoh, T. Hihara and H. Furuta, Inorg. Chem., 2010, 49, 8182–8184 CrossRef CAS PubMed.
- X. Zhu, W.-K. Wong, W.-K. Lo and W.-Y. Wong, Chem. Commun., 2005, 1022–1024 RSC.
- X.-J. Zhu, F.-L. Jiang, C.-T. Poon, W.-K. Wong and W.-Y. Wong, Eur. J. Inorg. Chem., 2008, 3151–3162 CrossRef CAS.
- A. Młodzianowska, L. Latos-Grażyński, L. Szterenberg and M. Stępień, Inorg. Chem., 2007, 46, 6950–6957 CrossRef PubMed.
- A. Młodzianowska, L. Latos-Grażyński and L. Szterenberg, Inorg. Chem., 2008, 47, 6364–6374 CrossRef PubMed.
- J. Skonieczny, L. Latos-Grażyński and L. Szterenberg, Inorg. Chem., 2009, 48, 7394–7407 CrossRef CAS PubMed.
- J. D. Harvey, J. L. Shaw, R. S. Herrick and C. J. Ziegler, Chem. Commun., 2005, 4663–4665 RSC.
- B. Babu, J. Mack and T. Nyokong, Dalton Trans., 2020, 49, 15180–15183 RSC.
- Y. Xie, T. Morimoto and H. Furuta, Angew. Chem., Int. Ed., 2006, 45, 6907–6910 CrossRef CAS PubMed.
- J.-C. Liu, T. Ishizuka, A. Osuka and H. Furuta, Chem. Commun., 2003, 1908–1909 RSC.
- T. Ogawa, H. Furuta, M. Takahashi, A. Morino and H. Uno, J. Organomet. Chem., 2000, 611, 551–557 CrossRef CAS.
- S. Sripothongnak and C. J. Ziegler, Inorg. Chem., 2010, 49, 5789–5791 CrossRef CAS PubMed.
- B. Szyszko, M. J. Białek, E. Pacholska-Dudziak and L. Latos-Grażyński, Chem. Rev., 2017, 117, 2839–2909 CrossRef CAS PubMed.
- N. Sprutta and L. Latos-Grażyński, Tetrahedron Lett., 1999, 40, 8457–8460 CrossRef CAS.
- M. Pawlicki and L. Latos-Grażyński, Chem. – Eur. J., 2003, 9, 4650–4660 CrossRef CAS PubMed.
- M. Pawlicki and L. Latos-Grażyński, J. Org. Chem., 2005, 70, 9123–9130 CrossRef CAS PubMed.
- T. D. Lash, in Advances in Heterocyclic Chemistry, ed. E. F. V. Scriven and C. A. Ramsden, Academic Press, 2022, vol. 138, pp. 243–334 Search PubMed.
- I. Schmidt and P. J. Chmielewski, Tetrahedron Lett., 2001, 42, 1151–1154 CrossRef CAS.
- M. Pawlicki, L. Latos-Grażyński and L. Szterenberg, Inorg. Chem., 2005, 44, 9779–9786 CrossRef CAS PubMed.
- T. D. Lash, A. M. Young, A. L. Von Ruden and G. M. Ferrence, Chem. Commun., 2008, 6309–6311 RSC.
- M. Ishida, H. Fujimoto, T. Morimoto, S. Mori, M. Toganoh, S. Shimizu and H. Furuta, Supramol. Chem., 2017, 29, 8–16 CrossRef CAS.
- A. M. Young, A. L. Von Ruden and T. D. Lash, Org. Biomol. Chem., 2011, 9, 6293–6305 RSC.
- T. D. Lash, A. D. Lammer and G. M. Ferrence, Angew. Chem., Int. Ed., 2011, 50, 9718–9721 CrossRef CAS PubMed.
- A. S. Almejbel and T. D. Lash, Org. Biomol. Chem., 2020, 18, 7336–7344 RSC.
- R. Li, G. M. Ferrence and T. D. Lash, Chem. Commun., 2013, 49, 7537–7539 RSC.
- M. J. Chmielewski, M. Pawlicki, N. Sprutta, L. Szterenberg and L. Latos-Grażyński, Inorg. Chem., 2006, 45, 8664–8671 CrossRef CAS PubMed.
- M. Pawlicki, I. Kańska and L. Latos-Grażyński, Inorg. Chem., 2007, 46, 6575–6584 CrossRef CAS PubMed.
- R. Li, A. D. Lammer, G. M. Ferrence and T. D. Lash, J. Org. Chem., 2014, 79, 4078–4093 CrossRef CAS PubMed.
- K. Berlin, C. Steinbeck and E. Breitmaier, Synthesis, 1996, 336–340 CrossRef CAS.
- T. D. Lash, Chem. Commun., 1998, 1683–1684 RSC.
- L. M. Stateman and T. D. Lash, Org. Lett., 2015, 17, 4522–4525 CrossRef CAS PubMed.
- T. Okujima, T. Kikkawa, H. Nakano, H. Kubota, N. Fukugami, N. Ono, H. Yamada and H. Uno, Chem. – Eur. J., 2012, 18, 12854–12863 CrossRef CAS PubMed.
- J. S. D. Moriones, A. N. Latham and T. D. Lash, J. Porphyrins Phthalocyanines, 2020, 24, 817–829 CrossRef CAS.
- S. Venkatraman, V. G. Anand, V. PrabhuRaja, H. Rath, J. Sankar, T. K. Chandrashekar, W. J. Teng and K. R. Senge, Chem. Commun., 2002, 1660–1661 RSC.
- M. J. Białek and L. Latos-Grażyński, Inorg. Chem., 2016, 55, 1758–1769 CrossRef PubMed.
- T. D. Lash, D. I. AbuSalim and G. M. Ferrence, Inorg. Chem., 2021, 60, 9833–9847 CrossRef CAS PubMed.
- T. J. Smolczyk and T. D. Lash, Chem. Commun., 2018, 54, 9003–9006 RSC.
- D. A. Colby and T. D. Lash, Chem. – Eur. J., 2002, 8, 5397–5402 CrossRef CAS PubMed.
- T. D. Lash, D. A. Colby and G. M. Ferrence, Eur. J. Org. Chem., 2003, 4533–4548 CrossRef CAS.
- T. D. Lash and S. T. Chaney, Angew. Chem., Int. Ed. Engl., 1997, 36, 839–840 CrossRef CAS.
- A. Ghosh, S. Larsen, J. Conradie and C. Foroutan-Nejad, Org. Biomol. Chem., 2018, 16, 7964–7970 RSC.
- V. A. K. Adiraju, G. M. Ferrence and T. D. Lash, Org. Biomol. Chem., 2016, 14, 10523–10533 RSC.
- T. D. Lash, J. A. El-Beck and G. M. Ferrence, Org. Biomol. Chem., 2014, 12, 316–329 RSC.
- T. D. Lash, D. A. Colby, S. R. Graham and S. T. Chaney, J. Org. Chem., 2004, 69, 8851–8864 CrossRef CAS PubMed.
- T. D. Lash, G. C. Gilot and D. I. AbuSalim, J. Org. Chem., 2014, 79, 9704–9716 CrossRef CAS PubMed.
- M. J. Hayes, J. D. Spence and T. D. Lash, Chem. Commun., 1998, 2409–2410 RSC.
- A. N. Latham, G. M. Ferrence and T. D. Lash, Organometallics, 2019, 38, 575–585 CrossRef CAS.
- T. D. Lash, D. A. Colby and L. F. Szczepura, Inorg. Chem., 2004, 43, 5258–5267 CrossRef CAS PubMed.
- M. A. Muckey, L. F. Szczepura, G. M. Ferrence and T. D. Lash, Inorg. Chem., 2002, 41, 4840–4842 CrossRef CAS PubMed.
- E. Y. Grabowski, D. I. AbuSalim and T. D. Lash, J. Org. Chem., 2018, 83, 11825–11838 CrossRef CAS PubMed.
- D. A. Colby, G. M. Ferrence and T. D. Lash, Angew. Chem., Int. Ed., 2004, 43, 1346–1349 CrossRef CAS PubMed.
- M. J. Białek, P. J. Chmielewski and L. Latos-Grażyński, Chem. – Eur. J., 2019, 25, 14536–14545 CrossRef PubMed.
- T. D. Lash, D. A. Colby, J. A. El-Beck, D. I. AbuSalim and G. M. Ferrence, Inorg. Chem., 2015, 54, 9174–9187 CrossRef CAS PubMed.
- T. D. Lash, D. A. Colby, S. R. Graham, G. M. Ferrence and L. F. Szczepura, Inorg. Chem., 2003, 42, 7326–7338 CrossRef CAS PubMed.
- T. D. Lash and G. M. Ferrence, Inorg. Chem., 2017, 56, 11426–11434 CrossRef CAS PubMed.
- L. Latos-Grażyński, E. Pacholska, P. J. Chmielewski, M. M. Olmstead and A. L. Balch, Angew. Chem., Int. Ed. Engl., 1995, 34, 2252–2254 CrossRef.
- T. D. Lash, Org. Lett., 2011, 13, 4632–4635 CrossRef CAS PubMed.
- M. J. Białek, A. Białońska and L. Latos-Grażyński, Inorg. Chem., 2015, 54, 6184–6194 CrossRef PubMed.
- V. A. K. Adiraju, G. M. Ferrence and T. D. Lash, Dalton Trans., 2016, 45, 13691–13694 RSC.
- T. D. Lash, W. T. Darrow, A. N. Latham, N. Sahota and G. M. Ferrence, Inorg. Chem., 2019, 58, 7511–7526 CrossRef CAS PubMed.
- L. M. Stateman, G. M. Ferrence and T. D. Lash, Organometallics, 2015, 34, 3842–3848 CrossRef CAS.
- K. Berlin, Angew. Chem., Int. Ed. Engl., 1996, 35, 1820–1822 CrossRef CAS.
- T. D. Lash and M. J. Hayes, Angew. Chem., Int. Ed. Engl., 1997, 36, 840–842 CrossRef CAS.
- M. J. Hayes and T. D. Lash, Chem. – Eur. J., 1998, 4, 508–511 CrossRef CAS.
- D. Li and T. D. Lash, J. Org. Chem., 2014, 79, 7112–7121 CrossRef CAS PubMed.
- D. Li and T. D. Lash, Eur. J. Org. Chem., 2017, 6775–6780 CrossRef CAS.
- A. Berlicka, P. Dutka, L. Szterenberg and L. Latos-Grażyński, Angew. Chem., Int. Ed., 2014, 53, 4885–4889 CrossRef CAS PubMed.
- A. Berlicka, M. J. Białek and L. Latos-Grażyński, Angew. Chem., Int. Ed., 2016, 55, 11231–11236 CrossRef CAS PubMed.
- M. Garbicz and L. Latos-Grażyński, Angew. Chem., Int. Ed., 2019, 58, 6089–6093 CrossRef CAS PubMed.
- N. Sahota, G. M. Ferrence and T. D. Lash, J. Org. Chem., 2017, 82, 9715–9730 CrossRef CAS PubMed.
- K. Hurej, M. Pawlicki and L. Latos-Grażyński, Chem. – Eur. J., 2017, 23, 2059–2066 CrossRef CAS PubMed.
- K. Hurej, M. Pawlicki and L. Latos-Grażyński, Chem. – Eur. J., 2018, 24, 115–126 CrossRef CAS PubMed.
- B. Szyszko, K. Kupietz, L. Szterenberg and L. Latos-Grażyński, Chem. – Eur. J., 2014, 20, 1376–1382 CrossRef CAS PubMed.
- B. Szyszko and L. Latos-Grażyński, Organometallics, 2011, 30, 4354–4363 CrossRef CAS.
- A. Idec, L. Szterenberg and L. Latos-Grażyński, Chem. – Eur. J., 2015, 21, 12481–12487 CrossRef CAS PubMed.
- H. Furuta, H. Maeda and A. Osuka, J. Am. Chem. Soc., 2000, 122, 803–807 CrossRef CAS.
- H. Uno, K. Muramatsu, S. Hiraoka, H. Tahara, M. Hirose, E. Tamura, T. Shiraishi, J. Mack, N. Kobayashi, S. Mori, T. Okujima and M. Takase, Chem. – Eur. J., 2020, 26, 5701–5708 CrossRef CAS PubMed.
- H. Maeda, A. Osuka and H. Furuta, J. Am. Chem. Soc., 2003, 125, 15690–15691 CrossRef CAS PubMed.
- J. Yan, M. Takakusaki, Y. Yang, S. Mori, B. Zhang, Y. Feng, M. Ishida and H. Furuta, Chem. Commun., 2014, 50, 14593–14596 RSC.
- N. Halder, M. Sangeetha, D. Usharani and H. Rath, J. Org. Chem., 2020, 85, 2059–2067 CrossRef CAS PubMed.
- N. Halder, R. Naryanasamy, D. Usharani and H. Rath, Org. Chem. Front., 2022, 9, 2333–2342 RSC.
- Z. J. Zhang, G. M. Ferrence and T. D. Lash, Org. Lett., 2009, 11, 101–104 CrossRef CAS PubMed.
- D. I. AbuSalim, G. M. Ferrence and T. D. Lash, J. Am. Chem. Soc., 2014, 136, 6763–6772 CrossRef CAS PubMed.
- T. D. Lash, D. A. Colby, A. S. Idate and R. N. Davis, J. Am. Chem. Soc., 2007, 129, 13800–13801 CrossRef CAS PubMed.
- T. D. Lash, A. D. Lammer, A. S. Idate, D. A. Colby and K. White, J. Org. Chem., 2012, 77, 2368–2381 CrossRef CAS PubMed.
- T. D. Lash, D. I. AbuSalim and G. M. Ferrence, Chem. Commun., 2015, 51, 15952–15955 RSC.
- T. D. Lash, J. L. Romanic, M. J. Hayes and J. D. Spence, Chem. Commun., 1999, 819–820 RSC.
- T. D. Lash, S. C. Fosu, T. J. Smolczyk and D. I. AbuSalim, J. Org. Chem., 2018, 83, 12619–12631 CrossRef CAS PubMed.
- S. R. Graham, D. A. Colby and T. D. Lash, Angew. Chem., Int. Ed., 2002, 41, 1371–1374 CrossRef CAS PubMed.
- N. Sprutta, M. Świderska and L. Latos-Grażyński, J. Am. Chem. Soc., 2005, 127, 13108–13109 CrossRef CAS PubMed.
- N. Sprutta, M. Siczek, L. Latos-Grażyński, M. Pawlicki, L. Szterenberg and T. Lis, J. Org. Chem., 2007, 72, 9501–9509 CrossRef CAS PubMed.
- S. Ahmad, A. Singhal, K. Nisa and S. M. S. Chauhan, Inorg. Chem., 2018, 57, 11333–11340 CrossRef CAS PubMed.
- T. Maulbetsch and D. Kunz, Angew. Chem., Int. Ed., 2021, 60, 2007–2012 CrossRef CAS PubMed.
- T. D. Lash, Synlett, 2000, 279–295 CAS.
- N. Sprutta, M. Wełnic, M. J. Białek, T. Lis, L. Szterenberg and L. Latos-Grażyński, Chem. – Eur. J., 2016, 22, 6974–6980 CrossRef CAS PubMed.
- D. A. Colby and T. D. Lash, J. Org. Chem., 2002, 67, 1031–1033 CrossRef CAS PubMed.
- N. Sprutta, S. Mackowiak, M. Kocik, L. Szterenberg, T. Lis and L. Latos-Grażyński, Angew. Chem., Int. Ed., 2009, 48, 3337–3341 CrossRef CAS PubMed.
- G. G. Qiao, M. W. Wong and C. Wentrup, J. Org. Chem., 1996, 61, 8125–8131 CrossRef CAS PubMed.
- Y. Chun, N. J. Singh, I.-C. Hwang, J. W. Lee, S. U. Yu and K. S. Kim, Nat. Commun., 2013, 4, 1797 CrossRef PubMed.
- M. R. Anneser, S. Haslinger, A. Pöthig, M. Cokoja, J.-M. Basset and F. E. Kühn, Inorg. Chem., 2015, 54, 3797–3804 CrossRef CAS PubMed.
- E. B. Bauer, M. A. Bernd, M. Schütz, J. Oberkofler, A. Pöthig, R. M. Reich and F. E. Kühn, Dalton Trans., 2019, 48, 16615–16625 RSC.
- H. M. Bass, S. A. Cramer, A. S. McCullough, K. J. Bernstein, C. R. Murdock and D. M. Jenkins, Organometallics, 2013, 32, 2160–2167 CrossRef CAS.
- M. R. Anneser, G. R. Elpitiya, X. B. Powers and D. M. Jenkins, Organometallics, 2019, 38, 981–987 CrossRef CAS.
- A. Weiss, H. Pritzkow and W. Siebert, Angew. Chem., Int. Ed., 2000, 39, 547–549 CrossRef CAS PubMed.
- X. B. Carroll, D. Errulat, M. Murugesu and D. M. Jenkins, Inorg. Chem., 2022, 61, 1611–1619 CrossRef CAS PubMed.
- J. F. DeJesus, R. W. F. Kerr, D. A. Penchoff, X. B. Carroll, C. C. Peterson, P. L. Arnold and D. M. Jenkins, Chem. Sci., 2021, 12, 7882–7887 RSC.
- M. R. Anneser, X. B. Powers, K. M. Peck, I. M. Jensen and D. M. Jenkins, Organometallics, 2019, 38, 3369–3376 CrossRef CAS PubMed.
- H. Furuta, H. Maeda, A. Osuka, M. Yasutake, T. Shinmyozu and Y. Ishikawa, Chem. Commun., 2000, 1143–1144 RSC.
- J. Yan, Y. Yang, M. Ishida, S. Mori, B. Zhang, Y. Feng and H. Furuta, Chem. – Eur. J., 2017, 23, 11375–11384 CrossRef CAS PubMed.
- M. J. Białek, N. Sprutta and L. Latos-Grażyński, Inorg. Chem., 2016, 55, 12061–12073 CrossRef PubMed.
- P. J. Altmann, D. T. Weiss, C. Jandl and F. E. Kühn, Chem. – Asian J., 2016, 11, 1597–1605 CrossRef CAS PubMed.
- T. P. Schlachta, M. R. Anneser, J. F. Schlagintweit, C. H. G. Jako, C. Hintermeier, A. D. Böth, S. Haslinger, R. M. Reich and F. E. Kühn, Chem. Commun., 2021, 57, 6644–6647 RSC.
- J. F. Schlagintweit, C. Hintermeier, M. R. Anneser, E.-M. H. J. Esslinger, S. Haslinger and F. E. Kühn, Chem. – Asian J., 2020, 15, 1896–1902 CrossRef CAS PubMed.
- M. R. Anneser, S. Haslinger, A. Pöthig, M. Cokoja, V. D'Elia, M. P. Högerl, J.-M. Basset and F. E. Kühn, Dalton Trans., 2016, 45, 6449–6455 RSC.
- J. W. Kück, M. R. Anneser, B. Hofmann, A. Pöthig, M. Cokoja and F. E. Kühn, ChemSusChem, 2015, 8, 4056–4063 CrossRef PubMed.
- S. Meyer, I. Klawitter, S. Demeshko, E. Bill and F. Meyer, Angew. Chem., Int. Ed., 2013, 52, 901–905 CrossRef CAS PubMed.
- C. Cordes, M. Morganti, I. Klawitter, C. Schremmer, S. Dechert and F. Meyer, Angew. Chem., Int. Ed., 2019, 58, 10855–10858 CrossRef CAS PubMed.
- J. F. Schlagintweit, P. J. Altmann, A. D. Böth, B. J. Hofmann, C. Jandl, C. Kaußler, L. Nguyen, R. M. Reich, A. Pöthig and F. E. Kühn, Chem. – Eur. J., 2021, 27, 1311–1315 CrossRef CAS PubMed.
- Z. S. Ghavami, M. R. Anneser, F. Kaiser, P. J. Altmann, B. J. Hofmann, J. F. Schlagintweit, G. Grivani and F. E. Kühn, Chem. Sci., 2018, 9, 8307–8314 RSC.
- F. E. Hahn, V. Langenhahn, T. Lügger, T. Pape and D. Le Van, Angew. Chem., Int. Ed., 2005, 44, 3759–3763 CrossRef CAS PubMed.
- H. Furuta, T. Ishizuka, A. Osuka, Y. Uwatoko and Y. Ishikawa, Angew. Chem., Int. Ed., 2001, 40, 2323–2325 CrossRef CAS PubMed.
- H. Furuta, T. Ishizuka and A. Osuka, Inorg. Chem. Commun., 2003, 6, 398–401 CrossRef CAS.
- P. Pushpanandan, Y. K. Maurya, T. Omagari, R. Hirosawa, M. Ishida, S. Mori, Y. Yasutake, S. Fukatsu, J. Mack, T. Nyokong and H. Furuta, Inorg. Chem., 2017, 56, 12572–12580 CrossRef CAS PubMed.
- N. Halder, M. H. Mahadavaiah, D. Usharani and H. Rath, J. Porphyrins Phthalocyanines, 2021, 25, 966–974 CrossRef CAS.
- R. Nishiyabu, M. A. Palacios, W. Dehaen and P. Anzenbacher, J. Am. Chem. Soc., 2006, 128, 11496–11504 CrossRef CAS PubMed.
- P. Anzenbacher, R. Nishiyabu and M. A. Palacios, Coord. Chem. Rev., 2006, 250, 2929–2938 CrossRef CAS.
- Q. Chen, T. Wang, Y. Zhang, Q. Wang and J. Ma, Synth. Commun., 2002, 32, 1051–1058 CrossRef CAS.
- J. Skonieczny, L. Latos-Grażyński and L. Szterenberg, Chem. – Eur. J., 2008, 14, 4861–4874 CrossRef CAS PubMed.
- Z. Duan, M. Clochard, B. Donnadieu, F. Mathey and F. S. Tham, Organometallics, 2007, 26, 3617–3620 CrossRef CAS.
- K. Fujino, Y. Hirata, Y. Kawabe, T. Morimoto, A. Srinivasan, M. Toganoh, Y. Miseki, A. Kudo and H. Furuta, Angew. Chem., Int. Ed., 2011, 50, 6855–6859 CrossRef CAS PubMed.
- Y. K. Maurya, K. Noda, K. Yamasumi, S. Mori, T. Uchiyama, K. Kamitani, T. Hirai, K. Ninomiya, M. Nishibori, Y. Hori, Y. Shiota, K. Yoshizawa, M. Ishida and H. Furuta, J. Am. Chem. Soc., 2018, 140, 6883–6892 CrossRef CAS PubMed.
- P. Pushpanandan, V. Grover and M. Ravikanth, Org. Lett., 2022, 24, 3184–3188 CrossRef CAS PubMed.
- M. Toganoh, Y. Kawabe, H. Uno and H. Furuta, Angew. Chem., Int. Ed., 2012, 51, 8753–8756 CrossRef CAS PubMed.
- B. Basumatary, I. Hashiguchi, S. Mori, S. Shimizu, M. Ishida and H. Furuta, Angew. Chem., Int. Ed., 2020, 59, 15897–15901 CrossRef CAS PubMed.
- S. Larsen, L. J. McCormick-McPherson, S. J. Teat and A. Ghosh, ACS Omega, 2019, 4, 6737–6745 CrossRef CAS.
- E. Pacholska, L. Latos-Grażyński and Z. Ciunik, Chem. – Eur. J., 2002, 8, 5403–5405 CrossRef CAS PubMed.
- E. Pacholska-Dudziak, J. Skonieczny, M. Pawlicki, L. Szterenberg, Z. Ciunik and L. Latos-Grażyński, J. Am. Chem. Soc., 2008, 130, 6182–6195 CrossRef CAS PubMed.
- T. D. Lash, S. A. Jones and G. M. Ferrence, J. Am. Chem. Soc., 2010, 132, 12786–12787 CrossRef CAS PubMed.
- E. Pacholska-Dudziak, L. Szterenberg and L. Latos-Grażyński, Chem. – Eur. J., 2011, 17, 3500–3511 CrossRef CAS PubMed.
- J.-H. Hong, A. S. Aslam, M.-S. Ko, J. Choi, Y. Lee and D.-G. Cho, Chem. – Eur. J., 2018, 24, 10054–10058 CrossRef CAS PubMed.
- E. Vogel, Pure Appl. Chem., 1990, 62, 557–564 CrossRef CAS.
- E. Vogel, Pure Appl. Chem., 1993, 65, 143–152 CrossRef CAS.
- E. Vogel, Pure Appl. Chem., 1996, 68, 1355–1360 CrossRef CAS.
- T. D. Lash, J. Porphyrins Phthalocyanines, 2012, 16, 423–433 CrossRef CAS.
- H. Furuta, H. Maeda and A. Osuka, J. Org. Chem., 2001, 66, 8563–8572 CrossRef CAS PubMed.
- D. I. AbuSalim and T. D. Lash, J. Org. Chem., 2013, 78, 11535–11548 CrossRef CAS PubMed.
- D. I. AbuSalim and T. D. Lash, J. Phys. Chem. A, 2015, 119, 11440–11453 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |