
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Designing biodegradable alternatives to commodity polymers†
Emanuella F.
Fiandra
 ,
Lloyd
Shaw
,
Matthieu
Starck
,
Christopher J.
McGurk
and
Clare S.
Mahon
*
,
Lloyd
Shaw
,
Matthieu
Starck
,
Christopher J.
McGurk
and
Clare S.
Mahon
*
Department of Chemistry, Durham University, South Road, Durham, DH1 3LE, UK. E-mail: clare.mahon@durham.ac.uk
First published on 27th October 2023
Abstract
The development and widespread adoption of commodity polymers changed societal landscapes on a global scale. Without the everyday materials used in packaging, textiles, construction and medicine, our lives would be unrecognisable. Through decades of use, however, the environmental impact of waste plastics has become grimly apparent, leading to sustained pressure from environmentalists, consumers and scientists to deliver replacement materials. The need to reduce the environmental impact of commodity polymers is beyond question, yet the reality of replacing these ubiquitous materials with sustainable alternatives is complex. In this tutorial review, we will explore the concepts of sustainable design and biodegradability, as applied to the design of synthetic polymers intended for use at scale. We will provide an overview of the potential biodegradation pathways available to polymers in different environments, and highlight the importance of considering these pathways when designing new materials. We will identify gaps in our collective understanding of the production, use and fate of biodegradable polymers: from identifying appropriate feedstock materials, to considering changes needed to production and recycling practices, and to improving our understanding of the environmental fate of the materials we produce. We will discuss the current standard methods for the determination of biodegradability, where lengthy experimental timescales often frustrate the development of new materials, and highlight the need to develop better tools and models to assess the degradation rate of polymers in different environments.
 Emanuella F. Fiandra | Emanuella F. Fiandra gained her MChem in 2021 from Durham University, with her final year masters project under the supervision of Dr Matthew Kitching, focusing on the enantioselective synthesis of propargylated ammonium cations. Emanuella began her PhD in 2021 as part of the EPSRC CDT in Soft Matter for Formulation and Industrial Innovation, focusing on the development of biodegradable polymers under the supervision of Dr Clare Mahon. |
 Lloyd Shaw | Lloyd Shaw began his academic career at Durham University in 2014 where he obtained an MChem with Honours in 2018. Lloyd continued his research at Durham University with a PhD under the supervision of Professor Lian Hutchings, investigating the use of bio-derived monomers for the synthesis of functional polymers synthesised by living anionic polymerisation. Lloyd now progresses his career with Dr Clare Mahon (Durham University) investigating the (bio)degradation of polymers with the aim of designing high-throughput methodologies for determining the rate of their degradation. |
 Matthieu Starck | Matthieu Starck was awarded his PhD in 2010 working on the synthesis and photophysical properties of luminescent lanthanide complexes, and their use as biological labels. After several post-doctoral positions focused on organic and inorganic chemistry, peptide synthesis and photophysical studies from 2010 to 2021, Matthieu joined Dr Clare Mahon's group at Durham University to work on the synthesis and characterisation of bio-sourced polymers. |
 Christopher J. McGurk | Christopher J. McGurk was awarded his PhD degree in 2016 from Newcastle University (UK) working under the supervision of Prof. Andrew Houlton on the development of functional crystal surfaces with applications in nanoscale electronics. After a postdoctoral position preparing and characterising dynamic polymer films with Dr David Fulton, also at Newcastle University, he has now taken up a position as a scientific advisor within the Department for Environment, Food and Rural Affairs in the UK Civil Service. His current research interests focus on the location and species of UK tree planting and climate change resilience strategies. |
 Clare S. Mahon | Clare S. Mahon was awarded a PhD in 2014 from Newcastle University. After postdoctoral work at the University of Leeds and the University of Sydney, she began her independent career at Durham University in 2019. Clare's research interests focus on the applications of synthetic polymers within biological systems. In 2021 she was awarded a UKRI Future Leaders fellowship, to design new biodegradable polymers and develop rapid assays to measure polymer (bio)degradation. |
Key learning points1. Choose appropriate monomer feedstocks: use of sustainably sourced monomers extracted from biomass will promote the biodegradation of polymers and build towards a circular economy.2. Consider microscopic characteristics of the polymer: biodegradation is promoted by incorporating hydrolysable bonds within the backbone of a polymer, using known biodegradable monomers in copolymers or blends, and designing polymers to include amorphous regions. 3. Consider macroscopic characteristics of the polymer: for solid polymer materials, biodegradation is enhanced by increasing the surface area, which promotes abiotic and biotic degradation mechanisms. 4. Assess biodegradation rigourously: standardised procedures to determine the stability of the polymer during the use-phase and biodegradability at its end-of-life phase should be used to ensure the material meets the requirements for a given application and does not remain persistent in the environment, and the material or its degradation products do no exert harmful effects in the environment. 5. Complete full life cycle assessments for new polymers: a cradle-to-grave approach to evaluate the potential direct and indirect effects associated with the sourcing, design, use and disposal of the polymer should be adopted. |
Polymers are a diverse and versatile group of materials, synthesised both in nature and through a multitude of different synthetic polymerisation techniques. Polymers are found in almost every facet of everyday life – from packaging, construction, medical devices to clothing.1 In combination with their versatile nature and the range of physical and chemical properties that they can offer, historically-low costs for raw materials and energy have encouraged the use of polymers within our everyday lives, to the point where a world without polymers is difficult to imagine.2 However, low production costs and mass usage of commodity polymers have taken an environmental toll, resulting in a ‘throw-away-culture’ and the rise of the single-use-plastic industry.3,4 The majority of plastic bags, plastic films and plastic straws, for example, are used once before disposal, resulting in a large accumulation of plastics in landfill sites and marine environments. Since 1950, it is estimated that less than 10% of plastic waste has been recycled, with the remainder deposited in landfill sites or released to the environment.5 In addition to problems around land use and greenhouse gas emissions, run-off from these sites into marine sources, along with direct disposal into aquatic environments, poses hazards to marine wildlife through suffocation, entanglement and digestive disruption.6,7
Governments and policymakers have made attempts to regulate polymer waste and establish methodology to assess biodegradation (Fig. 1). In 1981, the Organisation for Economic Co-operation and Development (OECD) published the first series of standardised tests, the OECD Guidelines for Testing of Chemicals,8 a set of testing methods used internationally by laboratories to identify hazards associated with chemicals (ESI,† Table S1). The American Society for Testing and Materials (ASTM) has also developed standardised test methods to assess the biodegradability of polymers (ESI,† Tables S2 and S3).9 In Europe, other standardised methods are used, for example British standards (BS) in the UK,10 along with methods defined by the German Institute for Standardisation (DIN)11 or Technischer Überwachungsverein (TÜV)12 (ESI,† Table S4). While attempts have been made to harmonise these standards, notably leading to the introduction of the International Organisation for Standardisation (ISO) standards (ESI,† Tables S5 and S6),13 no set of methods has been universally adopted, frustrating efforts to directly compare the biodegradability of polymers.
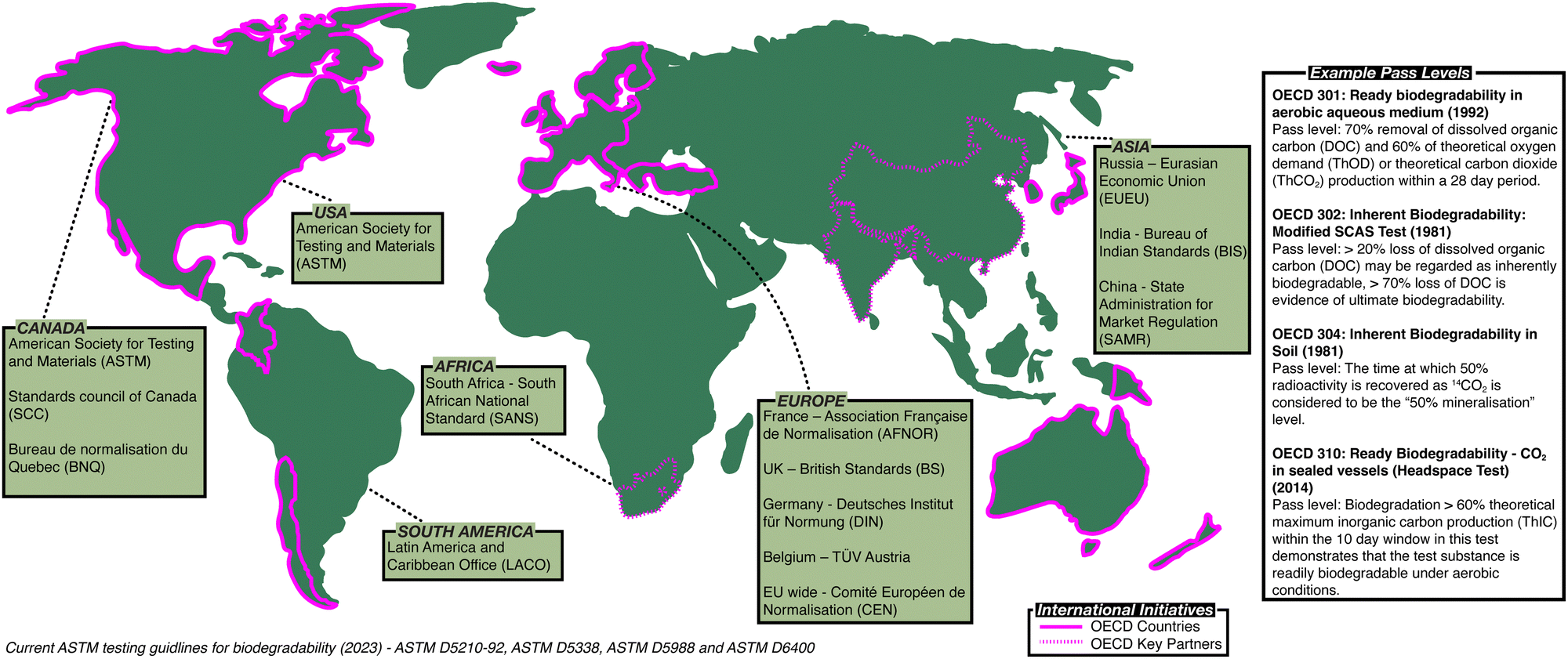 | ||
| Fig. 1 The global regulatory landscape for assessment of biodegradation. | ||
After the discovery of the Great Pacific Garbage Patch,14,15 a collection of marine debris estimated to have a surface area of approximately 1.6 million km2 as of 2018, changes in the public perception of polymers, particularly plastics, led to a drive to decrease environmental impacts of plastic consumption.16 This shift in public perception towards polymers resulted both in behavioural changes in how individuals use polymers and plastics (e.g. opting to reuse plastic bags) and policy changes from governments (e.g. introducing taxes on single-use plastics).17,18 The ‘Reduce, Reuse, Recycle’ campaign, first popularised in the early 1970s demonstrated that plastic waste was already a concern before many of the most serious consequences were seen. While in theory this approach could be a feasible long-term solution to much of the plastic waste being produced, the high infrastructure costs required for recycling, the ever-increasing reliance on polymer materials, the lack of public understanding that recycling generally produces lower-grade materials because of thermal degradation,19 and the lack of public engagement with the ‘reduce’ and ‘reuse’ components of the campaign, in favour of the practically more simplistic ‘recycling’ component, meant that this approach has not sufficiently reduced the amount of waste that is being produced and therefore new strategies must be employed.20–22 Additionally, while there has been a great deal of research into enhancing the recyclability of polymers through improvements in mechanical recycling practices19 and the development of tailored organocatalysts for chemical recycling,23,24 scaling up these practices can be difficult and effective recycling is still limited by the requirement for separation of the plastics and the overall downgrade in mechanical properties upon recycling. Although there is much debate as to how to best prevent this reduction in mechanical strength, several experts in the field have suggested that practices such as depolymerisation,25 establishing break points in any new materials,26 using selective catalysts so pure polymers can be synthesised from impure feedstocks27 and improvements in waste collection and separation,28 could be used in the future to reduce polymer waste by allowing polymers to be infinitely recyclable without any loss of performance.
An alternative approach, where recycling may not be feasible, is to produce commodity polymers that degrade rapidly at the end of their use phase.29 Some interventions to accelerate polymer biodegradation have been limited in their success, however. One strategy aimed at improving the rates of polyolefin degradation involved the addition of ‘prodegradants’30 such as complexes of Fe, Co and Mn, to assist in the generation of radicals and hence accelerate abiotic degradation. While these ‘oxo-degradable’ polymers do display more rapid degradation under laboratory conditions, the evidence for enhanced biodegradation in environmental conditions is sparse,31 and in 2021 the European Commission restricted the use of oxo-degradable plastics along with other single-use items.32
Many of the polymers in mass production (Fig. 2) are not as visible as those which cause the obvious problem of plastic waste. Polymers are used extensively in coatings, adhesives and consumer goods including shampoos, shower gels and detergent formulations. Many such polymers increase the effectiveness of the products they are found in, which has helped to reduce the overall carbon footprint of many of these sectors – either through the removal of ‘carbon-emission-heavy’ active compounds or through the reduction of the energy requirements for the products to work effectively.33,34 Whilst many polymers have a positive effect on reducing the carbon emissions of consumer products or activities, polymers themselves may find their way into marine environments where their degradation and subsequent removal from the environment may be very slow and potentially incomplete. Microplastics (Box 1) may be formed through the degradation of polymers and plastic fragments. Since the discovery of these small plastic fragments by Edward Carpenter and Kenneth Smith Jr. in 1972,35,36 and the popularisation of the term “microplastic” by Richard C. Thompson et al. in 2004,37 there has been global concern about their impact on human health and the environment. Microplastics are of growing ecological concern due to their ability to pass through epithelial and endothelial cells after inhalation or ingestion.38 Recently, a study by Lamoree et al. reported the presence of microplastics (including poly(ethylene terephthalate) (PET), polystyrene and polyethylene in over 75% of healthy blood donors tested,39 with microplastics previously identified in lung,40–42 liver,43,44 placenta,45 and faeces samples.46–49 While the long-term evidence around the dangers and toxicity of microplastics is still emerging, there is growing concern that microplastics are accumulating in the body where the rates of removal of microplastics by the kidneys and liver is slower than the rate of uptake through inhalation and ingestion.50,51 Microplastics have also been shown to harbour microbes that can then be transported over vast distances, which could result in the transportation of invasive species or pathogens from one part of the world to another.52–54
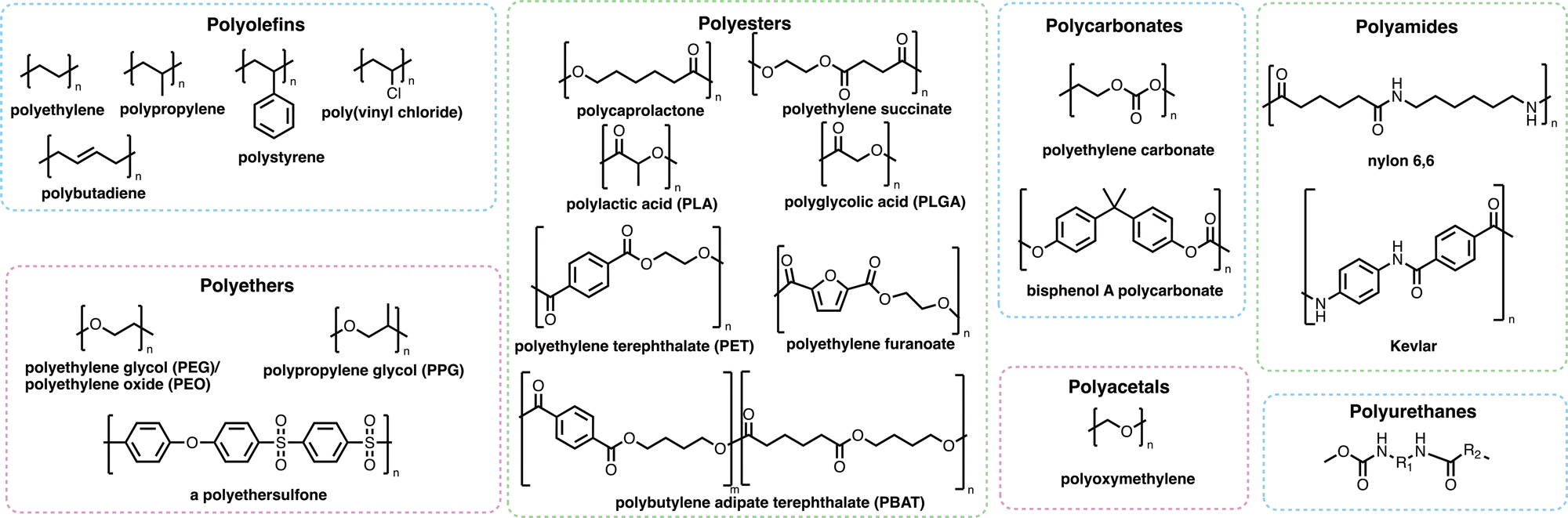 | ||
| Fig. 2 A selection of commodity polymers discussed in this review. | ||
It is clear that these environmental and health concerns must be considered throughout the process of developing new polymer materials. One important approach is to design polymers that will biodegrade efficiently at the end of their use phase (Box 1). Literature searches reveal a high level of activity in the field of biodegradable polymers, with a sustained increase in the number of reports from 2000 to 2023 (ESI,† Fig. S1). It is environmentally important to consider a sustainable approach to the sourcing of materials used in the synthesis of commodity polymers, but also a sustainable approach for the disposal of these materials.55Ultimate biodegradation, which is the degradation of a natural or anthropogenic material to CO2, H2O, biomass and inorganic substances such as NH3,56 needs to be achieved within a limited lifetime from an end-of-life perspective. However, polymeric materials have often been designed to have long lifetimes, enhanced by the addition of complex stabiliser blends to prevent degradation. Commodity polymers including PET, polyvinylchloride (PVC) and polypropylene display environmental lifetimes which typically range from 10–20 years or 500–1000 years.57–59 The best way to ensure that plastic waste does not accumulate is to ensure that the lifetime of any new material represents a lifetime of approximately the same length of the material's use (e.g. the PVC in window frames has a required lifespan of 20–30 years whereas polyethylene food packaging is only required to be stable for periods of months). To address this criterion, the material can either be reusable within the required application space, recyclable or susceptible to removal by biodegradation. Long-term sustainability requires materials which undergo ultimate biodegradation,56 which will therefore represent the focus of this tutorial review.
Life cycle assessments
To comprehensively evaluate the environmental impact of a material, life cycle assessments (LCAs)60 increasingly adopt a ‘cradle-to-grave’ approach, which involves the consideration of the extraction of feedstock, processing, manufacturing and distribution, product service, recycling or final disposal. Until recently, end-of-life products were absent from LCAs, but they have become a topic of interest in recent years,72–74 with special consideration towards the aquatic environment.29 In striving for a sustainable circular economy, recycled and biobased feedstocks offer the opportunity to transition from petroleum-derived raw materials in the design of polymers, offering potential for biodegradation whilst aiding in the drive for carbon neutrality (Box 1).75 A factor which cannot be ignored is cost. A primary driving force for the mass use of polymers is their relatively low cost, with global supply chains relying upon ready access to affordable building blocks. The EU Waste Framework Directive (2008/98/EC)76 defines a five-step waste hierarchy aiming to conserve resources, at the top of which is the prevention and minimisation of waste – this can be achieved in the design of polymers that are biobased, ideally from organic waste streams, and biodegradable. For example, the UKBioChem10 list77 highlights biobased chemicals that can aid in the transition to a sustainable chemical industry. Six of these platform chemicals: lactic acid,78 2,5-furandicarboxylic acid,60 5-hydroxymethyl furfural,79 muconic acid,80 1,3-butanediol81 and n-butanol82,83 have already found use in polymer synthesis.
Box 1: Key definitionsAtom economy: the ratio of atoms that can be found in the product of a reaction compared to the number of those atoms found in the reactants.61,62Biobased: composed or derived in whole or in part of biological products issued from biomass (including plant, animal, and marine or forestry materials). This definition may apply to polymers that are not biodegradable.63 Biodegradable: macromolecules or polymeric substances susceptible to degradation through biological activity involving decreases in the molar masses of constituent macromolecules.64 Biodegradation: the breakdown of a substance catalysed by enzymes in vitro or in vivo. Biodegradation may be further characterised as: Primary: alteration of the chemical structure of a substance, resulting in loss of a specific property of that substance. Environmentally acceptable: biodegradation to such an extent as to remove undesirable properties of the compound. This often corresponds to primary biodegradation, but it depends on the circumstances under which the products are discharged into the environment. Ultimate (also termed “mineralisation”): the complete breakdown of a compound to either fully oxidised or reduced simple molecules (such as carbon dioxide/methane, nitrate/ammonium and water) and inorganic matter. It should be noted that the products of biodegradation can be more harmful than the substance degraded.65,66 Biomass: material produced by the growth of microorganisms, plants or animals.66 Circular economy: an industrial economy that is restorative or regenerative by intention and design.67 Degradation: physical and/or chemical deleterious changes of the polymer through chain scissions, resulting in a decrease of molar mass and progressive loss of performance/characteristics of the polymer.63,64 Depolymerisation: process of converting a macromolecule into monomer or a mixture of monomers.63 Life cycle assessment: compilation and evaluation of the inputs, outputs and the potential environmental impacts of a product system throughout its life cycle.68 Microplastics: terminology used to refer to plastic particles smaller than 5 mm.37 Sustainable chemistry: the design, manufacture, and use of environmentally benign chemical products and processes that prevent pollution, reduce or eliminate the use and generation of hazardous waste, and reduce risk to human health and the environment.69,70 Reusable: materials that can be reused within their application space where there is no significant loss in performance and recollection is facile. Recyclable: materials that can be recycled through multiple cycles without any performance degradation or material loss. Polymer blend: macroscopically homogeneous mixture of two or more different species of polymer71 |
Undertaking a comprehensive LCA for a polymer can be challenging, owing to the complexities of their synthesis, use and disposal. These difficulties were highlighted by Walker and Rothman,72 who compared the LCAs of biobased and petroleum-derived plastics to the European Union Product Environmental Footprint (EU PEF) standards.84 It was found that no published articles were able to fully meet these standards – with only 25 publications between 2000 and 2019 partially meeting the requirements. The study evaluated 89 polymers, of which 50 were biobased and 39 petroleum based. Only seven of these polymers could be used for comparison across all seven impact categories: energy use, ecotoxicity, acidification, eutrophication, climate change, particulate matter formation and ozone depletion. Importantly, significant variations were reported between LCAs for the same polymer, which may be attributed to the variation of LCA methodologies used, especially when regarding the end-of-life treatment. It was proposed that LCAs reported should include a detailed account of each section of the EU PEF method and the relevant ISO standards, a principle that, if widely adopted, would allow for more meaningful comparisons to be made between LCAs for different polymers.
Looking to nature for solutions
In the search for new biodegradable polymers, much inspiration can be gained through the examination of biodegradable polymers which can already be sourced from nature. Polymers in nature can be largely divided into three main classes: poly(saccharides), poly(peptides) and poly(nucleic acids).85,86 In biological systems, these polymers fulfil both the structural functions we typically associate with commodity polymers, but also play key roles in the storage of energy and the transfer of information, and can adapt and respond to changes in their environment, presenting inspiration for new polymers with functions elevated beyond mere replacement of existing materials.87 These natural biodegradable polymers provide the foundation for all life on earth: with polysaccharides playing important structural roles in addition to functions in energy storage and cellular communication; poly(peptides) playing numerous structural and catalytic roles in addition to underpinning immune response; and poly(nucleic acids) enabling information storage and transmission.88,89 These highly functional polymers are all processed by endogenous or exogenous enzymes to return the initial monomer building blocks or simple compounds such as water, carbon dioxide and ammonia.90,91 While it is not possible to extract all required polymeric materials from nature due to limitations on production time and cost, and due to specific property requirements, design principles for new biodegradable synthetic polymers can be drawn from nature. By ensuring that the products of polymer degradation can be readily used by microbes or other organisms, the rate of ultimate biodegradation of the material can be greatly increased.In addition to ensuring polymers display a lifespan comparable to their use phase, and can either be readily recycled or otherwise removed from the environment, it is also important that the monomers and processes used to synthesise this new generation of polymers are sustainable. The principles of green chemistry suggest the design of syntheses that are high yielding, have a high atom efficiency, low energy requirements and involve minimal use of toxic solvents or reagents, and the use of raw materials from renewable sources where practicable.29,92–94 Nature provides an abundance of polymer precursors, from olefins to amino acids and carbohydrates.95 These sources should be considered by polymer chemists in the design of new materials. It is, however, important to note that polymers constructed using raw materials accessed from biomass may not necessarily be biodegradable. Many synthetic routes to conventional monomers have been developed,96 and while polymers made through these routes may be more sustainable, they will ultimately suffer the same limitations to their end-of-life environmental profile as conventional materials.
A key feature identified within natural biodegradable polymers is the production of degradation products which can be used by nature, guaranteeing ultimate biodegradation. The highly interconnected nature of ecosystems, whereby if an organism synthesises a compound, either that organism will use the compound or it will be used by another organism, can be harnessed to produce sustainable materials through the use of biobased monomers; with examples as diverse as dienes such as farnescene,97 presenting a potential alternative to polybutadiene, and diols such as 2,5-bis(hydroxymethyl)furan,98 which may find use in replacements to PET. These biobased monomers can be extracted directly from biomass or synthesised through the chemical reactions of natural products.95,99–102 Biobased monomers can also be produced in high yields and on large scales through the fermentation of genetically modified yeast and bacteria, with these approaches often presenting simpler purification processes compared to biomass extraction.103–105 By using naturally occurring sources for raw materials rather than petroleum-derived chemicals, sustainability may be greatly increased as the monomers themselves can be renewably sourced and any degradation products will be much more likely to be completely mineralised.
Mechanisms of biodegradation
The biodegradation process is grouped into four key stages: biodeterioration, biofragmentation, bioassimilation and mineralisation (Fig. 3).106 A combination of abiotic and biotic mechanisms can influence the rates of degradation of polymers, with the predominant abiotic mechanisms consisting of mechanical, thermal (or thermo-oxidative), photo (photo-oxidative) and hydrolytic degradation.107Biodeterioration results in the alteration of the physical and chemical characteristics of a material due to superficial degradation by external environmental factors, and the action of microbial communities and other decomposer organisms. External forces such as those exerted by wind, rain and waves cause superficial degradation, resulting in pores and cracks along the surface of the material, or breakage into smaller pieces, increasing the surface area for microbial attachment. Superficial degradation by light, termed photo-oxidation, also causes micro-lesions and cracks on surfaces which facilitate microbial attachment and subsequent biofilm formation. Oxidative degradation by ozone is another, albeit less common, chemical mechanism for the degradation of polymers.108 Polymer chains which have been oxidised generally display increased hydrophilicity due to the incorporation of hydroxyl and carboxylic acid functional groups, aiding microbial attachment. Biofragmentation is the cleavage of polymers into oligomers, dimers or monomers by enzymes or free radicals generated by microorganisms. Assimilation of water-soluble intermediates can then occur, with molecules transported into the cytoplasm of microorganisms and hence, subjected to metabolism. The complete degradation process ends with mineralisation: the excretion of metabolites, such as CO2, H2O, CH4 and N2.109,110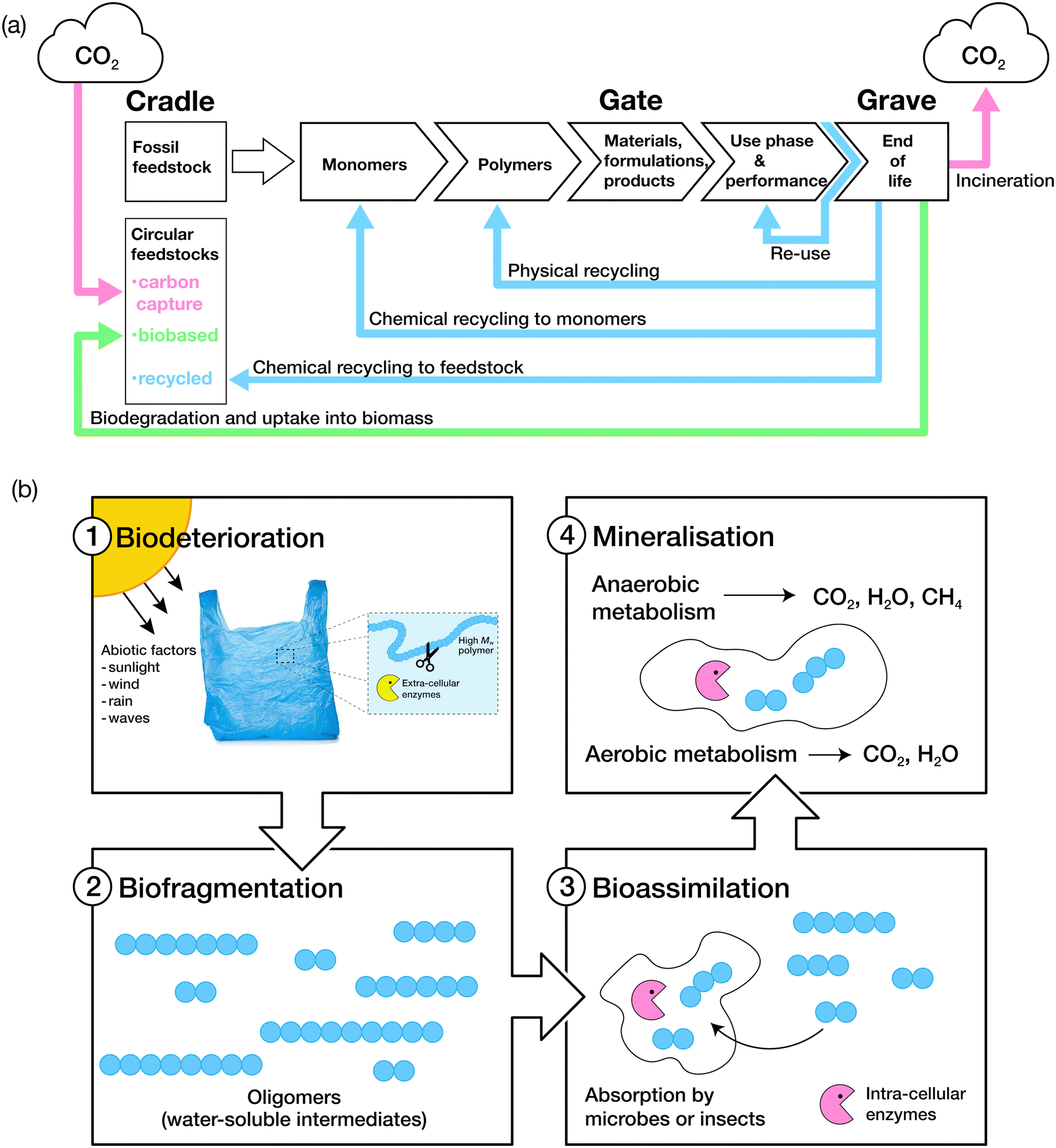 | ||
| Fig. 3 (a) The life cycle of consumer-use polymers. Adapted from ref. 75 with permission from John Wiley and Sons, copyright 2022. (b) The four stages of polymer biodegradation: biodeterioration, biofragmentation, bioassimilation, and mineralisation. Adapted from ref. 109 with permission from the American Chemical Society, copyright 2022. | ||
The predominant mechanism of polymer biodegradation consists of the enzymatic breakdown of polymer chains, via chain scission or oxidation, into oligomers and small molecules which can then be ingested or assimilated by microbes.109 The process of biodegradation is typically slower for synthetic polymers compared to other compounds due to their high molecular weight and typically limited water solubility, hindering their transport through the cell wall of microorganisms.109 Instead, biodegradation is usually achieved by the action of extracellular enzymes to give rise to water-soluble intermediates that can then be further metabolised.111 Some backbone polymer linkages can be enzymatically hydrolysed, with lipases known to cleave ester linkages, and proteases enabling hydrolysis of amide bonds, resulting in the generation of end products by either an aerobic or anaerobic degradation pathway (Fig. 3 and ESI† Table S7). Several excellent reviews detail the key microorganisms and enzymes involved in the biodegradation of polymers and the pathways through which degradation can be realised.106,112,113 However, further research is required to identify enzymes and microorganisms which may act on high molecular weight polymers such as polystyrene, polyamides, PVC, polypropylene, ether-based polyurethanes and polyethylene,114 which comprise of more than 80% of the annual plastic production.115
Ultimate biodegradation can be influenced by a myriad of factors including structural features of the polymer, specifically, carbon chain length, functional group variation and charge density.116 To further add complexity to the challenge of predicting polymer biodegradation, the impact of the end-of-life environment of the material can be profound, with large differences in degradation kinetics for some polymers depending on the enviromment.117 Poly(lactic acid) (PLA), for instance, is recognised to degrade rapidly under aerobic composting conditions but solid samples of PLA incubated in seawater display little to no degradation over timescales of over one year.118 In aqueous environments, it has been demonstrated that in the absence of extracellular degradation, polymers greater than 500–1000 Da are unlikely to degrade significantly under aerobic conditions as a result of poor bioassimilation.119 Even within the same types of environment, local conditions can affect the kinetics of degradation significantly, with studies highlighting that the degradation of conventional materials such as polyethylene, PET and PLA, in a terrestrial environment such as soil are dependent on temperature and water availability to modulate polymer degradation.120 Weight losses of less than 2% were reported for samples of these polymers after evaluation of one year of in situ manipulative experiments simulating different soil conditions.
Another important environmental consideration is the impact of the degradation products and the biota found in an environment after polymer biodegradation. Examination of plastic debris using scanning electron microscopy (SEM) has revealed complex and diverse microbial communities on surfaces, termed the ‘plastisphere’.121 The bacteria that make up the plastisphere are often distinct from those found in the surrounding environment. The plastisphere associated with biodegradable polymers including polybutylene adipate terephthalate (PBAT) and PLA has been shown to contain a less diverse range of bacteria than that associated with conventional polymers such as polyethylene,122 suggesting that the introduction of even biodegradable polymers to the soil environment may influence the ecosystem. Similarly, in aquatic environments, microplastics have been shown to provide a distinct ecological niche for microorganisms, leading to differences in the populations of species observed.123
Analytical approaches to monitor biodegradation
In order to fully understand the processes involved in biodegradation, analytical methods are required to monitor changes in the molecular weight and chemical composition of polymers.124,125 In many cases, polymers can present challenging cases for the study of biodegradation, owing to the complex, multi-step nature of the process, and their inherent structural heterogeneity. Often, however, valuable information can be obtained using simple methodology such as monitoring changes in the mass or dimensions of a solid polymer, in addition to quantifying changes in surface roughness. A range of complementary bioanalytical methods can be combined to overcome the limitations of individual methods and to gain a comprehensive understanding of biodegradation processes (ESI,† Table S8). Respirometric methods such as monitoring CO2 evolution118 or O2 consumption126 are commonly used to indirectly estimate the respiration rates of microorganisms and hence elucidate their activity. Although these provide a straightforward, economically viable and non-labour-intensive route to determining rates of biodegradation, the approach is not suitable for field studies and provides no detailed insight into degradation pathways. Ideally, standardised, widely-applicable methods to monitor polymer composition would provide more detailed information on the process of degradation and reduce variability in studies.127Despite a range of techniques providing kinetic information around biodegradation, a key bottleneck in the identification of biodegradable replacements for commodity polymers is establishing that their biodegradation kinetics are compatible with relevant OECD or equivalent standards (ESI,† Table S1 and Fig. S1), with testing processes typically requiring weeks to months. There is a pressing need for rapid, high-throughput assays to determine the biodegradation profile of a polymer in the early stages of its development. These assays will be complementary to advances in the predictive modelling of biodegradation,75,128,129 together presenting a route to the streamlined design of new polymers to fulfil human needs whilst minimising environmental impact.
Strategies for the design of biodegradable polymers
Designing polymers that are more susceptible to biodegradation is critical to ensure their continued economic and societal value. A holistic approach in the design of polymers is needed, considering a range of parameters including the sourcing of feedstock, the chemical and physical properties required of the material, and the desired biodegradation profile. Von Vacano et al. have evaluated the macromolecular design of materials and the possible impacts of various factors on the safety, the environmental impact and the circularity of a material.75 Amongst the features evaluated, including monomer chemistry, polymer architecture, molecular weight distribution and crystallinity, the chemical nature of the backbone linkage was reported to be the most important parameter in determining the biodegradation profile of a polymer. Separately, a literature-driven cluster analysis130 was performed using the PlasticDB database to identify a relationship between various polymers and their biodegradation profiles. Curated from 471 publications, the results demonstrated that the chemical classification, and hence the structure of the polymer backbone, has a direct relationship to its susceptibility to biodegradation. After refining the data in a local database containing the taxonomical identification of microorganisms, plastic type and literature reference, only 20 plastic types remained, which were used for hierarchical clustering and multidimensional scaling (MDS) analysis. Polymers were initially clustered within two broad groups: C–C (non-hydrolysable) and C–X (hydrolysable). A statistically significant difference in biodegradation patterns at the microbial genus level was identified, with Bacilli reported to degrade polymers grouped in the C–C class, and Pseudomonas and Bacillus in addition to fungal species Aspergillus and Penicillium involved in degradation of the C–X class of polymers. The seven C–C polymers including high- and low-density polyethylenes, polypropylene, polystyrene, polyvinylalcohol (PVA) and PVC all consist of a hydrocarbon backbone, whilst the remaining 13 (C–X) polymers, including polycaprolactone, polyurethane, poly(ethersulfone), PLA, PET and PBAT contain a heteroatom in their backbone, which are further grouped based on the nature of the bond: ester, amide and ether. One notable exception was highlighted in the hierarchical clustering plot as nylon, classed as C–X due to the presence of heteroatoms, is clustered with non-hydrolysable polymers, implying that a similar genus level biodegradation pattern is present. This work highlights the importance of the polymeric backbone linkage, amongst other factors, in determining the likely biodegradation profile of a polymer. The authors also highlighted the lack of uniformity in reporting of microbial degradation of plastics and that by addressing this issue, a wider search could be more easily performed to aid in the identification of key trends and thus potential solutions.Molecular features of polymers and their effects on biodegradation
Generally, the presence of heteroatoms in a polymer backbone has been observed to render the polymer more susceptible to biodegradation, as cleavable bonds are typically introduced. Farveen et al. compared the degradation of the main families of polymer (polyolefins, polyesters, polyurethanes and polyamides) in an in vitro study using bacteria isolated from soil.131 Preliminary studies using 1% low-density polyethylene resulted in the identification of isolates P. aeruginosa O1-P and B. cereus O2-B as highly active due to their production of laccases,132 a class of multi-copper oxidases which act on a versatile range of substrates. These laccases perform the critical role of installing hydroxyl functional groups on polyethylene chains, which promotes biodegradation, as confirmed by FTIR and GC-MS analysis.131 Homology modelling and molecular docking studies suggested that theoretically higher rates of biodegradation could be achieved for other families of polymers when compared to polyethylene, in line with experimental observations. This in silico approach has allowed for a greater understanding of the mechanism through which laccases can promote biodegradation, and highlights the potential of these enzymes to facilitate biodegradation of otherwise slow-degrading polymers.The polyolefin class of polymers includes common commodity polymers such as polyethylene and polystyrene, with a combined global annual production of 260.2 million metric tons in 2022.133 Biodegradation pathways for polyolefins are limited by the lack of readily cleavable linkages within the polymer backbone, and therefore molecular design is often relied upon to improve the recyclability of these polymers.75,134 Identifying enzymes involved in the degradation of polyolefins and elucidating their mechanisms remains a challenge, with limited literature reports in this area. Microplastic biodegradation via fungal biofilm formation, however, has been reported for several polyolefins with the identification of fungal species that act on polyethylene, polystyrene and PVC detailed in a review by Solanki et al.57 The Plastics-Active Enzyme Database (PAZy), reports only two enzymes135 with the ability to degrade polyolefins, with both enzymes acting upon polyethylene. Despite many publications referring to the degradation of polyolefins, a PAZy search does not lead to convincing biochemical data which clearly identifies enzymes and pathways involved in this process.133,136,137
Although the mechanisms through which polyolefins are biodegraded are not fully understood, some microbial species have been found to interact with polyethylene, with several genera of bacteria and a some fungi shown to degrade polyethylene.138 To initiate the initial (bio)degradation of polyethylene, a combination of environmental factors and the action of enzymes results in the degradation of polymeric chains into hydrocarbon fragments of 10 to 50 carbons in length that are more susceptible to oxidation and subsequent biodegradation. The degradation of polyethylene by the saliva of Galleria mellonella (wax worm) larvae has been reported by Sanluis-Verdes et al.,135 identifying phenol oxidases able to act on polyethylene. These enzymes produce small molecules that may be accessible to the insect and its microbiome, allowing for further metabolism within the insect digestive system. In this study, polyethylene was shown to be oxidised and degraded under physiological conditions within a few hours. Réjasse et al.139 used isotopic labelling and infrared microspectroscopy to investigate the ability of G. mellonella larvae to bioassimilate polyethylene, revealing micrometre-sized polyethylene particles within the larval digestive tract cavities. No bioassimilation was detected within 19 days when larvae were fed deuterated polyethylene, suggesting that G. mellonella larvae may degrade polyethylene, but ultimate biodegradation of the polymer is limited.
Polymers with a heteroatoms adjacent to their C–C backbone may display different biodegradation profiles to those which do not contain heteroatoms. A biodegradation pathway for PVC was proposed by Zhang et al.140 with the use of genomic, transcriptomic, proteomic and metabolomic analysis to identify the genes and enzymes potentially involved in the process. Abiotic mechanisms initially induce degradation by initiating C–C and C–H scission. A range of enzymes including catalase-peroxidases, dehalogenases, enolases, aldehyde dehydrogenases and oxygenases were found to contribute to the biodegradation of PVC, highlighting the complexity of the biochemical pathways required to effectively degrade a relatively structurally simple polymer. Work by Giacomucci et al.141 identified Pseudomonas citronellolis and Bacillus flexus as potential degraders of PVC films, with both strains shown to form biofilms on the PVC film surface, and induce fragmentation. After incubation for 45 days in the presence of P. citronellolis, a 10% reduction in the average molecular weight of a PVC film as determined by size-exclusion chromatography was observed. For waste PVC plastic, however, a lower extent of biodegradation was observed. Instead, bacterial strains were shown to act primarily on the additives present in the waste PVC plastic sample rather than the polymer, with a 19% gravimetric weight loss after 30 days recorded.
PVA is a water-soluble polyolefin with applications in adhesives, within laundry and dish detergent pods, and as a finishing agent in the textile industry. The end-of-life environment has a significant impact on the rate of biodegradation of PVA, with only 8–9% degradation achieved in a simulated soil burial biodegradation test after 74 days, with the microbial inoculum isolated from the sewage sludge of a papermill.142 The rate of biodegradation of PVA is enhanced in aqueous environments, as in wastewater and sewage sludge, with the bacterial species often associated with PVA degradation belonging to the genus Pseudomonas.143 PVA is biodegradable under both aerobic and anaerobic conditions,144 but the process may be slow, relying upon pyrroloquinoline quinone (PQQ) dependent enzymes and hence availability of this cofactor.145 von Haugwitz et al.146 have reported a PQQ-independent enzymatic cascade reaction using commercially-available enzymes which can partially biodegrade PVA, presenting scope to incorporate the process into wastewater treatment.
A major family of commodity polymers which may present favourable biodegradation profiles are the polyesters,117 with key examples including PLA,147 polycaprolactone148 and polyglycolide (PGLA).149 For these aliphatic polyesters, bulk degradation by chemical hydrolysis is the predominant mechanism at play, for which Bher et al.150 details methods on accelerating this process to give access to units that are able to enter microbial metabolic pathways. Adding plasticisers, like polyethylene glycol (PEG), can increase chain flexibility and reduce the glass transition temperature of the polymeric material, enabling hydrolysis by bulk erosion to occur more readily. Copolymerisation is another strategy which may be employed to increase the rate of degradation of polyesters, exemplified by polybutylene terephthalate, which is considered as non-biodegradable, and its copolymer polybutylene adipate terephthalate (PBAT). PBAT is more susceptible to enzymatic attack due to the increased flexibility of polymeric chains achieved through the introduction of adipic acid in place of some terephthalic acid units along the polymer backbone.151 Engineered cutinases from Thermobifida fusca have been demonstrated to completely degrade samples of PBAT in 48 h.59 Biodegradation is initiated by endo-cutinate-mediated hydrolysis, resulting in random cleavage of PBAT macromolecules.152 PBAT presents good biodegradability when less than 55 mol% of the aromatic moiety is incorporated in the backbone structure,153 but generally lower rates of hydrolytic degradation are observed compared to aliphatic polyesters like PLA and PGLA.
PET is a particularly important commodity polymer, with applications in packaging and in the textile industry. Although readily recyclable, often PET materials including plastic bottles or textiles arrive in landfill sites at the end of their use phase, and options for their biodegradation may be limited. In 2016, however, a novel strain of bacteria, Idenonella sakaiensis,154 was discovered close to a plastic recycling facility, and was shown to degrade PET, using the polymer as its primary source of both energy and carbon (Fig. 4). Initially, an extracellular PET hydrolase cleaves the polymer into the intermediates bis- and mono-(2-hydroxyethyl) terephthalic acid (BHET and MHET). A second enzyme, MHETase, then hydrolyses these fragments into the environmentally benign monomer units: ethylene glycol and terephthalic acid. Jerves et al.155 studied PETase with density functional theory and molecular dynamic simulations to confirm the mechanistic pathway for the biodegradation of PET, and as a result other polyesters that present a similar structure such as polyethylene furanoate,156 an emerging biosourced polymer that can replace the petroleum-derived PET for many applications. The use of enzymes such as PETases could also enable the controlled degradation of other polyesters, presenting a circular approach to their synthesis, use and removal from the environment. Orlando et al. have published a detailed review on enzyme-based biotechnological approaches to PET degradation, highlighting the potential for this approach to be expanded to polyesters in general, including polyester-based polyurethanes.157 Similar bacterial hydrolases that can cleave polyamide oligomers have also been reported.158
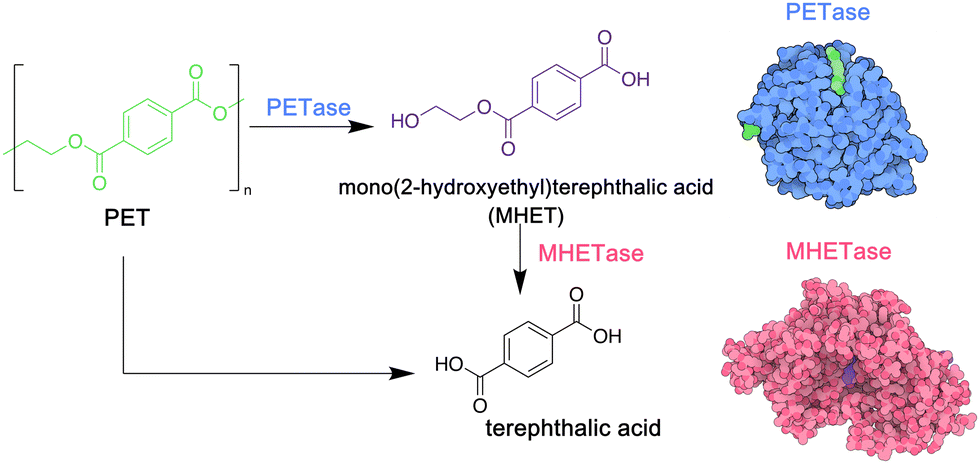 | ||
| Fig. 4 Biodegradation of PET by enzymes PETase and MHETase, isolated from Idenonella sakaiensis (5hx3.pdb; 6qga.pdb). Adapted from ref. 154 with permission from the American Association for the Advancement, copyright 2016. | ||
Advances in protein engineering present the opportunity to computationally redesign enzymes to enhance polymer degradation. If demonstrated to be effective, modified enzymes could be introduced to an environment to promote the depolymerisation of desired polymers, or enzymes could be added during processing stages as dispersion agents. Cui et al. used greedy accumulated strategy for protein engineering (GRAPE) to improve the robustness of I. sakaiensis PETase.159 The crystal structure of the engineered enzyme revealed the mutation responsible for enhanced degradation, confirming predictions. A library of redesigned PETases enabled the improved degradation of semicrystalline PET films, and complete biodegradation of microplastic suspensions after 10 days at 37 °C. Additionally, an engineered cutinase was reported to efficiently degrade PET at moderate temperatures,160 enabling the degradation of 1.3 g of untreated post-consumer PET waste within 3 days at 55 °C using only 1.25 mg of the enzyme.
Some polyesters may be degraded by the action of insects, presenting alternative opportunities for their removal from the environment. Shah et al. investigated the biodegradation of PLA blocks by larvae of the greater wax moth (G. mellonella), which are already known to biodegrade natural polymer bee waxes.161 Changes in the metabolites and lipids of G. mellonella larvae were monitored to gain insights into the biochemical process of PLA degradation using insects. It was reported that whilst PLA could be ingested, this resulted in metabolic stress for the host, along with a reduction in lipid reserves and ceramide levels which could be due to apoptosis and inflammation. A greater understanding of the biodegradation pathway may precipitate a strategy that reduces the stress imposed on the organism, whilst increasing the amount of plastic digested by the insect.
Polycarbonates are widely used commodity polymers because of their attractive physical, chemical and mechanical properties,162 with widespread use contributing to their environmental accumulation. Artham and Doble163 highlighted the important distinction between aliphatic and aromatic polycarbonates in determining their potential for biodegradation. Aliphatic polycarbonates include poly(ethylene carbonate), poly(1,3-trimethylene carbonate), poly(butylene carbonate) and poly(hexamethylene carbonate), with aromatic polycarbonates primarily comprised of bisphenol A polycarbonates. Whilst there are published studies of the biodegradation of aliphatic polycarbonates by microorganisms including bacteria164 and fungi,165 or enzymes including lipases166 or cholesterol esterases,167 there is limited information in the literature on the biodegradation of bisphenol A polycarbonates, with studies limited to polymer blends which include bisphenol A polycarbonates.168,169 The limited biodegradability of aromatic polycarbonates is primarily a consequence of their poor water solubility and bulk amorphous morphology, which prevents their effective bioassimilation.
Polyurethanes are used in a broad range of applications such as adhesives, coatings and personal care products, with increasing demand over the past 50 years leading to a bottleneck as they accumulate in both terrestrial and aquatic environments.170 Determining the environmental fate of polyurethanes is crucial because of the ability of both the polymer itself, and the additives typically present, to leach hazardous compounds into the environment, leading to the identification of polyurethanes as one of the most toxic classes of polymers.171 Pfohl et al. investigated the biodegradation of polyurethanes in compost to construct a structure-degradation relationship and investigate the mechanism by which fragmentation and subsequent biodegradation is achieved.172 The rate of biodegradation was shown to be dependent on the cross-linking density and the content of the ‘hard segment’ containing urethane linkages which promote crystallinity, compared to the ‘soft segment’ which form amorphous regions.153,173 Depending on the isocyanate and polyol used, polyester or polyether type polyurethanes can be synthesised, with enzymes of differing activities required for their degradation. The ‘soft segments’ of the polymer, i.e., the amorphous regions where polyol moieties are found, determine the biodegradability of the overall polymeric material, as ester and ether bonds are more susceptible to microbial action. The PAZy database133 has identified 26 biochemically characterised enzymes that have the capability to degrade polyurethanes, in particular polyester-based polyurethanes. Pantelic et al. identified the novel polyurethane-degrading bacterium Amycolatopsis mediterranei ISP5501 during a study to assess the toxicity and suitability of eight synthetic model compounds that represented partial polyurethane hydrolysis products. This urethane-degrading strain was found to act on polyether and polyether-based polyurethanes, with a reduction of up to 13.5% in the number average molecular weight of the polymer.174 Some cutinases, lipases and carboxylesterases have been found to act on the soft segments of ester-based polyurethanes, but as yet no enzymes which cleave ether linkages have been reported. Bhavsar et al. have discussed the challenges associated with biodegradation of polyurethanes, and evaluates the potential of employing bacteria and fungi to accelerate biodegradation.170
Polyamides include the important commodity polymers nylon and Kevlar. In the search to identify alternative biobased starting materials for polymer production, polyamides initially present an attractive option, with a wealth of readily available monomer units, notably amino acids. Polypeptides and proteins are capable of performing structural roles in biological systems in addition to fulfilling high-level functionality including precise molecular recognition and catalysis. Most synthetic polyamides, however, have been demonstrated to display limited biodegradation profiles,175 with enzymes only known to act on oligomers of polyamides reported.133 Considering nylon, for example, Flavobacterium sp. and Pseudomonas sp. (NK87) have been reported to degrade oligo(amides) but no reduction in molecular weight reduction was observed when a 20 kDa sample of nylon 4, a linear polymer of γ-aminobutyric acid, was exposed to the same bacteria. Polyamides are presumed to display limited biodegradability due to their stable, highly crystalline structures that arise through hydrogen bonding. Current approaches to the end-of-life management of polyamides rely heavily on chemical or mechanical recycling.
An emerging group of biodegradable polymers used in biomedical applications are the polyesteramides, which contain a backbone comprised of both ester and amide linkages.176,177 Some of the limitations displayed by polyamides can be overcome through this approach, with ester linkages presenting alternative sites for enzymatic attack, and hence presenting a promising solution for the redesign of commodity polyamides. Soleimani et al. used both solution and interfacial approaches to synthesise a range of polyesteramides, using different combinations of diols, dicarboxylic acids and α-amino acids to elucidate structure–property relationships,178 enabling the thermal, rheological and mechanical properties of the resultant polymers to be tuned.
Within the literature there has been debate, and some confusion, as to whether poly(ethers) such as poly(ethylene glycol) (PEG) and poly(propylene glycol) are biodegradable. Many within the polymer science community have generally considered the broad class of poly(ethers) to be biodegradable which may not be correct in many important cases.179–182 PEG, also termed poly(ethylene oxide) or poly(oxyethylene), is synthesised by the polymerisation of ethylene oxide and has many important applications ranging from drug delivery and formulation, to use as an anti-foaming agent within the food and drink industry.183–185 PEG has been shown to biodegrade through a degradation-biodegradation mechanism (Fig. 5) whereby the initial stages of the polymer degradation occur through oxidative degradation, either through thermo-oxidation, photo-oxidation or enzymatic oxidation. While there are many different oxidative degradation pathways, degradation usually proceeds via a radical hydrogen abstraction followed by reaction of the resultant radical with environmental oxygen to yield a hydroperoxide-ether. Finally, β-scission of the hydroperoxide-ether results in the splitting of the polymer chain and the subsequent reduction in molar mass of the remaining polymer fragments.181,186,187 Upon oxidative degradation to polymer units of approximately 500–1000 Da, PEG has been reported to be assimilated by a number of different microbial species, including P. aeruginosa,188P. stutzeri189 and several Sphingomonas strains190 such as S. terrae and S. macrogoltabidus. Upon microbial ingestion, oligomeric fragments are metabolised to CO2, H2O and basic minerals, enabling ultimate biodegradation.191 Mineralisation commonly proceeds through the successive oxidative-cleavage of individual glycol units at oligomer termini.179,180 The nature of the microbial species responsible for assimilating PEG oligomers is dependent on the environmental conditions the polymers are found in, the molecular weight of the oligomers and which degradation product can be found at the terminal end of the oligomers, with most of the microbes responsible for assimilating PEG requiring at least one chain terminal hydroxyl group.192
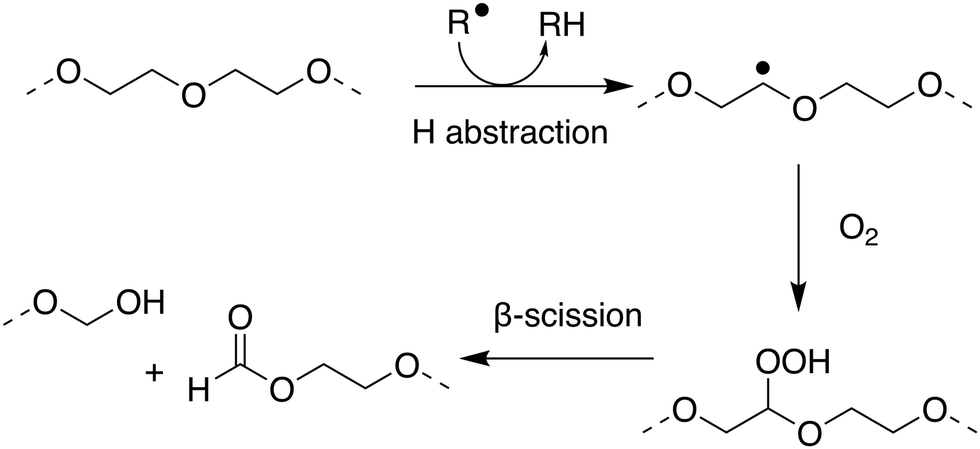 | ||
| Fig. 5 Proposed mechanism for oxidative degradation of PEG. Adapted from ref. 181 with permission from the Elsevier, copyright 2014. | ||
Due to the fact that the rate of (bio)degradation of PEG is largely limited by the abiotic oxidation and subsequent β-scission of the hydroperoxide-ether into short oligomeric units, PEGs can display sufficiently rapid rates of biodegradation to qualify as readily biodegradable under OECD guidelines. Satisfying this classification, however, is dependent on the initial molecular weight of the polymer and the environmental conditions under which biodegradation proceeds. At lower molecular weights, PEGs can generally be considered readily biodegradable, through oxidative degradation and mineralisation of resulting oligomers by microbes.182 At higher molecular weights, the extent of oxidative degradation required to produce fragments short enough for bioassimilation can result in lengthy degradation periods.
Another source of confusion regarding the biodegradation of PEG derives from the usage of differing nomenclature for PEGs, depending on their application space. The term poly(ethylene glycol) has been mostly used by pharmaceutical and biological chemists to describe polymers synthesised by the polymerisation of ethylene oxide. Within biochemistry and pharmaceutical chemistry, PEG has been extensively used for drug delivery and formulation owing to its excellent biocompatibility and reported ‘stealth’ properties, generally requiring low molecular weights (<20 kDa) so that polymers can cross cell membranes.193–195 As such, some within the polymer chemistry community have started to use the term PEG to define polymers of ethylene oxide with a molecular weight of 20 kDa or less, using poly(ethylene oxide) or poly(oxyethylene) to describe polymers of ethylene oxide with molecular weights of 20 kDa or more. Using this pseudo-definition of PEG, which has not been approved by IUPAC, longer PEGs could be mistakenly perceived as being readily biodegradable. This discrepancy has led to miscommunication between polymer scientists, mistakenly propagating the belief that all poly(ethylene oxide)s are readily biodegradable.
Further misconceptions surrounding the biodegradability of PEG stem from the development of testing standards such as ASTM D6868,196 ASTM D6954,197 or ISO 17556198 (ESI† Tables S2 and S5), which allow for the material being tested to be subjected to a period of either thermal- or photo-oxidative degradation prior to testing its biodegradability. The oxidative-degradation mechanism for PEG allows for the majority of the polymer to be degraded during this pre-treatment, yielding oligomeric fragments which are readily biodegradable in a number of different culture media utilised in biodegradation studies. This methodology allows high molecular weight PEGs to be classified as being readily biodegradable, when in reality the material would be classified as having low biodegradability by other testing standards and would take significant amounts of time within the open environment to achieve the same level of biodegradation.
Poly(propylene glycol) (PPG) has a polymer backbone that is very similar in structure to PEG bar the addition of one methyl branch on one of the carbons next to the oxygen in the repeat unit (Fig. 2). PPG is synthesised through the ring-opening polymerisation of propylene oxide and whilst its use is not as widespread as PEG, it has found applications as an anti-freeze solvent, and as a preservative and thickener in the food and drink industry.199 Due to the addition of this methyl group, the rate of abiotic oxidative-degradation of PPG is greater than that of PEG, as tertiary carbons display greater radical stability compared to secondary carbons.200 This enhanced stability results in a lower energy transition state leading to the free radical intermediate, meaning that the activation energy required to oxidise PPG is lower than the corresponding activation energy for PEG,201,202 and hence a greater rate of thermal- and photo-oxidative degradation. While the addition of the backbone methyl group can increase the rate of abiotic oxidative degradation in PPG compared to PEG, it exerts an opposing effect on the rate of biotic degradation, whereby the biotic-oxidative degradation of PEG is much greater than the rate for PPG.203 As will be discussed, monomeric branching next to or close to a polymer backbone unit which is cleaved enzymatically typically reduces the accessibility for enzymatic attack due to an increase in steric hindrance and hydrophobicity. This effect means that during the biotic degradation of PPG, enzymes generally only oxidise and cleave single glycol units from the primary-alcohol chain end, and the rate of biotic-degradation at the primary alcohol is much greater than the rate of biotic-degradation of the secondary alcohol. Conversely, in the biotic degradation of PEG, enzymes can initiate biodegradation at both ends of the polymer chain.204 As the abiotic and biotic degradation pathways of PEG and PPG are affected differently by the addition of the methyl group next to one of the oxygens, literature studies of the biodegradation of these polymers report different observations depending on the conditions used for the degradation experiments, and the initial molecular weight of the polymers.205 The rate at which each poly(ether) is removed from the environment will likely depend on whether its degradation is limited by abiotic or biotic pathways, a distinction based primarily on the initial molecular weight of the polymer.
Epoxy resins, also known as poly(epoxides), are another class of polymer which display a poly(ether) backbone and can therefore degrade through oxidative degradation. These polymers are, however, generally synthesised from large hydrophobic monomers which are based on bulky substituents to provide a large steric hindrance to increase rotational stiffness of the polymer and are typically crosslinked or cured with hardening agents to improve the mechanical stiffness, required for most applications of epoxy resins.206,207 The large degree of crosslinking generally found within epoxy resins means that for any significant loss in molecular weight of the polymer chains, oxidative degradation must occur at several locations along each polymer chain. Degradation of many epoxy resins leads to the release of the endocrine disruptor bisphenol A,208 further limiting the environmental profile of these polymers. Recently, attempts have been made to generate replacements for these materials, with the development of epoxy resins based on bio-sourced monomeric units, which may increase the potential for biodegradation. Shen and Robertson209 have reported epoxy resins based on epoxidised vanillic acid and epoxidised soybean oil that can be degraded under acidic conditions through ester hydrolysis to yield oligomer units which offer increased scope for biodegradation and chemical degradation.
Structural features of polymers and their effects on biodegradation
Beyond the chemical linkages that constitute a polymer, the rate of biodegradation is also dependant on the structural properties of the polymer, both in terms of the macroscopic properties of the material, and the microscopic structural features of its constituent polymer chains.The surface area of polymeric materials in relation to their volume may have a significant influence on their rates of biodegradation. Biodeterioration involves superficial degradation by microbes, decomposer organisms and external physical forces. This initial degradation occurs explicitly on the surface of the material, with microbial attachment leading to the formation of cracks and pores across the surface. This superficial degradation is accompanied by biofilm formation across the surface, leading to biofragmentation. Larger surface areas encourage greater microbial attachment and biofilm formation, leading to an increased rate of biodegradation.59 The effect of a polymer surface area on the rate of biodegradation was demonstrated by Degli-Innocenti et al. in 2018 using pellets of poly(butylene sebacate) with different specific surface areas.210 The rate of biodegradation was demonstrated to be proportional to the material's surface area, with kinetic data allowing estimation of the theoretical maximum rate of biodegradation for a material, where surface area is not the limiting factor.
In addition to the surface area, surface topology also plays a vital role in the attachment of microbes, and therefore the rate of biodegradation. Microbes generally attach onto surfaces along defects or rough areas before they can start to degrade the material and form biofilms. Smoother surfaces present reduced scope for microbial attachment and are typically associated with lower rates of biodegradation.211–213 This relationship between surface roughness and the rate of biodegradation was demonstrated by Kim et al. in 1999,214 when they showed that the rate of biodegradation of a poly(3-hydroxybutyrate) film containing microscopic cracks and pores was much greater than an equivalent film which had been annealed, despite the rough film displaying a higher degree of crystallinity, typically associated with slower degradation.
Often, polymeric materials undergo surface treatment through superficial chemical modification, UV photolysis or through the use of polymeric films added to the surface of a bulk material.215 This surface treatment results in a multitude of effects and can be used to reduce the permeability of the material to gaseous and liquid chemicals, to alter the wetting properties of the material or prevent microbial attachment to the surface.216–219 These chemical modifications generally reduce the rate of biodegradation because they typically frustrate microbial attachment through changes in the hydrophobic–hydrophilic balance of the surface, although there have also been many examples where surface modification has been used to increase the rate of biodegradation by enabling enhanced microbial attachment.220–222 Surface modification may mean that different microbes or enzymes are required to degrade surface components, compared to the bulk of the material, resulting in a slower, multistage biodegradation process. This effect on the rate of biodegradation can clearly be seen in the biodegradation of acetylated cellulose fibres and films. In 1993, Buchanan et al. demonstrated that increasing degrees of acetylation of cellulose fibres and films were associated with slower rates of biodegradation in both wastewater treatments and in vitro experiments.223
In general, increasing the hydrophilicity of a polymer increases microbial attachment and therefore rate of biodegradation, demonstrated with polyesteramides of varying monomer composition and increasing hydrophilicity,224,225 although the reverse trend has been observed in cellulose nanofibre films that are rendered hydrophobic through treatment with triethoxymethylsilane.226 Here, authors concluded that the hydrophobic silane layer had the effect of reducing the moisture absorption capacity and water permeability of the film, which reduced the number of swelling-induced fractures on the surface of the material, resulting in untreated films displaying a greater degree of roughness compared to treated films. The hydrophobicity or hydrophilicity of the microenvironment surrounding the hydrolysable bond in the backbone of polyesters may exert a more significant effect than the hydrophilicity of the bulk material on the rate of biodegradation. While demonstrating the effect of methyl branching on the properties of a series of furandioate-adipate copolyesters,227 Farmer et al. reported that while the rate of the biodegradation of the furandioate-adipate copolyesters was expected to follow the trend in water contact angles across the series, as would be expected for polymers where the biodegradation is limited by surface hydrophobicity, the position of methyl groups adjacent to the ester linkages exerted a more significant effect on the rate of biodegradation than the overall hydrophobicity of the polymer.
The use of polymer blends71 has become a major area of interest, with many research groups proving that the biodegradation of a slowly degrading polymer can be improved by blending with a more rapidly biodegradable polymer. Blending compatible polymers can disrupt chain alignment and alter both the thermal and mechanical properties of the resultant material and its crystallinity,228,229 with reduction in crystallinity typically leading to increased rates of biodegradation. By blending one polymer with a very high rate of biodegradation with a compatible polymer with a much lower rate of biodegradation, the overall rate of biodegradation for the whole polymeric material can be greatly increased with respect to the material with the lower rate of biodegradation.230–232 In addition to reductions in crystallinity, enzymes may biodegrade the more readily degradable components of the blend first, increasing the heterogeneity of the remaining material, and promoting mechanical breakage, increasing surface area and promoting microbial attachment.233 Polyformaldehyde or polyoxymethylene (POM), for instance, is an important engineering polymer with excellent mechanical properties and electrical resistance, with applications in construction, electronics and the automotive industry. While recyclable,234 POM is not biodegradable. Within the environment, it may be degraded through thermal oxidation,235,236 photo-oxidation or hydrolysis,237 all of which could lead to secondary pollution. Efforts to improve the environmental footprint of POM have largely focused on blending POMs with readily biodegradable polymers, usually PLA, to retain or enhance the physical and chemical properties of the polymer enhancing the rate of biodegradation of the material.238
Concerns have, however been raised about the potential negative effects that biodegradation of polymer blends may have on the environment, despite an increase in the overall rate of biodegradation. Peng et al. demonstrated that when blended polymers are aged in air, deionised water and seawater, large quantities of microplastics may be released into the surrounding environment – a consequence of the mechanical breakdown associated with differing rates of biodegradation.239 Despite the fact that, as mentioned previously, the ecological and the physiological effects of microplastics are still yet to be determined, care must be taken when considering polymer blending as a route to enhance biodegradation, as the potential effects of microplastics cannot be ignored.
Beyond the macroscopic characteristics of the polymer, molecular features of polymer chains exert profound influence on their biodegradation. The molecular weight of a polymer can have a large impact on its rate of biodegradation, especially with regards to bioassimilation. As the molecular weight of the polymer chain increases, the flexibility of the polymer backbone decreases,240 which reduces the rate of biodegradation due to increased chain entanglement and hence is expected to lead to a decrease in enzyme-substrate binding efficiency. Where enzymatic degradation requires action at the chain terminus, increased molecular weight effectively corresponds to a dilution of the possible reactive sites.241 High molecular weight synthetic polymers including polystyrene, polyethylene and polypropylene do not present easily accessible sources of carbon for microbes, with extensive degradation via abiotic routes required before surfaces can be effectively colonised.242,243 Whilst the dependence of the rate of biodegradation on molecular weight has been well known for several decades,244,245 many of the articles which have led to this conclusion were based solely on the results of linear polymers. Results published by Lei et al.246 suggest that the effect of molecular weight may be correlated with the number of chain ends available for (bio)degradation, which often occurs preferentially at chain ends. Using a series of PLA/PGLA copolymers of varying architecture, which are known to be hydrolysed sequentially from chain termini, the rate of hydrolytic degradation was shown to increase with the increase of arm number or with the decrease of arm length. These observations suggest that for (bio)degradation pathways which occur via elimination of single units at chain ends, the availability of chain ends in relation to the molecular weight exerts a greater effect on the rate of biodegradation than the frequency of backbone scission events.
Different chain arrangements arise due to differences in the spatial separation, interaction, and alignment of the polymer chains, resulting in morphologies along the surface and within the bulk of the material, ranging from amorphous to crystalline. These morphologies may have a direct impact on the rate of biodegradation. Within amorphous regions, intra-chain interactions are limited, providing greater access for extracellular enzymes to degrade polymer chains. Within crystalline materials or regions, polymer chains interact strongly, resulting in low spatial separation and a high degree of chain alignment, reducing the access of enzymes and hence a large reduction in the rate of biodegradation compared to amorphous materials or regions.247 Within semi-crystalline materials where both amorphous and crystalline regions are present, it has been shown that extracellular enzymes preferentially attack the amorphous regions.248,249 The effect of morphology on the rate of biodegradation is evident when the rate of biodegradation of natural proteins and synthetic polymers, with the same hydrolysable linkages, are compared. Naturally occurring proteins do not typically display repeating monomer sequences, whereas synthetic poly(peptides) often contain short repeating units due to the limitations of their synthesis. Within synthetic poly(peptides) there is typically little complexity within the monomer sequence compared to proteins, meaning that large sections of the monomer sequence are repeating. This feature allows large sections of synthetic poly(peptide) chains to align, resulting in high degrees of crystallinity, and reduced free volume space for access of enzymes. These factors lead to large reductions in the rate of biodegradation for synthetic poly(peptide)s compared to relatively similar natural equivalents. It has been shown that by increasing the complexity of the monomer sequence within a series of poly(amide-urethanes), the degree of crystallinity within the polymer can be reduced and the rate of biodegradation increased.250
The glass transition temperature (Tg) of a polymer has also been shown to correlate with its rate of biodegradation. Mathers et al. demonstrated that, after separating polymers into classes based on whether their biodegradation is limited by abiotic or biotic factors, as the Tg of a polymer increases within the same polymer class, the rate of biodegradation decreases.128 While this correlated relationship has been demonstrated more than once,251 it may not be a direct causal effect. Rather than the Tg directly influencing the rate of the biodegradation, it has been suggested that differences in chain flexibility impact both the Tg252 and the associated rate of biodegradation. In general, as a polymer chain becomes less flexible through increased steric hindrance, the movement of chains is restricted due to rotational stiffness, allowing chains to pack closely together. This close packing increases the interaction between chains, meaning that that the energy required to separate them is increased, increasing the Tg. Much like the described effect of crystallisation, close packing and increased intra-chain interaction may frustrate the access of enzymes, slowing biodegradation.
Other structural characteristics which influence the accessibility of the hydrolysable bonds by enzymes include polymer branching or crosslinking. Branching points may arise within monomer units themselves, or as a consequence of backbone branching. Due to the complex nature of the interplay between abiotic and biotic factors on the rate of biodegradation, the effects of structural branching within monomer units can be complex and varied depending on the monomer system under investigation. The effect of this branching on the rate of biodegradation, however, can be more easily understood when polymers are divided into classes based on the mechanism of their biodegradation (abiotic-dominant biodegradation and biotic-dominant biodegradation). Polymers which degrade primarily through the action of biotic factors e.g. PGLA, PLA, poly(hydroxybutyrate) and poly(butylene succinate), degrade via processes sensitive to both the hydrophilicity and sterics of the local environment surrounding the hydrolysable bond within their backbone. In these cases, the addition of a short hydrocarbon branch in the monomer unit can lead to a greatly reduced rate of biodegradation, especially when the branch is situated at or adjacent to the hydrolysable bonds. This effect of monomer branching on the rate of biodegradation of polyesters was demonstrated recently227 through comparison of the rates of biodegradation of a series of furandioate-adipate copolyesters based on 1,4-butanediol, 1,4-pentanediol, 1,6-hexanediol, 2,5-hexanediol and 2,7-octanediol. Enzymatic degradation studies revealed an 80% weight loss for copolymers based on 1,4-butanediol over a 48 h period, while copolymers based on 2,5-hexanediol were shown to undergo weight loss of only 19% under the same conditions. Conversely, polymers which degrade primarily through abiotic factors, including PEG, PPG, polyethylene and polypropylene, which are generally considered to display limited biodegradability, have been shown to have improved rates of biodegradation when short hydrocarbon branches are present within monomer units. Abiotic degradation of these polymers usually involves either the thermo-oxidation or photo-oxidation of the polymer backbone. As described for PEG, oxidative degradation processes are promoted by the presence of tertiary carbons, so this type of branching leads to an increase in the rate of polymer degradation and subsequent biodegradation.
Chain branching may occur where the polymer backbone features points where the chain splits into two, with branches of varying lengths and branching density possible. Due to the fact that at least three polymer chains are effectively connected at each branch point, branched polymers generally have a high chain density, with chain density being proportional to the amount of branching. This factor reduces the ability of enzymes to access and cleave any hydrolysable linkages, meaning that the rate of biodegradation is reduced, especially at points on the polymer chain close to branching points, due to significant steric hindrance that frustrates enzymatic attack.227,247,253 Where branching may have a positive impact towards the rate of biodegradation is in reducing the extent of polymer crystallinity. Despite branched chains generally displaying a high chain density, the interchain distance of non-connected chains is rather large, reducing the ability of the polymer chains to align. This lack of alignment reduces the extent of crystallinity within the polymer, which may increase the rate of biodegradation provided that the increase caused by the reduction in crystallinity exceeds the reduction in the rate of biodegradation caused by the increase in steric hindrance associated with enzymatic attack.
The effects of crosslinking are similar to branching, whereby along the polymer backbone there are points where the chain splits into two, however, within crosslinked polymers these chains interconnect with each other in one continuous polymer network. Crosslinking has a similar effect on polymer chain density to branching, whereby the chain density is proportional to the amount of crosslinking. Similar to branching, the presence of crosslinks reduces the ability of enzymes to access hydrolysable linkages, reducing the rate of biodegradation.254,255 Within a crosslinked polymer network, all chains are interconnected, leading to a number of other resulting effects which impact the rate of biodegradation. Crosslinking typically results in very high molecular weights, requiring a large degree of degradation by extracellular enzymes and external forces before bioassimilation can occur. Secondly, crosslinking typically renders a polymer insoluble, further reducing the rate of biodegradation, in line with observations on hydrophobicity discussed earlier.
Conclusions
The widespread adoption of commodity polymers has presented major advantages, yet significant challenges to society. In an effort to limit the impact of human activity on the environment, a combined effort from governments, industry, academia and wider public is required. The reuse and recycling of polymers presents the ideal route to their circular use, and should be pursued where possible, necessitating improvements in the infrastructure for recycling, improvements in recycling practices which conserve material performance, new materials with improved physical properties that are designed to persist as long as their lifetime requires and no longer, and greater public engagement to ensure effective sorting of plastic waste. In cases where discharge of polymers to the environment cannot easily be avoided e.g. polymers in formulated consumer products and within pharmaceuticals, rapid biodegradation at the end-of-life phase presents an attractive approach to limiting their environmental impact. In the process of designing new materials, undertaking a full LCA is key to determining the suitability of the material as potential replacements, with the incorporation of biobased feedstocks, the use of sustainable processes and environmental impacts during the use phase and end-of-life phase requiring consideration.Although there is much still to be learned about the specific biodegradation pathways of the wide range of polymers in extensive use, a number of factors have been found to influence the biodegradation profile of polymers. Effective strategies to enhance the biodegradation of polymers include the incorporation of readily hydrolysable bonds or other ‘weak links’ in the polymer backbone that undergo abiotic hydrolysis faster than the rest of the plastic, the generation of blends with water-soluble or more readily biodegradable polymers, and the addition of additives that promote photo-initiated oxidation.
Further work is required to establish truly ‘universal’ standardised testing methods to allow for easy comparison of the biodegradation profile of a particular polymer, across a range of environments. This standardisation, along with greater transparency on experimental testing conditions, would allow for direct comparisons to be made between studies and expediate the design of the commodity polymers of the future.
Author contributions
EFF, LS, MS: data curation, writing – original draft. CJM; data curation, visualisation, writing – review and editing. CSM: conceptualisation, funding acquisition, supervision, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Engineering and Physical Sciences Research Council [UKRI Future Leaders Fellowship MR/V027018/1], Innovate UK [KTP:10008174] and the SOFI2 CDT [EP/S023631/1].Notes and references
-
V. K. Patel, R. Kant, P. S. Chauhan and S. Bhattacharya, in Trends in Applications of Polymers and Polymer Composites, ed. V. K. Patel, R. Kant, P. S. Chauhan and S. Bhattacharya, AIP Publishing LLC, 2022, ch. 1, pp. 1–6 Search PubMed
.
- A. L. Andrady and M. A. Neal, Philos. Trans. R. Soc., B, 2009, 364, 1977–1984 CrossRef CAS PubMed
- WRAP, Eliminating Problem Plastics, 2022.
- H. Ritchie and M. Roser, Our World in Data, 2018.
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed
- I. E. Napper and R. C. Thompson, Global Challenges, 2020, 4, 1900081 CrossRef PubMed
- D. W. Laist, Mar. Pollut. Bull., 1987, 18, 319 CrossRef
- Organisation for Economic Co-operation and Development, OECD Guidelines for the Testing of Chemicals, OECD Publishing, Paris, 1981.
- American Society for Testing and Materials, Standards & Publications.
- A. Kjeldsen, M. Price, C. Lilley, E. Guzniczak and I. Archer, A Review of Standards for Biodegradable Plastics, Industrial Biotechnology Innovation Centre, 2018.
- Deutsches Institut für Normung, https://www.din.de/en.
- TÜV Austria Belgium, https://www.tuv-at.be/home/.
- The International Organization for Standardization, ISO - Standards.
- J. Kaiser, Science, 2010, 328, 1506 CrossRef PubMed
- E. A. Howell, S. J. Bograd, C. Morishige, M. P. Seki and J. J. Polovina, Mar. Pollut. Bull., 2012, 65, 16–22 CrossRef CAS PubMed
- L. Lebreton, B. Slat, F. Ferrari, B. Sainte-Rose, J. Aitken, R. Marthouse, S. Hajbane, S. Cunsolo, A. Schwarz, A. Levivier, K. Noble, P. Debeljak, H. Maral, R. Schoeneich-Argent, R. Brambini and J. Reisser, Sci. Rep., 2018, 8, 4666 CrossRef CAS PubMed
- DEFRA, The price of plastic: ending the toll of plastic waste, UK Parliament, 2022.
- N. Rivers, S. Shenstone-Harris and N. Young, J. Environ. Manage., 2017, 188, 153–162 CrossRef PubMed
- Z. O. G. Schyns and M. P. Shaver, Macromol. Rapid Commun., 2021, 42, 2000415 CrossRef CAS PubMed
- P. Vicente and E. Reis, Waste Manag. Res., 2008, 26, 140–146 CrossRef PubMed
- S. Oluwadipe, H. Garelick, S. McCarthy and D. Purchase, Waste Manag. Res., 2021, 40, 905–918 CrossRef PubMed
- J. Hopewell, R. Dvorak and E. Kosior, Philos. Trans. R. Soc., B, 2009, 364, 2115–2126 CrossRef CAS PubMed
- C. Jehanno, J. Demarteau, D. Mantione, M. C. Arno, F. Ruipérez, J. L. Hedrick, A. P. Dove and H. Sardon, Angew. Chem., Int. Ed., 2021, 60, 6710–6717 CrossRef CAS PubMed
- F. N. Singer, A. C. Deacy, T. M. McGuire, C. K. Williams and A. Buchard, Angew. Chem., Int. Ed., 2022, 61, e202201785 CrossRef CAS PubMed
- H. S. Wang, N. P. Truong, Z. Pei, M. L. Coote and A. Anastasaki, J. Am. Chem. Soc., 2022, 144, 4678–4684 CrossRef CAS PubMed
- C. K. Williams and G. L. Gregory, Nature, 2021, 590, 391–392 CrossRef CAS PubMed
- H. Sardon and A. P. Dove, Science, 2018, 360, 380–381 CrossRef CAS PubMed
- M. Burgess, H. Holmes, M. Sharmina and M. P. Shaver, Resour., Conserv. Recycl., 2021, 164, 105191 CrossRef CAS
- S. A. Backer and L. Leal, Acc. Chem. Res., 2022, 55, 2011–2018 CrossRef CAS PubMed
- A. Ammala, S. Bateman, K. Dean, E. Petinakis, P. Sangwan, S. Wong, Q. Yuan, L. Yu, C. Patrick and K. H. Leong, Prog. Polym. Sci., 2011, 36, 1015–1049 CrossRef CAS
- DEFRA, HSAC Review of Oxo-degradable Plastics, UK Parliament, 2019.
- Directive (EU) 2019/904 of the European Parliament and of the Council on the reduction of the impact of certain plastic products on the environment, The European Parliament and the Council of the European Union, 2019.
-
V. Marturano, P. Cerruti and V. Ambrogi, in Polymer Engineering, ed. B. Tylkowski, K. Wieszczycka and R. Jastrzab, De Gruyter, 2017, ch. 5, pp. 139–170 Search PubMed
- G. Singh, M. Esmaeilpour and A. Ratner, Combust. Sci. Technol., 2023, 195, 1299–1327 CrossRef CAS
- E. J. Carpenter, S. J. Anderson, G. R. Harvey, H. P. Miklas and B. B. Peck, Science, 1972, 178, 749–750 CrossRef CAS PubMed
- E. J. Carpenter and K. L. Smith, Science, 1972, 175, 1240–1241 CrossRef CAS PubMed
- R. C. Thompson, Y. Olsen, R. P. Mitchell, A. Davis, S. J. Rowland, A. W. G. John, D. McGonigle and A. E. Russell, Science, 2004, 304, 838 CrossRef CAS PubMed
- N. von Moos, P. Burkhardt-Holm and A. Köhler, Environ. Sci. Technol., 2012, 46, 11327–11335 CrossRef CAS PubMed
- H. A. Leslie, M. J. M. van Velzen, S. H. Brandsma, A. D. Vethaak, J. J. Garcia-Vallejo and M. H. Lamoree, Environ. Int., 2022, 163, 107199 CrossRef CAS PubMed
- L. C. Jenner, J. M. Rotchell, R. T. Bennett, M. Cowen, V. Tentzeris and L. R. Sadofsky, Sci. Total Environ, 2022, 831, 154907 CrossRef CAS PubMed
- L. F. Amato-Lourenço, R. Carvalho-Oliveira, G. R. Júnior, L. dos Santos Galvão, R. A. Ando and T. Mauad, J. Hazard. Mater., 2021, 416, 126124 CrossRef PubMed
- Q. Chen, J. Gao, H. Yu, H. Su, Y. Yang, Y. Cao, Q. Zhang, Y. Ren, H. Hollert, H. Shi, C. Chen and H. Liu, Environ. Sci. Eur., 2022, 34, 25 CrossRef CAS
- R. Shen, K. Yang, X. Cheng, C. Guo, X. Xing, H. Sun, D. Liu, X. Liu and D. Wang, Environ. Pollut., 2022, 300, 118986 CrossRef CAS PubMed
- T. Horvatits, M. Tamminga, B. Liu, M. Sebode, A. Carambia, L. Fischer, K. Püschel, S. Huber and E. K. Fischer, eBioMedicine, 2022, 82, 104147 CrossRef CAS PubMed
- A. Ragusa, A. Svelato, C. Santacroce, P. Catalano, V. Notarstefano, O. Carnevali, F. Papa, M. C. A. Rongioletti, F. Baiocco, S. Draghi, E. D'Amore, D. Rinaldo, M. Matta and E. Giorgini, Environ. Int., 2021, 146, 106274 CrossRef CAS PubMed
- G. Kutralam-Muniasamy, V. C. Shruti, F. Pérez-Guevara and P. D. Roy, Sci. Total Environ, 2023, 856, 159164 CrossRef CAS PubMed
- Z. Yan, H. Zhao, Y. Zhao, Q. Zhu, R. Qiao, H. Ren and Y. Zhang, J. Hazard. Mater., 2020, 384, 121489 CrossRef CAS PubMed
- J. Zhang, L. Wang, L. Trasande and K. Kannan, Environ. Sci. Technol. Lett., 2021, 8, 989–994 CrossRef CAS
- N. Zhang, Y. B. Li, H. R. He, J. F. Zhang and G. S. Ma, Sci. Total Environ, 2021, 767, 144345 CrossRef CAS PubMed
- J.-B. Fleury and V. A. Baulin, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2104610118 CrossRef CAS PubMed
- T. Roy, T. K. Dey and M. Jamal, Environ. Monit. Assess., 2022, 195, 27 CrossRef PubMed
- J. Bowley, C. Baker-Austin, A. Porter, R. Hartnell and C. Lewis, Trends Microbiol., 2021, 29, 107–116 CrossRef CAS PubMed
- M. K. Viršek, M. N. Lovšin, Š. Koren, A. Kržan and M. Peterlin, Mar. Pollut. Bull., 2017, 125, 301 CrossRef PubMed
- M. R. Gregory, Philos. Trans. R. Soc., B, 2009, 364, 2013–2025 CrossRef PubMed
- D. E. Fagnani, C. Jehanno, H. Sardon and A. J. McNeil, Macromol. Rapid Commun., 2022, 43, 2200446 CrossRef CAS PubMed
- Organisation for Economic Co-operation and Development, OECD Guidelines for Testing of Chemicals, Section 3, OECD Publishing, Paris, 2006.
- S. Solanki, S. Sinha and R. Singh, Biodegradation, 2022, 33, 529–556 CrossRef CAS PubMed
- S. Kubowicz and A. M. Booth, Environ. Sci. Technol., 2017, 51, 12058–12060 CrossRef CAS PubMed
- A. Chamas, H. Moon, J. Zheng, Y. Qiu, T. Tabassum, J. H. Jang, M. Abu-Omar, S. L. Scott and S. Suh, ACS Sustainable Chem. Eng., 2020, 8, 3494–3511 CrossRef CAS
- A. F. Sousa, C. Vilela, A. C. Fonseca, M. Matos, C. S. R. Freire, G.-J. M. Gruter, J. F. J. Coelho and A. J. D. Silvestre, Polym. Chem., 2015, 6, 5961–5983 RSC
- B. M. Trost, Science, 1991, 254, 1471–1477 CrossRef CAS PubMed
-
A. P. Dicks and A. Hent, Green Chemistry Metrics: A Guide to Determining and Evaluating Process Greenness, Springer International Publishing, 2014 Search PubMed
- M. Vert, Y. Doi, K.-H. Hellwich, M. Hess, P. Hodge, P. Kubisa, M. Rinaudo and F. Schué, Pure Appl. Chem., 2012, 84, 377–410 CrossRef CAS
- K. Horie, M. Barón, R. B. Fox, J. He, M. Hess, J. Kahovec, T. Kitayama, P. Kubisa, E. Maréchal, W. Mormann, R. F. T. Stepto, D. Tabak, J. Vohlídal, E. S. Wilks and W. J. Work, Pure Appl. Chem., 2004, 76, 889–906 CrossRef CAS
- J. Duffus, Pure Appl. Chem., 1993, 65, 2003–2122 CrossRef CAS
- B. Nagel, H. Dellweg and L. M. Gierasch, Pure Appl. Chem., 1992, 64, 143–168 CrossRef CAS
- Ellen MacArthur Foundation, Towards a Circular Economy, 2015.
- The International Organization for Standardization, ISO 14040:2006: Environmental management - Life cycle assessment - Principles and framework, 2022.
- M. W. Holdgate, Environ. Conserv., 1987, 14, 282 CrossRef
- I. T. Horváth, Chem. Rev., 2018, 118, 369–371 CrossRef PubMed
- J. V. Alemán, A. V. Chadwick, J. He, M. Hess, K. Horie, R. G. Jones, P. Kratochvíl, I. Meisel, I. Mita, G. Moad, S. Penczek and R. F. T. Stepto, Pure Appl. Chem., 2007, 79, 1801–1829 CrossRef
- S. Walker and R. Rothman, J. Cleaner Prod., 2020, 261, 121158 CrossRef CAS
- E. C. Van Roijen and S. A. Miller, Resour., Conserv. Recycl., 2022, 181, 106236 CrossRef CAS
- D. Maga, C. Galafton, J. Blömer, N. Thonemann, A. Özdamar and J. Bertling, Int. J. Life Cycle Assess., 2022, 27, 469–491 CrossRef CAS
- B. Von Vacano, H. Mangold, G. W. M. Vandermeulen, G. Battagliarin, M. Hofmann, J. Bean and A. Künkel, Angew. Chem., Int. Ed., 2023, e202210823 CAS
- The EU Waste Framework Directive (2008/98/EC), The European Parliament and the Council of the European Union, 2008.
- The Lignocellulosic Biorefinery Network, UKBioChem10: The Ten Green Chemicals which can Create Growth, Jobs and Trade for the UK, 2019.
- A. P. Gupta and V. Kumar, Eur. Polym. J., 2007, 43, 4053–4074 CrossRef CAS
- H. Chang, A. H. Motagamwala, G. W. Huber and J. A. Dumesic, Green Chem., 2019, 21, 5532–5540 RSC
- D. R. Vardon, N. A. Rorrer, D. Salvachúa, A. E. Settle, C. W. Johnson, M. J. Menart, N. S. Cleveland, P. N. Ciesielski, K. X. Steirer, J. R. Dorgan and G. T. Beckham, Green Chem., 2016, 18, 3397–3413 RSC
- C. A. Derosa, X. Q. Kua, F. S. Bates and M. A. Hillmyer, Ind. Eng. Chem. Res., 2020, 59, 15598–15613 CrossRef CAS
- A. M. Ostaniewicz-Cydzik, C. S. M. Pereira, E. Molga and A. E. Rodrigues, Ind. Eng. Chem. Res., 2014, 53, 6647–6654 CrossRef CAS
- Z. Tian, X. He, W. Pu, C. Wan and C. Jiang, Electrochim. Acta, 2006, 52, 688–693 CrossRef CAS
- Commission recommendation on the use of common methods to measure and communicate the life cycle environmental performance of products and organisations, The European Parliament and the Council of the European Union, 2021.
-
S. Doppalapudi, S. Katiyar, A. J. Domb and W. Khan, in Advanced Polymers in Medicine, ed. F. Puoci, Springer International Publishing, 2015, ch. 2, pp. 33–66 Search PubMed
-
G. Babak and A. Hadi, in Biodegradation - Life of Science, ed. C. Rolando and R. Francisca, IntechOpen, Rijeka, 2013, ch. 6, pp. 141–186 Search PubMed
- J.-F. Lutz, J.-M. Lehn, E. W. Meijer and K. Matyjaszewski, Nat. Rev. Mater., 2016, 1, 16024 CrossRef CAS
- A. C. Q. Silva, A. J. D. Silvestre, C. Vilela and C. S. R. Freire, Molecules, 2022, 27, 94 CrossRef CAS PubMed
-
A. Ravve, Principles of Polymer Chemistry, Springer, US, Boston, MA, 2000, pp. 449–475 Search PubMed
- A. Samir, F. H. Ashour, A. A. A. Hakim and M. Bassyouni, npj Mater. Degrad., 2022, 6, 68 CrossRef CAS
- I. Vroman and L. Tighzert, Materials, 2009, 2, 307–344 CrossRef CAS
- F. G. Calvo-Flores, ChemSusChem, 2009, 2, 905–919 CrossRef CAS PubMed
- D. M. Day, T. J. Farmer, J. Granelli, J. H. Lofthouse, J. Lynch, C. R. McElroy, J. Sherwood, S. Shimizu and J. H. Clark, Molecules, 2022, 27, 8427 CrossRef CAS PubMed
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC
- R. M. Cywar, N. A. Rorrer, C. B. Hoyt, G. T. Beckham and E. Y. X. Chen, Nat. Rev. Mater., 2022, 7, 83–103 CrossRef CAS
- G. Hayes, M. Laurel, D. MacKinnon, T. Zhao, H. A. Houck and C. R. Becer, Chem. Rev., 2023, 123, 2609–2734 CrossRef CAS PubMed
- S. B. Luk and M. Maríc, ACS Omega, 2021, 6, 4939–4949 CrossRef CAS PubMed
- C. Post, D. Maniar, V. S. D. Voet, R. Folkersma and K. Loos, ACS Omega, 2023, 8, 8991–9003 CrossRef CAS PubMed
- N. A. Rorrer, D. R. Vardon, J. R. Dorgan, E. J. Gjersing and G. T. Beckham, Green Chem., 2017, 19, 2812–2825 RSC
- H. Nakajima, P. Dijkstra and K. Loos, Polymers, 2017, 9, 523 CrossRef PubMed
- S. Thakur, J. Chaudhary, P. Singh, W. F. Alsanie, S. A. Grammatikos and V. K. Thakur, Bioresour. Technol., 2022, 344, 126156 CrossRef CAS PubMed
- S. Miri, R. Saini, S. M. Davoodi, R. Pulicharla, S. K. Brar and S. Magdouli, Chemosphere, 2022, 286, 131670 CrossRef CAS PubMed
- C. Chen, X. Chen, L. Liu, J. Wu and C. Gao, Fermentation, 2023, 9, 137 CrossRef CAS
- E.-M. Kim, J.-H. Eom, Y. Um, Y. Kim and H. M. Woo, J. Agric. Food Chem., 2015, 63, 4606–4612 CrossRef CAS PubMed
- T. A. Ewing, N. Nouse, M. van Lint, J. van Haveren, J. Hugenholtz and D. S. van Es, Green Chem., 2022, 24, 6373–6405 RSC
- H. Sharma and D. K. Neelam, J. Basic Microbiol., 2023, 63, 292–307 CrossRef CAS PubMed
- A. Bher, P. C. Mayekar, R. A. Auras and C. E. Schvezov, Int. J. Mol. Sci., 2022, 23, 12165 CrossRef CAS PubMed
- E. Olewnik-Kruszkowska, J. Nowaczyk and K. Kadac, Polym. Test., 2017, 60, 283–292 CrossRef CAS
- P. Tyagi, S. Agate, O. D. Velev, L. Lucia and L. Pal, Environ. Sci. Technol., 2022, 56, 2071–2095 CrossRef CAS PubMed
- N. Delangiz, S. Aliyar, N. Pashapoor, K. Nobaharan, B. Asgari Lajayer and S. Rodríguez-Couto, Chemosphere, 2022, 294, 133709 CrossRef CAS PubMed
- R.-J. Mueller, Process Biochem., 2006, 41, 2124–2128 CrossRef CAS
- S. M. Emadian, T. T. Onay and B. Demirel, Waste Manag., 2017, 59, 526–536 CrossRef CAS PubMed
- Shilpa, N. Basak and S. S. Meena, Front. Environ. Sci. Eng., 2022, 16, 161 CrossRef CAS PubMed
- K. Gravouil, R. Ferru-Clément, S. Colas, R. Helye, L. Kadri, L. Bourdeau, B. Moumen, A. Mercier and T. Ferreira, Environ. Sci. Technol., 2017, 51, 5172–5181 CrossRef CAS PubMed
- D. Danso, J. Chow and W. R. Streit, Appl. Environ. Microbiol., 2019, 85, e01095 CrossRef CAS PubMed
-
L. M. Koh and S. M. Khor, Handbook of Biodegradable Materials, Springer International Publishing, 2023, pp. 19–56 Search PubMed
- G. Becker and F. R. Wurm, Chem. Soc. Rev., 2018, 47, 7739–7782 RSC
- J. Greene, PLA and PHA Biodegradation in the Marine Environment, Department of Mechanical Engineering Mechatronic Engineering and Manufacturing Technology California State University, Chico, 2012.
- S. M. Barbon, M. C. D. Carter, L. Yin, C. M. Whaley, V. C. Albright and R. E. Tecklenburg, Macromol. Rapid Commun., 2022, 43, 2100773 CrossRef CAS PubMed
- A. Beltrán-Sanahuja, A. Benito-Kaesbach, N. Sánchez-García and C. Sanz-Lázaro, Sci. Total Environ., 2021, 787, 147678 CrossRef
- E. R. Zettler, T. J. Mincer and L. A. Amaral-Zettler, Environ. Sci. Technol., 2013, 47, 7137–7146 CrossRef CAS PubMed
- K. Li, W. Jia, L. Xu, M. Zhang and Y. Huang, J. Hazard. Mater., 2023, 442, 130011 CrossRef CAS PubMed
- M. Arias-Andres, M. T. Kettner, T. Miki and H.-P. Grossart, Sci. Total Environ, 2018, 635, 1152–1159 CrossRef CAS PubMed
-
M. van der Zee, Handbook of Biodegradable Polymers, ed. B. Catia, De Gruyter, Berlin, Boston, 2020, ch. 1, pp. 1–22 Search PubMed
- S. Baidurah, Polymers, 2022, 14, 4928 CrossRef CAS PubMed
- I. Dupret, C. David and A. Daro, Polym. Degrad. Stab., 2000, 67, 505–513 CrossRef CAS
- V. C. Albright and Y. Chai, Environ. Sci. Technol., 2021, 55, 11476–11488 CrossRef PubMed
- K. Min, J. D. Cuiffi and R. T. Mathers, Nat. Commun., 2020, 11, 727 CrossRef CAS PubMed
- M. Lee and K. Min, ACS Omega, 2022, 7, 3649–3655 CrossRef CAS PubMed
- S. Malik, A. Maurya, S. K. Khare and K. R. Srivastava, Polymers, 2023, 15, 1540 CrossRef CAS PubMed
- M. Shafana Farveen, T. Madhavan and R. Narayanan, Biodegradation, 2023, 34, 383–403 CrossRef CAS PubMed
- L. Arregui, M. Ayala, X. Gómez-Gil, G. Gutiérrez-Soto, C. E. Hernández-Luna, M. Herrera de Los Santos, L. Levin, A. Rojo-Domínguez, D. Romero-Martínez, M. C. N. Saparrat, M. A. Trujillo-Roldán and N. A. Valdez-Cruz, Microb. Cell Fact., 2019, 18, 200 CrossRef CAS PubMed
- P. C. F. Buchholz, G. Feuerriegel, H. Zhang, P. Perez-Garcia, L.-L. Nover, J. Chow, W. R. Streit and J. Pleiss, Proteins: Struct., Funct., Bioinf., 2022, 90, 1443–1456 CrossRef CAS PubMed
- N. Tripathi, M. Misra and A. K. Mohanty, ACS Eng. Au, 2021, 1, 7–38 CrossRef CAS
- A. Sanluis-Verdes, P. Colomer-Vidal, F. Rodriguez-Ventura, M. Bello-Villarino, M. Spinola-Amilibia, E. Ruiz-Lopez, R. Illanes-Vicioso, P. Castroviejo, R. Aiese Cigliano, M. Montoya, P. Falabella, C. Pesquera, L. Gonzalez-Legarreta, E. Arias-Palomo, M. Solà, T. Torroba, C. F. Arias and F. Bertocchini, Nat. Commun., 2022, 13, 5568 CrossRef CAS PubMed
- N. Zhang, M. Ding and Y. Yuan, Microorganisms, 2022, 10, 1537 CrossRef CAS PubMed
- S. Ghatge, Y. Yang, J.-H. Ahn and H.-G. Hur, Appl. Biol. Chem., 2020, 63, 27 CrossRef CAS
- J.-M. Restrepo-Flórez, A. Bassi and M. R. Thompson, Int. Biodeterior. Biodegrad., 2014, 88, 83–90 CrossRef
- A. Réjasse, J. Waeytens, A. Deniset-Besseau, N. Crapart, C. Nielsen-Leroux and C. Sandt, Environ. Sci. Technol., 2022, 56, 525–534 CrossRef PubMed
- Z. Zhang, H. Peng, D. Yang, G. Zhang, J. Zhang and F. Ju, Nat. Commun., 2022, 13, 5360 CrossRef CAS PubMed
- L. Giacomucci, N. Raddadi, M. Soccio, N. Lotti and F. Fava, New Biotechnol., 2019, 52, 35–41 CrossRef CAS PubMed
- E. Chiellini, A. Corti and R. Solaro, Polym. Degrad. Stab., 1999, 64, 305–312 CrossRef CAS
- M. Shimao, Curr. Opin. Biotechnol, 2001, 12, 242–247 CrossRef CAS PubMed
- N. Ben Halima, RSC Adv., 2016, 6, 39823–39832 RSC
- M. Shimao, H. Yamamoto, K. Ninomiya, N. Kato, O. Adachi, M. Ameyama and C. Sakazawa, Agric. Biol. Chem., 1984, 48, 2873–2876 CAS
- G. von Haugwitz, K. Donnelly, M. Di Filippo, D. Breite, M. Phippard, A. Schulze, R. Wei, M. Baumann and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2023, 62, e202216962 CrossRef CAS PubMed
- K. Madhavan Nampoothiri, N. R. Nair and R. P. John, Bioresour. Technol., 2010, 101, 8493–8501 CrossRef CAS PubMed
- Y. Yoon, H. Park, S. An, J.-H. Ahn, B. Kim, J. Shin, Y.-E. Kim, J. Yeon, J.-H. Chung, D. Kim and M. Cho, J. Environ. Manage., 2023, 335, 117493 CrossRef CAS PubMed
- P. K. Samantaray, A. Little, D. M. Haddleton, T. Mcnally, B. Tan, Z. Sun, W. Huang, Y. Ji and C. Wan, Green Chem., 2020, 22, 4055–4081 RSC
- A. Bher, Y. Cho and R. Auras, Macromol. Rapid Commun., 2023, 44, 2200769 CrossRef CAS PubMed
- A. Larrañaga and E. Lizundia, Eur. Polym. J., 2019, 121, 109296–109327 CrossRef
- Y. Yang, J. Min, T. Xue, P. Jiang, X. Liu, R. Peng, J.-W. Huang, Y. Qu, X. Li, N. Ma, F.-C. Tsai, L. Dai, Q. Zhang, Y. Liu, C.-C. Chen and R.-T. Guo, Nat. Commun., 2023, 14, 1645 CrossRef CAS PubMed
- F. Muroi, Y. Tachibana, P. Soulenthone, K. Yamamoto, T. Mizuno, T. Sakurai, Y. Kobayashi and K.-I. Kasuya, Polym. Degrad. Stab., 2017, 137, 11–22 CrossRef CAS
- S. Yoshida, K. Hiraga, T. Takehana, I. Taniguchi, H. Yamaji, Y. Maeda, K. Toyohara, K. Miyamoto, Y. Kimura and K. Oda, Science, 2016, 351, 1196–1199 CrossRef CAS PubMed
- C. Jerves, R. P. P. Neves, M. J. Ramos, S. da Silva and P. A. Fernandes, ACS Catal., 2021, 11, 11626–11638 CrossRef CAS
- S. K. Burgess, J. E. Leisen, B. E. Kraftschik, C. R. Mubarak, R. M. Kriegel and W. J. Koros, Macromolecules, 2014, 47, 1383–1391 CrossRef CAS
- M. Orlando, G. Molla, P. Castellani, V. Pirillo, V. Torretta and N. Ferronato, Int. J. Mol. Sci., 2023, 24, 3877 CrossRef CAS PubMed
- Y. Kawashima, T. Ohki, N. Shibata, Y. Higuchi, Y. Wakitani, Y. Matsuura, Y. Nakata, M. Takeo, D.-I. Kato and S. Negoro, FEBS J., 2009, 276, 2547–2556 CrossRef CAS PubMed
- Y. Cui, Y. Chen, X. Liu, S. Dong, Y. E. Tian, Y. Qiao, R. Mitra, J. Han, C. Li, X. Han, W. Liu, Q. Chen, W. Wei, X. Wang, W. Du, S. Tang, H. Xiang, H. Liu, Y. Liang, K. N. Houk and B. Wu, ACS Catal., 2021, 11, 1340–1350 CrossRef CAS
- V. Pirillo, M. Orlando, C. Battaglia, L. Pollegioni and G. Molla, FEBS J., 2023, 290, 3185–3202 CrossRef CAS PubMed
- R. Shah, T. V. Nguyen, A. Marcora, A. Ruffell, A. Hulthen, K. Pham, G. Wijffels, C. Paull and D. J. Beale, Environ. Res., 2023, 220, 115137 CrossRef CAS PubMed
-
K. Takeuchi, in Polymer Science: A Comprehensive Reference, ed. K. Matyjaszewski and M. Möller, Elsevier, Amsterdam, 2012, vol. 5, ch. 16, pp. 363–376 Search PubMed
- T. Artham and M. Doble, Macromol. Biosci., 2008, 8, 14–24 CrossRef CAS PubMed
- T. Suyama, H. Hosoya and Y. Tokiwa, FEMS Microbiol. Lett., 1998, 161, 255–261 CrossRef CAS PubMed
- M. Srikanth, T. S. R. S. Sandeep, K. Sucharitha and S. Godi, Bioresour. Bioprocess., 2022, 9, 42 CrossRef
- M. Arefian, A. Tahmourespour and M. Zia, Arch. Environ. Prot., 2020, 46, 14–20 CAS
- T. Suyama and Y. Tokiwa, Enzyme Microb. Technol., 1997, 20, 122–126 CrossRef CAS
- T. Artham and M. Doble, Biomacromolecules, 2010, 11, 20–28 CrossRef CAS PubMed
- W. Yue, C.-F. Yin, L. Sun, J. Zhang, Y. Xu and N.-Y. Zhou, J. Hazard. Mater., 2021, 416, 125775 CrossRef CAS PubMed
- P. Bhavsar, M. Bhave and H. K. Webb, World J. Microbiol. Biotechnol., 2023, 39, 122 CrossRef CAS PubMed
- L. Zimmermann, G. Dierkes, T. A. Ternes, C. Völker and M. Wagner, Environ. Sci. Technol., 2019, 53, 11467–11477 CrossRef CAS PubMed
- P. Pfohl, D. Bahl, M. Rückel, M. Wagner, L. Meyer, P. Bolduan, G. Battagliarin, T. Hüffer, M. Zumstein, T. Hofmann and W. Wohlleben, Environ. Sci. Technol., 2022, 56, 16873–16884 CrossRef CAS PubMed
- K. Skleničková, S. Abbrent, M. Halecký, V. Kočí and H. Beneš, Crit. Rev. Environ. Sci. Technol., 2022, 52, 157–202 CrossRef
- B. Pantelic, S. Skaro Bogojevic, D. Milivojevic, T. Ilic-Tomic, B. Lončarević, V. Beskoski, V. Maslak, M. Guzik, K. Makryniotis, G. Taxeidis, R. Siaperas, E. Topakas and J. Nikodinovic-Runic, Catalysts, 2023, 13, 278 CrossRef CAS
- B. D. Ulery, L. S. Nair and C. T. Laurencin, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 832–864 CrossRef CAS PubMed
- A. C. Fonseca, M. H. Gil and P. N. Simões, Prog. Polym. Sci., 2014, 39, 1291–1311 CrossRef CAS
- M. Winnacker and B. Rieger, Polym. Chem., 2016, 7, 7039–7046 RSC
- A. Soleimani, S. Drappel, R. Carlini, A. Goredema and E. R. Gillies, Ind. Eng. Chem. Res., 2014, 53, 1452–1460 CrossRef CAS
-
F. Kawai, in Biopolymers Online, ed. A. Steinbüchel, 2005 Search PubMed
-
F. Kawai, in Studies in Polymer Science, ed. Y. Doi and K. Fukuda, Elsevier, 1994, vol. 12, pp. 24–38 Search PubMed
- J. Ulbricht, R. Jordan and R. Luxenhofer, Biomaterials, 2014, 35, 4848–4861 CrossRef CAS PubMed
- J. Menzies, A. Wilcox, K. Casteel and K. McDonough, Sci. Total Environ, 2023, 858, 160006 CrossRef CAS PubMed
- L. Ma, L. Deng and J. Chen, Drug Dev. Ind. Pharm., 2014, 40, 845–851 CrossRef CAS PubMed
-
H. W. Leung, in Encyclopedia of Toxicology, ed. P. Wexler, Academic Press, Oxford, 2014, pp. 1043–1044 Search PubMed
- J. Herzberger, K. Niederer, H. Pohlit, J. Seiwert, M. Worm, F. R. Wurm and H. Frey, Chem. Rev., 2016, 116, 2170–2243 CrossRef CAS PubMed
- L. M. Smith, H. M. Aitken and M. L. Coote, Acc. Chem. Res., 2018, 51, 2006–2013 CrossRef CAS PubMed
- F. Kawai and H. Yamanaka, Arch. Microbiol., 1986, 146, 125–129 CrossRef CAS PubMed
- J. R. Haines and M. Alexander, Appl. Microbiol., 1975, 29, 621–625 CrossRef CAS PubMed
- N. Obradors and J. Aguilar, Appl. Environ. Microbiol., 1991, 57, 2383–2388 CrossRef CAS PubMed
- M. Takeuchi, F. Kawai, Y. Shimada and A. Yokota, Syst. Appl. Microbiol., 1993, 16, 227–238 CrossRef CAS
-
D. P. Cox, in Advances in Applied Microbiology, ed. D. Perlman, Academic Press, 1978, vol. 23, pp. 173–194 Search PubMed
- E. L. Fincher and W. J. Payne, Appl. Microbiol., 1962, 10, 542–547 CrossRef CAS PubMed
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288–6308 CrossRef CAS PubMed
- A. Bunker, Phys. Procedia, 2012, 34, 24–33 CrossRef
- A. Kolate, D. Baradia, S. Patil, I. Vhora, G. Kore and A. Misra, J. Controlled Release, 2014, 192, 67–81 CrossRef CAS PubMed
- American Society for Testing and Materials, ASTM D6868-21: Standard Specification for Labeling of End Items that Incorporate Plastics and Polymers as Coatings or Additives with Paper and Other Substrates Designed to be Aerobically Composted in Municipal or Industrial Facilities, 2021.
- American Society for Testing and Materials, ASTM D6954-18: Standard Guide for Exposing and Testing Plastics that Degrade in the Environment by a Combination of Oxidation and Biodegradation, 2018.
- The International Organization for Standardization, Plastics—Determination of the ultimate aerobic biodegradability of plastic materials in soil by measuring the oxygen demand in a respirometer or the amount of carbon dioxide evolved, 2019.
- J. A. Okolie, iScience, 2022, 25, 104903 CrossRef CAS PubMed
-
J. Clayden, N. Greeves and S. G. Warren, Organic Chemistry, Oxford University Press, Oxford, 2nd edn, 2012 Search PubMed
- L. Costa, G. Camino, M. P. Luda, G. G. Cameron and M. Y. Qureshi, Polym. Degrad. Stab., 1996, 53, 301–310 CrossRef CAS
- L. Costa, A. M. Gad, G. Camino, G. G. Cameron and M. Y. Qureshi, Macromolecules, 1992, 25, 5512–5518 CrossRef CAS
- E. Beran, S. Hull and M. Steininger, J. Polym. Environ., 2013, 21, 172–180 CrossRef CAS
- J. D. Rogers, E. M. Thurman, I. Ferrer, J. S. Rosenblum, M. V. Evans, P. J. Mouser and J. N. Ryan, Environ. Sci. Process. Impacts, 2019, 21, 256–268 RSC
- A. Zgoła-Grześkowiak, T. Grześkowiak, J. Zembrzuska and Z. Łukaszewski, Chemosphere, 2006, 64, 803–809 CrossRef PubMed
-
S. R. Sandler and W. Karo, in Polymer Syntheses, ed. S. R. Sandler and W. Karo, Academic Press, San Diego, 1994, ch. 3, pp. 87–128 Search PubMed
-
J. L. Massingill and R. S. Bauer, in Applied Polymer Science: 21st Century, ed. C. D. Craver and C. E. Carraher, Pergamon, Oxford, 2000, pp. 393–424 Search PubMed
- E. J. Eio, M. Kawai, K. Tsuchiya, S. Yamamoto and T. Toda, Int. Biodeterior. Biodegrad., 2014, 96, 166–173 CrossRef
- M. Shen and M. L. Robertson, ACS Sustainable Chem. Eng., 2021, 9, 438–447 CrossRef CAS
- S. Chinaglia, M. Tosin and F. Degli-Innocenti, Polym. Degrad. Stab., 2018, 147, 237–244 CrossRef CAS
- S. B. Chinnaraj, P. G. Jayathilake, J. Dawson, Y. Ammar, J. Portoles, N. Jakubovics and J. Chen, J. Mater. Sci. Technol., 2021, 81, 151–161 CrossRef
- S. Wu, B. Zhang, Y. Liu, X. Suo and H. Li, Biointerphases, 2018, 13, 060801 CrossRef CAS PubMed
- S. Zheng, M. Bawazir, A. Dhall, H.-E. Kim, L. He, J. Heo and G. Hwang, Front. Bioeng. Biotechnol., 2021, 9, 643722 CrossRef PubMed
- M.-N. Kim, A.-R. Lee, K.-H. Lee, I.-J. Chin and Y. Jin-San, Eur. Polym. J., 1999, 35, 1153–1158 CrossRef CAS
-
M. Gilliam, in Handbook of Manufacturing Engineering and Technology, ed. A. Y. C. Nee, Springer London, London, 2015, pp. 99–124 Search PubMed
- A. Uneputty, A. Dávila-Lezama, D. Garibo, A. Oknianska, N. Bogdanchikova, J. F. Hernández-Sánchez and A. Susarrey-Arce, Colloids Interface Sci. Commun., 2022, 46, 100560 CrossRef CAS
- A.-M. Holban, C. Farcasiu, O.-C. Andrei, A. M. Grumezescu and A.-T. Farcasiu, Materials, 2021, 14, 6994 CrossRef CAS PubMed
- E. K. Riga, M. Vöhringer, V. T. Widyaya and K. Lienkamp, Macromol. Rapid Commun., 2017, 38, 1700216 CrossRef PubMed
- N. Doroshenko, S. Rimmer, R. Hoskins, P. Garg, T. Swift, H. L. M. Spencer, R. M. Lord, M. Katsikogianni, D. Pownall, S. MacNeil, C. W. I. Douglas and J. Shepherd, Biomater. Sci., 2018, 6, 2101–2109 RSC
- D. Hegemann, H. Brunner and C. Oehr, Nucl. Instrum. Methods Phys. Res., Sect. B, 2003, 208, 281–286 CrossRef CAS
-
F. M. Michael, M. Khalid, R. Walvekar, H. Siddiqui and A. B. Balaji, in Biodegradable and Biocompatible Polymer Composites, ed. N. G. Shimpi, Woodhead Publishing, 2018, ch. 2, pp. 33–54 Search PubMed
- H. Turkoglu Sasmazel, M. Alazzawi and N. Kadim Abid Alsahib, Molecules, 2021, 26, 1665 CrossRef PubMed
- C. M. Buchanan, R. M. Gardner and R. J. Komarek, J. Appl. Polym. Sci., 1993, 47, 1709–1719 CrossRef CAS
- M. Nagata and T. Kiyotsukuri, Eur. Polym. J., 1994, 30, 1277–1281 CrossRef CAS
- C. Park, E. Y. Kim, Y. T. Yoo and S. S. Im, J. Appl. Polym. Sci., 2003, 90, 2708–2714 CrossRef CAS
- I. Surya, C. M. Hazwan, H. P. S. Abdul Khalil, E. B. Yahya, A. B. Suriani, M. Danish and A. Mohamed, Polymers, 2022, 14, 4147 CrossRef CAS PubMed
- A. Little, A. Pellis, J. W. Comerford, E. Naranjo-Valles, N. Hafezi, M. Mascal and T. J. Farmer, ACS Sustainable Chem. Eng., 2020, 8, 14471–14483 CrossRef CAS PubMed
-
J. Parameswaranpillai, S. Thomas and Y. Grohens, in Characterization of Polymer Blends, ed. J. Parameswaranpillai, S. Thomas and Y. Grohens, 2014, ch. 1, pp. 1–6 Search PubMed
-
L. A. Utracki, Introduction to Polymer Blends, Springer Netherlands, Dordrecht, 2003 Search PubMed
-
M. Yasin, A. J. Amass and B. J. Tighe, in Polymers and Other Advanced Materials: Emerging Technologies and Business Opportunities, ed. P. N. Prasad, J. E. Mark and T. J. Fai, Springer, US, Boston, MA, 1995, pp. 169–176 Search PubMed
- M. Jordá-Reolid, A. Ibáñez-García, L. Catani and A. Martínez-García, Polymers, 2022, 14, 5223 CrossRef PubMed
- T. Narancic, S. Verstichel, S. Reddy Chaganti, L. Morales-Gamez, S. T. Kenny, B. De Wilde, R. Babu Padamati and K. E. O’Connor, Environ. Sci. Technol., 2018, 52, 10441–10452 CrossRef CAS PubMed
- M. Nevoralová, M. Koutný, A. Ujčić, Z. Starý, J. Šerá, H. Vlková, M. Šlouf, I. Fortelný and Z. Kruliš, Front. Mater., 2020, 7, 141 CrossRef
- K. Beydoun and J. Klankermayer, ChemSusChem, 2020, 13, 488–492 CrossRef CAS PubMed
- T. R. Crompton, Thermo-oxidative degradation of polymers, Smithers, 2010.
-
G. P. Simon, Polymer blends and alloys, Routledge, 2019 Search PubMed
- V. V. Pchelintsev, A. Y. Sokolov and G. E. Zaikov, Polym. Degrad. Stab., 1988, 21, 285–310 CrossRef CAS
- J. Li, Y. Wang, X. Wang and D. Wu, Polymers, 2019, 11, 1516 CrossRef CAS PubMed
- R. Bao, J. Pu, C. Xie, T. Mehmood, W. Chen, L. Gao, W. Lin, Y. Su, X. Lin and L. Peng, J. Hazard. Mater., 2022, 434, 128891 CrossRef CAS PubMed
-
K. Balani, V. Verma, A. Agarwal and R. Narayan, in Biosurfaces, ed. K. Balani, V. Verma, A. Agarwal and R. Narayan, 2014, pp. 329–344 Search PubMed
- Y. Xie, J. Park and S. Kang, Int. J. Biosci. Biotechnol., 2016, 8, 315–322 Search PubMed
- C. Odobel, C. Dussud, L. Philip, G. Derippe, M. Lauters, B. Eyheraguibel, G. Burgaud, A. Ter Halle, A. L. Meistertzheim, S. Bruzaud, V. Barbe and J. F. Ghiglione, Front. Microbiol., 2021, 12, 734782 CrossRef PubMed
- G. Caruso, J. Mar. Sci. Eng., 2020, 8, 78 CrossRef
- M. Mochizuki and M. Hirami, Polym. Adv. Technol., 1997, 8, 203–209 CrossRef CAS
- J.-D. Gu, Int. Biodeterior. Biodegrad., 2003, 52, 69–91 CrossRef CAS
- D. D. Lu, J. C. Yuan, H. Li and Z.-Q. Lei, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3802–3812 CrossRef CAS
- M. C. Zambrano, J. J. Pawlak and R. A. Venditti, BioResources, 2020, 15, 9786–9833 Search PubMed
-
J. Pan, in Modelling Degradation of Bioresorbable Polymeric Medical Devices, ed. J. Pan, Woodhead Publishing Series in Biomaterials, 2015, ch. 4, pp. 53–69 Search PubMed
- X.-H. Zong, Z.-G. Wang, B. S. Hsiao, B. Chu, J. J. Zhou, D. D. Jamiolkowski, E. Muse and E. Dormier, Macromolecules, 1999, 32, 8107–8114 CrossRef CAS
- S. J. Huang and M. S. Roby, J. Bioact. Compatible Polym., 1986, 1, 61–71 CrossRef CAS
- L. Shang, Q. Fei, Y. H. Zhang, X. Z. Wang, D.-D. Fan and H. N. Chang, J. Polym. Environ., 2012, 20, 23–28 CrossRef CAS
- X. Yu, J. Jia, S. Xu, K. U. Lao, M. J. Sanford, R. K. Ramakrishnan, S. I. Nazarenko, T. R. Hoye, G. W. Coates and R. A. DiStasio, Nat. Commun., 2018, 9, 2880 CrossRef PubMed
- T. L. Schaeffer, S. G. Cantwell, J. L. Brown, D. S. Watt and R. R. Fall, Appl. Environ. Microbiol., 1979, 38, 742–746 CrossRef CAS PubMed
- L. Hossain, R. M. B. Ledesma, J. Tanner and G. Garnier, Carbohydr. Polym. Technol. Appl., 2022, 3, 100199 CAS
- Z. Yue-Hong, Z. Wu-Quan, G. Zhen-Hua and G. Ji-You, J. Appl. Polym. Sci., 2015, 132, 41387 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cs00556a |
| This journal is © The Royal Society of Chemistry 2023 |