
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDaisy chain architectures: from discrete molecular entities to polymer materials
Emilie
Moulin
 *a,
Christian C.
Carmona-Vargas
a and
Nicolas
Giuseppone
*ab
*a,
Christian C.
Carmona-Vargas
a and
Nicolas
Giuseppone
*ab
aSAMS Research Group, Université de Strasbourg, CNRS, Institut Charles Sadron UPR 22, 67000 Strasbourg, France. E-mail: emoulin@unistra.fr; giuseppone@unistra.fr
bInstitut Universitaire de France (IUF), France
First published on 18th October 2023
Abstract
Daisy chain architectures, made by the self-complementary threading of an axle covalently linked to a macrocycle, represent a particularly intriguing family of supramolecular and mechanically interlocked (macro)molecules. In this review, we discuss their recent history, their modular chemical structures, and the various synthetic strategies to access them. We also detail how their internal sliding motions can be controlled and how their integration within polymers can amplify that motions up to the macroscopic scale. This overview of the literature demonstrates that the peculiar structure and dynamics of daisy chains have already strongly influenced the research on artificial molecular machines, with the potential to be implemented from nanometric switchable devices to mechanically active soft-matter materials.
 Emilie Moulin | Emilie Moulin is a « Directrice de Recherche » CNRS at the Institut Charles Sadron in Strasbourg working in the group of Prof. Nicolas Giuseppone since 2008. In 2014, she completed her habilitation dedicated to the synthesis and characterization of complex supramolecular systems for materials science. Her research interests currently focus on combining polymers, supramolecular and organic chemistry to develop new functional materials. |
 Christian C. Carmona-Vargas | Christian C. Carmona Vargas earned his BSc in Chemistry with honours from Universidad del Valle in Cali, Colombia, and his MSc in chemistry in 2017 at Universidade Federal de São Carlos, Brazil. In 2022, he obtained his PhD degree from the Université de Strasbourg, working on [c2]daisy chain rotaxanes under the direction of Prof. Nicolas Giuseppone and Dr Émilie Moulin. His research interests encompass synthetic organic and supramolecular chemistry, aimed at obtaining compounds with potential applications in materials science and/or medicinal chemistry. |
 Nicolas Giuseppone | Nicolas Giuseppone obtained his MSc and PhD degrees from the University of Orsay, France. He is currently distinguished professor of chemistry (PRCE2) at the University of Strasbourg, and director of the Research Federation on Materials and Nanosciences – CNRS. In 2023, he was appointed a senior member of the Institut Universitaire de France (IUF). His current research interests are focused on supramolecular chemistry, molecular machines, and functional materials. |
Key learning points[1] The synthetic strategies, based on supramolecular host–guest recognition, to design linear or cyclic daisy chains.[2] The role of stoppers to topologically lock supramolecular daisy chains in mechanically bonded daisy chain rotaxanes. [3] The use of specific binding stations to access molecular bistability and to control mechanical motions. [4] The integration of these mechanical motions into polymer chains to produce collective actuations across length scales. [5] The opportunities to reach a new class of stimuli-responsive materials at macroscopic scale with unique functional properties. |
Introduction
Mechanically interlocked molecules (MIMs) represent a broad family of chemical objects, either comprised of one component with an entangled topology (such as knots),1–3 or of several individual components entangled in space by mechanical bonds (such as catenanes and rotaxanes).4–6 Interestingly, the rings in catenanes and rotaxanes can be potentially shuttled between well-defined binding sites, leading to their controlled molecular motion into bistable or multistable MIMs. These molecules have been at the foundation of a number of molecular machines by functioning as reversible switches, or as motors using ratchetting strategies to provide unidirectional motion in the presence of a fuel.7–17 Mechanical bonds have also been used to generate mechanically interlocked polymers (polycatenanes, polyrotaxanes), in which they provide substantially modified macroscopic properties compared to both their covalent and supramolecular counterparts.10,11,18–21 Among the various types of mechanical bonds, daisy chain rotaxanes present a particular topology in which several individual components – made of one ring and one axle each –, thread one another by host–guest complexations, thus forming cyclic (i.e. [cn]) or linear (i.e. acyclic [an]) oligomers and polymers (Fig. 1a and b), with n corresponding to the number of individual components in the interlocked structure. The origin of the term “daisy chain” relates to “garlands made of daisy chain flowers in which the stem of one daisy chain is made to breach a hole located in the stem of another (‘stoppered’ by the flower's head) to make a continuous chain”.4 Molecular daisy chains are therefore self-complementary, and such individual entities are able to spontaneously dimerize or polymerize without any additional components. They have been synthesized by different strategies, involving various molecular components and host–guest chemistries. Furthermore, their mechanical motions have been implemented toward different useful tasks going from nano- to macroscale. As an important subcategory of this family, [c2]daisy chains,22 also early named as Janus23 or hermaphrodite,24 have demonstrated their capacity to produce peculiar sliding motion between an extended state and a contracted state, reminiscent of the micrometric sliding motion between the myosin and actin filaments of sarcomeres in muscles (Fig. 1c).7,11,19,25–28 The objective of this review is to exemplify the main recent advances made on the synthesis of daisy chains having either acyclic [an] or cyclic [cn] topologies. We will discuss several situations depending on the absence or the presence of stoppers (i.e. bulky groups located at the end of the axle) on the daisy chains. In the absence of stoppers (Fig. 1a), daisy chains can unthread by dilution, and will be named hereafter “pseudo-daisy chain rotaxanes” or “supramolecular daisy chains”. Conversely, in the presence of stoppers, daisy chains cannot unthread under dilution because of their topologically locked mechanical bonds (Fig. 1b and c).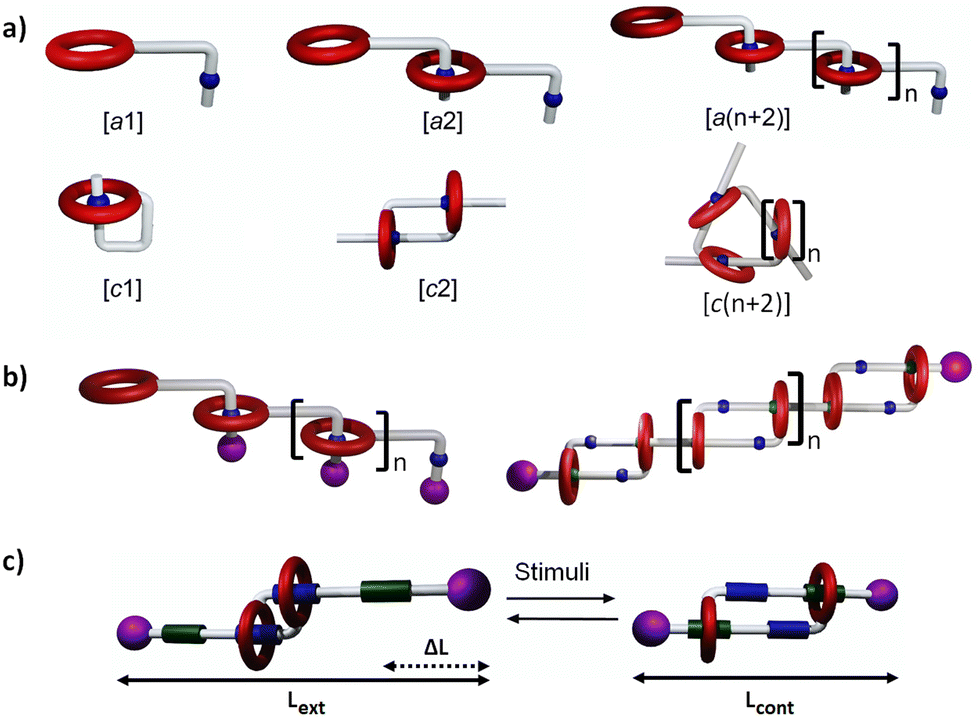 | ||
| Fig. 1 (a) Schematic representation of acyclic [an] and cyclic [cn] daisy chain architectures. The number “n” in brackets refers to the number of monomers involved in the daisy chain. The molecular component which is repeated in [an] and [cn] architectures consists in a macrocyclic unit (in red) and an axle (in grey) on which is located a station (in blue). The station binds to the macrocycle by host–guest chemistries. (b) Schematic representation of mechanically interlocked [a(n + 2)] and bistable [c2]n daisy chain polymers. (c) Schematic representation of a bistable [c2] daisy chain rotaxane in which the mechanical bond cannot unthread due to the presence of bulky stoppers (in purple), and in which the two distinct stations on the axle (binding sites in blue and green) can be specifically tuned by external stimuli to preferentially bind the macrocycle, leading to a controlled extension/contraction of the molecule. Lext and Lcont corresponds to the theoretical length of the [c2]daisy chain and ΔL = Lext − Lcont represents the difference in size between the extended and the contracted states. | ||
This review is organized in three main sections. The first one discusses both acyclic and cyclic molecular daisy chains, with a finite number of units ([a1–3] and [c1–4]). They can be made of purely synthetic organic molecules, but also of biomolecules such as peptides and DNA. We will discuss the synthetic strategies used to access molecular pseudo daisy chains, and to lock them in mechanically bonded daisy chain rotaxanes. When they exist, we will also highlight their mechanical actuation by internal switching upon the influence of external stimuli (photons, electrons, temperature, pH, metal ions, solvent, or even strand displacement in the case of DNA-based structures). The second and third sections will be dedicated to polymers made of acyclic and cyclic daisy chains, respectively. In both cases, we will discuss the synthetic strategies and the experimental conditions employed for their polymerization. We will also highlight the physico-chemical characterizations of their size, their structure, and their shape. Finally, we will discuss their physical (functional) properties as soft and potentially stimuli-responsive materials.
Molecular daisy chains
A large number of [a1] molecular units have been used to build cyclic and acyclic daisy chains.27 Most examples rely on either an α-cyclodextrin host which interacts with hydrophobic stilbene or azobenzene guests, or on a crown ether macrocycle capable of hosting a viologen rod or a secondary ammonium moiety. Alternative molecular hosts consist of pillar[5]arene, calix[4]arene or isophtalamide units. Typical chemical design to avoid the entropically favored and often undesired [c1] conformation is to locate the binding site of the axle sufficiently close to the macrocycle, to make use of relatively rigid axles, and to play with concentration. Among the various molecular daisy chains, some can be conformationally modified by using external stimuli such as light, pH, metal ions, and solvent effects, that trigger internal motions within the daisy chain structure.28 In order to precisely control the conformation of the mechanical bond, chemists have designed daisy chain architectures with stoppers and at least two binding stations on the thread, and whose affinity for the macrocyclic host can be fully differentiated upon external stimulation. A seminal example is the bistable [c2]daisy chain described by the group of Sauvage in 2000, which allows a reversible contraction motion of ∼27% by switching between metal ions of different coordination geometries.25 In this section, we want to highlight the molecular daisy chains which have been developed since the 2013 review article of Mayor and Rotzler dedicated to this topic.27 In particular, we will focus on the use of new building blocks and new synthetic approaches for daisy chain architectures, as well as on some new stimuli used to induce their motion.In 2019, the group of Mayor reported the first synthesis of a [c2]daisy chain in an aqueous solution.29 The monomer consisted in a “Diederich”-type cyclophane covalently connected by a Huisgen [3+2] cycloaddition reaction to an oligophenylene-ethynylene axle, which could be synthesized in ten steps (longest linear sequence) from commercially available compounds (entry 1, Table 1). Despite the presence of two quaternary ammonium on the macrocycle and two carboxylate anions on the axle, the addition of a co-organic solvent such as methanol was mandatory to enable solubility of the monomer in water. In order to isolate interlocked species possibly present in a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of water and methanol, the authors proposed to stopper the end of the axle by a bulky group using a Huisgen cycloaddition reaction. HPLC/ESI-MS analysis of the resulting mixture demonstrated the presence of three main species: the stoppered monomer, the [a2]daisy chain and the [c2]daisy chain as major component, which could be isolated in 30% yield after preparative reverse phase HPLC. Further 1D and 2D NMR experiments including DOSY NMR as well as optical spectroscopies and HRMS confirmed the self-threaded nature of the daisy chain. The presence of a short linker between the macrocycle and the axle entropically favored the formation of the cyclic architecture over the acyclic ones.
:1 mixture of water and methanol, the authors proposed to stopper the end of the axle by a bulky group using a Huisgen cycloaddition reaction. HPLC/ESI-MS analysis of the resulting mixture demonstrated the presence of three main species: the stoppered monomer, the [a2]daisy chain and the [c2]daisy chain as major component, which could be isolated in 30% yield after preparative reverse phase HPLC. Further 1D and 2D NMR experiments including DOSY NMR as well as optical spectroscopies and HRMS confirmed the self-threaded nature of the daisy chain. The presence of a short linker between the macrocycle and the axle entropically favored the formation of the cyclic architecture over the acyclic ones.
| Entry | Chemical structure of the molecular unit composing the (pseudo)daisy chain |
|
Formed daisy chains and corresponding yields | Stimuli responsiveness |
|---|---|---|---|---|
| 1 |

|
[a1]: 7.3% | No | |
| [a2]: 3.0% | ||||
| [c2]: 30% | ||||
| 2 |

|
[c2]: 53% | Yes (solvent) | |
| 3 |

|
[c2]/monomer 8:2 in CD3CN (20 mM) |
Yes [c2] to [c1] reversibility (hν) | |
| 4 |

|
[c1]: 22.1% and 4.4% at respectively 1.1 and 8.8 mM in CHCl3 | No | |
| [c2]: 3.1% at 8.8 mM in CHCl3 | ||||
| 5 |
|
[c2]: 32% | Yes (temperature) | |
| 6 |

|
[c2]: 7% | Yes (temperature, redox) | |
| 7 |

|
[c1]: 58% | Yes (redox) | |
| [c2]: 9% | ||||
| [c3]: 2% | ||||
| [a1]: 11% | ||||
| [a2]: 4% | ||||
| [a3]: 1% | ||||
| 8 |

|
Pseudo[c2] | Yes (pH, anion, temperature, solvent) | |
| 9 |
|
[c2]: 58% | Yes (pH) | |
| 10 |

|
[c2]: 82% | Yes (pH) | |
| 11 |

|
[c3]: 38% | No | |
| 12 |
|
X = -Ph- [c4]: 64% | Yes (metal ions) | |
| X = -Ph- ≡ -[c3]: 58% | ||||
| 13 |

|
[c3]: 91–93% | Yes (redox) | |

A few years later, the group of Nishiyama reported the formation of [c2]daisy chain rotaxanes from a permethylated α-cyclodextrin (PM α-CD) monomer in aqueous solution (entry 2, Table 1).30 The monomer which consists of a diarylacetylene axle covalently connected to a PM α-CD was synthesized in five steps from α-CD. In a 2:1 mixture of methanol and water, formation of a pseudo-[c2]daisy chain occurred which could be capped by an amidation reaction between the carboxylic acid located at the end of the axle and a bulky aniline. 1H NMR spectroscopy as well as optical spectroscopy experiments confirmed the threaded nature of the interlocked architecture as demonstrated, for instance, by the different absorption maximum between the [c2]daisy chain and the stoppered monomer. Depending on the nature of the solvents, fluorescence experiments further suggested the presence of extended and/or contracted architectures.
However, the absence of a second station precluded a precise control of this molecular motion. In 2021, the group of Schalley reported the synthesis of a pseudo-[c2]daisy chain rotaxane based on the ammonium/benzo-21-crown-7 recognition motif (entry 3, Table 1).31 The synthesis of the monomer was achieved in 8 steps from commercially available compounds. In acetonitrile, formation of the pseudo-[c2]daisy chain occurred almost completely as determined by 1D and 2D 1H NMR spectroscopy. Interestingly, in such polar solvents, irradiation at 365 nm of the arylazopyrazole photoswitch located on the axle between the crown ether macrocycle and the ammonium station was shown to induce a conversion from the pseudo-[c2] architecture to the pseudo-[c1]daisy chain (also known as lasso-type pseudo-[1]rotaxane). Irradiation of the lasso architecture at 530 nm allowed the recovery of the [c2]daisy chain architecture.
Such reversible process was also achieved in the gas phase as determined by tandem and ion-mobility mass spectrometry. In 2022, the group of Berna reported a templated approach to produce both a lasso compound and a pseudo-[c2]daisy chain architecture based on the fumaramide/tetralactam macrocycle recognition motif (entry 4, Table 1).32 By increasing the concentration for the templated reaction, the amount of [c2]daisy chain increased compared to the [c1]architecture (1/1.4 at 8.8 mM vs. 0/1 at 1.1 mM). 1H NMR spectroscopy demonstrated the cyclic nature of the dimeric structure (over the acyclic one) while DOSY NMR spectroscopy confirmed the higher hydrodynamic radius of the [c2]architecture over the [c1]. However, although two light-responsive stations (fumaramide and maleamide) were present on the axle of the daisy chain architecture, light irradiation under various conditions only led to the formation of a complex mixtures of isomers, and no contraction/extension event could be observed precisely. In 2019, our group reported the synthesis of the first and so far unique example of unsymmetric bistable [c2]daisy chain rotaxane (compound 1, Scheme 1).33 The design consisted in a pH-sensitive [c2]daisy chain blocked on one extremity by a triarylamine unit as electron donor and on the other extremity by a perylene acceptor. In order to reach the expected unsymmetric compound from the symmetric pseudo-[c2]daisy chain, the sequential approach was preferred over the one-pot procedure, as purification procedures were easier. Using a series of 1H and DOSY NMR, we demonstrated the reversible actuation of the daisy chain upon pH modulation. Interestingly, cyclic voltammetry experiments showed different electrochemical properties for the contracted and extended [c2]daisy chains, showing the potential of such mechanically interlocked molecules as pH-sensitive electrochemical sensors. Chemists have also recently designed molecular daisy chains made of biologically relevant building blocks, e.g. DNA and peptides. In 2016, the group of Famulok reported the first example of mechanically interlocked [c2]daisy chain rotaxane based on double-stranded DNA nanostructures (Fig. 2a).34,35 The synthesis consisted in two double-stranded macrocycle-axle macromolecules with complementary single-stranded gaps on each macrocycle and axle, so that, threading occurs through DNA hybridization. In addition, the presence of single-strand sticky ends at the end of each axle allowed to stopper the pseudo-[c2]daisy chain architecture with bulky DNA groups thus preventing its dethreading. Formation of the hybridized [c2]daisy chain rotaxane was confirmed by AFM experiments. Importantly, upon addition of release oligodeoxynucleotides (ROs), each macrocycle was released from the axle thus leading to the formation of a mechanically interlocked architecture, i.e. without hybridization between the two dumbbells which could slide one along the other. By combining fluorescence and AFM experiments, the authors showed that contraction and extension events occur over ∼6 nm.
 | ||
| Scheme 1 (a) One pot versus (b) sequential synthetic approach for the synthesis of the asymmetric [c2]daisy chain rotaxane 1. | ||
 | ||
| Fig. 2 (a) Schematic representation of the formation of a [c2]daisy chain rotaxane based on double-stranded DNA nanostructures, ROs: release oligodeoxynucleotides. Reproduced with permission from ref. 35. Copyright 2019 Springer Nature. (b) Schematic representation of the actuation of a [c2]daisy chain rotaxane based on DNA origamis using fuel and antifuel staples, bp: base pairs, (i) TEM image of the contracted NanoMuscle along with some unclosed monomers, (ii) TEM image of the DNA origami NanoMuscle after addition of Fuel staples, consisting mostly in extended NanoMuscle and unclosed monomers. Adapted with permission from ref. 36. Copyright 2020 the Royal Society of Chemistry; (c) Chemical structure of a 21-amino acid lasso peptide and its rearrangement into a [c2]daisy chain rotaxane after trypsin digestion and air oxidation. | ||
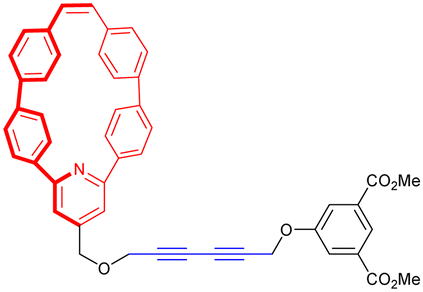
In 2020, the groups of Chern and Jiang reported the formation of mechanically interlocked [c2]daisy chain rotaxanes based on DNA origamis (Fig. 2b).36 Their design consisted in the dimerization of a small monomer and a large monomer by using 10-base pair complementary staples located close to the opened macrocycles. The large monomer had two recognition units on its axle which were blocked by staples during the synthesis, whereas the small monomer included two recognition sites on the macrocycle which could recognize each of the stations of the large monomer. After dimerization of the two monomers, closure of the macrocycles was achieved using ring-closing staples. Syntheses of the different units and of the [c2]daisy chain architecture were monitored by agarose gel electrophoresis as well as TEM experiments. In order to actuate the daisy chain, fuel or antifuel staples were added so that only one of the stations located on the axle of the large monomer was available for recognition by the macrocycle of the small monomer. TEM imaging showed that, using such staples, reversible sliding of the [c2]daisy chain occurred with a net dimension going from 310 nm in the extended state to 160 nm in the contracted one. This example describes the first controlled actuation of a [c2]daisy chain made of DNA origamis. In 2021, the group of Link reported the dynamic covalent synthesis of mechanically interlocked molecules including a [c2]daisy chain based on peptidic fragments (Fig. 2c).37 The synthesis started from a 21-amino acid lasso peptide which could be biosynthetically engineered with cysteine residues. In aqueous solution, a one-pot procedure including a trypsin cleavage followed by an air-driven oxidation afforded a molecular [c2]daisy chain in 80% yield. The preferential formation of the pseudo-[c2]daisy chain was ascribed to steric and conformational constraints related to the positioning of the cysteine residues. Depending on their position, various kinds of mechanically interlocked molecules, such as [3]rotaxane, [n]catenane (n = 2–5) or even double-lasso macrocycles could be synthesized. This work represents the first example of pseudo-[c2]daisy chain architecture made of peptidic fragments. It is noteworthy to mention that this evolution towards biologically based building blocks not only diversifies the nature of the used chemical structures, but also offers the opportunity to control motion by precisely encoded effectors, and to amplify the amplitude of the motions between the low nanometer regime to the hundreds of nanometers.
While most synthetic routes designed to access daisy chain architectures rely on a stepwise approach, the groups of Qu and Coutrot independently reported new self-sorting strategies to build such interlocked architectures (Fig. 3).38–40 In 2016, Qu and co-workers described a synthetic approach based on three building blocks: one DB24C8 macrocycle decorated with a dibenzylammonium recognition site and a terminal alkyne on the axle, one secondary ammonium axle decorated by a phenyl group on one side and by an azide unit on the other, and a B21C7 crown ether macrocycle (Fig. 3a(i)). Importantly, the dibenzylammonium unit was chosen as a selective station for the DB24C8 compared to B21C7 in order to favor the formation of the pseudo-[c2]daisy chain. The synthesis of [c2]daisy chain rotaxane 2, which includes two [2]rotaxane units on each side close to the stopper, was achieved in more than 50% yield starting from the three components in the presence of copper catalyst. HRMS complemented by 2D NOESY NMR experiments confirmed the interlocked nature of the [c2]daisy chain. Some years later, the authors designed a similar strategy to reach the same kind of [c2]daisy chain hetero[4]rotaxane architecture (Fig. 3a(ii)).40 In this example, the axle connected to the DB24C8 contained already the two recognition sites, namely a dibenzylammonium station and a benzylalkylammonium, while the other components were B21C7 and benzylazide. The synthesis of the targeted mechanically interlocked architecture 3 was achieved in a one-pot fashion with 43.1% overall yield. However, in both cases, no actuation of the daisy chain was reported. Shortly after the initial work of Qu, the group of Coutrot reported a self-sorting strategy to build either a [c2]daisy chain (4) or a hetero[4]rotaxane (5) architecture from the same building blocks.39 Their design consisted also in three building blocks: a DB24C8 macrocycle linked to a benzylalkylammonium station and terminated by an azide unit, a bulky di-tert-butylanilinium terminated by an alkyne moiety and a DB24C8 (Fig. 3b). The self-sorting strategy relied on the different affinities of the DB24C8 for benzylalkylammonium, anilinium, and triazolium moieties.41 In the absence of DB24C8, the one pot reaction between the benzylalkylammonium-containing DB24C8 and the anilinium moiety led, in the presence of copper catalyst, to the formation of the [c2]daisy chain 4. Methylation of the triazole moieties synthesized during the one-pot synthesis allowed the formation of the third station. Using pH modulation, actuation of the [c2]daisy chain was achieved between the benzylalkylammonium and the triazolium stations, as demonstrated by 1H NMR experiments. In the presence of DB24C8, the self-sorting synthetic strategy led to the formation of the [c2]daisy chain hetero[4]rotaxane architecture 5. After methylation of the triazole moieties, motion of the DB24C8 from the anilinium to the triazolium was achieved using pH modulation. However, in this kind of hetero[4]rotaxane architecture, no actuation of the daisy chain was possible regardless of the conditions used. Nevertheless, this work describes the first example of bistable [c2]daisy chain architectures synthesized using a self-sorting strategy. Such strategies, as already demonstrated in various supramolecular and dynamic covalent systems, are particularly useful to save time and compounds compared to stepwise approaches.42,43 Another alternative synthetic strategy developed recently by the group of Jasti consists in an active metal template synthesis of [c2]daisy chains (entry 5, Table 1).44 Such approach has been widely developed for the synthesis of mechanically interlocked architectures but was never applied to cyclic daisy chains.45,46 The synthesis of the daisy chain was achieved in one step by a Cadiot-Chodkiewicz templated reaction involving copper(I) under diluted conditions from a nanohoop macrocycle containing a pyridine unit functionalized by an alkyne moiety and a 3,5-diester functionalized bromo-alkyne. Unexpectedly, single X-ray crystallography experiments suggested that the conformation of the interlocked structure in the solid state is contracted. Furthermore, temperature-dependent 1H NMR experiments (1H and 1D ROESY) suggested that actuation of the [c2]daisy chain can be achieved thermally, from a contracted form at low temperatures to an extended one at higher ones. Although the distance of actuation was quite limited due to the short size of the axle, this work demonstrates the possibility to build [c2]daisy chains without particular affinity between the macrocycle and the axle thanks to the active template approach and to use temperature as a stimulus for their actuation.
 | ||
| Fig. 3 (a) One-pot self-sorting synthetic strategies developed by the group of Qu and Tian to reach [c2]daisy chain hetero[4]rotaxane architectures from three individual components; (b) (i) Individual building blocks used by the group of Coutrot to build (ii) either a [c2]daisy chain rotaxane or a [c2]daisy chain hetero[4]rotaxane using a one pot self-sorting strategy. | ||
Actually, the use of temperature to actuate [c2]daisy chains had already been shown by the group of Stoddart in 2014 (entry 6, Table 1).47 In this work, they reported the first example of bistable redox-responsive [c2]daisy chains consisting of napthlene diimide and triazole as stations for naphto[35]crown-10 macrocycles. The synthesis of the daisy chain was achieved in 6 steps from commercially available compounds albeit in low yield (7%), along with the non-interlocked monomer as main product (68%). The preferential formation of this molecule was attributed to stabilizing charge-transfer interactions between the NDI and the naphthyl groups, as determined by absorption spectroscopy. Variable-temperature NMR experiments demonstrated that, at temperature as low as 228 K, the chloroform solution consists of a 4:3 mixture of extended and contracted daisy chains while, at 335 K, only the extended architecture remains. Interestingly, using cyclic voltammetry, the authors showed that, upon electrochemical reduction, formation of the NDI diradical cation occurs leading to the motion of the macrocycles to the triazole stations and thus to a contraction of the daisy chain. Importantly, this process was reversible upon further oxidation. This example highlights the possibility to control the motion of a [c2]daisy chain by different stimuli. Few months later, the same group reported the formation of another redox switchable [c2]daisy chain involving viologen and 1,5-dinapthalene stations as guests for tetracationic cyclophane cyclobis(paraquat-p-phenylene) macrocyclic hosts (entry 7, Table 1).48 The monomer which consists of a cyclobisparaquat-p-phenylene so called Bluebox macrocycle linked to an axle incorporating the viologen and the naphthyl groups and terminated by an azide was synthesized in seven steps from commercially available 4,7-dimethylisoindoline-1,3-dione. Under copper-catalyzed Huisgen [3+2] cycloaddition reaction in acetone and in the presence of an alkyne-terminated bulky stopper, formation of a lasso-type compound ([c1]daisy chain) occurred in 58% yield. Importantly, this compound was accompanied by the formation of [a1], [a2] and [a3] as well as [c2] and [c3]daisy chains, which could be isolated in 11, 4, 1, 9 and 2% yield, respectively. Formation of the different daisy chains was confirmed by combining HRMS with spectroscopy and electrochemistry experiments. For instance, for [c1], [a2] and [c2], the authors observed the presence of an absorption band at ∼530 nm which is characteristic of a charge transfer event between the Bluebox and the 1,5-dialkoxynaphtalene as expected for these daisy chain architectures. The [c2]daisy chain, which was synthesized in its contracted state, could be reversibly extended in acetonitrile upon electrochemical reduction, which favors the formation of the Bluebox bis-radical cation that encircles viologen radical cations thanks to spin-pairing interactions. However, the limited amount of [c2]daisy chain obtained according to these pathways have precluded their applications so far.
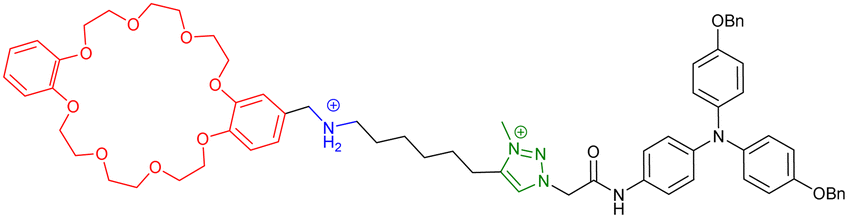
However, some groups reported the use of molecular [c2]daisy chain architectures as sensors for various physical and chemical effectors.33,49,50 In 2014, Yang and coworkers reported a fluorescent sensor based on a pillararene modified with an anthracene moiety (entry 8, Table 1). In moderately polar solvents such as chloroform, the pillararene derivative tends to form a [c2]daisy chain architecture which displays a strong fluorescence at ∼420 nm, due to limited motions of the anthracene moieties. Interestingly, upon heating or the addition of base such as DBU, the fluorescence decreases or is even turned off, as a result of the dissociation of the daisy chain architecture. Reversibility of the process was achieved upon cooling or addition of acid and characterized by 1D 1H and 2D DOSY NMR experiments. Shortly after, our group reported the self-assembly of [c2]daisy chain rotaxanes into bundled fibrils of several microns in length when decorated with supramolecular polymerizing units such as triarylamines (entry 9, Table 1).50 A pH-responsive pseudo-[c2]daisy chain rotaxane, initially developed by the group of Coutrot,51 was decorated with triarylamide monoamide as stopper thanks to a Huisgen cycloaddition reaction. Methylation of the resulting triazoles and further light irradiation of the mechanically interlocked molecule in a chlorinated solvent led to the formation of hierarchical micrometric structures, which could be imaged by TEM. Interestingly, when the mechanical bond was actuated with sodium hydroxide as base, disruption of the self-assembled structure occurred, because of steric hindrance caused by the shuttling of the macrocycle towards the amide function necessary for the self-assembly of the triarylamine moieties. Overall, this mechanically active self-assembled system behaves as a logic gate using a combination of pH and light as inputs. Finally, in 2018, the group of Qu and Tian demonstrated the possibility to control the plasmonic response of gold nanoparticles using individual [c2]daisy chain rotaxane molecules (entry 10, Table 1).52 For that, they decorated gold nanoparticles with pH-responsive [c2]daisy chain rotaxanes based on the secondary ammonium/triazolium recognition motifs for DB24C8 in order to control the distance between the nanoparticles. Interestingly, when the rotaxane was in a contracted state, a blue shift of the plasmonic response of ∼6.2 nm was monitored compared to the extended state. This process was shown to be reversible upon addition of acid to induce the extension of the mechanically interlocked molecules.
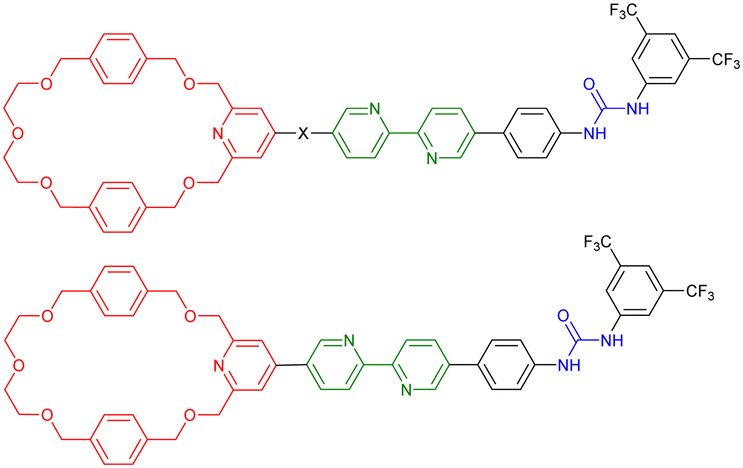
The formation in high yields of higher order cyclic daisy chains, i.e. with a number of monomeric units larger than 2, remains challenging although it allows motion in 2D and 3D unlike individual [c1] and [c2]daisy chains. In 2000, the group of Harada reported the first example of such a [c3]daisy chain architecture (entry 11, Table 1).53 The monomer consists of a 6-4-aminocynnamoyl α-CD which was synthesized in 2 steps. In the presence of 2,4,6-trinitrobenzenesulfonic acid sodium salt (TNBS) as bulky stoppers, formation of the [c3]daisy chain architecture occurred in aqueous solutions as determined by 1D and 2D NMR as well as MALDI-TOF experiments. However, this structure lacks the presence of a second station, necessary to reach bistable entities. Since this original work, only few groups focused on the synthesis of such interlocked architectures and to date, there are only two examples of bistable higher order [cn]daisy chains.54–56 The first example reported by the group of Chiu in 2017 was developed using a combination of covalent design and coordination chemistry (entry 12, Table 1).54 The chemical structure of the hermaphroditic monomers consists in a modified crown ether macrocycle covalently linked to a bipyridine-pyridine linker. In the presence of zinc ions, and depending on the distance between the macrocyclic unit and the linker, the selective formation of either [c4] or [c3]daisy chains was achieved after capping with bis(trifluoromethylphenyl) bulky stoppers. The formation of these higher order daisy chains structures was confirmed using a combination of 1D and 2D NMR spectroscopy, mass spectrometry and X-ray crystallography. Removal of the zinc ions using N,N,N′,N′-tetrakis(2-pyridylmethyl)ethylenediamine as competing ligand allows the reversible actuation of these [cn] architectures, with an increase in volume of ∼18–23% upon zinc complexation. In the absence of zinc, the presence of a diphenylurea station close to the stopper favors the presence of the macrocycle over the station due to hydrogen bonding interactions. On the other hand, the presence of zinc induces chelation at the bipyridine unit from one monomer and at the pyridine located within the macrocycle of the interlocked monomer, leading to a change in volume compared to the non-chelated structure. More recently, the group of Stoddart reported the selective formation of [c3]daisy chain architecture with more than 90% yield thanks to a template-directed strategy (entry 13, Table 1).55,56 They designed a monomer made of a cyclobis(paraquat) host linked to a bipyridinium guest by a single methylene unit. The rigidity of this molecule associated with the presence of hexafluorophosphate anions as template favor nearly exclusively the formation of the non-thermodynamically stable [c3]daisy chain. Further Huisgen [3+2] cycloaddition with a bulky 1,3-diisopropylbenzene stopper afforded the MIM which can be reversibly contracted and extended upon oxidation with nitrosonium hexafluorophosphate (NOPF6) and reduction with zinc dust, respectively. 1D and 2D NMR spectroscopy including diffusion-ordered NMR (DOSY) as well as mass spectrometry confirmed the exclusive formation of the [c3]daisy chain over possible acyclic constitutions and co-conformations. Importantly, the reversible actuation of this MIM was supported by 1H NMR and Vis/NIR spectroscopies which show a downfield shift of the proton of the triazole units and the appearance of an absorption band at 1070 nm upon reduction, respectively. These observations are in agreement with the presence of the macrocyclic hosts over the bipyridinium stations. Finally, the [c3]daisy chain system could be actuated reversibly using an electrochemical stimulus, i.e. a controlled potential electrolysis setup, up to five times without degradation.
Polymers made of acyclic daisy chains
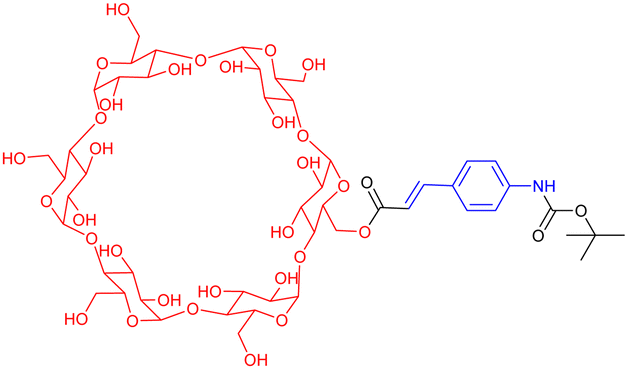
In the previous section, we have described well-defined entities made of discrete daisy chain architectures. Over the last twenty years, chemists have also considered the integration of these interlocked species by polymerization into macromolecules. Focusing on acyclic daisy chain polymers, two kinds of such architectures can be found in the literature: (a) the so-called supramolecular daisy chain polymers, which are indeed host–guest supramolecular polymers without mechanical bond but with an acyclic daisy chain structure and (b) acyclic daisy chain polymers with a mechanical bond at each repeating unit. The first supramolecular acyclic daisy chain polymers were reported in the late 90's concomitantly by the groups of Prof. Stoddart and Prof. Gibson (entries 1 and 2, Table 2).57,58 These examples involve viologen – crown ether complexes as the supramolecular polymerization motif. In the case of Stoddart, the use of liquid secondary ion (LSI) mass spectrometry confirmed the formation of oligomeric structures up to the pentamer for some rigid heteroditopic monomers. Interestingly, for a very similar structure of the monomer, and in agreement with the formation of a supramolecular polymer, the group of Gibson reported the evolution of the degree of polymerization (determine by 1H NMR spectroscopy) as a function of the concentration of the solution. They showed that at low concentration (below 1 mM in acetone), no polymer is formed. However, when increasing the concentration, the degree of polymerization increased from ∼2 units at 10 mM up to 50 units at 2 M in acetone. The formation of supramolecular acyclic daisy chain polymers was also achieved using the α-cyclodextrin-Boc-cinnamoyl host–guest recognition motif (entry 3, Table 2).59 The polymerization was performed at concentrations between 15 and 40 mM in water, reaching polymers with a number average molecular weight ranging from 16000 to 20000 (more than 15 monomers) as determined by VPO (vapor pressure osmometry) experiments. Furthermore, turbo ion spray TOF mass spectroscopy experiments confirmed the presence of a polymer with a distance between signals corresponding to the mass of the monomer. Using two complementary heteroditopic monomers, namely an α-cyclodextrin covalently linked to an adamantane group and a β-cyclodextrin covalently linked to a Boc-cinnamoyl group, Harada and co-workers managed to synthesize supramolecular copolymers made of α-CD and β-CD in a perfectly alternating manner (entry 4, Table 2).60 This is due to the high affinity between adamantane and β-CD on one hand and between α-cyclodextrin and Boc-cinnamoyl on the other hand. VPO and 1H NMR experiments confirmed the polymeric nature of the structure with a molecular weight of ∼10000 at 10 mM, corresponding to ∼5 AB alternating motifs. Some years later, the group of Huang reported the polymerization of heteroditopic monomers involving the bis(m-phenylene)-[32]crown-10-based cryptand – paraquat host guest recognition motif (entry 5, Table 2).61 The monomer was designed with a long flexible alkyl chain between that cryptand and the paraquat moieties. At low concentration (typically 2.7 mM in acetonitrile), the monomer formed a lasso-type ([c1]daisy chain) architecture. For concentrations between ∼17 and ∼89 mM, 1H NMR studies as well as viscosimetry experiments showed the presence of both cyclic oligomers and linear polymers. However, for high concentrations (i.e. more than 96.8 mM), the unique presence of linear polymers with high molecular weight was observed. The molecular weight of the synthesized polymers was not determined but the decrease in diffusion coefficient observed by DOSY NMR and high increase in viscosity for concentration higher than the critical polymerization concentration (CPC estimated to 75 mM) confirmed the high molecular weight of the supramolecular acyclic daisy chain polymer. As a step toward the formation of materials using these supramolecular acyclic daisy chain polymers (see below), the authors showed by using NOESY and DOSY NMR as well as viscosimetry that the linear supramolecular polymers could be reticulated using palladium as chelating agent for two triazole moieties from two different linear chains. The group of Huang also reported the formation of supramolecular acyclic daisy chain polymers using copillararene modified with a hexane side chain (entry 6, Table 2).62 Similarly to the previously mentioned study, the authors showed that at low concentrations, the formation of a lasso-like structure is preferred. However, as concentration increases, the formation of linear supramolecular polymers over cyclic oligomers prevails, reaching polymers with a molecular weight of ∼31.2 kDa, corresponding to ∼37 monomer units, at 768 mM in chloroform. The formation of the supramolecular polymers was confirmed by a strong decrease of the diffusion coefficient in DOSY NMR, a sharp rise of the viscosity for concentrations higher than the CPC as well as the observation of rod-like fibers with a large diameter by scanning electron microscopy. Furthermore, the interlocked nature of the polymer was confirmed by X-ray analysis of a single crystal of the polymer obtained by slow evaporation of a chloroform/methanol solution. However, although the topology of these supramolecular polymers is an acyclic daisy chain, they lack the presence of a mechanical bond. In fact, to date, only few examples of acyclic daisy chain polymers involving mechanical bonds have been reported. The first example dates back to 2003 by the group of Hadziioannou (entry 7, Table 2).63 This work is based on a stepwise polymerization of a bifunctional monomer. Following their initial work in which they reached a mechanically interlocked dimer,64 the authors proposed to sequentially desymmetrize their monomer so that esterification between the carboxylic acid located on the macrocycle and the phenol group on one stopper of the axle could take place selectively. Following this stepwise approach, the authors managed to synthesize a tetramer which already shows a molecular weight of ∼10000. In order to reach higher molecular weight polymer without encountering problems of cyclization of their monomer, the authors proposed to polymerize their acyclic daisy chain dimer with free phenol and carboxylic acid groups using a mixture of DCC, DMAP and p-toluenesulfonic acid in acetonitrile. Using this approach and static light scattering to determine the molecular weight using a Zimm plot analysis, the authors reported the formation of a polymer in high yield (80%) with a molecular weight of ∼400000, thus corresponding to the presence of ∼75 monomers. However, the stepwise approaches to make the [2]rotaxane monomer in low yield (0.6% over six steps (longest linear sequence)) and the dimer (59% yield over 2 steps from the [2]rotaxane monomer) precluded further use of this polymer. Some years later, the group of Takata achieved the synthesis of a mechanically interlocked acyclic daisy chain polymer by Sonogashira polycondensation of a bifunctional [2]rotaxane (entry 8, Table 2).65 This monomer was synthesized in eight steps from commercially available compounds. The crown ether macrocycle was obtained in five steps from catechol following a sequence of O-alkylation, tosylation, cesium-templated macrocyclisation, halogen exchange and Sonogashira coupling. Then, an axle containing a secondary ammonium, which has a high affinity for the dibenzo-24-crown-8 macrocycle (DB24C8), was threaded and capped with 3,5-dimethylphenyl isocyanate to reach the [2]rotaxane architecture. Further acylation of the secondary ammonium station with 4-iodobenzoyl chloride and deprotection of the acetylene unit reached the desired monomer in excellent yields. Sonogashira polycondensation of the [2]rotaxane monomer under classical Pd(II), Cu(I) conditions afforded a polymer which precipitates from methanol. 1H NMR experiments confirmed the disappearance of the acetylenic proton and an upfield shift of the proton of the phenyl ring involved in the Sonogashira coupling. The presence of mass signals with repeating units corresponding to the [2]rotaxane and a Mn estimated to 6850 (Đ = 1.59) confirmed the formation of a polymer, albeit with a limited number of repeating units (6–7). The authors also studied the thermal properties of the synthetic macromolecule with a glass transition temperature at ∼100 °C and a degradation temperature at 5% loss of 302 °C, indicating the thermal stability of the mechanical bonds. Shortly after, the group of Harada reported the formation of acyclic daisy chain polymers involving α-cyclodextrin derivatives (entry 9, Table 2).66 The 3-amino-α-cyclodextrin was modified in two steps at the amine position with para-amino-cinnamic acid. This compound was studied in water and the authors found that at 40 mM, the formation of a poly-pseudo-[2]rotaxane occurred, as determined by extensive 1D and 2D NMR studies. Further reaction with adamantane carboxylic
acid in the presence of DMTMM, a triazine-based activating agent, led to the formation of the corresponding acyclic daisy chain polymer with an estimated molecular weight up to ∼10000 and an interval between signals corresponding to the molecular weight of a monomer, as determined by MALDI-TOF. However, no further characterization of this polymer was reported to date. In 2011, the group of Huang succeeded in the formation of an acyclic daisy chain polymer using DB24C8 as host and viologen as guest with a much higher molecular weight than previously reported examples (entry 10, Table 2).67 The synthesis was achieved following a one-pot strategy consisting of a threading followed by polymerization. On one hand, the bis(pyridinium) guest was synthesized in three N-alkylation steps starting from 3,5-dimethylpyridine and using sequentially 1,2-dibromoethane, 2,2′-bipyridine and 2-iodoethanol. On the other hand, the DB24C8 macrocycle guest decorated with an alkyl chain terminated by a carboxylic acid moiety was reacted with thionyl chloride to form the corresponding acyl chloride. Host–guest complexation between these two units was performed at low temperature (−70 °C) to limit esterification before formation of the complex. Then the reaction temperature was raised and triethylamine was added, resulting in the formation of the acyclic daisy chain polymer which has a brown color arising from charge transfer interactions between DB24C8 and bis-pyridinium units. Characterization of the interlocked structure was achieved using 1H, COSY and NOESY NMR experiments. GPC experiments using polystyrene as standard indicated a molecular weight of 65 kDa, corresponding to ∼45 repeating units, and a dispersity of 1.5. Additional experiments using dynamic light scattering (DLS) and UV-Vis experiments confirmed the large size of the macromolecule, as indicated by a large hydrodynamic radius (∼250 nm) and a large increase in absorbance compared to a single inclusion complex. In 2019, the group of Yang reported the formation of dendrimers incorporating [an]-type daisy chains (Fig. 4a).68,69 The synthesis was achieved starting from a [2]rotaxane consisting of an axle containing an urea station and a pillar[5]arene wheel (Fig. 4a).68 The presence of a terminal alkyne unit on the axle allowed reaction with a platinum complex Pt(PEt3)2I2 to form the corresponding organometallic [2]rotaxane. Further copper-catalysed reaction with 1,3,5-triethynylbenzene allowed the formation of the first generation dendrimer which contains three [2]rotaxane units. The presence of two TIPS-protected alkyne units on each wheel of the [2]rotaxane allowed for the formation of dendrimers of higher generation with mechanical bonds reminiscent of [an]-type daisy chains. Characterization of the fourth-generation dendrimer by chromatographic, spectroscopic and microscopic techniques confirmed the monodisperse nature of the macromolecular entity with a molecular weight of ∼105 kDa, in agreement with the presence of 46 rotaxanes units on the scaffold. Interestingly, in THF, these dendrimers proved to be sensitive to acetate anions due to the presence of urea stations. Importantly, DOSY NMR, DLS and AFM experiments confirmed the anion-sensitive reversible extension/contraction of the dendrimers in THF with swelling ratio increasing for higher generation of dendrimers, in agreement with the collective integration of individual motion within the dendritic structure. More recently, the group of Stoddart reported the formation of mechanically interlocked acyclic daisy chain polymers using a molecular pump which relies on a redox-mediated energy ratchet mechanism (entry 11, Table 2).70 The design of the monomer was based on previous work involving the pumping of cyclobis(paraquat-p-phenylene) (CBPQT4+) units on an axle composed of a 3,5-dimethylpyridinium head and a neighboring bipyridinium station.17 For the formation of the acyclic daisy chain polymer, a monomer made of a CBPQT unit as one extremity covalently linked via a flexible linker to a bipyridinium terminated by a 3,5-dimethylpyridinium was designed. Synthesis of the 3,5-dimethylpyridinium – bipyridinium head was achieved in 3 steps from commercially available 3,5-dimethylpyridine while synthesis of the CBPQT derivatives functionalized with a terminal alkyne unit was obtained in five steps from 4,7-dimethyl-2,3-dihydro-1H-isoindole-1,3-dione. Further [3+2] Huisgen cycloaddition between these two molecules afforded the bifunctional monomer in a very good yield (81%). Polymerization of this monomer was then achieved in a 25 mM solution of acetonitrile using zinc dust as reducing agent, followed by the addition of silver sulfate as oxidizing agent to generate the mechanically interlocked polymer. The molecular mass was estimated to Mn ∼ 25 kDa based on 1H NMR experiments, in particular on the protons of the p-phenylene units close to the bipyridinium, which are shifted upfield when the BCPQT4+ host is located there (as in the case of the mechanically interlocked polymer). This mass corresponds to a polymerization degree (DP) of ∼11 units which is lower than the DP calculated from the association constant (∼15.4). This lower DP is probably due to a reduced efficiency of the molecular pumping with increasing size of the polymer. Interestingly, upon addition of zinc dust followed by slow diffusion of air in the reaction mixture after filtration, the monomer could be recycled almost purely. This work demonstrates the possibility to access mechanically interlocked polymers with high recycling efficiency. Very recently, the group of Yan reported that mechanically interlocked polymers based on acyclic daisy chains can be used to form materials with emergent dynamic and mechanical properties.71 The acyclic supramolecular daisy chain obtained by host guest complexation was stoppered using a thiol–ene click reaction with a mecapto-functionnd polydimethylsiloxane (PDMS). Interestingly, using a combination of advanced mechanical analysis and molecular modeling, the authors showed that the enhanced macroscopic mechanical performances of the material are directly related to the synergistic motion of the [an]daisy chain at the molecular level. This work represents the first example of the integration of mechanically interlocked [an]daisy chain in a material and paves the way to the development of polymer materials with emergent properties. Indeed, to date, mostly supramolecular acyclic daisy chain polymers have been reported to form gel-like structures with stimuli responsive properties.72–75 These examples rely on the formation of linear supramolecular polymers by host–guest complexation. Three host–guest systems have been reported: one based on the DB24C8 – dibenzylammonium complex (entry 12, Table 2),72 one on the benzo-21-crown-7 – secondary benzylalkylammonium recognition motif (entry 13, Table 3),73,74 and one on the viologen – pillar[5]arene interaction (entry 14, Table 2).75 In all cases, the formation of linear supramolecular polymers with high molecular weights was demonstrated using a combination of 1H and DOSY NMR spectroscopy, and viscosity measurements. For relatively high concentrations (see CPC in Table 2), solutions of the heteroditopic monomers formed physical gels, consisting in long entangled fibers made of bundles of the linear supramolecular polymers as established by optical and/or SEM microscopy. For the gel made of the DB24C8 – dibenzylammonium complex (entry 12, Table 2),72 reversible gel–sol transition have been observed by changing either the temperature, a well-known trigger for physical gels made of supramolecular polymers,76 or the pH. Indeed, secondary ammonium stations can be deprotonated upon addition of a base, leading to the disassembly of the polymer due to the absence of host–guest complexes, which can be restored by adding acid. In the case of the benzo-21-crown-7 – benzylalkylammonium host guest complex,74 reticulation of the linear supramolecular polymers using palladium ions was necessary to achieve the formation of a gel (entry 13, Table 2). Here, reversible gel–sol transitions could be triggered by four different stimuli: temperature, pH, cations and organic molecules. Indeed, when potassium hexafluorophosphate or triphenylphosphine is added, dissolution of the gel occurs as B21C7 macrocycles and palladium have a better affinity for potassium and triphenylphosphine, respectively. Overall, these examples highlight the possibility to use external triggers to influence the properties of materials. Similar responsiveness could be expected for materials made of purely acyclic daisy chain polymers involving mechanical bonds. We should however highlight the recent combination of permanently cross-linked dynamic polymers networks (so called vitrimers)77 with mechanically interlocked daisy chains.78 In this work, the group of Yan reports the synthesis of a polymer network starting from a [2]rotaxane monomer which is further reticulated with diamine and trisamine via vinylogous urethane linkages (Fig. 4b). One particularity of the rotaxane monomer is the presence of a side chain on the macrocycle which participates in the network formation. This material was compared with a control vitrimer based on a pseudo-[2]rotaxane which has no side chain but a macrocycle capable of sliding under load. Various mechanical experiments including stress–strain and cyclic tensile tests showed better properties for the vitrimer made of [2]rotaxanes compared to the control, including higher Young's modulus, toughness, maximum stress and strain and a fast deformation recovery ability. These enhanced properties have been attributed to the presence of mechanically interlocked motifs which can be sensitive to external physical force in a reversible manner. The conjugation of mechanical bonds with dynamic covalent ones provides these vitrimers with advanced reprocessing and recycling properties and open the door for the development of innovative materials based on sliding daisy chains.
| Entry | Chemical structure of the thread composing the polymer |
|
Experimental conditions | Nature of the polymer and degree of polymerization (DP) |
|---|---|---|---|---|
| 1 |

|
2.5 mM in acetonitrile | Supramolecular daisy chain | |
| DP: 5 (Liquid secondary ion mass spectrometry) | ||||
| 2 |

|
2.0 M in acetone | Supramolecular daisy chain | |
| DP: 50 (1H NMR) | ||||
| 3 |
|
15 to 40 mM in water | Supramolecular daisy chain | |
| DP: 16000 to 20000 (vapor pressure osmometry, Turbo-ion spray TOF) |
||||
| 4 |

|
10 mM in water | Supramolecular daisy chain | |
| DP: ∼ 10000 (vapor pressure osmometry) |
||||
| 5 |

|
>96.8 mM in acetonitrile | Supramolecular daisy chain | |
| n.d. | ||||
| 6 |
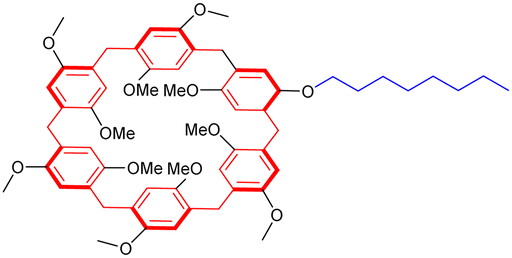
|
768 mM in CHCl3 | Supramolecular daisy chain | |
| ∼37 (1H NMR) | ||||
| 7 |

|
Stepwise esterification | Acyclic daisy chain | |
| ∼75 (Static light scattering) | ||||
| Đ: 2 (assumed considering a polycondensation) | ||||
| 8 |

|
Sonogashira polycondensation | Acyclic daisy chain | |
| 6–7, Đ = 1.59 (Gel permeation chromatography) | ||||
| 9 |

|
Amidation (capping of the supramolecular acyclic daisy chain) | Acyclic daisy chain | |
| Up to ∼9 (MALDI-TOF mass spectrometry), no dispersity | ||||
| 10 |

|
Threading followed by polycondensation | Acyclic daisy chain | |
| ∼45, Đ = 1.5 (Gel permeation chromatography) | ||||
| 11 |

|
Redox-mediated energy ratchet mechanism (molecular pumping) | Acyclic daisy chain | |
| ∼11 (1H NMR), no dispersity | ||||
| 12 |

|
Critical polymerization concentration (CPC): 60 mM in CH3CN | Supramolecular daisy chain | |
| n.d. | ||||
| 13 |

|
CPC: 55 mM in CHCl3 | Supramolecular daisy chain | |
| Critical gelation concentration (CGC): 30 mM | n.d. | |||
| 14 |

|
CPC: 15 mM in CH2Cl2 | Supramolecular daisy chain | |
| CGC > 25 mM in CH2Cl2 | n.d. | |||

 | ||
| Fig. 4 (a) Schematic representation of a dendrimer made of [an]-type daisy chains based on a pillar[5]arene wheel and its reversible actuation using acetate anions. Reproduced with permission from ref. 69. Copyright 2021 American Chemical Society; (b) schematic representation of mechanically interlocked vitrimers obtained by polymerizing a [2]rotaxane and diamine and trisamine monomers and their reprocessing properties under thermal stimulation. Reproduced with permission from ref. 75. Copyright 2022 American Chemical Society. | ||
| Entry | Chemical structure of [c2] daisy chain polymers |
|
Degree of polymerization and dispersity | Reversible contraction/extension |
|---|---|---|---|---|
| 1 |

|
1–5 (GPC, MALDI-TOF) | Yes (light) | |
| No dispersity (each oligomer was isolated as a single molecule) | ||||
| 2 |

|
2–5 (1H NMR, MALDI-TOF) | Yes (light) | |
| No dispersity (each oligomer was isolated as a single molecule) | ||||
| 3 |

|
6–7 (1H NMR) | No | |
| No dispersity (poor GPC results precluded the determination of Mn) | ||||
| 4 |

|
∼22, Đ = 2.94 (GPC) | No | |
| 5 |

|
∼11, Đ = 1.84 (SEC-MALLS) | Yes (pH) | |
| 6 |
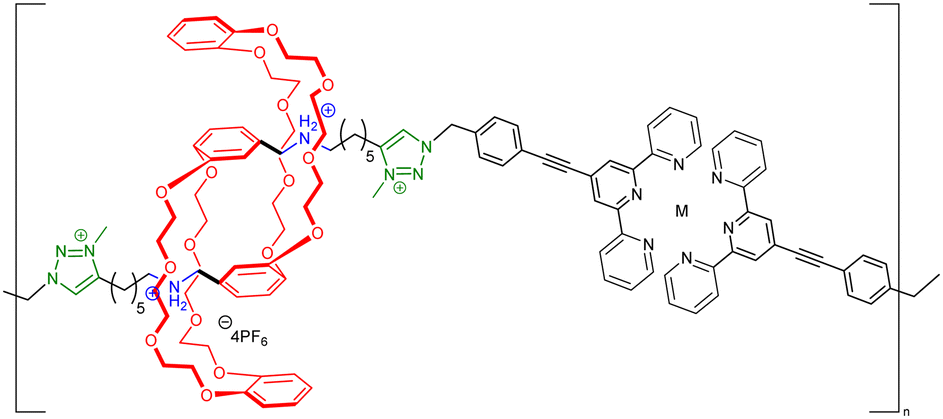
|
∼2000 for M = Zn | Yes (pH) | |
| ∼3000 for M = Fe | ||||
| (Static light scattering/Small angle neutron scattering) | ||||
| No dispersity (not available with these techniques) | ||||
| 7 |

|
n.d. | Yes (solvent) | |
| 8 |
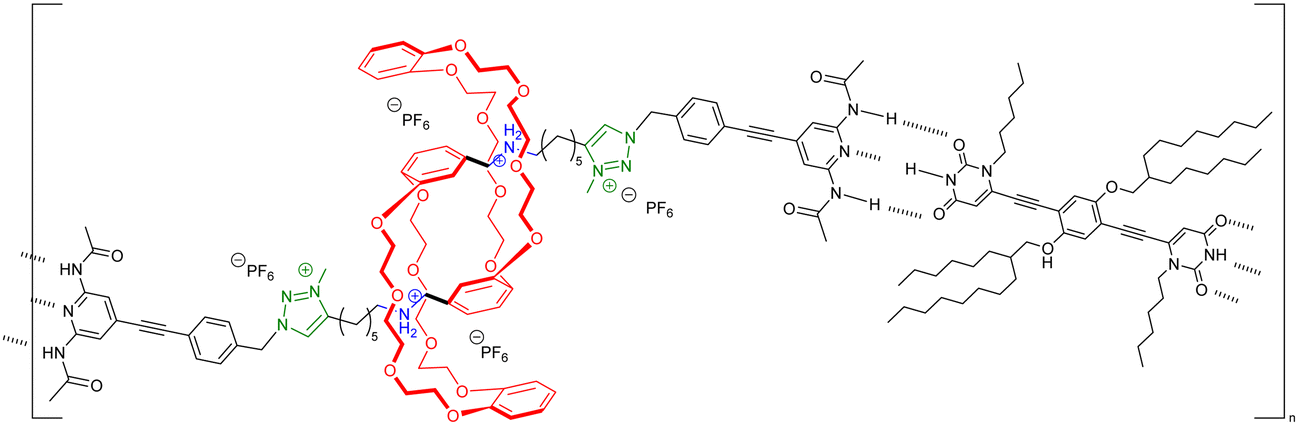
|
n.d. | Yes (pH)) | |
| 9 |

|
n.d. | No | |
| 10 |

|
22 for the contracted polymer (SANS) | Yes (pH) | |
| No dispersity (not available with this technique) | ||||

Polymers made of cyclic daisy chains
Before considering the polymerization of [c2]daisy chain rotaxanes, we want to highlight the work on Takata which consists in incorporating one individual [c2]daisy chain architecture into long polymer strands to produce cyclic polymers in an efficient way.79 In 2017, this group designed and synthesized a pseudo-[c2]daisy chain unit in which the axles could be elongated by ring opening polymerization of ε-caprolactone and capped using 3,5-dimethylphenyl isocyanate (Fig. 5a). Interestingly, after deprotonation of the secondary ammonium stations which hold the [c2]daisy chain architecture, and acetylation of the resulting secondary amine, sliding of the crown ether macrocycle over the urethane station close to the linker occurred, resulting in the formation of a cyclic polymer in high yield. The cyclic nature of the polymer incorporating one [c2]daisy chain unit at its center was confirmed by MALDI-TOF spectrometry and GPC experiments, which showed a lower hydrodynamic radius for the cyclic macromolecule compared to the linear one, in agreement with precedents from the literature.80 Interestingly, this synthetic strategy afforded multigram-scale quantities of the cyclic polymer with a very good dispersity (Đ = 1.1). Subsequently, the same group reported the reversible linear to cyclic transformation of their polymer using pH modulation in solution (Fig. 5b).81 The same linear polymer with the [c2]daisy chain at its center was N-methylated at the secondary station in order to reach its cyclic counterpart. Interestingly, upon addition of ammonium hexafluorophosphate, formation of a tertiary N-methyl ammonium occurs, resulting in the formation of a new station which has a higher affinity for the crown ether macrocycle compared to the urethane linkage, thus displaying a cyclic to linear transformation. The topological transformation of the macromolecule could be repeated up to 5 times without noticeable degradation and without loss of dispersity (Đ = 1.07 over the 5 cycles) as supported by a series of NMR and GPC experiments. In particular, 19F NMR spectroscopy shows a downfield shift of the fluorine signal when the macrocycle is located on the urethane station, and DOSY NMR spectroscopy which confirms the higher diffusion coefficient for the cyclic polymer compared to the linear one. In addition, the topological change at the molecular level due to the actuation of the mechanical bond was visible after drying the polymer, as determined by DSC experiments. Indeed, the cyclic structure showed a glass transition temperature at −46 °C and a melting temperature at ∼28 °C while its linear counterpart showed a slightly lower Tg (−52 °C) and no melting peak up to 150 °C. This difference in the material originates from a lower crystallinity of the polycaprolactone part in the linear topology compared to the cyclic one. This work shows how conformation of one single daisy chain unit can induce changes at the mesoscopic level. | ||
| Fig. 5 (a) Synthetic route towards cyclic polymers built on a pseudo-[c2]daisy chain unit, (b) reversible pH-triggered actuation of a cyclic polymer based on a [c2]daisy chain unit and corresponding diffusion coefficient (D) and hydrodynamic radius (RH) of the polymers as determined by DOSY NMR experiments (500 MHz, CDCl3, 298 K). | ||
To the best of our knowledge, the first example reporting the polymerization of [c2]daisy chain rotaxanes dates back to 2006. In this work, Kaneda and co-workers reported the formation of [c2]daisy chain oligomers (up to the pentamer) by polycondensation between p-bis(bromomethyl)benzene and a light-responsive [c2]daisy chain made of α-cyclodextrin and containing an azobenzene station (entry 1, Table 3).82 Upon UV irradiation, isomerization of the azobenzene moiety to the Z isomer lead to its exclusion from the cyclodextrin cavity thus inducing a contraction of the oligomers. However, the efficiency of the light-induced contraction was shown to decrease with increasing size of the oligomer due to the presence of only 5% of Z,Z-isomers for the monomer at the photostationary state. More recently, using identical host and guest moieties, the group of Easton reported the one-pot synthesis of [c2]daisy chain oligomers (up to the pentamer) from an α-cyclodextrin/azobenzene conjugate in the presence of an activated ester of suberic acid (entry 2, Table 3).83 Interestingly, this one-pot strategy also afforded cyclized [c2]daisy chain architectures composed of one or two interlocked molecules. For all isolated species, light-induced contraction and extension occurred, albeit with lower degree of isomerization at the photostationary state in water compared to methanol solutions. Besides interlocked architectures based on α-cyclodextrin and azobenzene, most examples reporting the formation of [c2]daisy chain macromolecules involve the crown ether/secondary ammonium host/guest recognition motif. For instance, in 2007, the groups of Stoddart and Grubbs reported the formation of a [c2]daisy chain monomer by complexation of a secondary ammonium station with an open crown ether macrocycle which was then closed by ring closing metathesis using Grubbs I catalyst (entry 3, Table 3).84 The resulting double bond was then reduced using platinum oxide and hydrogen, and the axle was modified with 4-pentenoic acid at the end chains. The terminal alkene units present on the daisy chain axles were then engaged in an ADMET polymerization process leading to the formation of an oligomer with a molecular weight estimated by 1H NMR spectroscopy to 13000 g mol−1, i.e. 6 to 7 monomer units. However, no actuation of this synthetic macromolecule was reported, probably because of the absence of a second station on the monomer. Further efforts to actuate polymers made of cyclic daisy chains based on the same host–guest recognition motif were pursued concomitantly by the groups of Stoddart and Grubbs. In 2009, Grubbs and co-workers reported the synthesis of a [c2]daisy chain monomer according to a procedure similar to the former one (entry 4, Table 3).85 In that case, the end groups were modified by azide units instead of ethylene. Polymerization was achieved by copper-catalyzed [3+2] Huisgen cycloaddition with 1,4-diethynylbenzene, providing a macromolecule with a molecular weight (Mw) of ∼48000 g mol−1 and a large dispersity of 2.94 as determined by GPC experiments. Interestingly, deprotonation of the ammonium station resulted in a macromolecule with a slightly lower radius of gyration (13.5 nm vs. 14.8 nm), confirming the interlocked nature of the macromolecule. Acetylation of the secondary ammonium station led to an increase of the size of the polymer as determined by MALLS. However, similarly to the former example, the reversible actuation of the macromolecule was not possible due to the lack of a second binding station. The same year, the group of Stoddart reported the synthesis of a bistable [c2]daisy chain monomer with a bipyridinium and a secondary ammonium unit as stations (entry 5, Table 3).86 The synthesis of this interlocked monomer was achieved by reacting 4-pyridylpyridinium moieties functionalized with an alkyne end chain and a pseudo-[c2]daisy chain bearing benzyl bromide end groups. The corresponding [c2]daisy chain monomer was then polymerized with 1,4-bis(2-azidoethoxy)benzene in the presence of copper iodide in DMF at 60 °C. The resulting mechanically interlocked polymer was characterized by chronocoulometry and DSC experiments, confirming the formation of a macromolecule compared to the monomer. SEC-MALLS analysis provided information on the size and dispersity of the polymer, i.e. 32.9 kDa, corresponding to ∼11 monomer units, and 1.85, respectively. The reversible switching of the polymer between extended and contracted states using pH modulation was achieved over more than 10 cycles as demonstrated by UV/Vis absorption spectroscopies. Using stopped-flow spectrophotometry measurements, the authors evidenced the faster actuation of the polymer compared to the monomer, suggesting cooperative kinetic effects for the shuttling of the macrocyclic host within the macromolecule. However, in that case, despite the bistability of the [c2]daisy chain, the low degree of polymerization precluded the amplification of motion to higher length scale. To achieve such objective, our group reported the polymerization of bistable [c2]daisy chain rotaxanes using supramolecular metal–ligand interactions (entry 6, Table 3).87 The monomer consisted in a pH-responsive [c2]daisy chain architecture with terpyridine end groups on each axle for further polymerization using metal ions. A pseudo-[c2]daisy chain consisting of a DB24C8 macrocycle threaded with a secondary ammonium station51 was functionalized on its terminal alkyne units with terpyridine ligands by copper-catalyzed [3+2] Huisgen cycloaddition, leading to the formation of triazole moieties on the daisy chain axle, which could be used as second station after methylation. The reversible switching of the [c2]daisy chain monomer using base for contraction and acid for extension was confirmed using 1H NMR spectroscopy. Polymerization occurred using either Zinc(II) or Iron(II) metal ions as monitored by UV-Vis absorption experiments which show the appearance of metal-to-ligand charge transfer bands characteristic of terpyridine complexes. Determination of the characteristic structural parameters of the polymers was achieved by combining neutron and light scattering experiments. For the polymerization using zinc(II) ions, we manage to synthesize a contracted polymer consisting of ∼1970 units, corresponding to a contour length of ∼8.9 μm. However, upon addition of acid to induce the extension of the [c2]daisy chain, partial depolymerization occurred, because of the weak association constant between zinc and terpyridine ligands. However, in the case of iron(II) ions, scattering experiments proved the formation of single worm-like chain polymers for both the extended and the contracted states. In both cases, a degree of polymerization of ∼2940 units was observed corresponding to a theoretical contour length of ∼10.5 and ∼14.1 μm for the contracted and extended polymers, respectively. These values are in good agreement with the experimental contour length of ∼9.4 and ∼15.8 μm for the contracted and extended polymers, respectively. The difference between the theoretical and experimental values can be explained by the coordinated telescopic motion of all the [c2]daisy chain monomers as well as a possible folding of the terpyridine stoppers, as determined theoretically by quantum chemical and molecular dynamics calculations.88 In addition, DFT calculations by the group of Zhao confirmed the superiority of iron(II) ions over other divalent transition metal ions to reach a high polymerization degree.89 Overall, this work demonstrated the first amplification of the nanoscale motion of [c2]daisy chain monomers to the micrometric scale by the integration of these molecular machines within a polymer. Using the same Fe2+/terpyridine coordination chemistry to induce polymerization, the group of Huang reported the formation of [c2]daisy chain polymers made of copillar[5]arene units as macrocyclic hosts (entry 7, Table 3).90 The synthesis of the [c2]daisy chain monomer was achieved from a copillar[5]arene functionalized by a decylamine chain, carbonyl diimidazole and an amine-functionalized terpyridine in chloroform. The formation of the self-included dimeric structure is favored by CH⋯π interactions between the alkyl chain and the aromatic units of the pillar[5]arene as well as van der Waals interactions between the decyl chain of each monomer involved in the formation of the interlocked structure. Interestingly, formation of the pseudo-[c2]daisy chain architecture unusually occurs in the contracted state and subsequent capping with terpyridine afford the desired monomer. Contraction and extension of the monomer as a function of solvent polarity was carried out using 1H NMR spectroscopy. In non-polar solvent such as chloroform, the monomer stays in its contracted state, while the addition of a polar
solvent such as DMSO leads to the formation of the extended state. Scattering experiments on the polymer in solvents of increasing polarity showed increasing hydrodynamic radius, in agreement with an extension of the [c2]daisy chain monomer in polar solvents such as DMSO. TEM and SEM imaging of the polymers in various ratio of solvents showed in all cases the presence of large fibrillar aggregates but without noticeable changes related to the polarity and thus to the actuation of the interlocked monomer. To achieve changes at the microscopic level as a function of the state of the [c2]daisy chain monomer, our group reported the copolymerization of a bistable [c2]daisy chain monomer functionalized with 2,6-bisacetamidopyridine stoppers and a complementary dumbbell consisting of phenylene-1,4-ethynylene modified with uracil derivatives as end groups (entry 8, Table 3).91 The synthesis of the [c2]daisy chain monomer was achieved according to a procedure similar to the one previously described for terpyridine-modified pH-responsive [c2]daisy chain. Synthesis of the complementary dumbbell was performed in five steps from commercially available 2-hexyl-1-decanol involving mostly Sonogashira couplings as key steps. Copolymerization of the two monomers in a 1:1 ratio occurred in a 4:1 mixture of chloroform and acetonitrile. Formation of an alternated polymer was confirmed by 1H NMR experiments which show a downfield shift of the protons involved in hydrogen-bonding interactions. Interestingly, AFM and TEM imaging experiments indicated the presence of different morphologies at the micrometric scale between the extended and the contracted polymers. The extended ones consisted of micrometer long soft entangled fibers while the contracted ones led to discrete coffee bean-like objects with a much smaller length (200–400 nm), albeit both were made of laterally aggregated single chain polymers. In both cases, the formation of hierarchical structures arises from non-covalent interactions between the single-chain polymers due to the presence of the conjugated linker which can be involved in π–π stacking and van der Waals interactions. The change in morphology for the contracted state is probably due to a higher rigidity of the [c2]daisy chain monomer as well as an increased steric hindrance close to the linker which disfavors lateral aggregation. Thus, the difference of morphology between the two states at the microscopic level is a direct consequence of the actuation of the mechanical bond at the molecular scale. As an alternative to the use of a chemical effector such as metal ions or a complementary linker to induce the polymerization of [c2]daisy chain monomers, the group of Qu and Tian reported the use of light as physical trigger (entry 9, Table 3).92 For that, they designed a [c2]daisy chain monomer based on DB24C8 hosts and secondary ammonium guests with ureidopyrimidinone (UPy) end groups which were protected by photocleavable coumarin protecting groups. Its synthesis was achieved by Huisgen cycloaddition between a pseudo-[c2]daisy chain terminated with alkyne groups and a protected UPy functionalized by an azide group. Upon photodeprotection of the [c2]daisy chain monomer at 365 nm, polymerization occurred as confirmed by 1H NMR spectroscopy, DLS experiments, viscosity measurements as well as microscopy experiments. Interestingly, the presence of triazole units on the polymer allowed coordination of palladium ions, which induces the formation of a polymer network leading to a sol-to-gel transition in acetonitrile. An increase of the temperature or the use of triphenylphosphine as a competitive ligand could trigger the recovery of a solution state. However, in this example, the monomer presented only one station, thus preventing actuation at the molecular scale due to the lack of bistability. Shortly after, our group reported that sol-to-gel transition can be achieved by actuating the mechanical bond of [c2]daisy chain polymers (entry 10, Table 3).93 The molecular system consisted in the previously mentioned bistable pH-responsive [c2]daisy chain capped with UPy end groups. Importantly, UPy end groups had two important features: (1) their functionalization with gallic acid moieties incorporating long alkyl chains in order to improve solubility and control aggregation, and (2) their protection with a 2-nitrobenzyl photolabile group to control the polymerization of the mechanically interlocked molecules. The synthesis of the [c2]daisy chain monomer consisted first in the synthesis of the azide-functionalized UPy derivative which was achieved in ten steps from methyl gallate at the gram scale. This compound was then engaged in a copper-catalyzed [3+2] Huisgen cycloaddition with the already used pseudo-[c2]daisy chain functionalized with terminal alkyne units. Actuation of the protected [c2]daisy chain was confirmed by 1H NMR spectroscopy which shows a downfield shift of the peaks close to the triazolium ring upon deprotonation. Polymerization of the daisy chain monomer in aromatic solvents such as toluene or xylene occurred after shining UV light with a 150 W xenon-Mercury lamp having a 320–375 nm filter. Importantly, this photodeprotection led to the formation of a gel for the extended monomer while a solution was obtained for the contracted one. Furthermore, when the gel was treated with a base such as triethylamine, formation of a solution occurred. This process was fully reversible upon addition of acid. To rationalize changes observed at high length scale, we performed a series of physical characterization including calorimetry and scattering experiments. Isothermal titration calorimetry experiments on a solution of the contracted polymer in toluene allowed us to determine an association constant of ∼1.1 × 104 M−1, which is almost 3 orders of magnitude lower than the expected association constant for UPy motifs in this solvent.94 This low association constant is in agreement with the low degree of polymerization (22 units) of the single chain polymer determined by small angle neutron scattering experiments. For the extended polymer, the scattering profile is in agreement with the formation of a highly organized gel as demonstrated by the presence of structural peaks and the intensity increase at high and low q values, respectively. The main structural peaks were assigned to the inter-reticulating distance (7.3 nm) and the average size of the reticulating units (2.7 nm), which consists in helical stacks of UPy dimers. Importantly, the absence of these structural peaks in the scattering profile of the contracted polymer suggests that the presence of the crown ether macrocycle close to the UPy moieties in the contracted state precludes their stacking and thus the reticulation of the single-chain polymers which lead to the formation of a gel in the extended state. Thus, the sol-to-gel transition observed at the macroscopic scale for the polymer material is a direct consequence of the actuation of the mechanical bond at the molecular level upon pH modulation. However, once more, integrity of the supramolecular polymer could not be maintained when actuating the [c2]daisy chain rotaxane. To circumvent this problem, our group and others considered the formation of a covalent network made of these molecular machines to achieve motion at the macroscopic scale. Two types of [c2]daisy chain rotaxane units have been employed to build soft materials which could sustain the actuation of individual molecular machines. In 2016, the group of Harada reported the formation of hydrogels and xerogels based on light-responsive [c2]daisy chain rotaxanes (Fig. 6a).95 They first described the synthesis in four steps of an α-cyclodextrin (α-CD) monomer modified on its large rim at the C-3 position by an (aminopropyl diethylene glycol)-4-azobenzene unit. Interestingly, this molecule forms spontaneously a [c2]daisy chain architecture in water and could be polymerized with a tetra-PEG polymer by a polycondensation reaction between the activated succinimidyl ester end groups of the polymer and the amine end group of the [c2]daisy chain. Mechanical characterization of the corresponding hydrogel confirmed its chemically crosslinked nature. Irradiation of a cubic piece of the hydrogel with UV light led to its contraction while visible light irradiation induced its expansion and recovery of its initial volume (Fig. 6a(ii)). Stress–strain measurements showed similar rupture stress, increased rupture strain and decreased Young's modulus for the contracted hydrogel compared to the extended one. These observations agree with a sliding of the cyclodextrin moieties on the PEG units when the trans azobenzene cannot fit in the α-CD after UV irradiation, confirming that the macroscopic motion is directly related to the molecular actuation of the [c2]daisy chain units. Reversible actuation of thin plates of hydrogel was achieved over at least 10 cycles without hysteresis (Fig. 6a(iii)). Interestingly, while maximum bending of the gel required over 3 hours under UV irradiation, return to the initial state occurred almost instantaneously under visible light irradiation. Such difference was attributed to the enhanced molecular recognition
of the α-CD for the trans-isomer and the weight of the gel itself. Similar bending experiments were performed on the xerogel (Fig. 6a(iv)). However, in that case, although actuation using UV light was more than 10000 times faster than for the corresponding hydrogel, visible light irradiation was ineffective to restore the initial state. Nevertheless, UV irradiation from the opposite side compared to the first irradiation allowed to recover the initial shape in a pseudo-reversible process. The different behavior compared to the hydrogel is attributed to the weak hydrophobic interaction between α-CD and azobenzene in the dry state which is one driving force for the actuation mechanism in solution. Finally, the authors demonstrated the use of this xerogel to lift a weight and determined a mechanical work of ∼0.12 μJ. To overcome the pseudo-reversibility issue encountered with the xerogel, the same group reported the use of stilbene as station for the α-CD instead of azobenzene (Fig. 6b).96 In that case, the α-CD monomer consisted in an α-CD modified on its small rim at the C-6 position by a 4,4′-stilbenedicarboxylic acid, which was synthesized in one step from commercially available compounds. Similarly to the azobenzene monomer, this modified α-CD readily formed in water a pseudo-[c2]daisy chain, which was polymerized via a polycondensation reaction with a tetra-arm PEG modified on its end chains by amino groups. The resulting hydrogel could be reversibly contracted and expanded using UV-A (λ = 350 nm) and UV-C (λ = 280 nm) respectively (Fig. 6b(ii)). Importantly, bending occurred much faster than with the azobenzene monomer. Indeed, for a thin plate of hydrogel, maximum bending occurred within ∼7 s vs. 3 h for previous work (Fig. 6a(iii)). Overall, upon UV-A irradiation, the stilbene switched mostly to its cis form which lead to its removal from the α-CD cavity which slides onto the PEG chains, resulting in a contraction event. Further irradiation with UV-C induces expansion of the hydrogel, resulting from the trans isomerization of the stilbene which can sit again within the α-CD. The mechanical properties of the hydrogel were similar to the ones observed in their former work. Interestingly, when longer PEG chains were used to form the hydrogel, UV-A irradiation led to higher contraction volume as well as higher Young modulus and fracture energy. These observations agree with the proposed mechanism related to the sliding of the α-CD over the PEG chains upon irradiation at 350 nm. Furthermore, reversible binding of the dry gel was achieved in a similar manner to the hydrogel and could be repeated up to 15 times with a maximum efficiency observed for the longer PEG chains (Fig. 6b(iv)). This reversibility was ascribed to the hydrophobic property of the stilbene compared to the azobenzene which facilitates the host–guest complexation in the dry state. Finally, a 150 time larger mechanical work was determined for this system compared to the previous one, as demonstrated by the ability of the gel to lift a weight that is 15 times larger than its own. In parallel to light-responsive materials, our group reported the covalent polymerization of pH-responsive [c2]daisy chain rotaxanes (Fig. 7a).97 The previously mentioned bistable pH-responsive [c2]daisy chain with terminal alkyne groups was polymerized with azide-terminated ditopic and tritopic polyethylene oxide molecules by Huisgen cycloaddition, leading to the formation of a chemical gel in acetonitrile. It was then soaked in a 1:1 mixture of acetonitrile/methyl iodide, allowing methylation of the triazole groups and thus providing bistability to the mechanically interlocked architecture. After treatment with sodium hydroxide as a base, contraction of the gel down to 60% of its original volume at the macroscopic scale occurred, arising from the molecular motion of the crown ether macrocycles from the secondary ammonium to the triazolium groups (Fig. 7a(ii and iii)). Addition of ammonium hexafluorophosphate induced protonation of the secondary amine and led to the recovery of the size of the gel as the crown ethers switch back to their original position. The reversible actuation could be repeated several times without noticeable degradation (Fig. 7a(iv)). Importantly, a gel lacking triazolium stations showed only negligible variations of its volume upon pH modulation, demonstrating the importance of the bistability of the [c2]daisy chain for the macroscopic motion. To understand the macroscopic motion at the molecular scale, we performed high-resolution magic angle spinning (HR-MAS) NMR and small angle neutron scattering (SANS) experiments on both the contracted and the extended gels. On the one hand, actuation of the molecular [c2]daisy chain unit was characterized by an upfield shift of the triazolium protons in the extended gel compared to the contracted one, in agreement with 1H NMR data recorded in solution for similar systems.91,93 On the other hand, SANS experiments showed two similar scattering profile for both the extended and the contracted gel, except a difference in the scattered intensity in the mid-q region (Fig. 7a(v)). This observation is particularly important as a higher intensity in this q-range correspond to a higher linear mass density of the polymer chains. Experimentally, in this q-region, the scattered intensity was higher for the contracted gel than for the extended one, in agreement with a higher linear mass density for the contracted gel compared to the extended one, as already observed for single chain polymers.87 Overall, all these data demonstrate that the pH-responsive macroscopic actuation is a direct consequence of the molecular motion of the [c2]daisy chain embedded in a polymer network. Very recently, the group of Yan reported the formation of polymer materials consisting of [c2]daisy chains in order to correlate microscopic responsiveness with macroscopic mechanical properties (Fig. 7b).98 In order to decouple the effect of either the contracted or the extended state, polymer networks were lacking the second station necessary for bistability. A common [c2]daisy chain monomer was synthesized on a multi-gram scale by a Huisgen cycloaddition reaction from the pH-responsive pseudo-[c2]daisy chain terminated with alkyne groups and an azide derivative terminated on the other end by an alkene group. For the extended monomer, the resulting triazole was not methylated while, for the contracted one, the secondary ammonium was acetylated. Formation of the polymer networks occurred by thiol–ene reaction between each of these monomers and trimethylolpropane tris(3-mercaptopropionate). By combining simulation with large-amplitude-oscillatory shear experiments, the authors demonstrated that, at the microscopic scale, the polymer network made of extended monomers undergoes an elastic deformation associated with a short-range sliding motion which can recover quickly after release, while a long-range sliding motion associated with a slow recovery of the deformation is measured for the polymer network with a contracted conformation. These observations result in materials with enhanced ductility and ability for energy dissipation for the contracted one while the extended polymer network displayed higher strength, elasticity and creep resistance, as determined by tensile and creep experiments. This study provides a better understanding of the mechanical properties of contractile materials made of [c2]daisy chain architectures. Recently, the groups of Yang and Qian described the first polymer material made of DNA-based daisy chain rotaxanes (DNA-DCR).99 After confirming the nanoscale motion of the DNA-DCR by fluorescence experiments, a polyacrylamide hydrogel with DNA-DCR as cross-links was synthesized. However, although the swelling degree of the hydrogel and its stiffness could be controlled by the addition of hairpin DNA strands which can initiate hybridization chain reaction (HCR) on the axles of the DNA-DCR, the changes observed at the macroscopic level were not related to a motion of the mechanical bond but to an elongation of the axles by HCR.
 | ||
| Fig. 6 (a) (i) Chemical structure of the polymer network [c2]AzoCD2 arising from the polycondensation of [c2]daisy chain monomers based on α-cyclodextrin with a tetraPEG polymer in an aqueous solution, (ii) photographs of the [c2]AzoCD2 hydrogel before irradiation, after irradiation with UV (λ = 365 nm) and with visible light (λ = 430 nm) for 3 hours, (iii) photographs of thin plates of the [c2]AzoCD2 hydrogel before irradiation, after irradiation with UV (λ = 365 nm) and with visible light (λ = 430 nm) for 3 hours, (iv) photographs of thin plates of the [c2]AzoCD2 xerogel before irradiation and after irradiation with UV (λ = 365 nm) for 3.6 seconds and evolution with time of the flexion angle of the material upon UV irradiation (blue areas). Panels (ii) and (iv) adapted with permission from ref. 95. Copyright 2016 Springer Nature; (b) (i) Chemical structure of the polymer network [c2]αCD-Sti arising from the polycondensation of [c2]daisy chain monomers based on α-cyclodextrin with stilbene stations and a tetraPEG polymer in an aqueous solution, (ii) photographs of the [c2]αCD-Sti hydrogel (10k) before irradiation, after irradiation with UV-A (λ = 350 nm) and with UV-C (λ = 280 nm) for 30 seconds, (iii) evolution with time of the flexion angle of this hydrogel upon sequential UV-A and UV-C irradiation (blue and purple areas, respectively), (iv) evolution with time of the flexion angle of three [c2]αCD-Sti dry gels with different polymer chain lengths (10, 15, 20k) upon sequential UV-A and UV-C irradiation (blue and purple areas, respectively). Panels (ii) and (iv) adapted with permission from ref. 96. Copyright 2018 American Chemical Society. | ||
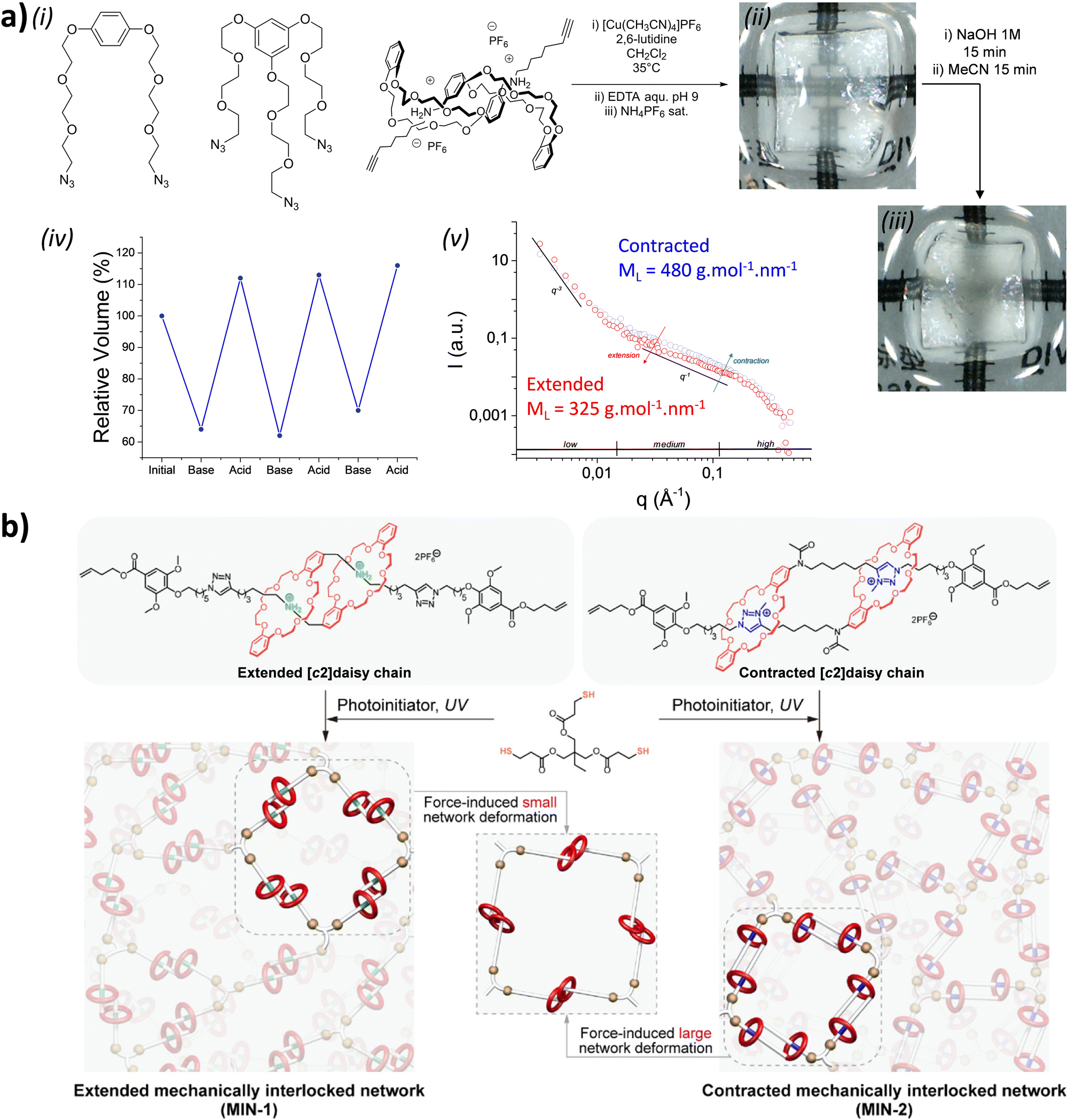 | ||
| Fig. 7 (a) (i) Chemical structure of the monomers used to form a chemical gel which can be swollen in acetonitrile and reversibly actuated by modulating the pH; (ii) and (iii) Corresponding images of the chemical gel (ii) before and (iii) after addition of base to induce contraction; (iv) Evolution of the volume of the chemical gel upon several pH modulation demonstrating the reversibility of the actuation; (v) Small angle neutron scattering (SANS) spectrum of the contracted and extended chemical gels. The difference between the scattering curves in the medium q region demonstrates a difference in linear mass density between the two gels. Adapted with permission from ref. 97. Copyright 2017 American Chemical Society. (b) (i) chemical structures of extended and contracted [c2]daisy chain monomers used for thiol–ene click photopolymerization with trimethylolpropane tris(3-mercaptopropionate) and leading to extended and contracted mechanically interlocked networks (MIN-1 and MIN-2), respectively. Adapted with permission from ref. 98. Copyright 2023 American Chemical Society. | ||
Besides polymers made of repeating [c2]daisy chain monomers, the group of Yang described the formation of [c2]daisy chain dendrimers and their actuation using a chemical stimuli (Fig. 8b).100 The [c2]daisy chain precursor consisted in a pillar[5]arene ring as wheel and either an urea or an alkyl chain as stations, while platinum-acetylide complexes served as stopper. In order to reach higher generation dendrimers, the [c2]daisy chain precursor was mono-functionalized by a 1,3,5-trisethynylbenzene derivative. This bi-functional mechanically interlocked molecule was then reacted in the presence of copper iodide with 1,3,5-tris(4-phenylethynyl)benzene to reach the first generation of [c2]daisy chain dendrimer. Similarly to the formation of [an]-type daisy chain dendrimers, further TIPS deprotection followed by copper-catalysed alkyne coupling of bi-functional [c2]daisy chain monomer allowed the synthesis of higher generation [c2]daisy chain dendrimers. While MALDI-TOF and GPC experiments were sufficient to confirm the formation of the first- and second-generation dendrimer, the third-generation dendrimer could not be characterized by mass spectrometry. Considering that platinum-acetylide units displays two main absorption bands by UV-vis spectroscopy, corresponding to the intra-ligand and metal-to-ligand charge transfer transitions, UV-Vis experiments confirmed the presence of 21 [c2]daisy chain monomers on the third generation dendrimer. Characterization of the reversible actuation of the macromolecular architecture using either acetate anions or DMSO as stimulus was achieved by a combination of DOSY, DLS and SANS experiments, revealing a shrinking ratio as high as 46% for the third-generation dendrimer. This dendrimer was also successfully inserted in a polyvinylidene difluoride matrix leading to a composite polymer film which could be folded quickly and reversibly using solvent modulation. Importantly, changes at the macroscopic level were associated with changes of the radius of gyration of the dendrimer at the nanometric level as determined by SANS experiments.
 | ||
| Fig. 8 (a) Chemical structure and corresponding schematic representation of a [c2]daisy chain which corresponds to the repetition unit of the daisy chain dendrimer. (b) Schematic representation of a third generation [c2]daisy chain dendrimer and its reversible shrinkage using solvent as stimuli. Adapted with permission from ref. 100. Copyright 2020 American Chemical Society. | ||
Conclusion and outlook
As exemplified in this review article, by exploiting the rules and tools of supramolecular chemistry, the synthesis of a large family of daisy chains has been achieved at the molecular and macromolecular levels. The control over these complex architectures is reachable by playing with a number of self-assembly processes, primarily involving classical host–guest recognition, but also more recently by using advanced preorganisation and self-sorting strategies. This review shows that such strategies can be performed not only in organic solvents but also in water, and that all types of recognition units can be exploited, including hydrogen bonds, ion pairing, ion dipole, π-stacking, and hydrophobic interactions. A few number of recent examples, such as non-symmetric [c2]daisy chains, [c3] topologies, heterorotaxanes, lasso and dendritic structures, highlight synthetic progresses made by researchers in the field. It is also of major interest to notice recent advances using peptidic or DNA building blocks (including origamis). The pure fundamental interest of these endeavours opens the way to access biocompatible daisy chain architectures, to change their typical size of actuation from the typical 1 nm scale to the 100 nm scale, and to specifically control their actuation by the displacement of encoded strands. One can expect many variations around – and developments from – these approaches in the future. The achieved threading of various monomers into advanced mechanically interlocked daisy chains is not only a demonstration of the power of supramolecular chemistry to generate complex topologies, but also the opportunity to design nanoscale objects capable of precise internal molecular motion, and which can therefore serve as individual elements for the construction of molecular machines and nanodevices. In that directions, many works have been dedicated to the control of internal mechanical motion by using various stimuli, including solvent composition, temperature, metal ions, pH or light. In addition, recent examples show the use of red-ox potential to drive actuation of the mechanical bond. As for light, electrochemical triggering is particularly relevant because it does not generate waste but these stimuli remain rarely used. As demonstrated recently, such triggers can also be instrumental in introducing ratchetting elements in daisy chain architectures in order to confer them with potentially out-of-equilibrium threading behaviours, capable of pumping and of actively actuating molecules when supplied in energy. This will certainly represent a rich domain of investigation in the coming years.In the second part of this review, we discussed a number of recent demonstrations showing that molecular sliding and molecular actuation of daisy chains can be amplified by the collective movements of these tiny machines when associated in polymer chains and materials therefrom. This global approach confers to these systems fundamentally novel mechanical properties across length scales, with a multitude of possibilities to generate stimuli-responsive soft materials. When these polymers do not contain mechanical bonds, they mainly follow the physical behaviour of classical supramolecular polymers, with their degree of polymerization being dependent on the concentration, temperature, and on the nature of the solvent. They also share with supramolecular polymers their sharp Tg and melting temperature transitions, as well as their self-healing and recycling character due to their equilibration between monomers, rings, and linear chains. Mechanically bonded daisy chain polymers are more original, because they are thermally stable but still integrate sliding and/or actuation properties at the level of the mechanical bonds, a property not contained in regular covalent polymers. This peculiarity confers them with enhanced deformation properties under load. Parameters such as Young modulus, toughness, maximum stress and strain, ability to recover after deformation, ductility and creep resistance have been shown to potentially benefit from daisy chain architectures, possibly in conjugation with vitrimers or in composites. A striking line of research is also exemplified by the polymerization of [c2]daisy chains in linear polymers and dendrimers, leading to the integration of nanomechanical contractions and extensions across length scales up to the formation of macroscopic movements. Such results hold promise to access very innovative responsive materials, and potentially to reach artificial muscles. In this direction, a number of challenges still remain and it is certainly timely to tackle them. In particular, amplification of motions in materials of interest will ideally require to reversibly actuate daisy chains in dry polymer materials such as elastomers. This will require to preserve some mobility of the chains and to use triggers that can diffuse fast enough in the bulk. In that direction, light suffers from a limited depth penetration in the materials, and chemicals generate undesired wastes in the systems. Therefore, electrochemistry seems an interesting perspective if coupled to conducting media, but developments remain to be done in this direction. An important physical understanding of the chain mobility in the dry state, above Tg, and their potential integration with molecular machines also remains to be studied. In principle, the combination of the current knowledge described in the present review article with the state-of-the-art techniques developed in materials engineering will be instrumental to achieve these goals. For instance, the possibility to align polymer chains and to bundle them in hierarchical structures will certainly play a crucial role to obtain optimized materials such as artificial fascicles and muscles. On this way, a combination of complementary analytical tools should be employed to characterize the dynamic structure–function relationship involving the controlled molecular motions and their effects across length scales. It represents a difficult task, but it is also one of the most interesting part of the research to be developed, combining spectroscopic, microscopic, scattering, mechanical and thermal analyses to decipher the multiple interacting behaviours linking the molecular world to the macroscopic one. Our feeling is that these challenges will produce a lot of fundamental knowledge as they will be tackled, with the promise to access advanced responsive materials with improved dynamic capacities and efficiency. Again, further insights in this direction using out-of-equilibrium ratchetting mechanisms will certainly complement this approach in the years to come, with the objective to reach artificial materials that can rival with the complexity and efficiency of mechanically active biological systems.101
Conflicts of interest
There are no conflicts to declare.Note added after first publication
This article replaces the version published on 18th October 2023, which contained incorrect versions of Fig. 3 and 5.Acknowledgements
The authors thank the European Commission's Horizon 2020 Programme as part of the MSCA-ITN project ArtMoMa under grant no. 860434 and the FET-Open project MAGNIFY under grant no. 801378 (fellowship to C. C. C.-V.), the ITI CSC, the CNRS, and the University of Strasbourg. C. C. C.-V. also wish to acknowledge the icFRC (Fondation Jean-Marie Lehn) and the Solvay Company for an excellence doctoral fellowship.Notes and references
- Molecular Catenanes, Rotaxanes and Knots, ed. J.-P. Sauvage and C. Dietrich-Buchecker, Wiley, 1999 Search PubMed.
- S. D. P. Fielden, D. A. Leigh and S. L. Woltering, Angew. Chem., Int. Ed., 2017, 56, 11166–11194 CrossRef CAS PubMed.
- Z. Ashbridge, S. D. P. Fielden, D. A. Leigh, L. Pirvu, F. Schaufelberger and L. Zhang, Chem. Soc. Rev., 2022, 51, 7779–7809 RSC.
- C. J. Bruns and J. F. Stoddart, The Nature of the Mechanical Bond: From Molecules to Machines, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2016 Search PubMed.
- S. A. Nepogodiev and J. F. Stoddart, Chem. Rev., 1998, 98, 1959–1976 CrossRef CAS PubMed.
- E. M. G. Jamieson, F. Modicom and S. M. Goldup, Chem. Soc. Rev., 2018, 47, 5266–5311 RSC.
- E. Moulin, L. Faour, C. C. Carmona-Vargas and N. Giuseppone, Adv. Mater., 2020, 32, 1906036 CrossRef CAS PubMed.
- A. Perrot, E. Moulin and N. Giuseppone, Trends Chem., 2021, 3, 926–942 CrossRef CAS.
- Y. Feng, M. Ovalle, J. S. W. Seale, C. K. Lee, D. J. Kim, R. D. Astumian and J. F. Stoddart, J. Am. Chem. Soc., 2021, 143, 5569–5591 CrossRef CAS PubMed.
- D. Dattler, G. Fuks, J. Heiser, E. Moulin, A. Perrot, X. Yao and N. Giuseppone, Chem. Rev., 2020, 120, 310–433 CrossRef CAS PubMed.
- L. F. Hart, J. E. Hertzog, P. M. Rauscher, B. W. Rawe, M. M. Tranquilli and S. J. Rowan, Nat. Rev. Mater., 2021, 6, 508–530 CrossRef CAS.
- S. Kassem, T. van Leeuwen, A. S. Lubbe, M. R. Wilson, B. L. Feringa and D. A. Leigh, Chem. Soc. Rev., 2017, 46, 2592–2621 RSC.
- M. Baroncini, S. Silvi and A. Credi, Chem. Rev., 2020, 120, 200–268 CrossRef CAS PubMed.
- D. Sluysmans and J. F. Stoddart, Trends Chem., 2019, 1, 185–197 CrossRef CAS.
- D. Thomas, D. J. Tetlow, Y. Ren, S. Kassem, U. Karaca and D. A. Leigh, Nat. Nanotechnol., 2022, 17, 701–707 CrossRef CAS PubMed.
- G. Ragazzon, M. Baroncini, S. Silvi, M. Venturi and A. Credi, Nat. Nanotechnol., 2015, 10, 70–75 CrossRef CAS PubMed.
- C. Cheng, P. R. McGonigal, S. T. Schneebeli, H. Li, N. A. Vermeulen, C. Ke and J. F. Stoddart, Nat. Nanotechnol., 2015, 10, 547–553 CrossRef CAS PubMed.
- S. Mena-Hernando and E. M. Pérez, Chem. Soc. Rev., 2019, 48, 5016–5032 RSC.
- T. Takata, ACS Cent. Sci., 2020, 6, 129–143 CrossRef CAS PubMed.
- G. Liu, P. M. Rauscher, B. W. Rawe, M. M. Tranquilli and S. J. Rowan, Chem. Soc. Rev., 2022, 51, 4928–4948 RSC.
- L. Chen, X. Sheng, G. Li and F. Huang, Chem. Soc. Rev., 2022, 51, 7046–7065 RSC.
- S. J. Rowan, S. J. Cantrill, J. F. Stoddart, A. J. P. White and D. J. Williams, Org. Lett., 2000, 2, 759–762 CrossRef CAS PubMed.
- T. Fujimoto, Y. Sakata and T. Kaneda, Chem. Commun., 2000, 2143–2144 RSC.
- S.-H. Chiu, S. J. Rowan, S. J. Cantrill, J. F. Stoddart, A. J. P. White and D. J. Williams, Chem. Commun., 2002, 2948–2949 RSC.
- M. C. Jiménez, C. Dietrich-Buchecker and J.-P. Sauvage, Angew. Chem., Int. Ed., 2000, 39, 3284–3287 CrossRef.
- A. Goujon, E. Moulin, G. Fuks and N. Giuseppone, CCS Chem., 2019, 1, 83–96 CAS.
- J. Rotzler and M. Mayor, Chem. Soc. Rev., 2013, 42, 44–62 RSC.
- C. J. Bruns and J. F. Stoddart, Acc. Chem. Res., 2014, 47, 2186–2199 CrossRef CAS PubMed.
- Y. Aeschi, S. Drayss-Orth, M. Valášek, D. Häussinger and M. Mayor, Chem. – Eur. J., 2019, 25, 285–295 CrossRef CAS PubMed.
- S. Tsuda, Y. Komai, S. Fujiwara and Y. Nishiyama, Chem. – Eur. J., 2021, 27, 1966–1969 CrossRef CAS PubMed.
- C.-W. Chu, D. L. Stares and C. A. Schalley, Chem. Commun., 2021, 57, 12317–12320 RSC.
- A. Saura-Sanmartin, A. Pastor, A. Martinez-Cuezva and J. Berna, Chem. Commun., 2022, 58, 290–293 RSC.
- A. Wolf, J.-J. Cid, E. Moulin, F. Niess, G. Du, A. Goujon, E. Busseron, A. Ruff, S. Ludwigs and N. Giuseppone, Eur. J. Org. Chem., 2019, 3421–3432 CrossRef CAS.
- J. Weigandt, C. Chung, S. Jester and M. Famulok, Angew. Chem., Int. Ed., 2016, 55, 5512–5516 CrossRef CAS PubMed.
- J. Valero, M. Centola, Y. Ma, M. Škugor, Z. Yu, M. W. Haydell, D. Keppner and M. Famulok, Nat. Protoc., 2019, 14, 2818–2855 CrossRef CAS PubMed.
- Y.-C. Chao, Y.-J. Hong, C.-Y. Lee, S.-C. Zhuang, M.-T. Wu, Y.-Y. Lee, H.-Y. Lee, Y.-S. He, H.-Y. Yu, Y.-Z. Huang, E. Chern and H.-R. Jiang, Nanoscale, 2020, 12, 2992–2998 RSC.
- H. V. Schröder, Y. Zhang and A. J. Link, Nat. Chem., 2021, 13, 850–857 CrossRef PubMed.
- X. Fu, Q. Zhang, S.-J. Rao, D.-H. Qu and H. Tian, Chem. Sci., 2016, 7, 1696–1701 RSC.
- P. Waelès, B. Riss-Yaw and F. Coutrot, Chem. – Eur. J., 2016, 22, 6837–6845 CrossRef PubMed.
- S.-J. Rao, Q. Zhang, X.-H. Ye, C. Gao and D.-H. Qu, Chem. – Asian J., 2018, 13, 815–821 CrossRef CAS PubMed.
- F. Coutrot, ChemistryOpen, 2015, 4, 556–576 CrossRef CAS PubMed.
- M. M. Safont-Sempere, G. Fernández and F. Würthner, Chem. Rev., 2011, 111, 5784–5814 CrossRef CAS PubMed.
- Z. He, W. Jiang and C. A. Schalley, Chem. Soc. Rev., 2015, 44, 779–789 RSC.
- J. M. Van Raden, N. N. Jarenwattananon, L. N. Zakharov and R. Jasti, Chem. – Eur. J., 2020, 26, 10205–10209 CrossRef CAS PubMed.
- J. D. Crowley, S. M. Goldup, A.-L. Lee, D. A. Leigh and R. T. McBurney, Chem. Soc. Rev., 2009, 38, 1530–1541 RSC.
- M. Denis and S. M. Goldup, Nat. Rev. Chem., 2017, 1, 0061 CrossRef CAS.
- C. J. Bruns, J. Li, M. Frasconi, S. T. Schneebeli, J. Iehl, H.-P. Jacquot de Rouville, S. I. Stupp, G. A. Voth and J. F. Stoddart, Angew. Chem., Int. Ed., 2014, 53, 1953–1958 CrossRef CAS PubMed.
- C. J. Bruns, M. Frasconi, J. Iehl, K. J. Hartlieb, S. T. Schneebeli, C. Cheng, S. I. Stupp and J. F. Stoddart, J. Am. Chem. Soc., 2014, 136, 4714–4723 CrossRef CAS PubMed.
- K. Wang, C.-Y. Wang, Y. Zhang, S. X.-A. Zhang, B. Yang and Y.-W. Yang, Chem. Commun., 2014, 50, 9458–9461 RSC.
- A. Wolf, E. Moulin, J. J. Cid Martín, A. Goujon, G. Du, E. Busseron, G. Fuks and N. Giuseppone, Chem. Commun., 2015, 51, 4212–4215 RSC.
- F. Coutrot, C. Romuald and E. Busseron, Org. Lett., 2008, 10, 3741–3744 CrossRef CAS PubMed.
- Q. Zhang, S.-J. Rao, T. Xie, X. Li, T.-Y. Xu, D.-W. Li, D.-H. Qu, Y.-T. Long and H. Tian, Chem, 2018, 4, 2670–2684 CAS.
- T. Hoshino, M. Miyauchi, Y. Kawaguchi, H. Yamaguchi and A. Harada, J. Am. Chem. Soc., 2000, 122, 9876–9877 CrossRef CAS.
- J.-C. Chang, S.-H. Tseng, C.-C. Lai, Y.-H. Liu, S.-M. Peng and S.-H. Chiu, Nat. Chem., 2017, 9, 128–134 CrossRef CAS.
- K. Cai, B. Cui, B. Song, H. Wang, Y. Qiu, L. O. Jones, W. Liu, Y. Shi, S. Vemuri, D. Shen, T. Jiao, L. Zhang, H. Wu, H. Chen, Y. Jiao, Y. Wang, C. L. Stern, H. Li, G. C. Schatz, X. Li and J. F. Stoddart, Chem, 2021, 7, 174–189 CAS.
- C. C. Carmona-Vargas and N. Giuseppone, Chem, 2021, 7, 11–13 CAS.
- N. Yamaguchi, D. S. Nagvekar and H. W. Gibson, Angew. Chem., Int. Ed., 1998, 37, 2361–2364 CrossRef CAS PubMed.
- P. R. Ashton, I. W. Parsons, F. M. Raymo, J. F. Stoddart, A. J. P. White, D. J. Williams and R. Wolf, Angew. Chem., Int. Ed., 1998, 37, 1913–1916 CrossRef CAS.
- M. Miyauchi, Y. Takashima, H. Yamaguchi and A. Harada, J. Am. Chem. Soc., 2005, 127, 2984–2989 CrossRef CAS PubMed.
- M. Miyauchi and A. Harada, J. Am. Chem. Soc., 2004, 126, 11418–11419 CrossRef CAS PubMed.
- F. Wang, J. Zhang, X. Ding, S. Dong, M. Liu, B. Zheng, S. Li, L. Wu, Y. Yu, H. W. Gibson and F. Huang, Angew. Chem., Int. Ed., 2010, 49, 1090–1094 CrossRef CAS PubMed.
- Z. Zhang, Y. Luo, J. Chen, S. Dong, Y. Yu, Z. Ma and F. Huang, Angew. Chem., Int. Ed., 2011, 50, 1397–1401 CrossRef CAS PubMed.
- M. P. L. Werts, M. van den Boogaard, G. M. Tsivgoulis and G. Hadziioannou, Macromolecules, 2003, 36, 7004–7013 CrossRef CAS.
- M. P. L. Werts, M. van den Boogaard, G. Hadziioannou and G. M. Tsivgoulis, Chem. Commun., 1999, 623–624 RSC.
- H. Sasabe, N. Inomoto, N. Kihara, Y. Suzuki, A. Ogawa and T. Takata, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 4154–4160 CrossRef CAS.
- A. Miyawaki, M. Miyauchi, Y. Takashima, H. Yamaguchi and A. Harada, Chem. Commun., 2008, 456–458 RSC.
- M. Zhang, S. Li, S. Dong, J. Chen, B. Zheng and F. Huang, Macromolecules, 2011, 44, 9629–9634 CrossRef CAS.
- X.-Q. Wang, W.-J. Li, W. Wang, J. Wen, Y. Zhang, H. Tan and H.-B. Yang, J. Am. Chem. Soc., 2019, 141, 13923–13930 CrossRef CAS PubMed.
- X.-Q. Wang, W.-J. Li, W. Wang and H.-B. Yang, Acc. Chem. Res., 2021, 54, 4091–4106 CrossRef CAS PubMed.
- K. Cai, Y. Shi, G.-W. Zhuang, L. Zhang, Y. Qiu, D. Shen, H. Chen, Y. Jiao, H. Wu, C. Cheng and J. F. Stoddart, J. Am. Chem. Soc., 2020, 142, 10308–10313 CrossRef CAS PubMed.
- Y. Wang, Z. Zhang, H. Zhang, J. Zhao, G. Liu, R. Bai, Y. Liu, W. You, W. Yu and X. Yan, Chem, 2023, 9, 2206–2221 CAS.
- S. Dong, Y. Luo, X. Yan, B. Zheng, X. Ding, Y. Yu, Z. Ma, Q. Zhao and F. Huang, Angew. Chem., Int. Ed., 2011, 50, 1905–1909 CrossRef CAS PubMed.
- X. Yan, M. Zhou, J. Chen, X. Chi, S. Dong, M. Zhang, X. Ding, Y. Yu, S. Shao and F. Huang, Chem. Commun., 2011, 47, 7086–7088 RSC.
- X. Yan, D. Xu, X. Chi, J. Chen, S. Dong, X. Ding, Y. Yu and F. Huang, Adv. Mater., 2012, 24, 362–369 CrossRef CAS PubMed.
- N. L. Strutt, H. Zhang, M. A. Giesener, J. Lei and J. F. Stoddart, Chem. Commun., 2012, 48, 1647–1649 RSC.
- S. Xian and M. J. Webber, J. Mater. Chem. B, 2020, 8, 9197–9211 RSC.
- N. J. Van Zee and R. Nicolaÿ, Prog. Polym. Sci., 2020, 104, 101233 CrossRef CAS.
- J. Zhao, Z. Zhang, L. Cheng, R. Bai, D. Zhao, Y. Wang, W. Yu and X. Yan, J. Am. Chem. Soc., 2022, 144, 872–882 CrossRef CAS PubMed.
- D. Aoki, G. Aibara, S. Uchida and T. Takata, J. Am. Chem. Soc., 2017, 139, 6791–6794 CrossRef CAS PubMed.
- B. A. Laurent and S. M. Grayson, Chem. Soc. Rev., 2009, 38, 2202 RSC.
- D. Aoki, G. Aibara and T. Takata, Polym. Chem., 2021, 12, 6381–6385 RSC.
- S. Tsuda, Y. Aso and T. Kaneda, Chem. Commun., 2006, 3072–3074 RSC.
- L. Randone, H. Onagi, S. F. Lincoln and C. J. Easton, Eur. J. Org. Chem., 2019, 3495–3502 CrossRef CAS.
- E. N. Guidry, J. Li, J. F. Stoddart and R. H. Grubbs, J. Am. Chem. Soc., 2007, 129, 8944–8945 CrossRef CAS PubMed.
- P. G. Clark, M. W. Day and R. H. Grubbs, J. Am. Chem. Soc., 2009, 131, 13631–13633 CrossRef CAS PubMed.
- L. Fang, M. Hmadeh, J. Wu, M. A. Olson, J. M. Spruell, A. Trabolsi, Y.-W. Yang, M. Elhabiri, A.-M. Albrecht-Gary and J. F. Stoddart, J. Am. Chem. Soc., 2009, 131, 7126–7134 CrossRef CAS PubMed.
- G. Du, E. Moulin, N. Jouault, E. Buhler and N. Giuseppone, Angew. Chem., Int. Ed., 2012, 51, 12504–12508 CrossRef CAS PubMed.
- Y.-L. Zhao, R.-Q. Zhang, C. Minot, K. Hermann and M. A. Van Hove, Phys. Chem. Chem. Phys., 2015, 17, 18318–18326 RSC.
- Y.-L. Zhao, R.-Q. Zhang, C. Minot, K. Hermann and M. A. Van Hove, Phys. Chem. Chem. Phys., 2016, 18, 7419–7426 RSC.
- L. Gao, Z. Zhang, B. Zheng and F. Huang, Polym. Chem., 2014, 5, 5734–5739 RSC.
- A. Goujon, G. Du, E. Moulin, G. Fuks, M. Maaloum, E. Buhler and N. Giuseppone, Angew. Chem., Int. Ed., 2016, 55, 703–707 CrossRef CAS PubMed.
- X. Fu, R.-R. Gu, Q. Zhang, S.-J. Rao, X.-L. Zheng, D.-H. Qu and H. Tian, Polym. Chem., 2016, 7, 2166–2170 RSC.
- A. Goujon, G. Mariani, T. Lang, E. Moulin, M. Rawiso, E. Buhler and N. Giuseppone, J. Am. Chem. Soc., 2017, 139, 4923–4928 CrossRef CAS PubMed.
- S. H. M. Söntjens, R. P. Sijbesma, M. H. P. van Genderen and E. W. Meijer, J. Am. Chem. Soc., 2000, 122, 7487–7493 CrossRef.
- K. Iwaso, Y. Takashima and A. Harada, Nat. Chem., 2016, 8, 625–632 CrossRef CAS PubMed.
- S. Ikejiri, Y. Takashima, M. Osaki, H. Yamaguchi and A. Harada, J. Am. Chem. Soc., 2018, 140, 17308–17315 CrossRef CAS PubMed.
- A. Goujon, T. Lang, G. Mariani, E. Moulin, G. Fuks, J. Raya, E. Buhler and N. Giuseppone, J. Am. Chem. Soc., 2017, 139, 14825–14828 CrossRef CAS PubMed.
- Z. Zhang, W. You, P. Li, J. Zhao, Z. Guo, T. Xu, J. Chen, W. Yu and X. Yan, J. Am. Chem. Soc., 2023, 145, 567–578 CrossRef CAS PubMed.
- S. Yao, Y. Chang, Z. Zhai, H. Sugiyama, M. Endo, W. Zhu, Y. Xu, Y. Yang and X. Qian, ACS Appl. Mater. Interfaces, 2022, 14, 20739–20748 CrossRef CAS PubMed.
- W.-J. Li, W. Wang, X.-Q. Wang, M. Li, Y. Ke, R. Yao, J. Wen, G.-Q. Yin, B. Jiang, X. Li, P. Yin and H.-B. Yang, J. Am. Chem. Soc., 2020, 142, 8473–8482 CrossRef CAS PubMed.
- N. Giuseppone and A. Walther, Out-of-equilibrium (supra)molecular systems and materials, Wiley-VC. Press, Weinheim, 2021 Search PubMed.
| This journal is © The Royal Society of Chemistry 2023 |