
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sustainable routes to alkenes: applications of homogeneous catalysis to the dehydration of alcohols to alkenes†
Daniel J.
Ward
 a,
Daniel J.
Saccomando
b,
Gary
Walker
b and
Stephen M.
Mansell
*a
a,
Daniel J.
Saccomando
b,
Gary
Walker
b and
Stephen M.
Mansell
*a
aInstitute of Chemical Sciences, School of Engineering and Physical Sciences, Heriot-Watt University, Edinburgh, EH14 4AS, UK. E-mail: s.mansell@hw.ac.uk; Web: http://mansellresearch.org.uk
bLubrizol Limited, The Knowle, Nether Lane Hazelwood, Derby, Derbyshire DE56 4AN, UK
First published on 23rd March 2023
Abstract
With the growing scientific and social awareness of environmental issues, there is an increasing demand for renewable alkene feedstocks used to make the products we rely on. Alkenes are typically synthesised through high temperature ‘cracking’ processes, but a more sustainable option is available through the dehydration of alcohols. This minireview compares the dehydration of alcohols to alkenes using homogeneous catalysts with well-established Brønsted acid and heterogeneous catalysts. Metal triflates M(OTf)4 (M = Ti, Hf) are catalysts for the dehydration of primary, secondary and tertiary alcohols at decreasing temperatures with increasing substitution of the alcohol. The rhenium catalysts methylrhenium trioxide (MTO) and Re2O7 are efficient catalysts at 100 °C for dehydrating benzylic, allylic and tertiary alcohols. A rhodium catalyst has been developed that utilises a catalytic quantity of HI to generate reactive alkyl halides that can undergo oxidative addition and β-H elimination. These expensive precious metal catalysts are contrasted by recent results with iron triflate catalysts that promise more cost-effective processes in the near future.
Introduction
Alkenes, and ethylene in particular, are amongst the most important starting materials for the chemical industry,1 with the annual production of ethylene estimated to be about 140 million tons.2 Alkenes are not present in crude oil or natural gas3–5 so they need to be synthesised, and this is currently achieved through high temperature ‘cracking’ processes that break C–C bonds and also result in dehydrogenation.1,6 Cracking processes are energy-intensive, often requiring temperatures of 500–900 °C,6 and rely on non-renewable starting materials so are potentially detrimental to the environment and non-sustainable. Finding direct, sustainable replacements to the currently used non-renewable fossil-fuel-sourced feedstocks, such as alkenes,7 is beneficial because these processes could be slotted into existing chemical plants without requiring large changes to downstream chemical processes that produce the commercial products, allowing more rapid reduction of CO2 than a complete overhaul of the entire chemical industry.Current technologies for the production of alkenes
Fluid catalytic cracking (FCC) makes up approximately 60% of olefin production (Scheme 1a).8 FCC is the process of converting high-boiling point (330–550 °C)9 high molecular weight hydrocarbons from crude oil into gasoline and olefinic gases at high reaction temperatures of approximately 550 °C.9 The remaining 40% of olefins are produced from steam cracking that involves the breakdown of larger chain alkanes present in naphtha into a mixture of products that are predominantly alkenes, or ethane and propane into ethene and propene respectively (Scheme 1b).8 These processes are highly energy intensive accounting for approximately 8% of the chemical sector's total primary energy demand,10 with the cost of energy accounting for 70% of the production costs in an ethane- or naphtha-based olefin plant.11 90% of CO2 produced by the chemical plant is directly related to the energy consumption of this endothermic reaction, and cracking accounts for more than 300 million tonnes of CO2 p.a.12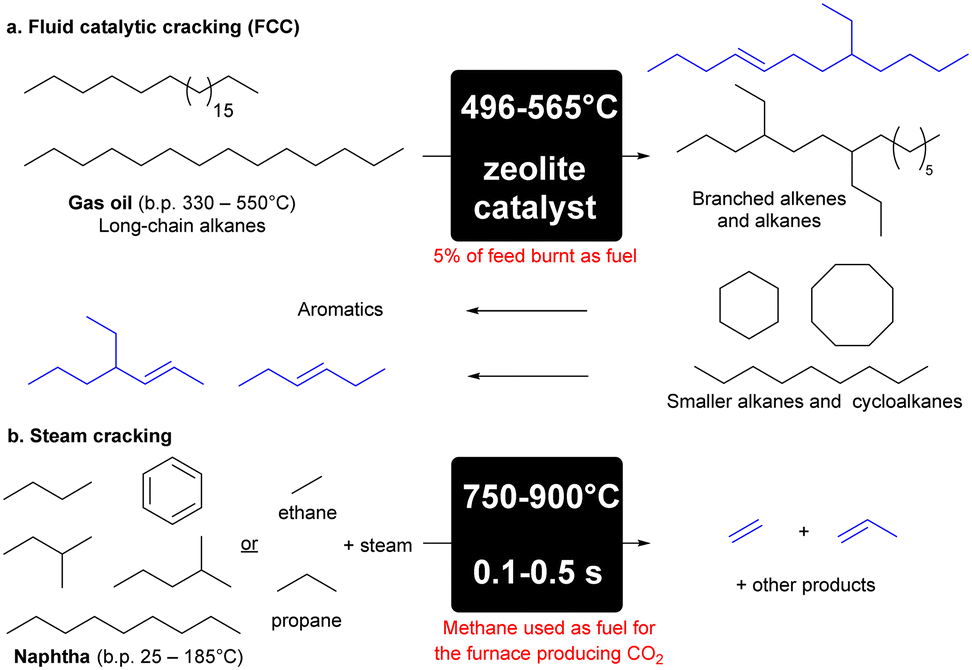 | ||
| Scheme 1 Cracking of hydrocarbons to produce alkenes via FCC (a) or steam cracking (b).8,9 | ||
Alcohols as starting materials
In order to limit our dependence on fossil fuels, alternative renewable starting materials are required. If their conversion is less energy-intense, a reduction in CO2 emissions will also be achieved. A sustainable alternative to crude-oil feedstocks could derive from biologically sourced alcohols.2,13,14 Small chain bioalcohols (alcohols produced from biological resources or biomass)15 such as bioethanol and biomethanol are currently produced in a number of ways including from the fermentation of sugar cane, corn and wheat (first generation fuels) or from waste-derived biomass (second generation).15 With the growing demand for bioalcohols, the market is set to expand with a consequent improvement in the scale and efficiency of their production. Renewable alcohols could be used as starting materials to produce olefins and other feedstocks for industrial use.16,17 The dehydration of alcohols at relatively low temperatures would allow for a new pathway to industrially important olefins with a potential reduction in the amount of CO2 released. One example is the dehydration of 1-phenyl ethanol that is an important pathway to styrene, accounting for 15% of the world's styrene production,18 which has been commercialised by LyondellBasell and Shell.19,20Alcohol dehydration is a well-known and documented reaction21 and can be achieved in several ways: by heat alone,22 catalysed by Brønsted acids,23 by heterogeneous metal oxides (typically alumina), by zeolites21,24–27 and by homogeneous metal complexes.28,29 Previous literature reviews on the subject have focussed on the transformation of ethanol to ethene,2,13,30–35 butene,36 butadiene37,38 or other hydrocarbons.39,40 A review has recently covered the dehydration of natural products containing alcohol functionalities.41
This minireview covers the dehydration of a variety of alcohols to ethene and longer chain alkenes primarily using homogeneous catalysts, but with suitable comparisons to the other approaches as well. Easily synthesised and cheap metal catalysts that have the potential to be industrially viable will be highlighted. The related deoxydehydration reaction, which converts a 1,2-diol into an alkene with the help of a sacrificial reductant, will not be covered as it has been reviewed recently.42
Alcohols to alkenes: Brønsted acid catalysis
Acids have been used for many years to dehydrate alcohols to alkenes. Primary alcohols undergo dehydration reactions to form alkenes via the concerted E2 mechanism43 due to the instability of primary carbocations,9 however, the pathways for the dehydration of secondary alcohols are not as clear.43 E1 and E2 mechanisms are both possible (Scheme 2) and yields can often be lower as well.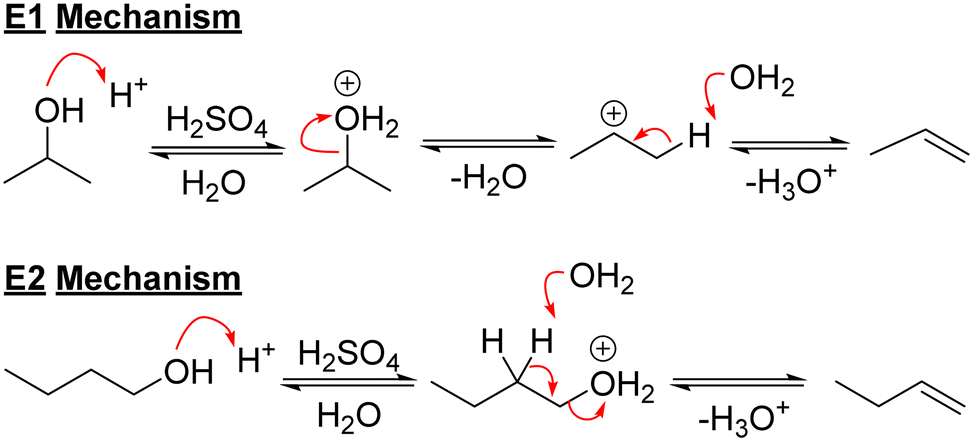 | ||
| Scheme 2 E1 and E2 mechanisms for alcohol dehydration. | ||
The dehydration of cyclohexanol with concentrated sulfuric acid led to alkene products in less than 20% yield, whereas use of hydrochloric acid gave a mixture of cyclohexene and methylcyclopentenes (14% of the product mixture) due to isomerisation.22 The choice of acid is clearly important. Sulfuric acid is one of the most common ways to dehydrate alcohols in a lab, however, it is also a strong oxidising agent and oxidises some of the alcohol into carbon dioxide, simultaneously generating sulfur dioxide,44 and so both gases need to be removed.45 For the dehydration of 1-phenyl ethanol at 100 °C, sulfuric acid (2.5 mol%) was more active than p-toluenesulfonic acid (pTSA, 5 mol%), but the yield of styrene was much worse (39% and 85%, respectively).46 Brønsted acids are known to cause skeletal isomerisation and double bond shifts.47,48
Phosphoric acid catalysts have been used industrially from the 1930s to the 1950s and were the first industrial catalysts for the dehydration of ethanol to ethylene. Polyphosphoric acid49 has been investigated in detail as a catalyst for this process.44 The process was first developed by Imperial Chemical Industries (ICI) which used phosphate loaded onto clay or coke.30 Ethylene production from ethanol was found to give high purity products but the catalyst was deactivated easily by coke deposition and required regeneration.44 Modern processes now use alumina-based catalysts,30 and can include promoters such as metal oxides and organic or inorganic acids.31 Bio-based polyethylene is now a commercial reality – the price of 1 kg of bio-PE has been estimated to be only 30% higher than petrochemical PE – with Brazilian company Braskem producing the most bio-PE, but with others also involved.50
In addition to alkenes, ethers can also be formed.43,51–53 The production of ethers from alcohols requires the presence of an acid, and concentrated sulfuric acid is used to produce diethyl ether from ethanol in 95% yield.52 Diethyl ether is an important solvent and is primarily manufactured by the chemical industry as a by-product of the vapour phase hydration of ethene to ethanol using a supported phosphoric acid catalyst.54,55 Brønsted acid catalysis works for both the hydration of ethene and the dehydration of ethanol, which is in line with the principle of microscopic reversibility. Ethers can also act as starting materials or intermediates in the production of alkenes, although very strong bases are required to induce hydro-alkoxy elimination at ambient conditions.53 Altogether, a network of reactions link alcohols, ethers and alkenes (Scheme 3).
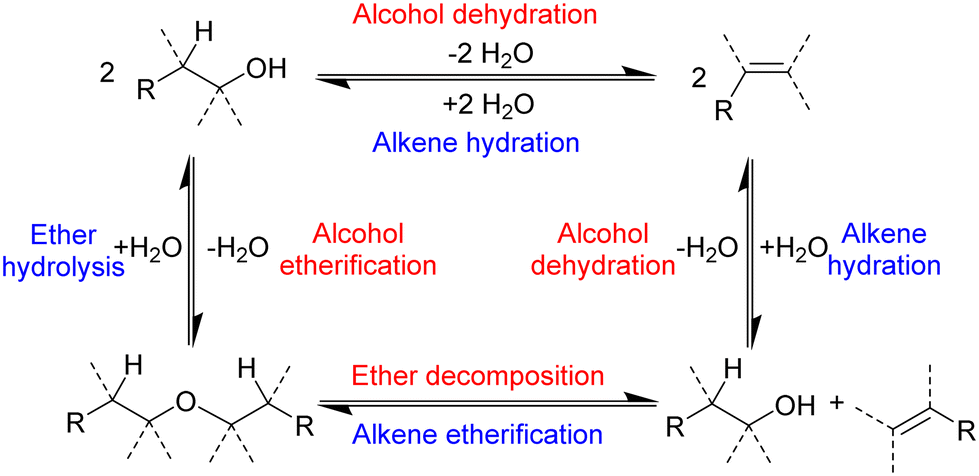 | ||
| Scheme 3 Reactions that interconvert alcohols, ethers and alkenes.55 | ||
Cationic ion-exchange resins
Cationic-exchange resins have been used to obtain ethers from primary alcohols56–58 whereas alkenes were formed in reactions with secondary and tertiary alcohols.59 A strongly acidic cation exchange resin containing sulfonic acid groups (Ar–SO2OH) was used to catalyse the dehydration of ethanol to diethyl ether at temperatures between 80 °C and 120 °C with no ethylene formed.60 It is known that ethers are formed when the process is run too cool as typical temperatures are 300 °C for the endothermic formation of alkenes.44 The dehydration of 1-pentanol to di-n-pentyl ether was achieved in 90% yield at 130–155 °C, with alkenes formed in only 5–10% yield.61 For isopropanol, both propylene and diisopropyl ether were obtained at temperatures between 90 °C and 110 °C using an ion exchange resin.62 At temperatures between 84 °C and 105 °C, 2-butanol was dehydrated to give predominantly trans-2-butene whereas isobutanol gave butenes, isobutane and tar.63 Overall, acidic resins are best used at low temperatures (<150 °C) where etherification can be favoured.55Main group Lewis acids
BF3·OEt2 has been shown to promote the dehydration of tertiary alcohols to alkenes under mild conditions in good yields.64 Tertiary benzylic alcohols were also dehydrated using Me3SiCl or ‘silica-chloride’ (generated from the reaction of thionyl chloride with silica), but saturated tertiary alcohols did not react.65 Boric acid (H3BO3) is a weak Lewis acid capable of dehydrating alcohols including menthol and cyclohexanol proceeding via metaborate esters, B3O3(OR)3 with a 6-membered boroxine ring, however, it was shown that no reaction took place below 260 °C.66A catalyst system involving a Lewis base and Lewis acid was used in the dehydration of 1-arylethanols to produce substituted styrenes (Scheme 4). AlCl3 (5 mol%) as the Lewis acid and triphenylphosphine (PPh3, 5 mol%) as the Lewis base at 80 °C gave a 95% conversion of (methoxyphenyl)ethanol with 93% yield of methoxystyrene after 2 hours.18 Although multiple combinations of Lewis acid and bases were tested, AlCl3 and PPh3 gave the best combination of high conversion and high yield to the desired product, inhibiting nucleophilic attack of the product styrene.18
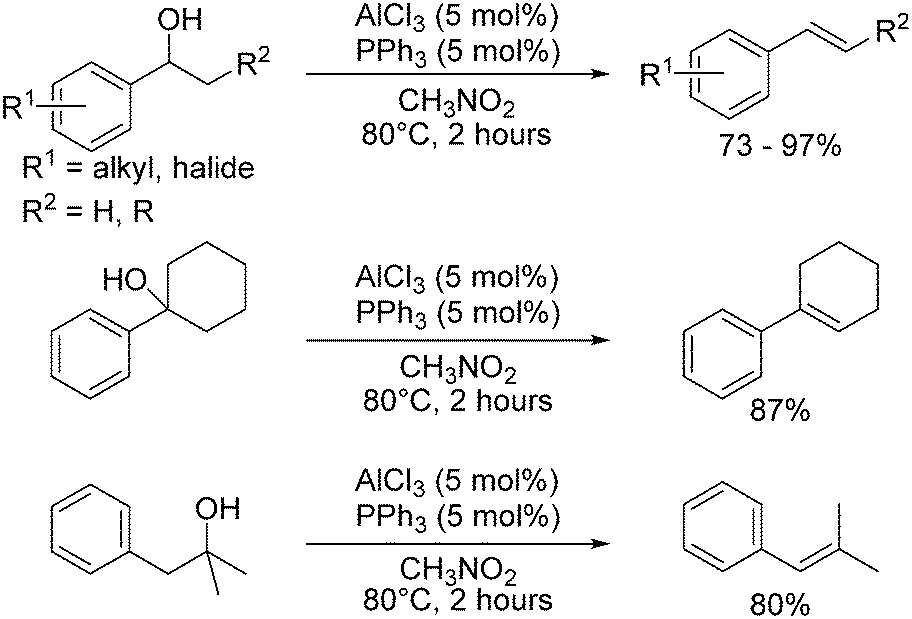 | ||
| Scheme 4 Lewis acid/Lewis base catalysed dehydration reactions. | ||
Heterogeneous catalysis
There is extensive literature on the use of heterogeneous catalysts for dehydration30,67,68 that is beyond the range of this minireview; only a brief summary is included here. For ethanol, dehydration to ethene is favoured at high temperatures (starting at 300 °C) as this is an endothermic reaction,30,69 whereas the exothermic formation of diethyl ether is favoured at lower temperatures.Alumina
γ-Alumina is a very important industrial catalyst and catalyst support, and yet its structure is still under discussion.70 Alumina has long been known to catalyse the dehydration of alkenes to alcohols,71 such as 1-hexanol to hexene at 450 °C,72 but the product and extent of isomerisation is highly dependent on the nature of the alumina catalyst.73 Older ethanol to ethylene industrial processes successfully utilised alumina catalysts,32 but unmodified γ-alumina required temperatures of 450 °C and the yield of ethylene is relatively low (80%).74 The formation of ethers is also a problem,75 and commercial catalysts for producing dimethyl ether from MeOH are well known.76 The mechanism of dehydration has been under investigation for some time, and it has been well documented that the alcohol binds to the γ-alumina via hydrogen bonding.77,78 γ-Alumina is thought to contain both Lewis and Brønsted acid sites,79 with the Lewis acidic sites responsible for minimising the unwanted side reactions such as skeletal isomerisations and double bond shifts caused by Brønsted acids.47,59,80Other metal oxides
Titanium dioxide has been shown to be both capable of dehydration and dehydrogenation depending on the reaction temperature. Anatase was very selective for dehydration forming alkenes in a mixture of 1-, trans-2- and cis-2 isomers at temperatures between 200 °C and 265 °C.81 Tertiary alcohols were dehydrated faster than secondary alcohols. Using anatase, the dehydration of 2-octanol to octene was achieved at 265 °C in one hour with 88% conversion.81 Zirconium oxide and hafnium oxide have also been investigated,82,83 but were less active than γ-alumina for the dehydration of isopropanol and tert-butanol.84 Addition of WO3 was needed to improve activity.85 Ga2O3 was shown to resemble Al2O3 as a selective dehydration catalyst for secondary alcohols at 170–222 °C, albeit forming a mixture of isomers,86 whereas In2O3 promoted the dehydrogenation of alcohols.87 Overall, although metal oxides are capable of dehydrating alcohols directly to alkenes, they often do not produce the highest yields or have the best selectivities, especially with larger alcohols with four or more carbons. The reaction temperatures are typically high, however, the catalysts are usually cheap and robust.Zeolites
Zeolites are hydrated, crystalline microporous aluminosilicates; their structures include channels and/or cavities of molecular dimensions featuring Brønsted and Lewis acidic sites making them ideal for catalysis.88–90 Zeolites are most widely implemented in FCC, but they are also very active for dehydration reactions,91–93 particularly the dehydration of methanol to give olefins.94 ZSM-5 has often been identified as the catalyst of choice for a variety of alcohol dehydrations due to its good performance at lower reaction temperatures (for example, ZSM-5 was found to be more active for ethanol dehydration at lower temperatures than alumina or silica containing 13 wt% alumina),69 and with higher alkene yields,25,30,93,95–100 but stability is an issue.96,101 Balancing activity, stability and selectivity with other factors, such as nature of the alcohol, can lead to other zeolites being favoured, such as H-MOR (mordenite)69,95 or silicoaluminophosphates, such as SAPO-34.27,102Homogeneous catalysis using molecular inorganic species
Stoichiometric reactions: Zeise's salt
Zeise's salt, K[PtCl3(C2H4)], was first isolated by Danish pharmacologist William Christopher Zeise and is considered to be the first organometallic complex.103,104 Zeise originally synthesised this complex by reacting platinum(IV) chloride103 (or a mixture of PtCl2 and PtCl4)105 with ethanol then adding KCl to crystallise the product (Scheme 5),103,105 thus demonstrating ethanol dehydration by a molecular species. Acetaldehyde is formed as ethanol reduces Pt(IV) to Pt(II).106 The related procedure of boiling Na2[PtCl6] in ethanol has been described as tedious and resulting in variable yields depending on impurities in the platinum salt, and with maximum yields of 75% only obtained after much experience.107 This contrasts with the facile coordination of ethene to Pt(II).107,108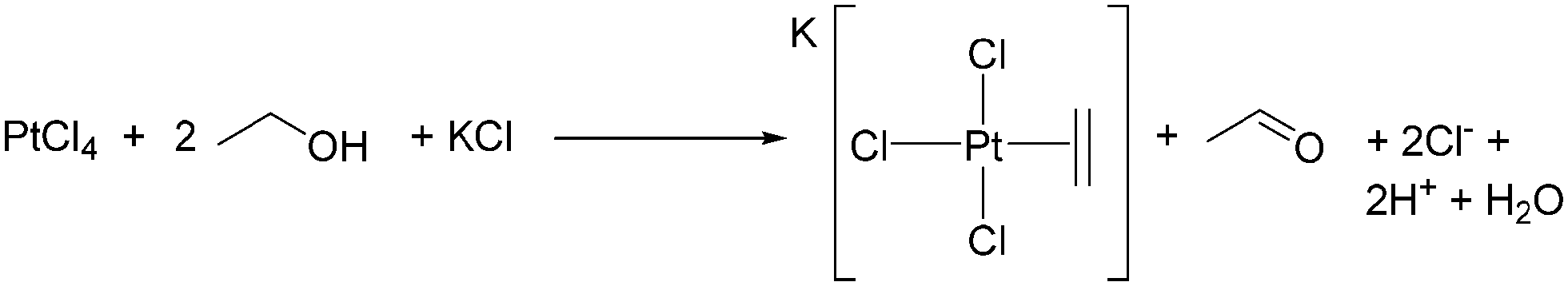 | ||
| Scheme 5 The original synthesis of Zeise's salt. | ||
Dissolving Na2[PtCl4] in ethanol, 1-propanol or 1-butanol followed by cation exchange gave a 20% yield of [NBu4][PtCl3(C2H4)], 32% yield of [NBu4][PtCl3(H2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCH3)] and 40% yield of [NBu4][PtCl3(H2CCHCH2CH3)], respectively. Branched alcohols only precipitated Pt metal,109 and it was proposed that branched alcohols do not give [PtCl3(alcohol)]− complexes, the necessary first step of the reaction to produce an alkene complex.109 For Zeise's salt, ethanol is the source of the ethene ligand, but for such an important reaction, only a few studies considering its mechanism of formation have been published.110–113 The dehydration of EtOH by Pt compounds was overtaken in interest by Pt-catalysed alkane C–H activation, and the C–H reactivity of EtOH gained more attention.114–116
CHCH3)] and 40% yield of [NBu4][PtCl3(H2CCHCH2CH3)], respectively. Branched alcohols only precipitated Pt metal,109 and it was proposed that branched alcohols do not give [PtCl3(alcohol)]− complexes, the necessary first step of the reaction to produce an alkene complex.109 For Zeise's salt, ethanol is the source of the ethene ligand, but for such an important reaction, only a few studies considering its mechanism of formation have been published.110–113 The dehydration of EtOH by Pt compounds was overtaken in interest by Pt-catalysed alkane C–H activation, and the C–H reactivity of EtOH gained more attention.114–116
Group 4 complexes and catalysts converting alcohols to alkenes
In 1991, Schobert described the use of dimethyl titanocene to stoichiometrically convert four 2° and one 3° alcohol to their corresponding alkenes favouring the E isomer.117 Addition of alcohol to [TiCp2Me2] gave the alkoxide [TiCp2(OR)Me], and heating this eliminated methane, (Cp2TiO)n and the alkene in yields of 71–82%.117 Repo and co-workers have investigated metal triflates (OSO2CF3; OTf) due to the lower reaction temperatures for alcohol dehydration that can be achieved. Dehydrations of 2-octanol using 0.5 mol% Hf(OT)4, Ti(OTf)4 or HOTf at 150 °C for 3 h gave conversions of over 99%, with octene yields of 93%, 71% and 84% observed, respectively, as a mixture of isomers (Scheme 6).118 The results correlated very well with oxophilicity, assessed in terms of the dissociation energy of the M–O bond.118 At 150 °C, conversion rates have a linear correlation with time suggesting zero order kinetics, commonly encountered when a catalyst is in a large excess of substrate.118 The primary alcohol 1-octanol was more difficult to dehydrate than the secondary alcohols, whereas a tertiary alcohol reacted at lower temperatures (Scheme 6). Overall, Hf(OTf)4 was singled out for being robust under solvent-free conditions while producing high yields of product.118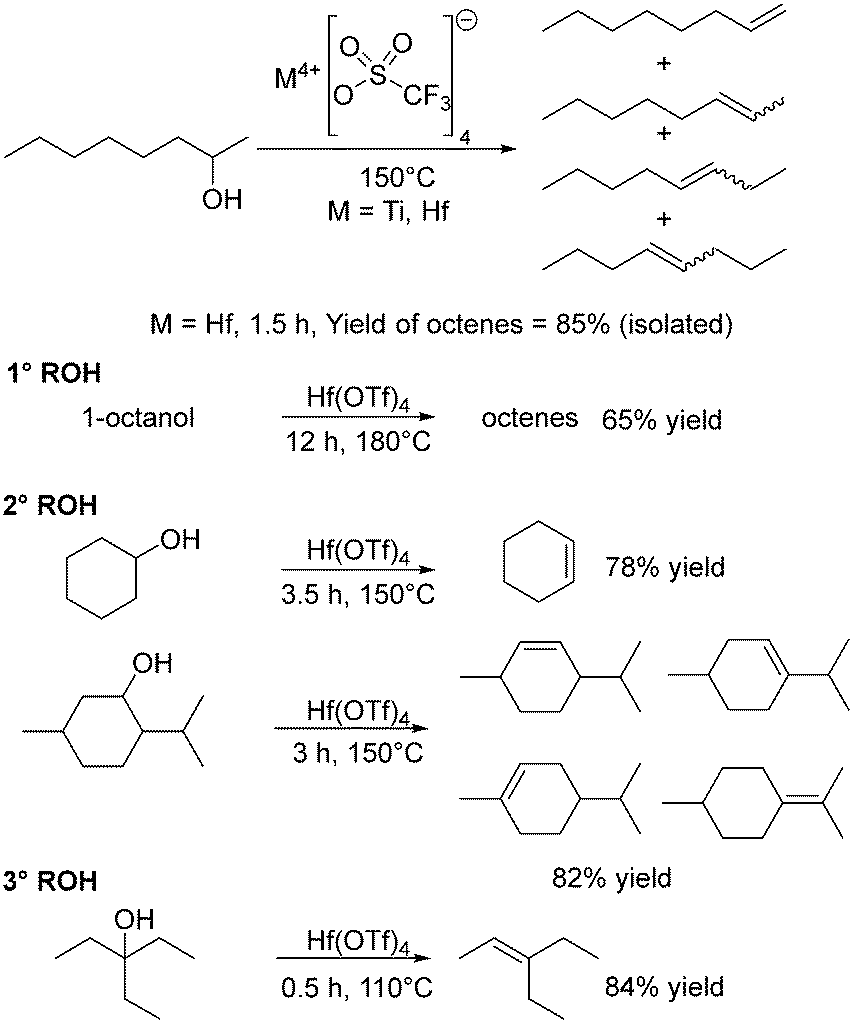 | ||
| Scheme 6 Alcohol dehydration using 0.5 mol% M(OTf)4. | ||
Group 6: chromium and molybdenum
Cr(OTf)3 is moderately Lewis acidic and gave 73% conversion for the dehydration of 2-octanol, but only a 35% yield of octene,118 making it an average catalyst for dehydration reactions. Mo complexes have been shown to be active in the dehydration of tertiary alcohols to alkenes.119 Mo(VI) oxide showed little activity in dehydration reactions of 1-phenylethanol to form styrene, with a conversion of 32% and selectivity of 10% (100 °C, 5 mol% catalyst loading, 22 h), whereas [MoO2(acac)2] at 100 °C and 1 mol% catalyst loading gave 97% conversion with 30% selectivity and a turnover number of 97.120 More active was a complex that featured the tert-butyl substituted ligand (Scheme 7), which in the same conditions gave >99% conversion and 28% selectivity with a turnover number of 99. In comparison, sulfuric acid (2.5 mol% catalyst loading) gave 99% conversion with 39% selectivity and a turnover number of 25.120 Various acac-derivatives were tested, but all showed very similar activity and selectivity for 1-phenylethanol dehydration. Dehydration of 2-octanol at 150 °C was best achieved with the tert-butyl-acac complex and gave a conversion of 88% to give a mixture of octene isomers (Scheme 7). A heterogeneous, high-valent carbon-supported molybdenum-dioxo catalyst (AC/MoO2) has been described recently, which resembles the molecular [MoO2(acac)2] systems, capable of dehydrating tertiary alcohols at 100–125 °C and secondary alcohols at 150 °C, but was less efficient for primary aliphatic alcohols (Scheme 7).121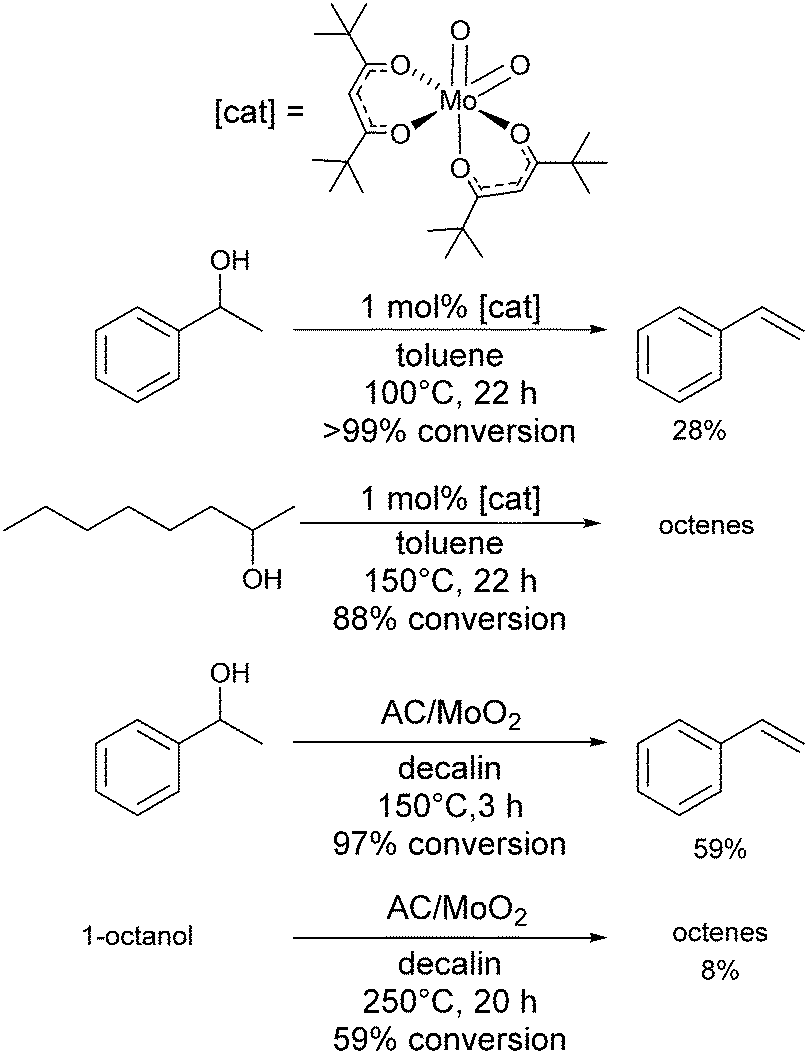 | ||
| Scheme 7 Molybdenum catalysts for dehydration reactions. | ||
Rhenium
In 1996, Zhu and Espenson used methylrhenium trioxide (MTO) as a catalyst for the direct conversion of alcohols to ethers, alcohols to alkenes and the disproportionation of alcohols to alkanes and carbonyl compounds, notably at room temperature.122 The outcome of the reaction is highly dependent on the structure of the alcohol. The reaction of primary aliphatic alcohols only gave low yields of the corresponding ether over the course of several days at room temperature (e.g. 7% nBuOnBu from nBuOH using 0.5 mol% MTO), however, PhCH(OH)R gave the corresponding ether in higher yields as the size of the R group increased (R = H, 36%; Me, 86%; Et, 89%; Ph, 100%), and the formation of unsymmetrical ethers between aliphatic and benzylic alcohols proceeded with similarly excellent yields.122 MTO dehydrated PhCH(OH)CH3 to styrene in 16% yield using 0.16 mol% of MTO at room temperature for 72 hours. 3-Octanol was converted into 3-octene with a turnover number of 40 after 3 d (0.2 mmol MTO in 15 mL of alcohol).122Klein Gebbink and co-workers have explored many dehydration reactions using rhenium catalysts, with particular focus on Re2O7 (Scheme 8).29 The dehydration of 1-phenylethanol at 100 °C gave the highest yield of styrene using Re2O7 as the catalyst (0.5 mol%: 98%, full conversion in just one hour), higher than MTO (1 mol%: 89% yield), H2SO4 (2.5 mol%: 39%) or pTSA (5 mol%: 85%) over 24 h.46 Contrasting with the success of Re2O7, Re complexes with PNP pincer ligands were not effective for the dehydration of 1-phenylethanol to styrene due to the reduced Lewis acidity of the Re centre.123 A variety of benzylic alcohols were successfully dehydrated using Re2O7 (Scheme 8), but there was no reaction in the presence of para-nitro or nitrile substituents nor with the primary non-benzylic alcohol 2-phenylpropan-1-ol.
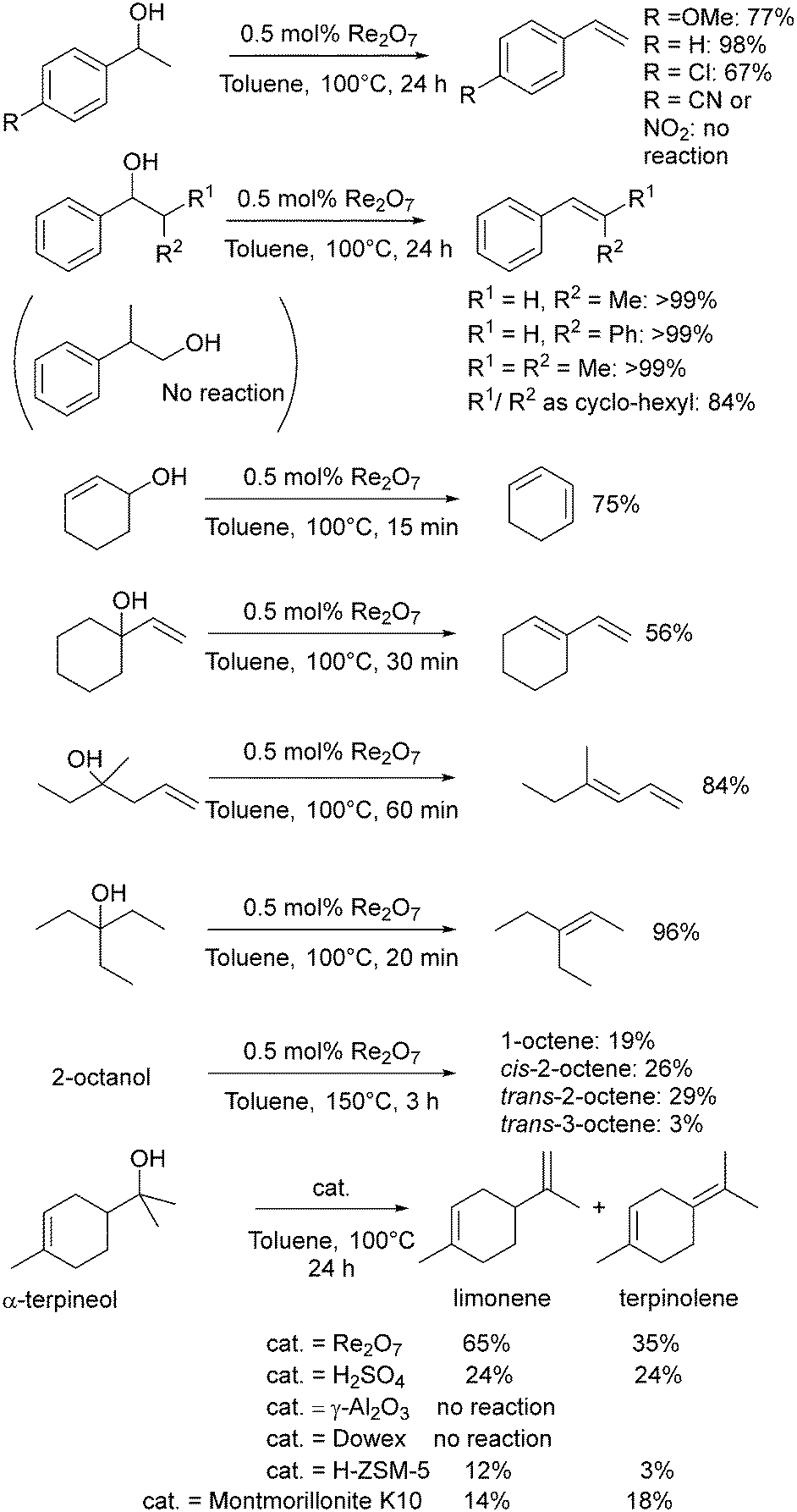 | ||
| Scheme 8 Summary of Re2O7 catalysed dehydration. | ||
Re2O7 was more active than sulfuric acid without sacrificing selectivity.46 An extension to this study showed good catalytic activity for Re2O7 across a range of substrates, with secondary aliphatic and homoallylic alcohols requiring higher temperatures (150 °C) than tertiary analogues (Scheme 8). Re2O7 gave superior catalytic activity to several heterogeneous catalysts for the dehydration of α-terpineol (Scheme 8).124
Either an ionic (cf. E1 mechanism) or concerted pathway (cf. E2) was considered for MTO-catalysed dehydration reactions (Scheme 9).29,122 An in-depth mechanistic study was conducted,125 noting that both air and moisture is essential for this reaction, which is likely to complicate computational investigations.29 High activation energies for the concerted pathway were found whereas both computational and experimental observations pointed towards the presence of carbocations.126
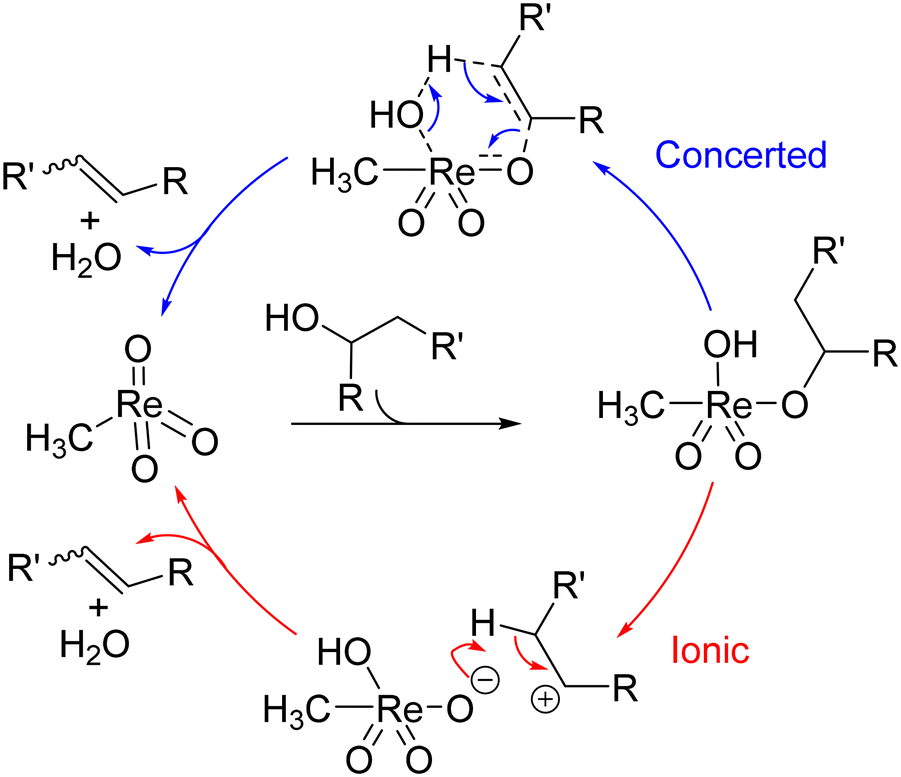 | ||
| Scheme 9 Two potential mechanisms for dehydration with MTO.125 | ||
Group 8: iron and ruthenium
Repo and co-workers assessed the use of iron triflates in the dehydration of alcohols. The group highlighted Fe(OTf)3 for its durability and robustness under solvent free conditions. At 150 °C for 3 h, 0.5 mol% Fe(OTf)3 catalysed the dehydration of 2-octanol (48% conversion) giving octene in 30% yield; increasing the temperature to 165 °C increased the yield to 91% yield (Scheme 10), similar to that of the more active hafnium triflate catalyst (1.5 h, 150 °C, 85% yield; Scheme 6), albeit at higher temperature and longer reaction time.118 Iron triflate was also capable of catalysing the dehydration of vicinal alcohols and a range of branched alcohols with reasonably good yields. The catalyst was not very effective in the dehydration of aromatic substrates such as phenylethanol only yielding 3% of styrene at 130 °C.118 Gunanathan and co-workers have also investigated Fe(OTf)3 as a dehydration catalyst. They found that the use of an ammonium chloride additive inhibited C–C coupling and instead made an efficient direct esterification system for benzylic primary and secondary alcohols.127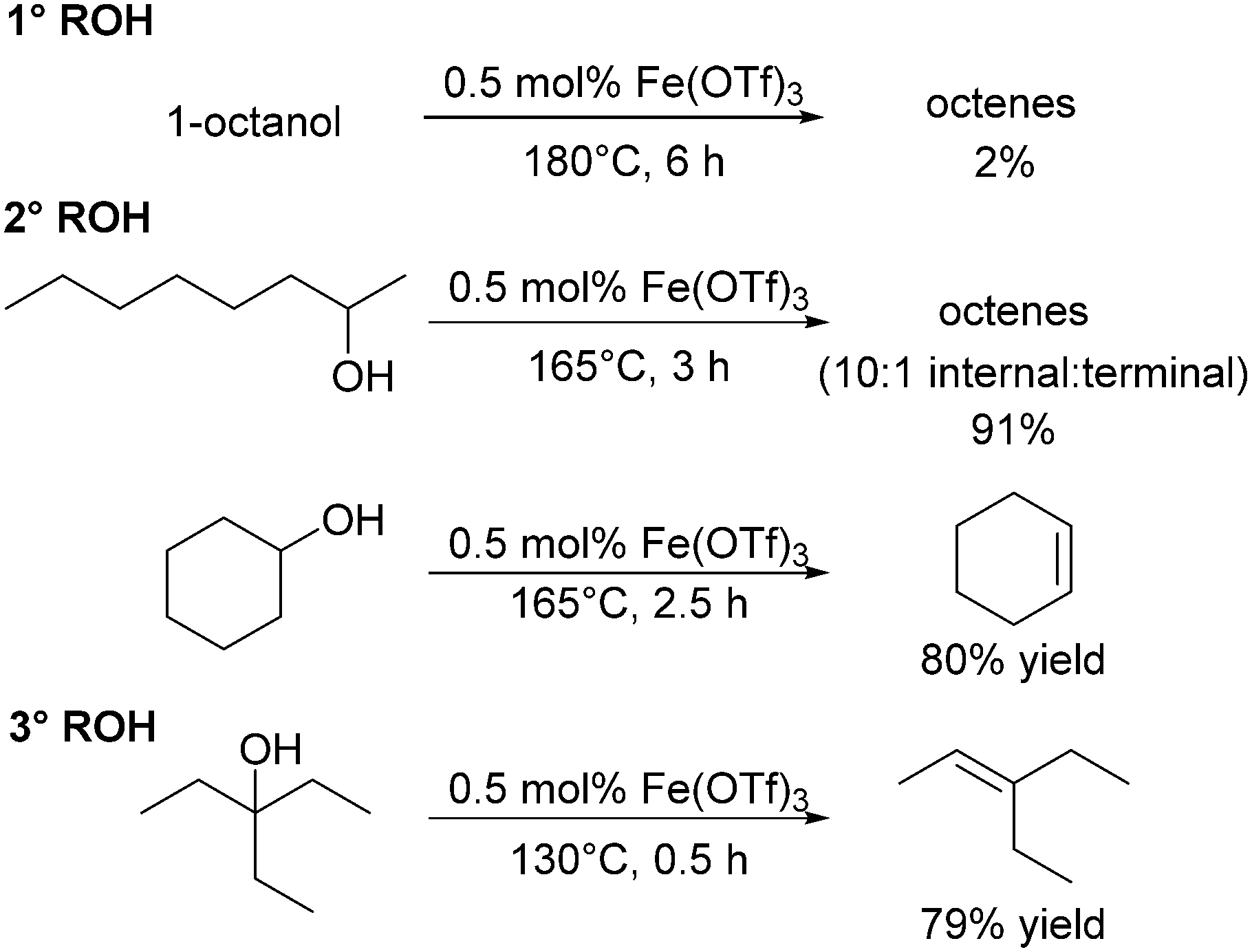 | ||
| Scheme 10 Dehydration reactions catalysed by Fe(OTf)3. | ||
Recently, Jones and co-workers have shown that the iron complex [Fe(OTf)2(FOX)] could catalyse the dehydration of 1-phenylethanol to styrene (Scheme 11).128 The complex showed high catalytic efficiencies at moderate temperatures without the use of Brønsted acids. At 100 °C after 24 hours in 1,2-dichlorobenzene, the yield of styrene was 74%, and using reactive distillation to remove the lower boiling styrene, quantitative conversions were achieved at 150 °C in >60% isolated yield.128
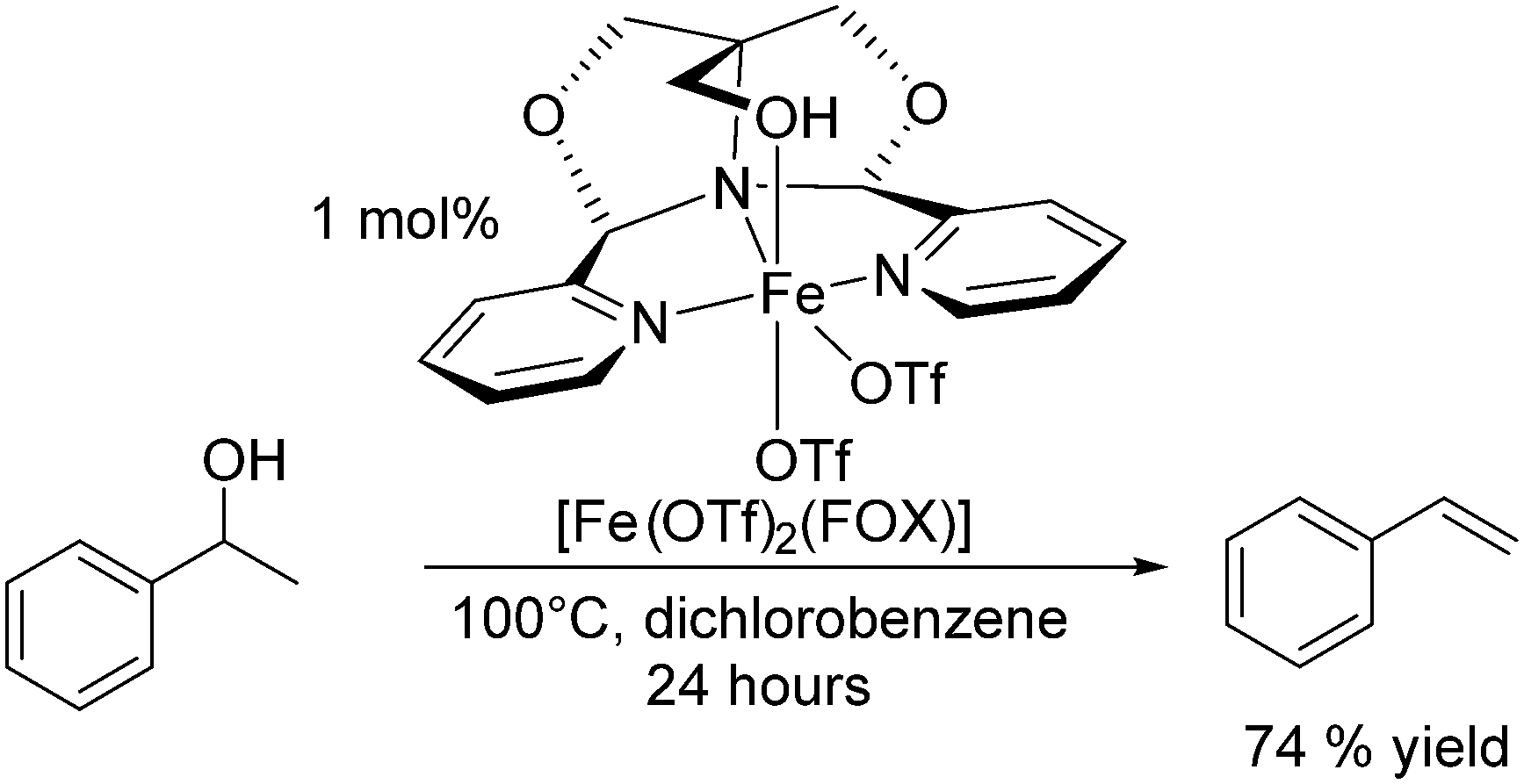 | ||
| Scheme 11 [Fe(OTf)2(FOX)] catalysed dehydration of styrene.128 | ||
Group 9: rhodium
In 2010, Wass and co-workers reasoned that a homogeneous catalysis approach to the dehydration of alcohols could allow for greater selectivity and milder reaction conditions than conventional high temperature heterogeneous approaches.129 They proposed a similar mechanism to the well-known Monsanto process.130 The alcohol initially reacts with a hydrogen halide acid forming the alkyl halide, which undergoes oxidative addition to Rh followed by β-elimination forming the alkene (Scheme 12). The chosen catalyst system was [Rh(μ-Cl)(CO)2]2 (0.065 mol%) with HI (6 mol% of 57% aqueous solution) as the acid, PMes3 (0.13 mol%) and either 1-butyl-3-methylimidazolium chloride (for good selectivity to alkenes) or NBu4Br (for higher conversions) at 110 °C for 48 h. Of the alcohols tested, n-butanol had the highest conversion and gave the highest yield (95% conversion and 99% selectivity). For ethanol, selectivity to ethene was more challenging, but a 68% conversion with 88% selectivity was achieved by doubling the amount of Rh salt and phosphine compared to the standard conditions.129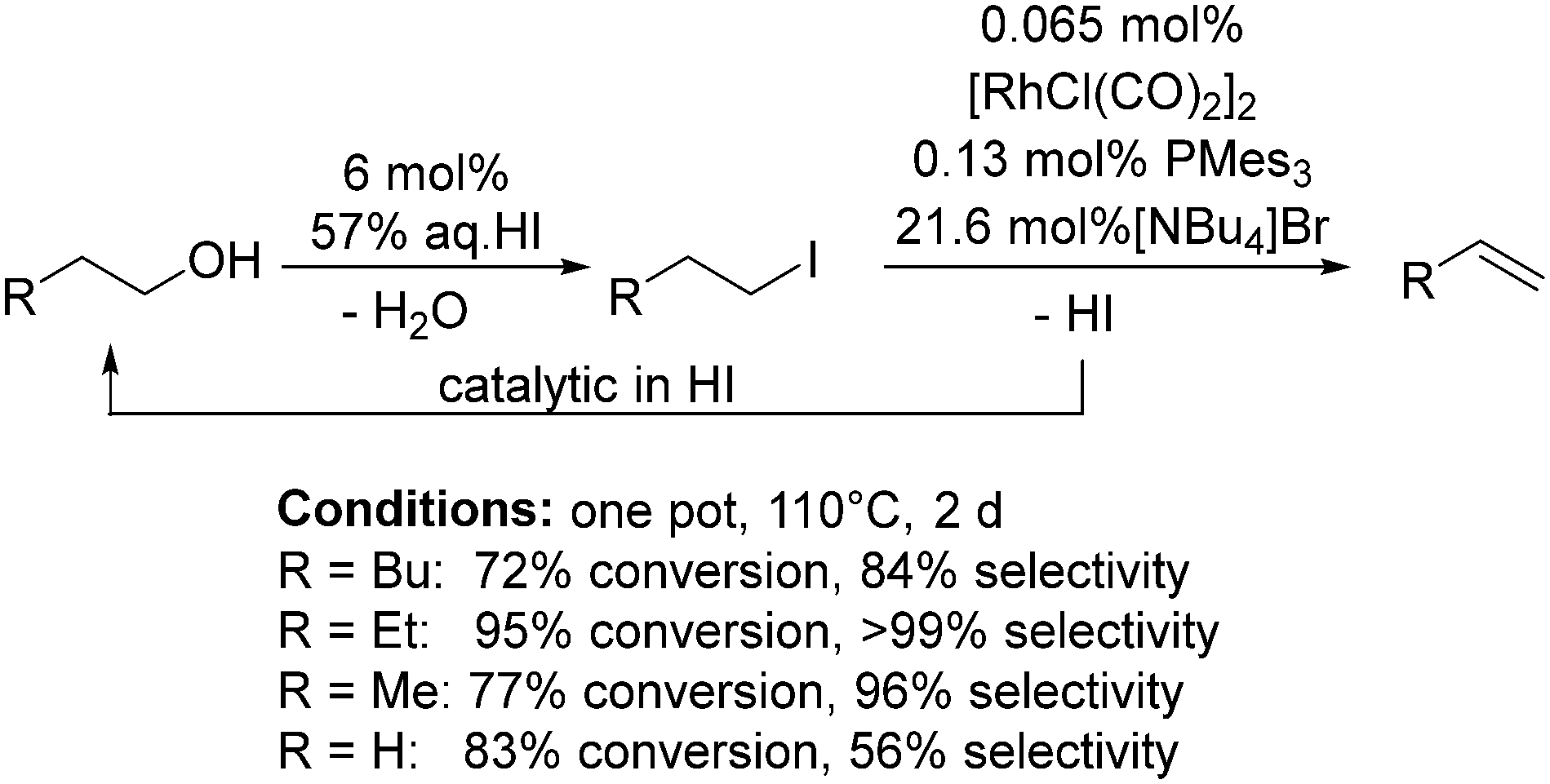 | ||
| Scheme 12 HI-promoted Rh-catalysed dehydration of alcohols. | ||
Group 11: copper
Dombroski and co-workers have investigated the use of copper(II) triflate for the dehydration of alcohols. They found that 0.1 mol% Cu(OTf)2 was an efficient catalyst for the dehydration of primary, secondary or tertiary alcohols and diols in decalin or heptane, or in the absence of solvent, at temperatures of less than 160 °C. The product alkene and water were distilled out of the reaction mixture and gave alkenes in yields ranging from 30–92%.131 1-Hexanol gave a 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 ratio of E-hex-2-ene:hex-1-ene in 38% yield, whereas 3-octanol was dehydrated to a mixture of octenes in 85% yield. In comparison, dehydration of 1-hexanol by H2SO4 or POCl3/pyridine only gave traces of alkene products.131
:3 ratio of E-hex-2-ene:hex-1-ene in 38% yield, whereas 3-octanol was dehydrated to a mixture of octenes in 85% yield. In comparison, dehydration of 1-hexanol by H2SO4 or POCl3/pyridine only gave traces of alkene products.131
Group 12: zinc
A kinetic resolution of β-hydroxy esters has been developed by selective dehydration of one enantiomer using stoichiometric BrZnCH2CO2tBu and 5 mol% of a chiral ligand. This system was able to dehydrate β-alkenyl-, β-alkynyl- and β-flavenyl-substituted β-hydroxy esters, with conversions necessarily limited to 50% as only one enantiomer of the product was produced, but with excellent enantiomeric excesses.132 The limitations of this reaction include the need for a conjugated β-substituent, no α-substituent and the efficiency of the chiral ligand (five were tested) was very substrate dependent.Group 13: aluminium
Aluminium triflate was also tested in the dehydration of alcohols by Repo and co-workers. Al(OTf)3 was the most Lewis acidic metal triflate tested, however, the conversion of 2-octanol (3 h, 150 °C, 0.5 mol% of catalyst: 75% conversion), the standard substrate, was lower than for Hf(OTf)4 (>99%) with a disappointing yield of octenes (34%). Full conversion could be achieved at 165 °C (3 h) giving a higher yield of octenes (85%).118Group 15: bismuth
Moreira and co-workers have investigated Bi(OTf)3·xH2O for its dehydrative properties on tertiary alcohols. It was found that in apolar solvents alkenes were formed in yields of up to 93% with 0.1 mol% catalyst loading. In the polar solvent nitromethane, 2-phenyl-2-propanol was instead dimerised forming a new C–C bond in a yield of 96% with 0.1 mol% catalyst loading (Scheme 13).133 It was demonstrated that the activity is derived from Brønsted acidity rather than Lewis acidity, which suggests that triflic acid is produced in situ and it is in fact the triflic acid that drives the reaction rather than the bismuth salt.133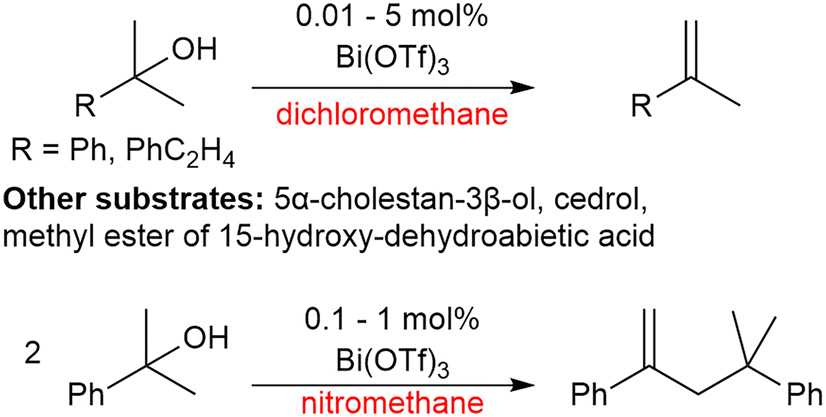 | ||
| Scheme 13 Effect of solvent on bismuth triflate catalysed dehydration.133 | ||
Ionic liquids
Another approach to homogeneous catalysis is to use ionic liquids as the reaction medium.134 Kerton and co-workers investigated the use of phosphonium and imidazolium ionic liquids as reaction media for the dehydration of benzylic alcohols to ethers in the presence of a Pd catalyst.135 However, the group then discovered the Pd catalyst was not necessary.136 If β-hydrogens were present on the substrate, dehydrations to alkenes were observed. 1-Phenylethanol was converted to styrene in 85% yield using trihexyl(tetradecyl)phosphonium bromide in a microwave reactor at 160 °C for 45 minutes.136 The dehydration of 1-indanol gave indene in higher yield (120 °C, 30 min, 96%) due to the more stable internal alkene that was formed.Conclusion
This literature review has identified a variety of catalysts for the dehydration of alcohols to alkenes. Well established catalysts for dehydration are simple Brønsted acids and heterogeneous systems using γ-alumina or the zeolite ZSM-5, with the order of reactivity for alcohols typically following the trend 3° > 2° > 1°. In comparison, homogeneous catalysts were often found to have lower temperature pathways for dehydration. Some of the most important catalysts for the direct dehydration of alcohols to alkenes are transition metal triflate catalysts, specifically Ti(OTf)4, Hf(OTf)4 and Fe(OTf)3. Other key homogeneous catalysts include rhenium complexes such as MTO and Re2O7 which showed the efficient dehydration of a wide variety of alcohols. The identification of molecular iron catalysts for dehydration reactions is potentially important for producing a cost-effective process. Utilising Rh organometallic reactivity for alcohol dehydration has also been successful, via oxidative addition of in situ formed alkyl halides followed by β-hydride elimination, but required HI and an imidazolium or ammonium halide additive. This reactivity is reminiscent of Pt chemistry in the formation of Zeise's salt, however, reactions of Pt complexes remain stoichiometric for the time being. The dehydration of ethanol to ethene is already a commercially successful process, but to expand this to other alcohols, simple catalysts with high yields and selectivity will be required at low reaction temperatures in order to make a less energy intensive process.Conflicts of interest
There are no conflicts to declare.Acknowledgements
DJW thanks Lubrizol and EaSI-cat for funding a PhD studentship. DJW and SMM thank the Royal Society of Chemistry for an RSC Research Enablement Grant (E21-8488203428).Notes and references
- C. W. Fernelius, H. Wittcoff and R. E. Varnerin, J. Chem. Educ., 1979, 56, 385 CrossRef.
- V. Hulea, ACS Catal., 2018, 8, 3263–3279 CrossRef CAS.
- J. Kiefer, Energies, 2015, 8, 3165–3197 CrossRef CAS.
- R. P. Philp, J. High Resolut. Chromatogr., 1994, 17, 398–406 CrossRef CAS.
- A. Akar and E. Ekinci, Fuel, 1995, 74, 1113–1117 CrossRef CAS.
- P. Wiseman, J. Chem. Educ., 1977, 54, 154 CrossRef CAS.
- B. M. Stadler, C. Wulf, T. Werner, S. Tin and J. G. de Vries, ACS Catal., 2019, 9, 8012–8067 CrossRef CAS.
- M. Bender, ChemBioEng Rev., 2014, 1, 136–147 CrossRef CAS.
- R. Sadeghbeigi, in Fluid Catalytic Cracking Handbook (Fourth Edition), ed. R. Sadeghbeigi, Butterworth-Heinemann, 2020, pp. 1–22 Search PubMed.
- T. Ren, M. K. Patel and K. Blok, Energy, 2008, 33, 817–833 CAS.
- T. Ren, M. Patel and K. Blok, Energy, 2006, 31, 425–451 CrossRef CAS.
- I. Amghizar, J. N. Dedeyne, D. J. Brown, G. B. Marin and K. M. Van Geem, React. Chem. Eng., 2020, 5, 239–257 RSC.
- A. Mohsenzadeh, A. Zamani and M. J. Taherzadeh, ChemBioEng Rev., 2017, 4, 75–91 CrossRef.
- A. G. Dedov, A. A. Karavaev, A. S. Loktev and A. K. Osipov, Pet. Chem., 2021, 61, 1139–1157 CrossRef CAS.
- M. Melikoglu, V. Singh, S. Y. Leu, C. Webb and C. S. K. Lin, in Handbook of Biofuels Production (Second Edition), ed. R. Luque, C. S. K. Lin, K. Wilson and J. Clark, Woodhead Publishing, 2016, pp. 237–258 Search PubMed.
- H. Aitchison, R. L. Wingad and D. F. Wass, ACS Catal., 2016, 6, 7125–7132 CrossRef CAS.
- M. Dryzhakov, E. Richmond and J. Moran, Synthesis, 2016, 48, 935–959 CrossRef CAS.
- C. Liu, B. Pan and Y. Gu, Chin. J. Catal., 2016, 37, 979–986 CrossRef CAS.
- J. K. F. Buijink, J.-P. Lange, A. N. R. Bos, A. D. Horton and F. G. M. Niele, in Mechanisms in Homogeneous and Heterogeneous Epoxidation Catalysis, ed. S. T. Oyama, Elsevier, Amsterdam, 2008, pp. 355–371 Search PubMed.
- J.-P. Lange and V. Otten, Ind. Eng. Chem. Res., 2007, 46, 6899–6903 CrossRef CAS.
- H. Adkins and P. P. Perkins, J. Am. Chem. Soc., 1925, 47, 1163–1167 CrossRef CAS.
- C. Bockisch, E. D. Lorance, H. E. Hartnett, E. L. Shock and I. R. Gould, ACS Earth Space Chem., 2018, 2, 821–832 CrossRef CAS.
- S.-Y. Park, Y. M. Lee, K. Kwac, Y. Jung and O.-H. Kwon, Chem. – Eur. J., 2016, 22, 4340–4344 CrossRef CAS PubMed.
- V. A. Chumachenko and E. V. Ovchinnikova, Katal. Prom-sti., 2016, 8, 134–138 Search PubMed.
- C. B. Phillips and R. Datta, Ind. Eng. Chem. Res., 1997, 36, 4466–4475 CrossRef CAS.
- Y. Huang, X. Dong, M. Li and Y. Yu, Catal. Sci. Technol., 2015, 5, 1093–1105 RSC.
- V. Zacharopoulou and A. A. Lemonidou, Catalysts, 2018, 8, 2 CrossRef.
- A. R. Petersen and P. Fristrup, Chem. – Eur. J., 2017, 23, 10235–10243 CrossRef CAS PubMed.
- S. Raju, M.-E. Moret and R. J. M. Klein Gebbink, ACS Catal., 2015, 5, 281–300 CrossRef CAS.
- M. Zhang and Y. Yu, Ind. Eng. Chem. Res., 2013, 52, 9505–9514 CrossRef CAS.
- A. Morschbacker, Polym. Rev., 2009, 49, 79–84 CrossRef CAS.
- A. Tripodi, M. Belotti and I. Rossetti, ACS Sustainable Chem. Eng., 2019, 7, 13333–13350 CrossRef CAS.
- N. Tabassum and S. S. Ali, Catal. Surv. Asia, 2022, 26, 261–280 CrossRef CAS.
- F. Jamil, M. Aslam, A. A. H. Al-Muhtaseb, A. Bokhari, S. Rafiq, Z. Khan, A. Inayat, A. Ahmed, S. Hossain, M. S. Khurram and M. S. A. Bakar, Rev. Chem. Eng., 2022, 38, 185–207 CrossRef CAS.
- D. Fan, D.-J. Dai and H.-S. Wu, Materials, 2013, 6, 101–115 CrossRef CAS PubMed.
- V. L. Dagle, A. D. Winkelman, N. R. Jaegers, J. Saavedra-Lopez, J. Hu, M. H. Engelhard, S. E. Habas, S. A. Akhade, L. Kovarik, V.-A. Glezakou, R. Rousseau, Y. Wang and R. A. Dagle, ACS Catal., 2020, 10, 10602–10613 CrossRef CAS.
- G. Pomalaza, P. Arango Ponton, M. Capron and F. Dumeignil, Catal. Sci. Technol., 2020, 10, 4860–4911 RSC.
- K. Wang, L. Guo, W. Gao, B. Zhang, H. Zhao, J. Liang, N. Liu, Y. He, P. Zhang, G. Yang and N. Tsubaki, ACS Sustainable Chem. Eng., 2021, 9, 10569–10578 CrossRef CAS.
- A. D. Chowdhury, A. Lucini Paioni, G. T. Whiting, D. Fu, M. Baldus and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2019, 58, 3908–3912 CrossRef CAS PubMed.
- N. M. Eagan, M. D. Kumbhalkar, J. S. Buchanan, J. A. Dumesic and G. W. Huber, Nat. Rev. Chem., 2019, 3, 223–249 CrossRef CAS.
- P. Hjerrild, T. Tørring and T. B. Poulsen, Nat. Prod. Rep., 2020, 37, 1043–1064 RSC.
- F. C. Jentoft, Catal. Sci. Technol., 2022, 12, 6308–6358 RSC.
- M. J. Antal, M. Carlsson, X. Xu and D. G. M. Anderson, Ind. Eng. Chem. Res., 1998, 37, 3820–3829 CrossRef CAS.
- D. E. Pearson, R. D. Tanner, I. D. Picciotto, J. S. Sawyer and J. H. Cleveland Jr, Ind. Eng. Chem. Prod. Res. Dev., 1981, 20, 734–740 CrossRef CAS.
- J. Clark, Dehydrating Alcohols to Make Alkenes, https://chem.libretexts.org/@go/page/3901, (accessed 11th March, 2022).
- T. J. Korstanje, J. T. B. H. Jastrzebski and R. J. M. Klein Gebbink, ChemSusChem, 2010, 3, 695–697 CrossRef CAS PubMed.
- N. P. Makgoba, T. M. Sakuneka, J. G. Koortzen, C. van Schalkwyk, J. M. Botha and C. P. Nicolaides, Appl. Catal., A, 2006, 297, 145–150 CrossRef CAS.
- J. Manassen and F. S. Klein, J. Chem. Soc., 1960, 4203–4213 RSC.
- N. N. Greenwood and A. Earnshaw, Chemistry of the Elements, Pergamon Press Ltd., Oxford, 1984, p. 601 Search PubMed.
- V. Siracusa and I. Blanco, Polymers, 2020, 12, 1641 CrossRef CAS PubMed.
- L. Brandsma and J. F. Arens, in The Ether Linkage (1967), 1967, pp. 553–615 Search PubMed.
- H. Feuer and J. Hooz, in The Ether Linkage (1967), 1967, pp. 445–498 Search PubMed.
- M. Smith and J. March, March's advanced organic chemistry: Reactions, mechanisms, and structure, Wiley, New York, 5th edn, 2001 Search PubMed.
- L. Karas and W. J. Piel, Ethers in Kirk-Othmer Encyclopedia of Chemical Technology, 2004, pp. 567–583 Search PubMed.
- A. de Klerk, in Fischer-Tropsch Refining, ed. A. de Klerk, John Wiley & Sons, Incorporated, 2011, ch. 17, Dehydration, Etherification, and Hydration, pp. 335–352 Search PubMed.
- B. C. Gates and L. N. Johanson, J. Catal., 1969, 14, 69–76 CrossRef CAS.
- S. M. Hsu and R. L. Kabel, J. Catal., 1974, 33, 74–82 CrossRef CAS.
- J. Guilera, E. Ramírez, C. Fité, M. Iborra and J. Tejero, Appl. Catal., A, 2013, 467, 301–309 CrossRef CAS.
- L. M. T. Frija and C. A. M. Afonso, Tetrahedron, 2012, 68, 7414–7421 CrossRef CAS.
- R. L. Kabel and L. N. Johanson, AIChE J., 1962, 8, 621–628 CrossRef CAS.
- J. Tejero, C. Fité, M. Iborra, J. F. Izquierdo, R. Bringué and F. Cunill, Appl. Catal., A, 2006, 308, 223–230 CrossRef CAS.
- J. C. Gottifredi, A. A. Yeramian and R. E. Cunningham, J. Catal., 1968, 12, 245–256 CrossRef CAS.
- R. Thornton and B. C. Gates, J. Catal., 1974, 34, 275–287 CrossRef CAS.
- G. H. Posner, E. M. Shulman-Roskes, C. H. Oh, J.-C. Carry, J. V. Green, A. B. Clark, H. Dai and T. E. N. Anjeh, Tetrahedron Lett., 1991, 32, 6489–6492 CrossRef CAS.
- H. Firouzabadi, N. Iranpoor, H. Hazarkhani and B. Karimi, Synth. Commun., 2003, 33, 3653–3660 CrossRef CAS.
- G. L. O'Connor and H. R. Nace, J. Am. Chem. Soc., 1955, 77, 1578–1581 CrossRef.
- P. Sudarsanam, E. Peeters, E. V. Makshina, V. I. Parvulescu and B. F. Sels, Chem. Soc. Rev., 2019, 48, 2366–2421 RSC.
- T. Ennaert, J. Van Aelst, J. Dijkmans, R. De Clercq, W. Schutyser, M. Dusselier, D. Verboekend and B. F. Sels, Chem. Soc. Rev., 2016, 45, 584–611 RSC.
- T. K. Phung, L. Proietti Hernández, A. Lagazzo and G. Busca, Appl. Catal., A, 2015, 493, 77–89 CrossRef CAS.
- R. Prins, J. Catal., 2020, 392, 336–346 CrossRef CAS.
- R. Wang, Z. Zhao, P. Gao, K. Chen, Z. Gan, Q. Fu and G. Hou, J. Phys. Chem. C, 2022, 126, 10073–10080 CrossRef CAS.
- H. Adkins and S. H. Watkins, J. Am. Chem. Soc., 1951, 73, 2184–2188 CrossRef CAS.
- H. Pines and W. O. Haag, J. Am. Chem. Soc., 1961, 83, 2847–2852 CrossRef CAS.
- C.-Y. Wu and H.-S. Wu, ACS Omega, 2017, 2, 4287–4296 CrossRef CAS PubMed.
- J. H. de Boer, R. B. Fahim, B. G. Linsen, W. J. Visseren and W. F. N. M. de Vleesschauwer, J. Catal., 1967, 7, 163–172 CrossRef CAS.
- T. K. Phung, C. Herrera, M. Á. Larrubia, M. García-Diéguez, E. Finocchio, L. J. Alemany and G. Busca, Appl. Catal., A, 2014, 483, 41–51 CrossRef CAS.
- H. Knözinger, H. Bühl and K. Kochloefl, J. Catal., 1972, 24, 57–68 CrossRef.
- B. Shi, H. A. Dabbagh and B. H. Davis, J. Mol. Catal. A: Chem., 1999, 141, 257–262 CrossRef CAS.
- Z. Wang, L. A. O'Dell, X. Zeng, C. Liu, S. Zhao, W. Zhang, M. Gaborieau, Y. Jiang and J. Huang, Angew. Chem., Int. Ed., 2019, 58, 18061–18068 CrossRef CAS PubMed.
- M. Digne, P. Sautet, P. Raybaud, P. Euzen and H. Toulhoat, J. Catal., 2002, 211, 1–5 CrossRef CAS.
- D. J. Collins, J. C. Watters and B. H. Davis, Ind. Eng. Chem. Prod. Res. Dev., 1979, 18, 202–205 CrossRef CAS.
- A. Auroux, P. Artizzu, I. Ferino, V. Solinas, G. Leofanti, M. Padovan, G. Messina and R. Mansani, J. Chem. Soc., Faraday Trans., 1995, 91, 3263–3267 RSC.
- F. Al-Bahar, D. J. Collins, J. C. Watters and B. H. Davis, Ind. Eng. Chem. Prod. Res. Dev., 1979, 18, 199–202 CrossRef CAS.
- P. Kostestkyy, J. Yu, R. J. Gorte and G. Mpourmpakis, Catal. Sci. Technol., 2014, 4, 3861–3869 RSC.
- E. Hong, H.-I. Sim and C.-H. Shin, Chem. Eng. J., 2016, 292, 156–162 CrossRef CAS.
- B. H. Davis, S. Cook and R. W. Naylor, J. Org. Chem., 1979, 44, 2142–2145 CrossRef CAS.
- B. H. Davis, J. Catal., 1978, 52, 435–444 CrossRef CAS.
- C. Martínez and A. Corma, in Comprehensive Inorganic Chemistry II (Second Edition), ed. J. Reedijk and K. Poeppelmeier, Elsevier, Amsterdam, 2013, pp. 103–131 Search PubMed.
- M. Ravi, V. L. Sushkevich and J. A. van Bokhoven, Nat. Mater., 2020, 19, 1047–1056 CrossRef CAS PubMed.
- D. Gabriëls, W. Y. Hernández, B. Sels, P. Van Der Voort and A. Verberckmoes, Catal. Sci. Technol., 2015, 5, 3876–3902 RSC.
- J. M. Müller, G. C. Mesquita, S. M. Franco, L. D. Borges, J. L. de Macedo, J. A. Dias and S. C. L. Dias, Microporous Mesoporous Mater., 2015, 204, 50–57 CrossRef.
- Y. Tavan, S. H. Hosseini, M. Ghavipour, M. R. Khosravi Nikou and A. Shariati, Chem. Eng. Process., 2013, 73, 144–150 CrossRef CAS.
- Q. Sheng, K. Ling, Z. Li and L. Zhao, Fuel Process. Technol., 2013, 110, 73–78 CrossRef CAS.
- P. Tian, Y. Wei, M. Ye and Z. Liu, ACS Catal., 2015, 5, 1922–1938 CrossRef CAS.
- I. Takahara, M. Saito, M. Inaba and K. Murata, Catal. Lett., 2005, 105, 249–252 CrossRef CAS.
- X. Zhang, R. Wang, X. Yang and F. Zhang, Microporous Mesoporous Mater., 2008, 116, 210–215 CrossRef CAS.
- J. Ouyang, F. Kong, G. Su, Y. Hu and Q. Song, Catal. Lett., 2009, 132, 64–74 CrossRef CAS.
- R. V. Ermakov and V. A. Plakhotnik, Pet. Chem., 2008, 48, 1–5 CrossRef.
- K. K. Ramasamy and Y. Wang, J. Energy Chem., 2013, 22, 65–71 CrossRef CAS.
- Y. Shi, A. S. Weller, A. J. Blacker and P. W. Dyer, Catal. Commun., 2022, 164, 106421 CrossRef CAS.
- A. Styskalik, V. Vykoukal, L. Fusaro, C. Aprile and D. P. Debecker, Appl. Catal., B, 2020, 271, 118926 CrossRef CAS.
- C. P. Nash, A. Ramanathan, D. A. Ruddy, M. Behl, E. Gjersing, M. Griffin, H. Zhu, B. Subramaniam, J. A. Schaidle and J. E. Hensley, Appl. Catal., A, 2016, 510, 110–124 CrossRef CAS.
- D. Seyferth, Organometallics, 2001, 20, 2–6 CrossRef CAS.
- W. C. Zeise, Ann. Phys., 1831, 97, 497–541 CrossRef.
- J. S. Thayer, J. Chem. Educ., 1969, 46, 442 CrossRef CAS.
- J. S. Anderson, J. Chem. Soc., 1934, 971–974 RSC.
- J. Chatt and M. L. Searle, Inorg. Synth., 1957, 5, 210–215 CAS.
- P. B. Chock, J. Halpern, F. E. Paulik, S. I. Shupack and T. P. Deangelis, in Inorg. Synth., 1990, pp. 349–351 Search PubMed.
- F. R. Hartley, Inorg. Chim. Acta, 1971, 5, 197–198 CrossRef CAS.
- S. A. Mitchenko, A. N. Vdovichenko, T. V. Bezbozhnaya and L. M. Kapkan, Theor. Exp. Chem., 2005, 41, 203–206 CrossRef CAS.
- A. C. Hutson, M. Lin, N. Basickes and A. Sen, J. Organomet. Chem., 1995, 504, 69–74 CrossRef CAS.
- A. Sen, M. Lin, L. C. Kao and A. C. Hutson, J. Am. Chem. Soc., 1992, 114, 6385–6392 CrossRef CAS.
- O. V. Khazipov, D. V. Nykytenko, T. V. Krasnyakova, A. N. Vdovichenko, D. A. Fuentes Frias and S. A. Mitchenko, J. Mol. Catal. A: Chem., 2017, 426, 490–498 CrossRef CAS.
- J. A. Labinger, A. M. Herring, D. K. Lyon, G. A. Luinstra, J. E. Bercaw, I. T. Horvath and K. Eller, Organometallics, 1993, 12, 895–905 CrossRef CAS.
- J. A. Labinger, A. M. Herring and J. E. Bercaw, J. Am. Chem. Soc., 1990, 112, 5628–5629 CrossRef CAS.
- A. Sen, M. A. Benvenuto, M. Lin, A. C. Hutson and N. Basickes, J. Am. Chem. Soc., 1994, 116, 998–1003 CrossRef CAS.
- R. Schobert, J. Organomet. Chem., 1991, 405, 201–205 CrossRef CAS.
- J. Keskiväli, A. Parviainen, K. Lagerblom and T. Repo, RSC Adv., 2018, 8, 15111–15118 RSC.
- M. L. Kantam, A. D. Prasad and P. L. Santhi, Synth. Commun., 1993, 23, 45–48 CrossRef CAS.
- T. J. Korstanje, E. Folkertsma, M. Lutz, J. T. B. H. Jastrzebski and R. J. M. Klein Gebbink, Eur. J. Inorg. Chem., 2013, 2013, 2195–2204 CrossRef CAS.
- J. Li, A. Das, Q. Ma, M. J. Bedzyk, Y. Kratish and T. J. Marks, ACS Catal., 2022, 12, 1247–1257 CrossRef CAS.
- Z. Zhu and J. H. Espenson, J. Org. Chem., 1996, 61, 324–328 CrossRef CAS.
- T. J. Korstanje, M. Lutz, J. T. B. H. Jastrzebski and R. J. M. Klein Gebbink, Organometallics, 2014, 33, 2201–2209 CrossRef CAS.
- T. J. Korstanje, E. F. de Waard, J. T. B. H. Jastrzebski and R. J. M. Klein Gebbink, ACS Catal., 2012, 2, 2173–2181 CrossRef CAS.
- T. J. Korstanje, J. T. B. H. Jastrzebski and R. J. M. Klein Gebbink, Chem. – Eur. J., 2013, 19, 13224–13234 CrossRef CAS PubMed.
- S. Raju, J. T. B. H. Jastrzebski, M. Lutz and R. J. M. Klein Gebbink, ChemSusChem, 2013, 6, 1673–1680 CrossRef CAS PubMed.
- P. K. Sahoo, S. S. Gawali and C. Gunanathan, ACS Omega, 2018, 3, 124–136 CrossRef CAS PubMed.
- O. Nachtigall, A. I. VanderWeide, W. W. Brennessel and W. D. Jones, ACS Catal., 2021, 11, 10885–10891 CrossRef CAS.
- G. R. M. Dowson, I. V. Shishkov and D. F. Wass, Organometallics, 2010, 29, 4001–4003 CrossRef CAS.
- N. Yoneda, S. Kusano, M. Yasui, P. Pujado and S. Wilcher, Appl. Catal., A, 2001, 221, 253–265 CrossRef CAS.
- K. Laali, R. J. Gerzina, C. M. Flajnik, C. M. Geric and A. M. Dombroski, Helv. Chim. Acta, 1987, 70, 607–611 CrossRef CAS.
- M. H. Lee, E. T. Choi, D. Kim, Y. M. Lee and Y. S. Park, Eur. J. Org. Chem., 2008, 2008, 5630–5637 CrossRef.
- L. E. Kolsi, J. Yli-Kauhaluoma and V. M. Moreira, ACS Omega, 2018, 3, 8836–8842 CrossRef CAS PubMed.
- Z. Lei, B. Chen, Y.-M. Koo and D. R. MacFarlane, Chem. Rev., 2017, 117, 6633–6635 CrossRef PubMed.
- H. A. Kalviri, C. F. Petten and F. M. Kerton, Chem. Commun., 2009, 5171–5173 RSC.
- H. A. Kalviri and F. M. Kerton, Adv. Synth. Catal., 2011, 353, 3178–3186 CrossRef CAS.
Footnote |
| † Dedicated to Prof. Paul R. Raithby, celebrating a career in inorganic and organometallic chemistry, on the occasion of his 70th birthday. |
| This journal is © The Royal Society of Chemistry 2023 |