
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluence and stability of the surface density of MoOx on TiO2 in deoxydehydration: structure–activity correlations†
Joby
Sebastian
a,
Chalachew
Mebrahtu
 a and
Regina
Palkovits
*ab
a and
Regina
Palkovits
*ab
aInstitut für Technische und Makromolekulare Chemie, RWTH Aachen University, Worringerweg 2, 52074 Aachen, Germany. E-mail: palkovits@itmc.rwth-aachen.de
bMax-Planck-Institut for Chemical Energy Conversion, Stiftstr. 34-36, 45470 Mülheim an der Ruhr, Germany
First published on 4th January 2023
Abstract
Titania supported MoOx catalysts containing varying surface densities of Mo were prepared and well-characterized to identify the most effective MoOx species for the deoxydehydration (DODH) reaction of 1,4-anhydroerythritol (1,4-AHE), and eventually close the gap between experimental observations and theoretical predictions. Structure–activity correlations revealed isolated MoOx species to be more active than oligomeric/polymeric/crystalline MoOx species. Decreasing activity with increasing Mo surface density proved to be a result of the increase in strong acidity of the catalysts. These acid sites favored side reactions involving dehydration reaction and ketal formation. Comparative studies with dried and calcined catalysts showed that calcination moderately improves the selectivity of isolated MoOx species while it decreases the selectivity of polymerized MoOx species. This was due to the increased Brønsted acidity of the catalyst, a consequence of calcination. Brønsted acid sites were not present on isolated MoOx species. Regarding leaching, the calcination only benefited the isolated species as the leached species were DODH inactive while for polymerized species, due to their higher loading, the leached species were DODH active. The use of a merely dried catalyst exposed significant leaching of catalytically active species into the reaction medium in the early hours of the reaction. Recycle studies with the most active catalyst demonstrated the stable loading of Mo on TiO2, carbon deposition, and possible acidity regeneration during heat treatments.
1. Introduction
Producing fuels and chemicals from renewable lignocellulosic biomass is an important way to address sustainability in chemical production, accelerating the transition from a linear economy to a circular economy, and regulate net CO2 emissions.1–4 However, the high oxygen content of lignocellulosic biomass feedstocks such as carbohydrates and lignin presents a major challenge for selective transformation to valuable products. Chemical deoxygenation strategies, like dehydration5,6 and hydrogenolysis,7–10 are the conventional solutions to this hurdle; however, lately, deoxydehydration (DODH) has significantly gained attention.11,12 DODH involves the effective removal of two oxygen atoms of a vicinal diol to a double bond by making use of a sacrificial reducing agent. The resulting alkene can serve as a platform chemical for the synthesis of monomers, specialty chemicals, and pharmaceuticals. Sugar alcohols, e.g. glycerol, and a variety of vicinal diols containing aliphatic, alicyclic, and aromatic substituents can be used as substrates for DODH. Concerning the sacrificial reducing agent, various alcohols, H2, CO, PPh3, etc. can be used.11Homogeneous organometallic complexes containing active centers Re, Mo, and V are most prevalent for DODH, thanks to their variable oxidation states.11 As compared to homogeneous catalysts, heterogeneous, solid catalysts are advantageous on the road to industrial implementation. The first solid catalyst for DODH, a carbon-supported ReOx catalyst, was reported by the group of Jentoft.13 Later, many supported ReOx catalysts on various supports such as TiO2,14 CeO2,15–18 ZrO2,19,20 Al2O3,20,21 and zeolites22 ensued. Though ReOx-based catalysts were highly effective for DODH, they hold the disadvantage of high price and leaching of Re.19 To circumvent these shortcomings, our group has reported a Mo-based solid catalyst (MoOx/TiO2) as a cost-effective alternative to ReOx systems.23 MoOx/TiO2 showed fairly good activity and stability in DODH of 1,4-anhydroerythritol (1,4-AHE) to 2,5-dihydrofuran (2,5-DHF) with 3-octanol as a reducing agent. Polymerized MoOx species owing to the high loading of Mo (4.9 wt%) on TiO2 were identified as the active species for DODH. Following this, Nicholas et al. compared the activities of MoOx on various supports such as TiO2, ZrO2, SiO2, Al2O3, and α-Fe2O3 in the DODH of 1,2-decanediol.24 The MoOx coverage on Al2O3, SiO2, and ZrO2 was nearly monolayers and on α-Fe2O3 and TiO2, it was multilayers. The most active catalysts (TOF = 1.2 × 10−4 s−1) were MoOx/TiO2 and MoOx/SiO2, revealing that both oligomeric and polymeric MoOx species are equally active for DODH. Recently, Tomishige et al. reported MoOx cluster species in their MoOx–Au/TiO2 (1 wt% of Mo) catalyst to be the active species for the DODH of 1,4-AHE using H2 as the reducing agent.25 In contrast to these experimental observations, through density functional theory (DFT) calculations, Fristrup et al. suggested mononuclear MoOx species to be more active in the DODH of 1,2-propanediol than its next higher homologue, the dinuclear MoOx species.26 Consistent with this finding, a first principles calculation on anatase TiO2(101) by Hayden et al. also demonstrated the effectiveness of monomeric MoOx species for the DODH of 1,4-AHE.27 Their obtained TOF was 4.8 times higher than that obtained by Nicholas et al.24 implying a higher activity of monomeric MoOx species compared with oligomeric/polymeric species. According to these reports, MoOx species of different nuclearities/surface densities appear to be active for DODH. Theoretical calculations concluded that mononuclear MoOx species possess superior activity; however, this hypothesis still requires experimental validation. Moreover, comparing the activities of MoOx species of increasing nuclearities starting from monomeric to oligomeric to polymerized to crystallites appears necessary to establish solid structure–activity correlations on the most active MoOx species as a basis of future catalyst design. Along this line, MoOx catalysts of previous experimental studies were employed either in the as-synthesized form, where the active species were proposed to be formed in situ,25 or after reduction treatment using H2,23 or after calcination in N2 or air.24 The importance of a prior temperature treatment of the catalyst for its activity and stability is not well understood; though, this step is pivotal for generating highly active MoOx species for DODH.
In this study, we report the effect of MoOx surface density on TiO2 for the model DODH reaction of 1,4-AHE using 3-octanol as the reducing agent (Scheme 1). Comprehensive structure–activity relations allow revealing the most effective MoOx species for DODH. Therefore, a set of catalysts possessing increasing surface densities of MoOx on TiO2 ranging from isolated species to highly polymerized/crystallized MoOx species were prepared and systematically characterized. Activity comparison of these catalysts was performed in the kinetic regime to ensure the identification of the most effective MoOx species for DODH. The consequences of a calcination step on MoOx species of different surface densities on catalytic activity and stability were examined through comparative studies involving dried catalysts, leaching experiments, and recycle studies. Correlations were made with regard to the effect of Mo surface density and catalyst acidity on catalytic activity and selectivity aiming to identify the critical features of an optimum Mo-based solid catalyst for DODH.
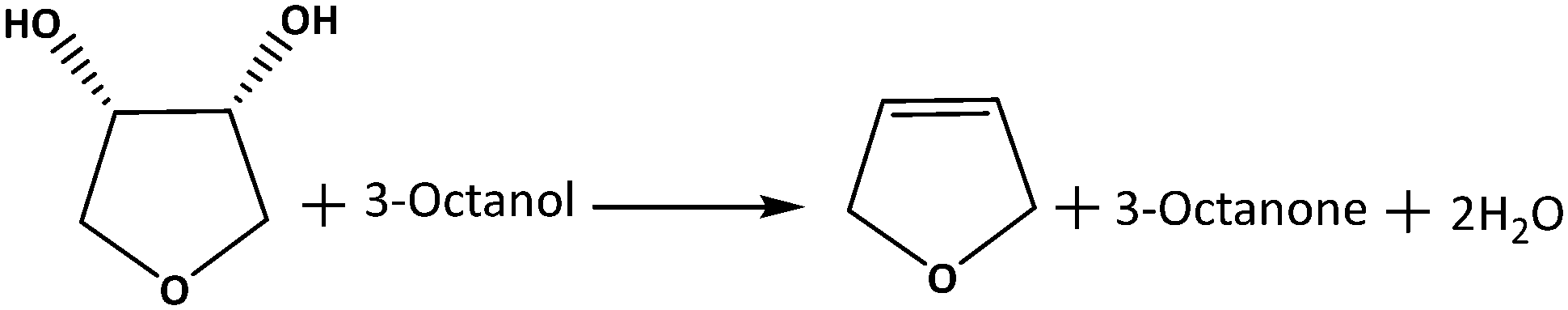 | ||
| Scheme 1 DODH of 1,4-AHE to 2,5-DHF using 3-octanol as the reducing agent. | ||
2. Experimental
2.1. Materials and reagents
AEROXIDE® TiO2 P25 and n-dodecane were procured from Acros Organics. Ammonium molybdate [(NH4)2MoO4, 99.98% trace metal basis] and 3-octanol (99%) were obtained from Sigma Aldrich. 1,4-Anhydroerythritol (>98%) was purchased from TCI. Ethanol (99.5%) was received from Merck.2.2. Catalyst synthesis
All the catalysts were prepared by the wetness impregnation method. A typical synthesis of 3.1 wt% Mo on TiO2 is as follows. First, the TiO2 powder was calcined at 500 °C for 4 h (2 °C min−1). Then, 1 g of calcined TiO2 was added to a solution of 0.0633 g of ammonium molybdate in 20 mL of deionized water and stirred for 24 h at room temperature. At the end of the impregnation, the water in the slurry was removed by rotary evaporation. The solid sample obtained was then dried at 80 °C under air for 3 days. Finally, the dried solid sample was calcined in a muffle furnace at 500 °C for 4 h (2 °C min−1) for characterization and activity studies.Catalysts containing 0.44, 0.88, 1.32, 1.75, and 3.95 wt% Mo were also prepared following the same procedure as above but varying the amounts of ammonium molybdate precursors in the impregnating solution.
2.3. Catalyst characterization
Powder X-ray diffraction patterns of the catalysts were recorded on a Bruker (D2 Phaser) diffractometer with a Lynxeye XE-T detector in the 2θ range of 10–90° using Cu-Kα radiation (λ = 0.154184 nm). The elemental composition of the catalysts was measured by the inductively coupled plasma-optical emission spectroscopy (ICP-OES) method by using a SpectroBlue ICP-OES analyzer instrument. An acid mixture comprised of concentrated H2SO4, HNO3, and HF was used as the digestion medium for the catalysts. Textural properties of the catalysts were analyzed using a Micromeritics ASAP 2060 instrument. The specific surface areas of the catalysts were calculated by applying the Brunauer, Emmett and Teller (BET) method. The total amount of acid sites on the catalysts was quantified by temperature-programmed desorption of ammonia (NH3-TPD) using a Micromeritics Autochem 2950 HP chemisorption analyzer instrument. Ammonia adsorption and desorption on the thermally activated catalysts (500 °C for 0.5 h) were conducted at 80 °C. Pyridine adsorbed Fourier transform infrared spectroscopy (Py-FTIR) measurements were done on a Bruker Optics Vertex 70 instrument with a homemade stainless-steel IR cell furnished with CaF2 windows. The catalyst samples (20 mg) were pressed on top of a KBr pellet and used for the analysis. Pyridine adsorption was done at 80 °C. After equilibration at 80 °C for 30 min and desorption at 150 °C for 30 min, the final spectra containing strongly adsorbed pyridine were recorded at 80 °C under vacuum (0.006 mbar). Thermogravimetric analysis of the catalyst after the reaction was performed on NETZSCH STA 409 equipment in an air atmosphere (5 °C min−1). Raman analyses of the catalysts under ambient conditions were performed on a Bruker RFS 100/S instrument operating at a wavelength of 1064 nm. The spectral resolution was 4 cm−1 and the number of scans was 700.2.4. Catalytic reaction
The DODH reactions of 1,4-AHE using 3-octanol as the sacrificial reducing agent over various catalysts were conducted in a glass reactor (Ace Druck glass pressure tube, 9 mL capacity). A stock solution containing 1,4-AHE and 3-octanol with a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 (mol:mol) was used for the activity studies. In a typical catalytic reaction, 0.050 g of 0.5Mo catalyst was placed in the glass reactor followed by 0.900 g of the reactant stock solution. The air inside the tube was purged with N2 and was sealed. The reactor was then placed inside a pre-heated (200 °C) aluminum block with a conical well. The stirring (600 rpm) was initiated and the reaction was conducted for a period of 18 h. At the end of the reaction time, the tube was taken out of the heating block and was immediately cooled down by immersing it in an ice bath. The product mixture was then filtered through a polyamide filter (pore diameter = 0.20 μm) and used for gas chromatography (GC) analysis.
:9 (mol:mol) was used for the activity studies. In a typical catalytic reaction, 0.050 g of 0.5Mo catalyst was placed in the glass reactor followed by 0.900 g of the reactant stock solution. The air inside the tube was purged with N2 and was sealed. The reactor was then placed inside a pre-heated (200 °C) aluminum block with a conical well. The stirring (600 rpm) was initiated and the reaction was conducted for a period of 18 h. At the end of the reaction time, the tube was taken out of the heating block and was immediately cooled down by immersing it in an ice bath. The product mixture was then filtered through a polyamide filter (pore diameter = 0.20 μm) and used for gas chromatography (GC) analysis.
2.5. Cold and warm filtration tests
The cold filtration test was conducted after 1 h of the reaction. After the designated reaction time, the reactor was cooled down to room temperature using a water bath, the catalyst was separated by centrifugation and the supernatant was filtered through a polyamide filter (pore diameter = 0.20 μm). The filtrate was then allowed to react for 17 h at 200 °C.The warm filtration test was conducted after 24 h of the reaction. Here, the reactor was cooled down to a temperature of 50 °C and the reaction mixture was directly filtered through a polyamide filter (pore diameter = 0.20 μm). The filtrate was then allowed to react for 18 h at 200 °C.
2.6. Recycle experiments
The catalyst after the reaction was separated by centrifugation and washed with absolute ethanol (4 times with 15 mL of ethanol each time). It was then dried under air at 80 °C for 48 h and directly used for the first and second recycle runs. After the second and third recycle runs, an additional calcination step (500 °C for 4 h, 2 °C min−1) of the dried catalyst was performed before their consecutive runs.2.7. Product analysis
Identification of the reaction products was achieved through a gas chromatography–mass spectrometry (GC-MS) instrument (Trace 1310 from Thermo Fisher Scientific coupled with an ISQ single quadrupole MS detector). An Optima 1701 column having a 60 m length and 0.25 mm internal diameter was used for the analysis. The injector temperature was set at 250 °C and the detector temperature at 250 °C. The injection volume was 1 μL. Quantification of the various products was done using a Trace GC Ultra instrument (Thermo Fisher Scientific) using the same column (Optima 1701, 60 m, 0.25 mm) and at the same injector and detector temperatures (250 °C). For the quantification, dodecane and absolute ethanol were used as the internal standard and the diluent, respectively. The conversion and the product yield using the corresponding response factors of the different product molecules were calculated by using the following equations: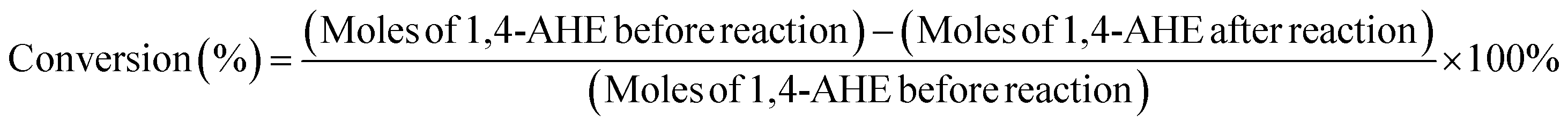

For the quantification of ketals, the effective carbon number method, using the response factor of dodecane, was adopted.
3. Results and discussion
3.1. Catalyst characterization
A series of catalysts with increasing surface densities of Mo atoms per nm2 surface area of TiO2 was designed to derive isolated, oligomerized, and polymerized MoO3 species. This was accomplished by considering the area occupied by a single [MoO6] octahedra (22 Å)28 and the specific surface area of the TiO2 support (58 m2 g−1). The lowest surface density was set at 0.5Mo atoms per nm2 and the highest at 4.5Mo atoms per nm2. Table 1 summarizes the Mo loadings and corresponding Mo surface densities on TiO2. Only a marginal change in the specific surface area of the catalysts with an increase in Mo loading was observed, still, it showed a trend that is decreasing with an increase in Mo loading.| Catalyst | Mo loading (wt%) | BET surface area (m2 g−1) | Actual Mo surface density (Mo nm−2) | Total acidity (μmol NH3 gcat−1) |
|---|---|---|---|---|
| TiO2 | 0.0 | 58 | 0.00 | 796 |
| 0.5Mo | 0.4 | 57 | 0.48 | 327 |
| 1.0Mo | 0.9 | 56 | 0.99 | 310 |
| 1.5Mo | 1.3 | 54 | 1.50 | 300 |
| 2.0Mo | 1.8 | 52 | 2.10 | 270 |
| 3.5Mo | 3.1 | 55 | 3.50 | 251 |
| 4.5Mo | 4.0 | 52 | 4.80 | 244 |
Fig. S1 in the ESI† shows the powder X-ray diffraction (XRD) patterns of the calcined MoOx/TiO2 catalysts. Every catalyst showed reflections attributable purely to the TiO2 support. No additional reflexes indicative of MoO3 crystallites were observable even at a higher Mo loading of 4.0 wt% (4.8 Mo nm−2) which is very close to the theoretical monolayer coverage of 4.2 wt%. The XRD analysis suggested highly dispersed and amorphous MoOx species on TiO2; nonetheless, the presence of X-ray amorphous MoO3 crystallites of very small size (<4 nm) could not be ruled out.29
Raman spectroscopy is extensively used to scrutinize the coordination behavior of Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O moieties of surface MoOx species.28,30–32 Since TiO2 has no dominant Raman features in the range of 850–1100 cm−1, bands in this region are exclusively attributed to the coordination status of the MoO moieties. Fig. 1 displays the Raman spectra of the catalysts in the MoO stretching region (850–1100 cm−1). For the catalyst containing 0.5Mo nm−2, a recognizable MoO stretching band is observed at 939 cm−1. A gradual shift of the maximum of this band to higher Raman shifts occurred with increasing surface densities of Mo on TiO2, and for the 4.5Mo nm−2 catalyst, this band possessed the maximum at 972 cm−1 (Fig. S2† provides the Raman spectrum of the monomeric precursor (NH4)2MoO4 and the representative spectrum of the polymeric (NH4)6Mo7O24 compound. The MoO stretching frequencies of these compounds appear at 903 cm−1 for (NH4)2MoO4 and 933 cm−1 for (NH4)6Mo7O24. The observed shift in the band position with an increase in Mo surface density can be ascribed to an increasing degree of oligomerization of the MoOx species from isolated species to oligomers and further to polymerized (2D) species.33–35 In comparison, MoO3 crystallites (3D-polymerized) exhibited an MoO stretching band at 996 cm−1 (Fig. 1, inset). A close inspection of the Raman spectra of 3.5Mo and 4.5Mo catalysts revealed the presence of a band at 996 cm−1 with very low intensity, which increased with increasing Mo surface density, suggesting the presence of XRD amorphous nano MoO3 crystallites. For the remaining catalysts (0.5Mo, 1.0Mo, 1.5Mo, and 2.0Mo), this finding implied the presence of XRD amorphous 2-dimensional MoOx species on TiO2. In addition, the MO peaks of MoO3 crystallites are always accompanied by the Mo–O–Mo stretching band at around 820 cm−1 (Fig. S3†). Indeed, the 4.5Mo catalyst showed the incidence of this band with a very low intensity (Fig. S3†) indicating the very little contribution of MoO3 crystallites in it. The 820 cm−1 band for 0.5, 1.0, and 1.5Mo was indistinguishable from the TiO2 background validating the absence of MoOx crystallites in these catalyst compositions. The synthetic approach was effective in modulating the coordination behavior of MoOx species on TiO2 from isolated species to oligomers to polymerized (2D) species, and to crystallites, respectively.
O moieties of surface MoOx species.28,30–32 Since TiO2 has no dominant Raman features in the range of 850–1100 cm−1, bands in this region are exclusively attributed to the coordination status of the MoO moieties. Fig. 1 displays the Raman spectra of the catalysts in the MoO stretching region (850–1100 cm−1). For the catalyst containing 0.5Mo nm−2, a recognizable MoO stretching band is observed at 939 cm−1. A gradual shift of the maximum of this band to higher Raman shifts occurred with increasing surface densities of Mo on TiO2, and for the 4.5Mo nm−2 catalyst, this band possessed the maximum at 972 cm−1 (Fig. S2† provides the Raman spectrum of the monomeric precursor (NH4)2MoO4 and the representative spectrum of the polymeric (NH4)6Mo7O24 compound. The MoO stretching frequencies of these compounds appear at 903 cm−1 for (NH4)2MoO4 and 933 cm−1 for (NH4)6Mo7O24. The observed shift in the band position with an increase in Mo surface density can be ascribed to an increasing degree of oligomerization of the MoOx species from isolated species to oligomers and further to polymerized (2D) species.33–35 In comparison, MoO3 crystallites (3D-polymerized) exhibited an MoO stretching band at 996 cm−1 (Fig. 1, inset). A close inspection of the Raman spectra of 3.5Mo and 4.5Mo catalysts revealed the presence of a band at 996 cm−1 with very low intensity, which increased with increasing Mo surface density, suggesting the presence of XRD amorphous nano MoO3 crystallites. For the remaining catalysts (0.5Mo, 1.0Mo, 1.5Mo, and 2.0Mo), this finding implied the presence of XRD amorphous 2-dimensional MoOx species on TiO2. In addition, the MO peaks of MoO3 crystallites are always accompanied by the Mo–O–Mo stretching band at around 820 cm−1 (Fig. S3†). Indeed, the 4.5Mo catalyst showed the incidence of this band with a very low intensity (Fig. S3†) indicating the very little contribution of MoO3 crystallites in it. The 820 cm−1 band for 0.5, 1.0, and 1.5Mo was indistinguishable from the TiO2 background validating the absence of MoOx crystallites in these catalyst compositions. The synthetic approach was effective in modulating the coordination behavior of MoOx species on TiO2 from isolated species to oligomers to polymerized (2D) species, and to crystallites, respectively.
 | ||
| Fig. 1 Raman spectra of the catalysts containing different surface densities of MoOx. | ||
The presence of Mo on TiO2 can modify the acidic properties of the final catalyst.36 The variation in the total acidity of the catalysts with an increase in Mo loading was monitored and quantified by using temperature-programmed desorption of NH3 (NH3-TPD). The blank TiO2 support possessed the highest total acidity (796 μmol NH3 gcat−1). The addition of 0.4 wt% Mo on the support (0.5Mo) decreased its acidity to 327 μmol NH3 gcat−1. With a further increase in Mo loading, a gradual decrease of the total acidity of the catalysts occurred (Table 1). This observation is in agreement with previous studies on MoOx supported on TiO2,35,37 Al2O3,38 and ZrO228 wherein an increase in loading of MoOx caused a decrease of the total acidity of the supports. The origin of this effect presents the coverage of acid sites of TiO2 by Mo surface species during Mo-impregnation, ultimately leading to the generation of new acid sites, which are less strong than those found on TiO2.35,37
The NH3-TPD profiles of the different catalysts possessed very broad desorption peaks (Fig. 2). Accordingly, the desorption peaks with maxima at around 150, 260, and 360 °C are discernible in all TPD profiles characteristic of the co-existence of acids sites of varying strengths. The desorption at 150 °C was assigned to weak acid sites (mostly physisorbed) and desorption at 260 and 360 °C was assigned to strong acid sites of slightly varying acid strengths. The relative contribution of these acid sites, especially the strong acid sites, to the total acidity was then calculated by signal deconvolution (Fig. S4†). The analysis showed an increase in the contribution of strong acid sites with increasing surface density of Mo on TiO2 (Fig. 3).
 | ||
| Fig. 2 NH3-TPD profiles of the catalysts. | ||
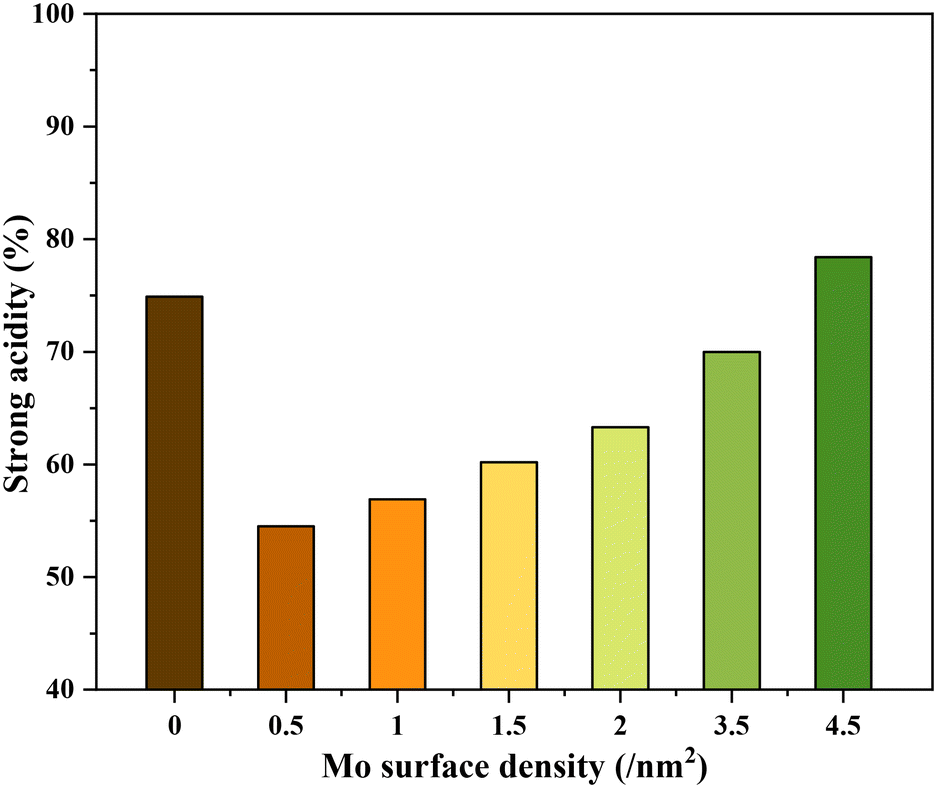 | ||
| Fig. 3 The contribution of strong acidity (%) to the total acidity of the catalysts. | ||
Since NH3-TPD does not allow differentiating Lewis and Brønsted acid sites, Py-FTIR analysis on the representative catalysts (TiO2, 0.5Mo, 2.0Mo, and 3.5Mo) was performed (Fig. 4). The Py-FTIR spectrum of TiO2 confirmed the sole presence of Lewis acid sites (bands at 1445, 1490, 1575, and 1609 cm−1).39,40 Brønsted acid sites were also absent on the 0.5Mo catalyst. However, at a higher loading of Mo (2.0Mo and 3.5Mo), Brønsted acid sites started to occur (Fig. 4, designated bands due to Brønsted acid sites at 1540 and 1637 cm−1) in the catalysts.40,41 The origin of these Brønsted acid sites is the generation of surface Mo6+–OH groups during the impregnation step.42–44 Moreover, the intensity of these bands increased with the increasing surface density of MoOx on TiO2 (I1540/I1445 ratios are 0.11 and 0.33 for 2.0Mo and 3.5Mo, respectively). Therefore, the increase in the amount of strong acid sites on the catalysts with an increase in Mo loading/surface density is attributed to the increase in the amounts of Brønsted acid sites.
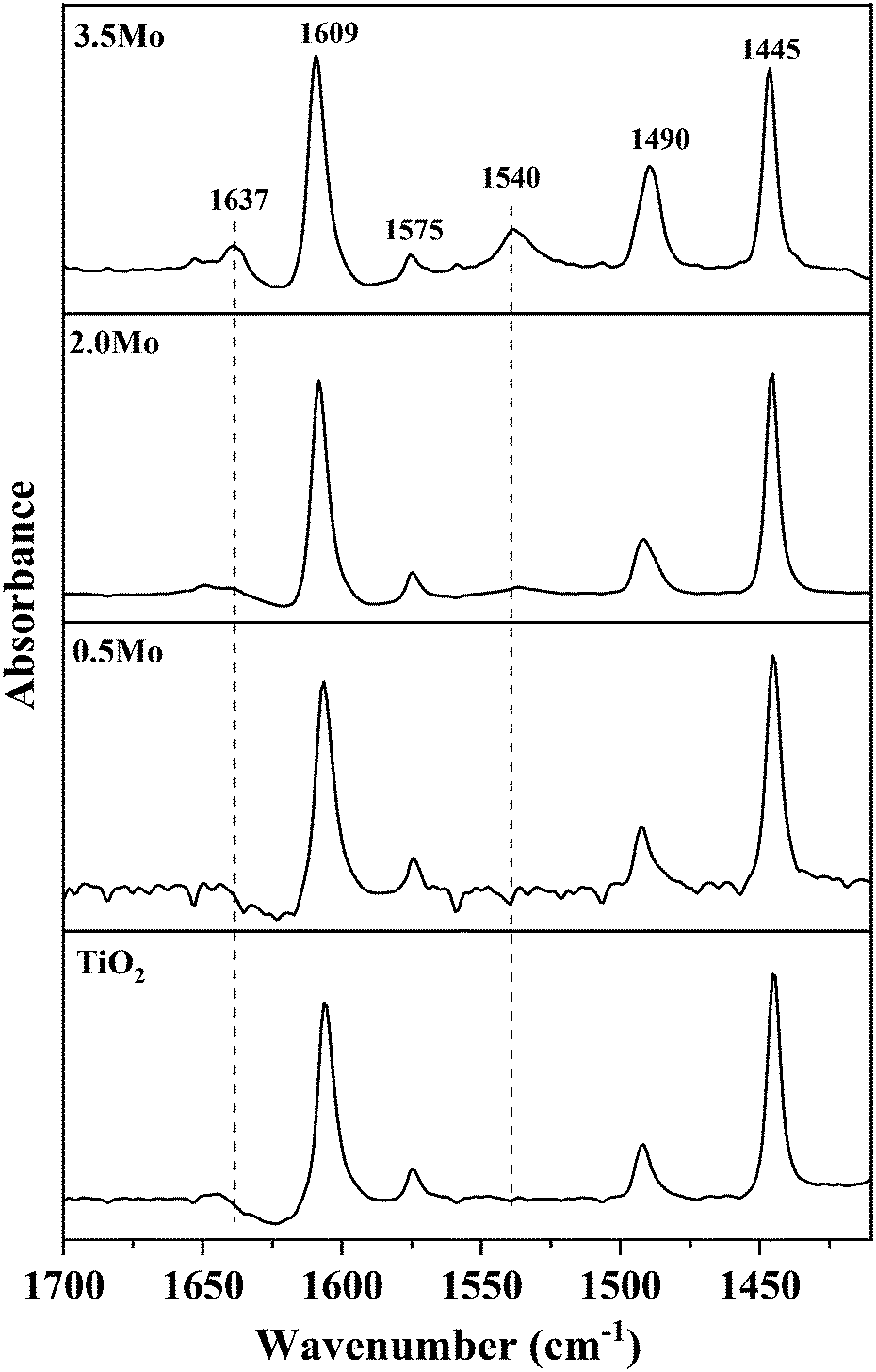 | ||
| Fig. 4 Py-FTIR spectra of TiO2, 0.5Mo, 2.0Mo, and 3.5Mo catalysts. | ||
3.2. Catalytic activity
Fig. 5 shows the correlation of Mo surface densities on TiO2versus the reaction rates for the desired product 2,5-DHF in the kinetic regime (Fig. S5†). The reaction rate decreased with an increasing Mo surface density on TiO2 and followed the trend: isolated species > oligomers > polymerized species (2D) > crystallites (3D). This observation unequivocally verifies the results from literature experimental observations that all kinds of MoOx species are active for the DODH reaction, and the predictions from theoretical calculations that monomeric MoOx species are the most effective as compared to oligomeric and polymeric MoOx species.26,27 The decrease in the reaction rate with an increasing Mo surface density also correlates to the acidity of the catalysts. The Brønsted acidity of the catalysts increased with an increasing Mo surface density on TiO2 (Fig. 3 and 4). Acidity appears to promote side reactions including dehydration reactions and ketal formation from 1,4-AHE and 3-octanone (a by-product of the reducing agent 3-octanol).22 The amount of octenes (2 and 3-octenes via dehydration of 3-octanol) and ketals increased with an increasing surface density of MoOx on TiO2 (Fig. 6). In parallel with the evolving Brønsted acidity of the catalysts, the yield of octenes and ketals increased (Fig. 6), thereby decreasing the reaction rate of the desired 1,4-AHE DODH to 2,5-DHF.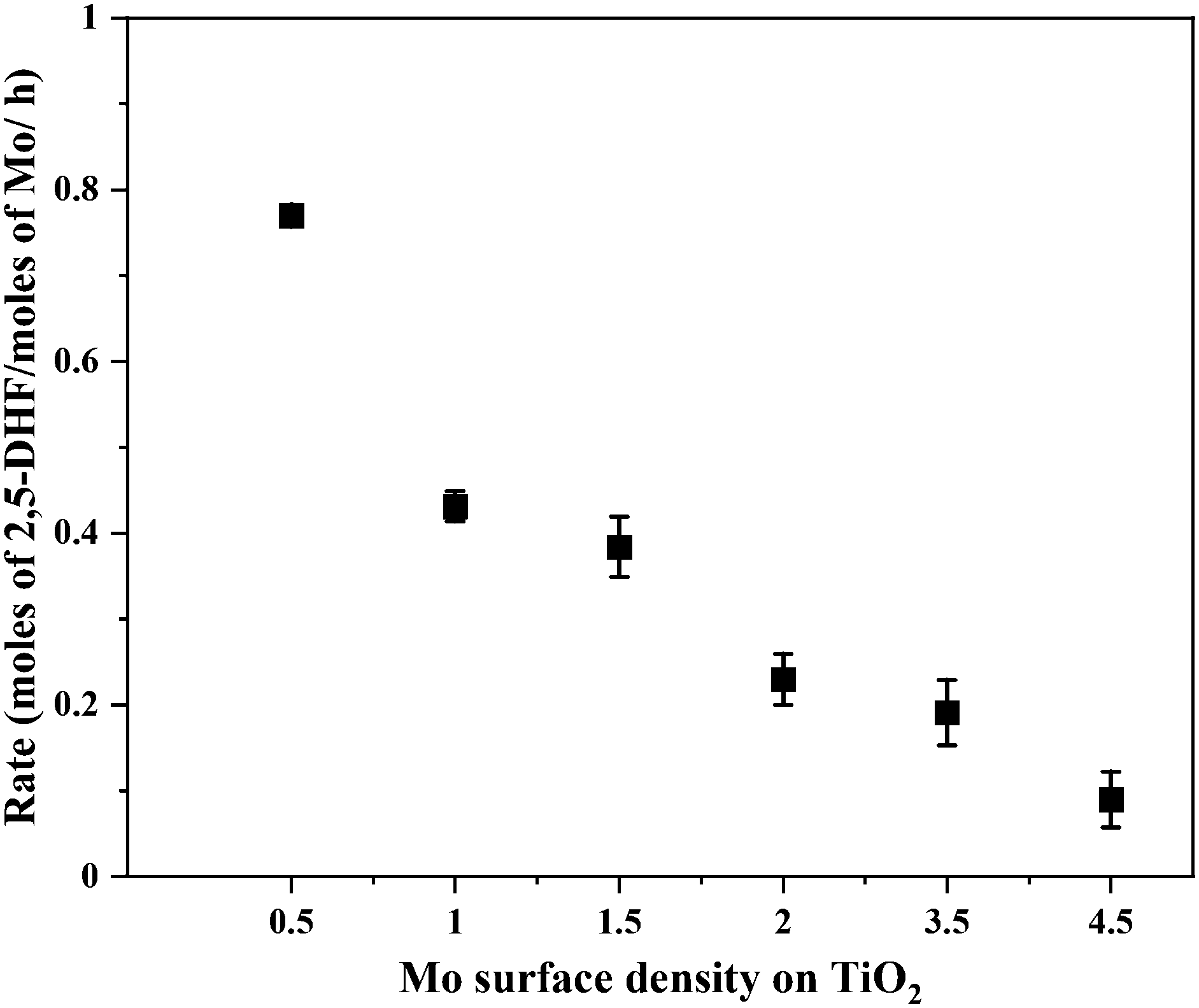 | ||
| Fig. 5 The correlation of the DODH reaction rate versus MoOx surface density on TiO2. Reaction conditions: 1,4-AHE = 0.6 mmol, 3-octanol = 6 mmol, Mo = 0.4–0.8 mol% w.r.t 1,4-AHE, T = 200 °C, t = 18 h. | ||
 | ||
| Fig. 6 Correlation of the yields of octenes and ketals with the content of strong acid sites. | ||
Next, the role and consequences of a calcination step on the activity were scrutinized using the dried and calcined catalysts. The experiments were carried out for the 0.5Mo and 3.5Mo catalysts to include the role of Mo surface density. Fig. 7 compares the performances of the dried and calcined catalysts. Interestingly, for the 0.5Mo catalyst, the calcination step improved the 1,4- AHE conversion to 2,5-DHF. A modest increase in the selectivity to 2,5-DHF also occurred. Nonetheless, an increase in the yields of octenes from <1 mol% to 2.9 mol% after calcination suggested an increasing acidity of the catalyst. For the 3.5Mo catalyst, though calcination improved the conversion of 1,4-AHE, the treatment reduced the selectivity to 2,5-DHF. A substantial increase in the yield of octenes rising from <1 mol% to 6.6 mol% accentuated a large increase in the acidity of the catalyst after calcination. At a lower surface density of Mo, where isolated species of MoOx exists, the negative role of acidity is overpowered by the increase in the selectivity of the catalyst. However, at higher Mo surface density, the acidity controls the selectivity of the catalyst. The main advantage of using a dried catalyst containing a high loading of Mo appears to circumvent the involvement of acidity in reducing the selectivity of the reaction.
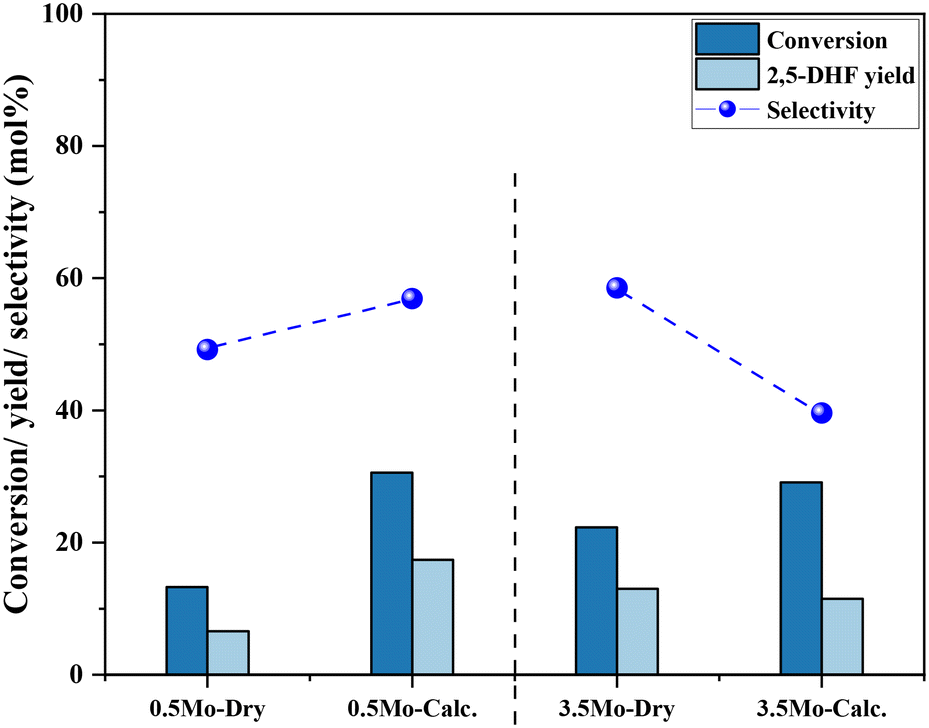 | ||
| Fig. 7 The effects of calcination of 0.5Mo and 3.5Mo on the 1,4-AHE conversion and 2,5-DHF yield. Reaction conditions: 1,4-AHE = 0.6 mmol, 3-octanol = 6 mmol, T = 200 °C, t = 18 h. | ||
Thereafter, the stability of MoOx species on the 0.5Mo catalysts (calcined and dried) during DODH was investigated through leaching studies. Initially, a cold filtration test after the first hour of the reaction was performed. A significant increase in the conversion of 1,4-AHE was observed (Fig. 8(a)). Though this increase in conversion was only half as compared to the conversion in the presence of the catalyst, the test revealed the leaching of Mo during the early stages of the reaction. Surprisingly, no considerable increase in the yield of 2,5-DHF was recorded after the cold filtration test (Fig. 8(b)). The leached Mo species from the 0.5Mo catalyst do not catalyze the selective conversion of 1,4-AHE to 2,5-DHF. Subsequently, a separate warm filtration test after 24 h of the reaction was performed (Fig. 8(a)). An increase in the 1,4-AHE conversion with an insignificant increase in the yield of 2,5-DHF (Fig. 8(b)) further confirmed the non-selective nature of leached Mo during the reaction. Afterwards, the same cold filtration test was repeated with the uncalcined, dried 0.5Mo catalyst. The test showed an increase in the yield (1.2 mol%) of 2,5-DHF (Fig. S6†). The use of a merely dried catalyst resulted in leaching of catalytically active Mo species into the reaction medium emphasizing the usefulness of calcination for the 0.5Mo catalyst. This characteristic behavior of leached Mo species can be correlated to the nature of Mo species existing on the dried and calcined catalysts. Some Mo species in the precursor form are expected to be present on the catalyst after the drying step. The calcination step decomposes the precursor Mo species to MoOx species. Conditional experiments with the precursor (NH4)2MoO4 and MoO3 crystallites were then performed to correlate the catalytic nature of the precursor MoOx species. The reaction with (NH4)2MoO4 established the catalytic nature of the precursor (5.5 mol% yields to 2,5-DHF at 10 mol% conversions of 1,4-AHE). Likewise, the reaction with MoO3 crystallites verified its catalytic nature too (6.9 mol% yields to 2,5-DHF at 8.7 mol% conversions of 1,4-AHE). The performances of the precursor and MoO3 catalysts are inferior to that of the MoOx/TiO2 catalysts. The leached Mo species from dried 0.5Mo can be presumed to originate from the supported (NH4)2MoO4 precursor. On the other hand, leached Mo species from calcined 0.5Mo are plausibly different in chemical nature as they originate from MoO3 (note that no MoO3 crystallites were found on the 0.5Mo catalyst, Fig. S1† and Fig. 1).
 | ||
| Fig. 8 Leaching studies of the 0.5Mo catalyst. (a) Reaction time versus 1,4-AHE conversion and (b) reaction time versus 2,5-DHF yield. Reaction conditions: 1,4-AHE = 0.6 mmol, 3-octanol = 6 mmol, Mo = 0.4 mol% w.r.t 1,4-AHE, T = 200 °C. | ||
An identical cold filtration test with the calcined 3.5Mo catalyst was conducted to include the effect of higher Mo surface density on leaching behaviors. The test confirmed (Fig. 9) leaching of catalytically active Mo species into the reaction medium. Their likely origin is in MoO3 crystallites as the catalyst constituted a small fraction of MoO3 crystallites (Fig. 1, Raman spectrum). The uncalcined, dried 3.5Mo catalyst is expected to leach more Mo species of the precursor origin due to its higher loading on the catalyst; therefore, it will be equally unstable as the 0.5Mo dried catalyst. At large, calcination has the benefit of improving the interaction between the TiO2 support and the Mo species through the generation of Ti–O–Mo bonds,30 but, it fosters the acidity of the final catalyst. At lower surface densities of Mo, such interactions are maximum while at higher surface densities, due to polymerization and crystal formation of MoOx species, interactions are lower leading to leaching of catalytically active species. A low surface density of Mo and a calcination step are identified to be the design parameters for a more active MoOx/TiO2 catalyst for the DODH of 1,4-AHE to 2,5-DHF.
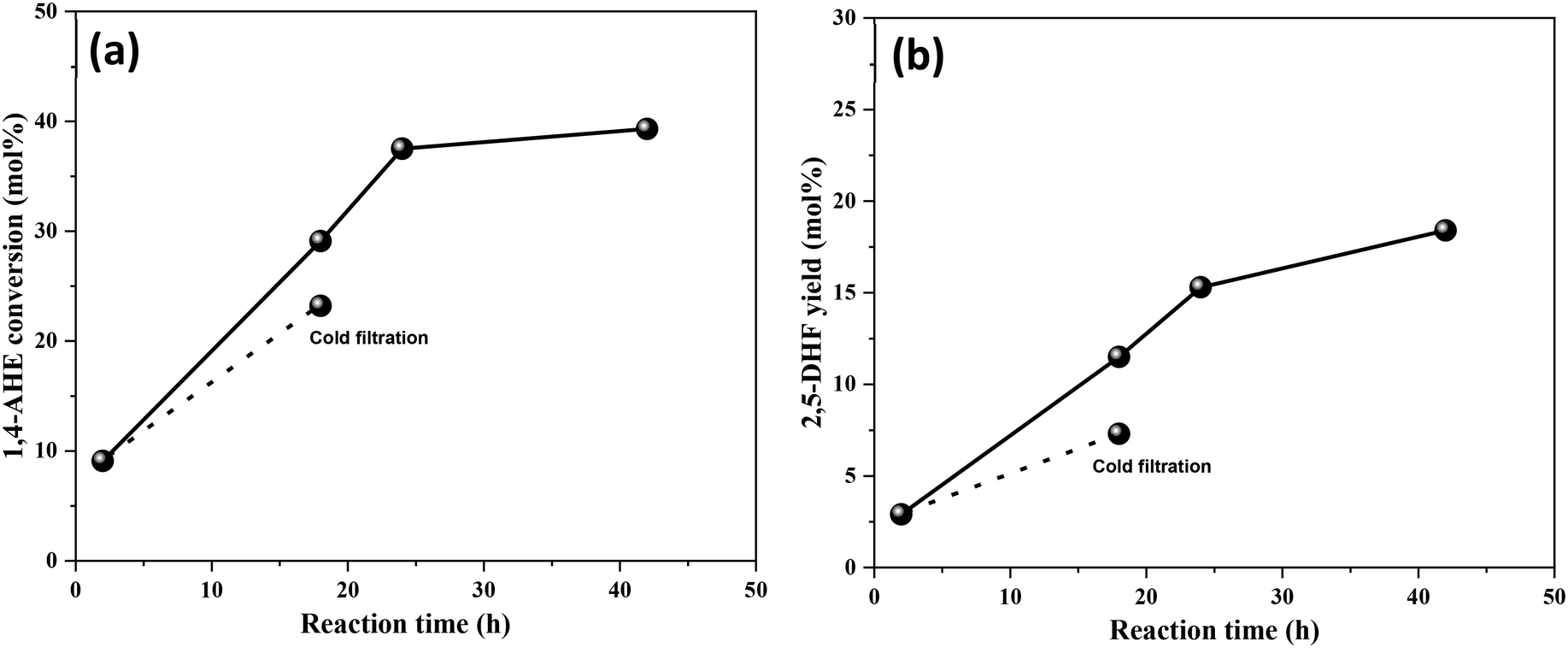 | ||
| Fig. 9 Leaching studies of the 3.5Mo catalyst. (a) Reaction time versus 1,4-AHE conversion and (b) reaction time versus 2,5-DHF yield. Reaction conditions: 1,4-AHE = 0.6 mmol, 3-octanol = 6 mmol, Mo = 0.86 mol% w.r.t 1,4-AHE, T = 200 °C. | ||
Having established the leaching of Mo and the inefficiency of leached Mo species from the 0.5Mo catalyst towards the DODH reaction, the stability of the remaining unleached Mo species on the 0.5Mo catalyst was investigated through recycle studies. Since higher conversions and yields were observed at higher temperatures (Fig. S7†), the recycle studies were conducted at a higher reaction temperature of 210 °C. Fig. 10 shows 4 recycle runs of the 0.5Mo catalyst without and with regeneration steps. A substantial decrease (18 mol%) in the conversion of 1,4-AHE was observed in the first recycle (recycle 1). The catalyst was only subjected to a simple drying treatment before its use in recycle 1. ICP analysis of the catalyst after the reaction showed a Mo loading of 0.3 wt% which is ∼0.29 wt% lower than the fresh catalyst. This was expected due to the leaching of some Mo during the fresh run as revealed by cold and warm filtration studies (Fig. 8). The decrease in the activity of the catalyst can thus be ascribed to the lower amount of Mo in recycle 1 as compared to the fresh run. Surprisingly, the selectivity to 2,5-DHF displayed an increment from ∼55 mol% in the fresh run to 79 mol% in recycle 1. Possibly, the catalyst at this stage constituted solely highly selective active species. This observation is similar to that found from the cold and warm filtration tests of the catalysts where leached species were non-selective for 2,5-DHF production. The fresh run might have resulted in the removal of most of the easily leachable non-selective Mo species from the support. The high selectivity of the catalyst could also be correlated to its decreased acidity as uncovered by the significantly reduced yields of octenes and ketals (Fig. 11). For the second recycle run (recycle 2), the same drying pretreatment step for the recycled catalyst was carried out. Recycle 2 displayed a similar 1,4-AHE conversion and 72 mol% 2,5-DHF selectivity as compared to recycle 1. ICP analysis of the catalyst after recycle 2 showed an Mo loading of ∼0.3 wt% similar to recycle 1. However, although less significant, a 7 mol% decrease in the selectivity towards 2,5-DHF compared to recycle 1 was noticed. No substantial change in the yields of octenes and ketals as compared to recycle 1 occurred excluding a significant effect of acidity on the selectivity. The catalyst after recycle 2 was analyzed by thermogravimetry (Fig. S8†) to check for carbon deposition impacting selectivity. A large weight loss (∼8 wt%) in the region of 200–600 °C was pointed at the presence of adsorbed organic matter on the catalyst.25 The presence of carbonaceous species on the catalyst could be the reason for the slight decrease in selectivity. Thus, for recycle 3, the recycled catalyst was subjected to a calcination step at 500 °C for 4 h to partially remove the carbonaceous matter. In recycle 3, the conversion remained almost the same as compared to recycles 1 and 2 (Fig. 10); however, the selectivity of the catalyst decreased by almost 10 mol%. Yet, in comparison to the fresh run, the selectivity of the catalyst in recycle 3 was ∼7.2 mol% higher. Py-FTIR analysis of the catalyst did not show indications of Brønsted acid sites (Fig. S9†) affecting the selectivity. No considerable decrease in the amount of Mo on the catalyst as compared to recycle 2 was detected by ICP analysis. For recycle 4, the catalyst was once again calcined before use to discern the after-effects of successive calcination. This resulted in a further increase in the conversion (3.7 mol%) with a small decrease (2 mol%) in the selectivity as compared to recycle 3. The minor increase in octene selectivity (Fig. 11) suggested a likely trend of a gradual increase in the acidity of the catalyst after consecutive calcination between recycles as it might have exposed the acid sites covered by the carbonaceous matter. An almost similar amount of ketals was observed on recycle 3 and recycle 4. A similar amount of Mo on the catalyst after the reaction as compared to recycle 1, recycle 2 and recycle 3 pointed out the stability of the unleached Mo species on the catalyst. An additional comparison was made on the rates of the reaction in the fresh run, recycle 2, and recycle 3. The rate showed a trend towards moderate improvement with recycles (0.85 ± 0.11, 0.94 ± 0.03, and 1.1 ± 0.15 moles of 2,5-DHF per moles of Mo per h, respectively, for the fresh, recycle 2, and recycle 3 catalysts). The catalyst was found to be stable in its performance. Both the XRD and Raman analysis of the catalyst after recycle 4 did not show any major change in the crystallinity and the coordination behavior of the MoOx species (Fig. S10 and S11†). Overall, the recycle studies demonstrated the supported MoOx to be stable and selective for multiple runs, the accumulation of carbonaceous materials on the catalyst during successive runs, and the possibility of an increase in acidity with regenerations involving calcination.
 | ||
| Fig. 10 Recycle runs of the 0.5Mo catalyst. Reaction conditions: 1,4-AHE = 0.6 mmol, 3-octanol = 6 mmol, T = 210 °C, t = 18 h. | ||
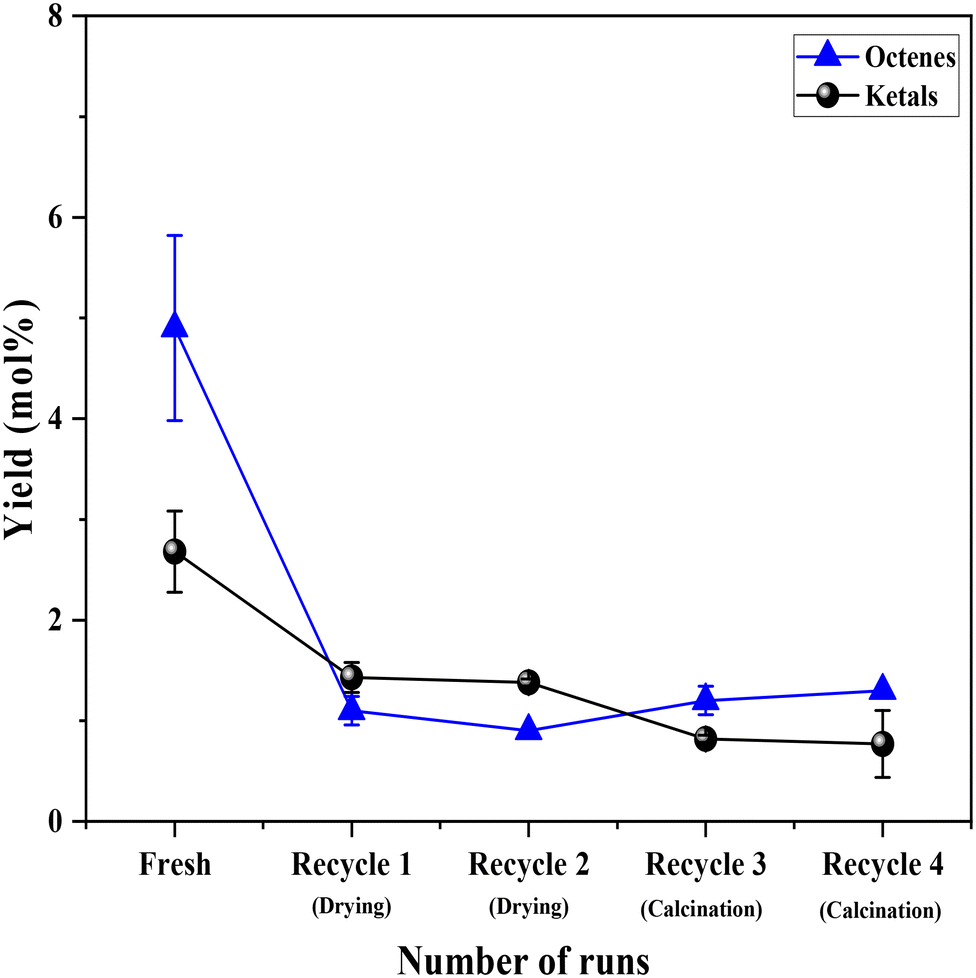 | ||
| Fig. 11 The yields of octenes and ketals during the recycling of the 0.5Mo catalyst. | ||
Finally, the carbon balance of the reaction was closely analyzed. In particular, the carbon balance for DODH reactions with sacrificial reducing agents is difficult to account for completely. This is because of (a) the vicinal diol can itself act as a reducing agent and most of the corresponding by-products are undetectable by GC due to their high boiling points, (b) the formation of ketals from the dehydrogenated product of the reducing agent and the vicinal diol, and (c) competing dehydrating reactions of reducing agents at higher acidity (Scheme S1† provides the various reaction pathways). The existence of octenes and ketals was already confirmed in our previous study through the 1H NMR analysis of the product mixture.23 To verify the coexistence of these side reaction pathways, conditional experiments with the 0.5Mo catalyst were performed. An experiment with 1,4-AHE alone without 3-octanol yielded 14 mol% 2,5-DHF at complete conversion of 1,4-AHE highlighting the possible contribution of route (a) to the desired product. The ketone formed from 1,4-AHE would produce a ketal with another molecule of 1,4-AHE with a high boiling point which is undetectable by GC. Similarly, another conditional experiment with 1,4-AHE and 3-octanone yielded 8.4 mol% 2,5-DHF and ketals at full conversion of 1,4-AHE verifying the possibility of route (b) contributing to the carbon balance. At higher acidity, the dehydration reactions of 3-octanol are dominant as discussed before. Therefore, designing a highly selective catalyst necessitates identifying specific active species for the main reaction as well as the different side reactions.
4. Conclusions
A series of MoOx/TiO2 catalysts containing varying surface densities of MoOx from isolated species to oligomeric species to polymeric MoOx species were successfully synthesized and characterized. Raman analysis of the catalysts confirmed the polymerization of MoOx species from isolated species to 3D crystallites with an increase in the surface density of Mo on TiO2. Supporting Mo on TiO2 introduced Brønsted acidity to the catalysts and its amount increased with the increase in Mo surface density. A correlation of the reaction rate with the surface density displayed a decreasing rate with an increasing Mo surface density and was correlated to the increase in strong acid sites, especially the Brønsted acid sites of the catalysts. Isolated MoOx species were found to be the most effective species for the DODH reaction. A calcination step has an advantage that can improve the interaction of the MoOx species with the TiO2 support but with a drawback of increasing the acidity and this increase in acidity is more prominent in catalysts with higher Mo surface densities. Leaching studies of the catalysts revealed the non-catalytic nature of leached species for the catalyst containing isolated MoOx species and the catalytic nature of polymerized MoOx species. Finally, recycle studies of the most effective catalyst indicated the stable nature of the unleached MoOx species on the catalyst, its selective nature, deposition of organic matter in the continuous use of the catalyst without calcination, and the regeneration of acid sites with successive calcination steps. The results discussed here provide insights into the critical design parameters for the future design of a highly effective and stable Mo-based catalyst for DODH reactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge support from the German Research Foundation (DFG, PA 1689/10-1). The authors thank Bea Becker, DWI – Leibniz-Institut für Interaktive Materialien, RWTH Aachen, for the Raman analysis. The authors also express their gratitude to Noah Avraham, Jens Heller, and Heike Bergstien for XRD, BET, TGA, and ICP analyses. We thank Riccardo Bacchiocchi for the help with Py-FTIR measurement.References
- B. Song, R. Lin, C. H. Lam, H. Wu, T.-H. Tsui and Y. Yu, Renewable Sustainable Energy Rev., 2021, 135, 110370 CrossRef CAS.
- J. A. Okolie, E. I. Epelle, M. E. Tabat, U. Orivri, A. N. Amenaghawon, P. U. Okoye and B. Gunes, Process Saf. Environ. Prot., 2022, 159, 323–344 CrossRef CAS.
- C. O. Tuck, E. Pérez, I. T. Horváth, R. A. Sheldon and M. Poliakoff, Science, 2012, 337, 695–699 CrossRef CAS PubMed.
- A. Jain, S. Sarsaiya, M. Kumar Awasthi, R. Singh, R. Rajput, U. C. Mishra, J. Chen and J. Shi, Fuel, 2022, 307, 121859 CrossRef CAS.
- F. H. Richter, K. Pupovac, R. Palkovits and F. Schüth, ACS Catal., 2013, 3, 123–127 CrossRef CAS.
- R. Weingarten, G. A. Tompsett, W. C. Conner and G. W. Huber, J. Catal., 2011, 279, 174–182 CrossRef CAS.
- A. M. Ruppert, K. Weinberg and R. Palkovits, Angew. Chem., Int. Ed., 2012, 51, 2564–2601 CrossRef CAS PubMed.
- K. Tajvidi, P. J. C. Hausoul and R. Palkovits, ChemSusChem, 2014, 7, 1311–1317 CrossRef CAS PubMed.
- J. Sebastian, Y. W. Cheah, D. Bernin, D. Creaser and L. Olsson, Catalysts, 2021, 11, 874 CrossRef CAS.
- Y. W. Cheah, M. A. Salam, J. Sebastian, S. Ghosh, O. Öhrman, D. Creaser and L. Olsson, Sustainable Energy Fuels, 2021, 5, 5270–5286 RSC.
- N. N. Tshibalonza and J.-C. M. Monbaliu, Green Chem., 2020, 22, 4801–4848 RSC.
- G. K. Cook and M. A. Andrews, J. Am. Chem. Soc., 1996, 118, 9448–9449 CrossRef CAS.
- A. L. Denning, H. Dang, Z. Liu, K. M. Nicholas and F. C. Jentoft, ChemCatChem, 2013, 5, 3567–3570 CrossRef CAS.
- L. Sandbrink, E. Klindtworth, H.-U. Islam, A. M. Beale and R. Palkovits, ACS Catal., 2016, 6, 677–680 CrossRef CAS.
- R. Hosaka, D. Asada, J. Cao, M. Tamura, Y. Nakagawa, K. Tomishige, J.-Y. Hasegawa and A. Nakayama, J. Phys. Chem. C, 2022, 126, 11566–11573 CrossRef CAS.
- N. Ota, M. Tamura, Y. Nakagawa, K. Okumura and K. Tomishige, Angew. Chem., Int. Ed., 2015, 54, 1897–1900 CrossRef CAS PubMed.
- N. Ota, M. Tamura, Y. Nakagawa, K. Okumura and K. Tomishige, ACS Catal., 2016, 6, 3213–3226 CrossRef CAS.
- K. Yamaguchi, J. Cao, M. Betchaku, Y. Nakagawa, M. Tamura, A. Nakayama, M. Yabushita and K. Tomishige, ChemSusChem, 2022, 15, e202102663 CrossRef CAS PubMed.
- B. E. Sharkey and F. C. Jentoft, ACS Catal., 2019, 9, 11317–11328 CrossRef CAS.
- J. Lin, H. Song, X. Shen, B. Wang, S. Xie, W. Deng, D. Wu, Q. Zhang and Y. Wang, Chem. Commun., 2019, 55, 11017–11020 RSC.
- Y. Kon, M. Araque, T. Nakashima, S. Paul, F. Dumeignil and B. Katryniok, ChemistrySelect, 2017, 2, 9864–9868 CrossRef CAS.
- I. Meiners, Y. Louven and R. Palkovits, ChemCatChem, 2021, 13, 2393–2397 CrossRef CAS.
- L. Sandbrink, K. Beckerle, I. Meiners, R. Liffmann, K. Rahimi, J. Okuda and R. Palkovits, ChemSusChem, 2017, 10, 1375–1379 CrossRef CAS PubMed.
- B. E. Sharkey, A. L. Denning, F. C. Jentoft, R. Gangadhara, T. V. Gopaladasu and K. M. Nicholas, Catal. Today, 2018, 310, 86–93 CrossRef CAS.
- S. Hacatrjan, L. Liu, J. Gan, Y. Nakagawa, J. Cao, M. Yabushita, M. Tamura and K. Tomishige, Catal. Sci. Technol., 2022, 12, 2146–2161 RSC.
- D. Lupp, N. J. Christensen, J. R. Dethlefsen and P. Fristrup, Chem. – Eur. J., 2015, 21, 3435–3442 CrossRef CAS PubMed.
- Y. Xi, J. Lauterbach, Y. Pagan-Torres and A. Heyden, Catal. Sci. Technol., 2020, 10, 3731–3738 RSC.
- M. Shetty, K. Murugappan, W. H. Green and Y. Román-Leshkov, ACS Sustainable Chem. Eng., 2017, 5, 5293–5301 CrossRef CAS.
- K. V. R. Chary, T. Bhaskar, G. Kishan and V. Vijayakumar, J. Phys. Chem. B, 1998, 102, 3936–3940 CrossRef CAS.
- G. Tsilomelekis, G. D. Panagiotou, P. Stathi, A. G. Kalampounias, K. Bourikas, C. Kordulis, Y. Deligiannakis, S. Boghosian and A. Lycourghiotis, Phys. Chem. Chem. Phys., 2016, 18, 23980–23989 RSC.
- K. Hamraoui, S. Cristol, E. Payen and J.-F. Paul, J. Phys. Chem. C, 2007, 111, 3963–3972 CrossRef CAS.
- G. D. Panagiotou, T. Petsi, K. Bourikas, A. G. Kalampounias, S. Boghosian, C. Kordulis and A. Lycourghiotis, J. Phys. Chem. C, 2010, 114, 11868–11879 CrossRef CAS.
- K. Liu, X. Huang, E. A. Pidko and E. J. M. Hensen, Green Chem., 2017, 19, 3014–3022 RSC.
- H. Hu, I. E. Wachs and S. R. Bare, J. Phys. Chem., 1995, 99, 10897–10910 CrossRef CAS.
- J. Faye, M. Capron, A. Takahashi, S. Paul, B. Katryniok, T. Fujitani and F. Dumeignil, Energy Sci. Eng., 2015, 3, 115–125 CrossRef CAS.
- P. Sudarsanam, N. K. Gupta, B. Mallesham, N. Singh, P. N. Kalbande, B. M. Reddy and B. F. Sels, ACS Catal., 2021, 11, 13603–13648 CrossRef CAS.
- I. Nova, L. Lietti, L. Casagrande, L. Dall'Acqua, E. Giamello and P. Forzatti, Appl. Catal., B, 1998, 17, 245–258 CrossRef CAS.
- A. Navajas, I. Reyero, E. Jiménez-Barrera, F. Romero-Sarria, J. Llorca and L. M. Gandía, Catalysts, 2020, 10, 158 CrossRef CAS.
- J. Henych, M. Šťastný, Z. Němečková, K. Mazanec, J. Tolasz, M. Kormunda, J. Ederer and P. Janoš, Chem. Eng. J., 2021, 414, 128822 CrossRef CAS.
- J. Sebastian, M. Zheng, X. Li, J. Pang, C. Wang and T. Zhang, J. Energy Chem., 2019, 34, 88–95 CrossRef.
- C. Mebrahtu, R. Sun, C. H. Gierlich and R. Palkovits, Appl. Catal., B, 2021, 287, 119964 CrossRef CAS.
- Y. Liu, X. Ma, S. Wang and J. Gong, Appl. Catal., B, 2007, 77, 125–134 CrossRef CAS.
- N. Jada, K. J. Sankaran, R. Sakthivel, D. Sethi and P. Mohapatra, Bull. Mater. Sci., 2021, 44, 167 CrossRef CAS.
- C. Martin, I. Martin, C. Delmoral and V. Rives, J. Catal., 1994, 146, 415–421 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cy01854c |
| This journal is © The Royal Society of Chemistry 2023 |