
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bifunctional catalysts for the conversion of CO2 into value-added products – distance as a design parameter for new catalysts
Maik Alexander
Rudolph
,
Philipp
Isbrücker
and
Reinhard
Schomäcker
 *
*
Technische Universität Berlin, Institut für Chemie – Technische Chemie, Straße des 17. Juni 124, D-10623 Berlin, Germany. E-mail: schomaecker@tu-berlin.de
First published on 1st May 2023
Abstract
More than 1000 CO2 chemistry publications within the last five years have featured the application of bifunctional catalysts. The majority of these articles investigate hydrogenation reactions of CO2 for producing alkanes, alcohols, and ethers. Reactions using CO2 and epoxides as reactants for producing cyclic carbonates or linear polycarbonates have also been extensively researched. For all of these CO2 chemistries, an informed choice of the combined materials and their arrangement as a bifunctional catalyst is critical to their performance. Herein, we identify the distance between active sites in bifunctional catalysts as an important control parameter for the system performance. We show that a range of optimal distances between the active sites can be identified for each of the interaction mechanisms enabled by bifunctional catalysts, namely steric and electrostatic interactions as well as concentration gradients of intermediate products. For the design of bifunctional core–shell catalysts, a model-based workflow is suggested.
1. Introduction
The catalytic conversion of CO2 into valuable products has become one of the largest and fastest-growing research areas in recent years.1–3 The motivation for this is the climate crisis caused by the increasing emission of CO2 that results from the use of fossil fuels. Although chemical product development is often cited as the primary motivation for research in CO2 conversion, the reduction of CO2 emissions needed to avoid major climate changes cannot be achieved solely in this way.4 Even by producing all chemical products from CO2, only a small fraction of the CO2 emitted by energy production from fossil fuels would be converted.4 In addition to the energy sector, significant emissions of CO2 also come from the steel, cement, and chemical industries.1 Many climate protection goals require a substantial reduction in the use of fossil raw materials, which is referred to as the “defossilization” of industry. Using CO2 as a building block for chemical synthesis contributes to this process, as it enables the creation of production cycles of carbon-based chemical products that are no longer totally dependent on finite fossil raw materials, as is the case with today's linear value chains.5The wide range of modern chemical value chains stems from a small number of basic chemicals produced from petroleum. Efforts to replace these core petrochemicals with renewable raw materials have led to the development of an assortment of base chemicals that can be fed into existing value chains and supply the market with familiar end products.6 To develop cycles of products with CO2 as an intermediate, its conversion into compounds that can be seamlessly introduced into value chains via short routes is necessary to replace the base petrochemicals produced from fossil feedstocks. The simplest concept for a CO2-centered carbon cycle would be the conversion of CO2 to CO combined with green hydrogen to produce syngas and subsequent syngas-based chemical industry. However, life cycle analyses and techno-economic analyses show that the associated energy demands render this option economically inviable.3 As an alternative, the direct conversion of CO2 into higher molecular weight compounds is being investigated. In many publications and review articles, the diverse range of possibilities for synthesizing several different products from CO2 is often depicted in circular diagrams symbolizing a wheel or a sun, where each ray represents a reaction of CO2 to form a valuable product.7 This diversity is somewhat deceiving to the viewer, as multiple products of the same reaction type are often shown. Since the carbon in the CO2 molecule is in the highest possible oxidation state, it is a very low-energy molecule that only acquires the necessary thermodynamic driving force for the reaction when combined with a high-energy reactant. In this low-energy state, very efficient activation of the CO2 by catalysts is required to achieve appreciable reaction rates with the respective reaction partners.
Leitner et al. devised a very descriptive representation in their review using a matrix visualized as a chessboard, which demonstrates the challenges associated with CO2 chemistry.8 In the matrix, the first column represents the C1 compounds of carbon in their different oxidation states. Here, CO2 is found at the bottom of the column, and methane at the top. The rows of the matrix show the products of the C1 compounds in their respective oxidation states, obtained by coupling reactions with different reactants. With the exception of products in the first row and the first column, all products within the matrix are produced directly from the simultaneous reduction of CO2 to a lower oxidation state intermediate and the coupling reaction of this reduced intermediate. This raises the question of whether direct synthesis is more useful than two separate reactions, each carried out at their optimum reaction conditions. If the process windows for the two reactions are very different, combining them into a tandem reaction can become an intractable task since too many degrees of freedom are lost in attempting to optimize them both. In many cases, this is not the case, however, and combining the reactions may even offer thermodynamic advantages since the reduction of CO2 is endothermic while the subsequent reactions are often exothermic. In this situation, the released energy can be used directly for the reduction reaction and shift its equilibrium in favor of product formation.
For the conversion of CO2 to value-added products, two groups of reaction partners can be used: one which first carries out the reduction followed by the linkage, and the other which acts as a single high-energy reaction partner (e.g., hydrogen, amines, or epoxides) capable of fulfilling both functions simultaneously. The choice of suitable catalysts for these reactions depends strongly on which reaction partner must be activated with the CO2. For the simultaneous activation of two very different reactants, or the combination of two reactions in one reactor, new bifunctional catalysts have been developed in recent years including homogeneous2,9,10 as well as heterogeneous.11,12 Homogeneous bifunctional catalysts combine soluble complexes with different functionalities and catalytic activity, which are either covalently linked or electrostatically bound.13 In heterogeneous bifunctional catalysts, the active sites are located on the surface of solids or support materials.14 Very close spacing at the molecular level or large mesoscopic spacing can occur, for example, when different nanoparticles are immobilized on support materials. Short distances between the different active sites, often referred to as proximity, can be highly beneficial for accelerating the second reaction by a high local concentration of the product of the first catalyst. On the contrary, adverse effects might occur when a product of the first catalyst inhibits the second or the catalytic material react with each other, causing deactivation. A trade-off between such effects will result in an optimal distance between the active sites that need to be adjusted by the catalyst preparation method. Three recent reviews summarized the synergetic effects offered by bifunctional catalysts for CO2 conversion to value-added products.15–17
In this mini-review, we discuss the use of bifunctional catalysts for the conversion of CO2 to valuable materials with a focus on reactions with epoxides and hydrogen. In this context, the catalysts for reactions with epoxides are examples of catalysts that activate both reaction partners simultaneously by at least two different active sites. In these reactions, no stable intermediates are formed that are released from the catalysts. On the surface of the bifunctional catalysts that react CO2 with hydrogen, often stable intermediates are formed that must be transported between the active sites by mass transfer mechanisms. We present a design concept for a bifunctional catalyst for the cooperative activation of two substrates and a workflow for active material selection, and a structural design of bifunctional catalysts for tandem reactions.
2. Value-added products from CO2 and epoxides
Generally, the catalysts for the coupling of CO2 and epoxides are binary systems, including a Lewis acid and a nucleophile.13 The Lewis acid activates the epoxide toward the ring opening due to the attack of the nucleophile on the less substituted carbon atom as illustrated in Fig. 1. Subsequently, a CO2 unit is inserted by the nucleophile, resulting in an alkyl semi-carbonate unit that can enter into different consecutive steps (Fig. 2a): i) a ring closure to a cyclic carbonate via a back-biting mechanism that leaves the active site ready for a new catalytic cycle, ii) an insertion of further epoxide and CO2 units to generate a linear alternating polycarbonate, and iii) the further insertion of additional epoxides leading to random polyethers or polyether carbonates.18 | ||
| Fig. 1 Mechanism of the formation of cyclic carbonates via a bifunctional catalyst system consisting of a Lewis acid and a nucleophile. | ||
 | ||
| Fig. 2 a) Competitive reaction pathways of cyclic carbonate and linear polycarbonate formation, b) energy profiles of reactions producing cyclic carbonates vs. linear polycarbonates. | ||
The formation rate ratio of these reactions not only depends on the used epoxides and the reaction conditions but also on the catalyst systems. Linear polycarbonates are thermodynamically favored over cyclic polycarbonates due to entropic reasons, as shown in Fig. 2b. The lower activation barrier for the copolymerization allows for the formation of polymers due to the kinetic control of the reaction. Since a mixture of cyclic carbonates and linear polycarbonates would be undesired, the objective of the catalyst and process design is a highly active and selective catalytic reaction for each separate reaction product, since both the cyclic carbonates and linear polycarbonates find a wide range of industrial applications.
Homogeneous catalysts for cyclic carbonate formation that have been intensively investigated include complexes of magnesium, aluminum, chromium, cobalt, zinc, zirconium, and other metal ions coupled to mono, bi-, or multidentate ligands like phenolates, porphyrins, phthalocyanines, salen, salphene or N-heterocyclic carbenes.7 These electron-withdrawing ligands tune the Lewis acidity of the metal ions for the activation of the epoxide. Halogen anions (Cl−, Br− or I−) assist this elementary step and control the insertion of CO2 and the subsequent steps to the cyclic or linear product. The binding strength of both the Lewis acid and the nucleophile to the intermediate is very sensitive to the formation rate ratios of the final products, making it challenging to develop a robust catalyst system with a broad substrate scope. Despite these challenges, highly active catalysts with Al and Fe salen complexes have been demonstrated.
Inoue et al. first described the copolymerization of carbon dioxide and epoxides in 1969.19 They copolymerized CO2 with propylene oxide by using dioxane as a solvent and a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 catalyst mixture of diethylzinc and water. As a result of this groundbreaking work, a variety of heterogeneous and homogeneous catalysts have been developed to copolymerize epoxides and CO2 with higher yields and higher efficiency. Interestingly, the majority of these catalysts contain both base metals (such as Mg, Na, or Al) and earth-abundant transition metals (such as Co, Cr, Zn, Ti, Fe, and Ni). A detailed mechanistic study has revealed that most of these catalysts, both homogeneous and heterogeneous, require intermediates of two or more metal centers to achieve high turnover numbers (TONs) and turnover frequencies (TOFs).20
:1 catalyst mixture of diethylzinc and water. As a result of this groundbreaking work, a variety of heterogeneous and homogeneous catalysts have been developed to copolymerize epoxides and CO2 with higher yields and higher efficiency. Interestingly, the majority of these catalysts contain both base metals (such as Mg, Na, or Al) and earth-abundant transition metals (such as Co, Cr, Zn, Ti, Fe, and Ni). A detailed mechanistic study has revealed that most of these catalysts, both homogeneous and heterogeneous, require intermediates of two or more metal centers to achieve high turnover numbers (TONs) and turnover frequencies (TOFs).20
For much better control of selectivity towards linear polycarbonates, bimetallic catalysts with two Lewis acid sites have been designed based on dimeric salen ligands bridged by linkers of different lengths.21 For these catalysts, one metal center activates the epoxide, and the other metal center coordinates with the alkoxide anion or carbonate anion of the polymer chain. The activated species takes the role of the nucleophile by promoting CO2 insertion to the epoxide, followed by internuclear migration. Linear polycarbonates are produced as long as this process is continued. These catalysts, developed by the groups of Williams,22,23 Rieger,24,25 Darensbourg,26 and Kim,27 show not only a high selectivity for linear polycarbonates but also a strong influence on the length of the linkers between the two sites. This effect is either caused by the distance between the sites controlled by the linker length or by the dynamics of the complexes caused by the linker flexibility.26 For distances between the sites that are too large, they act individually by promoting the formation of cyclic carbonates. In solutions, the cooperation of two Lewis acid sites is made possible with two individual monomeric complexes, enabling the formation of linear polycarbonates. By tuning the distance between the Lewis acid sites, selectivity either to cyclic or linear polycarbonates can be adjusted. In general, large distances increase the CO2/epoxide reaction selectivity to cyclic carbonates, while short distances facilitate cooperation of the Lewis acid sites for linear polycarbonate formation. These sorts of reactions can be realized with metal–organic frameworks (MOFs) whose large linkers provide adequate spacing between the Lewis acidic metal ions and catalytically expedite the highly selective production of cyclic carbonates.28 In contrast, so-called double metal catalysts use small ligands (e.g., cyanides) to form well-structured compounds with short distances between the metal sites, facilitating their cooperation within the catalytic cycle of CO2 as well as epoxide activation for copolymerization.20
Double metal catalysts were first introduced for the homopolymerization of epoxides in a 1966 patent by General Tire and Rubber. The usage of a double metal catalyst for ring-opening-copolymerization was later patented by the Dow Chemical Company in 1985. Since then, these catalysts have been utilized for the ring-opening of epoxides with amines as well as hydroamination, transesterification, hydrolysis, and condensation reactions.20 Among the reported heterogeneous catalysts, double metal cyanide (DMC) catalysts have been evaluated extensively for their use in the copolymerization of CO2 with epoxides, yielding high CO2 incorporation (up to 75 mol%) in the case of CO2/cyclohexene oxide copolymerization. DMC catalysts are generally prepared by reacting metal salts of the general formula M1Xn with cyanometalate compounds of the general formula M3a[M2(CN)b(A)c]d, where M1 is a metal ion selected from the group consisting of Zn2+, Fe2+, Fe3+, Co2+, and Co3+; M2 is a metal ion selected from the group consisting of Fe2+, Fe3+, Co2+ and Co3+, and Ir3+; M3 is an alkali metal or alkaline earth metal, and A is an anion.20 In a typical synthesis, aqueous solutions of an excess amount of ZnCl2 and K3Co(CN)6 are mixed, and tert-butyl alcohol (t-BuOH) is simultaneously added as a complexing agent (CA) to the produced slurry. After filtration purification of the product through repeated washing steps with aqueous t-BuOH, an active catalyst of the general formula Zn3[Co(CN)6]2·a(ZnCl2·b(H2O)·c(t-BuOH)) is prepared for copolymerization reactions. In this manner, DMC catalysts are the reaction products of four major compounds: (i) a water-soluble metal salt, (ii) a water-soluble metal cyanide salt, (iii) an electron-donating CA, and (iv) water. Generally, the activation of the DMC catalyst is induced by the CAs incorporated into the catalyst matrix. Extensive research has shown that DMC catalysts bearing water-soluble aliphatic alcohols, particularly t-BuOH, produce the best results in terms of both the polymerization activity and resulting polymer properties. The copolymers produced with these catalysts show versatile product properties, such as polyurethane foams29 or surface-active compounds.30
The highly versatile toolbox of MOF chemistry allows for the preparation of finely tuned MOF catalysts for CO2/epoxide copolymerization. The metal ion selected as the Lewis acidic active center can be incorporated into a solid framework using a variety of spacers with varying lattice constants, generating a broad range of structures and geometric properties. As a typical synthesis example, the preparation of MOF-5 is given in ref. 31.
The structural characterization of the two types of catalysts described above is represented in Fig. 3. A distinct difference becomes obvious. While the shortest metal–metal (Zn–Zn) distance for MOFs is in the range of 10 Å and tuneable by the selection of different spacers, the shortest Zn–Zn distance in DMC catalysts is only ∼7 Å.32–35 The data presented in Fig. 3 were reported by Mullica et al.32 and Allen et al., respectively.35 They were all obtained by XRD measurements. The crystallographic information files can be found in the supporting literature of the respective publication. It should be noted that the distance of the Zn atoms varies depending on the measurement method and metal atom under consideration and is thus only intended as an approximate classification. With this shortest difference in distance, the metal centers of the DMC catalyst can cooperate, while those of the MOF catalysts are too much separated from each other, thus favoring the interaction with the nucleophile as a co-catalyst. For the copolymerization reaction of CO2 with different epoxides, fine-tuning through the choice of different spacers or ligands for the catalyst synthesis has been detailed in the literature.20
 | ||
| Fig. 3 Schematic structures of solid bifunctional catalysts for CO2/epoxide conversion to cyclic carbonates or linear polycarbonates: a.) metal–organic framework (MOF),35 b.) double metal cyanide catalysts (DMC).32 | ||
3. Value-added products from CO2 and hydrogen
A variety of catalysts have been developed for the reduction of CO2 with hydrogen to C1 products, which produce their respective products with high selectivity and remain stable under the conditions required by thermodynamics. Similar developments have been made for the catalysts that convert the C1 synthesis building blocks into the downstream products of the various value chains. Particularly interesting material candidates for combination into bifunctional catalysts have been investigated for the production of CO or methanol, as well as for the conversion of CO or methanol into hydrocarbons, olefins, aromatics, ethers, and esters. Such material coupling possesses great optimization potential for syngas production where the energy of the exothermic Fischer–Tropsch (FT) reaction downstream can be used to drive the endothermic reverse water gas shift reaction (RWGS). At the same time, the consumption of CO in the FT reaction can advantageously shift the equilibrium of the RWGS, achieving high CO2 conversions at lower reaction temperatures. A similar situation arises for methanol synthesis from CO2 when its equilibrium is shifted by the formation of ethers, esters, or olefins from the methanol. In this context, there is the potential to operate at lower pressures than a pure methanol synthesis process. Both scenarios are depicted in Fig. 4.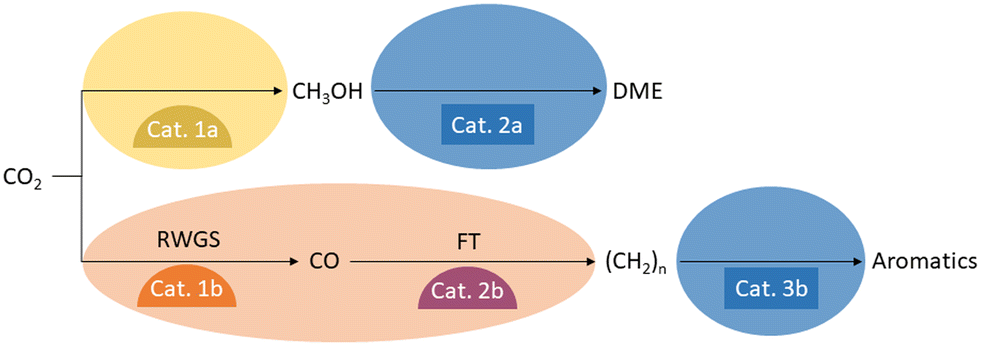 | ||
| Fig. 4 Hydrocarbon formation by CO2 hydrogenation via the syngas or methanol route. | ||
The compatibility of the materials must be considered when combining two catalysts into a bifunctional catalyst so that no unintended reaction between the materials diminishes their respective catalytic functions.36 In a bifunctional catalyst, both active centers must also have contact with the reactants and products of the other center. This can lead to inhibitory adsorption, side reactions, or even deactivation. Therefore, when choosing catalytic components for a bifunctional catalyst, such interactions must be considered in order to combine suitable catalysts in coordinated proportions to yield an optimal arrangement. Taking these considerations into account, researchers optimizing bifunctional catalysts for the reduction of CO2 to value-added products typically do not simply combine the best catalysts for the two individual reactions.
For the production of hydrocarbons from a CO2 Fischer–Tropsch synthesis (FTS), different combinations of catalysts have been developed for the RWGS and FTS reactions. Additionally, the reaction temperature must be matched to the FTS kinetics to produce products with molecular weight distributions suitable for use as fuels. Although the RWGS reaction does not achieve high conversion at these temperatures due to thermodynamics, the coinciding FTS reaction can shift the equilibrium to the product side.
Early publications on the direct hydrogenation of CO2 to hydrocarbons featured Fe- and Co-based catalysts supported on Al2O3 or SiO2.37,38 Since Fe had also been known as an RWGS catalyst, it was the preferred choice for a catalyst with bifunctional activity. Alkali metals were added during the catalyst synthesis by co-precipitation as promoters. Structural analysis revealed the formation of multi-phase systems under reaction conditions containing different oxides and carbides active for RWGS and FTS. By using the same precursor, these formed active sites, which are active for both reactions, are located in close proximity to each other at the surface of the support material. As a drawback, this preparation provides little control of the number and ratio of active sites for both reactions and, consequently, of their distance to each other as well as the rates of both reactions.
Typical experimental conditions fix the temperature between 300 and 400 °C and pressure around 10 bar for a feed mixture of CO2 and H2 with a molar ratio of 3:1. At moderate conversions of CO2 between 20 and 50%, product mixtures with light hydrocarbons were obtained, but with major fractions of methane (Table 1).
| Catalyst | Category | H2/CO2 ratio | T/°C | p/bar | X(CO2)/% | S/% | Ref. |
|---|---|---|---|---|---|---|---|
| Catalyst arrangement category: a: dual bed, non-mixing; b1: mechanical mixing of particles (without external forces); b2: mixing in aqueous dispersions; b3: pressure-mediated physical mixing (grinding, milling); c: chemical mixing (e.g. core–shell). | |||||||
| First works | |||||||
| Fe–K/Al2O3 | b3 | 3 | 400 | 20 | 70 | C2–C4 (48), C5+ (36) | 37 |
| Fe–K/Al2O3–MgO | b3 | 3 | 300 | 10 | 28 | C2–C5+ (59) | 38 |
| Fe–Co–K/Al2O3 | b3 | 3 | 300 | 11 | 36 | C2+ (71) | 39 |
| Co–Na–Mo/SiO2 | b3 | 3 | 370 | 1 | 44 | C2–C4 (44), C5+ (10) | 40 |
| Fe–La–Cu–K/TiO2 | b3 | 3 | 300 | 10 | 27 | C2–C4 (21), C5–C15 (40) | 41 |
| Fe–Ru–Zn–K/TiO2 | b3 | 3 | 300 | 10 | 27 | C2–C4 (23), C5–C15 (37) | 41 |
| Fe–Zr–Cu–K/TiO2 | b3 | 3 | 300 | 10 | 25 | C2–C4 (18), C5–C15 (30) | 41 |
| C2–C4 as target | |||||||
| Fe–Zn–K | b3 | 3 | 320 | 5 | 51 | C2–C4 (61), C5+ (4) | 42 |
| Pt/CeO2–Co/SiO2 | c | 3 | 250 | 25 | C2–C4 (40), C5+ (<5) | 43 | |
| Pt/SiO2–Co/SiO2 | c | 3 | 350 | 6 | 20 | C2–C4 (40), C5+ (<5) | 44 |
| C2–C5+ as target | |||||||
| 88.3Fe7.1K4.6Co | b3 | 3 | 300 | 25 | 55 | C2–C4 (32), C5+ (47) | 45 |
| 92.6Fe7.4K | b3 | 3 | 300 | 25 | 42 | C2–C4 (30), C5+ (59) | 45 |
| Na–Fe3O4/HZSM-5 | b3 | 3 | 320 | 30 | 34 | C2–C4 (18), C5+ (74) | 46 |
| Na–Zn–FeC | — | 3 | 340 | 25 | 38 | C2–C4 (47), C5+ (40) | 47 |
| Fe–K–(Co/Ru) | b3 | 3 | 300 | 25 | 55 | C2–C4 (8), C5+ (47) | 48 |
| FeAlOx–Na | b3 | 3 | 330 | 35 | 35 | C2–C4 (21), C5+ (75) | 49 |
| Aromatics as target | |||||||
| ZnFeO–nNa/HZSM-5 | b3 | 3 | 320 | 30 | 41 | C2–C4 (10), C5+ (10), Aro (76) | 50 |
| Na–Fe/HZSM-5 | b3 | 2 | 340 | 30 | 45 | C2–C4 (20), C5+ (10), Aro (65) | 51 |
With different catalyst optimization strategies, an improvement in CO2 conversion has been achieved along with the suppression of methane formation leading to an increase of higher molecular weight hydrocarbon products. One such strategy is the addition of alkali metal promoters that improves the adsorption of CO2 and increases the rate of the RWGS while at the same time suppressing methane formation by reducing the adsorption of hydrogen. These catalyst modifications, combined with lower reaction temperatures (300 to 340 °C) and increased pressures (25 to 30 bar), adjust the rates of RWGS and FTS to increase the conversion of CO2 as well as product selectivity to C5 to C15 species. To overcome the thermodynamic limitation of the RWGS, the rate of the FTS must be substantially higher than that of the RWGS reaction. Therefore, the selection of promoters is always guided by a compromise between RWGS conversion and FTS selectivity. The addition of Zn as a promoter modifies the FTS selectivity to enhance the production of lower olefins while suppressing the formation of higher hydrocarbons. Promotion with noble metals increases hydrogenation activity for the olefins but decreases the CO2 conversion slightly and reduces methane formation due to the increased consumption of hydrogen.40
Work conducted by Liu et al.48 on catalytic CO2 hydrogenation serves as an informative example of how to fine-tune a functional catalyst by adding promoters. Their results demonstrated that removing a major fraction of formed water from the reaction system drives the RWGS reaction and significantly improves the CO2 conversion efficiency. In addition, they highlighted the conversion of the CO intermediate during the FTS reaction as an important step for effectively boosting CO2 conversion. To achieve this boost in conversion, a K and/or Co (Ru) component was added to the precipitated Fe catalysts, directing the formation of iron carbide sites active for the FTS reaction. Specifically, the electron donation from K to Fe promotes chain growth of the hydrocarbons and prevents the direct hydrogenation of Fe–(CH2)n intermediates, which significantly increases the selectivity to C2+ hydrocarbons, particularly lower olefins. Since Co (Ru) has no WGS activity, it exclusively accelerates the FTS reaction's production of the CO intermediate without catalytically converting CO into CO2, thereby significantly improving CO2 conversion. The intimate relationship between the Co and Fe site effects has also been studied, from the nanoscale to the centimeter scale. The results show that the close contact between Fe and Co sites enhances their selectivity for C2+ hydrocarbons. This is because the CO intermediate easily spills over from Fe3O4 to Co sites, resulting in a higher CO concentration than at Co sites. On the other hand, the selectivity to CH4 increases dramatically as the distance between the Fe and Co sites increases. This could be due to the increased direct methanation of CO2 and the decreased chain growth possibility of the FTS reaction caused by the lower CO concentration over the Co sites.
As an example of the importance of material selection, Kim et al.49 reported a bifunctional iron aluminum oxide (FeAlOx) catalyst that directly converts CO2 into C5+ hydrocarbons with an overall selectivity of 77.0% and CO2 conversion of 20.2% at an H2/CO2 ratio of 1:1. Notably, the selectivity for linear α-olefins (LAOs) was 52.4%, accounting for 78.4% of the total C4+ olefins. At a higher H2/CO2 ratio of 3:1, the yield of C5+ hydrocarbons increased to 19.7%. The existence of crystalline-/amorphous-structured active sites in the single FeAlOx catalyst was proposed. The reducible magnetite (Fe3O4) phase, which contained surface oxygen vacancies, facilitated the RWGS reaction to form CO via CO2 hydrogenation, and subsequent C–C coupling over Hägg iron carbide afforded lower olefins (C2–C4=). Long-chain LAOs were then formed on the surface of amorphous aluminum oxide (AlOx) via the re-adsorption of C2–C4=. In addition, the amorphous AlOx phase enhanced CO2 and H2 adsorption, which facilitated the formation of carbonate, bicarbonate, and formate species via the RWGS reaction and the subsequent formation of long-chain hydrocarbons via the Fischer–Tropsch reaction. The bifunctional FeAlOx catalyst showed excellent stability for up to 450 h on stream, demonstrating its potential as a practical-scale catalyst for the conversion of CO2 into value-added liquid fuels and chemicals.
Catalyst preparation by single or multiple-step impregnation of a single support precursor combined with post-treatment and activation steps can efficiently develop multiple active sites well-dispersed on this support. Under reaction conditions, multi-phase systems are often established and controlled by their thermodynamic stability. Not only are the compositions of these phases uncontrollable, but neither is the number of active sites on each of these phases nor their arrangement. For better control of these properties, core–shell structures have been utilized for catalyst preparation using different precursors located in the core and the shell of these particles. This versatile approach for catalyst synthesis is comprehensively fleshed out in a review by Pérez-Ramírez et al.,52 and significantly broadens the range of design parameters for the optimization of bifunctional catalysts. The loading of the core and shell can be varied individually, and the distance between the catalysts can be adjusted by the inclusion of an inert separation layer that is permeable to the reactants and prevents the migration and interaction of the active phases. Core–shell catalysts for the combination of RWGS and FTS reactions have been published by Somorjai43 and Thomas.44 Through modification of FTS catalysts, comparable results to the systems described above were achieved at lower temperatures in the case of ref. 43 and lower pressure in the case of ref. 44. These results can be considered as a proof of concept with great potential for improvement by using the large space of design parameters, specifically the ratio of different active sites as well as their distance and arrangement.
To meet fuel specifications, even higher molecular weight products are required. These can be obtained by the addition of further active sites to the catalyst systems. Acidic sites provided by zeolites (e.g., ZSM-5) can catalyze the oligomerization and isomerization of lower olefins to fuel or aromatic hydrocarbons. Combinations of the tandem catalysts for CO2-FTS with different amounts and modifications of ZSM-5 can produce high yields of alkanes53 or aromatics.42,43
The methanol route for the conversion of CO2 into a value-added product offers even more degrees of freedom for the design of bifunctional catalysts since two catalysts that have been optimized individually can be combined in one reactor for the tandem reaction of methanol synthesis and conversion to dimethyl ether (DME), olefins, or carbonates. Here again, the second reaction can shift the equilibrium of the first reaction and obtain a higher conversion of CO2. The important question for the design of the catalyst system is the selection of two compatible catalysts, the ratio of the amounts of these catalysts, their arrangement, and the determination of optimal operating conditions for this system.
Several publications have described the synthesis of methanol and its simultaneous conversion to DME or olefin products. For the first step of this tandem reaction, different metal oxide-based methanol catalysts were selected with a focus on the chemical stability in presence of acidic zeolites.36,54–60 In many cases, the different catalysts were prepared with a mass ratio of 1:1 using differing degrees of mixing. Some authors only performed mechanical mixing of catalyst pellets,61 while others mixed powders and pelletized these powders.62 An even more intensive mixing was achieved by impregnation of a support with both active materials. Fig. 5 illustrates the different arrangements of the two catalysts for the tandem reactions that have been investigated by various groups. The performance of a dual bed arrangement (case a) is often compared to simply mechanically mixed catalyst particles (case b1), a mixture prepared in aqueous dispersions (case b2), or by applying external physical forces to the mixture of catalysts by grinding or milling (case b3) for reducing the distance between the active sites further. With layered systems, the layer thickness is a well-suitable tuning parameter for the distance between the different active sites. The best-defined arrangement is achieved by impregnation of core–shell particles with the active phases of the two catalysts in a way that keeps them separated between core and shell. Some chose one arrangement,63–65 while others compared all of them.66
 | ||
| Fig. 5 Arrangement of bifunctional catalysts for CO2 conversion. a) Dual catalyst bed without any mixing, b1) mechanical mixing of catalyst particles without applying external forces, b2) mixing in aqueous dispersions, b3) physical mixing of catalyst powders applying external forces (grinding, milling), c) chemical mixing by impregnation of core–shell particles. | ||
In some publications, catalysts have been filled into reactors in layers to demonstrate the advantages of tandem reactions with bifunctional catalysts with a summary of results and relevant citations provided in Table 2. Different combinations of copper-based methanol catalysts (Cu–ZnO, CZA, CZZA, Cu–Mo), indium, and chromium oxide catalysts combined with ZSM-5, H-ZSM5, and SAPO show good stability and performance in catalytic tests. With typical methanol synthesis conditions at temperatures between 200 and 300 °C and pressures around 30 bar, tandem systems with highly active acidic catalysts show CO2 conversions above the thermodynamic limit due to the fast removal of methanol from the reaction mixture by its conversion to DME. At the same time, this suppressed the formation of CO by the reverse water gas shift reaction.
| Catalyst | Preparation method | Catalyst mass ratio | H2/CO2 | Temp./°C | Pressure/bar | Selectivity DME/% | Conversion CO2/% | Ref. |
|---|---|---|---|---|---|---|---|---|
| DME as target | ||||||||
| Cu–ZnO/Al2O3 | b2 | 1:1 |
3:1 |
270 | 30 | 35 | 7.0 | 67 |
| CZA/HZSM-5 | b3 | 2:1 |
3:1 |
260 | 30 | 49.2 | 30.6 | 68 |
| CZA/HZSM-5 | b2 | 1.7:1.5 |
10:1 |
280 | 360 | 89.0 | 97.0 | 69 |
| CZZA/HZSM-5 | b1 | 1:1 |
3:1 |
240 | 28 | 17.5 > 14.3 | 26.8 > 24 | (Ref. 66) 100 hours on stream |
| CZA/NaHZSM-5 | b1 | 1:1 |
2:1 |
275 | 40 | 77.5 | 35.0 | 63 |
| CZZ/HZSM-5 | b3 | 10:1 C:Z:Z (0.5/0.2/0.3) |
3:1 |
250 | 30 | 67.6 | 22.2 | 70 |
| Cu–Mo/HZSM-5 | c | 1:1 |
3:1 |
240 | 20 | 77.2 | 12.4 | 71 |
| Cu–Fe–Ce/HZSM-5 | b1 | — | 4:1 |
260 | 30 | 63.1 | 20.9 | 72 |
| 0.5Pd-CZZA/HZSM-5 | b1 | 1:1 |
3.3:1 |
200 | 30 | 73.56 | 18.67 | 73 |
| CZA@silica alumina | c | Film coatings 4.7 and 7.1 on solid ilica core | 3:1 |
266 | 30 | 42.4 | 47.1 | 65 |
| CZA/SAPO-18 | b3 | 1:1 and 10:1 |
3:1 |
275 | 30 | ∼88 | ∼16 | 74 |
| CIZO–SAPO | b1/b2/a | 1:1 (weight distribution) |
3:1 |
250 | 30 | 60 | 4.3 | 62 |
| Pd/ZnO bio-ZSM-5 | c/a/b1 | 1:1 |
3:1 |
300 | 30 | 31 | ∼9 | 75 |
| Catalyst | Preparation method | Catalyst mass ratio | H2/CO2 | Temp./°C | Pressure/bar | Selectivity hydrocarbons/% | CO2 conversion/% | Ref. |
|---|---|---|---|---|---|---|---|---|
| Catalyst arrangement categories: a. dual bed non-mixing, b1. mechanical mixing of catalyst particles (without external forces), b2. mixing in aqueous dispersions, b3. pressure-mediated physical mixing (milling, grinding), c. chemical mixing (e.g. core–shell). | ||||||||
| Hydrocarbons as target | ||||||||
| In–Zr/SAPO-34 | b3 | 2:1 (In:Zr = 1:4) |
3:1 |
400 | 30 | C2–C4 (93), C5+ (3) | 36 | 56 |
| In2O3–ZnZrOx/SAPO-34 | b1 | 1:1 granule stacking |
3:1 |
380 | 30 | C2–C4 (96), C5+ (2) | 17 | 76 |
| ZnO-Y2O3/SAPO-34 | b1 | 1:1 |
4:1 |
390 | 40 | C2–C4 (97), C5+ (1) | 28 | 54 |
| In2O3/HZSM-5 | b3 | 2:1/1:2 and 4:1 best results 2:1 |
3:1 |
340 | 30 | C2–C4 (20), C5+ (64), Aro (15) | 13 | 36 |
| ZnO–ZrO2/ZSM-5 | b3 | 1:2 |
3:1 |
340 | 30 | C2–C4 (21), C5+ (8), Aro (70) | 9 | 77 |
| Cr2O3/Zn-ZSM-5@SiO2 | b3 | Different layer thicknesses tested | 2.7:1 |
350 | 30 | C2–C4 (23), C5+ (3), Aro (70) | 22 | 78 |
| No further data given | ||||||||
| Cr2O3/H-ZSM-5 | b3 | 1:1 |
3:1 |
350 | 30 | C2–C4 (16), C5+ (6), Aro (76) | 35 | 79 |
| ZnO–ZrO2/H-ZSM-5 | b1 | 1:2 |
3:1 |
340 | 40 | C2–C4 (16), C5+ (3), Aro (81) | 15 | 80 |
The operation of the tandem hydrogenation of CO2 at temperatures above 300 °C required a catalyst combination with improved stability such as the pairing of indium oxide or chromium oxide with HZSM-5 or SAPO-34. The increased activity of the zeolites at these temperatures results in the formation of lower olefins and aromatic hydrocarbons. Again, the equilibrium of methanol synthesis is shifted due to the higher rate of the consecutive reaction. Selectivities for aromatics of up to 80% have been obtained. Due to the complexity of the catalyst systems, only a few kinetic studies have been performed. Variations of the reaction conditions like temperature, pressure, or molar ratio of H2 to CO2 showed the expected impact on the conversion of CO2 and product selectivity.
For mechanically mixed catalysts, in general, variations of their mass ratios only weakly influence performance as long as the activity of the acidic catalyst is substantially higher than that of the methanol synthesis catalyst. Nevertheless, Gao et al.36 observed a synergetic effect of catalyst proximity when comparing different catalyst arrangements and degrees of mixing. For the combination of indium oxide as a methanol catalyst with HZSM-5, the hydrogenation of CO2 to hydrocarbons was studied under identical conditions with layered catalysts and differently treated mixtures of the catalyst. With a decreasing distance between the active sites, achieved by mixing smaller catalyst particles, the synergetic effect of the tandem catalyst was improved. But for a mixture of the catalyst powders, the selectivity shifted completely from C2 to C5 alkanes and olefins to methane. The same effect was shown later for other combinations of catalysts for CO2 hydrogenation to DME.75 This effect was also demonstrated at higher temperature ranges, where aromatic hydrocarbons are generated. Some representative results for a variation of the catalyst distance, as a measure of proximity are compiled in Table 3.
| Catalyst | Preparation method | H2/CO2 | Temp./°C | Pressure/bar | Selectivity DME/% | Conversion CO2/% | Ref. |
|---|---|---|---|---|---|---|---|
| Catalyst arrangement: a. dual bed non-mixing, b1. mechanical mixing of catalyst particles (without external forces), b2. mixing in aqueous dispersions, b3. pressure-mediated physical mixing (grinding, milling), c. chemical mixing (e.g. core–shell). | |||||||
| CIZO–SAPO | b1 | 3:1 |
250 | 30 | ∼60 | 4.3 | 62 |
| CIZO–SAPO | b3 | 3:1 |
250 | 30 | ∼60 | 3.7 | 62 |
| CIZO–SAPO | a | 3:1 |
250 | 30 | ∼60 | 2.9 | 62 |
| Pd/ZnO/bio-ZSM-5 | a | 3:1 |
300 | 30 | 4.5 | 8.9 | 75 |
| Pd/ZnO/bio-ZSM-5 | b1 | 3:1 |
300 | 30 | 31 | 10.6 | 75 |
| Pd/ZnO/bio-ZSM-5 | c | 3:1 |
300 | 30 | 2.9 | 9.1 | 75 |
4. General discussion
Similar findings related to the strong impact of structural features on bifunctional catalyst performance were also reported in a review by Roldán and Strasser for the electrochemical conversion of CO2.81 The influence on the performance of particle composition, nanostructure, size, and distance can finally be assigned to the electrostatic interactions within the overlapping electrical double layers at the catalyst surfaces, which tailor the adsorption of reactant and spectator molecules and thus activity and selectivity. The typical length scale at which these phenomena occur is on the order of several nanometers, depending on the ionic strength of the electrolyte that controls the thickness of the double layer.82,83 This length scale is well above the length scale for the site distances of traditional bifunctional catalysts that interact via steric interaction on the scale of one nanometer and below, as described above. In both cases, the interaction of the two sites causes simultaneous binding and activation of one or two substrates for conversion within one stoichiometric reaction.The role of proximity in tandem reactions was postulated by Weisz in his theoretical studies on mass transfer limitations within bifunctional catalysts for hydrocracking where he called it the “intimacy criterion”, which has often been interpreted simply as ‘the closer the better’ for positioning metal and acid sites.84 A lack of synthesis and material-characterization methods with nanometer precision has long prevented in-depth exploration of this postulate. Only as recently as 2015 did Martens et al. publish an experimental investigation of a model system that supports the postulate and indicates further potential for the optimization of such systems.85
This synergetic proximity effect on tandem reactions suggests a coupling of the two catalysts through a concentration gradient of the intermediate product in the environment of the methanol conversion catalyst. The closer the conversion catalyst is located to the methanol synthesis catalyst, the more it decreases the local concentration of methanol, thus shifting the equilibrium to higher conversion. This observation motivated Wang to increase the proximity of the catalysts further through the design of core–shell systems with the methanol catalyst as the core and the zeolite as the shell.36 A further increase in CO2 conversion and DME synthesis was reported for this catalyst structure. In the review by Pérez-Ramírez et al. the versatility of core–shell particles for catalyst design was presented,52 as well as in a perspective published in 2019 by Ma et al.86 The catalytic activity and transport processes within the catalyst particles can highly be tuned by varying the thickness of the core, shell, and inert separation layer between them, as well as using different loadings of core and shell catalyst material. The huge number and range of design parameters prevent empirical optimization. Furthermore, attempting to guide bifunctional catalyst optimization with kinetic investigations requires a considerable number of experiments due to the large number of model parameters that need to be quantified simultaneously. Assuming a formal kinetic model with four parameters and a two-parameter model for the mass transfer results in 10 unknown model parameters. A simple 3-step design of experiments would result in about 60000 measurements for parameter estimation. For example, an investigation of the impact of a design parameter on the catalyst like core–shell distance would be needed for each distance. This time and resource-intensive investigation should be replaced by a modeling-based approach, as shown by Brösigke et al. for core–shell systems.87 In this approach, illustrated in Fig. 6 a much smaller set of experimental data is required for completing and adjusting the model to the interactions between the two catalysts. For the given example, it would be a few hundred data points.
 | ||
| Fig. 6 Core–shell catalyst with catalyst A as core catalyzing the methanol reaction with rate R1. A shell with thickness δ separates catalyst B from catalyst A. At catalyst B methanol is converted to DME at a rate R2. The decaying curve represents the concentration profile of methanol as the intermediate product in this tandem reaction. | ||
For a postulated core–shell catalyst, a model was developed from kinetic models for the individual catalysts and a transport model based on diffusive transport.87 By applying numerical methods, the material balances for a single particle were solved to determine the performance parameters of CO2 conversion and DME yield. Simulations for catalysts with different separation distances between the active sites adjusted by the thickness of the shell layer confirmed the intimacy criterion, which is not supported by experimental results. The applied model for the methanol synthesis does not consider the adsorption of the water that is formed in the consecutive methanol conversion at the methanol catalyst, which causes an inhibition by blocking active sites. An extended model that considers this effect as feedback between the catalysts gives a different result for the distance variation. Only at a certain distance between the two catalysts can the diffusion of the water to the core be suppressed sufficiently for a good performance of the overall system. The modeling studies showed that for various “feedback scenarios” different distances of the active sites result in a synergetic performance. With further comparisons between model predictions and experimental results, the model can be extended and adjusted to the catalyst systems. With a validated model, there is a chance for catalyst optimization without extensive experimental effort. From this study described above, a workflow can be derived for the design of a core–shell catalyst. Instead of chemical intuition, a simulation with the mean-field model combining the kinetic models of the two catalyst candidates should be the starting point. With this, the operation window for the tandem reaction can be identified, especially the ratio of the two active materials. This model should be validated by kinetic studies with a powder mixture of the two catalysts at the identified conditions. An extension of the model by interaction and transport terms will provide a model for a more detailed simulation of a single core–shell catalyst, which allows a variation of the distance between the active sites and a search for an ideal value for this parameter. These results can guide the preparation of the desired bifunctional catalyst, which should need only a few iterations for optimization. From these considerations of the special aspects of CO2 activation and a knowledge transfer from the more mature field of bifunctional catalysts for hydrocarbon treatment88–93 a general workflow can be derived that is illustrated in Fig. 7.
 | ||
| Fig. 7 Proposal for a general workflow for the design of bifunctional catalysts for tandem reactions. | ||
After the identification of a possible operation window for a tandem reaction by thermodynamic calculations, two suitable catalysts can be selected on an empirical basis and tested for their material compatibility, e.g. by TGA. Kinetic studies of the separate catalysts will provide two kinetic models that can be combined for a simulation of the tandem reaction. Also, the influence of components involved in the second reaction on the individual catalyst needs attention in this step. The simulation of the tandem reaction will indicate the appropriate ratio of the two catalysts in the bifunctional catalyst and suitable operating conditions. The prepared bifunctional catalyst is tested and its performance is assessed against the predictions by the simulations. Strong deviations caused by interactions between the catalysts, the reactants, or mass transfer processes are accounted for in an extension of the model by appropriate terms. With the new model, an optimization of the ratio of the catalyst as well as their distance can be used for a further feedback loop to the catalyst preparation. The benefits of this modeling-supported catalyst design are nicely demonstrated by Ramirez et al.94
From a more general perspective, two catalysts can show a synergetic catalytic function in a tandem reaction if the operation window for the coupled reaction is close to the optimal individual operation windows. Still, tandem operation represents a loss of degrees of freedom in the process design that need to be matched with advantages in other aspects like overcoming thermodynamic limitation, omitting a separation step, or improving selectivity. If this is given, the mechanism of interaction of the two catalysts should be considered for choosing their arrangement. In general, the three different types of interaction involved in the mechanism of the reaction at bifunctional catalysts require distances of the active sites that allow for tuning and optimization of their interaction.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – EXC 2008/1 (UniSysCat) – 390540038. We thank Leo Brody for the intensive discussion and careful language editing of the manuscript.References
- P. Markewitz, W. Kuckshinrichs, W. Leitner, J. Linssen, P. Zapp, R. Bongartz, A. Schreiber and T. E. Müller, Worldwide innovations in the development of carbon capture technologies and the utilization of CO2, Energy Environ. Sci., 2012, 5, 7281–7305, 10.1039/c2ee03403d
.
- Q. W. Song, Z. H. Zhou and L. N. He, Efficient, selective and sustainable catalysis of carbon dioxide, Green Chem., 2017, 19, 3707–3728, 10.1039/c7gc00199a
- J. Artz, T. E. Müller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Sustainable Conversion of Carbon Dioxide: An Integrated Review of Catalysis and Life Cycle Assessment, Chem. Rev., 2018, 118, 434–504, DOI:10.1021/acs.chemrev.7b00435
- M. Peters, B. Köhler, W. Kuckshinrichs, W. Leitner, P. Markewitz and T. E. Müller, Chemical technologies for exploiting and recycling carbon dioxide into the value chain, ChemSusChem, 2011, 4, 1216–1240, DOI:10.1002/cssc.201000447
- E. Catizzone, G. Bonura, M. Migliori, F. Frusteri and G. Giordano, CO2 recycling to dimethyl ether: State-of-the-art and perspectives, Molecules, 2018, 23, 1–28, DOI:10.3390/molecules23010031
- W. Zhou, K. Cheng, J. Kang, C. Zhou, V. Subramanian, Q. Zhang and Y. Wang, New horizon in C1 chemistry: Breaking the selectivity limitation in transformation of syngas and hydrogenation of CO2 into hydrocarbon chemicals and fuels, Chem. Soc. Rev., 2019, 48, 3193–3228, 10.1039/c8cs00502h
- C. Maeda, Y. Miyazaki and T. Ema, Recent progress in catalytic conversions of carbon dioxide, Catal. Sci. Technol., 2014, 4, 1482–1497, 10.1039/c3cy00993a
- J. Klankermayer, S. Wesselbaum, K. Beydoun and W. Leitner, Selective Catalytic Synthesis Using the Combination of Carbon Dioxide and Hydrogen: Catalytic Chess at the Interface of Energy and Chemistry, Angew. Chem., Int. Ed., 2016, 55, 7296–7343, DOI:10.1002/anie.201507458
- H. Q. Liang, T. Beweries, R. Francke and M. Beller, Molecular Catalysts for the Reductive Homocoupling of CO2 towards C2+ Compounds, Angew. Chem., 2022, 61, e202200723, DOI:10.1002/anie.202200723
- J. H. Barnard, C. Wang, N. G. Berry and J. Xiao, Long-range metal-ligand bifunctional catalysis: Cyclometallated iridium catalysts for the mild and rapid dehydrogenation of formic acid, Chem. Sci., 2013, 4, 1234–1244, 10.1039/c2sc21923a
- T. Kimura, K. Kamata and N. Mizuno, A bifunctional tungstate catalyst for chemical fixation of CO 2 at atmospheric pressure, Angew. Chem., Int. Ed., 2012, 51, 6700–6703, DOI:10.1002/anie.201203189
- I. Sreedhar, Y. Varun, S. A. Singh, A. Venugopal and B. M. Reddy, Developmental trends in CO2 methanation using various catalysts, Catal. Sci. Technol., 2019, 9, 4478–4504, 10.1039/c9cy01234f
- T. Ema, Y. Miyazaki, S. Koyama, Y. Yano and T. Sakai, A bifunctional catalyst for carbon dioxide fixation: Cooperative double activation of epoxides for the synthesis of cyclic carbonates, Chem. Commun., 2012, 48, 4489–4491, 10.1039/c2cc30591g
- J. Song, Z. Zhang, S. Hu, T. Wu, T. Jiang and B. Han, MOF-5/n-Bu4NBr: an efficient catalyst system for the synthesis of cyclic carbonates from epoxides and CO2 under mild conditions, Green Chem., 2009, 11, 1031, 10.1039/b902550b
- K. B. Tan, G. Zhan, D. Sun, J. Huang and Q. Li, The development of bifunctional catalysts for carbon dioxide hydrogenation to hydrocarbonsviathe methanol route: From single component to integrated components, J. Mater. Chem. A, 2021, 9, 5197–5231, 10.1039/d0ta09607e
- Y. Gambo, S. Adamu, R. A. Lucky, M. S. Ba-Shammakh and M. M. Hossain, Tandem catalysis: A sustainable alternative for direct hydrogenation of CO2 to light olefins, Appl. Catal., A, 2022, 641, 118658, DOI:10.1016/j.apcata.2022.118658
- H. S. Whang, J. Lim, M. S. Choi, J. Lee and H. Lee, Heterogeneous catalysts for catalytic CO2 conversion into value-added chemicals, BMC Chem. Eng., 2019, 1, 1–19, DOI:10.1186/s42480-019-0007-7
- A. Buonerba, A. De Nisi, A. Grassi, S. Milione, C. Capacchione, S. Vagin and B. Rieger, Novel iron(iii) catalyst for the efficient and selective coupling of carbon dioxide and epoxides to form cyclic carbonates, Catal. Sci. Technol., 2015, 5, 118–123, 10.1039/c4cy01187b
- H. Sugimoto and S. Inoue, Copolymerization of carbon dioxide and epoxide, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5561–5573, DOI:10.1002/pola.20319
- L. Grefe and E. Mejía, Earth-abundant bimetallic and multimetallic catalysts for Epoxide/CO2 ring-opening copolymerization, Tetrahedron, 2021, 98, 132433, DOI:10.1016/j.tet.2021.132433
- Z. Duan, X. Wang, Q. Gao, L. Zhang, B. Liu and I. Kim, Highly active bifunctional cobalt-salen complexes for the synthesis of poly(ester-block-carbonate) copolymer via terpolymerization of carbon dioxide, propylene oxide, and norbornene anhydride isomer: Roles of anhydride conformation consideration, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 789–795, DOI:10.1002/pola.27057
- A. Buchard, M. R. Kember, K. G. Sandeman and C. K. Williams, A bimetallic iron(iii) catalyst for CO2/epoxide coupling, Chem. Commun., 2011, 47, 212–214, 10.1039/c0cc02205e
- M. R. Kember, A. Buchard and C. K. Williams, Catalysts for co2/epoxide copolymerisation, Chem. Commun., 2011, 47, 141–163, 10.1039/c0cc02207a
- S. Klaus, M. W. Lehenmeier, C. E. Anderson and B. Rieger, Recent advances in CO2/epoxide copolymerization-New strategies and cooperative mechanisms, Coord. Chem. Rev., 2011, 255, 1460–1479, DOI:10.1016/j.ccr.2010.12.002
- S. I. Vagin, R. Reichardt, S. Klaus and B. Rieger, Conformationally Flexible Dimeric Salphen Complexes for Bifunctional Catalysis, J. Am. Chem. Soc., 2010, 132, 14367–14369, DOI:10.1021/ja106484t
- D. J. Darensbourg and S. J. Kyran, Carbon Dioxide Copolymerization Study with a Sterically Encumbering Naphthalene-Derived Oxide, ACS Catal., 2015, 5, 5421–5430, DOI:10.1021/acscatal.5b01375
- J. Lim, S. H. Yun, M. R. Kim and I. Kim, Synthesis of polycarbonate polyols by double-metal cyanide catalyzed copolymerization of epoxide with carbon dioxide, J. Nanosci. Nanotechnol., 2017, 17, 7507–7514, DOI:10.1166/jnn.2017.14796
- A. Bavykina, N. Kolobov, I. S. Khan, J. A. Bau, A. Ramirez and J. Gascon, Metal-Organic Frameworks in Heterogeneous Catalysis: Recent Progress, New Trends, and Future Perspectives, Chem. Rev., 2020, 120, 8468–8535, DOI:10.1021/acs.chemrev.9b00685
- J. Langanke, A. Wolf, J. Hofmann, K. Böhm, M. A. Subhani, T. E. Müller, W. Leitner and C. Gürtler, Carbon dioxide (CO2) as sustainable feedstock for polyurethane production, Green Chem., 2014, 16, 1865–1870, 10.1039/c3gc41788c
- V. J. Spiering, A. Ciapetti, M. T. Lima, D. W. Hayward, L. Noirez, M. Appavou, R. Schomäcker and M. Gradzielski, Changes in Phase Behavior from the Substitution of Ethylene Oxide with Carbon Dioxide in the Head Group of Nonionic Surfactants, ChemSusChem, 2020, 13, 601–607, DOI:10.1002/cssc.201902855
- J. Song, Z. Zhang, S. Hu, T. Wu, T. Jiang and B. Han, MOF-5/n-Bu4NBr: An efficient catalyst system for the synthesis of cyclic carbonates from epoxides and CO2 under mild conditions, Green Chem., 2009, 11, 1031–1036, 10.1039/b902550b
- D. F. Mullica, W. O. Milligan, G. W. Beall and W. L. Reeves, Crystal structure of Zn 3 [Co(CN) 6] 2 .12H 2 O, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1978, 34, 3558–3561, DOI:10.1107/S0567740878011589
- J. Hafizovic, M. Bjørgen, U. Olsbye, P. D. C. Dietzel, S. Bordiga, C. Prestipino, C. Lamberti and K. P. Lillerud, The Inconsistency in Adsorption Properties and Powder XRD Data of MOF-5 Is Rationalized by Framework Interpenetration and the Presence of Organic and Inorganic Species in the Nanocavities, J. Am. Chem. Soc., 2007, 129, 3612–3620, DOI:10.1021/ja0675447
- C. A. Allen, J. A. Boissonnault, J. Cirera, R. Gulland, F. Paesani and S. M. Cohen, Chemically crosslinked isoreticular metal–organic frameworks, Chem. Commun., 2013, 49, 3200, 10.1039/c3cc40635k
- C. A. Allen and S. M. Cohen, Exploration of Chemically Cross-Linked Metal–Organic Frameworks, Inorg. Chem., 2014, 53, 7014–7019, DOI:10.1021/ic500951b
- P. Gao, S. Li, X. Bu, S. Dang, Z. Liu, H. Wang, L. Zhong, M. Qiu, C. Yang, J. Cai, W. Wei and Y. Sun, Direct conversion of CO2 into liquid fuels with high selectivity over a bifunctional catalyst, Nat. Chem., 2017, 9, 1019–1024, DOI:10.1038/nchem.2794
- P. H. Choi, K.-W. Jun, S.-J. Lee, M.-J. Choi and K.-W. Lee, Hydrogenation of carbon dioxide over alumina supported Fe-K catalysts, Catal. Lett., 1996, 40, 115–118, DOI:10.1007/BF00807467
- G. Kishan, M. W. Lee, S. S. Nam, M. J. Choi and K. W. Lee, The catalytic conversion of CO2 to hydrocarbons over Fe-K supported on Al2O3-MgO mixed oxides, Catal. Lett., 1998, 56, 215–219, DOI:10.1023/A:1019089919614
- R. Satthawong, N. Koizumi, C. Song and P. Prasassarakich, Bimetallic Fe–Co catalysts for CO2 hydrogenation to higher hydrocarbons, J. CO2 Util., 2013, 3–4, 102–106, DOI:10.1016/j.jcou.2013.10.002
- R. E. Owen, J. P. O'Byrne, D. Mattia, P. Plucinski, S. I. Pascu and M. D. Jones, Cobalt catalysts for the conversion of CO2 to light hydrocarbons at atmospheric pressure, Chem. Commun., 2013, 49, 11683, 10.1039/c3cc46791k
- U. Rodemerck, M. Holeňa, E. Wagner, Q. Smejkal, A. Barkschat and M. Baerns, Catalyst Development for CO 2 Hydrogenation to Fuels, ChemCatChem, 2013, 5, 1948–1955, DOI:10.1002/cctc.201200879
- J. Zhang, S. Lu, X. Su, S. Fan, Q. Ma and T. Zhao, Selective formation of light olefins from CO2 hydrogenation over Fe–Zn–K catalysts, J. CO2 Util., 2015, 12, 95–100, DOI:10.1016/j.jcou.2015.05.004
- C. Xie, C. Chen, Y. Yu, J. Su, Y. Li, G. A. Somorjai and P. Yang, Tandem Catalysis for CO2 Hydrogenation to C2-C4 Hydrocarbons, Nano Lett., 2017, 17, 3798–3802, DOI:10.1021/acs.nanolett.7b01139
- E. Gioria, L. Duarte-Correa, N. Bashiri, W. Hetaba, R. Schomaecker and A. Thomas, Rational design of tandem catalysts using a core–shell structure approach, Nanoscale Adv., 2021, 3, 3454–3459, 10.1039/D1NA00310K
- S. Geng, F. Jiang, Y. Xu and X. Liu, Iron-Based Fischer-Tropsch Synthesis for the Efficient Conversion of Carbon Dioxide into Isoparaffins, ChemCatChem, 2016, 8, 1303–1307, DOI:10.1002/cctc.201600058
- J. Wei, Q. Ge, R. Yao, Z. Wen, C. Fang, L. Guo, H. Xu and J. Sun, Directly converting CO2 into a gasoline fuel, Nat. Commun., 2017, 8, 15174, DOI:10.1038/ncomms15174
- Z. Zhang, H. Yin, G. Yu, S. He, J. Kang, Z. Liu, K. Cheng, Q. Zhang and Y. Wang, Selective hydrogenation of CO2 and CO into olefins over Sodium- and Zinc-Promoted iron carbide catalysts, J. Catal., 2021, 395, 350–361, DOI:10.1016/j.jcat.2021.01.036
- F. Jiang, B. Liu, S. Geng, Y. Xu and X. Liu, Hydrogenation of CO2 into hydrocarbons: Enhanced catalytic activity over Fe-based Fischer-Tropsch catalysts, Catal. Sci. Technol., 2018, 8, 4097–4107, 10.1039/c8cy00850g
- M. K. Khan, P. Butolia, H. Jo, M. Irshad, D. Han, K. W. Nam and J. Kim, Selective Conversion of Carbon Dioxide into Liquid Hydrocarbons and Long-Chain α-Olefins over Fe-Amorphous AlO xBifunctional Catalysts, ACS Catal., 2020, 10, 10325–10338, DOI:10.1021/acscatal.0c02611
- X. Cui, P. Gao, S. Li, C. Yang, Z. Liu, H. Wang, L. Zhong and Y. Sun, Selective Production of Aromatics Directly from Carbon Dioxide Hydrogenation, ACS Catal., 2019, 9, 3866–3876, DOI:10.1021/acscatal.9b00640
- C. Wen, J. Jiang, C. Chiliu, Z. Tian, X. Xu, J. Wu, C. Wang and L. Ma, Single-Step Selective Conversion of Carbon Dioxide to Aromatics over Na-Fe 3 O 4/Hierarchical HZSM-5 Zeolite Catalyst, Energy Fuels, 2020, 34, 11282–11289, DOI:10.1021/acs.energyfuels.0c02120
- S. Das, J. Pérez-Ramírez, J. Gong, N. Dewangan, K. Hidajat, B. C. Gates and S. Kawi, Core-shell structured catalysts for thermocatalytic, photocatalytic, and electrocatalytic conversion of CO2, Chem. Soc. Rev., 2020, 49, 2937–3004, 10.1039/c9cs00713j
- P. Gao, S. Li, X. Bu, S. Dang, Z. Liu, H. Wang, L. Zhong, M. Qiu, C. Yang, J. Cai, W. Wei and Y. Sun, Direct conversion of CO2 into liquid fuels with high selectivity over a bifunctional catalyst, Nat. Chem., 2017, 9, 1019–1024, DOI:10.1038/nchem.2794
- J. Li, T. Yu, D. Miao, X. Pan and X. Bao, Carbon dioxide hydrogenation to light olefins over ZnO-Y2O3 and SAPO-34 bifunctional catalysts, Catal. Commun., 2019, 129, 105711, DOI:10.1016/j.catcom.2019.105711
- J. Gao, C. Jia and B. Liu, Direct and selective hydrogenation of CO2 to ethylene and propene by bifunctional catalysts, Catal. Sci. Technol., 2017, 7, 5602–5607, 10.1039/c7cy01549f
- P. Gao, S. Dang, S. Li, X. Bu, Z. Liu, M. Qiu, C. Yang, H. Wang, L. Zhong, Y. Han, Q. Liu, W. Wei and Y. Sun, Direct Production of Lower Olefins from CO2 Conversion via Bifunctional Catalysis, ACS Catal., 2018, 8, 571–578, DOI:10.1021/acscatal.7b02649
- M. Huš, D. Kopač and B. Likozar, Catalytic Hydrogenation of Carbon Dioxide to Methanol: Synergistic Effect of Bifunctional Cu/Perovskite Catalysts, ACS Catal., 2019, 9, 105–116, DOI:10.1021/acscatal.8b03810
- E. Gomez, B. Yan, S. Kattel and J. G. Chen, Carbon dioxide reduction in tandem with light-alkane dehydrogenation, Nat. Rev. Chem., 2019, 3, 638–649, DOI:10.1038/s41570-019-0128-9
- X. Liu, M. Wang, C. Zhou, W. Zhou, K. Cheng, J. Kang, Q. Zhang, W. Deng and Y. Wang, Selective transformation of carbon dioxide into lower olefins with a bifunctional catalyst composed of ZnGa2O4 and SAPO-34, Chem. Commun., 2017, 54, 140–143, 10.1039/c7cc08642c
- K. Cheng, W. Zhou, J. Kang, S. He, S. Shi, Q. Zhang, Y. Pan, W. Wen and Y. Wang, Bifunctional Catalysts for One-Step Conversion of Syngas into Aromatics with Excellent Selectivity and Stability, Chem, 2017, 3, 334–347, DOI:10.1016/j.chempr.2017.05.007
- M. Stiefel, R. Ahmad, U. Arnold and M. Döring, Direct synthesis of dimethyl ether from carbon-monoxide-rich synthesis gas: Influence of dehydration catalysts and operating conditions, Fuel Process. Technol., 2011, 92, 1466–1474, DOI:10.1016/j.fuproc.2011.03.007
- L. Yao, X. Shen, Y. Pan and Z. Peng, Unravelling Proximity-Driven Synergetic Effect within CIZO-SAPO Bifunctional Catalyst for CO2Hydrogenation to DME, Energy Fuels, 2020, 34, 8635–8643, DOI:10.1021/acs.energyfuels.0c01256
- J. Ereña, R. Garoña, J. M. Arandes, A. T. Aguayo and J. Bilbao, Effect of operating conditions on the synthesis of dimethyl ether over a CuO-ZnO-Al2O3/NaHZSM-5 bifunctional catalyst, Catal. Today, 2005, 107–108, 467–473, DOI:10.1016/j.cattod.2005.07.116
- G. Bonura, M. Migliori, L. Frusteri, C. Cannilla, E. Catizzone, G. Giordano and F. Frusteri, Acidity control of zeolite functionality on activity and stability of hybrid catalysts during DME production via CO2 hydrogenation, J. CO2 Util., 2018, 24, 398–406, DOI:10.1016/j.jcou.2018.01.028
- F. Zha, J. Ding, Y. Chang, J. Ding, J. Wang and J. Ma, Cu-Zn-al oxide cores packed by metal-doped amorphous silica-alumina membrane for catalyzing the hydrogenation of carbon dioxide to dimethyl ether, Ind. Eng. Chem. Res., 2012, 51, 345–352, DOI:10.1021/ie202090f
- X. Fan, B. Jin, S. Ren, S. Li, M. Yu and X. Liang, Roles of interaction between components in CZZA/HZSM-5 catalyst for dimethyl ether synthesis via CO2 hydrogenation, AIChE J., 2021, 67, 1–13, DOI:10.1002/aic.17353
- R. J. Da Silva, A. F. Pimentel, R. S. Monteiro and C. J. A. Mota, Synthesis of methanol and dimethyl ether from the CO2 hydrogenation over Cu·ZnO supported on Al2 and Nb2, J. CO2 Util., 2016, 15, 83–88, DOI:10.1016/j.jcou.2016.01.006
- Y. Zhang, D. Li, S. Zhang, K. Wang and J. Wu, CO2 hydrogenation to dimethyl ether over CuO-ZnO-Al 2O3/HZSM-5 prepared by combustion route, RSC Adv., 2014, 4, 16391–16396, 10.1039/c4ra00825a
- A. Bansode and A. Urakawa, Towards full one-pass conversion of carbon dioxide to methanol and methanol-derived products, J. Catal., 2014, 309, 66–70, DOI:10.1016/j.jcat.2013.09.005
- L. Li, D. Mao, J. Xiao, L. Li, X. Guo and J. Yu, Facile preparation of highly efficient CuO-ZnO-ZrO2/HZSM-5 bifunctional catalyst for one-step CO2 hydrogenation to dimethyl ether: Influence of calcination temperature, Chem. Eng. Res. Des., 2016, 111, 100–108, DOI:10.1016/j.cherd.2016.04.018
- G. X. Qi, J. H. Fei, X. M. Zheng and Z. Y. Hou, DME synthesis from carbon dioxide and hydrogen over Cu-Mo/HZSM-5, Catal. Lett., 2001, 72, 121–124, DOI:10.1023/A:1009049513834
- X. Zhou, T. Su, Y. Jiang, Z. Qin, H. Ji and Z. Guo, CuO-Fe2O3-CeO2/HZSM-5 bifunctional catalyst hydrogenated CO2 for enhanced dimethyl ether synthesis, Chem. Eng. Sci., 2016, 153, 10–20, DOI:10.1016/j.ces.2016.07.007
- K. Sun, W. Lu, F. Qiu, S. Liu and X. Xu, Direct synthesis of DME over bifunctional catalyst: Surface properties and catalytic performance, Appl. Catal., A, 2003, 252, 243–249, DOI:10.1016/S0926-860X(03)00466-6
- A. Ateka, I. Sierra, J. Ereña, J. Bilbao and A. T. Aguayo, Performance of CuO–ZnO–ZrO2 and CuO–ZnO–MnO as metallic functions and SAPO-18 as acid function of the catalyst for the synthesis of DME co-feeding CO2, Fuel Process. Technol., 2016, 152, 34–45, DOI:10.1016/j.fuproc.2016.05.041
- W. Li, K. Wang, G. Zhan, J. Huang and Q. Li, Hydrogenation of CO2to Dimethyl Ether over Tandem Catalysts Based on Biotemplated Hierarchical ZSM-5 and Pd/ZnO, ACS Sustainable Chem. Eng., 2020, 8, 14058–14070, DOI:10.1021/acssuschemeng.0c04399
- S. Dang, H. Yang, P. Gao, H. Wang, X. Li, W. Wei and Y. Sun, A review of research progress on heterogeneous catalysts for methanol synthesis from carbon dioxide hydrogenation, Catal. Today, 2019, 61–75, DOI:10.1016/j.cattod.2018.04.021
- X. Zhang, A. Zhang, X. Jiang, J. Zhu, J. Liu, J. Li, G. Zhang, C. Song and X. Guo, Utilization of CO2 for aromatics production over ZnO/ZrO2-ZSM-5 tandem catalyst, J. CO2 Util., 2019, 29, 140–145, DOI:10.1016/j.jcou.2018.12.002
- Y. Wang, W. Gao, S. Kazumi, H. Li, G. Yang and N. Tsubaki, Direct and Oriented Conversion of CO 2 into Value-Added Aromatics, Chem. – Eur. J., 2019, 25, 5149–5153, DOI:10.1002/chem.201806165
- Y. Wang, L. Tan, M. Tan, P. Zhang, Y. Fang, Y. Yoneyama, G. Yang and N. Tsubaki, Rationally Designing Bifunctional Catalysts as an Efficient Strategy to Boost CO 2 Hydrogenation Producing Value-Added Aromatics, ACS Catal., 2019, 9, 895–901, DOI:10.1021/acscatal.8b01344
- C. Zhou, J. Shi, W. Zhou, K. Cheng, Q. Zhang, J. Kang and Y. Wang, Highly Active ZnO-ZrO 2 Aerogels Integrated with H-ZSM-5 for Aromatics Synthesis from Carbon Dioxide, ACS Catal., 2020, 10, 302–310, DOI:10.1021/acscatal.9b04309
- H. Mistry, A. S. Varela, S. Kühl, P. Strasser and B. Roldán Cuenya, Nanostructured electrocatalysts with tunable activity and selectivity, Nat. Rev. Mater., 2016, 1, 16009, DOI:10.1038/natrevmats.2016.9
- M. Nesselberger, M. Roefzaad, R. Fayçal Hamou, P. Ulrich Biedermann, F. F. Schweinberger, S. Kunz, K. Schloegl, G. K. H. Wiberg, S. Ashton, U. Heiz, K. J. J. Mayrhofer and M. Arenz, The effect of particle proximity on the oxygen reduction rate of size-selected platinum clusters, Nat. Mater., 2013, 12, 919–924, DOI:10.1038/nmat3712
- J. Speder, L. Altmann, M. Bäumer, J. J. K. Kirkensgaard, K. Mortensen and M. Arenz, The particle proximity effect: from model to high surface area fuel cell catalysts, RSC Adv., 2014, 4, 14971, 10.1039/c4ra00261j
-
P. B. Weisz, in Polyfunctional Heterogeneous Catalysis, 1962, pp. 137–190, DOI:10.1016/S0360-0564(08)60287-4
- J. Zecevic, G. Vanbutsele, K. P. De Jong and J. A. Martens, Nanoscale intimacy in bifunctional catalysts for selective conversion of hydrocarbons, Nature, 2015, 528, 245–254, DOI:10.1038/nature16173
- Z. Ma and M. D. Porosoff, Development of Tandem Catalysts for CO 2 Hydrogenation to Olefins, ACS Catal., 2019, 9, 2639–2656, DOI:10.1021/acscatal.8b05060
- G. Brösigke, J. U. Repke, R. Schomäcker and S. Matera, The closer the better? Theoretical assessment of the impact of catalytic site separation for bifunctional core–shell catalyst particles, Chem. Eng. J., 2022, 446, 136891, DOI:10.1016/j.cej.2022.136891
- P. G. Smirniotis, L. Davydov and E. Ruckenstein, Composite zeolite-based catalysts and sorbents, Catal. Rev.: Sci. Eng., 1999, 41, 43–113, DOI:10.1081/CR-100101949
- B. D. Vandegehuchte, J. W. Thybaut, J. A. Martens and G. B. Marin, Maximizing n-alkane hydroisomerization: The interplay of phase, feed complexity and zeolite catalyst mixing, Catal. Sci. Technol., 2015, 5, 2053–2058, 10.1039/c4cy01135j
- P. S. F. Mendes, F. M. Mota, J. M. Silva, M. F. Ribeiro, A. Daudin and C. Bouchy, A systematic study on mixtures of Pt/zeolite as hydroisomerization catalysts, Catal. Sci. Technol., 2017, 7, 1095–1107, 10.1039/c6cy02642g
- E. Gutierrez-Acebo, C. Leroux, C. Chizallet, Y. Schuurman and C. Bouchy, Metal/Acid Bifunctional Catalysis and Intimacy Criterion for Ethylcyclohexane Hydroconversion: When Proximity Does Not Matter, ACS Catal., 2018, 8, 6035–6046, DOI:10.1021/acscatal.8b00633
- J. I. Mirena, J. W. Thybaut, G. B. Marin, J. A. Martens and V. V. Galvita, Impact of the Spatial Distribution of Active Material on Bifunctional Hydrocracking, Ind. Eng. Chem. Res., 2021, 60, 6357–6378, DOI:10.1021/acs.iecr.0c05528
- N. Batalha, L. Pinard, C. Bouchy, E. Guillon and M. Guisnet, N-Hexadecane hydroisomerization over Pt-HBEA catalysts. Quantification and effect of the intimacy between metal and protonic sites, J. Catal., 2013, 307, 122–131, DOI:10.1016/j.jcat.2013.07.014
- A. Ramirez, P. Ticali, D. Salusso, T. Cordero-Lanzac, S. Ould-Chikh, C. Ahoba-Sam, A. L. Bugaev, E. Borfecchia, S. Morandi, M. Signorile, S. Bordiga, J. Gascon and U. Olsbye, Multifunctional Catalyst Combination for the Direct Conversion of CO2to Propane, JACS Au, 2021, 1, 1719–1732, DOI:10.1021/jacsau.1c00302
| This journal is © The Royal Society of Chemistry 2023 |