
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper-based metal–organic frameworks for CO2 reduction: selectivity trends, design paradigms, and perspectives†
Ugochukwu
Nwosu
 and
Samira
Siahrostami
*
and
Samira
Siahrostami
*
Department of Chemistry, University of Calgary, Calgary, Alberta T2N 1N4, Canada. E-mail: samira.siahrostami@ucalgary.ca
First published on 25th May 2023
Abstract
Can the carbon budget be balanced? Increasing greenhouse gas emissions and worsening environmental effects demand that humankind find a solution for anthropogenic climate change. As a carbon recycling strategy, the electrochemical carbon dioxide reduction reaction (CO2RR) represents a platform to convert CO2 to valuable chemicals. Despite the discovery that copper uniquely produces hydrocarbons, a lack of suitable catalysts prevents the realization of industrial-scale applications. Recently, metal–organic frameworks (MOFs), extended networks of organic ligands and metal nodes or clusters, have found application as electrocatalysts. Perhaps, this class of materials can be leveraged to tune the properties of copper and yield a suitable CO2RR catalyst. In this review, we present new developments in the application of copper-based MOFs (Cu MOFs) for CO2RR. Firstly, we highlight the potential of CO2RR as a solution for carbon neutrality and proceed by overviewing CO2RR mechanisms and catalysts. We then emphasize the role of copper which leads to our discussion of the trends in Cu MOFs for CO2RR. We conclude by presenting several challenges and perspectives relevant to Cu MOFs in the hope of spurring targeted research in the field.
1. Introduction
In lockstep with increasing atmospheric greenhouse gas levels, the pressure to advance CO2 utilization technologies continues to rise. Research on CO2 recycling via the electrochemical CO2 reduction reaction (CO2RR) has grown accordingly. As the sole pure metal electrocatalyst capable of producing valuable fuels and chemicals from CO2, copper features extensively in CO2RR research. In parallel, metal–organic frameworks (MOFs), extended networks of metal-containing nodes and organic ligands, have emerged as a class of materials with novel catalytic properties. Although numerous reviews exist on either the use of copper1–10 or MOFs for CO2RR,11–17 to the best of our knowledge, no such reviews exist focusing solely on the use of copper-based MOFs (Cu MOFs) for CO2RR. Herein, we aim to provide a critical review of the trends in this rapid developing field. Firstly, we motivate CO2RR as a CO2 recycling platform. We then describe CO2RR mechanisms on copper, summarize electrocatalysts used for CO2RR, and highlight the importance of copper. What follows is a critical analysis and summary of the developing trends regarding Cu MOFs used for CO2RR. Finally, we conclude by presenting challenges and perspectives for CO2RR on Cu MOFs.2. CO2 reduction for carbon neutrality
Owing largely to global energy and chemicals production, anthropogenic greenhouse gas (GHG) emissions constitute the vast majority of all GHG emissions on Earth.18Fig. 1 illustrates the global carbon cycle, highlighting the role of anthropogenic activities. In the decade beginning in 2012, fossil CO2 emissions and emissions from land-use change averaged an estimated 10.8 gigatons of carbon per year (GtC per year). These emissions resulted in rising atmospheric CO2 concentrations, 5.2 GtC per year, and uptake by ocean and terrestrial sinks, 2.9 GtC per year and 3.1 GtC per year, respectively.18 Assuming 2022 emissions levels, the remaining carbon budget for a 50% likelihood to limit global warming to 1.5 °C is estimated to last for nine years.18 As global warming is linked to a cascading list of environmental and socioeconomic problems,18 balancing the carbon budget – closing the carbon cycle – is of global concern. This is underscored by the widespread ratification of the 2015 Paris Agreement. As of the date of writing this paper, 195 members of the United Nations Framework Convention on Climate Change – all but three – have formally committed to reaching carbon neutrality by the year 2050.19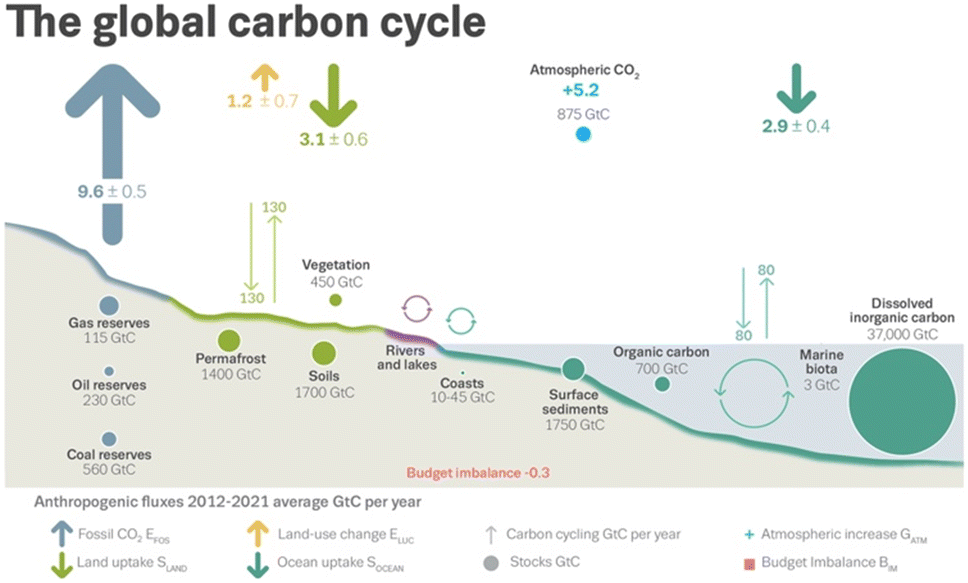 | ||
| Fig. 1 Schematic representation of the global carbon cycle, highlighting effects from anthropogenic activities averaged globally for the decade 2012–2021. Arrows designate flows. Circles designate reserves. Cyclic arrows designate cycles. Quantities are specified in gigatons of carbon (GtC). The budget imbalance is a measure of imperfect data and understanding of the contemporary carbon cycle. All uncertainties are reported as one standard deviation. Reproduced from Friedlingstein et al.18 | ||
As one of three strategies of closing the carbon cycle, in addition to decarbonization and carbon sequestration, CO2 recycling represents an important area of research requiring development. With the continued maturation and adoption of renewable energy, the attraction of CO2RR as a CO2 recycling platform has grown concomitantly. When powered by renewable energy sources, the mitigation of GHGs by CO2RR is two-fold. Firstly, carbon dioxide is converted to valuable chemicals and fuels, such as organic acids (e.g., formic acid, acetic acid), alcohols (e.g., methanol, ethanol), and/or hydrocarbons (e.g., methane, ethylene). Secondly, the use of renewable energy foregoes the GHG emissions that would otherwise be emitted while producing valuable chemicals by existing carbon-intensive technologies. Furthermore, since oxidized carbon constitutes more than 70% of anthropogenic GHG emissions,20 the chemical reduction of carbon sources represents an opposite solution. As vividly put by Nitopi et al., “carbon reduction is as of yet a missing piece of humanity's industrial metabolism”.5
3. CO2 reduction reaction (CO2RR) mechanisms
A great number of carbon products are possible from CO2RR. This variety is simultaneously an advantage, as it widens the range of potential CO2RR applications, and a disadvantage, as it hinders selectivity. An understanding of the mechanisms from which potential products arise is essential to navigating this duality to develop efficient catalysts. This section describes the potential chemical products from CO2RR as well as key insights into their reaction mechanisms. We prioritize insights relevant to copper due to the theme of the present review.3.1. CO2RR products
Table 1 lists reduction potentials for CO2RR as calculated by Nitopi and co-workers.5 The potentials are calculated from the Gibbs free energy of reaction using gas-phase thermochemistry and Henry's Law data (for aqueous species) from NIST.21| Reaction | E 0/[V vs. RHE] | Product/reaction name |
|---|---|---|
| 2H+ + 2e− → H2(g) | 0 | Hydrogen evolution reaction (HER) |
| 2CO2 + 2H+ + 2e− → (COOH)2(s) | −0.47 | Oxalic acid |
| CO2 + 2H+ + 2e− → CO(g) + 2H2O(l) | −0.10 | Carbon monoxide |
| CO2 + 2H+ + 2e− → HCOOH(aq) | −0.12 | Formic acid |
| CO2 + 4H+ + 4e− → C(s) + 2H2O(l) | 0.21 | Graphite |
| CO2 + 6H+ + 6e− → CH3OH(aq) + H2O(l) | 0.03 | Methanol |
| CO2 + 8H+ + 8e− → CH4(g) + 2H2O(l) | 0.17 | Methane |
| 2CO2 + 8H+ + 8e− → CH3COOH(aq) + 2H2O(l) | 0.11 | Acetic acid |
| 2CO2 + 10H+ + 10e− → CH3CHO(aq) | 0.06 | Acetaldehyde |
| 2CO2 + 12H+ + 12e− → C2H4(g) + 4H2O(l) | 0.08 | Ethylene |
| 2CO2 + 12H+ + 12e− → C2H5OH(aq) + 3H2O(l) | 0.09 | Ethanol |
| 2CO2 + 14H+ + 14e− → C2H6(g) + 4H2O(l) | 0.14 | Ethane |
| 3CO2 + 16H+ + 16e− → C2H5CHO(aq) + 5H2O(l) | 0.09 | Propionaldehyde |
| 3CO2 + 18H+ + 18e− → C3H7OH(aq) + 5H2O(l) | 0.10 | 1-Propanol |
The data in Table 1 highlights the inherent thermodynamic challenge to achieving selectivity in CO2RR. Formic acid and carbon monoxide have been identified as economically viable targets.22 The production of chemicals with two or more carbons (C2+ products) makes CO2RR especially attractive for fuel production.23 Yet, observe that the reduction potentials for all 14 products lie between −0.47 to 0.21 V and that five of the C2+ products listed exhibit reduction potentials in a 0.08 V range.
The reduction potentials do not tell the whole story, however. While thermodynamically, ethanol production should occur before hydrogen evolution, experimentally, hydrogen is typically the first reduction product observed. The experimentally observed kinetics can be related to the minimum energy path (MEP) for a given reaction mechanism. Specifically, the activation energies and free energy differences between intermediates along the MEP are relevant. In accordance with transition state theory and the computational hydrogen electrode model, large activation energies and free energy differences between reaction intermediates correlate with poor activities and prohibitive overpotentials.24,25 Determination of complete reaction mechanisms as well as the rate-limiting and potential-determining steps requires performing both computational studies and in situ/operando experimental measurements.
3.2. Key sub-pathways in CO2RR
It is important to note that CO2RR mechanisms are by no means concretized as much debate and work continues in the field. CO2RR mechanisms vary depending on the catalyst as well as the reaction conditions. Our aim here is not to give a definitive account of CO2RR mechanisms but to highlight important intermediates and reaction steps for CO2RR. Accordingly, we illustrate three sub-pathways (Fig. 2). The sub-pathways chosen feature frequently in reported reaction mechanisms and are relevant to alcohol and hydrocarbon formation. For a more extensive summary of CO2RR mechanisms, we direct the reader to Section 5 of the review by Nitopi and co-workers.5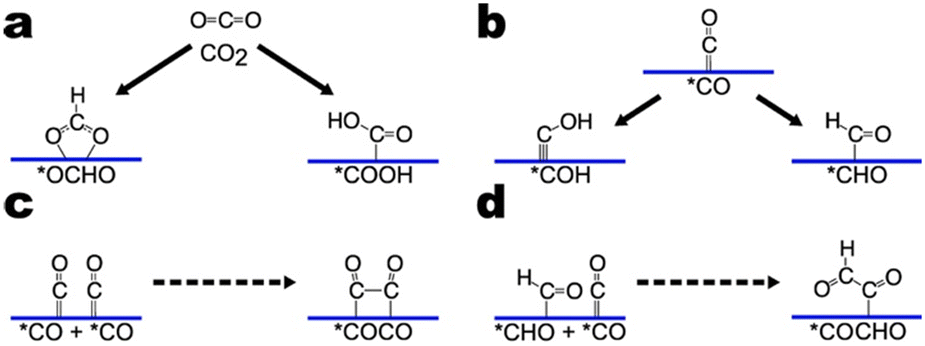 | ||
| Fig. 2 Selected sub-pathways of CO2RR. a) Proton-coupled electron transfer of CO2 to *OCHO or *COOH. b) Proton-coupled electron transfer of *CO to *COH or *CHO. c) *CO dimerization reaction. d) C–C coupling of *CHO and *CO. Solid lines denote proton-coupled electron transfers. Dashed arrows denote coupling reactions. | ||
The first sub-pathway pertains to the formation of two-electron products CO and HCOOH (Fig. 2a). Fig. 2a shows the mechanism for producing HCOOH. CO2 undergoes a proton-coupled electron transfer (PCET) to yield an *OCHO intermediate bound to the surface by two oxygen atoms. *OCHO is further reduced to HCOOH. Alternatively, the PCET can yield a *COOH intermediate which is further reduced to CO. In general, surface-bound oxygen species are common to the proposed mechanisms for HCOOH production while surface-bound carbon species are common to the proposed mechanisms for CO. Although DFT calculations suggest that *COOH formation may thermodynamically limit CO production,26 recent theoretical and experimental results suggest that CO2 adsorption is the rate-limiting step for both CO and HCOOH formation on a range of transition metals.27
The second sub-pathway goes through protonation of the *CO intermediate (Fig. 2b). *CO is ubiquitous in CO2RR as it has been reported as an intermediate in the proposed mechanisms for all CO2RR products but formate.5 Both experimental and theoretical studies indicate that hydrocarbon production on copper proceeds through a *CO intermediate.26,28–31 Protonation of the oxygen atom yields a *COH intermediate while protonation of the carbon atom yields a *CHO intermediate.32,33
Reports indicate that on copper surfaces, the set of favoured C1 products from the *COH and *CHO intermediates differ.33–36 For example, Nie et al. reported that on the Cu(111) surface, formation of *CHO favours methane production while the formation of *COH favours methane, methanol, and ethylene production.37 This implies that *CO protonation is a selectivity-determining step.33–36 Thus, stabilization of the subsequent protonated intermediate can enhance the selectivity of particular CO2RR products. Analogously, stabilization of the transition state of the rate-limiting step can improve CO2RR kinetics. However, as identification of the transition state can be challenging, DFT calculations can provide correlations for the activation energy.
For example, Liu et al. obtained linear scaling relations correlating the free energy of the *CO → *CHO transition state complex (H–CO*) with the adsorption energy of *CO (Fig. 3a) and *CHO (Fig. 3b) on (211) and (111) metal surfaces.33 Peterson and Nørskov obtained similar relations for the adsorption energy of carbon-bound intermediates (*COOH, *CHO, and *CH2O) vs. *CO (Fig. 3c) and oxygen-bound intermediates (*O and *OCH3) vs. *OH (Fig. 3d) on fcc(211) metal surfaces.26 Such relations suggest activity descriptors for ideal catalysts – for instance, CO adsorption energy for CH4 and C2+ production.26,38
 | ||
| Fig. 3 Linear scaling relations between the adsorption energies of important CO2RR intermediates on transition metal (211) and (111) surfaces a) GH–CO*vs. G*CO. b) GH–CO*vs. G*CHO. Adapted with permission from Liu et al.33 Copyright 2017 Nature Publishing. Linear scaling relations between the adsorption energies of important CO2RR intermediates on transition metal FCC(221) surfaces. c) EB [CH2O], [CHO], [COOH] vs. EB [CO] d) EB [OCH3] and [O] vs. EB [OH]. Adapted with permission from Peterson & Nørskov.26 Copyright 2012 American Chemical Society. | ||
The third sub-pathway pertains to C–C coupling. Numerous C–C coupling mechanisms are feasible depending on the nature of the catalyst employed. On catalysts with adjacent active sites, C–C coupling may occur through direct coupling of adsorbed intermediates, potentially mediated by an electron transfer.39 Alternatively, gaseous CO may react with adsorbed species to form similar intermediates. Fig. 2c and d illustrate two frequently reported C–C coupling mechanisms, CO dimerization and CO–CHO coupling.
Although the C–C coupling steps are not typically potential-determining, the observed pH dependence of C2+ yields suggests that the C–C coupling may be rate-determining on Cu.29,40–43 Kinetic barriers of C–C coupling steps that are not mediated by an electron transfer differ from PCET steps in that their energetics are not influenced in the same way by the chemical potential of aqueous protons. Field effects and solvent stabilization primarily influence the kinetic barriers of C–C coupling steps.40,44,45 DFT calculations on the Cu(211) surface indicate that C–C coupling proceeds more favourably for further hydrogenated intermediates.44 However, calculations on the Cu(211), Cu(111), and Cu(100) surfaces also support the feasibility of CO dimerization.39,40,45,46
Finally, it is important to note that the energetics of *OH and *H adsorption are also relevant for CO2RR performance. Strong OH binding can result in catalyst poisoning while *H adsorption dictates the kinetics of the hydrogen evolution reaction (HER). An ideal catalyst must suppress these side reactions yet still exhibit reasonable CO2RR kinetics at a low overpotential. The next section will discuss previous attempts to find such a material.
4. CO2RR catalysts
Broadly, CO2RR electrocatalysts can be classified as either homogeneous47 or heterogeneous,11 and they can be further categorized as metal or nonmetal catalysts. Nonmetal-doped carbon allotropes constitute the majority of nonmetal catalysts studied for the CO2RR. The presence of heteroatoms, such as boron, nitrogen, and sulfur, is required to endow carbon materials (graphene, carbon nanotubes (CNTs), and porous carbon) with CO2RR activity.48–50 Metal-containing CO2RR catalysts include pure transition metal catalysts51,52 as well as metal-doped carbon,49,50,53,54 metal oxides/nitrides and alloys,35,55–57 and organic–inorganic hybrid materials.41,52,55,58Fig. 4 illustrates the various categories of electrocatalysts employed for CO2RR. For a comprehensive overview of CO2RR catalysts, the reader is encouraged to consult other reviews and perspectives.5,6,9,59–64 | ||
| Fig. 4 Various types of CO2RR catalysts. | ||
Although metal-free carbon-based catalysts have shown great promise due to their favourable physical and electrical properties, experimental and theoretical investigations indicate that in order to reduce CO2 further than CO, the presence of a metal component is necessary.53 Of note, single atom catalysts (SACs), which contain isolated metal atoms, have been investigated for the CO2RR.53,54,65–67 The enhanced catalytic performance of SACs compared to bulk metal catalysts derives from the undercoordination of metal atoms in open metal sites (OMSs).54 The isolated catalytic sites offer fine-tuned control over the electronic structure of the metal. SACs also exhibit superior metal utilization compared to both bulk and nanoparticle catalysts due to the maximal dispersion of metal atoms in SACs.
Importantly, the choice of metal directly affects the CO2RR product such that metals can be separated into four groups based on their tendency to form CO2RR products. Formate-producing metals include Pb, Hg, Tl, In, Sn, Cd, and Bi; carbon monoxide-producing metals include Au, Ag, Zn, Pd, and Ga. Ni, Fe, Pt, and Ti display minimal activity for CO2 reduction. Copper is unique in that it is the only pure metal capable of producing C2+ products.5,41 This essential property makes copper the subject of immense interest for the CO2RR.
5. The importance of copper
5.1. The origin and challenge of C2+ selectivity
As previously mentioned, copper uniquely produces a variety of hydrocarbon and oxygenated products.30,41,68–72 From a thermodynamic standpoint, the trends in CO2RR activity among transition metals may be explained by the binding energies of *CO and *H. Fig. 5 illustrates the unique adsorption properties of copper relative to other transition metals. | ||
| Fig. 5 Two-parameter descriptor of the electrocatalytic activity of metal electrodes. The relative rate of CO2 reduction and H2 formation is shown as a function of the binding energy of an isolated CO molecule (horizontal axis) and differential adsorption energy of an H-adatom at an on-top site (vertical axis), occupied once the coverage exceeds one monolayer. Lines (a) and (b) demarcate the transition between CO poisoning/desorption and CO/HCOO− desorption. Reproduced with permission from Hussain et al.73 Copyright 2018 American Chemical Society. | ||
Using a two-parameter descriptor, Hussain et al. classified the electrocatalytic activity of various transition metal electrodes.73 Observe that copper is the sole metal lying within the H2/hydrocarbons and alcohols region. Copper exhibits an intermediate binding energy for CO and a positive binding energy for *H.34 In accordance with the Sabatier principle,74 the intermediate *CO binding energy on copper balances CO poisoning and activation, resulting in the ability to yield products of reductions of more than two electrons (>2e− products). The positive binding energy for *H explains copper's preference for CO2RR over HER.26,34,38,73,75
The unique ability of copper to produce >2e− products is unfortunately also a pitfall as copper can produce up to 14 different products (Table 1).76 This wide range of potential products augments the challenge of selectivity of copper catalysts.
5.2. Relevant strategies for improving the CO2RR performance of copper
Various strategies have been examined to improve the selectivity and activity of copper for CO2RR. Herein, we highlight trends in CO2RR performance on copper catalysts with counterparts in the field of copper-based MOFs to be discussed in the next section. Namely, we note trends involving the copper coordination environment and synergistic effects with non-copper atoms. Although there are additional factors affecting copper CO2RR performance, such as the oxidation state and electrolyte, we defer to existing reviews as discussion of these factors is beyond the scope of this review.5,77 However, investigation of these effects as they relate to copper-based MOFs would be beneficial to the field.Since the coordination of surface atoms is a principal difference between copper facets, the coordination number has been suggested as a key descriptor for catalytic performance. Indeed, that the generalized coordination number (GCN) proposed by Calle-Vallejo et al. correlates with catalytic performance underscores the relationship between site coordination and catalytic performance.80 However, although manipulation of the crystal facet can enhance selectivity for C2+ products, the use of single crystals as electrocatalysts is impractical due to their low geometric surface area and resulting low current densities.5 Nonetheless, the link between coordination and C2+ selectivity motivates the development of copper catalysts with undercoordinated sites by different means.
Nanostructured and single-atom catalysts attempt to leverage this phenomenon.65,81 As particle size decreases, the number of undercoordinated surface atoms increases.5 Additionally, the nature of single-atom catalysts can decouple adsorbate binding energies and unlock novel selectivity.67 In light of this, studies suggest that copper nanoparticles and mesocrystals may lead to high C2+ faradaic efficiencies.82 However, no clear connection between particle size and C2+ selectivity can be identified as studies indicate that peak hydrocarbon selectivity can be achieved at different particle sizes.83–85 All of this serves to highlight the possibility that a different paradigm for altering copper coordination could better take advantage of the connection between coordination and selectivity.
In terms of non-metal atoms, subsurface oxygen in oxide-derived copper can enhance *CO adsorption and promote H2O adsorption and subsequent electron transfer between CO2 and H2O.1,94,95 Boron doping increases the prevalence of catalytically active Cuδ+ species, increasing C2+ production.89 Finally, the functionalization of copper surfaces with ligands or dopants can also serve to stabilize key intermediates and enhance CO2RR performance.89,96–99 Despite the advances in CO2RR catalyst development, alcohol and hydrocarbon production still requires large overpotentials of up to 1 V and suffers from low selectivity.2,30,76 Further, the reported increases in current density for functionalized copper surfaces do not correspond to increases in intrinsic activity – that is, activity normalized by electrochemical surface area.100 Thus, the door is still open for increasing the CO2RR performance of copper.
6. Copper-based metal–organic frameworks
Metal–organic frameworks (MOFs), also referred to as porous coordination polymers, are extended networks of metal nodes or clusters bridged by simple organic ligands through metal–ligand coordination bonds.101 A metal centre or inorganic cluster constitutes the secondary building unit (SBU). Note that we use the terms “node” and “cluster” to denote SBUs featuring single and multiple metal atoms, respectively. The SBUs can be tuned to change the structure, gas adsorption, catalytic ability, electrical conductivity, and porosity of the MOF. Additionally, the choice of ligand and other factors such as counterions, pH, temperature, and solvents can influence MOF properties.102The two most widely used classes of ligands for MOF synthesis are N-donors and O-donors, composed primarily of pyridyl- and carboxylate-based ligands, respectively (Fig. 6). However, due to the requirement of strong metal–ligand bonds for retaining the structural integrity of MOFs after solvent removal, carboxylate ligands are more prevalent in MOF structures.101 Further, both pyridyl- and carboxylate-based ligands are dominated by rigid phenyl- or ethynyl-containing molecules, which are fundamental for the directional bonding in MOFs.101 It should be noted that although less studied, phosphonate and sulfonate ligands may also be used in MOFs.102 For a comprehensive treatment of the structure and synthesis of MOFs, we refer the reader to other works.101–105
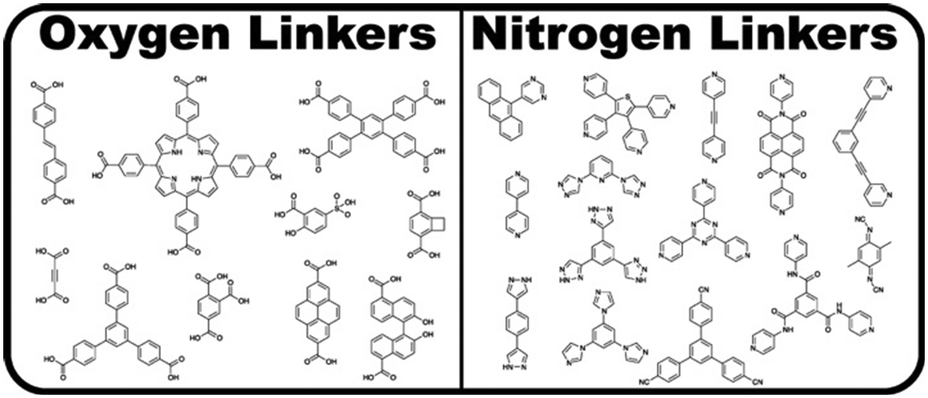 | ||
| Fig. 6 MOF ligands featuring oxygen and nitrogen linkers. Adapted with permission from Cook et al.101 Copyright 2013 American Chemical Society. | ||
Recently, MOFs have gained popularity due to their novel properties. MOFs exhibit high porosity, large surface areas, unique host–guest dynamics, thermal stability, and mechanical flexibility.102 In contrast to copper within pure metal surfaces, which primarily exists in the zero oxidation state, copper within MOFs also exists in the +1 and +2 oxidation states (Table S1†). Like SACs, MOFs feature isolated catalytic sites; however, their reticular nature enables more precise design of active sites than in SACs. Could metal–organic frameworks be the key to unlocking C2+ selectivity in copper catalysts? To this end, we limit the scope of our discussion to metal–organic frameworks featuring copper atoms within the secondary building unit, copper-based metal–organic frameworks (Cu MOFs). Specifically, our scope is limited to Cu MOFs reported as the catalytic material for CO2RR.
Beginning with formate production by copper rubeanate in 2012 (ref. 106) and oxalic acid production by HKUST-1 in the same year,107 copper-based metal–organic frameworks have been increasingly investigated for CO2RR. Although the goal of this review is not to merely summarize recent results of Cu MOFs, we have compiled comprehensive property and CO2RR performance data for Cu MOFs in the ESI† and the ESI† file for reference.
Herein, we reveal promising trends in Cu MOF design, assess proposed explanations for observed phenomena, and suggest promising new lines of inquiry regarding Cu MOFs for CO2RR. We focus on the secondary building unit as a determinant of selectivity, the effect of the MOF ligand, and strategies for improving selectivity by the incorporation of other transition metals and nanocluster–MOF composites. Finally, we conclude with a discussion of the stability of Cu MOFs.
6.1. The dependence of C2+ selectivity on the secondary building unit
As was mentioned as a motivation for single-atom catalysts and nanoparticles, modification of the copper coordination environment may be the key to designing efficient CO2RR catalysts. For bulk copper catalysts and SACs, there exists a great body of work presenting descriptors for CO2RR performance.53,108,109 In the spirit of said work, this section focuses on how the copper coordination environment in Cu MOFs relates to CO2RR performance. Specifically, we discuss how the type, number, and geometry of atoms which coordinate copper within the SBU affect the selectivity of CO2RR in Cu MOFs. For the present discussion, we will specify metal node SBUs by the constituting metal and the atoms bonded to the metal. Thus, a single copper atom coordinated by four oxygen atoms will be referred to as a CuO4 node.Liu et al. evaluated the performance of Cu-DBC111 and compared it to that of Cu-THQ112 and Cu-HHTP113 obtained from published data. Cu-DBC features square pyramidal CuO5 nodes. The CuO5 nodes are partially constituted by two dibenzo-[g,p]chrysene-2,3,6,7,10,11,14,15-octaol (DBC) ligands coordinating a copper atom. Additionally, an axially coordinating oxygen atom bridges each copper atom in the SBU to a single other copper atom in an adjacent sheet (Fig. 7d). The SBU of Cu-THQ consists of CuO4 nodes formed by two tetrahydroxy-1,4-quinone (THQ) ligands coordinating a square planar copper. Cu-HHTP also features square planar copper within CuO4 nodes; however, two 2,3,6,7,10,11-hexahydroxytriphenylene ligands coordinate the copper node. The structures of all three Cu MOFs are shown in Fig. 7.
 | ||
| Fig. 7 Structures of a) Cu-DBC, b) Cu-HHTP, and c) Cu-THQ. d) Close-up of the CuO5 node present in Cu-DBC. The blue, red, and white spheres denote copper, oxygen, and carbon atoms, respectively. Adapted with permission from Liu et al.111 Copyright 2022 American Chemical Society. | ||
According to the Dewar–Chatt–Duncanson model, the electronic structure of copper in a square pyramidal geometry features higher energy d orbitals than that of copper in a square planar geometry.114–116 This d orbital elevation may facilitate stronger interaction between Cu and CO via π-back-bonding, which is favourable for hydrogenation. Preliminary DFT calculations by Liu et al. showed that the adsorption energy of *CO on CuO5 sites was more than fifty percent larger than on the CuO4 sites (73.42 kJ mol−1vs. 48.6 kJ mol−1).111
Comparison of the CO2RR performance of the three Cu MOFs revealed stark differences in selectivity. While Cu-HHTP and Cu-THQ exhibited exclusively CO production (42% FECO and 91% FECO, respectively), Cu-DBC exhibited 56% FECH4.
DFT calculations explain the disparities in selectivity. The free energy diagram reveals that CO is more stable on the CuO4 nodes than on the CuO5 nodes relative to free CO (−0.23 eV vs. −0.18 eV) (Fig. 8). Notably, the difference in CH4 production derives from the stability of the *OCH2 intermediate relative to the preceding intermediate (*CHO). The free energy difference of this elementary step is 0.24 eV less on the CuO5 node than on the CuO4 node (0.35 eV vs. 0.59 eV, respectively).
 | ||
| Fig. 8 a) Free energy diagram of CH4 production on CuO5 and CuO4 nodes. Data from Liu et al.111 The free energies of CO adsorption were inferred from the free energy differences between steps presented in the original publication. Illustration of the cluster models of b) CuO5 and c) CuO4 nodes employed for calculation of the free energy diagrams in Fig. 8a by Liu et al.111 The orange, red, and grey spheres denote copper, oxygen, and carbon atoms, respectively. | ||
We note, however, that the experimental conditions for the comparison between Cu-DBC, Cu-THQ, and Cu-HHTP were not uniform. First, the CO2RR performance of Cu-THQ was tested in an alkaline choline chloride solution. Second, while the electrolytes used to test Cu-DBC and Cu-HHTP performance were the same (0.1 M KHCO3), the experimental setups used to test CO2RR performance differed, and the studies do not directly compare their results to benchmarks.
For comparison, we note the work by Zhang et al., which tested Cu-DBC and Cu-HHTP under alkaline conditions and reported CH4 production by both Cu-DBC (80% FECH4 at −0.9 V vs. RHE) and Cu-HHTP (42.6% FECH4 at −0.9 V vs. RHE). C2H4 production by Cu-HHTP (40.9% FEC2H4 at −0.8 V vs. RHE) was also reported.117 These results challenge the notion that the CuO5 node plays a definitive role in CH4 production. Thus, an analogous study under neutrally buffered conditions would more definitively illustrate the role of the CuO5 node. Nonetheless, the results by Liu et al. provide an important starting point for investigations of the effect of copper coordination geometry on CO2RR performance.
When copper resides within square planar CuN4 nodes, it appears that the copper sites either serve for CO production or are ineffective for CO2RR altogether.118 In fact, DFT calculations on various metallophthalocyanine-based Cu MOFs indicate that the free energy change for *COOH formation is consistently greater on CuN4 nodes compared to the analogous CuO4 nodes.115Fig. 9a and b indicate that for both sequential and concerted PCET mechanisms, *COOH formation proceeds more readily on PcCo–Cu–O and PcNi–Cu–O MOFs than on PcCo–Cu–NH and PcNi–Cu–NH MOFs, respectively. However, the work by Zhao and co-workers suggests that as edge sites, nitrogen-coordinated copper may be active for C2+ product formation. Cu-HITP, a structural analogue of Cu-HHTP featuring CuN4 nodes, exhibited 62% FE>2e− and 43% FEC2+ at −1.2 V vs. RHE. DFT calculations indicate an interplanar C–C coupling mechanism between *CO and *COH on adjacent edge sites.119
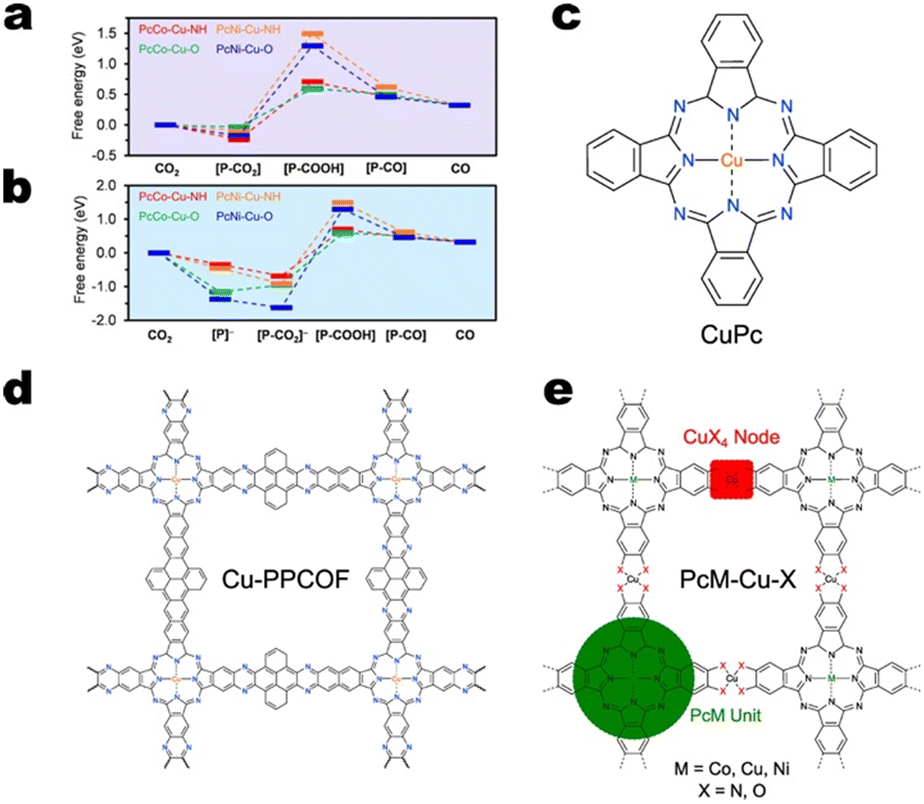 | ||
| Fig. 9 Free energy diagrams for CO production on metallophthalocyanine-based Cu MOFs considering the a) concerted PCET mechanism and b) sequential PCET mechanism. Free energies are relative to desorbed CO2. Adapted with permission from Meng et al.118 Copyright 2020 American Chemical Society. The structures of c) molecular PcCu, d) Cu-PPCOF, and e) PcM–Cu–X. | ||
Cu MOFs featuring CuO4 nodes and copper within polycyclic ligands may be effective for C2+ product formation.113,120 Consider the CO2RR selectivity of a series of phthalocyanine-based catalysts: PcCu–Cu–O, PcCo–Cu–O, Pc–Cu–O, Pc–Cu–NH, CuPc, and Cu-PPCOF. PcCu–Cu–O features two distinct copper sites – both with square planar geometry. One site resides within the SBU and features a copper ion coordinated by the oxygen atoms of two 2,3,9,10,16,17,23,24-octahydroxy-phthalo-cyaninato copper(II) ligands. The second copper site exists within the phthalocyanine (Pc) ligand. Here, the four pyrrolic nitrogens of phthalocyanine coordinate copper. If the copper within the ligand is replaced by cobalt, we obtain PcCo–Cu–O. If the cobalt within the ligand is removed, we obtain Pc–Cu–O. And if the oxygen atoms in the SBU are replaced with nitrogen atoms, we obtain Pc–Cu–NH. Copper phthalocyanine (CuPc) is essentially the ligand of PcCu–Cu–O. Finally, the covalent organic framework, Cu-PPCOF, also features copper within a polycyclic ligand, but instead of by a copper node, the phthalocyanine units are linked by pyrene units. The structures of the phthalocyanine-based catalysts are shown in Fig. 9c–e.
The major CO2RR product faradaic efficiencies for each of these catalysts are compiled in Fig. 10. Firstly, while PcCu–Cu–O exhibits 50% FEC2H4, PcCo–Cu–O exhibits primarily CO production (85% FECO). Pc–Cu–O and Pc–Cu–NH, which feature CuX4 nodes but do not feature copper within polycyclic ligands, display poor CO2RR performance (5.6% FECO and 4.5% FECO, respectively).118 Cu-PPCOF also exhibits poor CO2RR performance (8.6% FECO). Notably, CuPc does produce C2H4, albeit with lower selectivity (25% FEC2H4). Together, these observations suggest that both copper sites within PcCu–Cu–O are essential for C2H4 production.
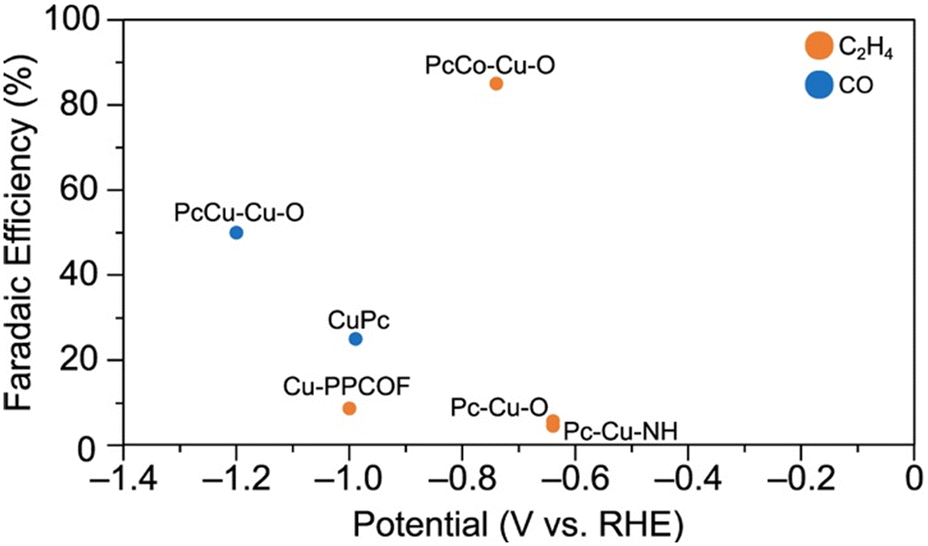 | ||
| Fig. 10 Major CO2RR product selectivity by metallophthalocyanine-based electrocatalysts. The data for PcCu–Cu–O are from Qiu et al.113 The data for CuPc are from Kusama et al.122 The data for PcCo–Cu–O, Pc–Cu–O, and Pc–Cu–NH are from Meng et al.118 The data for Cu-PPCOF is from Zhang et al.117 All data are reported for the conditions at which the highest single product FE is obtained. | ||
On the contrary, a study by Zhong et al. suggests that PcCu–Cu–O exhibits poor CO2RR selectivity altogether. Only CO production (11% FECO) was observed in the same 0.1 M HCO3 electrolyte. However, we note that the conductive additive used by Zhong et al. (CNTs) differs from that used by Qiu et al. (carbon black). Qiu et al. also tested the CO2RR performance of CNTs and PcCu–Cu–O mixed with CNTs in their setup. Under identical conditions to the experiments with PcCu–Cu–O, CNTs exhibited 100% FE for HER. Further, PcCu–Cu–O mixed with CNTs exhibited ∼90% FEHER and ∼10% FECO.110 Thus, we contend that in the study by Zhong et al., competition from the HER reduces C2+ product selectivity.
Given this explanation for the disparity reported in CO2RR products selectivity for PcCu–Cu–O, C2H4 production by the related single-site catalyst, CuPc, supports the argument presented by Qiu et al.:113 CO produced at the CuO4 node migrates to the copper within phthalocyanine where it couples with adsorbed CO2RR intermediates to yield C2H4.113 These results suggest the feasibility of the dual-site design paradigm for Cu MOFs.
Secondly, consider the implications of the calculations performed by Liu et al. to account for the differences in CH4 production by Cu-DBC, Cu-HHTP, and Cu-THQ (Fig. 8a).111 Note that since the same atomic model (Fig. 8c) is used for both Cu-HHTP and Cu-THQ, the calculations cannot possibly account for the fact that the CO selectivity exhibited by Cu-THQ is more than double that exhibited by Cu-HHTP. While this experimental observation may be attributed to bulk catalyst properties (e.g., pore size) due to their effects on the active site (e.g., local CO2 concentration), the inability of the CuO4 model to capture the differences in CO production suggests the importance of the ligand in tuning the electronic structure of the active site. This is underscored by the fact that Cu-HATNA-MOF, another Cu MOF featuring a single CuO4 node, exhibits greater CH4 selectivity than Cu-DBC (78% vs. 56% FECH4).123
Moreover, the importance of including the ligand can be seen when evaluating the C–C coupling mechanism proposed by Qiu and co-workers.113 Note that stronger CO adsorption on the Pc unit compared to on the CuO4 node supports the feasibility of the dual-site mechanism. The adsorption energies obtained by Qiu et al. are consistent with this requirement. On the contrary, DFT calculations performed by Zhang et al. suggest that CO is more stable on the CuO4 node than on the phthalocyanine unit.117
Fig. 11 plots ECuO4 − EPcCu, the difference in CO adsorption energy on the CuO4 node and the copper within the Pc unit, for each cluster model employed by Qiu et al.113 and Zhang et al.117 A negative value of ECuO4 − EPcCu indicates stronger adsorption on the CuO4 node. For adsorption on both the CuO4 node and the Pc unit, the immediate coordination environment is identical for both studies. However, observe that Zhang et al. employed a model featuring a DBC ligand for adsorption on the CuO4 node. Additionally, observe that their model for adsorption on the Pc unit is truncated with amine groups. The fact that only the ligands connected to the nodes differ suggests that changes away from the active site have a significant impact on the free energy of adsorbates. This point is further illustrated when comparing the CO2RR and HER overpotentials on the SBUs of PcM–Cu–X MOFs (Fig. 12a and b). Calculated CO2RR and HER overpotentials change by up to 0.43 eV and 0.19 eV, respectively, depending on the metal within the Pc unit. The upcoming section on ligand effects expands on the role that the ligand plays in modulating the reactivity of the SBU.
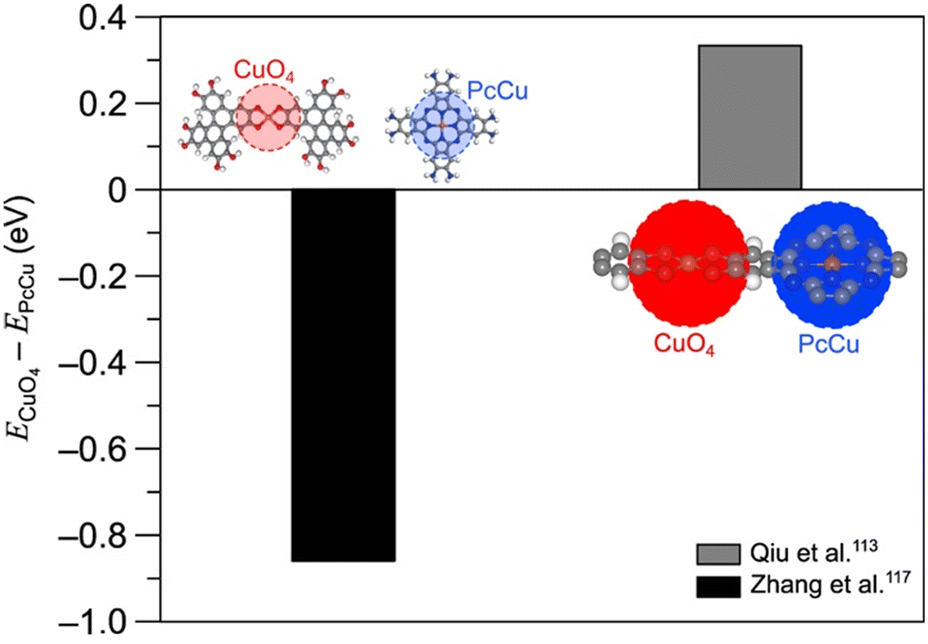 | ||
| Fig. 11 Differences between CO adsorption energies on CuO4 nodes and PcCu units as calculated by Zhang et al.117 and Qiu et al.113 Note that the energies by Zhang et al.117 are calculated relative to CO2 using the computational hydrogen electrode model,24 and the energies by Qiu et al. are calculated relative to CO. For each set of calculations, the cluster models are displayed adjacent to the data point. In all atomic models, the red, grey, blue, orange, and white spheres denote oxygen, carbon, nitrogen, copper, and hydrogen atoms, respectively. | ||

 | ||
| Fig. 13 a) Close-up of the copper dimer in the coordinatively unsaturated (anhydrous) structure of HKUST-1. The blue, red, and black spheres denote copper, oxygen, and carbon atoms, respectively. Adapted from Hendon & Walsh.124 b) The local structure of the {Cu4ZnCl4}6+ cluster, constituting the Cu4II-MFU-4l SBU. Adapted with permission from Zhu et al.134 Copyright 2021 American Chemical Society. | ||
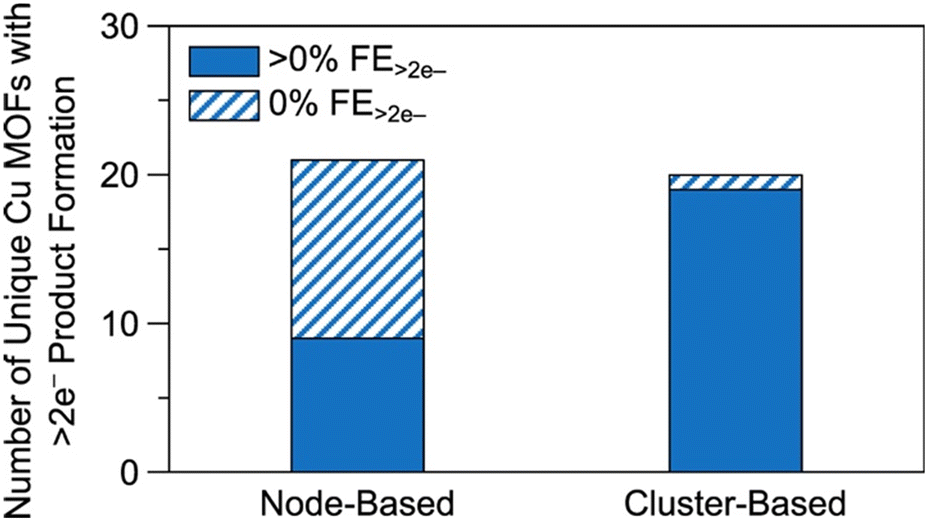 | ||
| Fig. 14 Comparison of the number of unique node- and cluster-based Cu MOFs and Cu MOF composites. The solid regions denote the number of unique Cu MOFs reported with greater than 0% FE for >2e− products. The striped regions denote the number of unique Cu MOFs reported with 0% FE for >2e− products. Raw data available in Table S2.† For details on methodology, see the ESI.† | ||
CO2RR studies on Cu MOFs containing SBUs with more than two copper atoms are scarce. To the best of our knowledge, only three such studies exist.133–135 Zhu et al. examined the CO2RR performance of Cu4II-MFU-4l, a Cu MOF consisting of a {Cu4ZnCl4}6+ cluster SBU and bis(1H-1,2,3-triazolo-[4,5-b],[4′,5′-i])dibenzo-[1,4]-dioxin (btdd) ligands.134 The {Cu4ZnCl4}6+ cluster can be visualized as four Cu2+ ions positioned at the corners of a tetrahedron featuring a single Zn2+ ion at its centroid. Further, the Zn2+ ion is octahedrally coordinated by six N atoms from six different btdd ligands, and each Cu2+ ion is tetrahedrally coordinated by three N atoms from three different btdd ligands and a single Cl− ion. Each btdd unit is a hexadentate ligand, bridging two SBUs by coordinating two Cu2+ ions and one Zn2+ ion in each SBU. Fig. 13b illustrates the local structure of the {Cu4ZnCl4}6+ cluster.
Under reaction conditions, the Cu2+ ions are reduced to Cu+ ions, and the Cl− ions are eliminated. The resulting CuN3 active sites feature copper ions coordinated by three nitrogen atoms and one labile solvent H2O molecule, effectively yielding Cu+ ions with trigonal pyramidal geometries. The reduced catalyst with Cu+ ions is referred to as Cu4I-MFU-4l and exhibits a maximum CH4 selectivity of 92% at −1.2 V vs. RHE.
Based on in situ ATR-FTIR and DFT calculations, the authors contend that the observed CO2RR selectivity and HER suppression derives from the stabilization of CO2RR intermediates and destabilization of H2O. The calculated equilibrium positions suggest more favourable interactions between CO2RR intermediates and the active site than between H2O and the active site. For *COOH, this is illustrated in Fig. 15a wherein *COOH is stabilized via an H-bonding interaction with an aromatic hydrogen. For *CHO and *CO, this is illustrated in Fig. 15b–d wherein the Cu–C distances between copper and the carbon atom on CO2RR intermediates are shorter than the Cu–O distance between copper within the SBU and the oxygen atom in H2O. *CHO and *CO are also stabilized via H-bonding interactions. Moreover, Zhu et al. reasoned that the aromatic hydrogens on the btdd ligand destabilize H2O.
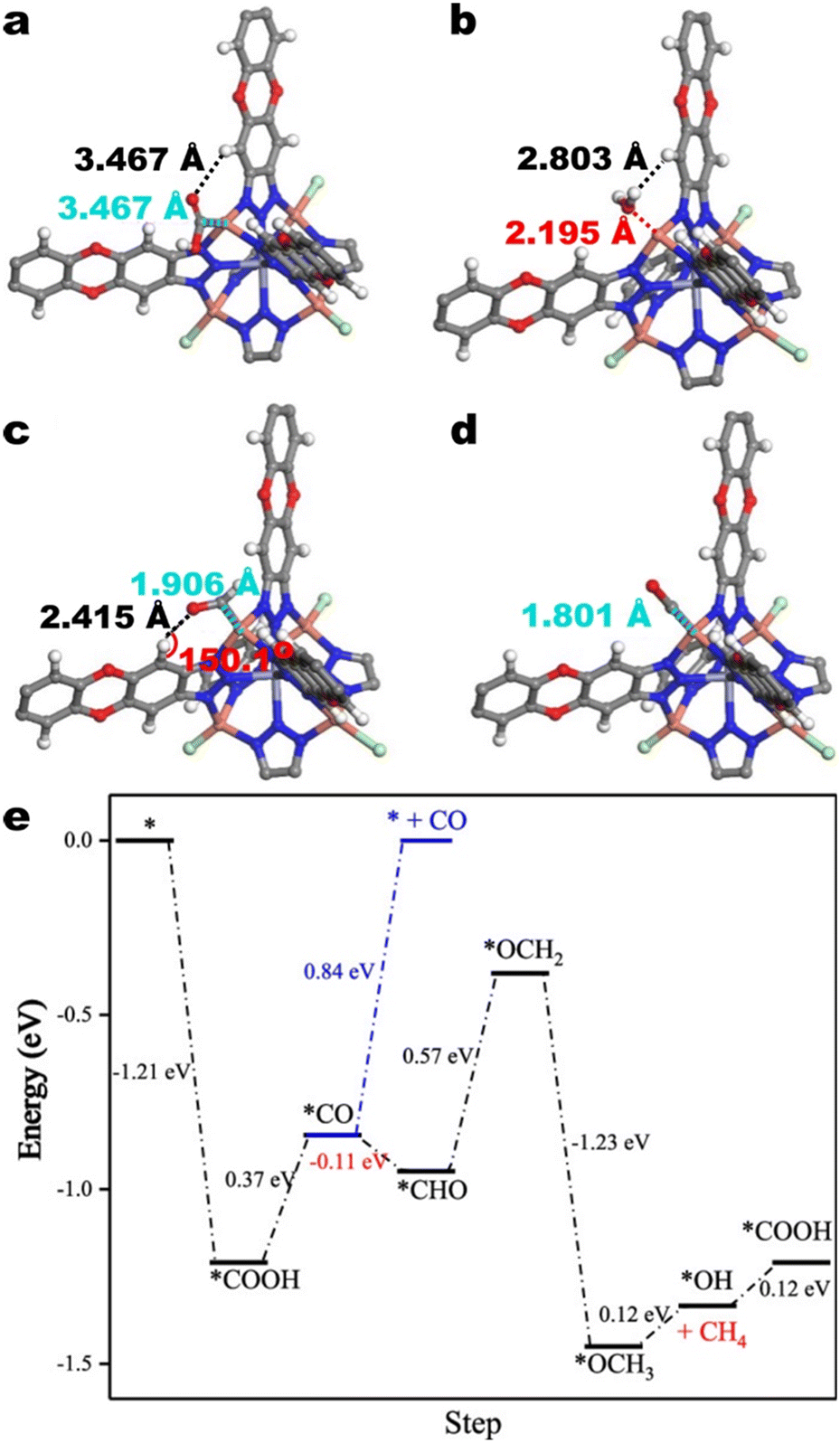 | ||
| Fig. 15 Atomic scale models of the interactions of a) *COOH, b) *H2O, c) *CHO, and d) *CO with a Cu atom in the SBU and aromatic hydrogens on the btdd ligand. The grey, white, red, green, blue, and orange spheres denote carbon, hydrogen, oxygen, chlorine, nitrogen, and copper atoms, respectively. e) Free energy diagram for CH4 formation on Cu4I-MFU-4l. Adapted with permission from Zhu et al.134 Copyright 2021 American Chemical Society. | ||
These effects manifest in the free energies of adsorbed CO2RR intermediates relative to that of adsorbed H2O (Fig. 15e). DFT calculations indicate that all detected CO2RR intermediates are more stable than H2O and that the selectivity for CH4 over non-hydrogenated product CO stems from the thermodynamic barrier for CO desorption over *CO hydrogenation. In addition to H-bonding, the authors identify σ–π back bonding between Cu+ and CO as a factor for the thermodynamic barrier for CO desorption.
The second study by van Phuc et al. investigates the CO2RR performance of a 2,5-dihydroxy-1,4-benzenedicarboxylate-based catalyst, Cu-MOF-74.135 Cu-MOF-74 features helical Cu–O–C rods constructed from 6-coordinated Cu2+ centres. Each copper atom is coordinated by three carboxyl groups, two hydroxy groups, and a solvent molecule. The rods consist of edge-sharing CuO6 octahedra and constitute the SBU (Fig. 16a).136 It is interesting to note that unlike the vast majority of cluster-based Cu MOFs (Fig. 14), Cu-MOF-74 exhibits only CO production (85% FECO). As such, an investigation of the CO selectivity of Cu-MOF-74 may illuminate the design principles underlying >2e− CO2RR.
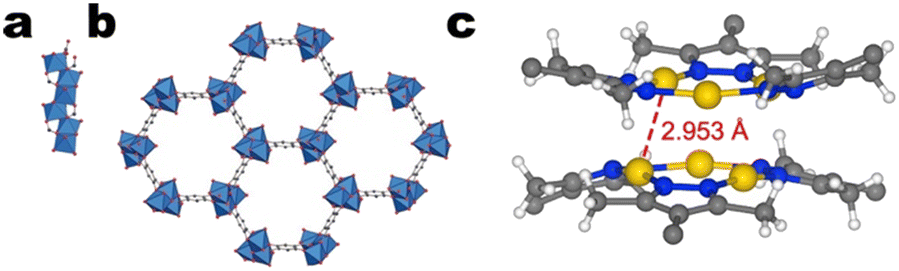 | ||
| Fig. 16 Representation of a) the helical Cu–O–C rods and b) the crystalline framework of Cu-MOF-74 with the SBUs linked by 2,5-dihydroxy-1,4-benzenedicarboxylate ligands. Red and grey spheres denote oxygen and carbon atoms, respectively. Copper atoms are represented by blue octahedra. Hydrogens are omitted for clarity. Reproduced with permission from Rosi et al.136 Copyright 2005 American Chemical Society. c) The hexanuclear cluster constituting the SBU of NNU-50. Yellow, blue, white, and grey spheres denote copper, nitrogen, hydrogen, and carbon atoms, respectively. | ||
Thirdly, we note the work of Dong et al. on NNU-50, which features a hexanuclear copper cluster SBU with strong cuprophilic interactions.133 NNU-50 is comprised of Cu3(Me4BPz)3/2 (Me4BPz = 3,3′,5,5′-tetramethyl-4-4′-bipyrazolyl) clusters; adjacent clusters form a trigonal antiprism (Fig. 16b). Like many CO2RR catalysts, NNU-50 switches selectivity from C2H4 (36% FEC2H4 at −0.8 V vs. RHE) to CH4 production (66.4% FECH4 −1.0 V vs. RHE) at more negative potentials. Further, NNU-50 maintains 61.4% FECH4 at total current densities of over 750 mA cm−2 (−1.2 V vs. RHE).138
The performance of Cu MOFs with multimetallic SBUs can also be modulated by reducing the extent to which the copper atoms are coordinated. Nam et al. showed that by thermally treating HKUST-1, the carboxylate moieties within the SBU sequentially detach, yielding further undercoordinated copper atoms.137 As a result, they achieved 45% FE for ethylene compared to the less than 15% FE for ethylene obtained by untreated HKUST-1 under the same conditions. In the same vein, Wei et al. generated low coordination copper sites by creating oxygen vacancies in Cu-DBC via high energy O2 plasma bombardment.132 After ten minutes of plasma activation, the so-called PA-Cu-DBC-1 catalyst still retains its crystallinity and avoids formation of metallic copper. Notably, PA-Cu-DBC-1 exhibits higher selectivity for CH4 (75.3%) than any other reported Cu-DBC catalyst in neutral conditions.123,138 These two examples highlight the possibility that defect engineering, frequently employed in the design of pure metal catalysts but seldom employed in the design of Cu MOFs, may improve CO2RR performance.
 | ||
| Fig. 17 Comparison of the number of unique node- and cluster-based Cu MOFs and Cu MOF composites. The solid regions denote the number of unique Cu MOFs reported with greater than 0% FE for C2+ products. The striped regions denote the number of unique Cu MOFs reported with 0% FE for C2+ products. Raw data available in Table S3.† For details on methodology, see the ESI.† | ||
Firstly, as the separation between active sites is too large for C–C coupling to occur between adsorbed species, the combination of weakly binding CO-producing sites and strongly binding CO-coupling sites may enhance C2+ product formation. Secondly, observe that eight of the nine node-based Cu MOFs exhibiting >2e− product formation also exhibit C2+ product formation and that only one node-based Cu MOF (CR-MOF) produces HCOOH (Tables S2 and S3†). Circumstantially, this suggests C2+ product formation via a CO intermediate, implying that either CO adsorption or protonation is a descriptor for C2+ selectivity in node-based Cu MOFs. CO adsorption studies may illuminate the more suitable of the two explanations or signal difficulties in CO dimerization. In the case of PcM–Cu–O catalysts, it appears that weak CO adsorption is responsible for poor C2+ selectivity.118,121
Finally, it is unclear to what extent modifications away from the active site influence the electronic structure of copper within the active site. Although we emphasize the importance of copper's immediate coordination environment for the CO2RR performance of MOFs, we note that ample evidence suggests that the order at longer length scales plays a role as well. In several examples, a given MOF exhibits higher CO2RR selectivity and/or activity than its molecular analogue (e.g., PcCu–Cu–O and CuPc,113 NNU-50 and Cu6MePz,133 Cu2(CuTCPP) and CuTCPP,120 HATNA-Cu-MOF, [(n-C3H7)4N]2[Cu(C6Cl4O2)2], and HATNA-6OH (ref. 123)). While this trend may be attributable to non-electronic effects, such as diffusion kinetics or catalyst surface area, we contend that the ligand may also modulate reactivity via the active site's electronic structure. That is, just as descriptors which account for second nearest neighbours (viz. the GCN) are insightful for transition metal catalysts, assessments of the relevant chemical environment for the active site of Cu MOFs may benefit from consideration of the ligand. Experimentally, Cu MOFs with the same CuO4 SBU exhibit a wide range of CO2RR reactivities. Moreover, theoretical calculations indicate that the chemical structure of the ligand affects the adsorption of intermediates on CuO4 nodes. While it may be difficult to assess the electronic effect of the ligand directly, determining the scale of non-electronic effects may provide insight into the degree of electronic structure modulation by the ligand. The following section expands on the effect of the ligand on CO2RR reactivity.
6.2. Ligand effects
The ligand is another major determinant of MOF properties. In addition to dictating the bulk structure, spacing of SBUs, and pore size, the ligand can host additional active sites for CO2RR and influence the electrical properties of the MOF. Furthermore, chemical species within the ligand may indirectly affect the electronic structure of the copper atom within the SBU. In this section, we present observations on the effect of the ligand on CO2RR by Cu MOFs. We specifically highlight correlations between CO2RR performance and ligand substituent electronegativity, the presence of framework nitrogen atoms, ligand flexibility, and active site hydrophobicity.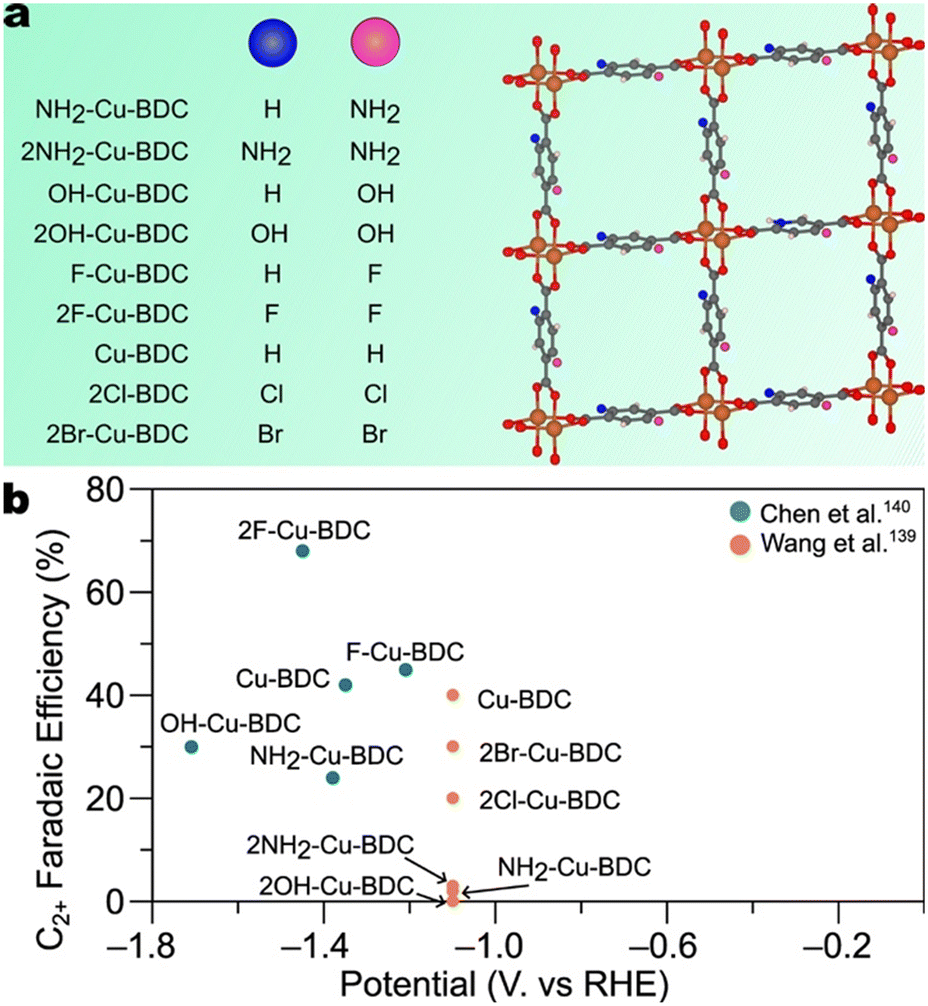 | ||
| Fig. 18 a) Depiction of the general structure of the series of substituted 1,4-benzene dicarboxylate-based MOFs studied by Chen et al.140 and Wang et al.139 Grey, orange, red, and white spheres denote carbon, copper, oxygen, and hydrogen atoms. Pink and blue spheres denote variable functional groups. b) Faradaic efficiencies for C2+ products by X–Cu-BDC MOFs studied by Chen et al.140 and Wang et al.139 | ||
The study by Chen et al. indicates that electron-withdrawing groups correlate with higher selectivity and lower onset potentials toward C2+ products than do electron-donating groups.140 The maximum faradaic efficiencies for C2+ products exhibited by 2F–Cu-BDC, F–Cu-BDC, Cu-BDC, OH-BDC, and NH2–Cu-BDC are shown in Fig. 18b. Further, when the C2+ products partial current is normalized by CO2 uptake, the trend is more pronounced. The C2+ product partial current density is two times greater than that of Cu-BDC and five times greater than that of NH2–Cu-BDC.
Chen et al. suggest two competing effects upon substitution: H2O dissociation and CO2 adsorption.140 The authors suggest that the electron-withdrawing ability of the substituted group correlates positively with the ease of H2O dissociation, thereby increasing the local *H concentration, facilitating *CO hydrogenation to *CHO, and enhancing C–C coupling. Secondly, substitution of electron-donating groups resulted in higher CO2 adsorption ability than did the substitution of electron-withdrawing groups. We note, however, that the trend observed by Chen et al. in alkaline conditions differs from that determined by Wang et al. in neutral conditions. Faradaic efficiencies reported by Wang et al. indicate that Cu-BDC exhibits higher C2+ selectivity than 2Cl–Cu-BDC, 2Br–Cu-BDC, NH2–Cu-BDC, 2OH–Cu-BDC, and 2NH2–Cu-BDC.139
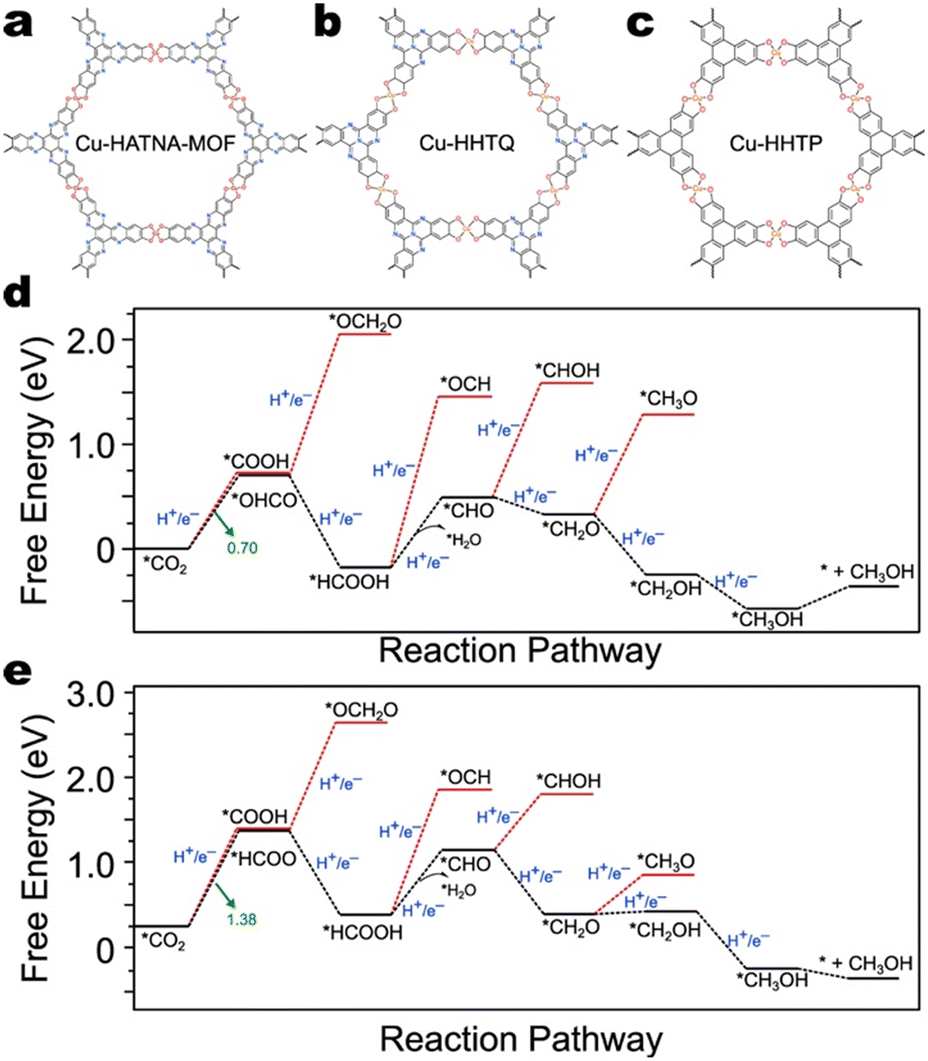 | ||
| Fig. 19 The structures of a) HATNA-Cu-MOF,123 b) Cu-HHTQ,141 and c) Cu-HHTP.117 Free energy diagrams for CH3OH production by d) Cu-HHTQ and e) Cu-HHTP.141 Data from Liu et al.141 | ||
HATNA-Cu-MOF exhibits 78% FE for CH4 production,123 and Cu-HHTQ exhibits 53.6% FE for CH3OH production.141 However, the highest reported single-product faradaic efficiency exhibited by Cu-HHTP under similar conditions is for CO (50%).113,118,141,142 There are no thorough DFT calculations to date studying the CO2RR mechanism on HATNA-Cu-MOF; however, DFT calculations performed by Liu et al. explain the disparity in CO2RR selectivity between Cu-HHTQ and Cu-HHTP (Fig. 19d and e).141 Specifically, on Cu-HHTP, the free energy difference for the first PCET is 0.68 eV higher than it is on Cu-HHTQ. As both catalysts possess CuO4 nodes, their results suggest that the ligands do, in fact, affect CO2RR selectivity.
 | ||
| Fig. 20 Structures of MAF-2 analogues a) the Cu(I) triazolate backbone with ligand side groups omitted. Local coordination environments of b) MAF-2ME, c) MAF-2E, and d) MAF-2P. Orange spheres denote copper atoms. Blue, grey, and white edges denote nitrogen, carbon, and hydrogen atoms. Orange spheres denote copper atoms. Free energy diagrams for CH4 and C2H4 formation on e) MAF-2ME, f) MAF-2E, and g) MAF-2P. Reproduced with permission from Zhuo et al.131 Copyright 2022 John Wiley & Sons, Inc. | ||
Zhuo et al. argue that by modifying the flexibility of groups adjacent to the active site, hydrocarbon production can be rationally tuned. The results indicate that as the ligand becomes bulkier, the selectivity switches from C2H4 to CH4 production. Based on in situ ATR-FTIR measurements and DFT calculations (Fig. 20e–g), two competing effects are suggested: ligand flexibility/steric hindrance and ligand hydrophobicity. The authors argue that bulky groups within the ligand prevent the accommodation of multiple intermediates and inhibit C–C coupling. Simultaneously, the bulkier, more hydrophobic ligands inhibit HER. Accordingly, MAF-2P exhibited the highest selectivity for CH4 (55.9% at −1.5 V vs. RHE), and MAF-2E exhibited the highest selectivity for C2H4 (51.2% at −1.3 V vs. RHE).
In summary, in addition to modulating CO2 adsorption, ligand substituents may modulate hydrophobicity, H2O dissociation, HER suppression and C–C coupling. The studies presented suggest that even when distant from the active site, ligand substituents affect the CO2RR reactivity of Cu MOFs. We continue to explore this theme in the following section in the context of active transition metals.
6.3. Inclusion of other transition metals
Although copper catalysts produce C2+ products, selectivity for a single CO2RR product is poor.30,41,76 On the other hand, other transition metals exhibit high selectivity, albeit for C1 products, such as CO and HCOOH.143 Since CO is an intermediate for C2+ products, one strategy to improve C2+ selectivity may be combining sites which produce CO with those that couple and reduce CO. In principle, one could tune the properties of the C–C coupling site without compromising the effectiveness of the CO-producing site. To this effect, the incorporation of active heteroatoms represents a strategy to design dual-site CO2RR catalysts for C2+ product formation. This section presents insights relevant to the implementation of this strategy.Firstly, we highlight the effect of incorporating cobalt, nickel, and zinc into metallophthalocyanine-based MOFs with square planar CuO4 nodes. Note that while cobalt and zinc are typically known to produce CO, nickel is known to facilitate HER.41,76,144 Studies indicate that the inclusion of these three metals into metallophthalocyanine-based Cu MOFs results exclusively in CO production. Cobalt results in the highest CO production (85% FECO),113,118 followed by nickel (56% FECO)118 and then zinc (9% FECO).121
As mentioned earlier, PcCu–Cu–O and PcCu both facilitate C2+ product formation. Thus, the copper sites within phthalocyanines have been regarded as C–C coupling sites.118,122 It stands to reason that replacing copper within the SBU of PcCu–Cu–O with a CO-producing metal, such as Zn, should improve C2+ product formation. However, the study by Zhong et al. indicates that the copper site of PcCu–Zn–O is not active for C2+ product formation, as PcCu–Zn–O only yields CO (88% FECO).121 Although this result implies that the reactivity of the two sites in metallophthalocyanine-based Cu MOFs cannot be tuned independently, we note again that in this study, PcCu–Cu–O exhibited only CO production (11% FECO). Indeed, the electrodes employed in this study were prepared by mixing the Cu MOFs with CNTs, which may suppress C2+ product formation.113 Nonetheless, DFT calculations support the notion that the SBU affects the reactivity of the Pc unit. Fig. 21a and b illustrate that the overpotentials for CO2RR and HER on the Pc unit of phthalocyanine-based MOFs can be changed by up to 0.36 eV and 0.22 eV, respectively, by modifying the SBU. These results underscore the value of a follow-up study with a different conductive additive. By controlling for the conductive additive, such a study would provide a clearer comparison with which to evaluate of the generalizability of the dual-site paradigm illustrated by Qiu and co-workers.113
 | ||
| Fig. 21 DFT-calculated overpotentials for a) CO and b) H2 production on the Pc units of various PcM1–M2–X MOFs. Data for Co- and Ni-bearing MOFs from Meng et al.118 Data for PcCu–Cu–O and PcZn–Cu–O from Zhong et al.121 | ||
Alternatively, copper within the SBUs may be partially replaced by transition metals to yield multimetallic MOFs. One such example of this paradigm was reported in a study by van Phuc et al., which featured a series of trimetallic Cu MOFs.145 The MOFs are designed as isostructural to HKUST-1 and host two CO-producing metals, Pd and Zn, in addition to copper. We note, however, that characterizations indicate that the metals may exist in metallic form, and clear evidence for incorporation into the MOF structure was not observed.145 These observations may account for the fact that the as-synthesized catalyst only exhibited selectivity for CO (95% FECO) whereas HKUST-1 typically exhibits C2+ selectivity.107,125,126,128,137
Transition metals may also be incorporated by simply physically mixing as-synthesized MOFs. Albo et al. prepared various bimetallic blends comprised of HKUST-1 and CAU-17, a bismuth-based MOF.126 Under the conditions tested, CAU-17 does not produce alcohols, and pure HKUST-1 exhibits maximum faradaic efficiencies for CH3OH and C2H5OH of about 3% and 10%, respectively. However, the optimal blend, CuBi12, consisting of 12% CAU-17, exhibited maximum MeOH and EtOH selectivities of 18.2% and 28.3%, respectively. This nonlinear enhancement of alcohol production suggests a synergistic effect of the two MOFs. The authors postulated that the enhanced production of alcohols by the MOF blends could be mainly ascribed to the formation of HCOO− at bismuth sites, which is then transferred to neighbouring copper active sites where further conversion toward alcohols takes place.
The incorporation of other transition metals offers flexibility in the design of Cu MOFs for CO2RR. Novel selectivity may derive from changes to the local concentrations of intermediates or the electronic structure of active sites. The design of dual-site Cu MOFs is a promising strategy to improve C2+ product selectivity; however, the electronic structure of the two active sites may be coupled, further complicating realization of this paradigm. Additional studies can illuminate the generalizability of this strategy and the potential synergy of multiple active sites. The next section examines synergistic effects between MOF and non-MOF phases.
6.4. Cu MOF composites
For several Cu MOFs, post-reaction characterizations indicate that the active catalyst may be a composite phase consisting of the Cu MOF and metallic aggregates. Such composite phases, herein denoted as Cu MOF composites, can also be derived from copper oxides. Fig. 22 depicts each of these paradigms for Cu MOF composite synthesis. In this section, we outline the potential advantages of and address issues with their implementation. We limit our scope to examples in which the Cu MOF is reportedly catalytically active and exclude reports of MOF pre-catalyst systems.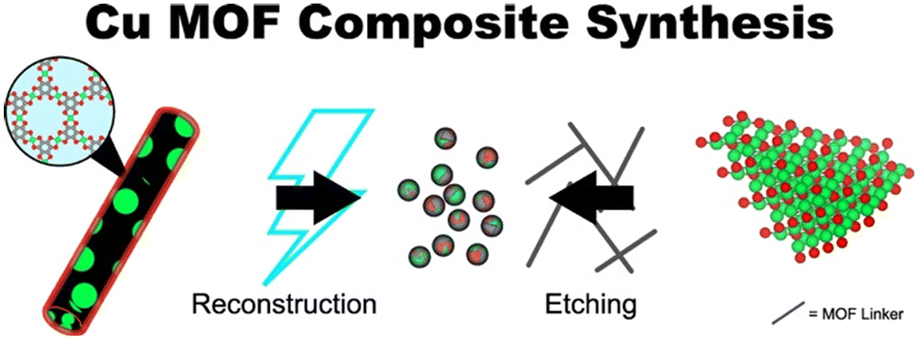 | ||
| Fig. 22 Representative syntheses for Cu MOF composites. The left-hand side depicts the synthesis of Cu(111)@Cu-THQ via electrochemical reconstruction of Cu-THQ reported by Zhao et al.146 The right-hand side depicts the synthesis of Cu2O@Cu-MOF via etching for Cu2O reported by Tan et al.128 Green, red, and grey spheres denotes copper, oxygen, and carbon atoms, respectively. | ||
Fig. 23 illustrates the potential of Cu MOF composites by comparing their CO2RR selectivity to that of their constituents in the same experimental conditions. Whereas Cu-HHTP produces CO with a maximum selectivity of 20% in the same conditions, Cu2O@Cu-HHTP exhibits 73% FECH4.142 Similarly, CuO@Cu-BDC exhibits 50% FEC2H4 compared to 40% by Cu-BDC.139 Cu2O@Cu-HHTP, Cu(111)@Cu-THQ, and CuO@Cu-BDC also exhibit higher CO2RR selectivities than their metal/metal-oxide constituents, Cu2O, Cu, and CuO, respectively (Fig. 23).
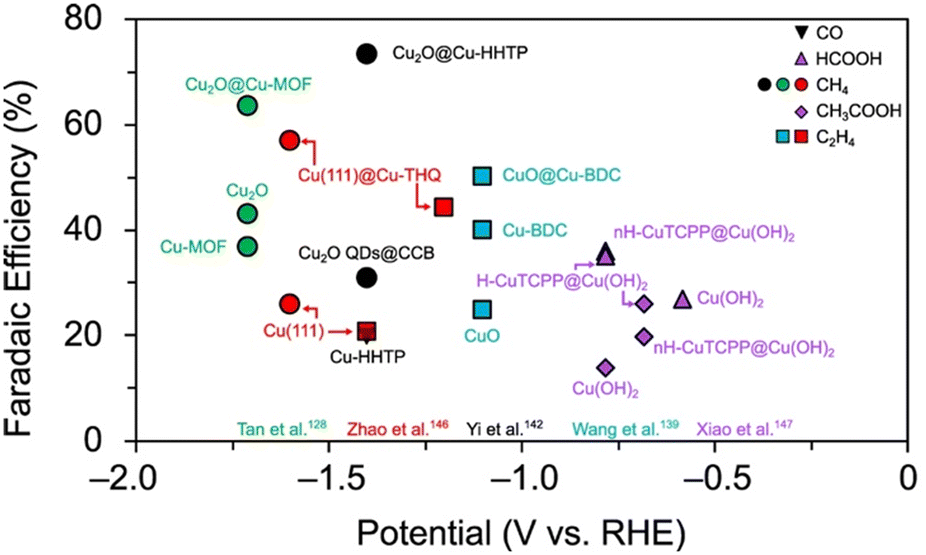 | ||
| Fig. 23 Maximum faradaic efficiencies for CO, HCOOH, CH4, CH3COOH, and C2H4 production for various Cu MOF composites compared to that of their constituent metallic and Cu MOF catalysts in the same experimental setup. QDs = quantum dots. CCB = conductive carbon black. | ||
Studies suggest that synergistic effects may arise from favourable interactions between exposed functional groups and intermediates142 or dual-site functionality.112 It is important to note, however, that the electrochemically constructed MOF composites lack stability. Additional facets and oxide phases are detectable at extended reaction times. Accordingly, unstable product selectivity and CO2RR performance is observed. CuO@Cu-BDC exists as an exception, exhibiting both structural and CO2RR performance stability in the conditions tested.139
As a final example, we highlight the work by Xiao and co-workers. In this study, a Cu2(CuTCPP) MOF phase was constructed from Cu(OH)2 nanoarrays to yield helical, H-CuTCPP@Cu(OH)2, and non-helical, nH-CuTCPP@Cu(OH)2, nanocomposites. The Cu MOF phase features a copper atom within a porphyrin ligand and a copper dimer within the SBU. Both H-CuTCPP@Cu(OH)2 and nH-CuTCPP@Cu(OH)2 exhibit higher maximum selectivities for HCOOH (35% and 35%, respectively) and CH3COOH (26.1% and 19.8%, respectively) than Cu(OH)2 (27% FEHCOOH and 14% FECH3COOH) in the conditions tested (Fig. 23).147
The syntheses of MOF-derived composites can be regarded as instances of favourable MOF transformations. We have shown that such transformations can enhance CO2RR performance such that the resultant composite catalyst outperforms its constituent catalysts. Oftentimes, however, these composite catalysts appear to be intermediate phases formed under reaction conditions. As such, without further understanding of these transformations, the use of MOF composites as effective CO2RR catalysts remains unreasonable. Also, note that despite reports of MOFs forming composites under CO2RR conditions, several of these MOFs have been reported elsewhere without mention of the formation of a composite phase (ESI† file). This point underlines the importance of our following discussion, which more generally addresses the topic of in situ MOF transformations.
6.5. In situ transformations and Cu MOF stability
The long-term stability of Cu MOFs remains a significant roadblock to their adoption for CO2RR. Significantly, Cu MOFs frequently exhibit decaying performance. Even when no performance drops are immediately obvious, Cu MOFs may undergo physical and chemical transformations. As such, studies elucidating the factors which dictate stability and govern degradation mechanisms are vital. Herein, we categorize the types of transformations observed for Cu MOFs in CO2RR and highlight useful design strategies for promoting stability.The second classification of transformation encompasses physical or chemical processes which may occur without electrode polarization, such as leaching of the catalyst into the electrolyte or inherent instability in the electrolyte. During this type of degradation, peeling off catalytic material from the electrode support may enhance HER due to easier access for H2O, leading to lower CO2RR selectivity.126 Identification of this form of degradation may consist of electron microscopy or X-ray diffraction of the catalyst or spectroscopic analysis of the electrolyte.148 The hydrolysis of HKUST-1 is a famous example for which there exists detailed investigation of the various stages of degradation.151
The final classification of transformation refers to reversible transformations which occur under CO2RR conditions and are only discernible by in situ or operando techniques. These transformations are suggested by changes in oxidation states or coordination numbers and can be identified by surface characterization techniques such as AFM, XPS, or XAS.148 For example, operando measurements by Majidi et al., 2021 revealed that under CO2RR conditions, copper is reduced to Cu+ nanoclusters (and potentially metallic copper) but rapidly re-oxidizes when the system is returned to the open circuit voltage.112 Studies suggest that the reversibility of copper reduction derives from the small size of copper particles formed in situ.112,129
In practice, a given MOF may undergo any combination of the above transformations. Nevertheless, stable Cu MOFs do exist, as some catalysts have been reported to exhibit unchanging performance with no evidence of a transformation for multiple hours (Table S1†). Based on these examples, we highlight a few strategies for designing stable Cu MOFs under CO2RR conditions. Specifically, we focus on design strategies for preventing electrochemically induced transformations. For a comprehensive overview of the strategies for designing stable MOFs in aqueous conditions, in different pHs, under mechanical stress, and in the presence of various coordinating anions, we suggest the excellent review by Yuan and co-workers.105
Finally, note that the aforementioned transformations are not inherently undesirable. Often, the resulting material exhibits favourable CO2RR performance.152 In such a case, it is more appropriate to refer to the as-synthesized MOF as a precatalyst. The strategies presently described address the case in which one desires to preserve the order afforded by as-synthesized MOFs.
Strong intermolecular interactions can also stabilize copper within the SBU. For example, the strong cuprophilic interactions within the hexanuclear cluster of NNU-50 stabilize Cu+ under CO2RR conditions.133 Additionally, Weng et al. suggested that the metal ion–ligand binding affinity of a copper complex influences the threshold potential as well as the reversibility of the reductive demetallation process. They also proposed that the restructuring process may involve other important factors such as the solubility of the demetallated ligand and the electronic structure of the complex.129 However, note that the presence of open metal sites is important for the catalytic activity of metal centres,154 The redox activity of the metal within the SBU contributes to MOF conductivity through charge-hopping transport mechanisms.103,155
Finally, as copper is often reduced to Cu+ under CO2RR conditions,94,150,156,157 coordination geometries preferred by Cu+, such as the tetrahedral geometry, may prevent demetallation.131,134 A notable example of this is Cu4II-MFU-4l134 in which the three nitrogen atoms and one Cl− ion form a tetrahedron around the copper ions within the SBU. Again, one should consider the existence of open metal sites when diverging from square planar copper coordination geometries.
7. Challenges & perspectives
In this review, we have focused on the aspects of copper-based MOF design that can be linked to selectivity in electrochemical carbon dioxide reduction. Several trends have emerged. Secondary building units featuring copper clusters commonly produce C2+ products. On the other hand, solitary, square planar nodes, frequently produce CO or HCOOH while other node-based Cu MOFs yield CO, HCOOH, and >2e− products. Regarding ligand design, modifications away from the SBU can significantly affect product selectivity. The incorporation of catalytic metal centres within the ligand can facilitate C–C coupling. Such catalysts show promise as a design paradigm for increasing C2+ product selectivity. Further, studies suggest that the reactivity of Cu MOFs can be tuned via adjacent functional groups and may be affected by more distant modifications. This point is especially relevant for computational work. Finally, the instability of Cu MOFs can be leveraged to yield CO2RR active MOF composites. As illustrated in Fig. 24, however, it is more often the case that such transformations represent one of several challenges to be addressed.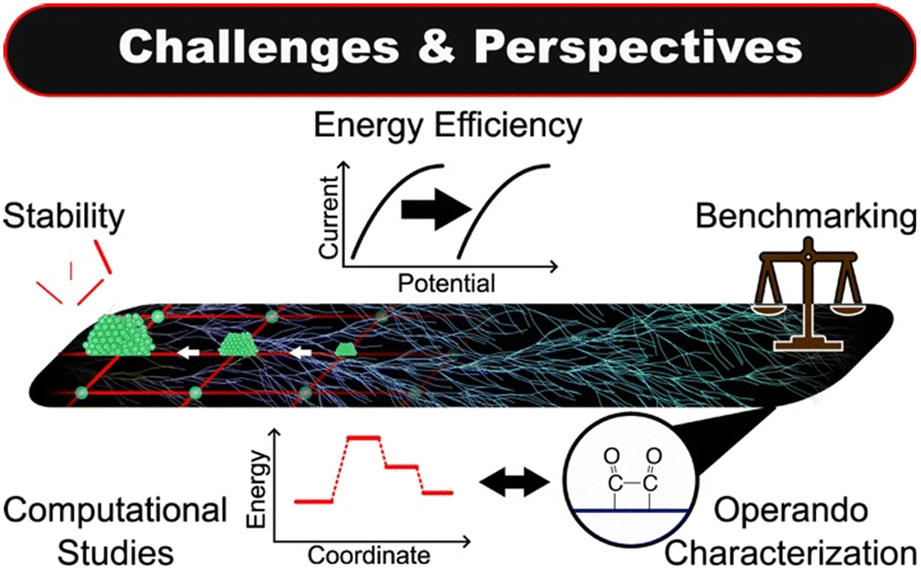 | ||
| Fig. 24 An illustration of the key challenges and perspectives for the development of copper-based metal–organic frameworks for CO2RR. | ||
1) The instability of copper-based metal–organic frameworks severely limits their application for industrial scale electrochemical carbon dioxide reduction. Atomic-scale design may improve the resilience of Cu MOFs to electrochemical degradation by stabilizing copper units and limiting nanocluster formation. The application of electrode synthesis techniques such as spray-drying or electro-deposition may aid in preserving the physical integrity of Cu MOF electrodes. In part, a fundamental understanding of instability motivates the next challenge.
2) The dynamic nature of Cu MOFs during CO2RR demands that their properties are characterized at operating conditions. We highlight the importance of in situ and operando methods, such as ATR-FTIR, SEIRA, DRIFTS, XPS, and XAS, which can illuminate how Cu MOF properties, as they manifest under CO2RR conditions, affect catalyst performance. Particularly, we note that such characterizations are prerequisite for probing electronic properties of copper, such as the proportions of Cu0, Cu+, and Cu2+ species, and for addressing the aforementioned challenge of stability. That the Cu2+ ion is reduced at more positive potentials than required for CO2RR highlights the relevance of the copper oxidation state. Regarding stability, we note that the time scales on which reconstruction or decomposition take place may prohibit their detection by implicit means such as chronoamperometry or ex situ characterizations.148 It should not be assumed that stable CO2RR performance implies a stable catalyst or that the structure of the catalyst after reaction is that of the catalyst during reaction. These assumptions can lead to incorrect characterizations of Cu MOF stability.
3) It is also important to understand the CO2RR mechanisms of Cu MOFs. Hence, the combination of theoretical and experimental studies will continue to play an important role in the development of the field. Density functional theory calculations can provide evidence for a proposed reaction mechanism and insight into how a given Cu MOF facilitates CO2RR selectivity. In situ and operando characterizations can provide experimental evidence for reaction intermediates as well as an accurate representation of the active site during CO2RR. This is especially important for theoretical calculations, the efficacy of which is predicated on reasonable atomic models.
We also emphasize the importance of using standard calculation conventions to determine the thermodynamics of CO2RR reaction mechanisms. Namely, we note that although adsorption energies of intermediates often mirror reactivity trends, these trends may diverge for intermediates of significantly different size, polarity, or binding atom. Proposed reaction mechanisms should be based on Gibbs free energy calculations, and the free energy of several potential intermediates for a given step should be compared to determine the minimum energy path for the reaction mechanism.
Further, that CO2RR mechanisms exhibit strong pH/potential-dependencies suggests that simple thermochemical models may be inadequate or unreliable. Considering this, one may need to employ more sophisticated models which account for solvation32,36,158–162 or the potential-dependence of transition states.32,33,37,73,144,158,163 For CO2RR on transition metals, the use of such models has yielded results which more closely align with experiment.37,144,164 However, to the best of our knowledge, only two works studying CO2RR on Cu MOFs have employed such methods.118,119
4) The energy efficiency of Cu MOFs must be improved through increases in activity and selectivity. Firstly, Cu MOFs still require large overpotentials for valuable products, and the activities reported for Cu MOFs are comparatively lower than those reported for state-of-the-art CO2RR catalysts. Although the selectivities of Cu MOFs are comparable to state-of-the-art catalysts, since Cu MOFs exhibit potential-dependent selectivity, valuable product yields cannot be increased by simply increasing the overpotential. As such, increasing the intrinsic activity of active sites is a priority. Notably, it appears that improving intrinsic activity may be a significant challenge.100
That the conductivities of Cu MOFs are low also poses a problem. In the case that conducting binders must be used, we note that care should be taken to control for the HER activity of the binder. As discussed in this review, CNTs may suppress C2+ selectivity.121
5) Finally, we emphasize the importance of benchmarking and current density reporting procedures. Frequently, publications do not include benchmarking data for their experimental setup. As such, it is impossible to deconvolve differences in performance due to the catalyst from variations in experimental setups. To facilitate meaningful comparisons of CO2RR performance, authors should report the performance of a standard CO2RR catalyst, such as copper foil, in their experimental setup. In the same vein, current densities are typically reported with respect to geometric surface area. However, ECSA-normalized current densities are more insightful for comparing the intrinsic activity of catalysts. Analysis of this metric in conjunction with others, such as CO2 adsorption capacity, may then elucidate design principles for increasing the intrinsic activity of Cu MOFs.
Adoption of the conventions may also aid in rationalizing the observation that the same Cu MOF may be reported as exhibiting large variations in CO2RR performance (activity and selectivity) under similar reported experimental conditions. For example, across the four studies which report CO2RR by Cu-HHTP in neutrally buffered H-cells, maximum CO selectivity varies from 0% to 50% faradaic efficiency.113,118,141,142 Similarly, in the two studies which report CO2RR by Cu-DBC in neutrally buffered H-cells, maximum CH4 selectivity varies from 10% to 34% faradaic efficiency.123,132
Nonetheless, the flexibility in design afforded by copper-based metal–organic frameworks makes them attractive as platforms for electrochemical carbon dioxide reduction. Further work is needed to understand the role of solvent, improve catalyst stability, and increase valuable product selectivity. The integrated design of Cu MOFs at the atomic scale and reaction conditions at the macroscale can potentially improve the CO2RR performance of Cu MOFs. Moreover, the modular nature of Cu MOFs lends itself to machine learning applications.165,166 With continued development, Cu MOFs may provide a solution to the challenge of C2+ selectivity in CO2RR.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the support received from the National Research Council of Canada's Materials for Clean Fuels (MCF) Challenge R&D Program (Grant number CH-MCF-115-1). This research in part was enabled by the support from the University of Calgary's Canada First Research Excellence Fund Program, the Global Research Initiative in Sustainable Low Carbon Unconventional Resources. UN acknowledges the support from Alberta Graduate Scholarship.References
- Z. Sun, Y. Hu, D. Zhou, M. Sun, S. Wang and W. Chen, ACS Energy Lett., 2021, 6, 3992–4022 CrossRef CAS.
- M. Gattrell, N. Gupta and A. Co, J. Electroanal. Chem., 2006, 594, 1–19 CrossRef CAS.
- Y. Wang, J. Liu, G. Zheng, Y. Wang, J. Liu and G. Zheng, Adv. Mater., 2021, 33, 2005798 CrossRef CAS PubMed.
- G. Zhang, L. Li, Z.-J. Zhao, T. Wang and J. Gong, Acc. Mater. Res., 2023, 4, 212–222 CrossRef CAS.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- D. Raciti and C. Wang, ACS Energy Lett., 2018, 3, 1545–1556 CrossRef CAS.
- Q. Zhou, W. Zhang, M. Qiu and Y. Yu, Mater. Today Phys., 2021, 20, 100443 CrossRef CAS.
- C. Zhu, S. Zhao, G. Shi and L. Zhang, ChemSusChem, 2022, 2022, e202200068 Search PubMed.
- H. J. Peng, M. T. Tang, J. Halldin Stenlid, X. Liu and F. Abild-Pedersen, Nat. Commun., 2022, 13, 1–11 Search PubMed.
- Y. Zheng, A. Vasileff, X. Zhou, Y. Jiao, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2019, 141, 7646–7659 CrossRef CAS.
- F. N. Al-Rowaili, A. Jamal, M. S. Ba Shammakh and A. Rana, ACS Sustainable Chem. Eng., 2018, 6, 15895–15914 CrossRef CAS.
- R. Wang, F. Kapteijn and J. Gascon, Chem. – Asian J., 2019, 14, 3452–3461 CrossRef CAS PubMed.
- H. He, J. A. Perman, G. Zhu and S. Ma, Small, 2016, 12, 6309–6324 CrossRef CAS PubMed.
- Y. Zhao, L. Zheng, D. Jiang, W. Xia, X. Xu, Y. Yamauchi, J. Ge, J. Tang, Y. Zhao, L. Zheng, D. Jiang, W. Xia, J. Ge, J. Tang, X. Xu and Y. Yamauchi, Small, 2021, 17, 2006590 CrossRef CAS PubMed.
- Q. Wang, Y. Zhang, H. Lin and J. Zhu, Chem. – Eur. J., 2019, 25, 14026–14035 CrossRef CAS PubMed.
- D. Narváez-Celada and A. S. Varela, J. Mater. Chem. A, 2022, 10, 5899–5917 RSC.
- C. Wang, Z. Lv, W. Yang, X. Feng and B. Wang, Chem. Soc. Rev., 2023, 52, 1382–1427 RSC.
- P. Friedlingstein, M. O'Sullivan, M. W. Jones, R. M. Andrew, L. Gregor, J. Hauck, C. le Quéré, I. T. Luijkx, A. Olsen, G. P. Peters, W. Peters, J. Pongratz, C. Schwingshackl, S. Sitch, J. G. Canadell, P. Ciais, R. B. Jackson, S. R. Alin, R. Alkama, A. Arneth, V. K. Arora, N. R. Bates, M. Becker, N. Bellouin, H. C. Bittig, L. Bopp, F. Chevallier, L. P. Chini, M. Cronin, W. Evans, S. Falk, R. A. Feely, T. Gasser, M. Gehlen, T. Gkritzalis, L. Gloege, G. Grassi, N. Gruber, Ö. Gürses, I. Harris, M. Hefner, R. A. Houghton, G. C. Hurtt, Y. Iida, T. Ilyina, A. K. Jain, A. Jersild, K. Kadono, E. Kato, D. Kennedy, K. Klein Goldewijk, J. Knauer, J. I. Korsbakken, P. Landschützer, N. Lefèvre, K. Lindsay, J. Liu, Z. Liu, G. Marland, N. Mayot, M. J. McGrath, N. Metzl, N. M. Monacci, D. R. Munro, S.-I. Nakaoka, Y. Niwa, K. O'Brien, T. Ono, P. I. Palmer, N. Pan, D. Pierrot, K. Pocock, B. Poulter, L. Resplandy, E. Robertson, C. Rödenbeck, C. Rodriguez, T. M. Rosan, J. Schwinger, R. Séférian, J. D. Shutler, I. Skjelvan, T. Steinhoff, Q. Sun, A. J. Sutton, C. Sweeney, S. Takao, T. Tanhua, P. P. Tans, X. Tian, H. Tian, B. Tilbrook, H. Tsujino, F. Tubiello, G. R. van der Werf, A. P. Walker, R. Wanninkhof, C. Whitehead, A. Willstrand Wranne, R. Wright, W. Yuan, C. Yue, X. Yue, S. Zaehle, J. Zeng and B. Zheng, Earth Syst. Sci. Data, 2022, 14, 4811–4900 CrossRef.
- V. UNFCCC, Conference of the Parties to the United Nations Framework Convention on Climate Change, 2015, pp. 1–32 Search PubMed.
- IPCC, 2022: Climate Change 2022: Mitigation of Climate Change. Contribution of Working Group III to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change, ed. P. R. Shukla, J. Skea, R. Slade, A. al Khourdajie, R. van Diemen, D. McCollum, M. Pathak, S. Some, P. Vyas, R. Fradera, M. Belkacemi, A. Hasija, G. Lisboa, S. Luz and J. Malley, Cambridge University Press, Cambridge, UK, 2022 Search PubMed.
- P. Linstorm, J. Phys. Chem. Ref. Data, Monogr., 1998, 9, 1–1951 Search PubMed.
- S. Verma, B. Kim, H. R. M. Jhong, S. Ma and P. J. A. Kenis, ChemSusChem, 2016, 9, 1972–1979 CrossRef CAS PubMed.
- O. S. Bushuyev, P. De Luna, C. T. Dinh, L. Tao, G. Saur, J. van de Lagemaat, S. O. Kelley and E. H. Sargent, Joule, 2018, 2, 825–832 CrossRef CAS.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- H. Eyring, J. Chem. Phys., 1935, 3, 107 CrossRef CAS.
- A. A. Peterson and J. K. Nørskov, J. Phys. Chem. Lett., 2012, 3, 251–258 CrossRef CAS.
- W. Deng, P. Zhang, B. Seger and J. Gong, Nat. Commun., 2022, 13, 1–9 Search PubMed.
- D. W. DeWulf, T. Jin and A. J. Bard, J. Electrochem. Soc., 1989, 136, 1686–1691 CrossRef CAS.
- K. J. P. Schouten, Y. Kwon, C. J. M. van der Ham, Z. Qin and M. T. M. Koper, Chem. Sci., 2011, 2, 1902–1909 RSC.
- Y. Hori, A. Murata and R. Takahashi, J. Chem. Soc., Faraday Trans. 1, 1989, 85, 2309–2326 RSC.
- R. L. Cook, R. C. MacDuff and A. F. Sammells, J. Electrochem. Soc., 1989, 136, 1982–1984 CrossRef CAS.
- H. Xiao, T. Cheng, W. A. Goddard and R. Sundararaman, J. Am. Chem. Soc., 2016, 138, 483–486 CrossRef CAS PubMed.
- X. Liu, J. Xiao, H. Peng, X. Hong, K. Chan and J. K. Nørskov, Nat. Commun., 2017, 8, 1–7 CrossRef PubMed.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, Energy Environ. Sci., 2010, 3, 1311–1315 RSC.
- P. Hirunsit, W. Soodsawang and J. Limtrakul, J. Phys. Chem. C, 2015, 119, 8238–8249 CrossRef CAS.
- W. Luo, X. Nie, M. J. Janik and A. Asthagiri, ACS Catal., 2016, 6, 219–229 CrossRef CAS.
- X. Nie, M. R. Esopi, M. J. Janik and A. Asthagiri, Angew. Chem., Int. Ed., 2013, 52, 2459–2462 CrossRef CAS PubMed.
- A. Bagger, W. Ju, A. S. Varela, P. Strasser and J. Rossmeisl, ChemPhysChem, 2017, 18, 3266–3273 CrossRef CAS.
- F. Calle-Vallejo and M. T. M. Koper, Angew. Chem., Int. Ed., 2013, 52, 7282–7285 CrossRef CAS PubMed.
- J. H. Montoya, C. Shi, K. Chan and J. K. Nørskov, J. Phys. Chem. Lett., 2015, 6, 2032–2037 CrossRef CAS PubMed.
- Y. Hori, in Modern Aspects of Electrochemistry, Springer, New York, 2008, pp. 89–189 Search PubMed.
- K. J. P. Schouten, Z. Qin, E. P. Gallent and M. T. M. Koper, J. Am. Chem. Soc., 2012, 134, 9864–9867 CrossRef CAS PubMed.
- K. J. P. Schouten, E. Pérez Gallent and M. T. M. Koper, ACS Catal., 2013, 3, 1292–1295 CrossRef CAS.
- J. H. Montoya, A. A. Peterson and J. K. Nørskov, ChemCatChem, 2013, 5, 737–742 CrossRef CAS.
- R. B. Sandberg, J. H. Montoya, K. Chan and J. K. Nørskov, Surf. Sci., 2016, 654, 56–62 CrossRef CAS.
- J. D. Goodpaster, A. T. Bell and M. Head-Gordon, J. Phys. Chem. Lett., 2016, 7, 1471–1477 CrossRef CAS PubMed.
- E. E. Benson, C. P. Kubiak, A. J. Sathrum and J. M. Smieja, Chem. Soc. Rev., 2008, 38, 89–99 RSC.
- H. Wan, Y. Jiao, A. Bagger and J. Rossmeisl, ACS Catal., 2020, 11, 533–541 CrossRef.
- K. Zhao and X. Quan, ACS Catal., 2021, 11, 2076–2097 CrossRef CAS.
- A. Vasileff, Y. Zheng and S. Z. Qiao, Adv. Energy Mater., 2017, 7, 1700759 CrossRef.
- Q. Lu, J. Rosen and F. Jiao, ChemCatChem, 2015, 7, 38–47 CrossRef CAS.
- D. Johnson, Z. Qiao and A. Djire, ACS Appl. Energy Mater., 2021, 4, 8661–8684 CrossRef CAS.
- S. Siahrostami, Ind. Eng. Chem. Res., 2018, 58, 879–885 CrossRef.
- Y. Cheng, S. Yang, P. Jiang, S. Wang, Y. Cheng, S. Wang, S. P. Jiang and S. Yang, Small Methods, 2019, 3, 1800440 CrossRef.
- J. He, N. J. J. Johnson, A. Huang and C. P. Berlinguette, ChemSusChem, 2018, 11, 48–57 CrossRef CAS PubMed.
- A. Vasileff, C. Xu, Y. Jiao, Y. Zheng and S. Z. Qiao, Chem, 2018, 4, 1809–1831 CAS.
- J. Christophe, T. Doneux and C. Buess-Herman, Electrocatalysis, 2012, 3, 139–146 CrossRef CAS.
- C. Yang, S. Li, Z. Zhang, H. Wang, H. Liu, F. Jiao, Z. Guo, X. Zhang and W. Hu, Small, 2020, 16, 2001847 CrossRef CAS PubMed.
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Nat. Energy, 2019, 4, 732–745 CrossRef CAS.
- M. Golam Kibria, J. P. Edwards, C. M. Gabardo, C.-T. Dinh, A. Seifitokaldani, D. Sinton, E. H. Sargent, M. G. Kibria, C. Dinh, A. Seifitokaldani, E. H. Sargent, J. P. Edwards, C. M. Gabardo and D. Sinton, Adv. Mater., 2019, 31, 1807166 CrossRef.
- J. Li, Y. Zhang and N. Kornienko, New J. Chem., 2020, 44, 4246–4252 RSC.
- S. N. Steinmann, C. Michel, R. Schwiedernoch and P. Sautet, Phys. Chem. Chem. Phys., 2015, 17, 13949–13963 RSC.
- J. Wang, H. Y. Tan, Y. Zhu, H. Chu and H. M. Chen, Angew. Chem., Int. Ed., 2021, 60, 17254–17267 CrossRef CAS PubMed.
- D. Gao, R. M. Arán-Ais, H. S. Jeon and B. Roldan Cuenya, Nat. Catal., 2019, 2, 198–210 CrossRef CAS.
- M. Li, H. Wang, W. Luo, P. C. Sherrell, J. Chen, J. Yang, M. H. Li, H. F. Wang, W. Luo, J. P. Yang, P. C. Sherrell and J. Chen, Adv. Mater., 2020, 32, 2001848 CrossRef CAS PubMed.
- Q. Zhang, J. Guan, Q. Zhang and J. Guan, Adv. Funct. Mater., 2020, 30, 2000768 CrossRef CAS.
- M. T. Darby, M. Stamatakis, A. Michaelides and E. C. H. Sykes, J. Phys. Chem. Lett., 2018, 9, 5636–5646 CrossRef CAS PubMed.
- Y. Hori, A. Murata and Y. Yoshinami, J. Chem. Soc., Faraday Trans., 1991, 87, 125–128 RSC.
- I. Takahashi, O. Koga, N. Hoshi and Y. Hori, J. Electroanal. Chem., 2002, 533, 135–143 CrossRef CAS.
- Y. Hori, I. Takahashi, O. Koga and N. Hoshi, J. Mol. Catal. A: Chem., 2003, 199, 39–47 CrossRef CAS.
- Y. Hori, H. Wakebe, T. Tsukamoto and O. Koga, Surf. Sci., 1995, 335, 258–263 CrossRef CAS.
- Y. Huang, A. D. Handoko, P. Hirunsit and B. S. Yeo, ACS Catal., 2017, 7, 1749–1756 CrossRef CAS.
- J. Hussain, H. Jónsson and E. Skúlason, ACS Catal., 2018, 8, 5240–5249 CrossRef CAS.
- P. Sabatier, Ber. Dtsch. Chem. Ges., 1911, 44, 1984–2001 CrossRef CAS.
- K. P. Kuhl, T. Hatsukade, E. R. Cave, D. N. Abram, J. Kibsgaard and T. F. Jaramillo, J. Am. Chem. Soc., 2014, 136, 14107–14113 CrossRef CAS PubMed.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 RSC.
- A. Xu, N. Govindarajan, G. Kastlunger, S. Vijay and K. Chan, Acc. Chem. Res., 2022, 55, 495–503 CrossRef CAS PubMed.
- K. W. Frese, in Electrochemical and Electrocatalytic Reactions of Carbon Dioxide, Elsevier, 1993, pp. 145–216 Search PubMed.
- C. Hahn, T. Hatsukade, Y. G. Kim, A. Vailionis, J. H. Baricuatro, D. C. Higgins, S. A. Nitopi, M. P. Soriaga and T. F. Jaramillo, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 5918–5923 CrossRef CAS PubMed.
- F. Calle-Vallejo, J. Tymoczko, V. Colic, Q. H. Vu, M. D. Pohl, K. Morgenstern, D. Loffreda, P. Sautet, W. Schuhmann and A. S. Bandarenka, Science, 2015, 350, 185–189 CrossRef CAS PubMed.
- H. Mistry, A. S. Varela, S. Kühl, P. Strasser and B. R. Cuenya, Nat. Rev. Mater., 2016, 1, 1–14 Search PubMed.
- T. Cheng, H. Xiao and W. A. Goddard, J. Am. Chem. Soc., 2017, 139, 11642–11645 CrossRef CAS PubMed.
- C. S. Chen, A. D. Handoko, J. H. Wan, L. Ma, D. Ren and B. S. Yeo, Catal. Sci. Technol., 2014, 5, 161–168 RSC.
- F. S. Roberts, K. P. Kuhl and A. Nilsson, Angew. Chem., 2015, 127, 5268–5271 CrossRef.
- G. L. de Gregorio, T. Burdyny, A. Loiudice, P. Iyengar, W. A. Smith and R. Buonsanti, ACS Catal., 2020, 10, 4854–4862 CrossRef CAS PubMed.
- P. Hirunsit, J. Phys. Chem. C, 2013, 117, 8262–8268 CrossRef CAS.
- D. Kim, J. Resasco, Y. Yu, A. M. Asiri and P. Yang, Nat. Commun., 2014, 5, 1–8 Search PubMed.
- M. Watanabe, M. Shibata, A. Kato, M. Azuma and T. Sakata, J. Electrochem. Soc., 1991, 138, 3382–3389 CrossRef CAS.
- Y. Zhou, F. Che, M. Liu, C. Zou, Z. Liang, P. de Luna, H. Yuan, J. Li, Z. Wang, H. Xie, H. Li, P. Chen, E. Bladt, R. Quintero-Bermudez, T. K. Sham, S. Bals, J. Hofkens, D. Sinton, G. Chen and E. H. Sargent, Nat. Chem., 2018, 10, 974–980 CrossRef CAS PubMed.
- S. Lee, G. Park and J. Lee, ACS Catal., 2017, 7, 8594–8604 CrossRef CAS.
- D. Ren, B. S. H. Ang and B. S. Yeo, ACS Catal., 2016, 6, 8239–8247 CrossRef CAS.
- E. L. Clark, C. Hahn, T. F. Jaramillo and A. T. Bell, J. Am. Chem. Soc., 2017, 139, 15848–15857 CrossRef CAS PubMed.
- D. Higgins, A. T. Landers, Y. Ji, S. Nitopi, C. G. Morales-Guio, L. Wang, K. Chan, C. Hahn and T. F. Jaramillo, ACS Energy Lett., 2018, 3, 2947–2955 CrossRef CAS.
- W. Zhang, P. He, C. Wang, T. Ding, T. Chen, X. Liu, L. Cao, T. Huang, X. Shen, O. A. Usoltsev, A. L. Bugaev, Y. Lin and T. Yao, J. Mater. Chem. A, 2020, 8, 25970–25977 RSC.
- M. Favaro, H. Xiao, T. Cheng, W. A. Goddard and E. J. Crumlin, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6706–6711 CrossRef CAS PubMed.
- W. Ma, S. Xie, T. Liu, Q. Fan, J. Ye, F. Sun, Z. Jiang, Q. Zhang, J. Cheng and Y. Wang, Nat. Catal., 2020, 3, 478–487 CrossRef CAS.
- C.-T. Dinh, T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. P. García de Arquer, A. Kiani, J. P. Edwards, P. de Luna, O. S. Bushuyev, C. Zou, R. Quintero-Bermudez, Y. Pang, D. Sinton and E. H. Sargent, Science, 2018, 360, 783–787 CrossRef CAS PubMed.
- M. Luo, Z. Wang, Y. C. Li, J. Li, F. Li, Y. Lum, D. H. Nam, B. Chen, J. Wicks, A. Xu, T. Zhuang, W. R. Leow, X. Wang, C. T. Dinh, Y. Wang, Y. Wang, D. Sinton and E. H. Sargent, Nat. Commun., 2019, 10, 1–7 CrossRef PubMed.
- F. Li, A. Thevenon, A. Rosas-Hernández, Z. Wang, Y. Li, C. M. Gabardo, A. Ozden, C. T. Dinh, J. Li, Y. Wang, J. P. Edwards, Y. Xu, C. McCallum, L. Tao, Z. Q. Liang, M. Luo, X. Wang, H. Li, C. P. O'Brien, C. S. Tan, D. H. Nam, R. Quintero-Bermudez, T. T. Zhuang, Y. C. Li, Z. Han, R. D. Britt, D. Sinton, T. Agapie, J. C. Peters and E. H. Sargent, Nature, 2019, 577, 509–513 CrossRef PubMed.
- O. Christensen, S. Zhao, Z. Sun, A. Bagger, J. V. Lauritsen, S. U. Pedersen, K. Daasbjerg and J. Rossmeisl, ACS Catal., 2022, 15737–15749 CrossRef CAS.
- T. R. Cook, Y. R. Zheng and P. J. Stang, Chem. Rev., 2013, 113, 734–777 CrossRef CAS PubMed.
- Y. Wen, X. Wu and Q. Zhu, in Advanced Structural Chemistry, Wiley, 2021, pp. 283–389 Search PubMed.
- L. S. Xie, G. Skorupskii and M. Dincǎ, Chem. Rev., 2020, 120, 8536–8580 CrossRef CAS PubMed.
- Q. He, F. Zhan, H. Wang, W. Xu, H. Wang and L. Chen, Mater. Today Sustain., 2022, 17, 100104 CrossRef.
- S. Yuan, L. Feng, K. Wang, J. Pang, M. Bosch, C. Lollar, Y. Sun, J. Qin, X. Yang, P. Zhang, Q. Wang, L. Zou, Y. Zhang, L. Zhang, Y. Fang, J. Li, H.-C. Zhou, S. Yuan, L. Feng, K. Wang, J. Pang, M. Bosch, C. Lollar, Y. Sun, J. Qin, X. Yang, P. Zhang, Q. Wang, L. Zou, Y. Zhang, L. Zhang, Y. Fang, J. Li and H. Zhou, Adv. Mater., 2018, 30, 1704303 CrossRef PubMed.
- R. Hinogami, S. Yotsuhashi, M. Deguchi, Y. Zenitani, H. Hashiba and Y. Yamada, ECS Electrochem. Lett., 2012, 1, H17 CrossRef CAS.
- R. Senthil Kumar, S. Senthil Kumar and M. Anbu Kulandainathan, Electrochem. Commun., 2012, 25, 70–73 CrossRef CAS.
- F. Calle-Vallejo, J. I. Martínez, J. M. García-Lastra, P. Sautet, D. Loffreda, F. Calle-Vallejo, P. Sautet, D. Loffreda and J. I. Martínez, Angew. Chem., 2014, 126, 8456–8459 CrossRef.
- Z. Zhao, Z. Chen, X. Zhang and G. Lu, J. Phys. Chem. C, 2016, 120, 28125–28130 CrossRef CAS.
- Ü. Kökçam-Demir, A. Goldman, L. Esrafili, M. Gharib, A. Morsali, O. Weingart and C. Janiak, Chem. Soc. Rev., 2020, 49, 2751–2798 RSC.
- Y.-Y. Liu, H.-L. Zhu, Z.-H. Zhao, N.-Y. Huang, P.-Q. Liao and X.-M. Chen, ACS Catal., 2022, 12, 2749–2755 CrossRef CAS.
- L. Majidi, A. Ahmadiparidari, N. Shan, S. N. Misal, K. Kumar, Z. Huang, S. Rastegar, Z. Hemmat, X. Zou, P. Zapol, J. Cabana, L. A. Curtiss, A. Salehi-Khojin, L. Majidi, A. Ahmadiparidari, S. N. Misal, S. Rastegar, Z. Hemmat, A. Salehi-Khojin, N. Shan, P. Zapol, L. A. Curtiss, K. Kumar, J. Cabana, Z. Huang and X. Zou, Adv. Mater., 2021, 33, 2004393 CrossRef CAS PubMed.
- X. F. Qiu, H. L. Zhu, J. R. Huang, P. Q. Liao and X. M. Chen, J. Am. Chem. Soc., 2021, 143, 7242–7246 CrossRef CAS PubMed.
- J. Chatt and L. A. Duncanson, J. Chem. Soc., 1953, 2939–2947 RSC.
- J. S. Dewar, Bull. Soc. Chim. Fr., 1951, 18, C71–C79 Search PubMed.
- M. Loipersberger, Y. Mao and M. Head-Gordon, J. Chem. Theory Comput., 2020, 16, 1073–1089 CrossRef CAS PubMed.
- Y. Zhang, L. Z. Dong, S. Li, X. Huang, J. N. Chang, J. H. Wang, J. Zhou, S. L. Li and Y. Q. Lan, Nat. Commun., 2021, 12, 1–9 CrossRef CAS PubMed.
- Z. Meng, J. Luo, W. Li and K. A. Mirica, J. Am. Chem. Soc., 2020, 142, 21656–21669 CrossRef CAS PubMed.
- Z. H. Zhao, H. L. Zhu, J. R. Huang, P. Q. Liao and X. M. Chen, ACS Catal., 2022, 12, 7986–7993 CrossRef CAS.
- Y. Zhou, S. Chen, S. Xi, Z. Wang, P. Deng, F. Yang, Y. Han, Y. Pang and B. Y. Xia, Cell Rep. Phys. Sci., 2020, 1, 100182 CrossRef.
- H. Zhong, M. Ghorbani-Asl, K. H. Ly, J. Zhang, J. Ge, M. Wang, Z. Liao, D. Makarov, E. Zschech, E. Brunner, I. M. Weidinger, J. Zhang, A. V. Krasheninnikov, S. Kaskel, R. Dong and X. Feng, Nat. Commun., 2020, 11, 1–10 CrossRef PubMed.
- S. Kusama, T. Saito, H. Hashiba, A. Sakai and S. Yotsuhashi, ACS Catal., 2017, 7, 8382–8385 CrossRef CAS.
- Y. Liu, S. Li, L. Dai, J. Li, J. Lv, Z. Zhu, A. Yin, P. Li and B. Wang, Angew. Chem., Int. Ed., 2021, 60, 16409–16415 CrossRef CAS PubMed.
- C. H. Hendon and A. Walsh, Chem. Sci., 2015, 6, 3674–3683 RSC.
- J. Albo, D. Vallejo, G. Beobide, O. Castillo, P. Castaño and A. Irabien, ChemSusChem, 2017, 10, 1100–1109 CrossRef CAS PubMed.
- J. Albo, M. Perfecto-Irigaray, G. Beobide and A. Irabien, J. CO2 Util., 2019, 33, 157–165 CrossRef CAS.
- Y.-L. Qiu, H.-X. Zhong, T.-T. Zhang, W.-B. Xu, P.-P. Su, X.-F. Li and H.-M. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 2480–2489 CrossRef CAS PubMed.
- X. Tan, C. Yu, C. Zhao, H. Huang, X. Yao, X. Han, W. Guo, S. Cui, H. Huang and J. Qiu, ACS Appl. Mater. Interfaces, 2019, 11, 9904–9910 CrossRef CAS PubMed.
- Z. Weng, Y. Wu, M. Wang, J. Jiang, K. Yang, S. Huo, X. F. Wang, Q. Ma, G. W. Brudvig, V. S. Batista, Y. Liang, Z. Feng and H. Wang, Nat. Commun., 2018, 9, 1–9 CrossRef CAS PubMed.
- J. X. Wu, S. Z. Hou, X. da Zhang, M. Xu, H. F. Yang, P. S. Cao and Z. Y. Gu, Chem. Sci., 2019, 10, 2199–2205 RSC.
- L. L. Zhuo, P. Chen, K. Zheng, X. W. Zhang, J. X. Wu, D. Y. Lin, S. Y. Liu, Z. S. Wang, J. Y. Liu, D. D. Zhou and J. P. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202204967 CrossRef CAS PubMed.
- S. Wei, X. Jiang, C. He, S. Wang, Q. Hu, X. Chai, X. Ren, H. Yang and C. He, J. Mater. Chem. A, 2022, 10, 6187–6192 RSC.
- L. Z. Dong, Y. F. Lu, R. Wang, J. Zhou, Y. Zhang, L. Zhang, J. Liu, S. L. Li and Y. Q. Lan, Nano Res., 2022, 15, 10185–10193 CrossRef CAS.
- H. L. Zhu, J. R. Huang, X. W. Zhang, C. Wang, N. Y. Huang, P. Q. Liao and X. M. Chen, ACS Catal., 2021, 11, 11786–11792 CrossRef CAS.
- T. Van Phuc, S. G. Kang, J. S. Chung and S. H. Hur, Mater. Res. Bull., 2021, 138, 111228 CrossRef CAS.
- N. L. Rosi, J. Kim, M. Eddaoudi, B. Chen, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 1504–1518 CrossRef CAS PubMed.
- D. H. Nam, O. S. Bushuyev, J. Li, P. De Luna, A. Seifitokaldani, C. T. Dinh, F. P. García De Arquer, Y. Wang, Z. Liang, A. H. Proppe, C. S. Tan, P. Todorović, O. Shekhah, C. M. Gabardo, J. W. Jo, J. Choi, M. J. Choi, S. W. Baek, J. Kim, D. Sinton, S. O. Kelley, M. Eddaoudi and E. H. Sargent, J. Am. Chem. Soc., 2018, 140, 11378–11386 CrossRef CAS PubMed.
- X. Zhou, J. Dong, Y. Zhu, L. Liu, Y. Jiao, H. Li, Y. Han, K. Davey, Q. Xu, Y. Zheng and S. Z. Qiao, J. Am. Chem. Soc., 2021, 143, 6681–6690 CrossRef CAS PubMed.
- L. Wang, X. Li, L. Hao, S. Hong, A. W. Robertson and Z. Sun, Chin. J. Catal., 2022, 43, 1049–1057 CrossRef CAS.
- R. Chen, L. Cheng, J. Liu, Y. Wang, W. Ge, C. Xiao, H. Jiang, Y. Li and C. Li, Small, 2022, 2200720 CrossRef CAS PubMed.
- J. Liu, D. Yang, Y. Zhou, G. Zhang, G. Xing, Y. Liu, Y. Ma, O. Terasaki, S. Yang and L. Chen, Angew. Chem., Int. Ed., 2021, 60, 14473–14479 CrossRef CAS PubMed.
- J. D. Yi, R. Xie, Z. L. Xie, G. L. Chai, T. F. Liu, R. P. Chen, Y. B. Huang and R. Cao, Angew. Chem., Int. Ed., 2020, 59, 23641–23648 CrossRef CAS PubMed.
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav3506 CrossRef CAS PubMed.
- J. Hussain, E. Skúlason and H. Jónsson, Procedia Comput. Sci., 2015, 51, 1865–1871 CrossRef.
- T. Van Phuc, J. S. Chung and S. H. Hur, Catalysts, 2021, 11, 537 CrossRef.
- Z. H. Zhao, K. Zheng, N. Y. Huang, H. L. Zhu, J. R. Huang, P. Q. Liao and X. M. Chen, Chem. Commun., 2021, 57, 12764–12767 RSC.
- Y. H. Xiao, Y. X. Zhang, R. Zhai, Z. G. Gu and J. Zhang, Sci. China Mater., 2021, 65, 1269–1275 CrossRef.
- W. Zheng and L. Y. S. Lee, ACS Energy Lett., 2021, 6, 2838–2843 CrossRef CAS.
- W. Zhang, C. Huang, J. Zhu, Q. Zhou, R. Yu, Y. Wang, P. An, J. Zhang, M. Qiu, L. Zhou, L. Mai, Z. Yi and Y. Yu, Angew. Chem., Int. Ed., 2022, 61, e202112116 CAS.
- L. Braglia, F. Tavani, S. Mauri, R. Edla, D. Krizmancic, A. Tofoni, V. Colombo, P. D'Angelo and P. Torelli, J. Phys. Chem. Lett., 2021, 12, 9182–9187 CrossRef CAS PubMed.
- M. Todaro, G. Buscarino, L. Sciortino, A. Alessi, F. Messina, M. Taddei, M. Ranocchiari, M. Cannas and F. M. Gelardi, J. Phys. Chem. C, 2016, 120, 12879–12889 CrossRef CAS.
- Y. Zhou, R. Abazari, J. Chen, M. Tahir, A. Kumar, R. R. Ikreedeegh, E. Rani, H. Singh and A. M. Kirillov, Coord. Chem. Rev., 2022, 451, 214264 CrossRef CAS.
- J. H. Dou, M. Q. Arguilla, Y. Luo, J. Li, W. Zhang, L. Sun, J. L. Mancuso, L. Yang, T. Chen, L. R. Parent, G. Skorupskii, N. J. Libretto, C. Sun, M. C. Yang, P. V. Dip, E. J. Brignole, J. T. Miller, J. Kong, C. H. Hendon, J. Sun and M. Dincă, Nat. Mater., 2020, 20, 222–228 CrossRef PubMed.
- A. Corma, H. García and F. X. Llabrés I Xamena, Chem. Rev., 2010, 110, 4606–4655 CrossRef CAS PubMed.
- L. Sun, M. G. Campbell and M. Dincă, Angew. Chem., Int. Ed., 2016, 55, 3566–3579 CrossRef CAS PubMed.
- S. H. Lee, J. C. Lin, M. Farmand, A. T. Landers, J. T. Feaster, J. E. Avilés Acosta, J. W. Beeman, Y. Ye, J. Yano, A. Mehta, R. C. Davis, T. F. Jaramillo, C. Hahn and W. S. Drisdell, J. Am. Chem. Soc., 2021, 143, 588–592 CrossRef CAS PubMed.
- A. D. Handoko, F. Wei, J. Lim, B. S. Yeo and Z. W. Seh, Nat. Catal., 2018, 1, 922–934 CrossRef CAS.
- X. Nie, W. Luo, M. J. Janik and A. Asthagiri, J. Catal., 2014, 312, 108–122 CrossRef CAS.
- M. Fishman, H. L. Zhuang, K. Mathew, W. Dirschka and R. G. Hennig, Phys. Rev. B: Condens. Matter, 2013, 87, 245402 CrossRef.
- K. Mathew, V. S. C. Kolluru, S. Mula, S. N. Steinmann and R. G. Hennig, J. Chem. Phys., 2019, 151, 234101 CrossRef PubMed.
- K. Mathew, R. Sundararaman, K. Letchworth-Weaver, T. A. Arias and R. G. Hennig, J. Chem. Phys., 2014, 140, 084106 CrossRef PubMed.
- T. Cheng, H. Xiao and W. A. Goddard, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1795–1800 CrossRef CAS PubMed.
- J. Hussain, H. Jónsson and E. Skúlason, Faraday Discuss., 2017, 195, 619–636 RSC.
- W. J. Durand, A. A. Peterson, F. Studt, F. Abild-Pedersen and J. K. Nørskov, Surf. Sci., 2011, 605, 1354–1359 CrossRef CAS.
- X. Wan, Z. Zhang, H. Niu, Y. Yin, C. Kuai, J. Wang, C. Shao and Y. Guo, J. Phys. Chem. Lett., 2021, 12, 6111–6118 CrossRef CAS PubMed.
- S. Siahrostami, S. R. Stoyanov, S. Gusarov, I. D. Gates and M. Karamad, in Accelerated materials discovery: How to use Artificial Intelligence to speed up development, ed. P. De Luna, De Gruyter, Berlin, 2022, pp. 27–64 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The ESI includes tabulated reaction and property data for all of the metal–organic frameworks discussed in the main text as well as a description of the methodology used to compile data. The ESI file contains all the raw data for the tables in the ESI as well as additional experimental details for each study. See DOI: https://doi.org/10.1039/d3cy00408b |
| This journal is © The Royal Society of Chemistry 2023 |