
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCyclometalated C^N diphosphine ruthenium catalysts for Oppenauer-type oxidation/transfer hydrogenation reactions and cytotoxic activity†
Dario
Alessi
 a,
Pierfrancesco
Del Mestre
b,
Eleonora
Aneggi
a,
Maurizio
Ballico
a,
Antonio P.
Beltrami
b,
Marta
Busato
a,
Daniela
Cesselli
b,
Alexandra A.
Heidecker
c,
Daniele
Zuccaccia
a and
Walter
Baratta
*a
a,
Pierfrancesco
Del Mestre
b,
Eleonora
Aneggi
a,
Maurizio
Ballico
a,
Antonio P.
Beltrami
b,
Marta
Busato
a,
Daniela
Cesselli
b,
Alexandra A.
Heidecker
c,
Daniele
Zuccaccia
a and
Walter
Baratta
*a
aDipartimento di Scienze Agroalimentari, Ambientali e Animali, Università di Udine, Via Cotonificio 108, I-33100 Udine, Italy. E-mail: walter.baratta@uniud.it
bDipartimento di Area Medica – Istituto di Anatomia Patologica, Università di Udine, Via Chiusaforte, F3, I-33100 Udine, Italy
cInorganic Chemistry/Molecular Catalysis, Department of Chemistry & Catalysis Research Center, TUM, Lichtenbergstraße 4, 85747 Garching b. München, Germany
First published on 16th August 2023
Abstract
The cyclometalated acetate ruthenium complexes [Ru(C^N)(η2-OAc)(dppb)] (dppb = 1,4-bis(diphenylphosphino)butane; HC^N = 2-phenylpyridine 1, benzo[h]quinoline 2, 1-phenylpyrazole 3, 2-phenyl-2-oxazoline 4) are easily obtained in 58–74% yield through a one-pot synthesis from [Ru(η2-OAc)2(dppb)] and the corresponding phenyl substituted N-heterocycle in methanol via elimination of HOAc. These complexes have been characterized by single crystal X-ray diffraction studies. Protonation of 2 with HCO2H (5 equiv.) in toluene affords the formate [Ru(C^N)(η2-HCO2)(dppb)] (5) isolated in 75% yield, without release of the HC^N ligand. The derivatives 1–4 display catalytic activity in the Oppenauer-type oxidation of secondary alcohols to ketones at S/C = 1000, using acetone or cyclohexanone as hydrogen acceptor and KOtBu as base in toluene, with TOF up to 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1 for 4. Complexes 1–4 at S/C = 1000 are also active in the TH of carbonyl compounds to alcohols in 2-propanol employing NaOiPr, with TOF up to 14300 h−1 for 4. The evaluation of the cytotoxic activity of these complexes against U87 glioblastoma cancer cell line via MTT test affords IC50 values ranging from 1.4 to 4.1 μM.
000 h−1 for 4. Complexes 1–4 at S/C = 1000 are also active in the TH of carbonyl compounds to alcohols in 2-propanol employing NaOiPr, with TOF up to 14300 h−1 for 4. The evaluation of the cytotoxic activity of these complexes against U87 glioblastoma cancer cell line via MTT test affords IC50 values ranging from 1.4 to 4.1 μM.
Introduction
Cyclometalated complexes containing a carbon–metal σ-bond have been deeply investigated in the last decades since these species have shown to play a crucial role as intermediates for catalytic coupling reactions, as well as robust metal fragments in metal-catalyzed organic transformations. The enhanced basicity at the metal center, combined with the chelate effect that allow increase in the stability of complexes, lead to derivatives that can find applications in several areas, including catalysis,1–7 photochemistry,8–11 and medicine.12,13 Particular attention has been devoted to palladium, platinum, rhodium, iridium and ruthenium complexes for the selective functionalization of C–H bonds, via cyclometalation promoted by neighbouring functional groups,14,15 and for the generation of fine-tuned catalysts for C–C and C–H forming reactions. Thus, cyclometalated ruthenium catalysts have been found active in C–H activation and functionalization, olefin metathesis, transfer hydrogenation (TH), cyclopropanation and cyclization reactions.16–18Hydrogenation19,20 and TH21–25 of carbonyl compounds to alcohols with H2 and 2-propanol as hydrogen donor, are fundamental processes for the syntheses of fine-chemicals and pharmaceutical products,26 a reaction which can be efficiently accomplished with diphosphine and arene type ruthenium bifunctional catalysts containing an amine N–H function.27–30 Conversely, the Oppenauer-type oxidation of alcohols with acetone, which is the reverse reaction of TH, allows the preparation of carbonyl compounds from alcohols, avoiding the use of hazardous high-valent metal oxides.31–35 These metal catalyzed equilibrium reactions, ruled by the redox potential of the carbonyl-alcohol couple,36 are benign and straightforward processes that prevent the tedious work-up steps of the original Al iso-propoxide procedures.37–41 It is worth noting that more recently, ruthenium TH catalysts have also been investigated in cancer therapy aiming to disturb the NADH/NAD+ and pyruvate/lactate redox homeostasis.42–45 Examples of cyclometalated ruthenium TH catalysts46 are [RuCl(C^P)(NN)(CO)],47,48 [RuCl(C^NCH)(NN)(PPh3)] (NCH = N-heterocyclic carbene),49 [(p-cymene)Ru(C^N)(MeCN)](PF6),50 [(p-cymene)RuCl(C^P)],51 [(p-cymene)RuX(C^NHC)],52 [RuCl(P^C^P)(PPh3)Cl],53 [RuCl(C^NN)(PP)],54–57 [Ru(C^NO)(CO)(PPh3)2],58 whereas only few Oppenauer-type oxidation catalysts have been described, namely [RuCl(C^N)(CO)2]2,59 [RuH(C^NCH)(CO)(PPh3)2].60 In addition, [(p-cymene)RuCl(C^N)],6 [RuCl(C^NN)(PP)],61 were found active in the N-alkylation of aromatic amines with alcohols via borrowing hydrogen. Interestingly, the derivatives [Ru(C^N)(η2-OAc)(PPh3)2], isolated by Ackermann, displays catalytic activity in the meta C–H functionalization of aromatic compounds, whereas the diphosphine [Ru(C^N)(η2-OAc)(DPEPhos)] appears not active.62 As regards the synthesis of cyclometalated ruthenium complexes, a general procedure entails the cleavage of a C–H bond of a potentially bidentate aromatic ligand in the presence of an external or internal base. Facile C–H metalation is observed by addition of NaOAc to ruthenium chloride derivatives or using acetate ruthenium precursors, via a concerted carboxylate-assisted deprotonation process.63–66
Herein, we report the straightforward preparation of cyclometalated ruthenium complexes [Ru(C^N)(η2-OAc)(dppb)] containing phenyl substituted N-heterocycles, their catalytic activity in Oppenauer-type oxidation of alcohols and TH of carbonyl compounds, and their cytotoxicity against glioblastoma U87 cancer cell line.
Discussion and results
Synthesis and characterization of cyclometalated C^N diphosphine complexes
The compounds of general formula [Ru(C^N)(η2-OAc)(dppb)] (C^N = a (1); b (2); c (3); d (4)) have been easily synthesized in high yield by reaction of the precursor [Ru(η2-OAc)2(dppb)]67 with a HC^N ligand in methanol at 55 °C via elimination of acetic acid (eqn (1)). | (1) |
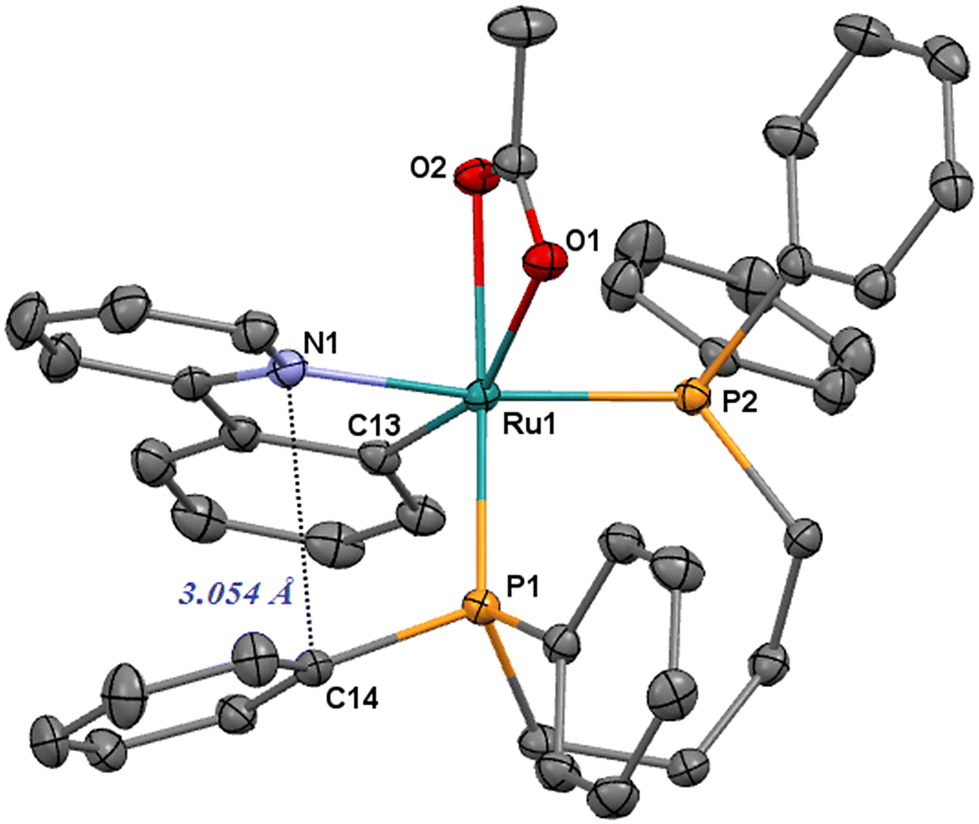 | ||
| Fig. 1 ORTEP style plot of compound 1 in the solid state (CCDC 2253559). Ellipsoids are drawn at the 50% probability level. Hydrogen atoms are omitted for clarity. | ||
The Ru–N1 bond length is 2.1030(16) Å and can be compared with that of the related terdentate [Ru(OAc)(CNN)(dppb)] (2.055(2) Å)68 and cis-[RuCl2(diphosphine)(ampy)] (2.138(2)–2.148(2) Å)28 complexes. The orthometalated ligand shows a Ru1–C13 distance of 2.0342(18) Å, in line with that observed for [Ru(OAc)(CNN)(dppb)] (2.060(3) Å), whereas the Ru1–P1 and Ru1–P2 distances (2.2292(5) and 2.2814(5) Å) are in the range of phosphine ruthenium compounds (2.26–2.41 Å).69–73 The η2 acetate displays Ru1–O1 and Ru1–O2 lengths of 2.2582(13) and 2.2242(13) with the C13–Ru–O1 and C13–Ru–O2 angles of 158.30° and 102.61°, respectively. Similarly to the CNN complexes, there is also a relatively short contact between the pyridine nitrogen atom and the ipso carbon C14 of a phosphine phenyl (3.054 Å), indicating a stacking between the Py and Ph rings (Fig. 1).
Complex [Ru(b)(η2-OAc)(dppb)] (2) was isolated as a bright orange precipitate in 71% yield (4 h), following the procedure used for 1, employing benzo[h]quinoline (Hb) in place of 2-phenylpyridine (eqn (1)). Despite the similar structural features of 1 and 2, the latter proven to be soluble in toluene and in heated 2-propanol or acetone and poorly soluble in CH2Cl2. The 31P{1H} NMR spectrum of 2 in toluene-d8 displays two doublets at δ = 57.9 and 54.1 ppm with a 2JPP of 37.2 Hz. The 1H NMR spectrum of 2 shows a π-stacking between a phenyl and the benzo[h]quinoline ligand, with well resolved diastereotopic CH2 signals of dppb. The proton in α position to the cyclometalated benzo[h]quinoline (H11 of Fig. 5) couples with two P atoms, in agreement with a fac RuCP2 arrangement. Conversely, the ortho pyridine proton (H2) gives a coupling only with the P atom (δ = 54.1 ppm) trans to N, as inferred from a 31P–1H 2D NMR HMBC experiment, whereas the doublet of doublets at δC = 181.8 ppm (2JCP of 18.2 and 8.8 Hz) is for the cyclometalated carbon. The X-ray structure of 2 is reported in Fig. S71 of ESI.†
Treatment of [Ru(η2-OAc)2(dppb)] with 1-phenylpyrazole (Hc) gives the derivative [Ru(c)(η2-OAc)(dppb)] (3) as a pale-yellow product in 12 h (eqn (1)). Also for 3, an interaction between one phenyl and the heterocyclic ring has been observed by NOE effect between the ortho phenyl protons at δ = 6.72 ppm and the pyrazole proton at δ = 6.11 ppm (H4 of Fig. 5). In addition, the resonance at δC = 161.8 ppm (dd, 2JCP = 18.2 and 8.4 Hz) is for the cyclometalated carbon, significantly upfield shifted compared to 1 and 2 (Δδ > 20 ppm). The X-ray structure of 3 is depicted in Fig. 2 and shows a ruthenium center in a distorted octahedral environment with Ru–N, –P and –O bond lengths similar to those of 1.
A stacking between the pyrazole and a phenyl ring is also observed, as confirmed by a short contact of the nitrogen atom and the ipso carbon atom C12 (3.072 Å, Fig. 2).
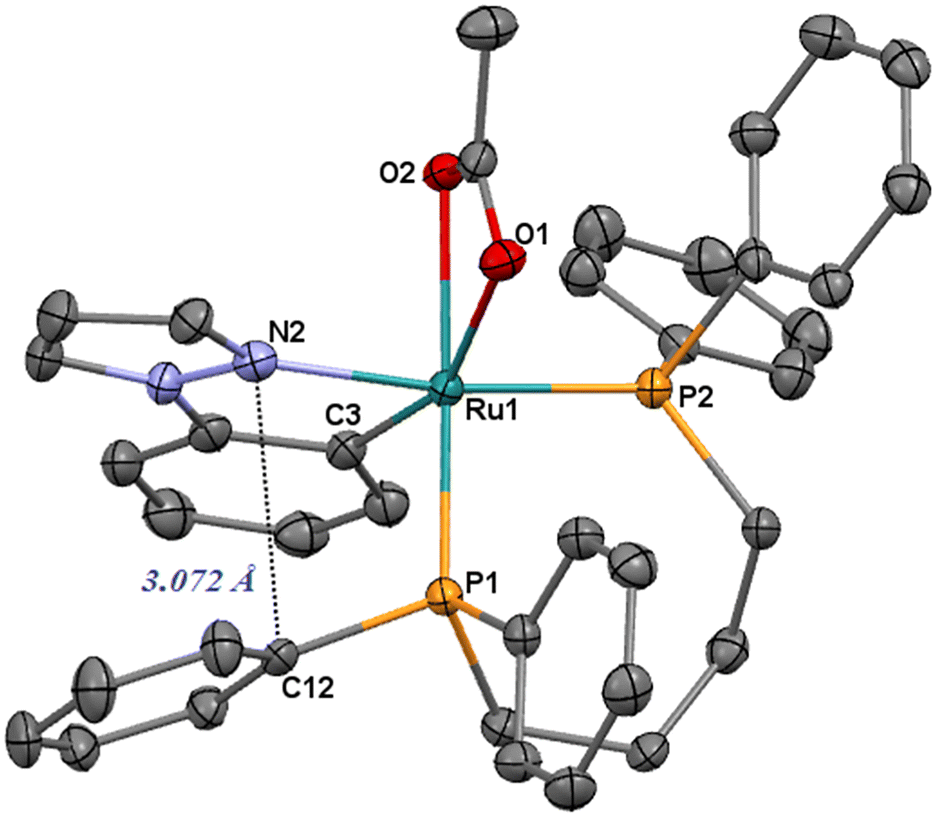 | ||
| Fig. 2 ORTEP style plot of compound 3 in the solid state (CCDC 2253560). Ellipsoids are drawn at the 50% probability level. Hydrogen atoms are omitted for clarity. | ||
Compound [Ru(d)(η2-OAc)(dppb)] (4) was prepared from [Ru(η2-OAc)2(dppb)] and 2-phenyl-2-oxazoline (Hd) in methanol at reflux for 12 h (eqn (1)). By difference from the previous syntheses, the addition of the oxazoline ligand leads to a soluble cyclometalated complex that has been isolated as a bright yellow product in 65% yield, by precipitation at −4 °C. The derivative 4 has proven to be remarkably soluble in a variety of solvents, including CH2Cl2, toluene, 2-propanol and slightly soluble in diethyl ether. The 31P{1H} NMR spectrum of 4 in CD2Cl2 shows two doublets at δ = 60.5 and 53.7 ppm (2JPP = 39.3 Hz). In the 1H NMR spectrum, two ortho protons of one phenyl are upfield shifted at δ = 6.28 ppm and display a NOE interaction with one CH2 proton (H5 of Fig. 5) of the oxazoline ring. A strong coupling between the proton in α position to the cyclometalated carbon (H10) with the two phosphorus atoms is inferred from a 31P–1H 2D NMR HMBC analysis. Finally, the 13C{1H} NMR signal of the cyclometalated carbon atom appears as a doublet of doublets at δ = 184.2 ppm (2JCP = 18.1 Hz, 2JCP = 9.5 Hz). These data are consistent with the X-ray analysis of 4 showing a distorted octahedral geometry with a contact (3.076 Å) between the nitrogen atom and the ipso carbon C25 (Fig. 3).
 | ||
| Fig. 3 ORTEP style plot of compound 4 in the solid state (CCDC 2253558). Ellipsoids are drawn at the 50% probability level. Hydrogen atoms are omitted for clarity. | ||
The facile cyclometalation of nitrogen-containing heterocycles on the ruthenium acetate phosphine precursor is in accordance with the studies of Dixneuf et al. on the arene ruthenium complexes where the acetate promotes the C–H bond activation/deprotonation.74
The formate complex [Ru(b)(η2-HCO2)(dppb)] (5) can be easily prepared in 75% isolated yield by protonation of 2 with formic acid (5 equiv.) in toluene at RT within 30 min and elimination of acetic acid (eqn (2)).
 | (2) |
Oppenauer-type oxidation of alcohols to ketones catalyzed by complexes 1–4
Secondary alcohols can be oxidized to ketones under mild reaction conditions using acetone, cyclohexanone, acetophenone and benzoquinone as hydrogen acceptors.36 The reaction is generally carried out in acetone, toluene or poor hydrogen donor alcohols such as t-BuOH or MeOH as solvents and in the presence of ruthenium catalysts. The nature of the base (KOtBu, K2CO3) required to generate ruthenium hydride species is crucial since carbonyl compounds are generally sensitive to basic media, leading to side reactions with concomitant detrimental effects on selectivity. Compounds 1–4 have been found active in the Oppenauer-type oxidation of secondary alcohols to ketones using acetone or cyclohexanone at S/C = 500–1000 (eqn (3)). | (3) |
| Entry | Oxidant | Solvent | T [°C] | S/C | Base | Time [h] | Conv.b [%] | Ketonec [%] | TOFd [h−1] |
|---|---|---|---|---|---|---|---|---|---|
| a Reaction conditions: α-tetralol (1.0 mmol), acetone (10 equiv.), solvent (10 mL total volume). b Yields are determined by GC analysis. c No by-products have been observed. d TOF turnover frequency (moles of alcohol converted to ketone per mole of catalyst per hour) at 50% conversion. e Cyclohexanone (6 equiv.) in place of acetone. | |||||||||
| 1 |
|
t-BuOH | 82 | 500 | KOtBu | 3 | 71 | 71 | 250 |
| 2 |
|
Toluene | 110 | 1000 | KOtBu | 1 | 98 | 98 | 2000 |
| 3 |
|
Toluene | 110 | 1000 | K2CO3 | 1 | 0 | 0 | — |
| 4e |
|
Toluene | 110 | 1000 | KOtBu | 4 | 97 | 97 | 200 |
Lowering the reaction temperature to 60 °C affords only 10% of conversion in 24 h, while no reaction has been observed with K2CO3 as weak base (entry 3). Employment of cyclohexanone (6 equiv.) leads to the oxidation of α-tetralol (97% conversion) in 4 h (entry 4), indicating that acetone and cyclohexanone are suitable hydrogen acceptors with 2. Under the optimal reaction conditions (i.e. acetone, KOtBu 5 mol%, toluene at reflux), complexes 1, 3 and 4 have been tested in the oxidation of several secondary alcohols. Thus, α-tetralol is efficiently oxidized with 1, 3, 4 within 15 and 20 min achieving a TOF values of 12000 h−1 for the oxazoline derivative 4 (S/C = 1000) (entries 1–3 of Table 2).
| Entry | Complex | Substrate | Oxidant | S/C | Time [min] | Conv.b [%] | Ketone (isolated yield) [%] | By-products [%] | TOFc [h−1] |
|---|---|---|---|---|---|---|---|---|---|
| a Reaction conditions: alcohol (1.0 mmol), acetone (10 equiv.), toluene (10 mL total volume). b Yields are determined by GC analysis. c TOF turnover frequency (moles of alcohol converted to ketone per mole of catalyst per hour) at 50% conversion. d Cyclohexanone (6 equiv.) in place of acetone. | |||||||||
| 1 | 1 |
|
|
1000 | 20 | 98 | 98 | — | 2400 |
| 2 | 3 |
|

|
1000 | 20 | 97 | 97 | — | 4000 |
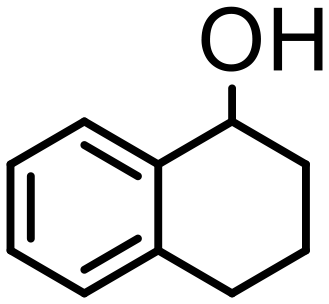
| 3 | 4 |
|
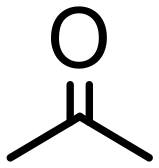
|
1000 | 15 | 98 | 98 (95) | — | 12000 |
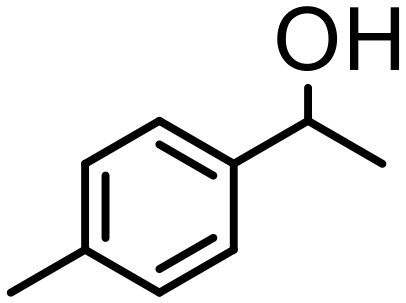
| 4 | 4 |
|
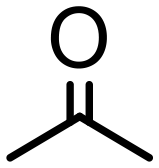
|
500 | 10 | 97 | 97 (94) | — | 4300 |
| 5 | 4 |
|
|
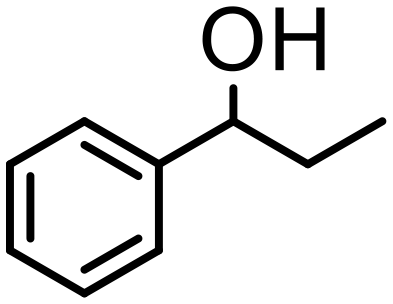
1000 | 8 h | 80 | 79 (75) | <1 | 180 |
| 6 | 4 |
|

|
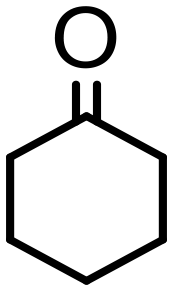
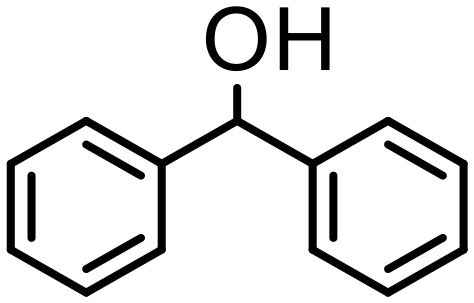
500 | 10 | 75 | 75 (71) | — | 3800 |
| 7 | 4 |
|
|
250 | 90 | 88 | 88 (83) | — | 200 |
| 8 | 4 |
|

|
250 | 120 | 52 | 50 (44) | 2 | 70 |
The most active oxazoline derivative 4 has been tested the oxidation of several alcohols. Thus, 1-(4-methy-phenyl)-ethanol is quantitatively converted in 10 min with S/4 = 500, with a TOF of 4300 h−1 (entry 4). In the presence of cyclohexanone, 1-phenylpropanol gives propiophenone (80% conv.) after 8 h (entry 5). Benzhydrol was converted to benzophenone in acetone with 75% yield at S/4 = 500 and a TOF of 3800 h−1 (entry 6), whereas 2-heptanol is oxidized to the corresponding ketone in 88% yield at S/4 = 250 in 90 min (entry 7). Finally, the bicycle (1R)-(+)-borneol is slowly oxidized to (1R)-(+)-camphor (52% conv., 2 h), on account of the high steric hindrance of the substrate that may prevent the approach at the metal center (entry 8). By using this protocol starting from 1.0 mmol of substrate and catalyst 4, the resulting ketones were purified, isolated in 44–95% yields (Table 2) and characterized by NMR (see Fig. S44–S55†).
TH of carbonyl compounds to alcohols catalyzed by complexes 1–4
Compounds 1–4 have been tested in the TH of carbonyl compounds to alcohols at S/C = 1000, using 2-propanol as hydrogen donor and solvent at reflux and in the presence of NaOiPr (2 mol%) (eqn (4)).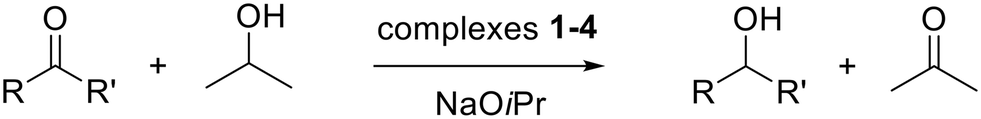 | (4) |
| Entry | Complex | Substrate | Time [h] | Conv.b [%] | Alcohol (isolated yield) [%] | By-products [%] | TOFc [h−1] |
|---|---|---|---|---|---|---|---|
| a Reaction conditions: carbonyl compound (1.0 mmol), NaOiPr (2 mol%), 2-propanol (10 mL total volume). b Yields are determined by GC analysis. c TOF turnover frequency (moles of carbonyl compound converted to alcohol per mole of catalyst per hour) at 50% conversion. d K2CO3 (5 mol%) in place of NaOiPr. e Small amounts of benzoic acid. | |||||||
| 1 | 1 |
|
8 | 75 | 74 | <1 | 250 |
| 2 | 2 |
|
2 | 96 | 96 | — | 4000 |
| 3 | 3 |
|
7 | 99 | 99 | — | 2500 |
| 4 | 4 |
|
2 | 97 | 97 (94) | — | 4300 |
| 5 | 4 |
|
0.5 | 96 | 96 (91) | — | 14000 |
| 6 | 4 |
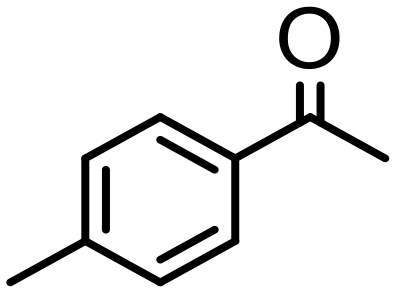
|
0.5 | 97 | 97 (89) | — | 14300 |
| 7 | 4 |
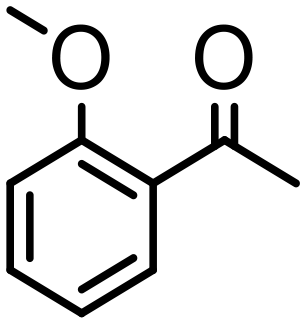
|
10 | 99 | 98 (92) | <1 | 350 |
| 8 | 4 |
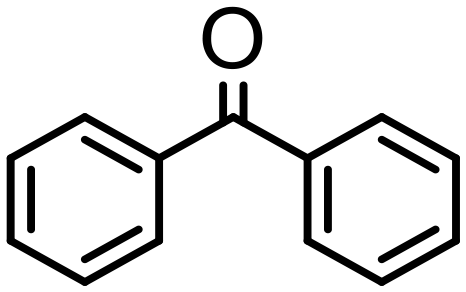
|
8 | 99 | 99 (90) | — | 1300 |
| 9 | 4 |
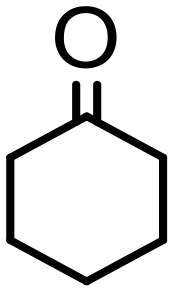
|
0.25 | 99 | 99 (95) | — | 4800 |
| 10 | 4 |
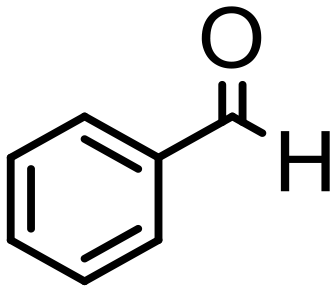
|
48 | 80 | 78 (72) | 2e | 25 |
Complex 2 containing the benzo[h]quinoline ligand gives complete conversion in 2 h with TOF of 4000 h−1, showing a significant higher activity with respect to 1, demonstrating that small changes in the C^N ligand can strongly affect the activity of the catalyst (entries 1–2). This may be ascribed to a possibly stronger π-stacking in solution between the benzo[h]quinoline and the acetophenone phenyl rings. As regards the pyrazole 3 and oxazoline 4 derivatives, both complexes afford quantitative reduction of acetophenone, 4 displaying a higher rate and shorter reaction time (TOF = 4300 h−1, 2 h) (entries 3 and 4). Under these catalytic conditions and in the absence of ruthenium complexes, poor reduction of acetophenone (<2%, 1 h) has been observed, in agreement with the data of Le Page and James, who showed complete formation of 1-phenyletanol with NaOH in high amount (34 mol%) after one day.75 Compound 4, which shows the highest activity in the Oppenauer-type oxidation, has been chosen to broaden the scope of the TH. With 4 (S/C = 1000), the substrates 2′- and 4′-methylacetophenone are rapidly reduced to the corresponding alcohols (96 and 97% conv.) in 30 min with TOF up to 14300 h−1 (entries 5 and 6). Conversely, the TH of 2′-methoxyacetophenone requires 10 h (TOF = 350 h−1), possibly due to the chelating effect of the methoxy group, which may retard the alkoxide dissociation from the ruthenium center (entry 7). Benzophenone is also quantitatively converted to benzhydrol in 8 h, whereas reduction of cyclohexanone is attained in 15 min with a TOF of 4800 h−1 (entries 8 and 9). Finally, benzaldehyde in the presence of the weak base to K2CO3 (5 mol%) gives benzyl alcohol in 80% yield in 2 days (entry 10). Following this protocol, starting from 1.0 mmol of substrate with 4, the obtained alcohols were purified, isolated in 72–94% yields (Table 3) and characterized by NMR (see Fig. S56–S69†).
Attempts to isolate hydride Ru complexes involved in catalysis failed. NMR experiments carried out using 4 with of NaOiPr (2 equiv.) in 2-propanol/toluene-d8 (4/1 in volume) reveal no reaction at RT after 30 min. Conversely, by heating this solution at reflux for 5 min, 4 is completely converted to several uncharacterized hydride species (i.e. triplet at δH = − 18.8 ppm with 2JHP = 31.4 Hz for a fac RuHP2 arrangement) (see Fig. S40†). Apparently, no dihydride ruthenium complexes have detected on the basis on 1H–1H 2D NMR COSY experiments, suggesting that the cyclometalated moiety is maintained. Thus, for the mechanism of the Oppenauer-type oxidation reaction it is likely that the catalysis follows an inner-sphere pathway.76 Thus, reaction of the cyclometalated ruthenium [Ru(C^N)(η2-OAc)(dppb)] complex with the alkoxide KOR, formed from the substrate (ROH) and KOtBu, affords the coordinatively unsaturated species [Ru(C^N)(OR)(dppb)]. Subsequent β-H elimination, involving a cis vacant site,77,78 gives the ruthenium hydride [Ru(C^N)(H)(dppb)] with extrusion of the ketone product.79,80 Coordination of acetone at the Ru center, followed by insertion into the Ru–H bond, affords [Ru(C^N)(OiPr)(dppb)]. Finally, protonation of the Ru(OiPr) species with ROH leads to 2-propanol and [Ru(C^N)(OR)(dppb)], closing the cycle. The catalytic TH of carbonyl compounds, which is the reverse of the Oppenauer-type oxidation of alcohols, is likely to occur through the same mechanism, based on the microscopic reversibility. Reaction of [Ru(C^N)(η2-OAc)(dppb)] with NaOiPr gives [Ru(C^N)(OiPr)(dppb)] that converts to the hydride [Ru(C^N)(H)(dppb)]. Subsequent reduction of the substrate leads to the alkoxide [Ru(C^N)(OR)(dppb)] that upon protonation by 2-propanol gives [Ru(C^N)(OiPr)(dppb)] and the alcohol product. It is worth pointing out that the related cyclometalated pincer complexes [Ru(C^NN)X(dppb)] (X = carboxylate),68 displaying a NH2 function, show extremely high activity and involve an outer sphere mechanism.55
Cytotoxic activity of complexes 1–4
The effects of complexes 1–4 on the metabolic activity has been evaluated against two cell lines, namely human glioblastoma U87 MG and immortalized human cortical astrocytes, via MTT assay. Glioblastoma is the most common primary malignant tumour of the central nervous system with an average median survival of less than one year.81 Glioblastoma, characterized by high invasiveness and proliferation, cellular and metabolic heterogeneity, development of resistance and recurrence,82 is one of the most aggressive and difficult to treat tumour. The current standard treatment includes debulking surgery, radiotherapy, and adjunct chemotherapy with temozolomide (TMZ), which acts by methylation of N7 and O6 guanine and on O3 adenine sites in genomic DNA, triggering cytotoxicity and apoptosis.83 However, this methylation is detected in only 45% of patients84 and given the poor survival rate of the current standardized treatment, new therapeutic approaches are needed. As shown in Fig. 4, compounds 1 and 2 display a significant high cytotoxic activity in U87 and astrocyte lines in the concentration range of 10 to 5 μM.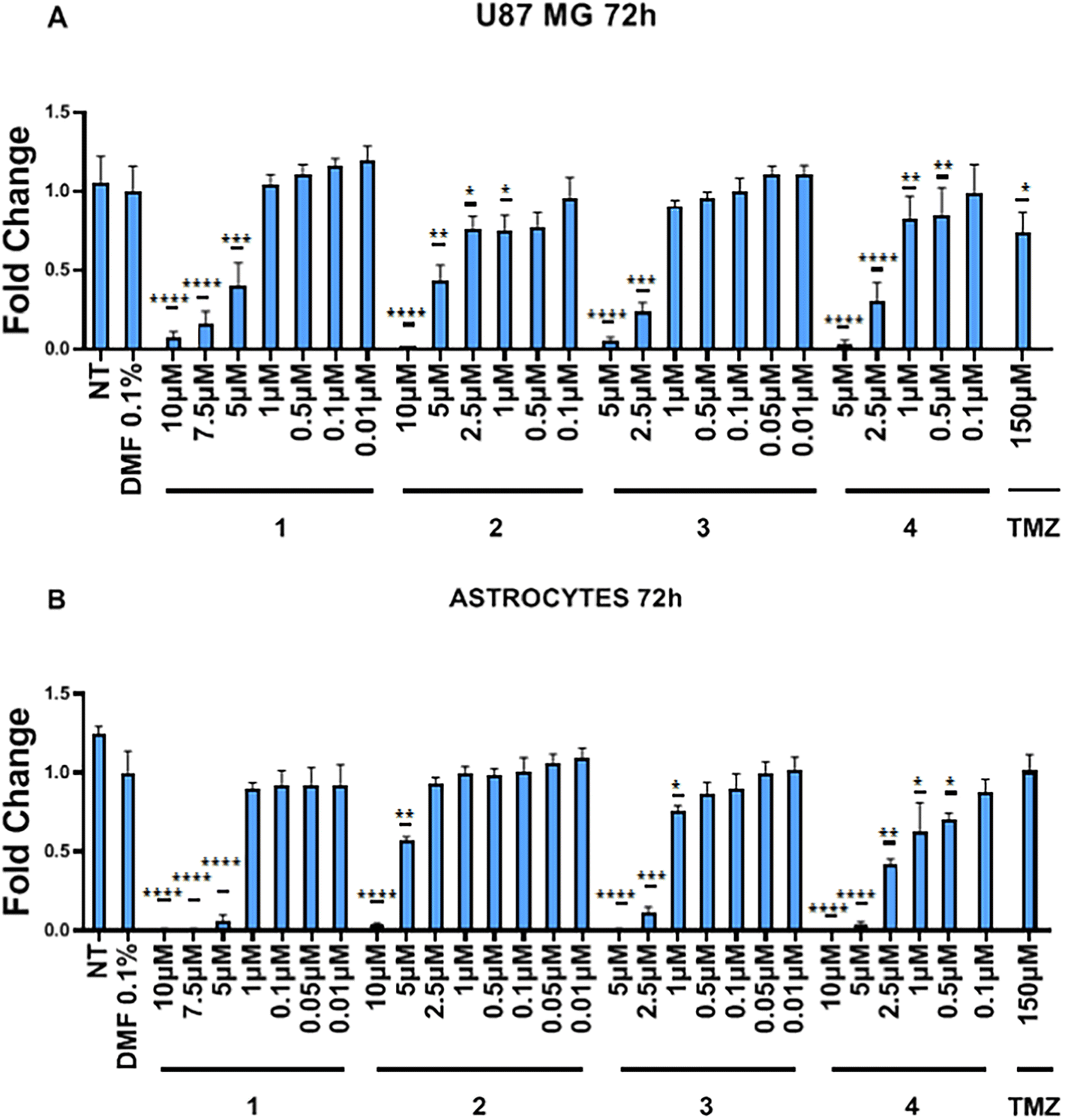 | ||
| Fig. 4 Effect of complexes 1–4 on cell metabolic activity in U87 MG (panel A) and astrocytes (panel B) cells. Cell metabolic activity was evaluated by MTT assay at 72 h and expressed as percentage of control (DMF 0.1%). Data are presented as mean ± SD (n = 6). * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001 by Student's T-test. | ||
Intriguingly, at lower concentrations (2.5–1 μM), complex 2 displays a certain degree of selectivity against neoplastic vs. astrocyte cells. The derivative 3 exhibits cytotoxicity in the concentration range from 5 to 2.5 μM for the two cell lines and shows a toxic effect on astrocytes even at 1 μM concentration. The derivative 4, which shows the highest cytotoxic activity for the two cell lines, significantly reduces the cell viability as low as to 0.5 μM concentration. Overall, the derivatives 1–4 exhibit toxic effects at concentrations ranging from 10 to 0.5 μM, with 2 showing a slight selectivity against neoplastic cells at concentrations lower than 2.5 μM. The IC50 values of these compounds on U87 and astrocytes are summarized in Table 4.
Treatment of the U87 with TMZ, which is the standard drug used for the glioblastoma treatment, affords a IC50 value significantly higher (>150 μM) compared to those for 1–4, whereas the astrocytes viability is apparently not affected at the same TMZ concentration (see Fig. S74†). These data can be compared with those reported in literature for the U87 cell lines viability in the presence of TMZ (100 μM)85 and cisplatin (54.1 μM),86 the latter suffering from a nonselective distribution between normal and tumor tissues and is associated to severe adverse side effects, including nephro- and neurotoxicity. The values obtained from the ruthenium acetate 1–4 can be compared to those of the related cationic acetate complexes [Ru(η1-OAc)(CO)(dppb)(phenanthroline)]OAc, which show cytotoxicity against anaplastic thyroid cancer cell lines with IC50 values ranging from 3.10 to 0.09 μM.87
Conclusions
In summary, we have reported a straightforward preparation of a series of cyclometalated acetate ruthenium complexes [Ru(C^N)(η2-OAc)(dppb)] obtained in high yield from the precursor [Ru(η2-OAc)2(dppb)] and phenyl substituted N-heterocycles (i.e. pyridine, pyrazole, oxazoline) via elimination of HOAc. Protonation with formic acid leads to substitution of the acetate without release of the HC^N ligand. These derivatives in the presence of base display both catalytic activity in the Oppenauer-type oxidation of secondary alcohols to ketones and in the TH of carbonyl compounds to alcohols at S/C = 1000 using acetone or cyclohexanone and 2-propanol, as hydrogen acceptor and donor, respectively. These complexes show cytotoxicity against U87 cancer cell line with IC50 values between 1.4 to 4.1 μM. Studies are ongoing to extend this class of cyclometalated ruthenium complexes for applications in catalytic organic reactions and in medicinal chemistry.Experimental
General
All reactions were carried out under an argon atmosphere by using standard Schlenk techniques. The precursor [Ru(η2-OAc)2(dppb)] was prepared according to literature procedures.67 Ligands such as dppb, 2-phenylpyridine, benzo[h]quinoline, 1-phenylpyrazole, 2-phenyl-2-oxazoline and all other chemicals were purchased from Sigma-Aldrich, Strem and Merck, and used without further purification. NMR measurements were recorded on a Bruker Advance III HD NMR 400 spectrometer and the chemical shifts, in ppm, are relative to TMS for 1H and 13C{1H} NMR and 85% H3PO4 for 31P{1H} NMR. The atom-numbering scheme for the NMR assignment of the C^N ligands a–d in the ruthenium complexes is presented in Fig. 5. Elemental analyses (C, H, N) were carried out with a Carlo Erba 1106 elemental analyzer. GC analyses were performed with a Varian CP-3380 gas chromatograph equipped with a 25 m length MEGADEX-ETTBDMS-β chiral column with hydrogen (5 psi) as the carrier gas and flame ionization detector (FID).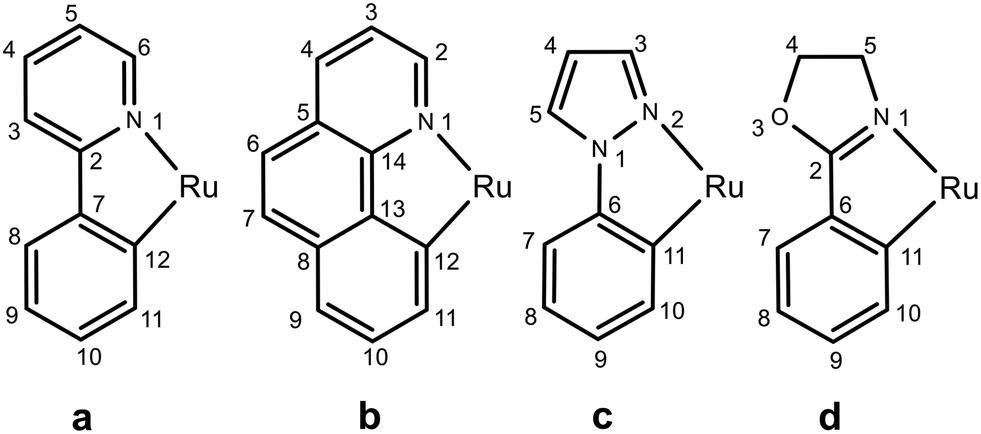 | ||
| Fig. 5 NMR numbering scheme of the C^N ligands a–d in the [Ru(C^N)(η2-OAc)(dppb)] complexes. | ||
Synthesis of [Ru(a)(η2-OAc)(dppb)] (1)
[Ru(η2-OAc)2(dppb)] (64.5 mg, 0.110 mmol) and 2-phenylpyridine (Ha) (18.8 mg, 0.121 mmol, 1.1 equiv.) were dissolved in 2.0 mL of methanol and the mixture was stirred at 55 °C for 4 h. The yellow precipitate obtained was filtered, washed with cold methanol (3 mL), diethyl ether (5 mL), n-pentane (2 mL) and dried under reduced pressure. Yield: 60.0 mg (74%). Elemental analysis calc. (%) for C41H39NO2P2Ru (740.79): C 66.48, H 5.31, N 1.89; found: C 66.61, H 5.40, N 1.88. 1H NMR (400.1 MHz, CD2Cl2, 298 K): δ = 8.47 (br d, 3JHH = 5.6 Hz, 1H; H6), 8.08 (br s, 2H; Ph), 7.85 (br t, 3JHH = 8.2 Hz, 2H; Ph), 7.57 (m, 3H; Ph), 7.44 (td, 3JHH = 7.4 Hz, 4JHH = 1.2 Hz, 2H; Ph), 7.40–7.31 (m, 4H; H5, H8 and Ph), 7.30–7.24 (m, 3H; H3 and Ph), 7.23–7.14 (m, 3H; H11 and Ph), 6.79 (m, 3H; H4, H9 and Ph), 6.57 (td, 3JHH = 8.7 Hz, 4JHH = 1.8 Hz, 2H; Ph), 6.55–6.49 (m, 1H; H10), 5.90 (t, 3JHH = 8.2 Hz, 2H; Ph), 2.98 (pseudo-q, J = 12.0 Hz, 1H; PCH2), 2.69 (tt, JHP = 13.6 Hz, JHH = 3.5 Hz, 1H; PCH2), 2.39–1.79 (m, 5H; PCH2CH2), 1.60–1.45 (m, 1H; CH2), 1.22 ppm (s, 3H; CH3CO). 13C{1H} NMR (100.6 MHz, CD2Cl2, 298 K): δ = 183.9 (s; COCH3), 183.8 (dd, 2JCP = 18.6 Hz, 2JCP = 6.7 Hz; C12-Ru), 164.5 (s; ipso-C2), 149.1 (s, C6), 147.6 (d; JCP = 4.4 Hz; C11), 147.0 (s; ipso-C7), 141.4 (d, 1JCP = 33.0 Hz; ipso-Ph), 139.6 (d, 1JCP = 43.1 Hz; ipso-Ph), 136.2 (d, 1JCP = 35.9 Hz; ipso-Ph), 135.6–125.9 (m; phenyl carbon atoms), 127.7 (s, C4), 125.5 (s; C10), 123.4 (s; C8), 120.2 (d, JCP = 2.2 Hz; C5), 118.7 (s; C9), 117.4 (d, JCP = 1.4 Hz; C3), 30.5 (d, 1JCP = 24.9 Hz; PCH2), 27.1 (d, 1JCP = 30.4 Hz; PCH2), 25.8 (s; CH2), 24.2 (s; OCOCH3), 22.1 ppm (s; CH2). 31P{1H} NMR (162.0 MHz, CD2Cl2, 298 K): δ = 57.7 (d, 2JPP = 37.4 Hz), 51.5 ppm (d, 2JPP = 37.4 Hz).Synthesis of [Ru(b)(η2-OAc)(dppb)] (2)
The complex 2 was prepared following the procedure described for 1, using benzo[h]quinoline (Hb) (21.7 mg, 0.121 mmol, 1.1 equiv.) in place of 2-phenylpyridine and affording an orange product. Yield: 59.6 mg (71%). Elemental analysis calc. (%) for C43H39NO2P2Ru (764.81): C 66.53, H 5.14, N 1.83; found: C 66.48, H 5.20, N 1.84. 1H NMR (400.1 MHz, toluene-d8, 298 K): δ = 8.95 (ddd, 3JHH = 4.9 Hz, 5JHH = 2.4 Hz, 4JHP = 1.5 Hz, 1H; H2), 8.26 (br s, 2H; Ph), 8.06 (td, 3JHH = 8.3 Hz, 4JHH = 1.0 Hz, 2H; Ph), 7.59 (d, 3JHH = 8.1 Hz, 1H; H11), 7.52 (td, 3JHH = 8.4 Hz, 4JHH = 1.4 Hz, 2H; Ph), 7.46 (d, 3JHH = 8.7 Hz, 1H; H6), 7.41 (br t, 3JHH = 7.5 Hz, 2H; Ph), 7.34–7.28 (m, 2H; H9 and Ph), 7.27–7.08 (m, 8H; H4, H10 and Ph), 6.93 (d, 3JHH = 8.7 Hz, 1H; H7), 6.68 (dd, 3JHH = 7.9 Hz, 4JHH = 5.2 Hz, 1H; H3), 6.29 (td, 3JHH = 7.7 Hz, 4JHH = 1.4 Hz, 1H; Ph), 6.07 (td, 3JHH = 8.5 Hz, 4JHH = 1.8 Hz, 2H; Ph), 5.62 (br t, 3JHH = 8.4 Hz, 2H; Ph), 2.83 (pseudo-q, J = 12.0 Hz, 1H; PCH2), 2.72 (tt, JHP = 13.6 Hz, JHH = 4.1 Hz, 1H; PCH2), 2.29 (m, 1H; PCH2), 2.08–1.60 (m, 4H; PCH2CH2), 1.48–1.40 (m, 1H; CH2), 1.38 ppm (s, 3H; CH3CO). 13C{1H} NMR (100.6 MHz, toluene-d8, 298 K): δ = 184.2 (s; COCH3), 181.8 (dd, 2JCP = 18.2 Hz, 2JCP = 8.8 Hz; C12–Ru), 154.5 (s; ipso-C14), 147.4 (s; C2), 145.2 (d, 3JCP = 3.4 Hz; C11), 144.0 (s; ipso-C13), 141.5 (d, 1JCP = 33.7 Hz; ipso-Ph), 138.8 (d, 1JCP = 43.0 Hz; ipso-Ph), 136.6 (d, 1JCP = 35.7 Hz; ipso-Ph), 134.8 (d, 1JCP = 36.6 Hz; ipso-Ph), 135.5–125.0 (m; phenyl carbon atoms), 133.6 (s; ipso-C8), 125.5 (s; ipso-C5), 122.3 (s; C7), 119.2 (d, 4JCP = 3.1 Hz; C3), 117.6 (s; C9), 30.8 (d, 1JCP = 25.0 Hz; PCH2), 26.6 (d, 1JCP = 30.4 Hz; PCH2), 25.8 (s; CH2), 24.3 (s; OCOCH3), 22.1 ppm (s; CH2). 31P{1H} NMR (162.0 MHz, toluene-d8, 298 K): δ = 57.9 (d, 2JPP = 37.2 Hz), 54.1 ppm (d, 2JPP = 37.2 Hz).Synthesis of [Ru(c)(η2-OAc)(dppb)] (3)
The complex 3 was prepared following the procedure described for 1, using 1-phenylpyrazole (Hc) (17.4 mg, 0.121 mmol, 1.1 equiv.) in place of 2-phenylpyridine, keeping the reaction mixture at 55 °C for 12 h and affording a pale-yellow product. Yield: 46.7 mg (58%). Elemental analysis calc. (%) for C39H38N2O2P2Ru (729.76): C 64.19 H 5.25, N 3.84; found: C 64.12, H 5.39, N 3.90. 1H NMR (400.1 MHz, CD2Cl2, 298 K): δ = 8.04 (br s, 2H; Ph), 7.88 (td, 3JHH = 8.5 Hz, 4JHH = 1.2 Hz, 2H; Ph), 7.56 (br m, 3H; Ph), 7.49–7.41 (m, 3H; H3, H5 and Ph), 7.38 (td, 3JHH = 7.8 Hz, 4JHH = 1.6 Hz, 2H; Ph), 7.30–7.25 (m, 3H; H7 and Ph), 7.24–7.17 (m, 2H; Ph), 7.06 (d, 3JHH = 8.4 Hz, 1H; H10), 6.89 (td, 3JHH = 7.3 Hz, 4JHH = 1.2 Hz, 1H; Ph), 6.86–6.77 (m, 2H; H8 and Ph), 6.71 (td, 3JHH = 7.9 Hz, 3JHH = 1.8 Hz, 2H; Ph), 6.55–6.47 (m, 1H; H9), 6.11 (br m, 1H; H4), 6.01 (t, 3JHH = 8.1 Hz, 2H; Ph), 2.98 (pseudo-q, J = 11.9 Hz, 1H; PCH2), 2.57 (tt, JHP = 13.8 Hz, JHH = 3.7 Hz, 1H; PCH2), 2.36–1.74 (m, 5H; PCH2CH2), 1.58–1.44 (m, 1H; CH2), 1.27 ppm (s, 3H; CH3CO). 13C{1H} NMR (100.6 MHz, CD2Cl2, 298 K): δ = 184.2 (s; COCH3), 161.8 (dd, 2JCP = 18.2 Hz, 2JCP = 8.4 Hz; C11–Ru), 148.0 (d; 3JCP = 3.6 Hz; C10), 145.4 (s, ipso-C6), 141.1–125.9 (m; phenyl carbon atoms), 137.0 (s; C5), 123.4 (s; C3), 122.6 (s; C9), 119.5 (s; C8), 110.7 (s; C7), 106.4 (d, JCP = 3.1 Hz; C4), 30.3 (d, 1JCP = 25.9 Hz; PCH2), 27.2 (d, 1JCP = 30.9 Hz; PCH2), 25.7 (s; CH2), 24.1 (s; OCOCH3), 22.0 ppm (s; CH2). 31P{1H}4 NMR (162.0 MHz, CD2Cl2, 298 K): δ = 58.0 (d, 2JPP = 39.1 Hz), 53.4 ppm (d, 2JPP = 39.1 Hz).Synthesis of [Ru(d)(η2-OAc)(dppb)] (4)
The complex 4 was prepared following the procedure described for 1, using 2-phenyl-2-oxazoline (Hd) (17.8 mg, 0.121 mmol, 1.1 equiv.) in place of 2-phenylpyridine and keeping the reaction mixture at 55 °C for 12 h. After cooling to −4 °C, the suspension was filtered, affording a bright yellow product, which was washed with n-heptane (2 × 3 mL), n-pentane (2 × 3 mL) and dried under reduced pressure at 55 °C for two days. Yield: 47.6 mg (59%). Elemental analysis calc. (%) for C39H39NO3P2Ru (732.76): C 63.93, H 5.36, N 1.91; found: C 63.81, H 5.40, N 1.88. 1H NMR (400.1 MHz, CD2Cl2, 298 K): δ = 8.04 (br s, 2H; Ph), 7.81 (t, 3JHH = 8.2 Hz, 2H; Ph), 7.58 (m, 3H; Ph), 7.42 (td, 3JHH = 7.2 Hz, 4JHH = 1.0 Hz, 1H; Ph), 7.34 (td, 3JHH = 7.3 Hz, 4JHH = 1.3 Hz, 2H; Ph), 7.30–7.25 (m, 3H; Ph), 7.22–7.12 (m, 3H; Ph), 7.09 (dd, 3JHH = 7.5 Hz, 4JHH = 1.3 Hz, 1H; H10), 7.03 (d, 3JHH = 7.7 Hz, 1H; H7), 6.98 (t, 3JHH = 7.0 Hz, 2H; Ph), 6.74 (t, 3JHH = 7.2 Hz, 1H; H8), 6.58 (td, 3JHH = 7.5 Hz, 4JHH = 1.4 Hz, 1H; H9), 6.24 (t, 3JHH = 7.9 Hz, 2H; Ph), 4.36 (dt, 1JHH = 10.2 Hz, 3JHH = 7.8 Hz, 1H; OCH2), 3.80–3.69 (m, 1H; OCH2), 3.74 (dt, 1JHH = 12.5 Hz, 3JHH = 10.2 Hz, 1H; NCH2), 3.55 (ddd, 1JHH = 12.7 Hz, 3JHH = 10.4 Hz, 3JHH = 6.3 Hz, 1H; NCH2), 3.03 (pseudo-q, J = 13.4 Hz, 1H; PCH2), 2.51 (tt, JHP = 13.3 Hz, JHH= 3.7 Hz, 1H; PCH2), 2.26 (pseudo-t, J = 15.3 Hz, 1H; PCH2), 2.20–1.91 (m, 3H; PCH2CH2), 1.91–1.74 (m, 1H; CH2), 1.57–1.44 (m, 1H; CH2), 1.30 ppm (s, 3H; CH3CO). 13C{1H} NMR (100.6 MHz, CD2Cl2, 298 K): δ = 184.2 (dd, 2JCP = 18.1 Hz, 2JCP = 9.5 Hz; C11–Ru), 183.6 (s; COCH3), 172.9 (s; ipso-C2), 147.8 (d; 3JCP = 4.0 Hz; C10), 143.0 (d, 1JCP = 41.1 Hz; ipso-Ph), 140.7 (d, 1JCP = 34.3 Hz; ipso-Ph), 135.9 (d, 1JCP = 37.4 Hz; ipso-Ph), 135.4 (s; ipso-C6), 134.6 (d, 1JCP = 37.9 Hz; ipso-Ph), 134.5–126.0 (m; phenyl carbon atoms), 127.1 (s; C9), 125.6 (s; C7), 118.4 (s; C8), 69.1 (s; OCH2), 49.7 (s; NCH2), 30.9 (d, 1JCP = 26.2 Hz; PCH2), 27.2 (d, 1JCP = 30.6 Hz; PCH2), 26.4 (s; CH2), 23.6 (s; OCOCH3), 22.0 ppm (s; CH2). 31P{1H} NMR (162.0 MHz, CD2Cl2, 298 K): δ = 60.5 (d, 2JPP = 39.2 Hz), 53.7 ppm (d, 2JPP = 39.2 Hz).Synthesis of [Ru(b)(η2-HCOO)(dppb)] (5)
Complex 2 (76.5 mg; 0.100 mmol) was dissolved in toluene (1 mL) and formic acid (23 mg; 0.500 mmol, 5 equiv.) was added. The orange-red solution turned promptly light orange and was stirred at room temperature for 30 min. The excess of acid was extracted from the organic phase by treatment with water (3 × 3 mL) in a separation funnel and the organic layer was dried with anhydrous Na2SO4 and concentrated (0.5 mL) under reduced pressure. Addition of diethyl ether (5 mL) afforded a yellow precipitate that was washed with n-pentane (2 × 3 mL) and dried under reduced pressure. Yield: 56.3 mg (75%). Elemental analysis calc. (%) for C42H37NO2P2Ru (750.78): C 67.19, H 4.97, N 1.87; found: C 67.01, H 5.17, N 1.86. 1H NMR (400.1 MHz, toluene-d8, 298 K): δ = 8.77 (br m, 1H; H2), 8.12 (br s, 2H; Ph), 7.91 (t, 3JHH = 8.2 Hz, 2H; Ph), 7.81 (dd, 3JHH = 8.0 Hz, JHH = 1.5 Hz, 1H; H4), 7.79 (dd, 4JHP = 3.9 Hz, 4JHP = 1.4 Hz, 1H; HCOO), 7.64 (d, 3JHH = 8.7 Hz, 1H; H6), 7.61–7.55 (m, 3H; Ph) 7.45 (td, 3JHH = 7.2 Hz, 4JHH = 1.5 Hz, 1H; Ph), 7.42–7.34 (m, 3H; H11, and Ph), 7.34–7.24 (m, 7H; H7, H9 and Ph), 7.19 (dd, 3JHH = 8.0 Hz, 4JHH = 1.5 Hz, 1H; H3) 6.99 (t, 3JHH = 7.6 Hz, 1H; H10), 6.44 (td, 3JHH = 7.4 Hz, 4JHH = 1.5 Hz, 1H; Ph), 6.15 (td, 3JHH = 7.8 Hz, 3JHH = 2.2 Hz, 2H; Ph), 5.46 (br t, 3JHH = 8.4 Hz, 2H; Ph), 3.08 (pseudo-q, J = 11.4 Hz, 1H; PCH2), 2.66 (tt, JHP = 13.3 Hz, JHH = 3.7 Hz, 1H; PCH2), 2.43 (m, 1H; PCH2), 2.26–1.86 (m, 4H; PCH2CH2), 1.66–1.55 ppm (m, 1H; CH2). 13C{1H} NMR (100.6 MHz, CD2Cl2, 298 K): δ = 179.1 (dd, 2JCP = 19.3 Hz, 2JCP = 9.0 Hz; C12–Ru), 173.5 (d, 3JCP = 18.2 Hz; HCOO), 153.7 (s; ipso-C14), 148.1 (s; C2), 144.7 (d, 3JCP = 3.9 Hz; C11), 143.4 (s; ipso-C13), 140.7 (d, 1JCP = 35.0 Hz; ipso-Ph), 138.6 (d, 1JCP = 44.2 Hz; ipso-Ph), 135.6 (d, 1JCP = 35.7 Hz; ipso-Ph), 134.8–125.1 (m; phenyl carbon atoms), 133.4 (s; ipso-C8), 125.6 (s; ipso-C5), 122.7 (s; C7), 120.0 (d, 4JCP = 2.8 Hz; C3), 117.6 (s; C9), 30.4 (d, 1JCP = 26.3 Hz; PCH2), 26.4 (d, 1JCP = 32.2 Hz; PCH2), 25.7 (s; CH2), 21.9 ppm (s; CH2). 31P{1H} NMR (162.0 MHz, CD2Cl2, 298 K): δ = 57.7 (d, 2JPP = 38.1 Hz), 51.6 ppm (d, 2JPP = 37.2 Hz).Typical procedure for the Oppenauer-type oxidation of alcohols
The ruthenium catalyst solutions used for these reactions were prepared by dissolving the complexes (1–4, 2 μmol) in toluene (2 mL). The alcohol substrate (1.0 mmol) was dissolved in 8.26 mL of toluene (when acetone was used as proton acceptor) or 8.38 mL (when cyclohexanone was used)), and the catalyst solution (1.0 mL, 1.0 μmol) and KOtBu (5.6 mg, 0.05 mmol) were added. After heating at reflux, acetone (740 μL, 580 mg, 10 mmol) or cyclohexanone (621 μL, 588.8 mg, 6.0 mmol) were added (final volume 10 mL). The reaction was sampled by removing an aliquot of the reaction mixture, which was quenched by addition of diethyl ether (1:1 v/v), filtered over a short silica pad and submitted to GC analysis. The ketone addition was considered as the start time of the reaction. The S/C molar ratio was 1000/1, whereas the base concentration was 5 mol% respect to the alcohol substrates (0.1 M). The same procedure was followed for the Oppenauer-type oxidation reactions with different S/C (250–1000), using the appropriate amount of catalyst.
For the isolation of ketones with 4, the final mixture was filtered over a short silica pad and the solvent was evaporated under reduced pressure. The crude residue was dissolved with diethyl ether (5 mL) and the organic layer washed with a diluted solution of HCl (0.1 M; 3 × 3 mL), dried over anhydrous Na2SO4, and the solvent gently evaporated, affording the ketone products. Further purification of the products was attained by flash silica gel column chromatography, using petroleum/ethyl acetate as eluent, (yields: 44–95%). Camphor, on the other hand, was purified through a sublimation process.
Typical procedure for the TH of aldehydes and ketones
The ruthenium catalyst solutions used for the catalytic TH were prepared by dissolving the complexes (1–4, 2 μmol) in 2-propanol (2 mL). The catalyst solution (1.0 mL, 1.0 μmol) and a 0.1 M solution of NaOiPr (200 μL, 20 μmol) in 2-propanol were added subsequently to the carbonyl substrate (1.0 mmol) dissolved in 2-propanol (final volume 10 mL), and the mixture was heated at reflux. The reaction was sampled by removing an aliquot of the reaction mixture, which was quenched by addition of diethyl ether (1:1 v/v), filtered over a short silica pad and submitted to GC analysis. The base addition was considered as the start time of the reaction. The S/C molar ratio was 1000/1, whereas the base concentration was 2 mol% respect to the ketone substrates (0.1 M). The same procedure was followed for the TH reactions with different S/C, using the appropriate amount of catalyst.
For the isolation of alcohols with 4, the final mixture was filtered over a short silica pad and the solvent was evaporated under reduced pressure. The crude residue was dissolved with diethyl ether (5 mL) and the organic layer washed with a diluted solution of HCl (0.1 M; 3 × 5 mL), dried over anhydrous Na2SO4, and the solvent gently evaporated, affording the alcohol products. Further purification was attained by flash silica gel column chromatography, using petroleum ether 40–60 °C/ethyl acetate or chloroform/methanol as eluents (yields: 72–94%).
Cell culture
Human immortalized astrocytes (Innoprot, P10251-IM) were seeded in poly-(L)-lysine (Merck) coated 100 mm dishes (Falcon) at 5000 cells per cm2 density. Cells were grown in astrocyte medium supplemented with 10% fetal bovine serum (FBS), 1% astrocyte growth factor and 1% penicillin/streptomycin (Innoprot) until they reached 70% confluency. Medium was changed every 2–3 days. Human glioblastoma line U87 MG (American Type Culture Collection, ATCC, HTB-14) were seeded at 10000 cells per cm2 density in 100 mm dishes. Cells were grown in DMEM (Euroclone) supplemented with 10% FBS (Gibco) and 1% penicillin/streptomycin (Merck) and were split when they reached 70% confluency. Medium was changed every 2–3 days. Both lines were grown in a 5% CO2 incubator with atmospheric oxygen pressure.
MTT assay
Cell lines were detached upon reaching 70% confluency with Tryple (Gibco). U87 MG and astrocytes were seeded in 200 μL transparent 96-well plates (Corning) at a density of 28000 and 42000 cells per cm2, respectively. After 24 hours, cells were treated either with compounds at different concentrations (final concentration in cell culture media ranging from 10 μM to 0.01 μM), the vehicle dimethylformamide, DMF, (stock concentration 100% down to final concentration 0.1% in cell culture media; Merck) and DMEM/astrocyte medium-only. After 72 h, 20 μL of a 4 mg mL−1 solution of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, Merck) were added to the plates and cells were incubated for another 4 h protected from light. Then, supernatant was removed and 100 μL of DMSO (PanReac AppliChem) were added. Absorbance was measured at 570 nm using an EMax Plus Microplate Reader (Molecular Devices) bundled with its software SoftMax Pro 5.2.
Statistical analysis
Statistical analysis was performed for all the experiments using GraphPAD Prism 8.4.2 software for Windows (GraphPAD software, San Diego, CA, USA). Data obtained from experiments were calculated as the mean ± SD, and significances were analysed with the Student's T test: p-values lower than 0.05 were considered statistically significant.X-ray crystallography
Single crystals of complexes 1 and 3 were obtained by slow evaporation of CH2Cl2 solutions, whereas 2 crystallizes from toluene. Crystals of 4 were formed by slow evaporation of a methanol, diethyl ether and n-pentane mixture solution. X-ray diffraction data were collected at 100 K on an X-ray single crystal diffractometer equipped with a CPAD detector (Bruker Photon-II CPAD), an IMS microsource with MoKα radiation (λ = 0.71073 Å) and a Helios optic. CCDC 2253558–2253561 contains the supplementary crystallographic data for this paper. For additional details on collection and refining of data, see the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the University of Udine. The authors thank Mr. Pierluigi Polese for the elemental analyses and Dr. Paolo Martinuzzi for NMR assistance.Notes and references
- F. Han, P. H. Choi, C. X. Ye, Y. Grell, X. L. Xie, S. I. Ivlev, S. M. Chen and E. Meggers, ACS Catal., 2022, 12, 10304–10312 CrossRef CAS
.
- I. Nakajima, M. Shimizu, Y. Okuda, R. Akiyama, R. Tadano, M. Nagaoka, N. Uemura, Y. Yoshida, T. Mino, H. Shinozaki and T. Yamamoto, Adv. Synth. Catal., 2022, 364, 1763–1768 CrossRef CAS
- Z. Wu, Z.-Q. Wang, H. Cheng, Z.-H. Zheng, Y. Yuan, C. Chen and F. Verpoort, Appl. Catal., A, 2022, 630, 118443 CrossRef CAS
- J. R. Zbieg, T. Fukuzumi and M. J. Krische, Adv. Synth. Catal., 2010, 352, 2416–2420 CrossRef CAS PubMed
- P. M. Illam and A. Rit, Catal. Sci. Technol., 2022, 12, 67–74 RSC
- P. Piehl, R. Amuso, A. Spannenberg, B. Gabriele, H. Neumann and M. Beller, Catal. Sci. Technol., 2021, 11, 2512–2517 RSC
- A. Dumas, R. Tarrieu, T. Vives, T. Roisnel, V. Dorcet, O. Baslé and M. Mauduit, ACS Catal., 2018, 8, 3257–3262 CrossRef CAS
- K. P. S. Cheung, S. Sarkar and V. Gevorgyan, Chem. Rev., 2022, 122, 1543–1625 CrossRef PubMed
- A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Chem. Rev., 2022, 122, 1485–1542 CrossRef CAS PubMed
- K. Korvorapun, J. Struwe, R. Kuniyil, A. Zangarelli, A. Casnati, M. Waeterschoot and L. Ackermann, Angew. Chem., Int. Ed., 2020, 59, 18103–18109 CrossRef CAS PubMed
- C. E. Housecroft and E. C. Constable, Coord. Chem. Rev., 2017, 350, 155–177 CrossRef CAS
- K. Yokoi, Y. Yasuda, A. Kanbe, T. Imura and S. Aoki, Molecules, 2023, 28, 1433 CrossRef CAS PubMed
- C. Licona, J.-B. Delhorme, G. Riegel, V. Vidimar, R. Cerón-Camacho, B. Boff, A. Venkatasamy, C. Tomasetto, P. da Silva Figueiredo Celestino Gomes, D. Rognan, J.-N. Freund, R. Le Lagadec, M. Pfeffer, I. Gross, G. Mellitzer and C. Gaiddon, Inorg. Chem. Front., 2020, 7, 678–688 RSC
- M. Albrecht, Chem. Rev., 2010, 110, 576–623 CrossRef CAS PubMed
- A. D. Ryabov, Chem. Rev., 1990, 90, 403–424 CrossRef CAS
- M. T. Findlay, P. Domingo-Legarda, G. McArthur, A. Yen and I. Larrosa, Chem. Sci., 2022, 13, 3335–3362 RSC
- P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879–5918 CrossRef CAS PubMed
- J. P. Djukic, J. B. Sortais, L. Barloy and M. Pfeffer, Eur. J. Inorg. Chem., 2009, 2009, 817–853 CrossRef
- C. S. G. Seo and R. H. Morris, Organometallics, 2019, 38, 47–65 CrossRef CAS
- X. Xie, B. Lu, W. Li and Z. Zhang, Coord. Chem. Rev., 2018, 355, 39–53 CrossRef CAS
- D. Wang and D. Astruc, Chem. Rev., 2015, 115, 6621–6686 CrossRef CAS PubMed
- F. Foubelo, C. Nájera and M. Yus, Tetrahedron: Asymmetry, 2015, 26, 769–790 CrossRef CAS
- G. Chelucci, S. Baldino and W. Baratta, Coord. Chem. Rev., 2015, 300, 29–85 CrossRef CAS
- J.-i. Ito and H. Nishiyama, Tetrahedron Lett., 2014, 55, 3153–3166 CAS
- W. Baratta and P. Rigo, Eur. J. Inorg. Chem., 2008, 2008, 4041–4053 CrossRef
- J. Magano and J. R. Dunetz, Org. Process Res. Dev., 2012, 16, 1156–1184 CrossRef CAS
- W. Baratta, G. Chelucci, S. Magnolia, K. Siega and P. Rigo, Chem. – Eur. J., 2009, 15, 726–732 CrossRef CAS PubMed
- W. Baratta, E. Herdtweck, K. Siega, M. Toniutti and P. Rigo, Organometallics, 2005, 24, 1660–1669 CrossRef CAS
- H. Doucet, T. Ohkuma, K. Murata, T. Yokozawa, M. Kozawa, E. Katayama, A. F. England, T. Ikariya and R. Noyori, Angew. Chem., Int. Ed., 1998, 37, 1703–1707 CrossRef CAS PubMed
- K.-J. Haack, S. Hashiguchi, A. Fujii, T. Ikariya and R. Noyori, Angew. Chem., Int. Ed. Engl., 1997, 36, 285–288 CrossRef CAS
- A. D. Böth, M. J. Sauer, W. Baratta and F. E. Kühn, Catal. Sci. Technol., 2022, 12, 5597–5603 RSC
- A. Mukherjee and S. Bhattacharya, RSC Adv., 2021, 11, 15617–15631 RSC
- L. Pardatscher, B. J. Hofmann, P. J. Fischer, S. M. Hölzl, R. M. Reich, F. E. Kühn and W. Baratta, ACS Catal., 2019, 9, 11302–11306 CrossRef CAS
- R. Labes, C. Battilocchio, C. Mateos, G. R. Cumming, O. de Frutos, J. A. Rincón, K. Binder and S. V. Ley, Org. Process Res. Dev., 2017, 21, 1419–1422 CrossRef CAS
- C. M. Nicklaus, P. H. Phua, T. Buntara, S. Noel, H. J. Heeres and J. G. de Vries, Adv. Synth. Catal., 2013, 355, 2839–2844 CrossRef CAS
- H. Adkins, R. M. Elofson, A. G. Rossow and C. C. Robinson, J. Am. Chem. Soc., 1949, 71, 3622–3629 CrossRef CAS
- J. Ballester, A.-M. Caminade, J.-P. Majoral, M. Taillefer and A. Ouali, Catal. Commun., 2014, 47, 58–62 CrossRef CAS
- C. R. Graves, B.-S. Zeng and S. T. Nguyen, J. Am. Chem. Soc., 2006, 128, 12596–12597 CrossRef CAS PubMed
- T. Ooi, T. Miura, Y. Itagaki, H. Ichikawa and K. Maruoka, Synthesis, 2002, 2002, 0279–0291 Search PubMed
- C. F. de Graauw, J. A. Peters, H. van Bekkum and J. Huskens, Synthesis, 1994, 1994, 1007–1017 CrossRef
- H. Rapoport, R. Naumann, E. R. Bissell and R. M. Bonner, J. Org. Chem., 1950, 15, 1103–1107 CrossRef CAS
- D. Lovison, D. Alessi, L. Allegri, F. Baldan, M. Ballico, G. Damante, M. Galasso, D. Guardavaccaro, S. Ruggieri, A. Melchior, D. Veclani, C. Nardon and W. Baratta, Chem. – Eur. J., 2022, 28, e202200200 CrossRef CAS PubMed
- K. Tyagi, T. Dixit and V. Venkatesh, Inorg. Chim. Acta, 2022, 533, 120754 CrossRef CAS
- S. Banerjee and P. J. Sadler, RSC Chem. Biol., 2021, 2, 12–29 RSC
- J. J. Soldevila-Barreda and P. J. Sadler, Curr. Opin. Chem. Biol., 2015, 25, 172–183 CrossRef CAS PubMed
- V. Ritleng and J. G. de Vries, Molecules, 2021, 26, 4076 CrossRef CAS PubMed
- W. Baratta, P. Da Ros, A. Del Zotto, A. Sechi, E. Zangrando and P. Rigo, Am. Ethnol., 2004, 116, 3668–3672 Search PubMed
- W. Baratta, A. Del Zotto, G. Esposito, A. Sechi, M. Toniutti, E. Zangrando and P. Rigo, Organometallics, 2004, 23, 6264–6272 CrossRef CAS
- W. Baratta, J. Schütz, E. Herdtweck, W. A. Herrmann and P. Rigo, J. Organomet. Chem., 2005, 690, 5570–5575 CrossRef CAS
- J.-B. Sortais, V. Ritleng, A. Voelklin, A. Holuigue, H. Smail, L. Barloy, C. Sirlin, G. K. M. Verzijl, J. A. F. Boogers, A. H. M. de Vries, J. G. de Vries and M. Pfeffer, Org. Lett., 2005, 7, 1247–1250 CrossRef CAS PubMed
- R. Sun, X. Chu, S. Zhang, T. Li, Z. Wang and B. Zhu, Eur. J. Inorg. Chem., 2017, 2017, 3174–3183 CrossRef CAS
- S. Bauri, S. N. R. Donthireddy, P. M. Illam and A. Rit, Inorg. Chem., 2018, 57, 14582–14593 CrossRef CAS PubMed
- P. Dani, T. Karlen, R. A. Gossage, S. Gladiali and G. van Koten, Angew. Chem., Int. Ed., 2000, 39, 743–745 CrossRef CAS PubMed
- S. Facchetti, V. Jurcik, S. Baldino, S. Giboulot, H. G. Nedden, A. Zanotti-Gerosa, A. Blackaby, R. Bryan, A. Boogaard, D. B. McLaren, E. Moya, S. Reynolds, K. S. Sandham, P. Martinuzzi and W. Baratta, Organometallics, 2016, 35, 277–287 CrossRef CAS
- W. Baratta, S. Baldino, M. J. Calhorda, P. J. Costa, G. Esposito, E. Herdtweck, S. Magnolia, C. Mealli, A. Messaoudi, S. A. Mason and L. F. Veiros, Chem. – Eur. J., 2014, 20, 13603–13617 CrossRef CAS PubMed
- W. Baratta, M. Ballico, S. Baldino, G. Chelucci, E. Herdtweck, K. Siega, S. Magnolia and P. Rigo, Chem. – Eur. J., 2008, 14, 9148–9160 CrossRef CAS PubMed
- W. Baratta, G. Chelucci, S. Gladiali, K. Siega, M. Toniutti, M. Zanette, E. Zangrando and P. Rigo, Angew. Chem., Int. Ed., 2005, 44, 6214–6219 CrossRef CAS PubMed
- J. Dutta, M. G. Richmond and S. Bhattacharya, Eur. J. Inorg. Chem., 2014, 2014, 4600–4610 CrossRef CAS
- A. Hijazi, K. Parkhomenko, J.-P. Djukic, A. Chemmi and M. Pfeffer, Adv. Synth. Catal., 2008, 350, 1493–1496 CrossRef CAS
- S. Burling, M. K. Whittlesey and J. M. J. Williams, Adv. Synth. Catal., 2005, 347, 591–594 CrossRef CAS
- S. Agrawal, M. Lenormand and B. Martín-Matute, Org. Lett., 2012, 14, 1456–1459 CrossRef CAS PubMed
- K. Korvorapun, R. Kuniyil and L. Ackermann, ACS Catal., 2020, 10, 435–440 CrossRef CAS
- Y. Boutadla, D. L. Davies, R. C. Jones and K. Singh, Chem. – Eur. J., 2011, 17, 3438–3448 CrossRef CAS PubMed
- L. Ackermann, Chem. Rev., 2011, 111, 1315–1345 CrossRef CAS PubMed
- L. Ackermann, R. Vicente, H. K. Potukuchi and V. Pirovano, Org. Lett., 2010, 12, 5032–5035 CrossRef CAS PubMed
- F. Požgan and P. H. Dixneuf, Adv. Synth. Catal., 2009, 351, 1737–1743 CrossRef
- W.-K. Wong, K.-K. Lai, M.-S. Tse, M.-C. Tse, J.-X. Gao, W.-T. Wong and S. Chan, Polyhedron, 1994, 13, 2751–2762 CrossRef CAS
- W. Baratta, M. Ballico, A. Del Zotto, E. Herdtweck, S. Magnolia, R. Peloso, K. Siega, M. Toniutti, E. Zangrando and P. Rigo, Organometallics, 2009, 28, 4421–4430 CrossRef CAS
- L. F. Szczepura, S. A. Kubow, R. A. Leising, W. J. Perez, M. H. Vo Huynh, C. H. Lake, D. G. Churchill, M. R. Churchill and K. J. Takeuchi, J. Chem. Soc., Dalton Trans., 1996, 1463–1470 RSC
- M. R. Churchill, R. F. See, C. A. Bessel and K. J. Takeuchi, J. Chem. Crystallogr., 1996, 26, 543–551 CrossRef CAS
- M. R. Churchill, L. M. Krajkowski, L. F. Szczepura and K. J. Takeuchi, J. Chem. Crystallogr., 1996, 26, 853–859 CrossRef CAS
- C. A. Bessel, R. F. See, D. L. Jameson, M. R. Churchill and K. J. Takeuchi, J. Chem. Soc., Dalton Trans., 1992, 3223–3228 RSC
- R. A. Leising, S. A. Kubow, M. R. Churchill, L. A. Buttrey, J. W. Ziller and K. J. Takeuchi, Inorg. Chem., 1990, 29, 1306–1312 CrossRef CAS
- B. Li, T. Roisnel, C. Darcel and P. H. Dixneuf, Dalton Trans., 2012, 41, 10934–10937 RSC
- M. D. Le Page and B. R. James, Chem. Commun., 2000, 1647–1648 RSC
-
S. Gladiali and R. Taras, in Modern Reduction Methods, ed. P. G. Andersson and I. J. Munslow, Wiley-VCH Verlag GmbH & Co KGaA, Weinheim, 2008, pp. 135–157 Search PubMed
- H. Itagaki, N. Koga, K. Morokuma and Y. Saito, Organometallics, 1993, 12, 1648–1654 CrossRef CAS
- H. E. Bryndza, J. C. Calabrese, M. Marsi, D. C. Roe, W. Tam and J. E. Bercaw, J. Am. Chem. Soc., 1986, 108, 4805–4813 CrossRef CAS
- Y. Matsubara, E. Fujita, M. D. Doherty, J. T. Muckerman and C. Creutz, J. Am. Chem. Soc., 2012, 134, 15743–15757 CrossRef CAS PubMed
- A. Aranyos, G. Csjernyik, K. J. Szabó and J.-E. Bäckvall, Chem. Commun., 1999, 351–352 RSC
- R. Stupp, W. P. Mason, M. J. van den Bent, M. Weller, B. Fisher, M. J. B. Taphoorn, K. Belanger, A. A. Brandes, C. Marosi, U. Bogdahn, J. Curschmann, R. C. Janzer, S. K. Ludwin, T. Gorlia, A. Allgeier, D. Lacombe, J. G. Cairncross, E. Eisenhauer and R. O. Mirimanoff, N. Engl. J. Med., 2005, 352, 987–996 CrossRef CAS PubMed
- I. Manini, F. Caponnetto, A. Bartolini, T. Ius, L. Mariuzzi, C. Di Loreto, A. P. Beltrami and D. Cesselli, Int. J. Mol. Sci., 2018, 19, 147 CrossRef PubMed
- S. Y. Lee, Genes Dis., 2016, 3, 198–210 CrossRef PubMed
- K. Zhang, X.-q. Wang, B. Zhou and L. Zhang, Fam. Cancer, 2013, 12, 449–458 CrossRef CAS PubMed
- A. A. Puca, V. Lopardo, F. Montella, P. Di Pietro, D. Cesselli, I. G. Rolle, M. Bulfoni, V. Di Sarno, G. Iaconetta, P. Campiglia, C. Vecchione, A. P. Beltrami and E. Ciaglia, Cells, 2022, 11, 294 CrossRef CAS PubMed
- G. Facchetti and I. Rimoldi, Bioorg. Med. Chem. Lett., 2019, 29, 1257–1263 CrossRef CAS PubMed
- D. Lovison, L. Allegri, F. Baldan, M. Ballico, G. Damante, C. Jandl and W. Baratta, Dalton Trans., 2020, 49, 8375–8388 RSC
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of the isolated complexes and the products of the catalysis, X-ray diffraction parameters and GC-FID chromatograms related to the catalytic reactions promoted by the ruthenium derivatives. CCDC 2253558–2253561. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cy00676j |
| This journal is © The Royal Society of Chemistry 2023 |