
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemical representation learning for toxicity prediction†‡
Jannis
Born§
 *ab,
Greta
Markert§
ac,
Nikita
Janakarajan
ad,
Talia B.
Kimber
e,
Andrea
Volkamer
ef,
María Rodríguez
Martínez¶
a and
Matteo
Manica¶
a
*ab,
Greta
Markert§
ac,
Nikita
Janakarajan
ad,
Talia B.
Kimber
e,
Andrea
Volkamer
ef,
María Rodríguez
Martínez¶
a and
Matteo
Manica¶
a
aIBM Research Europe, Zurich, Switzerland. E-mail: jab@zurich.ibm.com
bDepartment of Biosystems Science and Engineering, ETH Zürich, Switzerland
cDepartment of Chemistry and Applied Biosciences, ETH Zürich, Switzerland
dDepartment of Computer Science, ETH Zürich, Switzerland
eIn silico Toxicology and Structural Bioinformatics, Institute of Physiology, Charité-Universitätsmedizin Berlin, Charitéplatz 1, 10117, Berlin, Germany
fData Driven Drug Design, Saarland University, 66123 Saarbrücken, Germany
First published on 3rd April 2023
Abstract
Undesired toxicity is a major hindrance to drug discovery and largely responsible for high attrition rates in early stages. This calls for new, reliable, and interpretable molecular property prediction models that help prioritize compounds and thus reduce the high costs for development and the risk to humans, animals, and the environment. Here, we propose an interpretable chemical language model that combines attention with multiscale convolutions and relies on data augmentation. We first benchmark various molecular representations (e.g., fingerprints, different flavors of SMILES and SELFIES, as well as graph and graph kernel methods) revealing that SMILES coupled with augmentation overall yields the best performance. Despite its simplicity, our model is then shown to outperform existing approaches across a wide range of molecular property prediction tasks, including but not limited to toxicity. Moreover, the attention weights of the model allow for easy interpretation and show enrichment of known toxicophores even without explicit supervision. To introduce a notion of model reliability, we propose and combine two simple methods for uncertainty estimation (Monte-Carlo dropout and test-time-augmentation). These methods not only identify samples with high prediction uncertainty, but also allow formation of implicit model ensembles that improve accuracy. Last, we validate our model on a large-scale proprietary toxicity dataset and find that it outperforms previous work while giving similar insights into revealing cytotoxic substructures.
1 Introduction
The costs of research and development per new FDA-approved drug have been doubling every 9 years since 1950.1 A major bottleneck in this process is toxicity which is alone responsible for the failure of >30% of all clinical trials.2 A commonly utilized approach in lead compound design is to avoid molecules with toxicophores, i.e., substructures or chemical motifs that are likely to exert toxic effects.3 Alternatively, computational approaches can be used for tasks such as activity prediction in order to design more active and selective compounds.4 Empirically, these heuristics have proven to be limited—the success rates are steadily declining and oncological pharmaceuticals are particularly affected as only 3.4% of the clinical trials are successful.5 Even worse, oncology drugs in clinical trials often do not work by their proposed mechanism of action. Lin A. et al.6 found that when knocking out the target genes of 10 cancer drugs in clinical trials, all 10 drugs retained their efficacy through other mechanisms, suggesting that off-target toxicity is a common mechanism of action of anticancer drugs in clinical trials.Therefore, more precise computational approaches that help reduce the costs of development and the risks posed to humans and the environment throughout the process are desired. Machine learning (ML) methods have been applied in the field of quantitative structure–activity relationship (QSAR) prediction for decades.7,8 Lately, deep learning (DL) has promised a methodological turnaround toward data-driven approaches to combat the ever-growing need for new therapeutics9 and in some instances, replaced ML methods.10 Consequently, a considerable body of literature developed around molecular property and activity prediction11–15 with several studies focusing on toxicity prediction.16–21 It is widely accepted that the choice of the selected molecular representation for model building plays a crucial role in accurately predicting small molecule properties and bioactivities.22 Traditional chemoinformatics typically relied on 1D descriptors such as binary fingerprints;23 however in the past few years, chemical language models relying on the molecular inline notation SMILES24 have gained popularity for QSAR prediction models.22,25,26 But despite the ubiquitous usage of SMILES, there is no universal, canonical SMILES representation (e.g.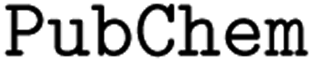 kekulizes “canonical” SMILES, whereas
kekulizes “canonical” SMILES, whereas  does not), even though several attempts toward unification have been proposed.27
does not), even though several attempts toward unification have been proposed.27
In this work, we systematically investigate the predictive power of models built on different flavors of SMILES and compare it to established chemical descriptors such as molecular fingerprints or more complex representations such as graphs. We propose a novel, uncertainty-aware and interpretable chemical language model. While this model has been developed mainly in the context of toxicity prediction, it is a generic molecular property prediction model that, as we demonstrate, exhibits excellent prediction performance across several datasets, focused on but not limited to toxicity.
Model interpretability is a heavily sought-after trait in QSAR modeling since most DL models are black-boxes. Considerable previous work has been invested to explain molecular property prediction models28 and contributions of individual parts of a molecule to its properties or effect.29,30 However, unlike previous work31,32 our model does not require any post-hoc workflow such as integrated gradients which itself suffers from limitations such as gradient saturation32 to become interpretable. Instead, we achieve high model explainability via a built-in-attention mechanism.33 This is an ante hoc interpretability method that produces attention maps as a byproduct of a prediction. Building upon our previous work26 where we demonstrated the utility of the attention mechanism in a regression task (drug sensitivity prediction) by reporting that (1) highly attended genes significantly enrich apoptotic processes and (2) molecular attention correlates strongly with similarity of functional fingerprints, we herein show how attention maps can be useful to understand the model's predictive process and find that the attention maps align, in many cases, with prior knowledge about toxicophores. Since model safety and reliability is a critical aspect of drug modeling, we borrow two simple uncertainty estimation methods proposed in related fields34,35 and provide quantitative evidence on how they can be used to identify probable misclassifications. Compared to graph-based models,13,36 our method is significantly simpler and solely relies on the SMILES chemical language. It exploits data augmentation – through the non-uniqueness of SMILES - to boost model performance and outperforms previous studies on this task.
2 Methods
2.1 Problem formulation
Let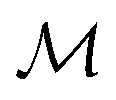 denote the molecular space and
denote the molecular space and  denote the QSAR property scores, e.g. measured toxicity of a molecule. We are interested in learning a function
denote the QSAR property scores, e.g. measured toxicity of a molecule. We are interested in learning a function  that maps a molecule to a property score. The function Φ is learned from a labelled dataset
that maps a molecule to a property score. The function Φ is learned from a labelled dataset 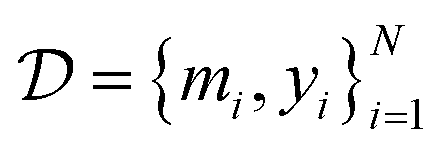 where
where  and
and 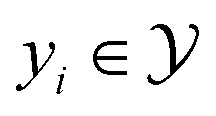 .
.
2.2 Molecular representations
Throughout this work we compare three classes of molecular representations:(1) Molecular fingerprints: molecular fingerprints are binary 1D vectors where the value at each position indicates the presence (or absence) of a certain substructure. Here, we use extended connectivity fingerprints (ECFPs23), that are based on the Morgan algorithm37 as a baseline. We use ECFP4, the most common choice in the literature,38 with 512 bits.
(2) Molecular graphs: each molecule is denoted by an undirected graph G = (V, E) where atoms denote vertices and bonds denote edges. Vertices are labelled with their atom identity (chemical element) and edges by the bond order (e.g. a single or double bond).
(3) String-based representations: we examine molecular inline notations, namely SMILES24 and SELFIES.39 Due to their recent success in QSAR prediction models26 and even epitope-related tasks,40 different types of string representations, in particular different SMILES flavors, are our main focus.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ) or triple (#). In a kekulized format (typically used by RDKit), capital letters represent non-aromatic atoms, whereas small letters represent aromatic ones. In a non-kekulized format (typically used by PubChem), aromatic atoms are not explicit, as they are represented by capital letters connected through alternating single and double bonds. Numbers are used to denote rings. One can think of it as an imaginary cut between bonds of two neighboring atoms in a ring. The ring-closing number occurs twice: once for the atom that ”opens” the ring and once for the atom that ”closes” it (e.g., cyclohexane is represented as
) or triple (#). In a kekulized format (typically used by RDKit), capital letters represent non-aromatic atoms, whereas small letters represent aromatic ones. In a non-kekulized format (typically used by PubChem), aromatic atoms are not explicit, as they are represented by capital letters connected through alternating single and double bonds. Numbers are used to denote rings. One can think of it as an imaginary cut between bonds of two neighboring atoms in a ring. The ring-closing number occurs twice: once for the atom that ”opens” the ring and once for the atom that ”closes” it (e.g., cyclohexane is represented as  ; it is important to notice that the first
; it is important to notice that the first 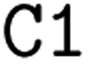 and the second
and the second  are not representing the same atom). Branches, i.e., when an atom has three or more bonds, are represented using parentheses. Hydrogen atoms are also not stated explicitly except if they are important for the stereo information of a tetrahedral center (e.g.,
are not representing the same atom). Branches, i.e., when an atom has three or more bonds, are represented using parentheses. Hydrogen atoms are also not stated explicitly except if they are important for the stereo information of a tetrahedral center (e.g.,  ). Stereoisomers have the same atomic sequence and the same molecular formula, but differ in the three-dimensional orientation of their atoms which can lead to different behaviours in chiral environments (e.g., enzymes). Stereoisomers used as drugs can have huge differences in toxicity and potencies.41 For the description of stereoisomers in SMILES, one considers the first token before the token containing
). Stereoisomers have the same atomic sequence and the same molecular formula, but differ in the three-dimensional orientation of their atoms which can lead to different behaviours in chiral environments (e.g., enzymes). Stereoisomers used as drugs can have huge differences in toxicity and potencies.41 For the description of stereoisomers in SMILES, one considers the first token before the token containing  or
or  as the chiral centre. The remaining atoms connected to the central atom are listed either by ordering them anticlockwise
as the chiral centre. The remaining atoms connected to the central atom are listed either by ordering them anticlockwise 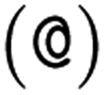 or clockwise
or clockwise 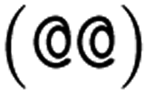 . The choice whether to use clockwise or anticlockwise notation is arbitrary (e.g.,
. The choice whether to use clockwise or anticlockwise notation is arbitrary (e.g.,  is equivalent to
is equivalent to 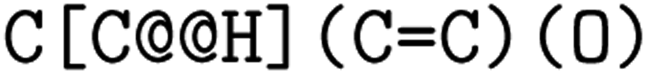 ). Generally, there are 2n possible stereoisomers, where n is the number of stereo centers. As a consequence, if the stereoinformation is removed from a molecule with two stereocenters, the SMILES string is ambiguous and could represent four different molecules. This is exemplarily depicted in Fig. A2, see ESI.‡ The same holds true for molecules with information about the bond direction (
). Generally, there are 2n possible stereoisomers, where n is the number of stereo centers. As a consequence, if the stereoinformation is removed from a molecule with two stereocenters, the SMILES string is ambiguous and could represent four different molecules. This is exemplarily depicted in Fig. A2, see ESI.‡ The same holds true for molecules with information about the bond direction ( or
or  for (E), respectively, and
for (E), respectively, and  or
or  for (Z), respectively). Here, the removal of the information about the bond direction can have greater consequences, as the SMILES string can represent molecules with very different molecular properties (again 2n, where n represents the number of explicit bond directions in the molecule). Fig. 1 gives an overview of the explored SMILES flavors. The starting point is always the raw SMILES representation as read from the data source.
for (Z), respectively). Here, the removal of the information about the bond direction can have greater consequences, as the SMILES string can represent molecules with very different molecular properties (again 2n, where n represents the number of explicit bond directions in the molecule). Fig. 1 gives an overview of the explored SMILES flavors. The starting point is always the raw SMILES representation as read from the data source.
 | ||
| Fig. 1 Explored SMILES flavors, exemplified on (S,E)-4-phenylbut-3-en-2-ol. Transforming a molecule into a string representation can be divided into four transformation groups which are executed sequentially. For visual clarity and to pronounce the relation between the SMILES sequence and the depiction, atoms are colored. | ||
Chemical transformations refer to semantic changes in the visibility of certain properties in the string and include:
(1) Canonicalization: since a molecular graph traversal is non-univocal, usually, traversed graphs start at a non-hydrogen atom and proceed in any user-defined direction. Therefore, SMILESs are non-unique representations of molecules. Canonicalization ensures that every molecule is represented by exactly one string. Here, we use canonicalization as defined in  .42 Canonicalization has the advantage of increased data uniformity.
.42 Canonicalization has the advantage of increased data uniformity.
(2) Kekulization: aromatic moieties can either be represented explicitly or implicitly. In the explicit (kekulized) version, the aromatic π-electrons are static between every second carbon. Instead, in the canonical form, the electrons are delocalized (cf.Fig. 1). The kekulized version is slightly longer but uses the same token to denote an atom, irrespective of its aromaticity.
(3) Removal of stereoinformation: to uniquely identify a molecule from a SMILES, information about the tetrahedral center or the double bond direction (E or Z) is sometimes needed. Since stereoinformation is not always explicitly denoted in datasets, it is often discarded in affected molecules for reasons of simplicity and uniformity. We experiment separately with removing chirality and bond direction. Once removed, the SMILES string is ambiguous and can represent different molecules (cf. Fig. A2, see ESI‡).
(4) Explicitness: we experimented with making hydrogen atoms or single bonds (or both) explicit in the SMILES. This not only increased sequence length but also better distributes the frequency of tokens in the vocabulary.
Randomized transformations refer to non-chemical changes in the syntax or grammar of the language. Since they are stochastic, they resemble a form of data augmentation.
(1) Augmentation: since SMILESs are non-unique, their multiplicity can be used for data augmentation and provably improves performance of predictive15,43 and generative44 models. Here, we use online augmentation which samples the graph traversal and generates the corresponding string at runtime, similar to random rotations or cropping of images. This makes it impossible to measure the actual inflation of training molecules; however as shown in Fig. A3, see ESI,‡ we empirically report the number of obtained augmentations for all Tox21 molecules. For the majority of molecules more unique SMILES can be obtained than the usual number of training epochs (100); some molecules even showed >100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 unique SMILESs.
000 unique SMILESs.
(2) Shuffling: Liu P. et al.45 observed that randomly shuffling the position of the SMILES tokens (i.e., destroying their local structure) does not significantly reduce performance in QSAR prediction tasks. Similar to augmentation, shuffling occurs as a stochastic transformation at runtime.
Language translations are optional. The default language is SMILES and the only alternative language explored in this work is SELFIES.
(1) SELFIES: SELFIES is a self-referencing chemical language that overcomes the validity problem of SMILES (i.e., random SMILES strings are not generally valid) and was devised for generative models.39
Note that most of the transformations can be combined (see appendix Fig. A1, see ESI‡ for a flowchart about their combination). The entire SMILES processing pipeline is implemented in the publicly available package  .||46
.||46
Tokenization: lastly, the obtained strings are split into tokens to ensure that each molecular entity (e.g., the atom  ) is represented as one feature vector to the model. SMILESs are split with the regular expression from Schwaller P.47 As an alternative tokenization method, we explored SMILES pair encodings (SPEs), a method inspired by byte-pair encodings that splits SMILES into substructures of varying lengths based on their occurrence in ChEMBL.48 They showed that SPE exhibits prediction accuracy comparable to that of atom-based tokenization in QSAR prediction tasks.48 SPE is ideal to handle larger molecules since it drastically reduces the number of tokens. For example, the SMILES shown in Fig. 1 is split into
) is represented as one feature vector to the model. SMILESs are split with the regular expression from Schwaller P.47 As an alternative tokenization method, we explored SMILES pair encodings (SPEs), a method inspired by byte-pair encodings that splits SMILES into substructures of varying lengths based on their occurrence in ChEMBL.48 They showed that SPE exhibits prediction accuracy comparable to that of atom-based tokenization in QSAR prediction tasks.48 SPE is ideal to handle larger molecules since it drastically reduces the number of tokens. For example, the SMILES shown in Fig. 1 is split into  whereas SPE splits into
whereas SPE splits into 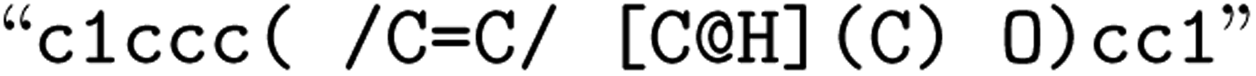 . In our experiments, we coupled the SMILES PE tokenizer with SMILES augmentation. For SELFIES, the split function built in the
. In our experiments, we coupled the SMILES PE tokenizer with SMILES augmentation. For SELFIES, the split function built in the 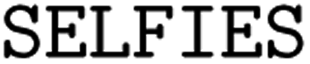 package is used. All sequences are enclosed by
package is used. All sequences are enclosed by  and
and  tokens and are left-padded to the longest sequence in the dataset, respectively.
tokens and are left-padded to the longest sequence in the dataset, respectively.
2.3 Models
Chemical language model (CLM). Our proposed CLM model is an attention-based multiscale convolutional neural network. It is inspired by a bimodal variant of this model, called
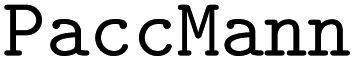 , which was originally developed for drug sensitivity prediction26,49 but has also inspired proteochemometric models for binding affinity prediction.50,51 The network architecture is shown in Fig. 2. In our canonical CLM, each token is represented as a learned embedding of dimensionality H = 256, such that the input matrix
, which was originally developed for drug sensitivity prediction26,49 but has also inspired proteochemometric models for binding affinity prediction.50,51 The network architecture is shown in Fig. 2. In our canonical CLM, each token is represented as a learned embedding of dimensionality H = 256, such that the input matrix  where T is the sequence length (i.e., padding size). Note that we performed ablation studies on the embedding type. The embeddings X are processed by three parallel 1D-convolutional layers with kernel sizes 3, 5, and 11. A fourth channel has a residual connection without convolutions (not shown in Fig. 2). For each of the four channels, we utilize a stack of m = 6 attention heads. In each head, an attention mechanism, similar to the one proposed by Bahdanau, D.33 and developed prior to the one by ref. 52 is used to enable the model to focus on relevant parts of the molecule. In each head, the attention weight αi of token i is computed using:
where T is the sequence length (i.e., padding size). Note that we performed ablation studies on the embedding type. The embeddings X are processed by three parallel 1D-convolutional layers with kernel sizes 3, 5, and 11. A fourth channel has a residual connection without convolutions (not shown in Fig. 2). For each of the four channels, we utilize a stack of m = 6 attention heads. In each head, an attention mechanism, similar to the one proposed by Bahdanau, D.33 and developed prior to the one by ref. 52 is used to enable the model to focus on relevant parts of the molecule. In each head, the attention weight αi of token i is computed using: | (1) |
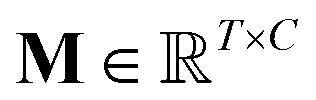 is the output of the convolutional layer with C = 128 filters,
is the output of the convolutional layer with C = 128 filters,  and
and 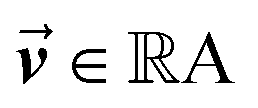 are learnable parameters, and A = 256 is the dimensionality of the attention space. For notational purposes,
are learnable parameters, and A = 256 is the dimensionality of the attention space. For notational purposes, 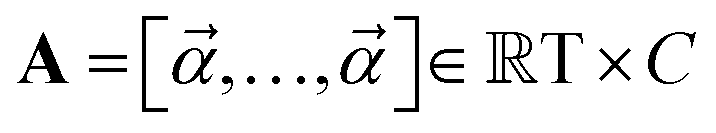 should be considered the attention matrix with the attention vector repeated C times. Then, the output vector
should be considered the attention matrix with the attention vector repeated C times. Then, the output vector  of each attention head is obtained by filtering M with the attention matrix (i.e., ”we attend”):
of each attention head is obtained by filtering M with the attention matrix (i.e., ”we attend”):![[x with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/b_i_char_0078_20d1.gif) out = 1T[M∘A] out = 1T[M∘A] | (2) |
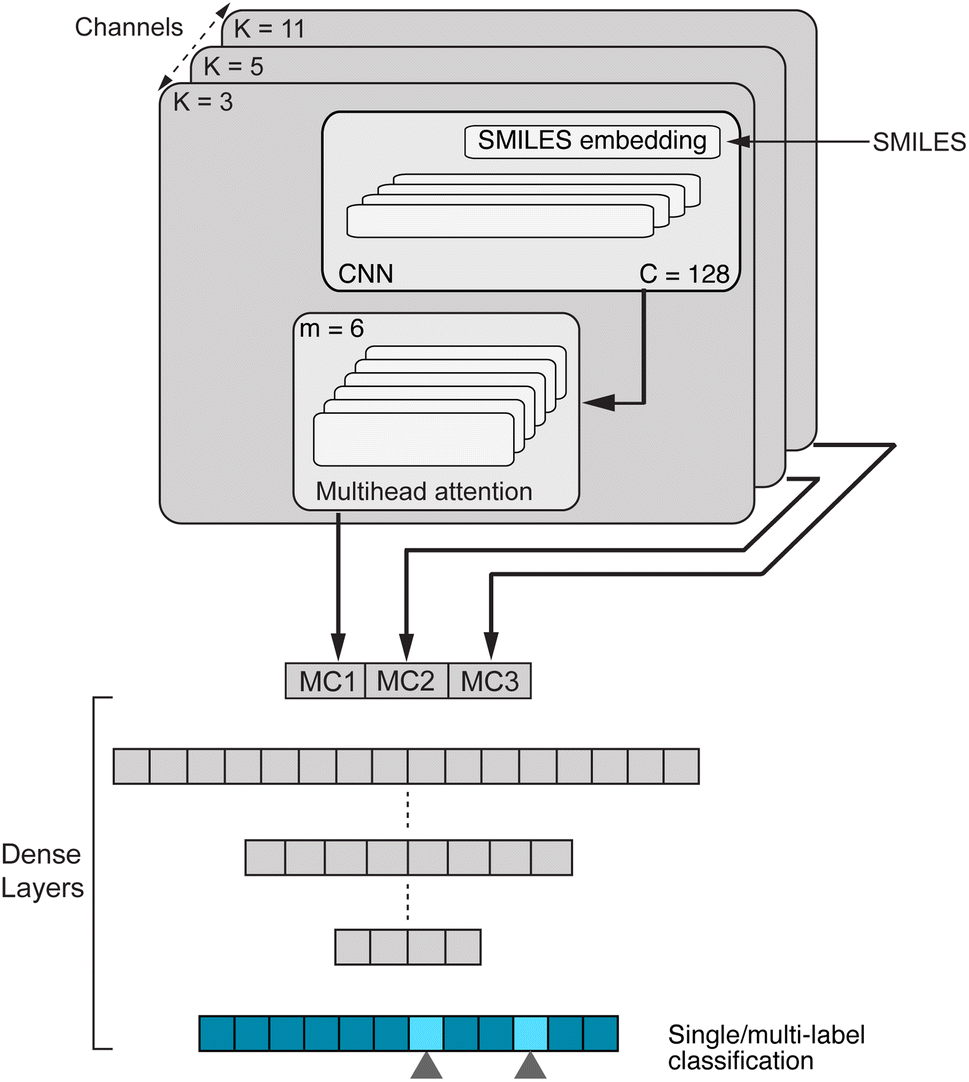 | ||
| Fig. 2 Our CLM, a convolutional, attention-based neural network for molecular property prediction. The first step is sequence embedding which is then used for three (parallel) 1D convolutional layers to aggregate local information. Next, the multi-head-attention mechanism calculates the attention weights and filters the inputs accordingly. The resulting outputs of all three multiscale convolutional (MC) blocks as well as one residual connection (not shown) are concatenated and processed by a set of dense layers, resulting in one (or multiple) output scores. C denotes the number of convolutional filters and m is the number of attention heads. | ||
Basically, we filter the output sequence from a given convolutional kernel with the attention scores and then we sum over the sequence dimension to obtain a single score for each filter. This is similar to the attention by ref. 33 with the difference that (1) we refrain from having an additional tanh non-linearity (it did not perform well in initial experiments) and (2) there is no need for additive attention because there are no output tokens to attend to. With three parallel convolutional layers (plus one residual connection), each with m attention heads, we obtain 4m output vectors out which are stacked to form a single large vector and processed by a stack of dense layers ([1024, 512] units) before a final layer with a sigmoid activation computes the class-wise predictions. The model is trained with a binary cross-entropy loss.
Recurrent networks. We examined two flavours of recurrent neural networks; the gated recurrent unit, GRU,53 and the neuromodulated bistable recurrent cell, nBRC.54 The GRU employs a gating mechanism to control the information flow, making it suitable for handling longer sequences. The nBRC is a biologically inspired modification of the GRU that is superior to the other cells in exact memorization and counting,54 crucial properties when handling SMILES sequences due to the presence of ring opening and closure symbols. The cell can switch between a monostable and a bistable state and can hold onto information for an arbitrarily long period of time. For both models, we use two bidirectional layers with 256 units. The last hidden states from both directions are processed by a 3-layered dense network with 1024, 1024, and 512 hidden units respectively (50% dropout). The final scores are returned by an ensemble of 5 linear networks acting on the 512-dimensional representation. The models were optimized by Adam55 at a constant learning rate of 1e − 4.
Random forests. Random forests56 are popular non-linear models that show competitive performances on most classification tasks.56,57 Owing to high class imbalance in the data, the class_weight parameter was set to balanced. We also performed hyperparameter optimisation on the number of trees in the forest and found 500 to be optimal in line with prior work.58,59
k-nearest-neighbor (k-NN). As a non-parametric baseline, we explored the k-NN algorithm and employed (inverted) Tanimoto similarity60 as a distance function. We set k = 23, based on the performance on the Tox21 test dataset (see subsection 2.6.1).
Dense neural network (DNN). This was a simple, four-layered, fully connected neural network with 512, 1024, 2048, and 1024 units with a sigmoid activation function.
Graph convolutional network (GCN). Following Duvenaud D. K. et al.,61 a GCN with two graph-convolutional layers (64 units) and one dense layer (128 units), no dropout, 75 atom features, and a sigmoid activation function was employed.
Graph kernels. Graph kernels rely on a kernel k(x,x′) that measures similarity between molecular graphs x and x′.62 We experimented with four different kernels.
(1) Shortest-path (SP): this path kernel63 first transforms the graphs G1 and G2 into shortest-path graphs S1 and S2 using the Floyd algorithm.64 Let S1 = (V1, E1) and S2 = (V2, E2), then our shortest-path kernel is:
 | (3) |
(2) Weisfeiler–Lehman (WL): This subtree kernel relies on the Weisfeiler–Lehman (WL) relabeling method.65 Let Gn = (V, E, ln) and 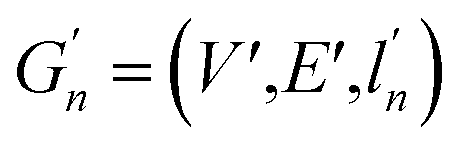 be the n-th iteration rewriting of the graphs G and G′. Then the WL kernel is defined as
be the n-th iteration rewriting of the graphs G and G′. Then the WL kernel is defined as
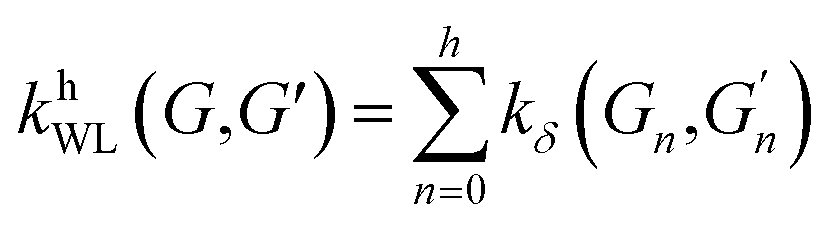 | (4) |
 | (5) |
(3) Message passing (MP): this subtree kernel66 extends the concept of message-passing67 from GNNs to graph kernels. It's a generalization of the WL kernel that uses a smoother definition of structural equivalence.
(4) Wasserstein-Weisfeiler Lehmen (WWL): this extension of the WL kernel relies on the Wasserstein distance between node feature vector distributions of the WL subgraphs. For details see Togninalli M. et al.68
In all cases, the graph kernels were used to measure sample similarity and a support vector machine (SVM)69 was employed for classification.
2.4 Attention analysis
As a byproduct of the forward-pass, our CLM produces attention scores assigning relevance to each token. We analysed the attention to assess whether the attention scores carry any meaning regarding the toxicity of the respective token (i.e., atom/bond). Therefore, we relied on so-called toxicophores, molecular substructures that are known to have toxic effects.70 Dependent on the dataset, specific toxicity alerts were employed (see details in the respective results section) and training molecules were excluded from the analysis. The attention scores were computed following eqn (1) which provides a single attention score αi for each token of the (SMILES) sequence. Atom and bond tokens were considered for the analysis whereas attention on other tokens (e.g., ring tokens) was discarded since it could not always be determined whether these tokens belong to a toxicophore or not. Next, all analyzed molecules (given in SMILES) were queried against the desired toxicity alerts (given in the SMARTS pattern71). Whenever a match was obtained (by a substructure match in ), the SMILES tokens affected by the alert were assigned as toxicophore tokens whereas the other tokens kept their non-toxic status. This resulted in a grouping of the attention weights as either belonging to a toxic or non-toxic substructure.
), the SMILES tokens affected by the alert were assigned as toxicophore tokens whereas the other tokens kept their non-toxic status. This resulted in a grouping of the attention weights as either belonging to a toxic or non-toxic substructure.
2.5 Uncertainty estimation
In an attempt to obtain a trustworthy model, we further employ two techniques to measure prediction uncertainty. Specifically, we assess epistemic (model) uncertainty with Monte Carlo dropout (MC dropout), a method that draws Monte Carlo samples from the approximate predictive posterior by performing repeated forward passes of an input sample while the network's dropout layers are turned on.34 Moreover, we assess aleatoric (data) uncertainty, an uncertainty measure independent of epistemic uncertainty, via test-time data augmentation.35 In our case, this amounted to performing repeated forward passes with different SMILES strings corresponding to the same molecule.43From the resulting prediction ensembles, the confidence estimate ci of sample i was obtained by scaling the sample's standard deviation to the range [0,1] and interpreting it as inverse precision:
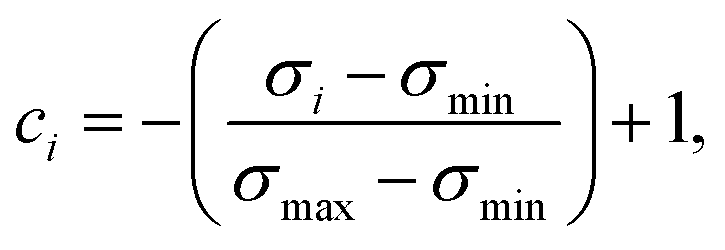 | (6) |
2.6 Datasets
707 compound entries of small molecules tested on 12 targets.72,73 The input data are binarily classified as toxic or non-toxic.
Five of the 12 targets are associated with hormones (such as the estrogen receptor (ER and the ER ligand binding domain), the androgen receptor (AR and the AR ligand binding domain), and aromatase). Both receptors, the ER and AR, regulate gene expression and are important in sexual maturation and gestation.74 Aromatase catalyzes the hormone reaction from testosterone to estradiol. Aromatase deficiency can lead to delayed puberty in females, osteoporosis in males and virilisation in pregnant mothers. Excess leads to the contrary: premature puberty and breast evolution in males.75 The other seven belong to stress response pathways (HSE, MMP, ATAD5, PPARγ, ARE, AhR, and p53). Cells activate the heat shock factor response element (HSE) in response to stressful conditions. The mitochondrial membrane potential (MMP) decreases during apoptotic cell death.76 A low ATPase family AAA domain containing 5 (ATAD5) expression leads to an extended lifespan which leads to an increase of inactive replication factories resulting in a delay in S-phase progression.77 Peroxisome proliferator-activated receptor γ (PPARγ) plays a major regulatory role in energy homeostasis.78 The antioxidant responsive element (ARE) regulates cytoprotective genes, which play a critical role in redox homeostasis.79 In addition to regulating metabolizing enzymes through gene expression, the aryl hydrocarbon receptor (AhR) has roles in regulating immunity, stem cell maintenance, and cellular differentiation. The induction of metabolizing enzymes can lead to the production of toxic metabolites.80
When cells undergo DNA damage, the tumor-suppressor protein p53 is expressed, in order to counteract the effects. It induces growth arrest, repairs the DNA or starts the cell death process.81 However, it is also linked to drug resistance in cancer cells.82
The dataset comes with a fixed split of 11764 training, 296 test, and 647 score molecules. Test labels were withheld from participants during the original Tox21-challenge but later made available so that participants could refine their models for the final evaluation on the molecules in the score set.
(1) BACE: this is a dataset of 1522 inhibitors against human β-secretase 1 (BACE-1), represented quantitatively by their IC50 values and qualitatively through binary labels indicating their inhibition success.83 The 2D structures of these compounds and IC50 values are gathered from the experimental scientific literature. Scaffold splitting is recommended for this dataset. This dataset has only one task.
(2). SIDER: Side Effect Resource (SIDER) is a database of 1427 approved drugs and their associated adverse drug reactions (ADR), grouped into 27 tasks (i.e., system organ classes84,85).
(3) ClinTox: this dataset comprises 1491 drug compounds and holds drugs approved by the FDA as well as those that have failed clinical trials due to toxicity. This dataset has two tasks: clinical trial toxicity status and FDA drug approval status.86,87
(4) BBBP: the blood–brain barrier penetration (BBBP) dataset contains information on the blood–brain barrier permeability properties of over 2000 compounds.88 This is a single binary classification and a scaffold split is recommended.
(5) HIV: the HIV dataset contains information about the ability of 40000 compounds to block HIV replication.89 This single binary classification task categorizes compounds as being active or inactive for HIV replication. A scaffold split is recommended.
(6) Tox21: this is a redistribution of the original Tox21 dataset.90 While there are still 12 tasks there are several important differences that are detailed below. A random split is recommended.
On each dataset, we trained ten models on repeated data splits according to the recommended strategy. For a comparison with Grover,13 who trained the models on scaffold splits for all datasets, we additionally trained ten models on scaffold splits for the affected datasets (SIDER, ClinTox, Tox21).
Difference between original Tox21 and MoleculeNet Tox21. Like all other datasets from the MoleculeNet that are distributed via DeepChem,91 their Tox21 dataset does not come with a fixed split, and thus, by convention, repeated splits with different random seeds are performed. The DeepChem distribution is significantly smaller, containing only 8014 molecules (SMILESs are largely canonicalized). A detailed analysis of the differences is available in Table A1, see ESI.‡ We believe that these differences are important to emphasize as they have been confounded/disregarded in some prior work.
848 measured compounds. Pre-processing of the data is performed in the same manner as in the original study.30 More specifically, it uses  and consists of a sanitization, a standardization, and a de-duplication step, resulting in 34366 compounds. Out of these molecules, only 4.65% are labeled cytotoxic, leading to a highly imbalanced, yet consistent, data set. The experiments with this data set were run using high-performance computer (HPC) services from the Freie Universität Berlin.93 To compare to the feed-forward neural network (FNN) by Webel H. E. et al.,30 we used a 10-fold stratified cross-validation split with 10% held-out data for testing, as performed in their work. Note that Webel H. E. et al.30 used 2048-bit ECFP4s.
and consists of a sanitization, a standardization, and a de-duplication step, resulting in 34366 compounds. Out of these molecules, only 4.65% are labeled cytotoxic, leading to a highly imbalanced, yet consistent, data set. The experiments with this data set were run using high-performance computer (HPC) services from the Freie Universität Berlin.93 To compare to the feed-forward neural network (FNN) by Webel H. E. et al.,30 we used a 10-fold stratified cross-validation split with 10% held-out data for testing, as performed in their work. Note that Webel H. E. et al.30 used 2048-bit ECFP4s.
2.7 Hyperparameter optimization
All models were trained for 200 epochs with early stopping, the Adam optimizer,55 and a cross-entropy loss. The learning rate varied across models, but unless otherwise specified, was set to 1e − 4.Emulating the original Tox21 challenge, the hyperparameters of the models were tuned using the test dataset using raw SMILESs as inputs. After the optimal configuration was found, 10 models were trained for each investigated dataset. For the original Tox21 dataset, the 10 models were obtained from identical training data with different weight initializations. For the remaining datasets, the random split was repeated for each run. Note that we refrained from further optimizing any hyperparameters on any of the remaining datasets.
2.8 Performance metrics
All models were evaluated on performance metrics that are in alignment with previous work on those datasets.11,30,90 The main metric is the area under the ROC curve (ROC-AUC). For the cytotoxicity dataset, we report the true positive rate (TPR, also called sensitivity), the true negative rate (TNR, also called specificity), and the balanced accuracy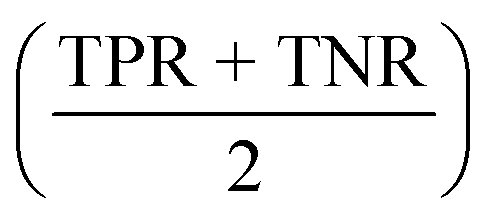 .
.
3 Results
In the following, we will first examine the impact of using different types of property prediction models as well molecular representations on the Tox21 dataset. Then, we will pick the best model and deepen our analysis by comparing it to a large body of related work on several datasets from the MoleculeNet benchmark. We will demonstrate the interpretability of our method by analzying the attention scores – a natural byproduct of every prediction – and also propose two simple uncertainty estimation techniques that improve robustness as well as model performance. Last, we show in a case-study how our CLM can be applied to a proprietary, large-scale dataset and achieve improved performance in virtual screening.3.1 Model & data representation comparison on Tox21
In Table 1, we display the performance of the different algorithms and representations as measured using ROC-AUC. The performances refer to the Tox21 score dataset which was used to determine the Tox21 challenge winners. Comparing the model classes shows that graph kernels generally yielded the worst performance. Since the complexity of graph kernels scales quadratically with the size of the dataset, reports on graph kernels on datasets with >10000 examples are scarce to absent.94 Thus, graph kernels are predominantly useful in small data regimes. The random forests, on the other hand, prove to be a strong baseline, second only to the GCN (0.828 ROC-AUC). We see that increasing the fingerprint bit-size improves the performance of the RF with an ROC-AUC score of 0.782 for the higher 1024-bit and 2048-bit ECFP4s. This suggests that while the random forest requires more information about the molecule to achieve higher classification accuracy, it saturates beyond a certain point when there is no new information to be gained.
The SMILES representations used to train the CLM (cf.Table 1) yielded a performance that statistically significantly surpassed (one-sided Mann–Whitney U test, U) the DNN trained on ECFP as well as all graph kernel techniques in all cases. By comparing the SMILES representations, it was found that the best results were not obtained by consistently formatting the SMILES, but rather by SMILES augmentation,43 which resembles a form of data augmentation by exploiting the multiplicity of SMILES for each molecule. This SMILES augmentation model outperforms all other models (p < 1e − 4, U) that do not use augmentation. This is in alignment with prior work reporting superiority of SMILES augmentation to canonical SMILES.15,43 Generally, the differences between different SMILES representations were minor. Stereochemistry information stemming from chirality tokens  or bond direction
or bond direction  tend to confuse the model as removing them yielded slightly better performance, which maybe explainable by their scarcity in the training data (18% and 7% respectively). Notably, even though SELFIESs were devised for generative tasks,39 they barely rank behind SMILES regarding their predictive power for toxicity prediction. A previously unreported finding is that the benefits of SMILES augmentation also extend to SELFIES. While, overall, the semantic (i.e., chemical) transformation of SMILES has a minor impact on performance, language transformations and the tokenization scheme can be critical. For example, SMILES pair-encoding48 gave a much worse performance than all other SMILES representations, maybe because the sequences are shorter and the vocabulary is much larger leading to sparsity. We hypothesize that more labelled data or pretraining on SMILES-PE could have closed this gap. A staggering finding is that shuffling the SMILES did not change the performance significantly. This result is in accordance with ref. 45 on other datasets and suggests that instead of aggregating local information in the SMILES sequences, the models predominantly make predictions similar to a bag-of-words model. If structural information is stripped off, the models can only rely on atom counts. However, the shuffling can be interpreted as another form of data augmentation since it is performed stochastically at runtime. In that sense, it is worth mentioning that the SMILES augmentation performed significantly better than shuffling. Last, the two RNN-based models operating on raw SMILES (GRU and nBRC) performed much worse than the CLM, suggesting the superiority of our architecture.
tend to confuse the model as removing them yielded slightly better performance, which maybe explainable by their scarcity in the training data (18% and 7% respectively). Notably, even though SELFIESs were devised for generative tasks,39 they barely rank behind SMILES regarding their predictive power for toxicity prediction. A previously unreported finding is that the benefits of SMILES augmentation also extend to SELFIES. While, overall, the semantic (i.e., chemical) transformation of SMILES has a minor impact on performance, language transformations and the tokenization scheme can be critical. For example, SMILES pair-encoding48 gave a much worse performance than all other SMILES representations, maybe because the sequences are shorter and the vocabulary is much larger leading to sparsity. We hypothesize that more labelled data or pretraining on SMILES-PE could have closed this gap. A staggering finding is that shuffling the SMILES did not change the performance significantly. This result is in accordance with ref. 45 on other datasets and suggests that instead of aggregating local information in the SMILES sequences, the models predominantly make predictions similar to a bag-of-words model. If structural information is stripped off, the models can only rely on atom counts. However, the shuffling can be interpreted as another form of data augmentation since it is performed stochastically at runtime. In that sense, it is worth mentioning that the SMILES augmentation performed significantly better than shuffling. Last, the two RNN-based models operating on raw SMILES (GRU and nBRC) performed much worse than the CLM, suggesting the superiority of our architecture.
| Representation | ROC-AUC |
|---|---|
| Raw SMILES | 0.832* ± 0.005 |
| Canonical SMILES | 0.830* ± 0.008 |
| Kekulized SMILES | 0.830* ± 0.006 |
| Augmented SMILES | 0.853 ± 0.003 |
| SMILES without bond direction | 0.834* ± 0.006 |
| SMILES without chirality | 0.834* ± 0.004 |
| SMILES w/o bond direction & chirality | 0.835* ± 0.006 |
| Kekulized w/o bond direction & chirality | 0.831* ± 0.004 |
| SMILES with explicit bonds | 0.834* ± 0.003 |
| SMILES with explicit hydrogen | 0.829* ± 0.007 |
| SELFIES | 0.827* ± 0.007 |
| Augmented SELFIES | 0.852 ± 0.004 |
| Shuffled SMILES | 0.830* ± 0.003 |
| SMILES pair encoding | 0.776* ± 0.01 |
| Augmented SMILES pair encoding | 0.825* ± 0.005 |
| Model | Repr. | ROC-AUC |
|---|---|---|
| Random forest (RF) | 512-bit ECFP4 | 0.774 ± 0.002 |
| 1024-bit ECFP4 | 0.782 ± 0.002 | |
| 2048-bit ECFP4 | 0.782 ± 0.002 | |
| KNN | 512-bit ECFP4 | 0.759 |
| DNN | 512-bit ECFP4 | 0.777 ± 0.004 |
| GRU (RNN) | Raw SMILES | 0.781 ± 0.003 |
| nBRC (RNN) | Raw SMILES | 0.756 ± 0.002 |
| Weisfeiler lehman (WL) | Graph kernels | 0.754 ± 0.019 |
| Message passing | 0.703 ± 0.040 | |
| Wasserstein-WL | 0.758 ± 0.023 | |
| Shortest path | 0.567 ± 0.108 | |
| GCN | Graphs | 0.828 ± 0.008 |
While we used learned embeddings throughout all experiments in this section, we conducted an ablation study comparing them to one-hot embeddings and pretrained embeddings from a variational autoencoder trained on >2M ChEMBL molecules.46 In the absence of augmentation, the pretrained embeddings significantly outperform the one-hot and especially the learned embeddings. As can be seen in Table A2, see ESI,‡ this holds especially true if the pretrained embeddings are allowed to be finetuned. The use of augmentation consistently improved performance across all embedding types and generally relaxed differences between embedding types.
Since this section demonstrated the superiority of (1) the use of SMILES augmentation, all remaining results are generated with this configuration.
3.2 Analysing attention scores in light of toxicophores
Our proposed CLM utilizes an attention mechanism (similar to the one proposed by Bahdanau D. et al.33) which naturally returns attention maps indicating relevance for each SMILES token. In contrast to the work by ref. 32, our method cannot provide signed attributions for each atom. However, as the attention scores are a natural byproduct of the forward pass, no postprocessing scheme (e.g., integrated gradients) has to be applied. In our previous work, a predecessor of this attention mechanism demonstrated positive quantitative evidence (for the extraction of genes related to apoptotic processes) in a drug sensitivity regression task.26 We sought to validate the attention scores with toxicophores (molecular substructures with known toxic effects), investigate whether the scores align with known toxicophores, and compare them quantitatively and qualitatively. In Fig. 3, we show the attention weights of the best model for all molecules from the Tox21 score dataset. We focus on two toxicity endpoints, acute aquatic toxicity (99 alerts, see ref. 95 and 96) and endocrine disruption (35 alerts, see ref. 97) that are most similar to the Tox21 tasks. The corresponding SMARTSs were extracted from ref. 98. Similar to the results by Nendza M. et al.97 we found that not all appearances of toxicophores lead to a toxic compound.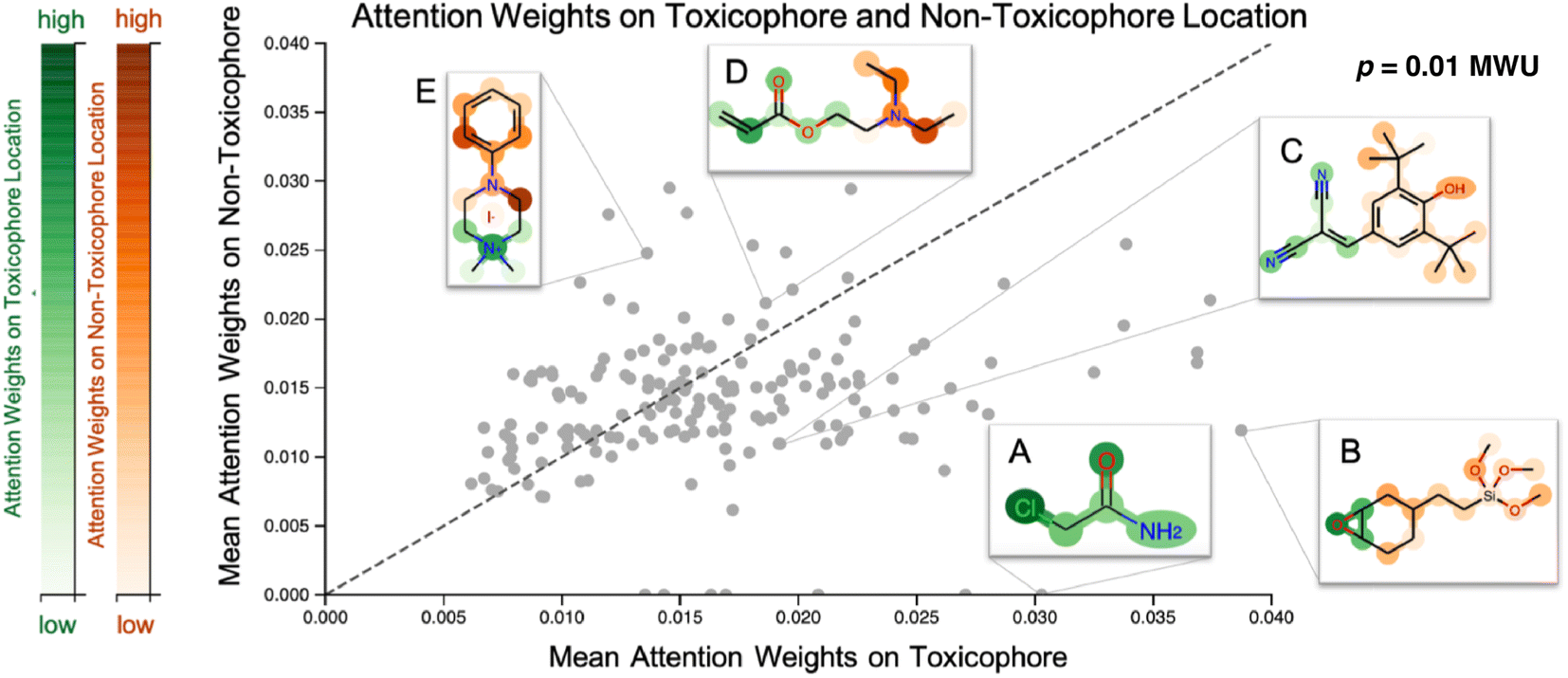 | ||
| Fig. 3 Visualization of attention maps of Tox21 compounds. Scatterplot of the mean attention of the toxicophore part of the molecule versus the mean attention of the remaining ones. Toxicophoric atoms are colored green, the remaining parts are colored red, and intensity encodes the attention weight. Toxicophores are assigned significantly higher attention weights. Interactive visualization is available at: https://ibm.biz/tox21_attention. MWU denotes a Mann–Whitney U test. | ||
To assess whether the model selectively focused on informative substructures, we compared the mean attention weight on the toxicophoric parts of all molecules to the mean attention weight of the remaining part. Molecules that exclusively consisted of toxicophores were excluded from the analysis. This revealed a significantly higher mean for toxicophore substructures (p = 0.011 in the two-sided MWU) showing that the model focused predominantly on toxic substructures. This is remarkable given that the attention scores were learned entirely unsupervised. While several related studies on proteochemometric modeling claimed via case studies that similar SMILES attention mechanisms could automatically re-discover biochemical concepts such as protein binding sites,99,100 Li S. et al.101 demonstrated later in a quantitative analysis that performance was not exceeding the chance level. Instead, on the Tox21 dataset previous work by Mayr A et al.16 and Preuer K et al.102 reported that the activation of a significant number of hidden neurons in ECFP-based DNNs could be associated with toxicophore features. However, both their analyses were performed on training molecules and involved significant post-hoc experimentation whereas our attention scores are produced en passant the forward pass. Moreover, we emphasize that the attention maps are global, i.e., not assay-specific, and thus, the task-specific inference is limited to single-task classifications. To further assess the usability of attention maps we enclose a case study where the attention highlighted an epoxide (cf. Fig. A5, see ESI‡).
3.3 Comparison to existing QSAR models
We sought to validate the proposed model on related tasks beyond toxicity prediction. The MoleculeNet benchmark11 is ideally suited for this since it comprises several molecular datasets ranging from biophysical and physiology tasks. To ensure fair comparability, we excluded previous work whenever the data splitting strategy was not clear or no repeated experiments were conducted. The results on all six datasets are shown in Table 2 and underline the superiority of our model to previous approaches, including graph convolutional networks11 and several variants of message-passing neural networks,67 in particular, the directed MPNN,12 attention-MPNN, edge-MPNN and SELU-MPNN.103 Even the work by Shen W. X. et al.104 who built a convolutional neural network based on a highly customized featurization pipeline including thirteen multidimensional descriptor classes and three fingerprint types was significantly outperformed by our method, a purely SMILES-based model that did not incorporate any topological or structural features directly. Note that the Tox21 dataset listed in this section differs from the original one by Huang R. et al.90 Since the derivative distribution by Wu Z. et al.11 is frequently used for benchmarking, we also trained our CLM on this flavor of Tox21.| Dataset | BACE | SIDER | Clintox | BBBP | Tox21 | HIV | Average |
|---|---|---|---|---|---|---|---|
| Split | Scaffold | Random | Random | Scaffold | Random | Scaffold | |
| Ours (CLM) | 0.861 ±0.04 | 0.659 ±0.04 | 0.878±0.00 | 0.915 ±0.02 | 0.858±0.05 | 0.813 ±0.03 | 0.831 |
| GraphConv (Wu Z. et al.)11 | 0.783±0.01 | 0.638±0.01 | 0.807±0.05 | 0.690±0.01 | 0.829±0.01 | 0.763±0.02 | 0.752 |
| Weave (Wu Z. et al.)11 | 0.806±0.00 | 0.581±0.03 | 0.832±0.04 | 0.671±0.01 | 0.820±0.01 | 0.703±0.04 | 0.736 |
| D-MPNN (Yang K. et al.)12 | 0.838±0.06 | 0.646±0.02 | 0.894 ±0.03 | 0.888±0.03 | 0.845 ±0.002 | 0.794 ±0.02 | 0.818 |
| SELU-MPNN (Withnall M. et al.)103 | — | 0.632±0.01 | — | 0.693±0.06 | 0.820±0.01 | 0.747±0.01 | — |
| AMPNN (Withnall M. et al.)103 | — | 0.639±0.01 | — | 0.709±0.04 | 0.812±0.02 | 0.742±0.02 | — |
| EMPNN (Withnall M. et al.)103 | — | 0.651±0.01 | — | 0.705±0.02 | 0.829±0.01 | 0.759±0.01 | — |
| MMNB (Shen W. X. et al.)104 | 0.849 | 0.680 | 0.888 | 0.739 | 0.842 | 0.777 | 0.796 |
In the analysis shown in Table 2, we relied on the data splitting strategy recommended by Wu Z. et al.11 for each dataset. However, splitting molecules randomly between training and testing often results in overly optimistic model performance (due to data collection biases such as limited diversity and sparse coverage of the chemical space). Instead, splitting the scaffolds rather than the molecules poses a more challenging task that might better approximate the generalization performance. We, therefore, re-assessed the performance on three datasets where a random split was recommended (SIDER, ClinTox, and Tox21). The scaffold split on those datasets enabled a fair, yet additional comparison to the benchmarking performed by Rong Y. et al.13 They evaluated a wide range of prediction models on scaffold splits of all MoleculeNet datasets and then proposed GROVER, a large-scale graph transformer that was pretrained with self-supervision on >10M molecules. The results are displayed in Fig. 4 and include comparisons to fully connected networks (TF_Robust91), three graph-convolutional networks (GraphConv,36 Weave105 and SchNet106), four message-passing graph neural networks (a vanilla GNN107 and the MPNN67 and its variants D-MPNN12 and MGCN108), an n-gram model,109 and two graph transformer networks (AttentiveFP14 and GROVER itself13). The results demonstrate that we consistently obtained superior performance compared to all flavors of fully connected, graph-convolutional or message-passing neural networks. Only the group of graph-transformer networks (AttentiveFP and GROVER) outperformed our model, on average by 0.7% and 3%, respectively. GROVER, the only model that consistently surpasses us, is significantly larger and more complex. Our CLM contains only ∼5M parameters (exact number depends on the dataset/vocabulary size), consists of vanilla convolutional layers coupled with plain Bahdanau-style attention, and was trained from scratch on SMILES sequences (with augmentation). AttentiveFP is conceptually similar to our CLM but relies on molecular graphs and graph attention rather than SMILES and sequence attention. Furthermore, AttentiveFP uses recurrent units that are slower than the highly parallelizable convolutions in our model. Note that for AttentiveFP no evidence is given on the utility of uncertainty estimation techniques to enhance model reliability. Unlike here, their attention analysis only provides qualitative but no quantitative arguments that support model interpretability. Instead, GROVER employs an order of magnitude more parameters (50M) and relied on large-scale pretraining on >11M molecules that utilized 250 GPUs.
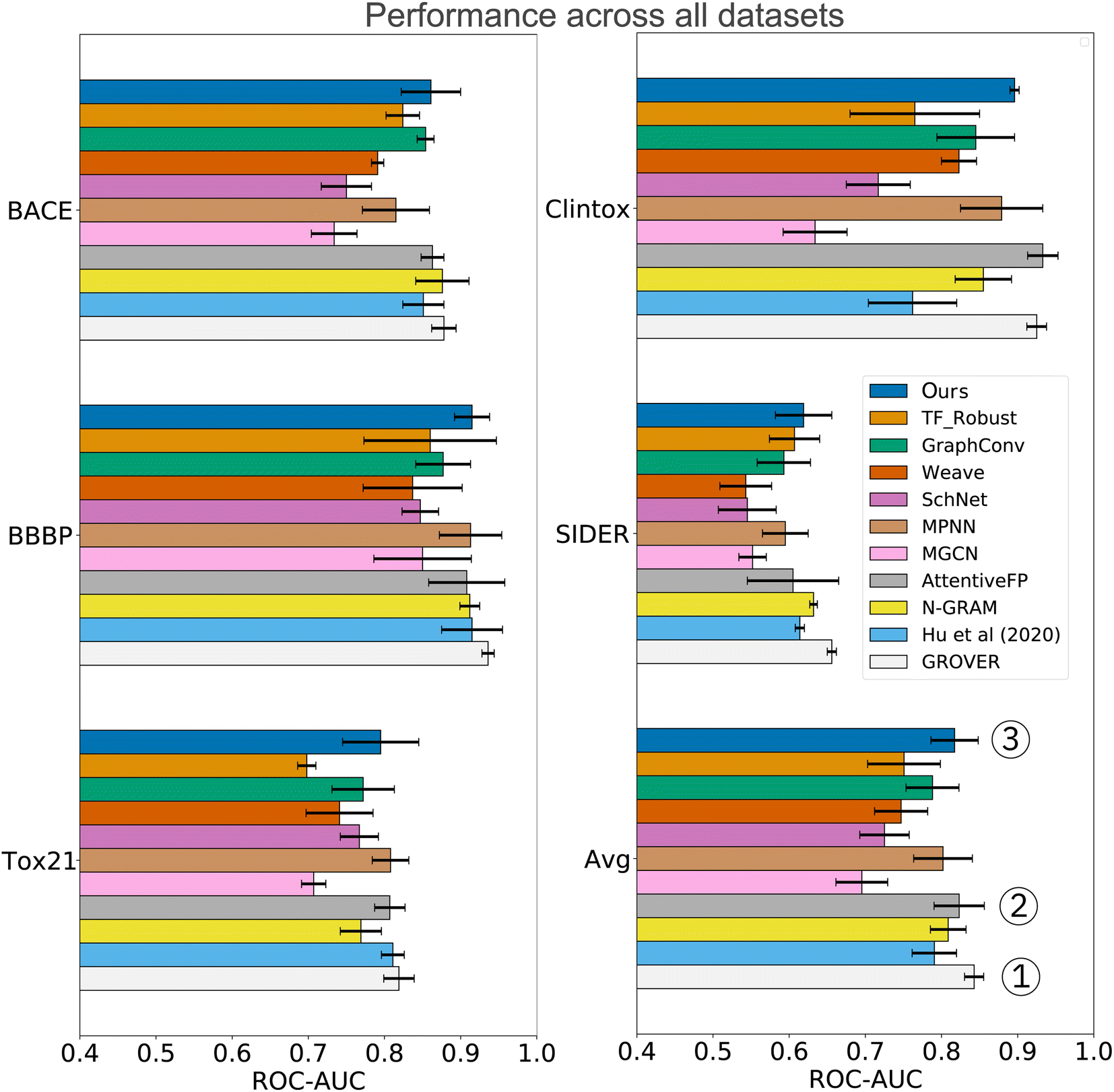 | ||
| Fig. 4 Comparison to previous work exclusively on scaffold splits of several MoleculeNet datasets. Overall, our CLM is the third best model, only surpassed by GROVER and AttentiveFP. For each dataset, the average ROC-AUC across all tasks is reported. The results were obtained by measuring test performance for 10 repeated scaffold splits. All other numbers are taken from Rong Y. et al.13 who trained all models on 3 repeated scaffold splits. The detailed numerical results can be found in Table A3, see ESI.‡ | ||
Lastly, we conducted an ablation study on the impact of the number of heads (m) in the multihead attention. For each m = 1, 3, 6 and 12, we computed the area under the ROC curve as the performance measure of the model on four datasets from MoleculeNet,11 namely, BACE, BBBP, Tox21 and Clintox. Fig. A4, see ESI‡ illustrates the variation in performance of each model across the four datasets. On visualising this performance and the 95% confidence interval associated with it, we conclude that m = 6 heads is the best choice in terms of performance and number of parameters to optimise. Please note that we refrained from conducting further ablation studies on the impact of the convolutional or the attention block as a whole because this has been performed in our previously developed  model [26, Table 1].
model [26, Table 1].
3.4 Uncertainty estimation & model ensembling
Deriving prediction confidences is an important topic in chemoinformatics and methods such as nested cross-validation or snapshot ensembling have been used in the past to understand model trustworthiness.17,110,111 In this section, we borrow two existing and efficient, yet much simpler methods to estimate model uncertainty in order to assess the reliability of our model's predictions, namely Monte Carlo (MC) dropout34 and TTA —test-time data augmentation.35 In both cases, the idea is to obtain a bag of predictions for one molecule (either by dropping out nodes or by passing different SMILES strings corresponding to the same molecule). These experiments were conducted on the Tox21 dataset (MoleculeNet flavor) and the results are shown in Fig. 5. Both—epistemic and aleatoric—model uncertainties were computed for each sample and each of the 12 toxicity assays and subsequently converted into a confidence estimate (cf.eqn (6)). Both confidence estimates are strongly negatively correlated with the residual of the prediction: the higher the error, the lower the confidence (Pearson's r = −0.558 and −0.536 for epistemic and aleatoric confidence respectively). When averaging both estimates (their correlation is ∼0.8), we obtain a single confidence estimate that is even more negatively correlated (see Fig. 5A). While the average confidence is relatively high with a value of 0.94, a known phenomenon,112 comparing the mean confidences of correctly and incorrectly classified samples reveals significant relative differences (0.96 versus 0.85). In a real-world scenario of screening large-scale virtual libraries, this difference could be used out-of-the-box to eliminate molecules where predictions are more likely to be incorrect. A specific example of the benefit of the confidence estimation is shown in Fig. 5C. While Bisphenol E was correctly predicted as toxic for the NR-ER assay, phenoxypropazine was incorrectly predicted as toxic. The model's internal class probabilities are 0.95 in both cases (1 means toxic and 0 non-toxic) and thus do not allow drawing conclusions**. However, investigating the respective prediction confidences (97% vs. 63%) can reveal that bisphenol E was a true positive while phenoxypropazine was a false positive.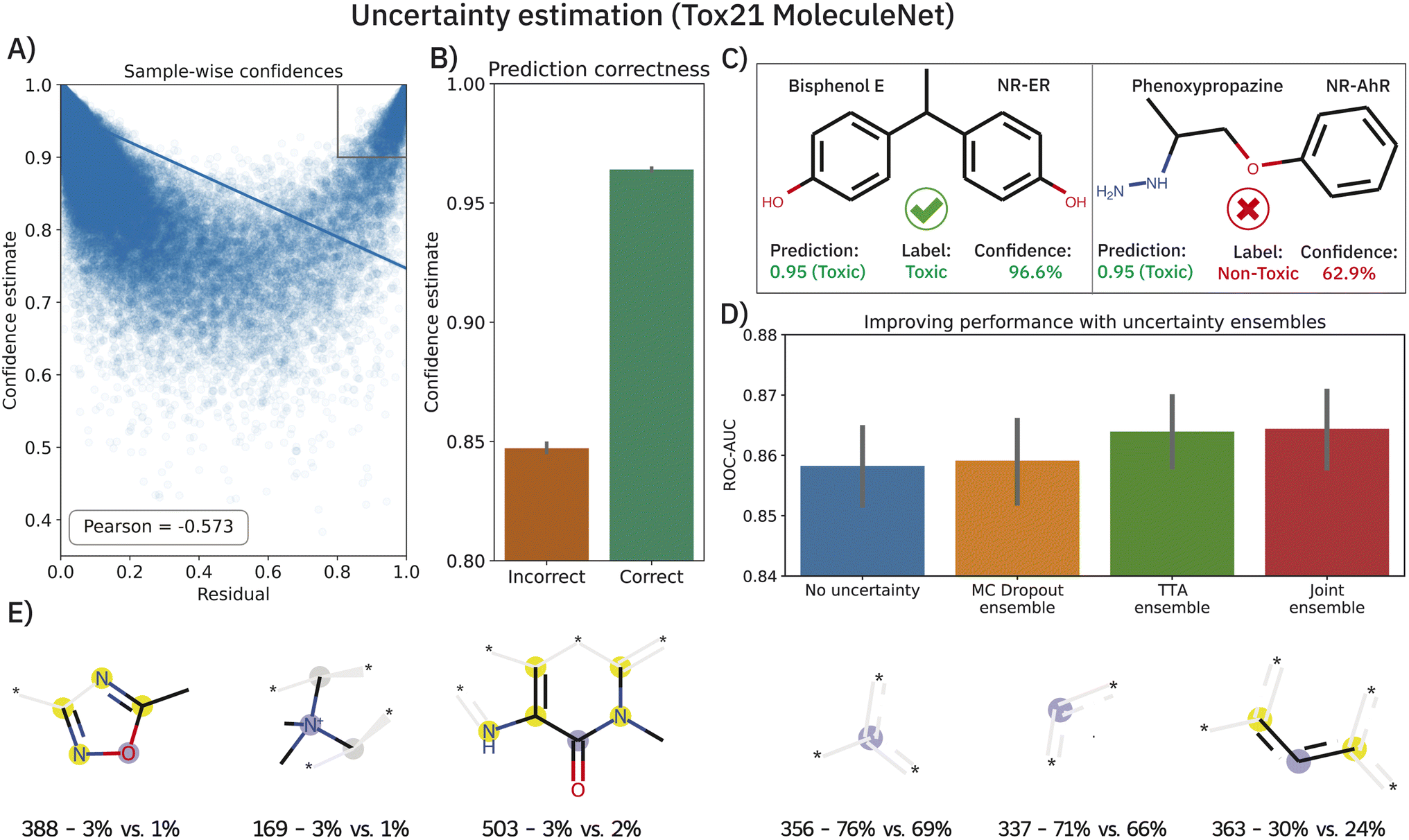 | ||
| Fig. 5 Uncertainty estimation analysis on the Tox21 MoleculeNet dataset. (A) Scatterplot of prediction residuals and confidences reveals a strong negative correlation. The gray box in the upper right marks molecules that were incorrectly classified with high confidence. (B) Confidence estimates are significantly lower for incorrectly classified samples. (C) Two exemplary molecules, both predicted as toxic, with an incorrect prediction identified from a low confidence estimate. All plots show the results across all 10 splits. (D) The prediction ensembles formed by MC dropout or TTA (test-time data augmentation) can significantly improve the prediction accuracy of the model. (E) The six fragments that were found most predominantly in incorrectly classified high-confidence (ICHC) molecules (see A), gray box. For each fragment, we display the ECFP4 bit and the percentage of ICHC molecules and remaining molecules where this fragment was present. Aromatic atoms are shown in yellow and central atoms in blue. | ||
The scatterplot in Fig. 5A reveals a small subset of incorrectly classified high-confidence (ICHC) molecules (see gray box). These incorrect predictions are particularly undesirable as they cannot be recognized and removed with our method. We inspected the molecules in the gray box (confidence >0.9 and residual >0.8) more closely, aiming to identify fragments that occur commonly in ICHC molecules but rarely in the remaining molecules. In Fig. 5E, we show the six ECFP4 bits that were most indicative for ICHC molecules. In a real-world scenario, such an analysis could easily increase robustness since molecules that include these bits could be removed from the screening library. The three bits shown in Fig. 5E (left) had the highest relative difference between ICHC and the remaining molecules, whereas the three bits shown on the right had the highest absolute difference. Some of these fragments can be linked to tremendous recent literature, for example bit 388 corresponds to a 1,2,4-oxadiazole ring. 1,2,4-Oxadiazole-derivatives have been largely neglected by medicinal chemistry until 2005, but in the past 15 years, research has grown exponentially113 and only in 2022 researchers reported cytotoxic,114 fungicidal,115 anti-inflammatory,114 antiparasitary and antiproliferative116 effects of 1,2,4-oxadiazole derivatives.
The benefits of using MC dropout and TTA are, however, not limited to confidence estimation. The prediction ensembles formed by both methods can be further used to improve the predictions. As demonstrated in Fig. 5D, replacing the baseline predictions (blue), with the mean of the 200 predictions obtained from MC dropout or TTA, improves the ROC-AUC on the Tox21 MoleculeNet benchmark from 0.858 ± 0.001 to 0.859 ± 0.001 (MC dropout) and 0.864 ± 0.001 (TTA). Lastly, a late-fusion average of both techniques yields the best performance (0.865 ± 0.001) which is significantly superior to that of the baseline model across 10 splits (p < 0.01,W+). Apart from one unpublished study,117 this performance is the best-reported one thus far on the highly benchmarked Tox21 dataset.††
3.5 External validation on cytotoxicity data
We validated the performance of our proposed CLM model on an external data set from the FMP.92 This dataset is comparably large (>34000 molecules) and indicates for each molecule whether it inhibited relative growth in a kidney and/or a liver cell line by at least 50% (for details see Section 2.6.3). Since this dataset is not generally available to the public, it can serve as an ideal tool to validate our method for potential proprietary use.
| Model | Source | Bal. Acc | TPR | TNR |
|---|---|---|---|---|
| FNN | (Webel HE et al.)30 | 68.89±1.46 | 61.57±7.39 | 76.22±6.62 |
| Ours | This study | 73.85 ±2.17 | 69.81 ±5.82 | 77.88 ±5.50 |
| Alert class | # alerts | # matches |
|---|---|---|
| Genotoxic carcino- & mutagenecity | 69 | 5 |
| Acute aquatic toxicity | 54 | 0 |
| Hepatotoxicity | 36 | 18 |
| Idiosyncratic toxicity | 32 | 9 |
| Mitochondrial toxicity (MT) | 17 | 0 |
| Developmental and MT | 12 | 0 |
| Non-genotoxic carcinogenicity | 5 | 4 |
| Kidney toxicity | 4 | 0 |
From the set of 17 compounds, three case studies are described here, of which the first two are also discussed by Webel H. E. et al.30 While molecule 1 (Fig. 6A) represents a false negative based on our prediction – a true positive example from Webel H. E. et al.30 – high attention is attributed to the tertiary substituted ethylendiamine. A similar toxicophore is identified in the study by Webel H. E. et al.30 and is caught by the given toxicophores from eMolTox,17 pointing to genotoxic carcinogenicity, mutagenicity, and hepatoxicity. The second molecule was correctly predicted as cytotoxic (ŷ = 0.96) and the prediction partly relied on a hepatotoxicity alert from 4-ethylphenol that was also identified by Webel H. E. et al.30 The third molecule (Fig. 6C) is especially interesting. While it was correctly predicted as toxic by our model and by the FNN from Webel H. E. et al.,30 both models did not highlight a particularly challenging substructure, namely the thiophene ring, which is sometimes associated with idiosyncratic drug reactions.17,119 However, the prediction is largely based on the sulfur tail, a hepatotoxicity toxicophore17 that was not identified by Webel H. E. et al.30
 | ||
| Fig. 6 Cytotoxicity case studies. Three molecules from the FMP dataset are visualized, using either their attention scores from our method (top) or their cytotoxicity maps using deep Taylor decomposition (bottom) following the original work.30 For our CLM, the color mapping is identical to Fig. 3: green for toxicophore atoms and orange for non-toxicophore atoms. Opacity corresponds to the attention score. | ||
Overall, it has to be emphasized that this analysis relied purely on unsupervised learning of toxicophores; neither ours nor the model by Webel H. E. et al.30 is aware of the notion of toxicophores. While their work mostly focused on the potential identification of new toxicophores, we validated our method in light of existing toxicophores. However, the dark red shaded areas in our attention maps might as well give a good starting point in the search for new toxicophores.
4 Discussion
In this work, we have conducted an extensive comparison of different molecular representations and machine learning models for toxicity and other molecular property prediction. The experiments revealed that competitive performance can be achieved with purely sequence-based chemical language models that do not rely on traditional molecular descriptors (such as fingerprints) or structure-based models (such as graph neural networks). We report evidence that SELFIES,39 a chemical language devised for generative modeling, exhibits similar predictive power for QSAR tasks as SMILES.Importantly, we presented a simple and interpretable model that relies solely on SMILES sequences. By coupling our CLM with SMILES augmentation, we surpassed a wide range of previous models and obtained state-of-the-art performance on several QSAR tasks, including but not limited to toxicity. Compared to competitive methods, an advantage of our method is that it does not require large-scale pretraining and is thus particularly suitable for low data/resource settings. A key feature of the proposed model is the attention mechanism, an ante hoc interpretability method that learns to extract the most important chemical motifs without explicit supervision. On the Tox21 dataset, we demonstrated that the attention on toxicophores is significantly enriched compared to remaining chemical motifs. These attention maps can not only be useful to validate existing toxicophores but also support the identification of so far unknown toxicophores. We validated our CLM on a proprietary toxicity dataset92 where we consistently outperformed a previous method while enabling similar interpretability analyses. Lastly, we evaluated two simple methods for uncertainty estimation that can not only help to identify misclassified samples but also form an implicit model ensemble that further boosts performance.
In light of the recent success of deep learning in multimodal settings (e.g., chemogenomics and proteochemometrics120), future work could explore approaches to featurize toxicity screening assays based on their experimental characteristics. Such models would be even more generic than the multi-task models presented herein because they could be transferred to novel assays and would allow the analysis of the attention scores for a specific assay.
5 Data and code availability
The source code used throughout the work for this paper is available at: https://github.com/paccmann/toxsmi. The models trained on the Tox21, SIDER and ClinTox datasets are available in GT4SD,121 the Generative Toolkit for Scientific Discovery and can be used via the properties submodule: https://github.com/GT4SD/gt4sd-core. Moreover, these three models are exposed through a Gradio web-app (https://huggingface.co/spaces/GT4SD/molecular_properties) where they can be used directly from a UI, either with single SMILES or in batch processing. We released the data preparation and SMILES processing pipeline in a separate PyPI package called pytoda: https://pypi.org/project/pytoda/.46,122 Similarly, the PyTorch implementation for the BRC is available in a separate PyPI package called brc_pytorch: https://pypi.org/project/brc-pytorch/.123 The data from the original Tox21 challenge is available from Huang R. et al.:90https://tripod.nih.gov/tox21/challenge/. The data from the MoleculeNet benchmark is available from Wu Z. et al.:11https://moleculenet.org/datasets. The data from the cytotoxicity case study is available from the FMP authors.17,92Author contributions
Conceptualization: JB, AV, and MM, data curation: JB, GM, and TBK, formal analysis: JB, GM, NJ, and MM, funding acquisition: AV and MRM, investigation: JB, GM, and NJ, methodology: JB, AV, and MM, software: JB, MM, GM, NJ, and TBK, supervision: JB, AV, and MM, validation: JB, NJ, and TBK, visualization: JB, GM, and NJ, writing – original draft: JB, GM, AV, NJ, and TBK, and writing – review and editing: JB, GM, AV, NJ, MRM, and TBK.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
JB, MRM, and MM acknowledge funding from the European Union's Horizon 2020 research and innovation programme [826121]. NJ acknowledges funding from the Swiss National Funding Agency [CRSII5_193832]. We would like to thank Bastian Rieck for useful discussions about graph kernels and the development of the LATEX-credit package (https://github.com/Pseudomanifold/latex-credits) for the CRediT contributions.124 TBK and AV thank their collaboration partners from FMP for providing the data to them not only for the original study but also for this case study.References
- J. W. Scannell, A. Blanckley, H. Boldon and B. Warrington, Diagnosing the decline in pharmaceutical r&d efficiency, Nat. Rev. Drug Discovery, 2012, 11(3), 191–200 CrossRef CAS PubMed.
- I. Kola and J. Landis, Can the pharmaceutical industry reduce attrition rates?, Nat. Rev. Drug Discovery, 2004, 3(8), 711–716 CrossRef CAS PubMed.
- P. K. Singh, A. Negi, P. K. Gupta, M. Chauhan and R. Kumar, Toxicophore exploration as a screening technology for drug design and discovery: techniques, scope and limitations, Arch. Toxicol., 2016, 90(8), 1785–1802 CrossRef CAS PubMed.
- E. Lounkine, M. J. Keiser, S. Whitebread, D. Mikhailov, J. Hamon and J. L. Jenkins, et al., Large-scale prediction and testing of drug activity on side-effect targets, Nature, 2012, 486(7403), 361–367 CrossRef CAS PubMed.
- C. H. Wong, K. W. Siah and A. W. Lo, Estimation of clinical trial success rates and related parameters, Biostatistics, 2019, 20(2), 273–286 CrossRef PubMed.
- A. Lin, C. J. Giuliano, A. Palladino, K. M. John, C. Abramowicz and M. L. Yuan, et al., Off-target toxicity is a common mechanism of action of cancer drugs undergoing clinical trials, Sci Transl Med., 2019, 11(509), eaaw8412 CrossRef CAS PubMed.
- E. Lo.Piparo, A. Worth, et al., Review of qsar models and software tools for predicting developmental and reproductive toxicity, JRC Rep EUR, 2010, p. 24522 Search PubMed.
- K. Mansouri, A. Abdelaziz, A. Rybacka, A. Roncaglioni, A. Tropsha and A. Varnek, et al., Cerapp: collaborative estrogen receptor activity prediction project, Environ. Health Perspect, 2016, 124(7), 1023–1033 CrossRef CAS PubMed.
- H. Chen, O. Engkvist, Y. Wang, M. Olivecrona and T. Blaschke, The rise of deep learning in drug discovery, Drug discovery today, 2018, 23(6), 1241–1250 CrossRef PubMed.
- L. Zhang, J. Tan, D. Han and H. Zhu, From machine learning to deep learning: progress in machine intelligence for rational drug discovery, Drug discovery today, 2017, 22(11), 1680–1685 CrossRef PubMed.
- Z. Wu, B. Ramsundar, E. N. Feinberg, J. Gomes, C. Geniesse and A. S. Pappu, et al., Moleculenet: a benchmark for molecular machine learning, Chem. Sci., 2018, 9(2), 513–530 RSC.
- K. Yang, K. Swanson, W. Jin, C. Coley, P. Eiden and H. Gao, et al., Analyzing learned molecular representations for property prediction, J. Chem. Inf. Model., 2019, 59(8), 3370–3388 CrossRef CAS PubMed.
- Y. Rong, Y. Bian, T. Xu, W. Xie, Y. Wei, W. Huang, et al., Self-supervised graph transformer on large-scale molecular data, Advances in Neural Information Processing Systems, 2020, vol. 33, pp. 12559–12571 Search PubMed.
- Z. Xiong, D. Wang, X. Liu, F. Zhong, X. Wan and X. Li, et al., Pushing the boundaries of molecular representation for drug discovery with the graph attention mechanism, J. Med. Chem., 2019, 63(16), 8749–8760 CrossRef PubMed.
- T. B. Kimber, M. Gagnebin and A. Volkamer, Maxsmi: maximizing molecular property prediction performance with confidence estimation using smiles augmentation and deep learning, Artif. Intell. Life Sci., 2021, 1, 100014 CAS.
- A. Mayr, G. Klambauer, T. Unterthiner and S. Hochreiter, Deeptox: toxicity prediction using deep learning, Front. Environ. Sci., 2016, 3, 80 Search PubMed.
- C. Ji, F. Svensson, A. Zoufir and A. Bender, Emoltox: prediction of molecular toxicity with confidence, Bioinformatics, 2018, 34(14), 2508–2509 CrossRef CAS PubMed.
- H. Yang, L. Sun, W. Li, G. Liu and Y. Tang, In silico prediction of chemical toxicity for drug design using machine learning methods and structural alerts, Front. Chem., 2018, 6, 30 CrossRef PubMed.
- Y. Peng, Z. Zhang, Q. Jiang, J. Guan and S. Zhou, Top: Towards better toxicity prediction by deep molecular representation learning, in 2019 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), IEEE, 2019, pp. 318–325 Search PubMed.
- M. Zaslavskiy, S. Jégou, E. W. Tramel and G. Wainrib, Toxicblend: Virtual screening of toxic compounds with ensemble predictors, Comput. Toxicol., 2019, 10, 81–88 CrossRef.
- A. Karim, A. Mishra, M. H. Newton and A. Sattar, Efficient toxicity prediction via simple features using shallow neural networks and decision trees, ACS Omega, 2019, 4(1), 1874–1888 CrossRef CAS.
- K. V. Chuang, L. Gunsalus and M. J. Keiser, Learning molecular representations for medicinal chemistry, J. Med. Chem., 2020, 63(16), 8705–68722 CrossRef CAS.
- D. Rogers and M. Hahn, Extended-connectivity fingerprints, J. Chem. Inf.Model., 2010, 50(5), 742–754 CrossRef CAS PubMed.
- D. Weininger, Smiles, a chemical language and information system 1 introduction to methodology and encoding rules, J. Chem. Inf. Comput., 1988, 28(1), 31–36 CrossRef CAS.
- E. N. Muratov, J. Bajorath, R. P. Sheridan, I. V. Tetko, D. Filimonov, V. Poroikov, et al., Qsar without borders, Chemical Society Reviews, 2020 Search PubMed.
- M. Manica, A. Oskooei, J. Born, V. Subramanian, J. Saez Rodriguez and M. Rodriguez Martinez, Toward explainable anticancer compound sensitivity prediction via multimodal attention-based convolutional encoders, Mol. Pharm., 2019, 16(12), 4797–4806 CrossRef CAS PubMed.
- N. M. O'Boyle, Towards a universal smiles representation-a standard method to generate canonical smiles based on the inchi, J. Cheminformatics, 2012, 4(1), 22 CrossRef PubMed.
- J. Jiménez Luna, F. Grisoni and G. Schneider, Drug discovery with explainable artificial intelligence, Nat. Mach. Intell., 2020, 2(10), 573–584 CrossRef.
- R. P. Sheridan, Interpretation of qsar models by coloring atoms according to changes in predicted activity: how robust is it?, J. Chem. Inf. Model., 2019, 59(4), 1324–1337 CrossRef CAS PubMed.
- H. E. Webel, T. B. Kimber, S. Radetzki, M. Neuenschwander, M. Nazaré and A. Volkamer, Revealing cytotoxic substructures in molecules using deep learning, J. Comput.-Aided Mol. Des., 2020, 34(7), 731–746, DOI:10.1007/s10822-020-00310-4.
- Q. Ding, S. Hou, S. Zu, Y. Zhang and S. Li, Visar: an interactive tool for dissecting chemical features learned by deep neural network qsar models, Bioinformatics, 2020, 36(11), 3610–3612 CrossRef CAS PubMed.
- J. Jiménez Luna, M. Skalic, N. Weskamp and G. Schneider, Coloring molecules with explainable artificial intelligence for preclinical relevance assessment, J. Chem. Inf. Model., 2021, 61(3), 1083–1094 CrossRef PubMed.
- D. Bahdanau, K. Cho and Y. Bengio, Neural machine translation by jointly learning to align and translate, in 3rd International Conference on Learning Representations, ICLR, 2015 Search PubMed.
- Y. Gal and Z. Ghahramani, Dropout as a bayesian approximation: Representing model uncertainty in deep learning, in International conference on machine learning (ICML), PMLR, 2016. pp. 1050–1059 Search PubMed.
- M. S. Ayhan and P. Berens, Test-time data augmentation for estimation of heteroscedastic aleatoric uncertainty in deep neural networks and Proceedings of the 1st Conference on Medical Imaging with Deep Learning, MIDL, 2018 Search PubMed.
- T. N. Kipf and M. Welling, Semi-supervised classification with graph convolutional networks, in J. International Conference on Learning Representations, ICLR 2017, 2016 Search PubMed.
- H. L. Morgan, The generation of a unique machine description for chemical structures-a technique developed at chemical abstracts service, J. Chem. Doc., 1965, 5(2), 107–113 CrossRef CAS.
- T. Le, R. Winter, F. Noé and D. A. Clevert, Neuraldecipher–reverse-engineering extended-connectivity fingerprints (ecfps) to their molecular structures, Chem. Sci., 2020, 11(38), 10378–10389 RSC.
- M. Krenn, F. Häse, A. Nigam, P. Friederich and A. Aspuru.Guzik, Self-referencing embedded strings (selfies): A 100% robust molecular string representation, Mach. Learn.: Sci. Technol., 2020, 1(4), 045024 Search PubMed.
- A. Weber, J. Born and M. Rodriguez Martínez, TITAN: T-cell receptor specificity prediction with bimodal attention networks, Bioinformatics, 2021, 37(Supplement_1), i237–i244, DOI:10.1093/bioinformatics/btab294.
- V. Höll, M. Kouba, M. Dietel and G. Vogt, Stereoisomers of calcium antagonists which differ markedly in their potencies as calcium blockers are equally effective in modulating drug transport by p-glycoprotein, Biochem. Pharmacol., 1992, 43(12), 2601–2608 CrossRef PubMed.
- G. Landrum, Rdkit: Open-source cheminformatics, v. 2019’, GitHub, 2019, https://github.com/rdkit/rdkit Search PubMed.
- E. J. Bjerrum, Smiles enumeration as data augmentation for neural network modeling of molecules, arXiv, 2017, preprint arXiv:170307076 Search PubMed.
- J. Arús Pous, S. V. Johansson, O. Prykhodko, E. J. Bjerrum, C. Tyrchan and J. L. Reymond, et al., Randomized smiles strings improve the quality of molecular generative models, J. Cheminformatics, 2019, 11(1), 1–13 CrossRef PubMed.
- P. Liu, H. Li, S. Li and K. S. Leung, Improving prediction of phenotypic drug response on cancer cell lines using deep convolutional network, BMC Bioinf., 2019, 20(1), 408 CrossRef PubMed.
- J. Born, M. Manica, A. Oskooei, J. Cadow, G. Markert and M. R. Martínez, Paccmannrl: De novo generation of hit-like anticancer molecules from transcriptomic data via reinforcement learning, Iscience, 2021, 24(4), 102269 CrossRef CAS PubMed.
- P. Schwaller, T. Gaudin, D. Lanyi, C. Bekas and T. Laino, found in translation”: predicting outcomes of complex organic chemistry reactions using neural sequence-to-sequence models, Chem. Sci., 2018, 9(28), 6091–6098 RSC.
- X. Li and D. Fourches, Smiles pair encoding: A data-driven substructure tokenization algorithm for deep learning, J. Chem. Inf. Model., 2021, 61(4), 1560–1569 CrossRef CAS PubMed.
- J. Cadow, J. Born, M. Manica, A. Oskooei and M. Rodríguez Martínez, Paccmann: a web service for interpretable anticancer compound sensitivity prediction, Nucleic Acids Res., 2020, 48(W1), W502–W508 CrossRef CAS PubMed.
- J. Born, Y. Shoshan, T. Huynh, W. D. Cornell, E. J. Martin and M. Manica, On the choice of active site sequences for kinase-ligand affinity prediction, J. Chem. Inf. Model., 2022, 62(18), 4295–4299, DOI:10.1021/acs.jcim.2c00840.
- J. Born, T. Huynh, A. Stroobants, W. D. Cornell and M. Manica, Active site sequence representations of human kinases outperform full sequence representations for affinity prediction and inhibitor generation: 3d effects in a 1d model, J. Chem. Inf. Model., 2022, 62(2), 240–257, DOI:10.1021/acs.jcim.1c00889.
- A. Vaswani, N. Shazeer, N. Parmar, J. Uszkoreit, et al., Attention is all you need in Advances in Neural Information Processing Systems, 2017. pp. 5998–6008 Search PubMed.
- J. Chung, C. Gulcehre, K. Cho and Y. Bengio, Empirical evaluation of gated recurrent neural networks on sequence modeling, arXiv, 2014, preprint arXiv:14123555 Search PubMed.
- N. Vecoven, D. Ernst and G. Drion, A bio-inspired bistable recurrent cell allows for long-lasting memory, PLoS One, 2021, 16(6), e0252676 CrossRef CAS PubMed.
- D. P. Kingma, B. J. Adam, A method for stochastic optimization, in 3rd International Conference on Learning Representations, ICLR, 2015 Search PubMed.
- L. Breiman: Random forests, Machine learning, 2001, vol. 45, pp. 5–32 Search PubMed.
- G. Biau, L. Devroye and G. Lugosi, Consistency of random forests and other averaging classifiers, J. Mach. Learn. Res., 2008, 9(9) Search PubMed.
- R. L. Marchese Robinson, A. Palczewska, J. Palczewski and N. Kidley, Comparison of the predictive performance and interpretability of random forest and linear models on benchmark data sets, J. Chem. Inf. Model., 2017, 57(8), 1773–1792 CrossRef CAS PubMed.
- V. Svetnik, A. Liaw, C. Tong, J. C. Culberson, R. P. Sheridan and B. P. Feuston, Random forest: a classification and regression tool for compound classification and qsar modeling, J. Chem. Inf. Comput. Sci., 2003, 43(6), 1947–1958 CrossRef CAS PubMed.
- T. Tanimoto. An elementary mathematical theory of classification and prediction, ibm report (november, 1958), cited in: G. salton, automatic information organization and retrieval, McGraw-HillNew York, 1968 Search PubMed.
- D. K. Duvenaud, D. Maclaurin, J. Iparraguirre, R. Bombarell, T. Hirzel and A. Aspuru.Guzik, et al., Convolutional networks on graphs for learning molecular fingerprints, Adv. Neural Inf. Process Syst., 2015, 28 Search PubMed.
- S. V. N. Vishwanathan, N. N. Schraudolph, R. Kondor and K. M. Borgwardt, Graph kernels, J. Mach. Learn. Res., 2010, 11, 1201–1242 Search PubMed.
- K. M. Borgwardt and H. P. Kriegel, Shortest-path kernels on graphs, in Fifth IEEE International Conference on Data Mining (ICDM’05), IEEE, 2005, p. 8 Search PubMed.
- R. W. Floyd, Algorithm 97: shortest path, Commun. ACM, 1962, 5(6), 345 CrossRef.
- N. Shervashidze, P. Schweitzer, E. J. Van Leeuwen, K. Mehlhorn and K. M. Borgwardt, Weisfeiler-Lehman graph kernels, J. Mach. Learn. Res., 2011, 12, 2539–2561 Search PubMed.
- G. Nikolentzos and M. Vazirgiannis, Message passing graph kernels, arXiv, 2018, preprint arXiv:180802510 Search PubMed.
- J. Gilmer, S. S. Schoenholz, P. F. Riley, O. Vinyals and G. E. Dahl, Neural message passing for quantum chemistry, in International conference on machine learning, PMLR, 2017, pp. 1263–1272 Search PubMed.
- M. Togninalli, E. Ghisu, F. Llinares López, B. Rieck and K. Borgwardt: ‘Wasserstein weisfeiler-lehman graph kernels’, Advances in Neural Information Processing Systems, 2019, p. 32 Search PubMed.
- M. A. Hearst, S. T. Dumais, E. Osuna, J. Platt and B. Scholkopf, Support vector machines, IEEE Intell. Syst., 1998, 13(4), 18–28 Search PubMed.
- J. Kazius, R. McGuire and R. Bursi, Derivation and validation of toxicophores for mutagenicity prediction, J. Med. Chem., 2005, 48(1), 312–320 CrossRef CAS PubMed.
- Daylight, Chemical.Information.Systems, I. ‘Smarts™—a language for describing molecular patterns, 2007 Search PubMed.
- N. T. Program, et al., A national toxicology program for the 21st century: A roadmap for the future, National Toxicology Program: Research Triangle Park, NC, USA, 2004 Search PubMed.
- R. R. Tice, C. P. Austin, R. J. Kavlock and J. R. Bucher, Improving the human hazard characterization of chemicals: a tox21 update, Environ. Health Perspect., 2013, 121(7), 756–765 CrossRef PubMed.
- G. Kerdivel, D. Habauzit and F. Pakdel, Assessment and molecular actions of endocrine-disrupting chemicals that interfere with estrogen receptor pathways, 2013 Search PubMed.
- C. Stocco, Tissue physiology and pathology of aromatase, Steroids, 2012, 77(1–2), 27–35 CrossRef CAS PubMed.
- E. Gottlieb, S. Armour, M. Harris and C. Thompson, Mitochondrial membrane potential regulates matrix configuration and cytochrome c release during apoptosis, Cell Death Differ., 2003, 10(6), 709–717 CrossRef CAS PubMed.
- K. y. Lee, H. Fu, M. I. Aladjem and K. Myung, Atad5 regulates the lifespan of dna replication factories by modulating pcna level on the chromatin, J. Cell Biol., 2013, 200(1), 31–44 CrossRef CAS PubMed.
- S. Tyagi, P. Gupta, A. S. Saini, C. Kaushal and S. Sharma, The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases, J. Adv. Pharm. Technol. Res., 2011, 2(4), 236 CrossRef CAS PubMed.
- A. Raghunath, K. Sundarraj, R. Nagarajan, F. Arfuso, J. Bian and A. P. Kumar, et al., Antioxidant response elements: discovery, classes, regulation and potential applications, Redox Biol., 2018, 17, 297–314 CrossRef CAS PubMed.
- I. A. Murray, A. D. Patterson and G. H. Perdew, Aryl hydrocarbon receptor ligands in cancer: friend and foe, Nat. Rev. Cancer, 2014, 14(12), 801–814 CrossRef CAS PubMed . Available from: https://www.nature.com/articles/nrc3846.
- K. M. Ryan, A. C. Phillips and K. H. Vousden, Regulation and function of the p53 tumor suppressor protein, Curr. Opin. Cell Biol., 2001, 13(3), 332–337 CrossRef CAS PubMed . Available from: http://www.sciencedirect.com/science/article/pii/S0955067400002167.
- K. Hientz, A. Mohr, D. Bhakta Guha and T. Efferth, The role of p53 in cancer drug resistance and targeted chemotherapy, Oncotarget, 2017, 8(5), 8921 CrossRef PubMed.
- G. Subramanian, B. Ramsundar, V. Pande and R. A. Denny, Computational modeling of β-secretase 1 (bace-1) inhibitors using ligand based approaches, J. Chem. Inf. Model., 2016, 56(10), 1936–1949 CrossRef CAS PubMed.
- M. Kuhn, I. Letunic, L. J. Jensen and P. Bork, The sider database of drugs and side effects, Nucleic Acids Res., 2016, 44(D1), D1075–D1079 CrossRef CAS PubMed.
- H. Altae Tran, B. Ramsundar, A. S. Pappu and V. Pande, Low data drug discovery with one-shot learning, ACS Cent. Sci., 2017, 3(4), 283–293 CrossRef CAS PubMed.
- K. M. Gayvert, N. S. Madhukar and O. Elemento, A data-driven approach to predicting successes and failures of clinical trials, Cell Chem. Biol., 2016, 23(10), 1294–1301 CrossRef CAS PubMed.
- A. V. Artemov, E. Putin, Q. Vanhaelen, A. Aliper, I. V. Ozerov and A. Zhavoronkov, Integrated deep learned transcriptomic and structure-based predictor of clinical trials outcomes, BioRxiv, 2016, p. 095653 Search PubMed.
- I. F. Martins, A. L. Teixeira, L. Pinheiro and A. O. Falcao, A bayesian approach to in silico blood–brain barrier penetration modeling, J. Chem. Inf. Model., 2012, 52(6), 1686–1697 CrossRef CAS PubMed.
- Aids antiviral screen data, 2017. http://wiki.nci.nih.gov/display/NCIDTPdata/AIDS+Antiviral+Screen+Data Search PubMed.
- R. Huang, M. Xia, D. T. Nguyen, T. Zhao, S. Sakamuru and J. Zhao, et al., Tox21challenge to build predictive models of nuclear receptor and stress response pathways as mediated by exposure to environmental chemicals and drugs, Frontiers in Environmental Science, 2016, 3, 85 CrossRef.
- B. Ramsundar, P. Eastman, P. Walters and V. Pande, Deep learning for the life sciences: applying deep learning to genomics, microscopy, drug discovery, and more, O'Reilly Media, 2019 Search PubMed.
- M. Lisurek, B. Rupp, J. Wichard, M. Neuenschwander, J. P. von Kries and R. Frank, et al., Design of chemical libraries with potentially bioactive molecules applying a maximum common substructure concept, Mol. Diversity, 2010, 14(2), 401–408 CrossRef CAS PubMed.
- L. Bennett, B. Melchers and B. Proppe, Curta: A general-purpose high-performance computer at ZEDAT, Freie Universität Berlin’., 2020, DOI:10.17169/refubium-26754.
- N. M. Kriege, F. D. Johansson and C. Morris, A survey on graph kernels, Appl. Netw. Sci., 2020, 5(1), 1–42 CrossRef.
- H. J. M. Verhaar, C. J. van Leeuwen and J. L. M. Hermens, Classifying environmental pollutants, Chemosphere, 1992, 25(4), 471–491 CrossRef CAS.
- J. L. M. Hermens, Electrophiles and acute toxicity to fish, Environ. Health Perspect., 1990, 87, 219–225 CrossRef CAS PubMed.
- M. Nendza, A. Wenzel, M. Müller, G. Lewin, N. Simetska and F. Stock, et al., Screening for potential endocrine disruptors in fish: evidence from structural alerts and in vitro and in vivo toxicological assays, Environ. Sci. Eur., 2016, 28(1), 26 CrossRef PubMed . Available from: http://enveurope.springeropen.com/articles/10.1186/s12302-016-0094-5.
- I. Sushko, E. Salmina, V. A. Potemkin, G. Poda and I. V. Tetko, Toxalerts: a web server of structural alerts for toxic chemicals and compounds with potential adverse reactions, J. Chem. Inf. Model., 2012, 52(8), 2310–2316 CrossRef CAS PubMed.
- M. Karimi, D. Wu, Z. Wang and Y. Shen, Deepaffinity: interpretable deep learning of compound–protein affinity through unified recurrent and convolutional neural networks, Bioinformatics, 2019, 35(18), 3329–3338 CrossRef CAS PubMed.
- M. Tsubaki, K. Tomii and J. Sese, Compound–protein interaction prediction with end-to-end learning of neural networks for graphs and sequences, Bioinformatics, 2019, 35(2), 309–318 CrossRef CAS PubMed.
- S. Li, F. Wan, H. Shu, T. Jiang, D. Zhao and J. Zeng, Monn: a multi-objective neural network for predicting compound-protein interactions and affinities, Cell Syst., 2020, 10(4), 308–322 CrossRef CAS.
- K. Preuer, G. Klambauer, F. Rippmann, S. Hochreiter and T. Unterthiner, ‘Interpretable deep learning in drug discovery’, Explainable AI: interpreting, explaining and visualizing deep learning, 2019, pp. 331–345 Search PubMed.
- M. Withnall, E. Lindelöf, O. Engkvist and H. Chen, Building attention and edge message passing neural networks for bioactivity and physical–chemical property prediction, J. Cheminformatics, 2020, 12(1), 1–18 CrossRef CAS PubMed.
- W. X. Shen, X. Zeng, F. Zhu, C. Qin, Y. Tan and Y. Y. Jiang, et al., Out-of-the-box deep learning prediction of pharmaceutical properties by broadly learned knowledge-based molecular representations, Nat. Mach. Intell., 2021, 3(4), 334–343 CrossRef.
- S. Kearnes, K. McCloskey, M. Berndl, V. Pande and P. Riley, Molecular graph convolutions: moving beyond fingerprints, J. Comput.-Aided Mol. Des., 2016, 30(8), 595–608 CrossRef CAS PubMed.
- K. Schütt, P. J. Kindermans, H. E. Sauceda Felix, S. Chmiela, A. Tkatchenko and K. R. Müller, Schnet: A continuous-filter convolutional neural network for modeling quantum interactions, Adv. Neural Inf. Process Syst., 2017, 30 Search PubMed.
- W. Hu, B. Liu, J. Gomes, M. Zitnik, P. Liang, V. S. Pande, et al., Strategies for pre-training graph neural networks, 8th International Conference on Learning Representations, ICLR 2020, 2020 Search PubMed.
- C. Lu, Q. Liu, C. Wang, Z. Huang, P. Lin and L. HeMolecular property prediction: A multilevel quantum interactions modeling perspective, in Proceedings of the AAAI Conference on Artificial Intelligence, 2019, vol. 33, pp. 1052–1060 Search PubMed.
- S. Liu, M. F. Demirel and Y. Liang, N-gram graph: Simple unsupervised representation for graphs, with applications to molecules, Adv. Neural Inf. Process Syst., 2019, 32 Search PubMed.
- D. Baumann and K. Baumann, Reliable estimation of prediction errors for qsar models under model uncertainty using double cross-validation, J. Cheminformatics, 2014, 6(1), 1–19 CrossRef PubMed.
- I. Cortés Ciriano and A. Bender, Deep confidence: a computationally efficient framework for calculating reliable prediction errors for deep neural networks, J. Chem. Inf. Model., 2018, 59(3), 1269–1281 CrossRef PubMed.
- C. Corbiere, N. Thome, A. Saporta, T. H. Vu, M. Cord and P. Perez, Confidence estimation via auxiliary models, IEEE Trans. Pattern Anal. Mach. Intell., 2021 Search PubMed.
- T. M. Dhameliya, S. J. Chudasma, T. M. Patel and B. P. Dave, A review on synthetic account of 1, 2, 4-oxadiazoles as anti-infective agents, Mol. Diversity, 2022, 1–14 Search PubMed.
- B. Ruan, X. Tang, W. Guo, Y. Hu and L. Chen, Synthesis and biological evaluation of novel phthalide analogs-1, 2, 4-oxadiazole hybrids as potential anti-inflammatory agents, Chem. Biodiversity, 2022, 19(8), e202200039 CrossRef CAS PubMed.
- B. L. Sun, Y. Y. Wang, S. Yang, M. T. Tu, Y. Y. Shao and Y. Hua, et al., Benzamides substituted with quinoline-linked 1, 2, 4-oxadiazole: Synthesis, biological activity and toxicity to zebrafish embryo, Molecules, 2022, 27(12), 3946 CrossRef CAS PubMed.
- Y. M. Rocha, E. P. Magalhães, M. de Medeiros Chaves, M. Machado Marinho, V. Nascimento e Melo de Oliveira and R. Nascimento de Oliveira, et al., Antiparasitary and antiproliferative activities in vitro of a 1, 2, 4-oxadiazole derivative on trypanosoma cruzi, Parasitol. Res., 2022, 1–16 Search PubMed.
- Z. Alperstein, A. Cherkasov and J. T. Rolfe, All smiles variational autoencoder, arXiv, 2019, preprint arXiv:190513343 Search PubMed.
- I. V. Tetko, P. Karpov, E. Bruno, T. B. Kimber and G. GodinAugmentation is what you need!, in International Conference on Artificial Neural Networks, Springer, 2019, pp. 831–835 Search PubMed.
- N. L. Dang, T. B. Hughes, G. P. Miller and S. J. Swamidass, Computational approach to structural alerts: furans, phenols, nitroaromatics, and thiophenes, Chem. Res. Toxicol., 2017, 30(4), 1046–1059 Search PubMed.
- B. J. Bongers, A. P. IJzerman and G. J. Van.Westen: ‘Proteochemometrics–recent developments in bioactivity and selectivity modeling’, Drug Discovery Today: Technologies, 2020 Search PubMed.
- M. Manica, J. Cadow, D. Christofidellis, A. Dave, J. Born, D. Clarke, et al., Gt4sd: Generative toolkit for scientific discovery, NPJ Computational Materials, in press Search PubMed.
- J. Born, M. Manica, J. Cadow, G. Markert, N. A. Mill and M. Filipavicius, et al., Data-driven molecular design for discovery and synthesis of novel ligands: a case study on SARS-CoV-2, Mach. Learn.: Sci. Technol., 2021, 2(2), 025024 Search PubMed . Available from: https://iopscience.iop.org/article/10.1088/2632-2153/abe808.
- N. Janakarajan, J. Born, M. Manica, A fully differentiable set autoencoder, in Proceedings of the 28th ACM SIGKDD Conference on Knowledge Discovery and Data Mining. KDD ’22., Association for Computing Machinery, New York, NY, USA, 2022. pp. 3061–3071. DOI:10.1145/3534678.3539153.
- B. Rieck, Latex-credits, BSD-3-Clause, https://github.com/Pseudomanifold/latex-credits Search PubMed.
Footnotes |
| † The code for reproduction of our experiments is available at https://github.com/PaccMann/toxsmi, and the trained models were integrated into the GT4SD library and can be used via a web-app: https://huggingface.co/spaces/GT4SD/molecular_properties. |
| ‡ Electronic supplementary information (ESI) available. Appendix A1, 1.1, 1.2 and 1.3. Fig. A1–A5. Tables A1–A3. See DOI: https://doi.org/10.1039/d2dd00099g |
| § Equal contribution. |
| ¶ Co-senior authors. |
| || https://pypi.org/project/pytoda |
| ** Even though class probabilities are generally insufficient confidence estimators,34 they are frequently misused in practice for this task. |
| †† See: https://paperswithcode.com/sota/drug-discovery-on-tox21 |
| This journal is © The Royal Society of Chemistry 2023 |