
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Automated patent extraction powers generative modeling in focused chemical spaces†
Akshay
Subramanian‡
 a,
Kevin
P. Greenman‡
b,
Alexis
Gervaix
c,
Tzuhsiung
Yang
d and
Rafael
Gómez-Bombarelli
*a
a,
Kevin
P. Greenman‡
b,
Alexis
Gervaix
c,
Tzuhsiung
Yang
d and
Rafael
Gómez-Bombarelli
*a
aDepartment of Materials Science and Engineering, Massachusetts Institute of Technology, Cambridge, MA, USA. E-mail: rafagb@mit.edu
bDepartment of Chemical Engineering, Massachussets Institute of Technology, Cambridge, MA, USA
cSwiss Airtainer SA, Yverdons-les-Bains, Vaud, Switzerland
dDepartment of Chemistry, National Tsing Hua University, Hsinchu City, Taiwan
First published on 15th June 2023
Abstract
Deep generative models have emerged as an exciting avenue for inverse molecular design, with progress coming from the interplay between training algorithms and molecular representations. One of the key challenges in their applicability to materials science and chemistry has been the lack of access to sizeable training datasets with property labels. Published patents contain the first disclosure of new materials prior to their publication in journals, and are a vast source of scientific knowledge that has remained relatively untapped in the field of data-driven molecular design. Because patents are filed seeking to protect specific uses, molecules in patents can be considered to be weakly labeled into application classes. Furthermore, patents published by the US Patent and Trademark Office (USPTO) are downloadable and have machine-readable text and molecular structures. In this work, we train domain-specific generative models using patent data sources by developing an automated pipeline to go from USPTO patent digital files to the generation of novel candidates with minimal human intervention. We test the approach on two in-class extracted datasets, one in organic electronics and another in tyrosine kinase inhibitors. We then evaluate the ability of generative models trained on these in-class datasets on two categories of tasks (distribution learning and property optimization), identify strengths and limitations, and suggest possible explanations and remedies that could be used to overcome these in practice.
1 Introduction
The efficient navigation of chemical space for the design of novel candidate molecules has long been of interest to chemists and materials scientists. With the rapid surge in interest for data-driven approaches, deep generative models have emerged as an exciting avenue for inverse molecular design.1,2 Progress in this field has come from the interplay between training algorithms and molecular representations. Over the last few years, approaches have used autoregressive, latent variable and reinforcement learning (RL) algorithms to generate string,3–7 and graph8–11 representations of molecules. While fully unsupervised models can be trained on large unlabeled data (for instance the 100+ million known, individually synthesized molecules from PubChem), inverse molecular design requires some form of supervision to steer generation towards high-performance molecules at the extremes of the property distribution.12 One of the key challenges in the applicability of such inverse design models to materials science and chemistry has been the lack of accessibility to sizeable labeled training datasets in these fields.13Published patents are an important source of scientific knowledge since the discovery of new materials and molecular candidates are disclosed in patents, years before their publication in scientific journals.14,15 Patent authorities such as the United States Patent and Trademark Office (USPTO), European Patent Office (EPO), Japanese Patent Office (JPO), and World Intellectual Property Organization (WIPO) make published patents accessible through their web interfaces. In the past decade, there has been significant progress in extracting and collating information from these sources programmatically to create large databases of chemical compounds,16 and reactions.17 This large body of extracted knowledge has immense potential in feeding ‘data hungry’ deep learning models, but has remained relatively untapped in the field of molecular design.
Since patents are filed seeking protection within a given application, they are thematically labeled into domains. This makes it relatively simple to extract domain-specific molecular structures. Moreover, they are likely to be high-performance since they merited the investment of a patent application, which allows us to create domain-specific generative models by training exclusively on molecules known to belong to the desired class. Our hypothesis is that training generative models on these smaller, but more meaningful datasets can automatically steer generation towards in-class high-performance molecules.
All post-2001 chemistry patents published by the USPTO contain ChemDraw CDX, MDL, and TIFF files of chemical structures, as required by the Complex Work Unit (CWU) Pilot Program.18 This makes chemical structures more accessible in a computer readable format for large scale mining and screening efforts. In our work, we attempt to bridge the gap between these bulk data sources and data-driven chemical design, by developing an automated pipeline to isolate chemical structures from USPTO patents based on relevance to user-defined keywords, and demonstrating their utility as training data for deep generative models for molecular design. We choose three model types JTVAE,9 RNN + SELFIES,19,20 and REINVENT + SELFIES7 to explore a variety of representations (graph, SELFIES,21 and SELFIES respectively) and training algorithms (latent variable, autoregressive, and RL respectively), and show their applicability to learn data distributions in two patent-mined datasets that explore very different areas of the chemical space, i.e., organic photodiodes (OPD) and tyrosine kinase inhibitors (TKI).
We then test the ability of these models to perform property optimization in each of the following cases: (1) the property being optimized can be predicted accurately and cheaply, (2) oracle property predictor is expensive, so we only have access to a proxy neural network predictor trained on oracle property data.22–24 In the TKI case, we optimize for high structural similarity to held-out, FDA-approved TKI molecules. This is a means to test the ability of models to optimize a robust, well-defined objective function with a relatively narrow solution space. This is an example of case 1 since we can calculate the similarity between molecules cheaply without the need for an approximator. In the OPD case, we choose our optimization objective to be the identification of organic molecules with low optical gaps. This is an example of case 2 since we approximate expensive DFT-computed optical gaps with a neural network predictor. Materials with low optical gaps, especially those that are sensitive to wavelengths of light in the near infrared (NIR) region of the spectrum have seen a growing interest due to their ability to utilize a larger portion of the solar spectral range which was previously difficult to access. Their applications are diverse ranging from military equipment to biomedical and semi-transparent devices.25–28
The key observations we make through our experiments are summarized as follows: (1) we identify that patent-mined datasets offer the ability to create focused in-domain datasets of high-performing molecular structures. Training generative models on these datasets allows us to create in-domain generators that can generate novel candidates that model property distributions of the training data well. This offers a way to bootstrap focused domains of chemical space with limited human intervention. (2) Property optimization towards the edges of the training data distribution can be effective if we have access to a cheap oracle predictor, but is challenging when proxy neural network approximators are used. Proxy predictors are brittle (have the tendency to be adversarially attacked in our RL experiments), and difficult to train accurately end-to-end (learning properties from compressed latent space in JTVAE is difficult).
2 Methods
2.1 Pipeline overview
Our overall pipeline consists of six steps: (1) download patents from USPTO, (2) parse chemistry patents, (3) shortlist patents based on keywords, (4) standardize data and add to our in-house database, (5) property labeling for supervised property optimization tasks (DFT calculated optical gaps for OPD, and similarity to FDA-approved drugs for TKI), and (6) generative modeling for distribution learning (unsupervised) and property optimization (semi-supervised). Fig. 1 shows a diagrammatic illustration of all steps involved. We make publicly available the code utilized in steps 1, 2, 3 and 6 along with this paper (URLs provided in Data availability section). Step 4 involved storage of all data in a database, followed by de-duplication of SMILES strings29 and simple post-processing steps as described in Section S2 in the ESI.† A detailed description of procedures used in step 5 are provided in Section 2.3. These steps can be replaced by any form of data storage and property labeling technique depending on the chosen domain. An open source database framework similar to the one we used can be found at ref. 30.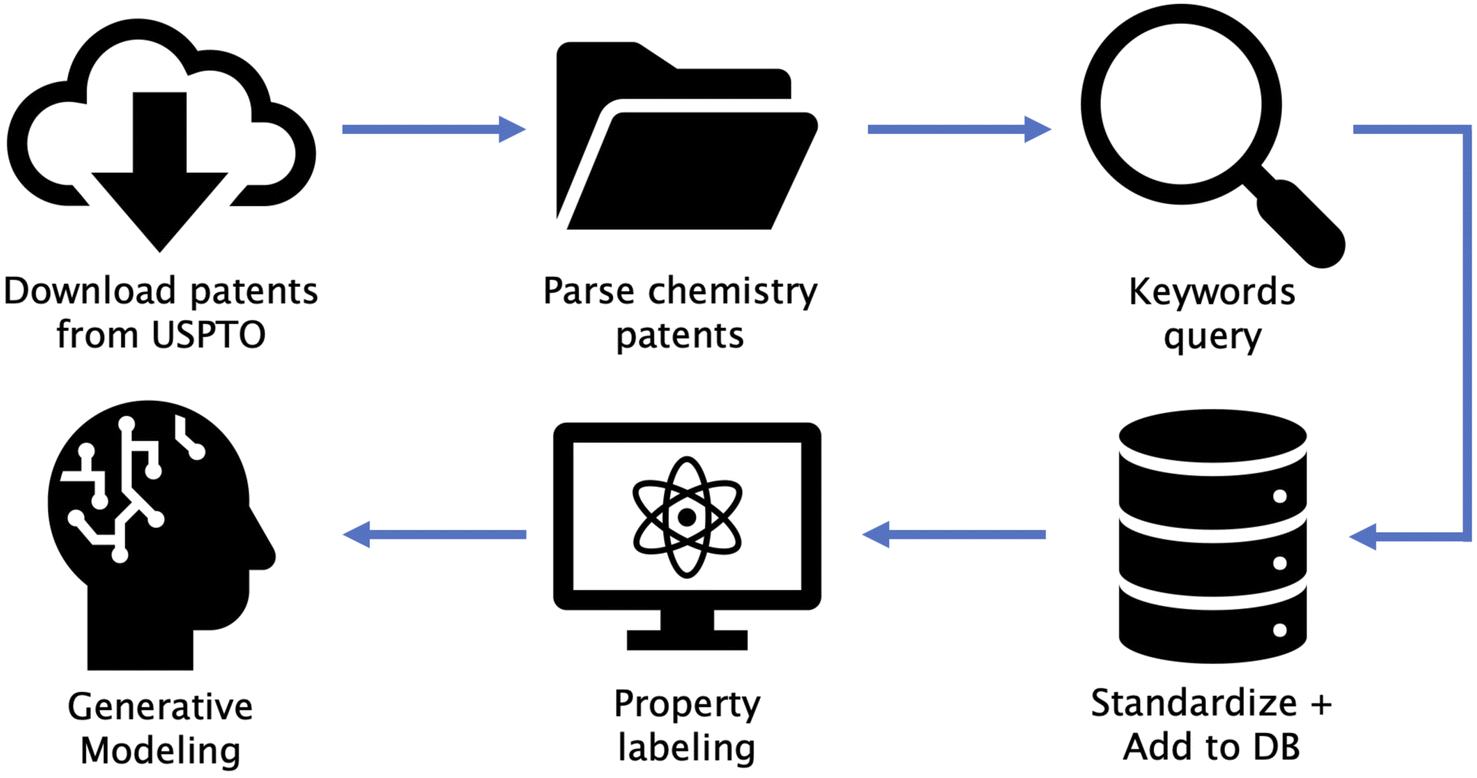 | ||
| Fig. 1 Diagram of the workflow. Patents are downloaded from USPTO, and chemistry patents are isolated. Keyword-based search is then performed to filter relevant patents and corresponding SMILES strings. A subset of molecules chosen based on computational budget are then labeled with properties. Generative models are trained to model the data distribution, which can be sampled to suggest novel candidates. | ||
2.2 Patent extraction
All granted USPTO patents from 2001 onward are available for download in weekly archives from the agency's Bulk Data Storage System (BDSS) at https://bulkdata.uspto.gov/data/patent/grant/redbook/%3cYEAR%3e/. We downloaded all of these archives from the BDSS using Python scripts by March 1, 2022. The compressed file size of all downloads was approximately 1.83 TB, including between 30 and 200 GB for each individual year. Next, we filtered out all patents that did not contain molecular structures in the form of CDX files. We encountered some difficulties in this filtering step with a subset of patent years due to inconsistent formatting and directory structures in the USPTO data (please refer to Section S1 for details†). For the remaining chemistry-related patents, we used RDKit31 to convert MOL files to SMILES strings. The number of new, unique SMILES strings extracted per year using this method are shown in Fig. 2(a). We queried all chemistry-related patents by searching for keywords in each XML file. The TKI molecules shown in Fig. 2(b) were found using the keywords “tyrosine kinase inhibitor”, and the OPD molecules in Fig. 2(c) are the result of querying for “organic electronic”, “photodiode”, and “organic photovoltaic”. Any Markush structures in the dataset were filled in with ethyl groups because the particular substituents for each core molecule are not stored in a structured format that could be accessed without natural language processing; this included 17% of molecules from the OPD query and 11% of molecules from the TKI query. Thus, we generated a list of domain-relevant SMILES strings related to each set of keywords. More details on post-processing/filtering applied to the data are provided in Section S2.†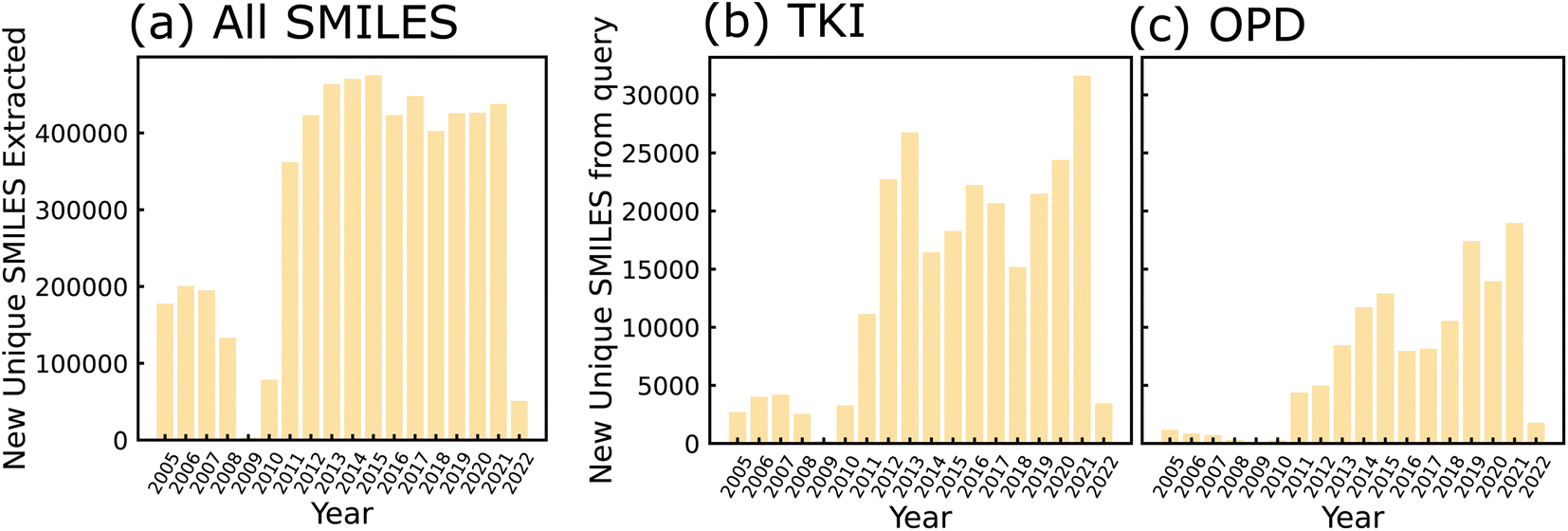 | ||
| Fig. 2 Bar charts depicting number of SMILES strings extracted as a function of publishing year. Strings extracted from patents published between 2005 and 2022 (a) before keyword-based filtering, (b) after application of TKI-based keyword search and (c) after application of OPD-based keyword search. SMILES were de-duplicated after sanitization by RDKit, such that all molecules within a given year are unique, and any molecule counted in a given year will not be counted in any future years. Years 2001–2004 are not shown and years 2008–2010 are incomplete due to inconsistencies in patent formatting (see Section S1 for details†). | ||
2.3 Property labeling
2.4 Generative modeling
For distribution-learning tasks, we evaluated models on the GuacaMol distribution learning benchmark metrics: validity, uniqueness, novelty, KL divergence and Frechet ChemNet distance.40,41 We also visualized the ground-truth property distribution of the sampled data and compared it with that of the training data. A close match between the two is an indicator of success in learning the training data distribution. For property optimization, we performed a similar visualization. Here, a shift in distribution towards higher values of the objective function is an indicator of good performance. Finally, to test the value of domain-focused training on property optimization, we compared the patent-trained models against baseline models that were trained on the ZINC dataset42 but optimized for OPD and TKI properties. It is considered good performance if the domain-trained models generate molecules with more optimal properties than the generic model trained on the ZINC dataset. This would suggest that the structural priors imposed on the models by training on the domain-specific patent datasets reflect in more optimal properties for that domain. More specifics on the task formulation for each dataset are given below.
For OPD tasks, the patent-mined OPD molecules were used as the training dataset. The property of interest in the distribution learning tasks was the DFT-computed optical gaps of sampled molecules. Since our aim was to generate molecular candidates with low optical gaps, the negative of the optical gaps as predicted by a proxy neural network predictor was used as the objective function which was maximized in the property-optimization tasks.
For TKI tasks, the patent-mined TKI dataset was used as the training dataset. The property of interest in the distribution learning tasks was the similarity between sampled molecules and Erlotinib, an FDA-approved inhibitor, to gauge the model's ability to optimize a robust, well-defined objective function with a relatively narrow solution space. This quantity was also used as the objective function which was maximized in the property-optimization tasks. In addition to the tasks described earlier in this section, an additional distribution learning task was introduced for this dataset. Molecules sampled from models trained on TKI and ZINC datasets, and 27 held-out FDA-approved TKI molecules were projected on a 2-dimensional space with Principal Component Analysis (PCA). Samples from TKI-trained models lying closer than the ZINC-trained samples to the held-out molecules, would indicate that the models have accurately learned information about molecular structure from the training dataset. It is a way to test the utility that training on domain-focused data (TKI-patents) has over training on publicly accessible large databases (ZINC) that have a similar chemical space (drug-like molecules) but are less-focused on the domain of interest. Morgan fingerprints of size 2048 and computed with radius 2 was the molecular representation used during PCA.
Recurrent Neural Networks (RNNs) have been shown to be simple but powerful text-based models for distribution modeling tasks in molecules.43 They are trained using an auto-regressive training strategy where the next token is predicted at every time-step. The implementation from the MOSES Benchmarking platform19 was used with some modifications pertaining to change in representation from SMILES to SELFIES. The trained RNN can be sampled by feeding a BOS (beginning of sentence) token, and sampling the probability distribution predicted by the model autoregressively. An LSTM20 network with 3 hidden layers and dropout probability of 0.2 between layers was used, with a final linear layer to transform the LSTM output into the required output sequence size. All LSTM hidden layers and the final linear layer were of size 768, and a learning rate of 1 × 10−3 was used for the Adam optimizer.
Junction Tree Variational Autoencoder (JTVAE) is a graph-based generative model that learns to sequentially decode graph substructures using Message Passing Neural Networks (MPNNs), and combine them to form complete molecular structures.9 It maintains a vocabulary of substructures decomposed from the training data, that are used during the decoding step to ensure validity of generated molecules. The model is trained by training the encoder, decoder and property predictors end-to-end with a multi-task loss function. Once trained, the latent space can be either randomly sampled or optimized by utilizing gradients from the property predictors. In both cases, the sampled latent vectors are passed through the decoder to obtain molecular candidates. A graph Message Passing Network (MPN) with 3 layers was used in the graph encoder, and a graph GRU44 network with 20 layers was used in the tree encoder, to form a concatenated latent vector of size 56. A learning rate of 1 × 10−3 that was set to decay exponentially during the course of training was used for the Adam optimizer.45 More details given in Section S4.†
REINVENT is a policy based Reinforcement Learning (RL) approach that learns to generate molecular structures optimized with a chosen objective function.7 Training is performed in two steps: (1) a Prior RNN is pre-trained on a language modeling task, i.e., learning to predict the next token of the sequence by maximizing the likelihood on the training dataset. (2) Then, an augmented likelihood function is defined to be the sum of the prior likelihood and a score indicating the desirability of the sequence. The agent, which is initialized with the prior RNN weights, is then fine-tuned to minimize the squared difference between the agent likelihood and the augmented likelihood on samples drawn from the agent RNN. Sampling from the trained model is performed in identical fashion to RNN (described in previous paragraph). We once again use SELFIES representations of molecules. The agent RNN was composed of three GRU cells,44 each of size 512, followed by a linear output layer. Pre-training and fine-tuning were carried out using an Adam optimizer with learning rates of 1 × 10−4 and 5 × 10−4 respectively. We retained the same architectural choices used by Olivecrona et al. since our task of similarity-based optimization is nearly identical to the similarity guided structure generation experiments described in their work.
3 Results and discussions
3.1 Distribution learning
Table 1 compares the scores of the RNN + SELFIES and JTVAE models on the GuacaMol distribution learning benchmarks. Both models were able to generate molecules with relatively high validity, uniqueness and KL divergence scores. We however found that JTVAE is superior to RNN + SELFIES in novelty scores, and both models perform relatively poorly on Frechet ChemNet distance scores. These observations may both be characteristics of the training datasets that we use being smaller and more domain-focused than the larger and more diverse drug datasets that have been benchmarked on these metrics in the past.| Model | Metric | Dataset | |
|---|---|---|---|
| TKI | OPD | ||
| a While rdkit does not process one generated molecule as valid, it is formally valid with bivalent lithium forming two covalent bonds. | |||
| RNN + SELFIES (random sample) | Validity | 1.00 | 0.99a |
| Uniqueness | 0.99 | 0.99 | |
| Novelty | 0.55 | 0.58 | |
| KL divergence | 0.98 | 0.96 | |
| Frechet ChemNet distance | 0.60 | 0.61 | |
| JTVAE (random sample) | Validity | 1.00 | 1.00 |
| Uniqueness | 1.00 | 0.99 | |
| Novelty | 1.00 | 0.89 | |
| KL divergence | 0.75 | 0.87 | |
| Frechet ChemNet distance | 0.32 | 0.28 | |
As can be seen in sub-figures (a), (b), (d), and (e) of Fig. 3, both models generated molecules whose properties matched well with the training dataset. It can also be observed that RNN + SELFIES is able to match the distributions better than the JTVAE, which conforms with the observations made by ref. 43. Additionally, sub-figures (c) and (f) show that samples from TKI-trained models lie closer to held-out FDA-approved inhibitors than ZINC-trained samples, which indicates that both models have been able to learn structural information from the training datasets.
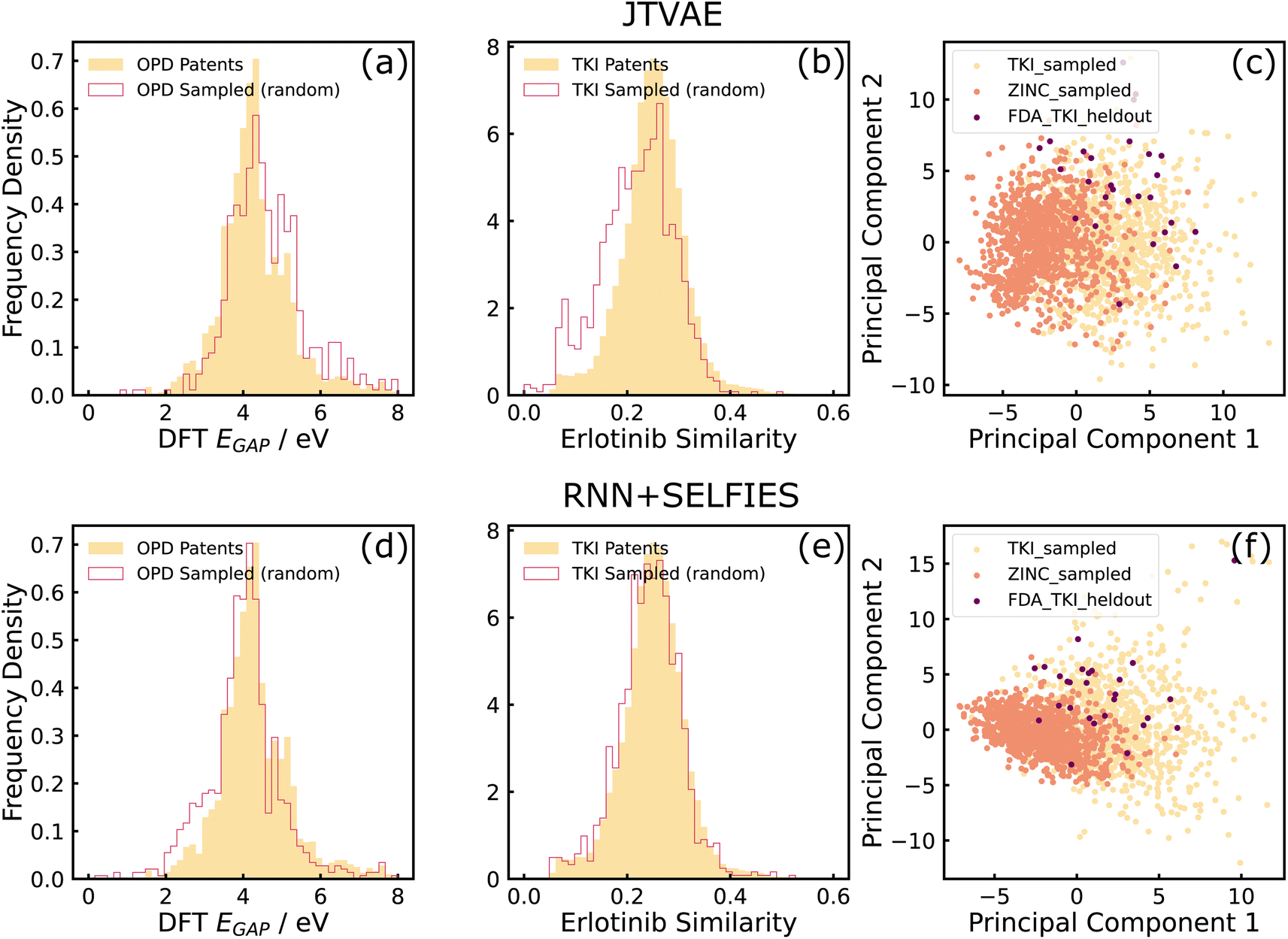 | ||
| Fig. 3 Results on distribution learning tasks. (a) and (b) show the property distributions of JTVAE-sampled molecules in comparison to training data properties, on OPD and TKI datasets respectively. (d) and (e) show the same distributions for molecules sampled from RNN + SELFIES. (c) and (f) show PCA projections of molecules randomly sampled from TKI-trained and ZINC-trained models, and held-out FDA approved inhibitors, for JTVAE and RNN + SELFIES respectively. | ||
From these results, we conclude that the deep generative models explored in this work are effective tools to model property distributions of arbitrary small, chemically focused, training datasets automatically extracted from the patent literature. The models can thus sample novel, in-distribution molecular structures that resemble the training data in terms of structure and properties. Furthermore, this suggests that domain-specific, focused chemical spaces can be boostrapped automatically from the literature without user-defined heuristics for the domain, as evidenced by the GuacaMol distribution learning benchmarks in two very distinct chemical spaces.
3.2 Property optimization
We evaluated generative models trained on patent-extracted, domain-focused datasets for property optimization. We evaluated REINVENT + SELFIES, which uses reinforcement learning and a string-based representation and JTVAE, which performs optimization in the latent space and decodes locally optimal molecules, under this category of tasks. We identified that property optimization tasks towards edges of the training data distribution are challenging for a variety of reasons. We observed from our RL experiments that optimizers may push the designs out of the training domain which was particularly acute when a neural network predictor was used as a proxy for the oracle property. Here, the generative model can be thought of as performing an adversarial attack on the poorly-covered areas of the predictor. From our VAE experiments, we observed that it is sometimes challenging for proxy predictors to learn properties from compressed latent representations, and the unreliable objective function thereby leads to challenges in latent space optimization.Both these challenges arise from coupling generation and property optimization end-to-end. By instead splitting these into two separate steps of random sampling and post hoc filtering, we observed better shifts in property histograms. More details on the post hoc filtering approach are provided below in Section 3.2.1.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20:20) was used to train, tune and evaluate the model.
:20:20) was used to train, tune and evaluate the model.
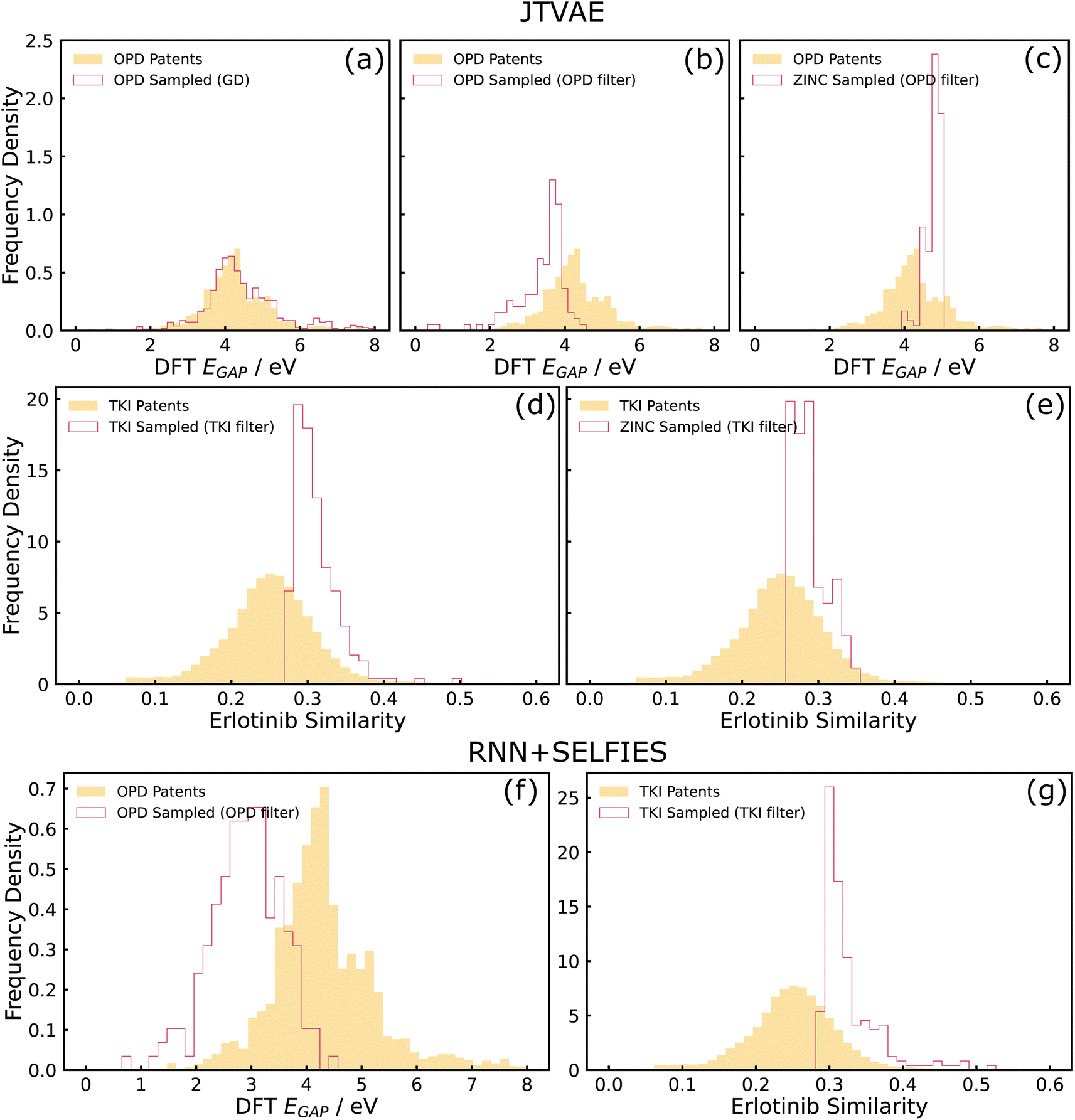 | ||
| Fig. 4 Results on property optimization tasks. (a)–(e) show results for JTVAE, while (f) and (g) show results for RNN + SELFIES. (a) and (b) show OPD property distributions of molecules sampled by gradient descent (GD), and post hoc filter respectively in comparison to training data properties. (c) shows the property distribution obtained by applying an OPD post hoc filter to samples drawn from a ZINC-trained model. (d) and (e) are analogous to (b) and (c) but on TKI instead of OPD. (f) and (g) are analogous to (b) and (d) but on RNN + SELFIES instead of JTVAE. | ||
Apart from the described issue pertaining to the poor predictive performance of the MLP, there could be other potential reasons for the failure of gradient descent on the latent space. One possibility is the presence of cascading effects. The unreliability of the MLP could have caused the points reached by gradient descent (on the latent space) to be outside the data distribution that the decoder saw during training, causing the decoder to be unreliable and collapse to a distribution more similar to the training data. One way to investigate this failure mode in the future could be the use of decoder uncertainty estimation techniques to identify such points and restrict samples to low-uncertainty regions of the decoder.50 Another possibility is that the latent space manifold of the trained model was “rough” with respect to the MLP-predicted property, rendering optimization techniques such as gradient descent challenging. This could be investigated in more depth by evaluating the ‘roughness' of the latent space with metrics such as roughness index (ROGI).23 Therefore, it should be noted that the coupled interactions between generators and property predictors is a complicated problem, and utilizing approaches such as the post hoc filter could be relatively simple remedies to these pitfalls even without a detailed knowledge of the failure mode. Demonstration of post hoc filter with another model (RNN + SELFIES) is shown in Fig. 4(f), which can be used as a remedy for the adversarial attack issues observed in the REINVENT example again arising from coupling of generators (RNN) and property optimizers (RL).
Finally, Fig. 4(c) is a baseline where training was performed on the ZINC dataset and post hoc filters on the OPD target was applied. It can be clearly seen that sub-figure (b) is more shifted towards optimal properties than the ZINC baselines which suggests that the structural priors imposed by training on the domain-specific OPD patent dataset offers significant value in achieving optimal properties for that domain. For example, molecules incorporating structural priors such as conjugated rings have more potential in achieving low optical gaps than drug-like structures.
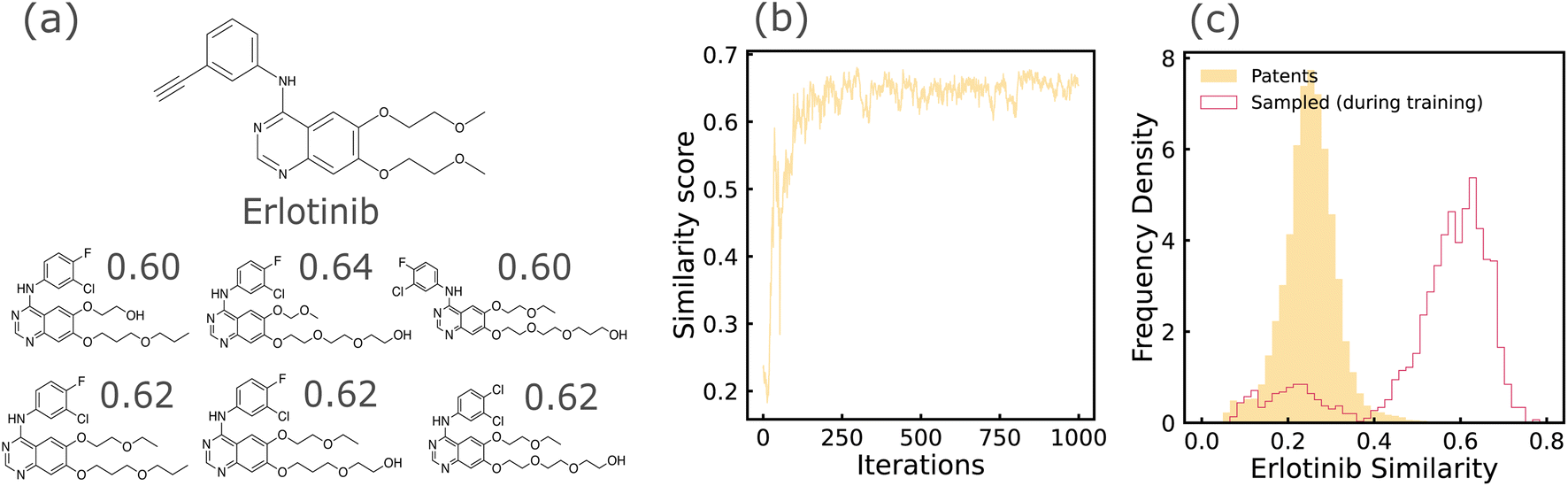 | ||
| Fig. 5 Results based on REINVENT + SELFIES model trained on the TKI dataset. (a) Candidates generated by REINVENT towards the end of training, with structural similarity to Erlotinib being the reward function. Similarity scores are indicated below each candidate. (b) Tanimoto similarity score computed between generated candidates and Erlotinib, as a function of training iterations. (c) Histograms showing properties of candidates sampled during agent training, in comparison with the training data distribution. | ||
A post hoc filter using the oracle predictor can also be utilized in this case as a way to generate a set of novel candidate molecules that are optimized in comparison to the training data (Fig. 4(d) and (g) for JTVAE and RNN + SELFIES respectively). Similar to the example described in Section 3.2.2, we also compared with a ZINC-trained baseline optimized for the TKI target, and observe minor improvements in shifts for the TKI-trained model in comparison to the ZINC-trained baseline (see Fig. 4(d) and (e)). This difference is not as significant as the OPD-ZINC baseline since the chemical spaces of ZINC and TKI datasets are fairly similar structurally.
4 Conclusions
In this work, we developed a framework to automatically extract molecular structures from the USPTO patent repository based on user-defined keyword searches, and generate datasets for machine learning in chemistry. We demonstrate the utility of the extracted datasets in training generative models for inverse molecular design tasks. We show that these datasets can be utilized to generate novel molecular structures with properties similar to the training dataset, in a completely unsupervised setting. We also evaluate model performance on supervised property optimization tasks, identify some limitations of existing models in shifting property distributions away from the training data regime, and suggest some possible explanations and remedies that could be used to overcome these in practice. The key observations we make through our experiments are summarized as follows: (1) we identify that patent-mined datasets offer the ability to create focused in-domain datasets of high-performing molecular structures and offers a way to bootstrap focused domains of chemical space with limited human intervention. (2) Property optimization towards the edges of the training data distribution can be effective if we have access to a cheap oracle predictor, but is challenging when proxy neural network approximators are used.Data availability
The code used to train models is publicly available. JTVAE: https://github.com/wengong-jin/icml18-jtnn, REINVENT: https://github.com/MarcusOlivecrona/REINVENT. The RNN models were trained using the char-rnn code from https://github.com/molecularsets/moses. A static version of the exact forks used is available at https://doi.org/10.5281/zenodo.7719958, and checkpoints of trained models and all training data including DFT-calculated properties are available at https://doi.org/10.5281/zenodo.7996464.52 Code for the patent mining and filtering pipeline can be found at https://github.com/learningmatter-mit/PatentChem. This patent code is archived at https://doi.org/10.5281/zenodo.7719675.53 GuacaMol benchmarking was performed using https://github.com/BenevolentAI/guacamol.Author contributions
A. S. trained the generative models and analyzed the distribution learning and property optimization results. K. P. G. updated and organized the patent code and ran the high-throughput physics-based calculation pipeline. A. G. wrote an initial version of the patent code. T. Y. trained initial versions of the generative models. A. S. and K. P. G. wrote the first manuscript draft. R. G.-B. conceived the project, supervised the research, and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. S. was supported by funding from Sumitomo Chemical. K. P. G. was supported by the National Science Foundation Graduate Research Fellowship Program under Grant No. 1745302. This work was also supported by the DARPA Accelerated Molecular Discovery (AMD) program under contract HR00111920025. We acknowledge the MIT Engaging cluster and MIT Lincoln Laboratory Supercloud cluster54 at the Massachusetts Green High Performance Computing Center (MGHPCC) for providing high-performance computing resources to run our TD-DFT calculations and train our deep learning models.References
- D. Schwalbe-Koda and R. Gómez-Bombarelli, Generative models for automatic chemical design, in Machine Learning Meets Quantum Physics, Springer, 2020, pp. 445–467 Search PubMed.
- D. C. Elton, Z. Boukouvalas, M. D. Fuge and P. W. Chung, Deep learning for molecular design—a review of the state of the art, Mol. Syst. Des. Eng., 2019, 4(4), 828–849 RSC.
- M. H. Segler, T. Kogej, C. Tyrchan and M. P. Waller, Generating focused molecule libraries for drug discovery with recurrent neural networks, ACS Cent. Sci., 2018, 4(1), 120–131 CrossRef CAS PubMed.
- E. J. Bjerrum and R. Threlfall, Molecular generation with recurrent neural networks (RNNs), arXiv, 2017, preprint, arXiv:1705.04612, DOI:10.48550/arXiv.1705.04612.
- R. Gómez-Bombarelli, J. N. Wei, D. Duvenaud, J. M. Hernández-Lobato, B. Sánchez-Lengeling, D. Sheberla, J. Aguilera-Iparraguirre, T. D. Hirzel, R. P. Adams and A. Aspuru-Guzik, Automatic chemical design using a data-driven continuous representation of molecules, ACS Cent. Sci., 2018, 4(2), 268–276 CrossRef.
- M. J. Kusner, B. Paige and J. M. Hernández-Lobato, “Grammar variational autoencoder,” in International Conference on Machine Learning, PMLR, 2017, pp. 1945–1954 Search PubMed.
- M. Olivecrona, T. Blaschke, O. Engkvist and H. Chen, Molecular de-novo design through deep reinforcement learning, J. Cheminf., 2017, 9(1), 1–14 Search PubMed.
- R. Mercado, T. Rastemo, E. Lindelöf, G. Klambauer, O. Engkvist, H. Chen and E. J. Bjerrum, Graph networks for molecular design, Mach. Learn.: Sci. Technol., 2021, 2(2), 025023 Search PubMed.
- W. Jin, R. Barzilay and T. Jaakkola, Junction tree variational autoencoder for molecular graph generation, in International Conference on Machine Learning, PMLR, 2018, pp. 2323–2332 Search PubMed.
- G. Simm, R. Pinsler and J. M. Hernández-Lobato, Reinforcement learning for molecular design guided by quantum mechanics, in International Conference on Machine Learning, PMLR, 2020, pp. 8959–8969 Search PubMed.
- D. Flam-Shepherd, T. C. Wu and A. Aspuru-Guzik, MPGVAE: improved generation of small organic molecules using message passing neural nets, Mach. Learn.: Sci. Technol., 2021, 2(4), 045010 Search PubMed.
- S. Kim, P. A. Thiessen, E. E. Bolton, J. Chen, G. Fu, A. Gindulyte, L. Han, J. He, S. He and B. A. Shoemaker, et al., PubChem substance and compound databases, Nucleic Acids Res., 2016, 44(D1), D1202–D1213 CrossRef CAS PubMed.
- Z. Wu, B. Ramsundar, E. N. Feinberg, J. Gomes, C. Geniesse, A. S. Pappu, K. Leswing and V. Pande, MoleculeNet: a benchmark for molecular machine learning, Chem. Sci., 2018, 9(2), 513–530 RSC.
- S. Senger, L. Bartek, G. Papadatos and A. Gaulton, Managing expectations: assessment of chemistry databases generated by automated extraction of chemical structures from patents, J. Cheminf., 2015, 7(1), 1–12 CAS.
- J. Ohms, Current methodologies for chemical compound searching in patents: A case study, World Pat. Inf., 2021, 66, 102055, DOI:10.1016/j.wpi.2021.102055.
- G. Papadatos, M. Davies, N. Dedman, J. Chambers, A. Gaulton, J. Siddle, R. Koks, S. A. Irvine, J. Pettersson and N. Goncharoff, et al., SureChEMBL: a large-scale, chemically annotated patent document database, Nucleic Acids Res., 2016, 44(D1), D1220–D1228 CrossRef CAS.
- D. M. Lowe, Extraction of chemical structures and reactions from the literature, PhD thesis, University of Cambridge, 2012.
- Complex Work Unit Pilot Program, https://www.uspto.gov/patents/initiatives/complex-work-unit-pilot-program Search PubMed.
- D. Polykovskiy, A. Zhebrak, B. Sanchez-Lengeling, S. Golovanov, O. Tatanov, S. Belyaev, R. Kurbanov, A. Artamonov, V. Aladinskiy and M. Veselov, et al., Molecular sets (MOSES): a benchmarking platform for molecular generation models, Front. Pharmacol., 2020, 11, 565644 CrossRef CAS PubMed.
- S. Hochreiter and J. Schmidhuber, Long short-term memory, Neural Comput., 1997, 9(8), 1735–1780 CrossRef CAS.
- M. Krenn, F. Häse, A. Nigam, P. Friederich and A. Aspuru-Guzik, Self-referencing embedded strings (SELFIES): A 100% robust molecular string representation, Mach. Learn.: Sci. Technol., 2020, 1(4), 045024 Search PubMed.
- W. Gao, T. Fu, J. Sun and C. W. Coley, Sample efficiency matters: a benchmark for practical molecular optimization, Adv. Neural Inf. Process. Syst., 2022, 35, 21342–21357 Search PubMed.
- M. Aldeghi, D. E. Graff, N. Frey, J. A. Morrone, E. O. Pyzer-Knapp, K. E. Jordan and C. W. Coley, Roughness of molecular property landscapes and its impact on modellability, J. Chem. Inf. Model., 2022, 62(19), 4660–4671 CrossRef CAS PubMed.
- J. Westermayr, J. Gilkes, R. Barrett and R. J. Maurer, High-throughput property-driven generative design of functional organic molecules, Nat. Comput. Sci., 2023, 1–10 Search PubMed.
- X. Xu, M. Davanco, X. Qi and S. R. Forrest, Direct transfer patterning on three dimensionally deformed surfaces at micrometer resolutions and its application to hemispherical focal plane detector arrays, Org. Electron., 2008, 9(6), 1122–1127 CrossRef CAS.
- H. Xu, J. Liu, J. Zhang, G. Zhou, N. Luo and N. Zhao, Flexible organic/inorganic hybrid near-infrared photoplethysmogram sensor for cardiovascular monitoring, Adv. Mater., 2017, 29(31), 1700975 CrossRef PubMed.
- F. Liu, Z. Zhou, C. Zhang, J. Zhang, Q. Hu, T. Vergote, F. Liu, T. P. Russell and X. Zhu, Efficient semitransparent solar cells with high NIR responsiveness enabled by a small-bandgap electron acceptor, Adv. Mater., 2017, 29(21), 1606574 CrossRef.
- Y. Li, J.-D. Lin, X. Che, Y. Qu, F. Liu, L.-S. Liao and S. R. Forrest, High efficiency near-infrared and semitransparent non-fullerene acceptor organic photovoltaic cells, J. Am. Chem. Soc., 2017, 139(47), 17114–17119 CrossRef CAS PubMed.
- D. Weininger, SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules, J. Chem. Inf. Comput. Sci., 1988, 28(1), 31–36 CrossRef CAS.
- D. Schwalbe-Koda, mkite: A distributed computing platform for high-throughput materials simulations, arXiv, 2023, preprint, arXiv:2301.08841, DOI:10.48550/arXiv.2301.08841.
- G. Landrum, et al., RDKit: A software suite for cheminformatics, computational chemistry, and predictive modeling, ed. G. Landrum, 2013 Search PubMed.
- S. Riniker and G. A. Landrum, Better informed distance geometry: Using what we know to improve conformation generation, J. Chem. Inf. Model., 2015, 55(12), 2562–2574 CrossRef CAS PubMed.
- C. Bannwarth, S. Ehlert and S. Grimme, GFN2-xTB - An Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-Dependent Dispersion Contributions, J. Chem. Theory Comput., 2019, 15(3), 1652–1671, DOI:10.1021/acs.jctc.8b01176.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, The ORCA quantum chemistry program package, J. Chem. Phys., 2020, 152(22), 224108, DOI:10.1063/5.0004608.
- A. D. Becke, Density-functional exchange-energy approximation with correct asymptotic behavior, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38(6), 3098–3100, DOI:10.1103/PhysRevA.38.3098 , http://www.ncbi.nlm.nih.gov/pubmed/<?ccdc_no 9900728?>9900728<?ccdc END?>.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32(7), 1456–1465, DOI:10.1002/jcc.21759.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7(18), 3297–3305, 10.1039/b508541a.
- S. Hirata and M. Head-Gordon, Time-dependent density functional theory within the Tamm–Dancoff approximation, Chem. Phys. Lett., 1999, 314(3–4), 291–299 CrossRef CAS.
- J.-D. Chai and M. Head-Gordon, Long-range corrected double-hybrid density functionals, J. Chem. Phys., 2009, 131(17), 174105 CrossRef PubMed.
- N. Brown, M. Fiscato, M. H. Segler and A. C. Vaucher, GuacaMol: benchmarking models for de novo molecular design, J. Chem. Inf. Model., 2019, 59(3), 1096–1108 CrossRef CAS PubMed.
- K. Preuer, P. Renz, T. Unterthiner, S. Hochreiter and G. Klambauer, Fréchet ChemNet distance: a metric for generative models for molecules in drug discovery, J. Chem. Inf. Model., 2018, 58(9), 1736–1741 CrossRef CAS PubMed.
- J. J. Irwin and B. K. Shoichet, ZINC - a free database of commercially available compounds for virtual screening, J. Chem. Inf. Model., 2005, 45(1), 177–182 CrossRef CAS PubMed.
- D. Flam-Shepherd, K. Zhu and A. Aspuru-Guzik, Language models can learn complex molecular distributions, Nat. Commun., 2022, 13(1), 1–10 Search PubMed.
- K. Cho, B. Van Merriënboer, D. Bahdanau and Y. Bengio, On the properties of neural machine translation: Encoder-decoder approaches, arXiv, 2014, preprint, arXiv:1409.1259, DOI:10.48550/arXiv.1409.1259.
- D. P. Kingma and J. Ba, Adam: A method for stochastic optimization, arXiv, 2014, preprint, arXiv:1412.6980, DOI:10.48550/arXiv.1412.6980.
- K. Yang, K. Swanson, W. Jin, C. Coley, P. Eiden, H. Gao, A. Guzman-Perez, T. Hopper, B. Kelley and M. Mathea, et al., Analyzing learned molecular representations for property prediction, J. Chem. Inf. Model., 2019, 59(8), 3370–3388 CrossRef CAS PubMed.
- U. Ekström, L. Visscher, R. Bast, A. J. Thorvaldsen and K. Ruud, Arbitrary-order density functional response theory from automatic differentiation, J. Chem. Theory Comput., 2010, 6(7), 1971–1980 CrossRef PubMed.
- T. Tamayo-Mendoza, C. Kreisbeck, R. Lindh and A. Aspuru-Guzik, Automatic differentiation in quantum chemistry with applications to fully variational Hartree–Fock, ACS Cent. Sci., 2018, 4(5), 559–566 CrossRef CAS PubMed.
- P. Eckmann, K. Sun, B. Zhao, M. Feng, M. K. Gilson and R. Yu, LIMO: Latent Inceptionism for Targeted Molecule Generation, arXiv, 2022, preprint, arXiv:2206.09010, DOI:10.48550/arXiv.2206.09010.
- P. Notin, J. M. Hernández-Lobato and Y. Gal, Improving black-box optimization in VAE latent space using decoder uncertainty, Adv. Neural Inf. Process. Syst., 2021, 34, 802–814 Search PubMed.
- H. Kajino, K. Miyaguchi and T. Osogami, Biases in In Silico Evaluation of Molecular Optimization Methods and Bias-Reduced Evaluation Methodology, arXiv, 2022, preprint, arXiv:2201.12163, DOI:10.48550/arXiv.2201.12163.
- A. Subramanian, K. Greenman, A. Gervaix, T. Yang and R. Gomez-Bombarelli, Automated patent extraction powers generative modeling in focused chemical spaces, Training data and code release, 2023, DOI:10.5281/zenodo.7719959.
- K. Greenman, A. Gervaix and R. Gómez-Bombarelli, learningmatter-mit/PatentChem: initial public release, version v0.0.1, 2023, DOI:10.5281/zenodo.7719676.
- A. Reuther, J. Kepner, C. Byun, S. Samsi, W. Arcand, D. Bestor, B. Bergeron, V. Gadepally, M. Houle, M. Hubbell, et al., Interactive supercomputing on 40,000 cores for machine learning and data analysis, in 2018 IEEE High Performance extreme Computing Conference (HPEC), IEEE, 2018, pp. 1–6 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3dd00041a |
| ‡ Contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |