
Improving the synthetic H2 production catalyst design strategy with the neurotransmitter dopamine†
Santanu
Ghorai
 a,
Shikha
Khandelwal
b,
Srewashi
Das
a,
Surabhi
Rai
ac,
Somnath
Guria
a,
Piyali
Majumder
c and
Arnab
Dutta
*acd
a,
Shikha
Khandelwal
b,
Srewashi
Das
a,
Surabhi
Rai
ac,
Somnath
Guria
a,
Piyali
Majumder
c and
Arnab
Dutta
*acd
aChemistry Department, Indian Institute of Technology Bombay, Powai, Mumbai, 400076 India. E-mail: arnab.dutta@iitb.ac.in
bChemistry Discipline, Indian Institute of Technology Gandhinagar, Palaj, 382355 India
cNational Centre of Excellence in CCU, Indian Institute of Technology Bombay, Powai, Mumbai, 400076 India
dInterdisciplinary Program in Climate Studies, Indian Institute of Technology Bombay, Powai, Mumbai, 400076 India
First published on 15th December 2022
Abstract
The strategic incorporation of the neurotransmitter dopamine around a cobaloxime core resulted in excellent electrocatalytic (rate 8400 s−1) and photocatalytic H2 production under neutral aqueous conditions. The influence of the synthetic outer coordination sphere features continues even with a phenylene–diimino–dioxime motif-coordinated cobalt core.
Adopting a viable carbon footprint-free energy infrastructure has emerged as one of the top priority sustainable development goals, where renewable resources (solar, wind, tidal, etc.) are reckoned as the best alternatives in the pursuit for a greener future.1 The use of hydrogen molecules (H2) as an ideal energy carrier is essential here, considering the intermittent nature of the renewables.2,3 Hence, renewable energy-driven H2 production from abundantly available water has been the crux of the green H2-driven energy strategy. Therefore, the development of efficient, economical, and eco-friendly catalysts driving H2 production from aqueous media has become a prime research focus.4 Hydrogenase enzyme structure-motivated catalyst design offers unique synthetic model complexes, which deploy first-row transition metals for rapid H2 production under electrocatalytic and photocatalytic conditions.5 Molecular catalysts act as a perfect template for testing multiple combinations of metals and ligands for catalytic small molecule activation. The unique structure–function relationship between the metal and ligand provides the leeway for finetuning the active site reactivity.6,7 The rational incorporation of the surrounding protein scaffold-inspired outer coordination sphere (OCS) features has emerged as a game changer in unravelling new facets of reactivity for functional mimics.8–17 Among the available models, cobaloxime [Co(dimethylglyoxime)2] derivatives have surfaced as a popular template owing to their oxygen tolerance during electrocatalytic H2 evolution and robust nature in near-neutral water.18,19 The abundant cobalt metal also provides the added advantage as any strategy involving cobaloxime can be scaled up later for industrial-scale applications. The cobaloxime reactivity is finely tuned with N-coordinating heteroaromatic molecules (pyridine or imidazole) that can also harbour divergent protic functionalities to assist the proton movement around the catalyst. To date, amino acid-derived functionalities have been deployed to probe such OCS effects on cobaloxime-driven H2 evolution reaction (HER) activity.20,21 The phenolic–OH containing tyrosine was found to be an excellent peripheral functionality leading to an ∼10 times enhancement in the HER activity of the cobaloxime core. In this study, we have expanded the possible pool of OCS feature groups and inserted a two-phenolic–OH group (catechol)-bound dopamine motif into the cobaloxime core. As a major monoamine neurotransmitter in biology, dopamine plays indispensable roles by modulating motor neurons,22 spatial memory function,23 motivation,24 arousal,25,26 and reward and pleasure,27,28 as well as in lactation,29 sexual behavior,30 and nausea.31 However, this molecule has rarely been deployed in synthetic catalyst design. Additionally, we have altered the widely used dimethylglyoxime motif with phenylene–diimino–dioxime (pdd) to elucidate the influence of the primary ligation geometry on catalysis. Both pdd and dmg ligands offer a square planar N4 coordination scaffold for cobalt ion binding. However, pdd acts as a tetradentate ligand (C2), while the bidentate nature of dmg warrants the presence of two separate dmg molecules for completing the N4 primary coordination (C1) (Fig. 1). The remaining axial ligation sites of C1 and C2 are capped with pyridine and the labile Cl− ion. This OCS-ligation strategy allowed us to decorate the OCS of the resultant molecule C3 with two phenolic–OH groups that can further improve the HER catalysis (Fig. 1). We have also included the pyridinyl–tyramine functionality around the newly developed Co(pdd) core (C4) to survey the OCS effect of the phenolic–OH functionality with this new primary coordination sphere combination (Fig. 1). The dopamine-bound C3 showcased the best catalytic performance in this series followed by the tyramine-bound C4. Hence, the present study highlights the unique dual effect of both primary and secondary coordination spheres on cobalt core-based bio-inspired molecular catalysts driving the hydrogen evolution reaction (HER) under both electrocatalytic and photocatalytic conditions. To date, such a differential effect of primary and secondary coordination sphere moieties on the catalytic properties of molecular catalysts has been observed for porphyrin and Ni(bis-phosphine) complexes.8,32–34 We have attempted to prepare the Co(pdd)–pyridinyl–dopamine compound; however, its fragile nature in aqueous media restricted us from recording any structural and reactivity data for the same.
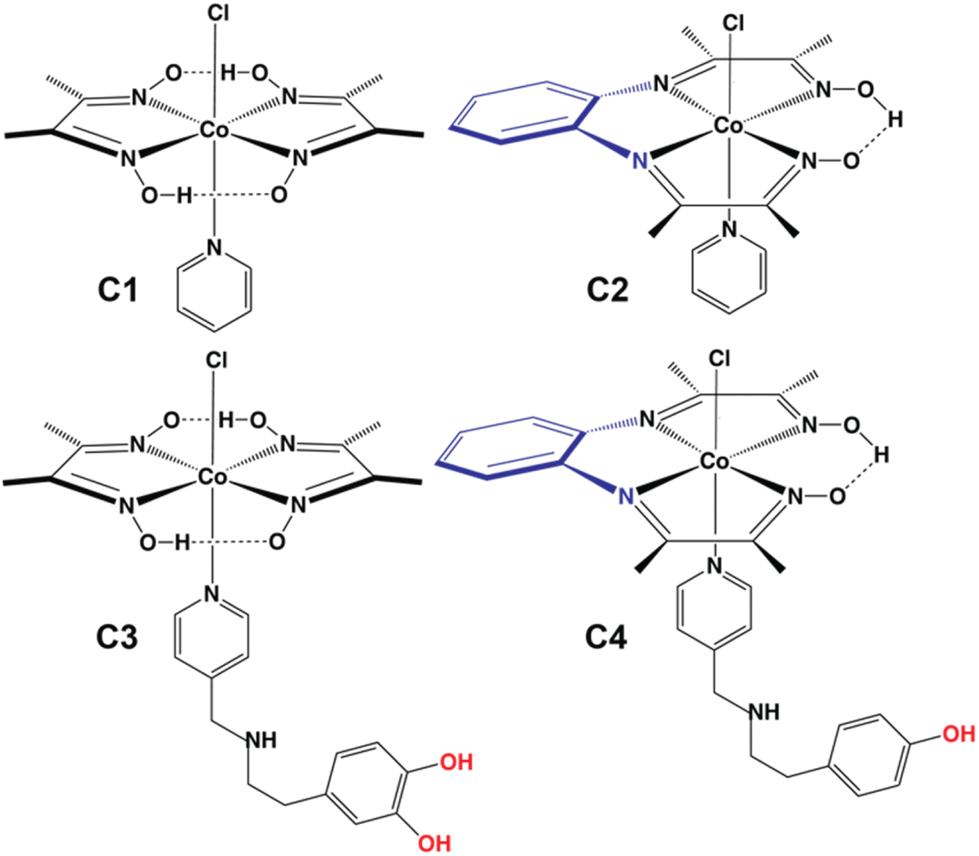 | ||
| Fig. 1 Chemical structures of complexes C1–C4 deployed for electrocatalytic and photocatalytic HER studies. | ||
All these complexes (C1–C4) were synthesized following the stoichiometric addition of pyridine or pyridyl derivatives to a pre-assembled Co(dmg)2 or Co(pdd) core in a methanolic solution (Scheme S1†). The resulting brown-coloured complexes were characterized via optical spectroscopy following their purification. The optical spectra of complexes C1–C4 were initially recorded in an organic medium (DMF). A strong feature was noticed for all complexes at ∼260 nm, which was attributed to the π–π* transition originating from the different aromatic motifs (pyridinyl and phenyl) present in the molecular framework.35 Next, relatively weaker twin bands were observed around 320 and 380 nm for these complexes (Fig. S1 and Table S1†). These peaks appear for the signature Noxime → Co(III) and Npyridinyl → Co(III) transitions, as observed for analogous cobalt complexes.19,20,36 The d–d transition is also active in these complexes, which is reflected by the appearance of a broad and weak band in the far visible region (∼550 nm) (Fig. S1 and Table S1†). These optical signatures are typical for a low-spin Co(III) core stabilized in a hexa-coordinated N5Cl environment. Here, the similarity between C1/C3 and C2/C4 also emphasized the resemblance between Co-pdd-pyridine and the cobaloxime motif (Fig. 1). A similar trend of optical bands for C1–C4 was recorded even in aqueous media, albeit with subtle shifts in their respective wavelengths (Fig. S2†). The successful incorporation of the axial pyridinyl motif in the cobalt core was further supported by FTIR spectroscopy, where all the complexes displayed the characteristic Co–Npyridinyl stretching band at the ∼480–500 cm−1 region along with the signature aromatic stretching peaks (Fig. S3†).14,20,35 A broad FTIR feature was also observed around ∼3450 cm−1 which was assigned to the water-assisted hydrogen bonding nexus around oxime. Next, the complexes were probed with 1H and 13C NMR spectroscopy, where the diamagnetic feature of the signal indicates the presence of a low-spin Co(III) (d6 system) core. The presence of aromatic phenyl (only in C2 and C4) and pyridine motifs (in C1–C4) was evident from the signature splitting patterns recorded in this experiment (Fig. S4–S9†). Interestingly, the 1H NMR spectra displayed a significantly down-fielded signal (δ ∼ 18 ppm) for all the complexes. This particular feature indicates the existence of a hydrogen bonded oxime proton (–N–O−⋯H–O–N) in the primary coordination sphere.14,20
Cyclic voltammetry (CV) experiments were performed with complexes C1–C4 to probe their HER activity. C2 showed two stoichiometric signals during a cathodic scan in organic media in the absence of any proton source. The first quasi-reversible reductive peak was noticed beyond −1.0 V before a reversible redox signal at −1.58 V (vs. FeCp2+/0) (Fig. S10A†). Similar behavior was noticed for C1 earlier and the signals were assigned to quasi-reversible Co(III/II) and reversible Co(II/I) steps, respectively.37 The OCS-appended complexes showed a similar trend, albeit with a significant anodic shift of the cobalt-based reduction signals. For C3 and C4, the Co(III/II) and Co(II/I) signals were observed at −0.3 V and −1.10 V (vs. FeCp2+/0), respectively (Fig. S10C and 10D†). This drift in the stoichiometric signals is typically attributed to the electronic effect of the axially ligated para-substituted pyridine derivatives (C3 and C4) compared to the unsubstituted pyridine derivatives (C1 and C2). The H2 production activity of the complexes was investigated in DMF following the serial addition of trifluoroacetic acid (TFA) as a proton source. Complex C2 displayed an anodic shift of the Co(III/II) reduction feature towards the anodic direction following the addition of the acid accompanied by the loss of the Co(II/III) oxidation feature. The reversible Co(II/I) signature was also influenced under similar conditions as it loses its reversibility. However, a stark increase in the magnitude of the reductive current was noticed with increasing acid concentration in the solution (Fig. S10B†). This change was attributed to the inception of the H2 production activity triggered by the Co(I) center. C3 and C4 showed similar responses to acid addition, as the HER responses were noticed around −1.0 and −1.2 V (vs. FeCp2+/0), respectively, with a relatively lower enhancement of the catalytic current compared to C2 (Fig. S10C–S10F†). Adding water to the reaction mixture improves the catalytic current marginally without significantly changing other reductive features (Fig. S10D and S10F†).
Next, the H2 production reactivity of these complexes was investigated in buffered aqueous media. Here, all the potential values are reported against the standard hydrogen electrode (SHE) unless otherwise mentioned. In neutral water (pH 7.0), the electrochemical behavior of C1–C4 altered significantly compared to organic media. C1 exhibited three distinct features during the cathodic scan, starting from a quasi-reversible Co(III/II) step (at −0.13 V). A minor catalytic response was observed at −0.75 V, which was attributed to the Co(II)-driven H2 production via an electron–chemical–electron–chemical (ECEC) transfer pathway.20 An enhanced catalytic response was recorded at −0.9 V, which was driven by a Co(I) centric electron–electron–chemical–chemical (EECC) mechanism (Fig. 2A and Table S2†).20,21 The presence of dopamine around the periphery of the Co(dmg)2 core in C3 visibly improved the electrocatalytic HER response, whereas the rest of the electrochemical signatures remained the same (Fig. 2A). The two critical catalytic parameters, turnover frequency (TOF or kobs) and overpotential (OP, η), were measured to compare the catalytic performance of C1–C4. The ratio of the current response of the final catalytic feature and the stoichiometric Co(III/II) signature was deployed for computing the TOF following eqn (S1).† On the other hand, the difference between the thermodynamic potential of H+/H2 conversion (EH+/H2) and the experimentally observed potential for achieving the half maxima of the catalytic response (Ecat/2) displays the overpotential. C3 demonstrated a sharp rise of ∼7-fold in the HER TOF at pH 7.0 (TOF 6370 s−1) compared to the control complex C1 (TOF ∼900 s−1), albeit with minimal change in overpotential values (η ∼ 465–475 mV) (Fig. 2C and D, Table S3†). The maximum catalytic rate (TOF 8400 s−1) was observed at pH 6.0, which was on par with the fastest reported cobaloxime catalyst.20 This observation indicated the influence of the dopamine motif as an extended OCS feature on electrocatalysis while retaining the intrinsic property of the cobalt core. The inclusion of pdd in place of dmg in the primary coordination sphere resulted in a substantial variation in the electrochemical behavior. Complexes C2 and C4 showcased an anodically shifted Co(III/II) signature compared to C1 and C3. Interestingly, the minor catalysis triggered by the ECEC pathway disappeared for the Co(pdd) derivatives, while the EECC-driven HER response moved towards the positive potential (Fig. 2A). A series of scan rate-dependent CV and spectroelectrochemistry experiments corroborated the assignment of these electrochemical signatures (Fig. S11 and S12†). The TOF for the HER improved ∼2.8 times for pdd-coordinated C2 compared to the native dmg-ligated C1. This contrast between C1 and C2 possibly originated from the fundamental difference between dmg and pdd ligands. Although both of them provide an N4-coordination geometry to the central cobalt ion, the presence of the benzene motif induces a redox-active character to the pdd ligand. Therefore, the π-accepting pdd ligand can facilitate the electron exchange during the reduction to instigate an anodically shifted redox behavior.38,39
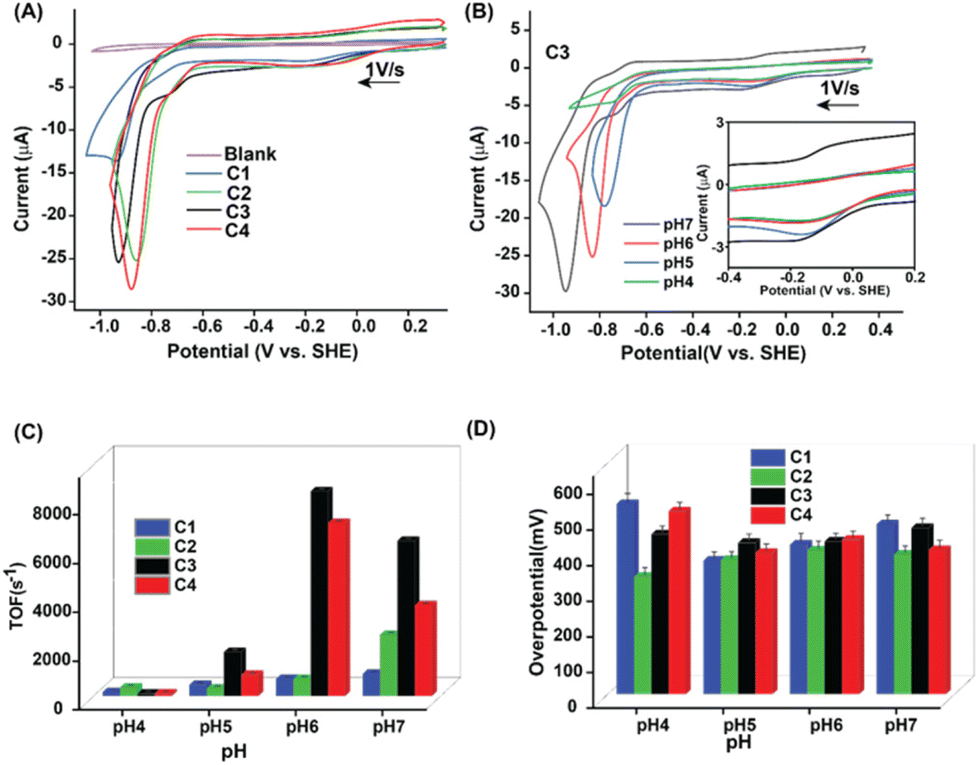 | ||
| Fig. 2 (A) The comparative cyclic voltammograms (CVs) of blank solution (purple trace) and complexes C1 (blue trace), C2 (green trace), C3 (black trace), and C4 (red trace) recorded at pH 7.0 (0.1 M MES buffer with 0.1 M Na2SO4 supporting electrolyte) under an inert atmosphere (Ar) at room temperature. (B) The comparative CVs of C3 recorded at pH 7.0 (black trace), 6.0 (red trace), pH 5.0 (blue trace), and pH 4.0 (green trace). All the CV data were recorded at a 1.0 V s−1 scan rate with a 1.0 mm diameter glassy carbon working electrode, a Pt wire counter electrode, and an Ag/AgCl (3.0 M KCl) reference electrode. The experimentally measured (C) turnover frequency (TOF) and (D) overpotential values for C1 (blue trace), C2 (green trace), C3 (black trace), and C4 (red trace) at variable pH conditions (pH 4.0–7.0). | ||
The inclusion of the tyramine motif in the OCS (C4) (TOF ∼3700 s−1) illustrated a 50% increase in the electrocatalytic HER response, while the rest of the CV resembled the behavior of C2 (TOF ∼2500 s−1) (Fig. 2A–C). Interestingly, C2 and C4 operated at a relatively low overpotential (η ∼ 390–405 mV) compared to Co(dmg)2 derived C1 and C3. These data indicated that the inherent electrochemical behavior is regulated by the primary coordination sphere, while the remotely positioned OCS features primarily modulate the catalytic response. The two-dimensional NMR experiment exchange spectroscopy (EXSY) was performed with C3 in a slightly wet d6-DMSO to capture the dynamic nature of the dopamine-curated OCS. The EXSY spectra revealed the rapid exchange of protons between phenolic–OH groups with solvent water, the pyridine linking secondary amine, and the hydrogen-bonded oxime present in the primary coordination sphere. The intramolecular proton exchange between twin phenolic groups was also visible from the same spectra (Fig. 3). All these data indicated a solvent water-mediated vibrant proton relay around the cobalt core in C3, which possibly escalates H+ movement to remotely accelerate the electrocatalytic H2 production.
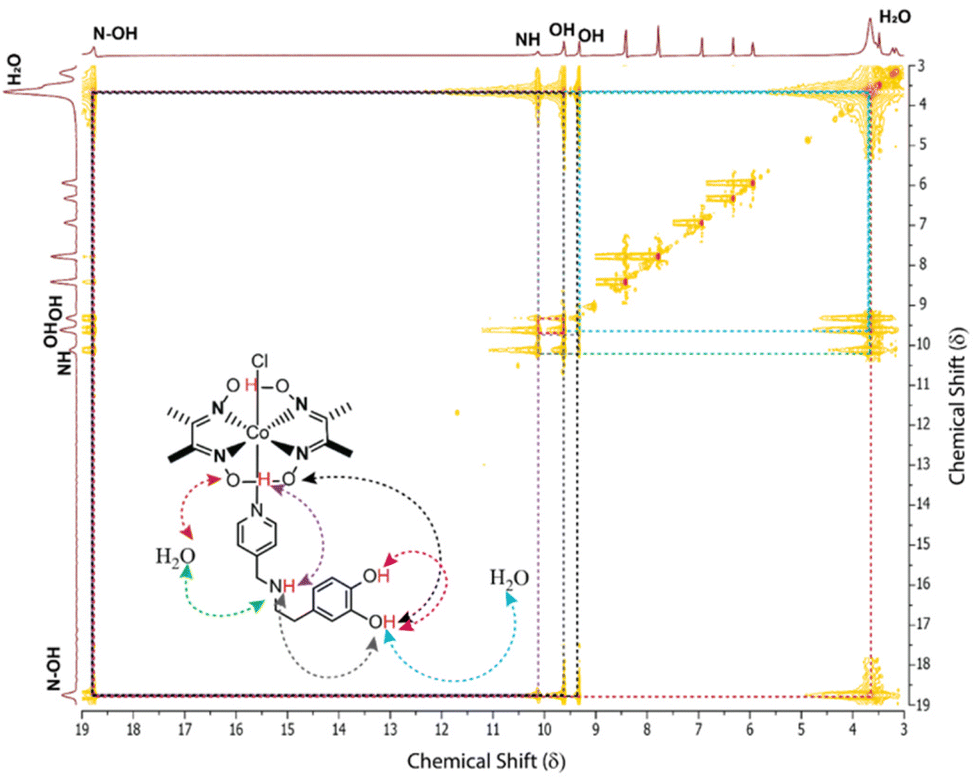 | ||
| Fig. 3 Two-dimensional NMR experiment exchange spectroscopy (EXSY) plot of C3 recorded in DMSO-d6 at 300 K with 400 ms mixing time. The rapid exchange between oxime and water (red trace), secondary amine and water (green trace), oxime and phenolic–OH groups (black and blue traces), and oxime and secondary amine (purple trace) are illustrated here. | ||
The complexes followed a similar catalytic performance trend as the pH of the solution was gradually shifted slightly to the acidic region. Interestingly, a sharp drop in the electrocatalytic HER signature was noticed below pH 5.0 before it completely disappeared beyond pH 4.0 (Fig. 2B and S13†). Such an abrupt change in the catalysis was attributed to the cleavage of the vital hydrogen-bonded oxime network in the primary coordination sphere (pKa ∼5.0).20,36 The vulnerability of the vital hydrogen-bonded oxime motif under acidic conditions was probed via a 1H NMR experiment. The addition of stoichiometric amounts of an acid (trifluoroacetic acid, TFA) in a d6-DMSO solution of C2 resulted in the disappearance of the sharp hydrogen-bonded oxime (–N–O–H⋯O−–N–) along with the emergence of a broad protonated oxime functionality (Fig. S14†). An array of chronocoulometry (bulk electrolysis) experiments was executed in an air-tight container with C1–C4 at the respective catalytic potentials. Here the production of H2 was verified following gas chromatography (GC) with the head-space gas injection (Fig. S13†). All complexes exhibited high faradaic efficiency (FE) (>60%), while the highest conversion was noticed for C3 (FE 93%) (Table S4†).
The homogeneous nature of the electrocatalytic H2 production caused by C1–C4 in an aqueous solution was probed in detail with the rinse test and surface analysis of the working electrode pre-and post-electrolysis. The rinse test was performed with a regular 1 mm diameter glassy carbon working electrode. Here, a partial reductive scan was recorded till the potential where the maximum catalytic current was observed, and then the working electrode was removed and placed in a blank solution (without catalyst) following rinsing but no polishing. Then a subsequent scan was recorded starting from the same highly reducing potential, which did not display any catalytic signature (Fig. S15†). These data indicated that no heterogeneous material was generated in situ during the catalysis. Parallel bulk electrolysis for complexes was performed at an H2 producing-potential with a plastic chip working electrode where no detectable amount of cobalt deposition was observed on the material during a scanning electron microscopy-energy dispersive spectroscopy (SEM-EDS) experiment (Fig. S16†). The optical spectra and cyclic voltammograms of C3 and C4 were also recorded after bulk electrolysis that exhibited minimal changes compared to the pre-electrolysis sample (Fig. S17†). The turnover number (TON) measurement of electrocatalytic H2 production for these complexes showcased a similar trend of C3 > C4 > C2 under neutral conditions. C3 showcased the maximum TON of ∼125 after 2 hours of bulk electrolysis at pH ∼6.0 (Table S5†).
Complexes C1–C4 was also found to be active for H2 production in neutral aqueous media under photocatalytic conditions (Fig. 4A). These experiments were conducted in an air-tight Schlenk tube containing appropriate amounts of catalysts along with Eosin-Y and triethanolamine (TEOA), which were deployed as the photosensitizer and sacrificial electron donor, respectively. Under exposure to an artificial light source (λmax 518 nm, power density = 50 mW cm−2, beam size = 3.8 cm2) continuous formation of H2 gas was detected over six hours (Fig. 4B). The direct influence of the redox-active pdd-ligand was evident from the ∼2 times higher activity of C2 (TON 42) under photocatalytic conditions compared to C1 (TON 21) (Fig. 4A and Table S3†). The extended effect of the OCS feature was noticed on photoirradiated H2 production in water as C3 (TON 330) and C2 (TON 280) showed superior activity compared to non-OCS-based complexes C1 and C2, respectively.36
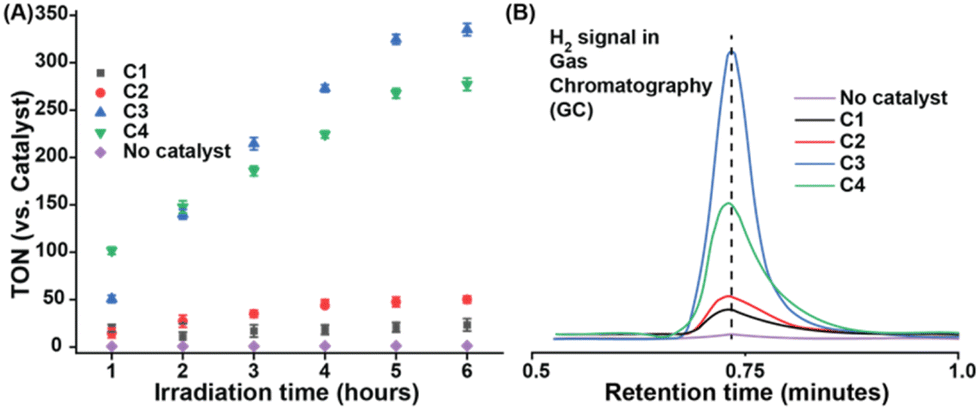 | ||
| Fig. 4 (A) Photo-irradiated H2 production data for C1 (black trace), C2 (red trace), C3 (blue trace), C4 (green trace), and blank (without catalyst) (violet trace) against time in an aqueous solution (0.1 M MES buffer, pH 7.0) containing EOSYN-Y PS, catalyst, 10% TEOA as the sacrificial electron donor under green laser exposure (λmax = 518 nm, power density = 40 mW cm−2, beam size = 3.8 cm2). (B) The comparative gas chromatography (GC) H2 detection signal recorded for the complexes C1–C4 under photocatalytic conditions. | ||
Conclusions
In this work, we showcased two different structural features with the series of four complexes C1–C4. C1 and C2 have different primary coordination spheres, where C1 has a dimethylglyoxime (dmg) ligand, while C2 has a phenylene–diimino–dioxime (pdd) ligand. The axial ligand remains identical (pyridine) in both these complexes. The inclusion of the tetracoordinated pdd ligand specifically improves the catalytic performance of C2, as it produces H2 at a ∼3 times higher rate compared to C1 (Table S3†). Additionally, a significant improvement of ∼85 mV was observed for the overpotential requirement.On the other hand, C3 and C4 not only differ in the primary coordination sphere (dmg vs. pdd), but they also contain dissimilar axial ligands. C3 has dopamine-linked pyridine, and C4 has tyramine-appended pyridine. As dopamine has two phenolic–OH groups compared to only one in tyramine, C3 is expected to have a better catalytic rate than C4. However, the extent of this change is subdued, possibly due to the presence of the pdd primary coordination site. The influence of this primary coordination sphere was also evident from the relatively energy efficient (by 60 mV) HER for C4 compared to C3 (Table S3†). Previously, we have observed that the Co(dmg)2 coordination geometry reached an HER rate of 8830 s−1 only when tyrosine is anchored to the axial pyridine (that contains a three-component proton relay: one amine, one carboxylic acid, and one phenolic–OH) (Table S6†). Almost a similar rate was observed for C3, which also has a three-component proton relay, albeit of a different variety (one amine, two phenolic–OH). Hence, this rational inclusion of catechol-based neurotransmitter dopamine around a cobaloxime core illustrated rapid and moderately energy-efficient electrocatalytic HER to extend the potential synthetic OCS feature options beyond natural amino acids. The active catalysis by the Co(pdd)-core emphasizes the presence of only one hydrogen-bonded oxime functionality to sustain H2 production from near-neutral aqueous solutions. Although these bio-inspired catalysts produce a relatively small amount of H2, they are active under ambient conditions, and they are durable even beyond the course of the catalysis. Hence, they can be assembled in an up-scaled assembly for industrially relevant H2 production.
Author contributions
SG and AD: conceptualization; SG, SK, and SR: experimentation; SG, SK, SR, and AD: data analysis; SG, SK, SR, PM, and AD: manuscript writing; and AD: supervision.Conflicts of interest
There are no conflicts to declare.References
- Climate Change 2022: Impacts, Adaptation and Vulnerability. Contribution of Working Group II to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change, ed. H.-O. Pörtner, D. C. Roberts, M. M. B. Tignor, E. S. Poloczanska, K. Mintenbeck, A. Alegría, M. Craig, S. Langsdorf, S. Löschke, V. Möller, A. Okem and B. Rama, 2022 Search PubMed.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS.
- D. K. Dogutan and D. G. Nocera, Acc. Chem. Res., 2019, 52, 3143–3148 CrossRef CAS PubMed.
- V. Artero, Nat. Energy, 2017, 2, 1–6 Search PubMed.
- J. M. Le and K. L. Bren, ACS Energy Lett., 2019, 4, 2168–2180 CrossRef CAS.
- S. Dey, B. Mondal, S. Chatterjee, A. Rana, S. Amanullah and A. Dey, Nat. Rev. Chem., 2017, 1, 1–20 CrossRef.
- A. Dutta, A. M. Appel and W. J. Shaw, Nat. Rev. Chem., 2018, 2, 244–252 CrossRef CAS.
- B. Ginovska-Pangovska, A. Dutta, M. L. Reback, J. C. Linehan and W. J. Shaw, Acc. Chem. Res., 2014, 47, 2621–2630 CrossRef CAS PubMed.
- J. A. Laureanti, M. O'Hagan and W. J. Shaw, Sustainable Energy Fuels, 2019, 3, 3260–3278 RSC.
- J. W. Slater, S. C. Marguet, H. A. Monaco and H. S. Shafaat, J. Am. Chem. Soc., 2018, 140, 10250–10262 CrossRef PubMed.
- J. W. Slater, S. C. Marguet, M. E. Gray, H. A. Monaco, M. Sotomayor and H. S. Shafaat, ACS Catal., 2019, 9, 8928–8942 CrossRef CAS.
- E. H. Edwards, J. M. Le, A. A. Salamatian, N. L. Peluso, L. Leone, A. Lombardi and K. L. Bren, J. Inorg. Biochem., 2022, 230, 111753 CrossRef CAS.
- S. Chattopadhyay, M. Mukherjee, B. Kandemir, S. E. J. Bowman, K. L. Bren and A. Dey, Chem. Sci., 2021, 12, 11894–11913 RSC.
- A. Panagiotopoulos, K. Ladomenou, D. Sun, V. Artero and A. G. Coutsolelos, Dalton Trans., 2016, 45, 6732–6738 RSC.
- X. Li, B. Lv, X.-P. Zhang, X. Jin, K. Guo, D. Zhou, H. Bian, W. Zhang, U.-P. Apfel and R. Cao, Angew. Chem., Int. Ed., 2022, 61, e202114310 CAS.
- I. K. Sideri, G. Charalambidis, A. G. Coutsolelos, R. Arenal and N. Tagmatarchis, Nanomaterials, 2022, 12, 3077 CrossRef CAS PubMed.
- E. Nikoloudakis, M. Pigiaki, M. N. Polychronaki, A. Margaritopoulou, G. Charalambidis, E. Serpetzoglou, A. Mitraki, P. A. Loukakos and A. G. Coutsolelos, ACS Sustainable Chem. Eng., 2021, 9, 7781–7791 CrossRef CAS.
- D. Dolui, S. Ghorai and A. Dutta, Coord. Chem. Rev., 2020, 416, 213335 CrossRef CAS.
- D. Dolui, A. Q. Mir and A. Dutta, Chem. Commun., 2020, 56, 14841–14844 RSC.
- D. Dolui, S. Khandelwal, A. Shaik, D. Gaat, V. Thiruvenkatam and A. Dutta, ACS Catal., 2019, 9, 10115–10125 CrossRef CAS.
- D. Dolui, S. Das, J. Bharti, S. Kumar, P. Kumar and A. Dutta, Cell Rep. Phys. Sci., 2020, 1, 100007 CrossRef.
- K. A. Harrington, S. J. Augood, A. E. Kingsbury, O. J. Foster and P. C. Emson, Brain Res. Mol. Brain Res., 1996, 36, 157–162 CrossRef CAS PubMed.
- M. Luciana, P. F. Collins and R. A. Depue, Cereb. Cortex, 1998, 8, 218–226 CrossRef CAS PubMed.
- J. D. Salamone and M. Correa, Neuron, 2012, 76, 470–485 CrossRef CAS.
- R. Andretic, B. van Swinderen and R. J. Greenspan, Curr. Biol., 2005, 15, 1165–1175 CrossRef CAS.
- I. Z. Ben Zion, R. Tessler, L. Cohen, E. Lerer, Y. Raz, R. Bachner-Melman, I. Gritsenko, L. Nemanov, A. H. Zohar, R. H. Belmaker, J. Benjamin and R. P. Ebstein, Mol. Psychiatry, 2006, 11, 782–786 CrossRef CAS PubMed.
- W. Schultz, J. Neurophysiol., 1998, 80, 1–27 CrossRef CAS PubMed.
- K. C. Berridge and M. L. Kringelbach, Psychopharmacology, 2008, 199, 457–480 CrossRef CAS PubMed.
- K. T. Demarest, G. D. Riegle and K. E. Moore, Neuroendocrinology, 1984, 38, 467–475 CrossRef CAS.
- T. H. C. Krüger, U. Hartmann and M. Schedlowski, World J. Urol., 2005, 23, 130–138 CrossRef PubMed.
- M. Nakagawa, M. Kuri, N. Kambara, H. Tanigami, H. Tanaka, Y. Kishi and N. Hamajima, J. Anesth., 2008, 22, 397–403 CrossRef PubMed.
- S. Bhunia, A. Ghatak and A. Dey, Chem. Rev., 2022, 122, 12370–12426 CrossRef CAS.
- M. E. Ahmed, S. Dey, M. Y. Darensbourg and A. Dey, J. Am. Chem. Soc., 2018, 140, 12457–12468 CrossRef CAS PubMed.
- D. L. DuBois, Inorg. Chem., 2014, 53, 3935–3960 CrossRef CAS PubMed.
- D. W. Wakerley and E. Reisner, Phys. Chem. Chem. Phys., 2014, 16, 5739–5746 RSC.
- A. Q. Mir, S. Saha, S. Mitra, S. Guria, P. Majumder, D. Dolui and A. Dutta, Sustainable Energy Fuels, 2022, 6, 4160–4168 RSC.
- M. Razavet, V. Artero and M. Fontecave, Inorg. Chem., 2005, 44, 4786–4795 CrossRef CAS.
- B. Sarkar, D. Schweinfurth, N. Deibel and F. Weisser, Coord. Chem. Rev., 2015, 293–294, 250–262 CrossRef CAS.
- L. Suntrup, F. Stein, J. Klein, A. Wilting, F. G. L. Parlane, C. M. Brown, J. Fiedler, C. P. Berlinguette, I. Siewert and B. Sarkar, Inorg. Chem., 2020, 59, 4215–4227 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2dt03509j |
| This journal is © The Royal Society of Chemistry 2023 |