
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The perfluoroadamantoxy aluminate as an ideal weakly coordinating anion? – synthesis and first applications†
Andreas
Billion
 a,
Marcel
Schorpp
a,
Rebecca
Feser
a,
Manuel
Schmitt
a,
Lea
Eisele
a,
Harald
Scherer
a,
Takaaki
Sonoda
b,
Hajimu
Kawa
c,
Burkhard
Butschke
a and
Ingo
Krossing
*a
a,
Marcel
Schorpp
a,
Rebecca
Feser
a,
Manuel
Schmitt
a,
Lea
Eisele
a,
Harald
Scherer
a,
Takaaki
Sonoda
b,
Hajimu
Kawa
c,
Burkhard
Butschke
a and
Ingo
Krossing
*a
aInstitut für Anorganische und Analytische Chemie and Freiburger Materialforschungszentrum FMF, Albert-Ludwigs-Universität Freiburg, Albertstr. 21, 79104 Freiburg, Germany. E-mail: krossing@uni-freiburg.de
bInstitute for Materials Chemistry and Engineering, Kyushu University, 6-1 Kasuga koen, Kasuga-shi, Fukuoka 816-8580, Japan
cExfluor Research Corporation, 2350 Double Creek Drive, Round Rock, Texas 78664, USA
First published on 10th March 2023
Abstract
Weakly coordinating anions (WCAs) facilitate the stabilization and isolation of highly reactive and almost “naked” cations. Alkoxyaluminate-based WCAs such as [Al(OC(CF3)3)4]− ([pf]−) are widely used due to their synthetic accessibility and their high stability. However, small cations are still able to coordinate the oxygen atoms of the [pf]− anion or even to abstract an alkoxy ligand. The novel WCA [Al(OC10F15)4]− ([pfAd]−; OC10F15 = perfluoro-1-adamantoxy) is characterized by a very rigid core framework, thus indicating a higher stability towards fluoride-ion abstraction (DFT calculations) and providing hope to generate less disordered crystal structures. The [pfAd]− anion was generated by the reaction of the highly acidic alcohol perfluoro-1-adamantanol C10F15OH with LiAlH4 in o-DFB. Li[pfAd] could not be synthesized free of impurities (and still contains unreacted alcohol). Yet, starting from contaminated Li[pfAd], the very useful pure salts Ag[pfAd], [Ph3C][pfAd] and [H(OEt2)2][pfAd] could be synthesized. The salts were characterized by NMR spectroscopy, single-crystal X-ray diffraction and IR spectroscopy. Additionally, [NO][pfAd] could be synthesized containing alcohol impurities but nonetheless enabled the synthesis of the salt P9+[pfAd]−. The synthesis of Tl[pfAd] in a mixture of H2O/acetone/o-DFB demonstrated the water stability of the [pfAd]− anion.
Introduction
Weakly coordinating anions (WCAs)1 enable the formation, isolation and structural characterization of reactive cations,2 the provision of truly free coordination sites during cationic catalysis (e.g. silylium ion based,3 olefin polymerization,4 asymmetric hydrogenation5 and hydrosilylation6) and inhibit the ion-pairing in ionic liquids7 or battery electrolytes (e.g. Li+,8 Mg2+ (ref. 9) or Ca2+ (ref. 10) ions). WCAs often have at least partially fluorinated structural units. Fluorination induces the delocalization of the negative charge over the entire entity, minimizing basic or nucleophilic sites as well as the polarizability of the anion. The size of a WCA also influences the delocalization of the negative charge, as such or in a material. Hence, the size impact of WCAs on charge transfer processes occurring in thin films was recently investigated.11 Interestingly, increased anion size led to an increase in the relative intensity of the stimulated emissions, finally leading to a decrease of self-quenching and increased photovoltaic efficiency.12Common WCA classes are halogenated alkyl- and arylborate anions (e.g. [B(C6F5)4]− (ref. 13)), triflimidates (e.g. [N(SO2CF3)2]− (ref. 14)), closo-carborate and closo-borate anions (e.g. [HCB11(CF3)nF11−n]− with n = 5, 6, 10, 1115) and aluminate anions [Al(ORF)4]− (RF = univalent fluorinated residue). The Lewis acidic aluminum center of the latter combined with a strong aluminum–oxygen bond place fluorinated alkoxyaluminate based anions among the most widely used WCAs.16 In addition, the [M(ORF)X]− (x = 4, 6) WCA-type is known for several central atoms M such as B,7,17 Al,7,18–20 Ta,21 Nb21,22 and ORF = OC(R)(CF3)2 with R = H, CH3, Ph and CF3. Further, aluminates with ORF = OC6F5 and OC5F4N are known.23,24
WCA-limitations
Highly electrophilic metal and non-metal cations as well as highly oxidizing cations are often part of the ionic systems under investigation and in part require tailor-made anions to withstand degradation. Three properties need to be considered when deciding on a type of WCA: (i) coordination ability of the anion towards the cation (ii) electrophilic stability and (iii) oxidation stability.1,2 Unfortunately, there is no single WCA that fulfills all requirements to stabilize the reactive cation (intermediates), which can be illustrated by example reactions of the very commonly used [B(C6F5)4]− anion: it suffices to stabilize the sterically demanding silylium ion [Si(Mes)3]+ and is heavily used in silylium ion catalysis,25 whereas being incompatible towards oxidizing agents exceeding the potential of mild [FeCp2]+ such as [NO]+ or [NO2]+.26,27 Similarly, its solvent free silver salt Ag[B(C6F5)4] is unknown and rather decomposes giving AgC6F5 and B(C6F5)3.28 The anions [B(C6F5)4]−/[B(ArF)4]− (ArF = C6H3-3,5-(CF3)2) even serve as a source for the C6F5/ArF residue in the mild electrochemical synthesis of perfluorobiphenyl/FAr-ArF.29 Consequently, current research still focuses on the development of novel and more resilient ‘ideal’ WCAs such as the recently published WCAs [Al(OTeF5)4]− and [Ga(C2F5)4]−.30,31WCAs of type [M(ORF)X]−
The water and acid stable [Al(OC(CF3)3)4]− anion (from now on [pf]−) is the most utilized WCA of this class and represents a very good compromise in terms of the three mentioned properties. The preparation of [Ag(C2H2)4]+[pf]− underlines the extremely low coordination ability.32 Further, the very high stability towards deelectronation was illustrated by a deelectronation potential of +5.0 V (with RF = CH(CF3)2, vs. Li+/Li).20 The synthetic accessibility of NO+, NO2+, PhenazinF or AnthraceneHal salts with fully reversible formal potentials of 0.87 to 1.42 V and in unpublished work even up to 1.89 V (vs. Fc+/Fc) demonstrates the stability towards strongly oxidizing agents.33,34 Achilles heel of the [pf]− anion is the still very good, but limited stability towards extraordinary strong Lewis acidic cations. Thus, solutions of the [CCl3]+ salt are stable at −20 °C and have a half-life time of 2–3 hours at RT. Large and bulky silylium ions such as [Si(C6Me5)3]+ are compatible with [pf]−. However, small cations such as [SiMe3]+ lead to anion decomposition induced by fluoride ion abstraction.35 Thus, in order to improve the properties of WCAs of the type [M(ORF)X]−, an improved (preferably water stable) version is necessary. It should at least reach the properties of the [pf]− anion in terms of stability towards deelectronation and coordination, but it should surpass its properties in terms of stability against extremely Lewis acidic cations. Another issue constitutes the strong tendency of the WCA [pf]− to lead to disordered crystal structures or superstructures. Indeed, single-crystal X-ray structural analysis is of paramount importance for the characterization of many reactive-cation salts. Although we have recently published a freely downloadable tool to assist in modelling disorder in such kind of crystal structures,36,37 intrinsically more ordered structures as induced by a suitable WCA would be helpful to facilitate structure solution/refinement and wider application.Here we report on the synthesis and first application of Li+, Ag+, Ph3C+, Tl+ and H(OEt2)2+ salts of the WCA [pfAd]− (=tetrakis-perfluoro-1-adamantoxy-aluminate, [Al(OC10F15)4]−, Fig. 1). In addition, we have reinvestigated the fluoride-ion affinities (FIA) as well as a series of related properties of various WCAs including the title compound [pfAd]−.38 D3(BJ) dispersion correction was included and a reliable approach delineated earlier by Greb et al. was further selected for the investigation.39–41
 | ||
| Fig. 1 Illustration of the novel WCA [pfAd]− = [Al(OC10F15)4]−. | ||
Results and discussion
We start with a general investigation underlining the favorable properties of the target WCA [pfAd]− based on the properties of the alcohol HOpfAd = HO-C10F15. Afterwards, the thermodynamic properties of the [pfAd]− anion are discussed along the lines of our 2004 publication38 and underlying its potential WCA performance. Subsequently, the focus is turned to synthetic aspects and the characterization of suitable starting materials to introduce the WCA [pfAd]− into a desired ionic system.Properties of several fluoroalcohols vs. HOpfAd
The acidity of fluorinated alcohols in solution (pKa(MeCN)) and in the gas phase (gas phase acidity GA) provides a first indication towards the stability of a resulting [Al(ORF)4]− WCA: the higher the acidity of the alcohol, the greater is the (expected) stability of the corresponding [Al(ORF)4]− anion (Table 1). Here, the immense influence of perfluorination (when compared with the properties of the non-fluorinated entities) may be underlined by the experimental gas-phase acidity difference of +209 kJ mol−1 for HOpfAd (C10F15OH, 1321 kJ mol−1) in comparison to HO-Ad (C10H15OH 1530 kJ mol−1).42,43 In addition, all HOpfAd values collected in Table 1 exceed those of HO-C(CF3)3 and are very favorable for the projected WCA [pfAd]−.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) KAHA values in acetonitrile; experimental and calculated (MP2/TZVPP) gas phase acidities (GA, kJ mol−1) of perfluorinated alcohols including C10F15OH as well as the non-fluorinated species
KAHA values in acetonitrile; experimental and calculated (MP2/TZVPP) gas phase acidities (GA, kJ mol−1) of perfluorinated alcohols including C10F15OH as well as the non-fluorinated species
In silico study of the properties of the WCA [pfAd]− in comparison to others
We use known38 theoretical concepts to evaluate the properties of our target WCA [pfAd]− and justify possible synthesis. Hence, the fluoride ion affinity (FIA) of the (typically hard) Lewis acid underlying the corresponding WCA is widely used to quantify the stability of a WCA,47 best calculated with an isodesmic SiMe3+-based reference system48 that uses the benchmark FIA(SiMe3+) of 952.5 kJ mol−1.41 Other ion affinities address further aspects: (i) the hydride ion affinity (HIA) takes soft interactions into account, (ii) the chloride (CIA) and (iii) methide ion affinities (MIA) both also consider steric contributions to Lewis acidity. Reliable ion affinity values were recently benchmarked for 183 p-block element Lewis acids by Greb et al.49 Yet, the full evaluation of WCA performance needs the calculation of further properties: the HOMO-/LUMO-energy, HOMO–LUMO-Gap, partial charges qsurf/qneg, the ligand affinity LA as well as the proton/copper decomposition reactions PD/CuD.38 These properties were assessed for a set of ten WCAs and are discussed in detail. First, we discuss the relevant FIA values before turning to the other indicators summarized in Table 2. Note: For backward compatibility, all thermodynamic values were (re-)calculated with the computationally less demanding BP86/def-SV(P) method but including D3(BJ) dispersion correction. It was shown earlier48 that the differences of BP86(D3BJ)/def-SV(P) calculations to benchmark DLPNO-CCSD(T)/cc-pVQZ calculations are typically small for this model chemistry and do not change the relative order. However, for forward compatibility, the thermodynamic values were additionally calculated at Greb's recommended reliable DSD-PBEP86(D3BJ)/def2-QZVPP//pbeh3-c/def2-mSVP level of theory (second, italic values in Table 2).| Anion | Sym. | FIA/kJ mol−1 | LA/kJ mol−1 | PD/kJ mol−1 | CuD/kJ mol−1 | HOMO/eV | Gap/eV | q neg | q surf |
|---|---|---|---|---|---|---|---|---|---|
| a 596 kJ mol−1 at the very reliable DLPNO-CCSD(T)/cc-pVQZ level. Abbreviations: fluoride ion affinity (FIA), ligand affinity (LA), proton induced decomposition (PD), copper induced decomposition (CuD), highest occupied molecular orbital (HOMO), Gap between HOMO and LUMO (lowest unoccupied molecular orbital), partial charges on the surface (qsurf) and most negative partial charges (qneg). | |||||||||
| [B(OCH(CF3)2)4]− | S 4 | 404/391 | 301/284 | −1140/−1164 | −483/−446 | −3.37 | 7.11 | −0.21 F | −0.21 F |
| [B(C6F5)4]− | S 4 | 449/441 | 329/341 | −1177/−1164 | −494/−442 | −3.11 | 4.22 | −0.95 B | −0.22 F |
| [Ga(C2F5)4]− | C 1 | 479/501 | 401/417 | −1174/−1154 | −466/−389 | −3.07 | 6.28 | −0.22 Ga | −0.20 F |
| [B(OC5F4N)4]− | C 1 | 494/465 | 263/246 | −1067/−1094 | −425/−400 | −3.64 | 4.17 | −0.21 N | −0.21 N |
| [Al(OC(CF3)3)4]− | S 4 | 535/547 | 352/366 | −1024/−1020 | −377/−317 | −4.11 | 6.86 | −0.24 O | −0.20 F |
| [Al(C6F5)4]− | S 4 | 539/546 | 367/373 | −1139/−1132 | −456/−410 | −3.35 | 4.29 | −0.21 F | −0.21 F |
| [Al(OTeF5)4]− | S 4 | 545/603 | 293/339 | −981/−950 | −394/−282 | −6.17 | 2.93 | −0.47 O | −0.40 F |
| [Ga(OTeF5)4]− | S 4 | 545/571 | 308/325 | −966/−964 | −379/−296 | −6.09 | 2.79 | −0.46 O | −0.40 F |
| [Al(OC5F4N)4]− | C 1 | 581/588 | 352/363 | −979/−977 | −337/−283 | −3.80 | 4.37 | −0.22 O | −0.21 F |
| [Al(OC10F15)4]− | C 1 | 585a/600 | 379/388 | −959/−963 | −325/−271 | −4.53 | 4.75 | −0.29 O | −0.24 F |
The Ligand affinity (LA) defines the abstraction of a ligand from the Lewis acidic central atom of the WCA and is always endothermic. The highest value collected in Table 2 (417 kJ mol−1, [Ga(C2F5)4]−) indicates good stability towards ligand abstraction. However, the LA also includes the intrinsic stability of the freed ligand. Consequently, released stable perfluorinated alcoholates result in lower LA values and thus the stability of the corresponding anion tends to be underestimated. Nevertheless, the [pfAd]− anion has with 388 kJ mol−1 the second highest entry in Table 2, further supporting its excellent stability.
The stability of the WCAs towards hard and soft electrophiles can be calculated isodesmically considering decomposition reactions with hard H+ for proton decomposition PD and soft Cu+ for copper decomposition CuD. Herein, the contribution of the intrinsic stability of the freed fragment is eliminated. PD and CuD are always exothermic due to the reaction of a gaseous anion and a gaseous cation under formation of two neutral molecules. The less negative the values obtained are, the more stable the corresponding WCA is towards an attack by electrophiles. Once again, promising values were obtained for the [pfAd]− anion, which improve on the frequently used [pf]− anion. The PD values amount to −963 kJ mol−1 compared to −1020 kJ mol−1 and the CuD values amount to −271 kJ mol−1 compared to −317 kJ mol−1 in favor of the [pfAd]− anion. Also, the HOMO energy is with −4.53 eV by 0.42 eV more favorable/lower than the HOMO of the [pf]− anion and thus less susceptible to oxidation. This oxidation stability goes along with an increased reduction sensitivity, as the HOMO–LUMO gap of the [pfAd]− anion is 2.11 eV lower compared to the [pf]− anion (see below). An indication of the coordination ability of an anion is provided by qneg and qsurf. In general, the most negatively charged atom (qneg) shows the greatest tendency towards coordination. However, the steric crowding around the most basic sites need to be considered. Therefore, the most negatively charged but accessible atom (qsurf) is of greater interest. The oxygen atoms carry the largest negative charges in the [pfAd]− anion but are not accessible due to the sterically demanding adamantoxy cages. The [pfAd]− anion tends to be slightly more coordinating compared to the [pf]− anion, but not as strongly coordinating as the OTeF5-based anions with much higher O- and F-charges in Table 2.
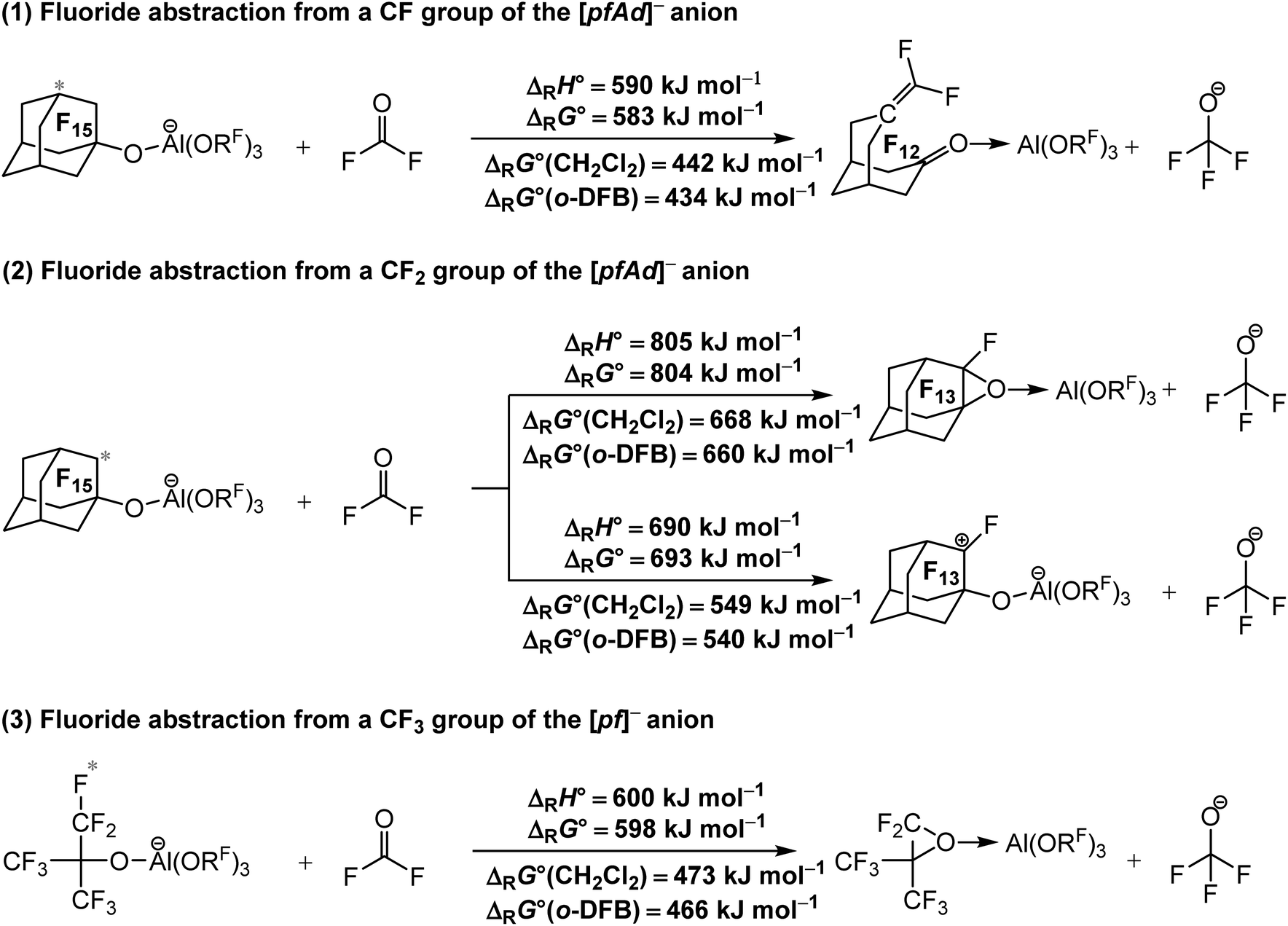 | ||
| Scheme 1 Calculated isodesmic reaction enthalpies ΔRH° and Gibbs free energies ΔRG° for the decomposition of the [Al(ORF)4]− WCAs ((1) and (2) RF = C10F15), (3) (RF = C(CF3)3) under fluoride ion abstraction (BP86/def-SV(P) level with D3(BJ) dispersion). Solvation effects on the Gibbs free energy ΔRG° were considered by using the COSMO solvation model for CH2Cl2 (ε = 8.9352) and o-DFB (ε = 13.3852). The red star marks the abstracted fluoride ion. The experimental FIA of COF2 of 209 kJ mol−1 was used as an anchor point for comparison. | ||
Towards the pure Li[pfAd] salt: unexpected synthetic challenges
The synthesis of Li[pfAd] (1) turned out to be non-trivial. Hence, straightforward reaction of LiAlH4 and C10F15OH in hexane, heptane, toluene, ether, THF etc. according to the established synthesis protocol for Li[Al(OC(CF3)3)4]18 does not work as expected. In order to prevent side reactions caused by strongly hydrogen-bonded water, the alcohol had to be dried in o-DFB solution over activated 3 Å molecular sieves prior to use. Since C10F15OH is one of the most acidic alcohols known, a non-negligible content of NaOC10F15 forms from reaction with the sieves. Hence, the dried alcohol must be sublimed at least twice afterwards to remove any alkoxide traces. Reaction between LiAlH4 and this dried purified alcohol C10F15OH in the weakly coordinating solvent toluene does not proceed even at reflux and under ultrasonic activation. However, reaction of 4.6 equiv. of C10F15OH with LiAlH4 in o-DFB results in an immediate evolution of gas (eqn (1)).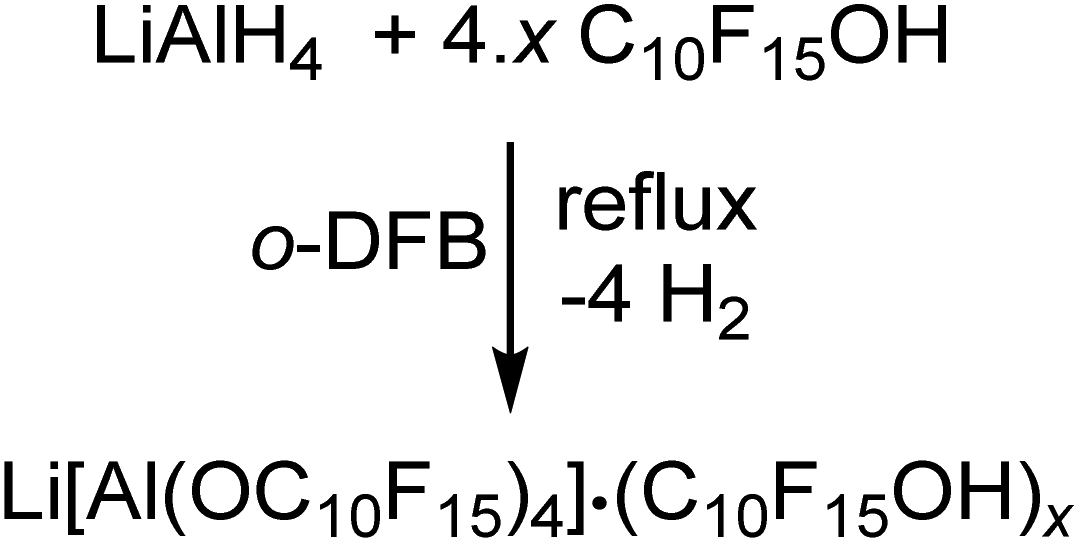 | (1) |
After the gas evolution was finished, indicating the complete turnover, and cooling down the reaction mixture, precipitation of a small amount of a colorless solid was observed. Multinuclear NMR spectroscopy of the precipitated solid and the reaction solution showed resonances caused by the target compound Li[pfAd] (1) as well as unreacted alcohol C10F15OH. Adding a small amount of C10F15OH to an already measured NMR sample leads to a distinct increase in the intensity of the alcohol resonances confirming the impurity as the alcohol. Furthermore, integration of the resonances of the alcohol and the solvent o-DFB in the 1H- and 19F NMR spectra yielded the same stoichiometric ratio providing evidence for the absence of alkoxide C10F15O− (for details see ESI†).
Purifying Li[pfAd]·(C10F15OH)x by derivatization to target salts Cat+[pfAd]−
Since we could only obtain Li[pfAd]·(C10F15OH)x with excess alcohol, we targeted converting the (impure) lithium salt into useful starting materials that are pure products and free of alcohol.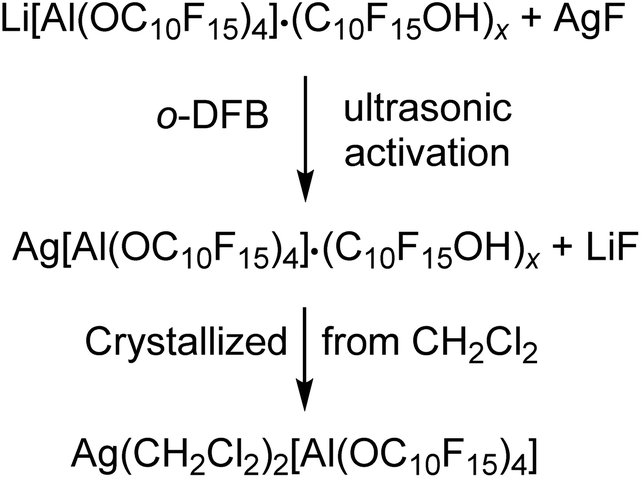 | (2) |
The analysis of the reaction solution of the first attempt showed resonances in the 19F NMR spectrum indicating impurities by the alkoxide C10F15O−, since the proton resonance of the alcohol was missing. This agrees with the fact that the alcohol was not sublimed prior to the synthesis of the Li[pfAd] (1) resulting in a contamination by NaOC10F15 (from the molecular sieves) and AgOC10F15, respectively, after derivatization with AgF. Subsequent gas phase diffusion crystallization with n-pentane resulted in the growth of two different kinds of crystals. The structures were determined as [Ag(o-DFB)2][pfAd] (2-oDFB) and [Ag3(OC10F15)2(o-DFB)3][pfAd] (3) confirming the presence of alkoxide as well as demonstrating the crucial need to sublime the alcohol prior to use. Yet, with these precautions, the silver salt was obtained free of impurities after extraction with CH2Cl2 and crystallization by gas phase diffusion with n-pentane (yield: 40%, each Ag+ coordinated with two CH2Cl2 molecules). Multinuclear NMR spectroscopy of 2 in CD2Cl2 shows three resonances in the 19F NMR spectrum located at δF = −121.6 and −122.0 ppm for the 48 CF2 fluorine atoms and at δF = −223.4 ppm for the 12 CF fluorine atoms. A major resonance in the 27Al NMR spectrum at δAl = 34.7 ppm, consistent with a fourfold coordinated aluminum center, completes the expected NMR resonances. The ESI† further contains the obtained IR spectroscopic data.
 | (3) |
However, the Li+ salt used was contaminated with alcohol and is also hardly soluble in CH2Cl2. The reaction started already in the solid state visualized by an immediate color change to yellow after mixing of the starting materials. Addition of the solvent CH2Cl2 resulted in an intense yellow color of the solution indicating the formation of some 4, yet the large majority remained insoluble at the bottom. The reaction solution was allowed to sediment after two days and analyzed by NMR spectroscopy, showing the presence of the target compound as well as the alcohol C10F15OH as main compound indicating an incomplete turnover or at least a poor solubility of the target compound in CH2Cl2. Suitable crystals for single-crystal X-ray diffraction (scXRD) were obtained from the reaction mixture after filtration and gas phase diffusion crystallization with n-pentane (see X-ray section below). With a yield of less than 3%, this synthesis route does not provide a useful access to [Ph3C][pfAd] (4). Due to the poor solubility of 4 in CH2Cl2, liquid SO2 was used, and the reaction time was extended to 5 days. Then, the SO2 was removed, the solid washed with CH2Cl2 and crystallized by diffusion crystallization from an o-DFB solution with n-pentane resulting in a yield of 55% pure 4. Multinuclear NMR spectroscopy of [Ph3C][pdAd] (4) in CD3CN shows two resonances in the 19F NMR spectrum at δF = −122.0 ppm for the 48 CF2 fluorine atoms and δF = −223.7 ppm for the 12 CF fluorine atoms (for details see ESI†). The 1H NMR spectrum of 4 in CD3CN shows the aromatic protons of the trityl cation with chemical shifts of δH = 7.76 (o-CH), δH = 7.92 (m-CH) and δH = 8.33 ppm (p-CH) (for details see ESI†). Interestingly, the trityl signals of the proton NMR spectrum in CD3CN are initially observed as one broad signal. After about 13 h, the signal begins to split and are cleanly resolved after 24 h. The 7Li- and 27Al NMR spectra as well as the IR spectroscopic data (for details see ESI†) confirm the successful synthesis of 4 free of impurities.
 | (4) |
NMR investigations of the isolated solid in CD2Cl2 showed the alcohol as main compound next to the anion. Based on this observation, the alcohol should be washed off with CH2Cl2 in analogy to the [Ph3C][pfAd] (4) synthesis. Several attempts were unsuccessful and further washing with C6F6 could not remove the alcohol C10F15OH. Due to the reactivity of the NO+ cation, the choice of potential solvents for purification is limited. To the best of our knowledge, it seems likely that there is a strong coordination of the alcohol towards 5. Furthermore, we cannot provide clear evidence for the presence of the cation NO+. The NMR spectra (see ESI†) show the already discussed resonances of the anion as well as the alcohol in the 19F NMR spectrum. The absence of a lithium signal confirms the successful metathesis reaction and removal of Li[BF4]. The 14N NMR spectrum shows no signal, which does not necessarily exclude the presence of the NO+ cation. In fact, the NMR spectra do also not show any signals that would indicate the presence of another cation than NO+. Typically, the NO+ cation can be detected by Raman spectroscopy. The Raman spectrum of 5 shows a broad vibrational band at 2332 cm−1 of low intensity, which can be assigned to the NO+ cation. However, the anion has a great influence on the intensity of the NO+ vibration band of around 2340 cm−1. Thus, NO[F-Al(OC(CF3)3)3] shows a very intense band, whereas the NO+ cation in the bridged [((CF3)3CO)3Al-F-Al(OC(CF3)3)3]− anion shows a vibration band of low intensity.56 Great efforts were therefore made to find suitable crystallization conditions for purification. However, these were unsuccessful. Since there is no clear structural evidence for the NO+ cation apart from the indications from the Raman spectrum, the obtained compound was investigated for its oxidation capacity. NO[pfAd] (5) containing impurities by alcohol and an excess of P4 were suspended in CH2Cl2 under ultrasonic activation resulting in the formation of an orange solution of the known P9+ cation in CH2Cl2 (for details see ESI†).26,57 Furthermore, a small amount of ferrocene Fc was added to a solution of 5 in o-DFB resulting in a change of color from orange to blue, indicative for Fc+ formation. The absence of Li+ and the further described observations allow the conclusion that the synthesis of NO[pfAd] (5) was successful (46%), albeit including a small impurity of the alcohol C10F15OH.
:1 stoichiometry resulted in the formation of [Tl3F2Al(ORF)3][Al(ORF)4] (ORF = O(CH)(CF3)2). Since Li[pfAd] (1) contained impurities of the corresponding alcohol and itself is resistant towards air and moisture, the synthesis in this work was carried out with an excess of TlNO3 in a solution of o-DFB, acetone and H2O (eqn (5)). Here, acetone served as solution intermediary and was allowed to evaporate after a reaction time of 5 d. | (5) |
Tl[pfAd] (6) is well soluble in o-DFB, whereas TlNO3 and LiNO3 show a good solubility in the aqueous phase and consequently could be separated. The obtained colorless solid (65%) showed resonances which indicate the presence of the alcoholate (no alcohol here, 1H NMR signal of the alcohol at 4.00 ppm is absent) and traces of lithium. Crystals suitable for scXRD were obtained from a solution of 6 in o-DFB solution by gas phase diffusion with n-pentane. While the diffraction data are of very good quality, the model remained unsatisfying (for details see ESI†) but supports the conclusion that Tl[pfAd] crystallized without any solvent molecules, neither co-crystallized, nor did the anion coordinate to the Tl+ cation. Also, the NMR and IR spectroscopic analyses of isolated crystals indicate the target compound to be free of any impurities (for details see ESI†). In addition, this reaction illustrates the high stability of the anion in aqueous environment.
An optimized procedure to clean M[pfAd] Salts
The preparation of salts containing the [pfAd]− anion free of impurities was only possible by derivatization into the [Ph3C]+ or the Ag+ salt so far. However, the introduction of the Li+ and potentially further metal salts was still a declared objective. Hence, we targeted the synthesis of [H(OEt2)2]+[pfAd]− (7) according to eqn (6). Acid 7 should serve as an appropriate starting material for subsequent reactions with M[N(Si(CH3)3)2] (with M = Li+, K+, 0.5 Mg2+, …) under formation of the respective M[pfAd] salts.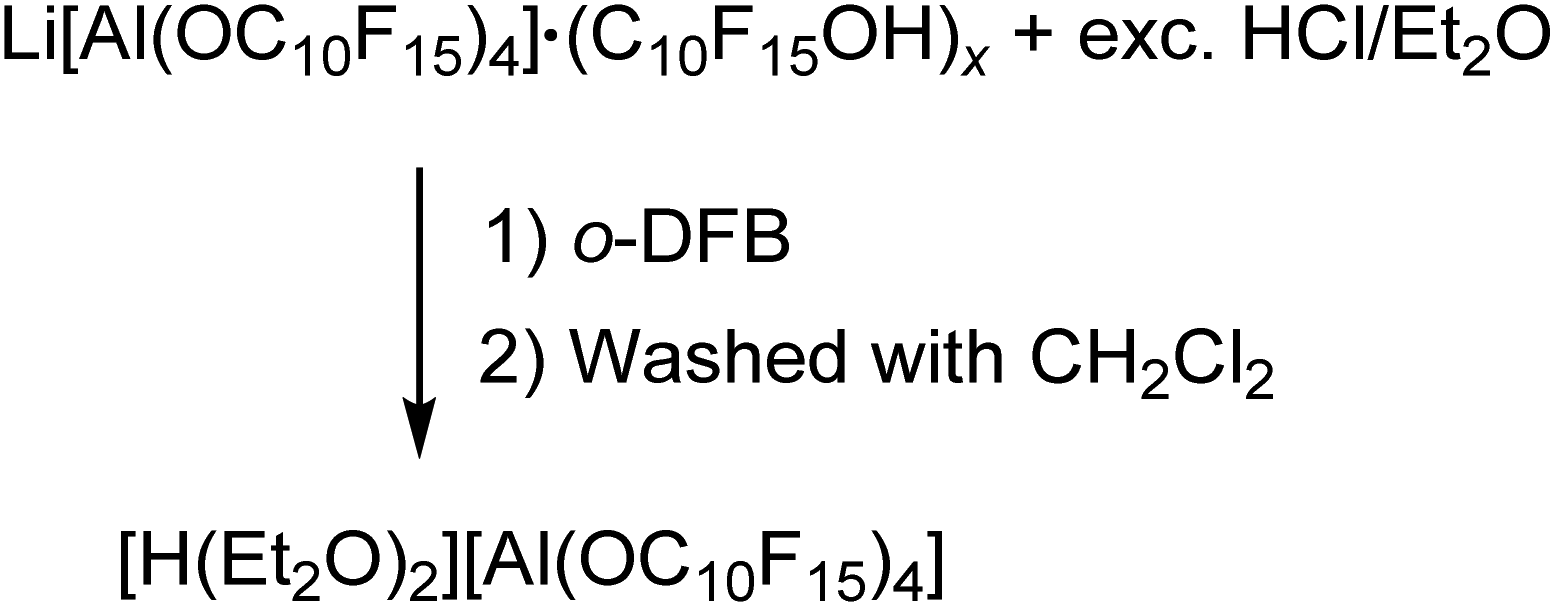 | (6) |
The synthesis of [H(OEt2)2][pfAd] (7) was realized by reaction of Li[pfAd] (1) containing impurities by alcohol with a solution of HCl in Et2O (2 M). This reaction is known for the [pf]− anion with the difference that the preparative effort for 7 was considerably lower due to the simple addition of an excess of etheric HCl solution.59 The target compound 7 is insoluble in CH2Cl2, by contrast to [H(OEt2)2][pf]. Hence, 7 was extracted with o-DFB. NMR investigations still showed the presence of some traces of C10F15OH, which could be washed off with CH2Cl2 after which the target compound was obtained pure (for details see ESI†). The 19F- and 27Al NMR spectra show the characteristic signals for the intact counterion, free of impurities. The 1H NMR spectrum shows the resonances resulting from the CH2 and CH3 protons of the coordinating diethyl ether molecules. A broad, downfield-shifted signal at δH = 16.20 ppm is observed and can be attributed to the coordinated acidic proton. However, the chemical shift of the acidic proton differs from sample to sample within 4 ppm and depending on the solvent used as already observed for [H(OEt2)2][pf].59 The broad signal exacerbates integration but within the limits of the expected inaccuracies, the constitution of two coordinated diethyl ether molecules per proton can be confirmed. Suitable crystals for scXRD investigations could be obtained from a solution of [H(OEt2)2][pfAd] (7) in o-DFB at −40 °C. Furthermore, we showed that this reaction also works with contaminated Na[pfAd] as starting material illustrating the versatility of this reaction (for details see ESI†). The ESI† further contains the obtained IR spectroscopic data for 7. Starting from 7, pure Li[pfAd] (1) was obtained as etherate by reaction with Li[N(Si(CH3)3)2] = LiHMDS (eqn (7)).
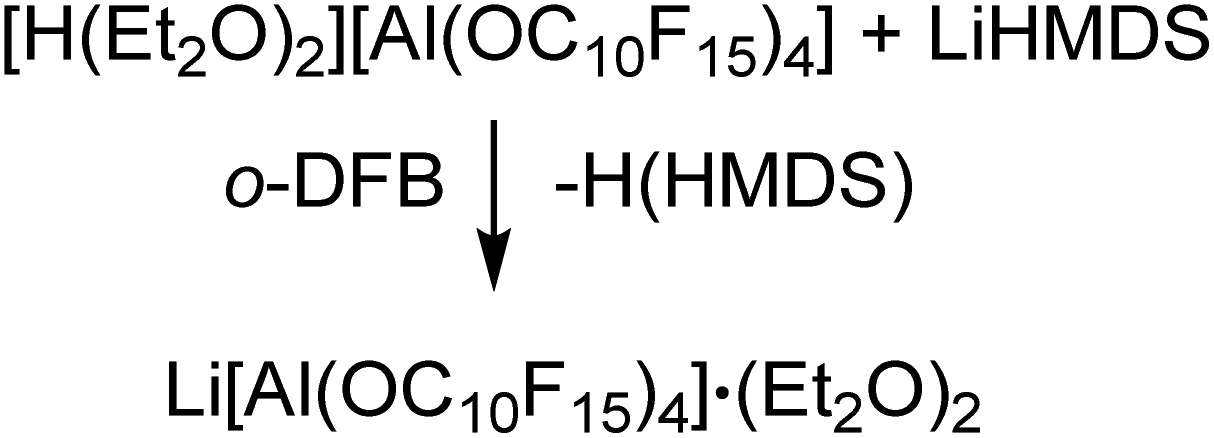 | (7) |
The 19F NMR spectrum only shows the expected resonances attributed to the anion. Nevertheless, the 1H NMR spectrum clearly shows that the obtained Li+-salt is not free of ether (approx. two ether molecules per Li+ salt) even after drying for several hours under reduced pressure at elevated temperatures. Overall, the protonated ether salt 7 can function as a useful starting material for the synthesis of numerous metal salts from commercially available M(HMDS)x sources.
The LUMO energy of the [pfAd]− anion is the lowest in Table 3 and consequently also the calculated  clearly shows that the electronation of the [pfAd]− anion (as well as [pf]−) are thermodynamically feasible in solution. Based on these results and the observed suppressed Mg0 deposition with the [pf]− anion, but not with the related [Al(OCH(CF3)2)4]− for which
clearly shows that the electronation of the [pfAd]− anion (as well as [pf]−) are thermodynamically feasible in solution. Based on these results and the observed suppressed Mg0 deposition with the [pf]− anion, but not with the related [Al(OCH(CF3)2)4]− for which  is endergonic,60 we suggest that Mg0 deposition will be unlikely with WCA-electrolytes containing major (per-)fluorinated entities such as the [pfAd]− anion, as their tendency towards electronation is too high.
is endergonic,60 we suggest that Mg0 deposition will be unlikely with WCA-electrolytes containing major (per-)fluorinated entities such as the [pfAd]− anion, as their tendency towards electronation is too high.
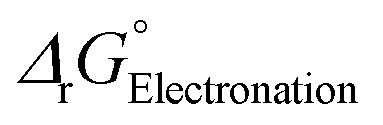 for the one-electron reduction of four WCAs and accepting LUMO energy levels for these WCAs in DME (εr = 7.30).52 Except the values for the [pfAd]− anion, taken from60 (RI-B3LYP(D3BJ)/def2-TZVPP)
for the one-electron reduction of four WCAs and accepting LUMO energy levels for these WCAs in DME (εr = 7.30).52 Except the values for the [pfAd]− anion, taken from60 (RI-B3LYP(D3BJ)/def2-TZVPP)

Molecular structures of the [pfAd] Salts
 | ||
| Fig. 2 Molecular structures of the structurally characterized [pfAd]− salts: (a) [Ag(o-DFB)2][Al(OC10F15)4] (2-oDFB), (b) [Ag3(OC10F15)2(o-DFB)3][Al(OC10F15)4] (3), (c) [Ph3C][Al(OC10F15)4] (4), (d) [H(OEt2)2][Al(OC10F15)4] (7). Hydrogen atoms are omitted for clarity with the exception for the acidic, bridging proton in (d) (purple). Disorders are omitted for clarity for (a) and (b) as well as two further o-DFB molecules with an occupation number of 4% in (b). The main occupation numbers are presented here. Thermal displacement ellipsoids were drawn at the 50% probability level. For details see ESI.† | ||
:36 ratio) and shows the typical O–O separation of 245.1(12) pm within the major PART (for details see ESI†).
The delocalization of the negative charge in WCAs should reduce the overall cation–anion interaction. Fig. 3 visualizes the coordination ability of the [pfAd]− anion in comparison to other well-known WCAs. The first row of Fig. 3 shows Hirshfeld surface analyses of salts of the [pfAd]− anion in comparison to salts of the WCAs [pf]− (ref. 59) and [Al(OTeF5)4]−.31Fig. 3 shows several red areas indicating cation–anion distances below the van the Waals radii, most visible for the salt Ph3C+[Al(OTeF5)4]−. Yet, overall the Hirshfeld surfaces of the WCAs indicate very well dispersed and only little interacting surface sites. The coordination ability of WCAs can further be visualized by a projection of the BP86(D3BJ)/def-SV(P) calculated electrostatic potential onto a 0.025 e− Bohr−3 isodensity surface, shown in the bottom row of Fig. 3. A red coloration indicates a high accumulation of negative charge. The charge in the [pfAd]− anion is strongly delocalized resulting in only faintly red colored oxygen and almost green fluorine atoms. In contrast, the oxygen atoms in the anions [pf]− and [Al(OC5F4N)4]− are more red colored and show a higher electron density. Furthermore, Fig. 3 shows that a lower number of fluorine atoms reduces the delocalization of the negative charge and results in a higher electron density on the fluorine atoms, see for example on [Al(OTeF5)4]−.
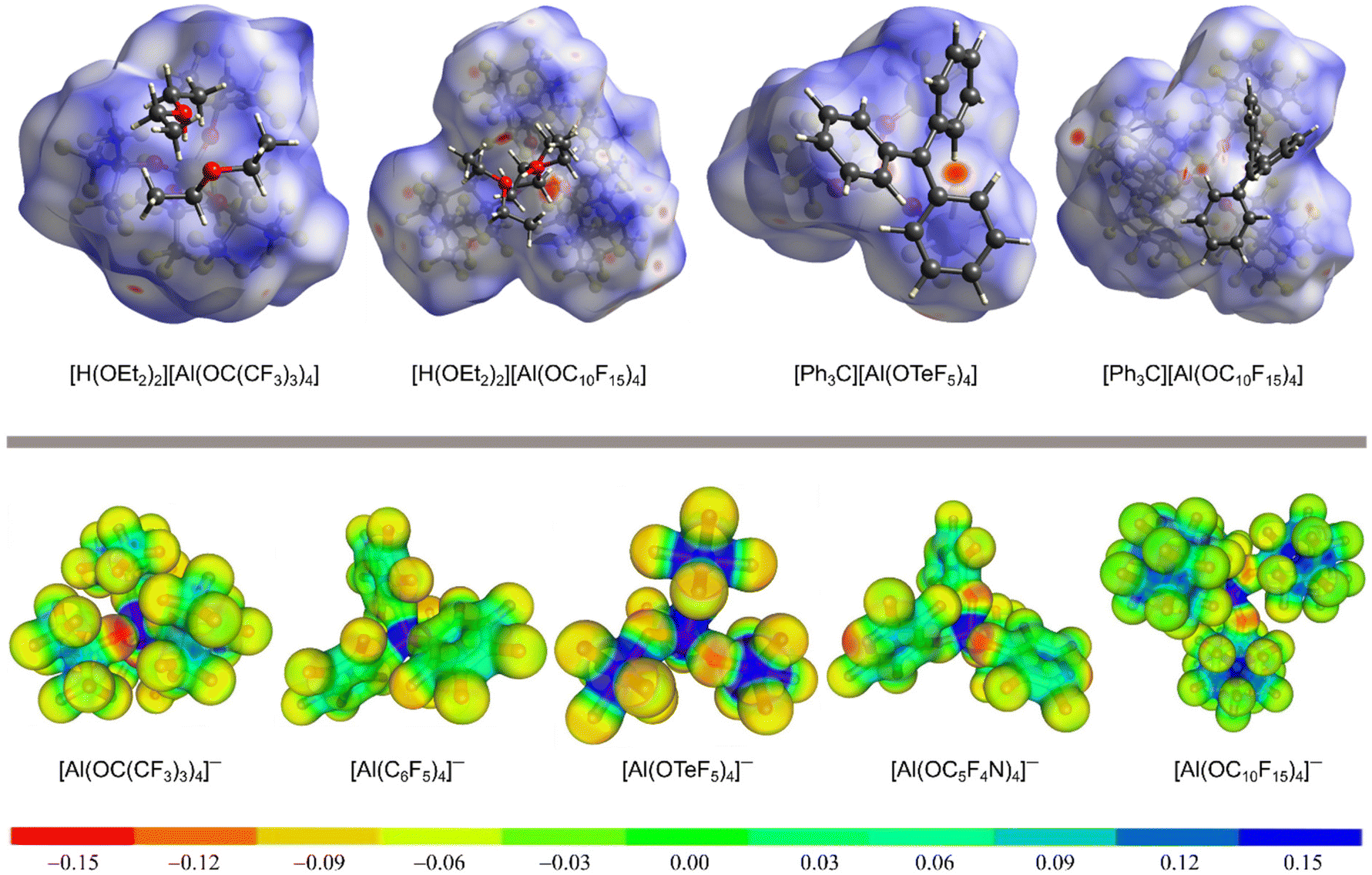 | ||
| Fig. 3 Top: Hirshfeld surface analysis of [H(OEt2)2][WCA] (with WCA = [pf]− (ref. 59) and [pfAd]−) and [Ph3C][WCA] (with WCA = [Al(OTeF5)4]− (ref. 31) and [pfAd]−).63 The red colored areas show close cation-anion contacts below the van der Waals radii, whereas blue colored areas indicated distances above the van der Waals radii. Bottom: Projection of the BB86(D3BJ)/def-SV(P) calculated electrostatic potential mapped onto a 0.025 e− Bohr−3 isodensity surface of the [pfAd]− anion in comparison to already known WCAs. | ||
Conclusions
Quantum chemical calculations of the novel weakly coordinating anion [Al(OC10F15)4]− [pfAd]− allude an improved stability towards reactive cations compared to the widely used [Al(OC(CF3)3)4]− [pf]− anion. The [pfAd]− anion is featured by a high fluoride ion affinity of the corresponding Lewis acid, ligand affinity as well as a high stability towards proton and copper induced decomposition. Unexpectedly, the development of rational synthesis routes to starting materials to introduce the anion was challenging. The synthesis of Li[pfAd] was always accompanied by the occurrence of unreacted alcohol, which cannot be removed, nor omitted. This crucial problem was solved in subsequent salt metathesis reactions enabling the syntheses and structural characterizations of the pure Ag+, Ph3C+ and [H(OEt2)]+ salts in good to excellent yields. Especially the protonated ether salt allows an easy access to a variety of useful materials by further reaction with M[N(Si(CH3)3)2], which was exemplarily demonstrated here by the reaction with Li[N(Si(CH3)3)2] finally providing access to a pure, but ether solvated Li[pfAd] salt. The useful water stability of the [pfAd]− anion is highlighted by the synthesis of Tl[pfAd] under aqueous reaction conditions. This facilitates further application enormously. The actual synthetic value of the [pfAd]− anion to stabilize strong electrophiles is currently under investigation.Experimental and method section
General considerations
All manipulations were carried out under an inert argon atmosphere, using standard vacuum and Schlenk techniques or a glovebox (box atmosphere kept below 1 ppm H2O/O2) unless stated otherwise. Glassware has been stored in an oven at 180 °C overnight and was further dried with a heat gun under vacuum prior to use. Unless stated otherwise all reactions were performed in special double-Schlenk tubes separated by a G3 or G4 frit and equipped with grease-free PTFE or glass valves. All solvents and reagents were dried using conventional drying agents, distilled afterwards and stored under argon atmosphere over activated 3 Å molecular sieves.NMR spectroscopy
NMR spectra were recorded on a Bruker Avance II+ 400 MHz WB, Bruker Avance III HD 300 MHz and a Bruker Avance DPX 200 MHz NMR spectrometer at room temperature (unless described otherwise). The software package Bruker TopSpin 4.0.7 was used for analysis. The spectra were calibrated based on the chemical shift of the solvents used. All relevant spectra can be found in the ESI.†Vibrational spectroscopy
Raman spectroscopy was performed at r.t. with a Bruker VERTEX 70 spectrometer equipped with a Bruker RAM II module (Nd/YAG 1064 nm laser) with a nitrogen cooled Ge detector. The samples were measured in sealed soda-lime glass Pasteur pipettes or in NMR tubes equipped with a gas-tight J. Young valve in the region of 4000 to 200 cm−1 with a resolution of 4 cm−1 with 1000 scans. ATR FTIR spectroscopy was performed at r.t. with a diamond crystal on a Bruker ALPHA spectrometer with a QuickSnap Platinum ATR sampling module inside an inert atmosphere glovebox. A KBr beam splitter was used for the spectral range from 4000 to 400 cm−1. The spectra were recorded with 64 scans and resolution of 2 cm−1. Data processing was carried out with the software package OPUS 7.5 and OriginPro 2019b. Unless stated differently, the usual processing procedure included a baseline correction with one iteration and a standardization of the signal intensities. The relative band intensities were described as follows: ≥0.8 = very strong (vs), ≥0.6 = strong (s), ≥0.4 = medium (m), ≥0.2 = weak (w), <0.2 = very weak (vw).Single crystal X-ray diffraction
Single-crystal X-ray data were collected on a Bruker SMART APEXII QUAZAR three-circle diffractometer with a microfocus sealed X-ray tube using Incoatec mirror optics as a monochromator. Mo-Kα radiation (λ = 0.71073 Å) was used for all measurements. The obtained crystals were coated with perfluoropolyalkylether oil (AB128330, ABCR GmbH & Co. KG), mounted on 0.1 to 0.3 mm micromounts and shock-cooled to 100 K on the diffractometer (using an Oxford Cryosystem open flow N2 cooling device).64 All data were integrated with SAINT,65 a multi-scan absorption-correction using SADABS-2016/2 was applied.66 The structures were solved by direct methods using SHELXT 2014/567 and refined by full-matrix least-squares methods against F2 by SHELXL-2018/368 employing the shelXle GUI (Revision 1449).69 All non-hydrogen atoms were refined with anisotropic displacement parameters. The hydrogen atoms were refined isotropically on calculated positions using a riding model with their Uiso values constrained to 1.5 times the Ueq of their pivot atoms for terminal sp3 carbon atoms and 1.2 times for all other carbon atoms. Disordered moieties were refined using bond-length restraints and displacement-parameter restraints. Some parts of the disorder model were introduced by the program DSR.36,37 The cif files were generated using FinalCif.37,70 Graphical representations were prepared using the software OLEX2 (version 1.3.0).71 Crystallographic data (including structure factors) for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre. CCDC 2224626 (for 2-oDFB), 2224627 (for 3), 2224628 (for 4), 2224629 (for 7)† contain the supplementary crystallographic data for this paper. Additionally, the cif file of Tl[Al(OC10F15)4] is attached (including structure factors) since it was not deposited with the CCDC.†Computational details
Unless stated otherwise, quantum chemical calculations were carried out with the TURBOMOLE software72 (v7.2 or v7.5) in the highest possible point group using the DFT functional BP8673 or pbeh-3c74 (the resolution-of-identity RI approximation75) in combination with the def-SV(P),76 def2-mSVP74 and def-TZVPP77 basis sets and D3 dispersion correction with Becke–Johnson (BJ) damping,39,40 a fine integration grid (m3 or m4) and the default SCF convergence criteria (10−6 a.u.). Every species presented herein was checked in terms of reasonable geometry and electronic occupation with the EIGER module. Vibrational frequencies were calculated analytically using the AOFORCE module.78 All structures represented true minima without imaginary frequencies on the respective hypersurface. Thermodynamic terms were calculated with inclusion of zero-point energy and thermal contributions to the enthalpy/entropy (FREEH tool; unscaled BP86 vibrational frequencies). Gibbs free energies of solvation were calculated with the COSMO model.79 Single point calculations at the DLPNO-CCSD(T)80/cc-pVQZ81 and DSD-PBEP86(D3BJ)82/def2-QZVPP83 level of theory were performed with ORCA (v4.1.2 or v5.0.2) based on the pbeh-3c structures. The ESI† contains a more detailed description of all performed calculations.Preparation of Li[Al(OC10F15)4]·(C10F15OH)x (with x = 0.43)
Purified and slightly ground colorless (!) LiAlH4 (0.100 g, 2.64 mmol) and perfluoro-1-adamantanol (5.005 g, 11.86 mmol, 4.50 eq.) were weighed into a Schlenk flask and suspended in o-DFB (40 mL). The reaction mixture was stirred for 3 h under reflux. The solvent was removed under reduced pressure, the obtained colorless solid was washed with toluene (3×, 20 mL), dried under reduced pressure at 100 °C for 3 h (4.80 g, 2.53 mmol, 96%) and analyzed by NMR spectroscopy showing the impurity of unreacted alcohol C10F15OH (Li[Al(OC10F15)4]:C10F15OH = 1.0:0.43 by integration of the CF groups) (Note: The chemical shift of the proton resonance of the OH group of C10F15OH shows a significant downfield shift in THF compared to the same signal in o-DFB. The yield was determined by use of the calculated molar mass of 1891.74 g mol−1 for Li[Al(OC10F15)4]·(C10F15OH)x with x = 0.43). 1H NMR (300.18 MHz, 298 K, THF): δ = 9.58 (br. s, 1H, C10F15OH) ppm. 7Li NMR (116.66 MHz, 298 K, THF): δ = −0.5 (s, 1Li, Li[Al(OC10F15)4]) ppm. 19F NMR (282.45 MHz, 298 K, THF): δ = −224.0 (m, CF, 12F, Li[Al(OC10F15)4]), −223.4 (m, CF, 3F, C10F15OH), −140.2 (m, CF, 2F, o-DFB), −122.0 (m, CF2, 48F, Li[Al(OC10F15)4]), −121.8 (chemical shift not exactly determinable due to overlapping, CF2, 12F, C10F15OH) ppm. 27Al NMR (78.22 MHz, 298 K, THF): δ = 34.6 (s, 1Al, Li[Al(OC10F15)4]) ppm.
Preparation of Na[Al(OC10F15)4] containing impurities by C10F15OH
Purified and slightly ground NaAlH4 (0.033 g, 0.61 mmol) and perfluoro-1-adamantanol (1.074 g, 2.544 mmol, 4.16 eq.) were weighed into a Schlenk flask and suspended in o-DFB (10 mL). The reaction mixture was stirred for two days under reflux and analyzed by NMR spectroscopy. The solvent was removed under reduced pressure, washed with toluene (3× 5.0 mL) and dried in vacuo at 90 °C. The obtained colorless solid (0.980 g, 0.565 mmol, 92.5%) was analyzed by NMR spectroscopy showing impurity of unreacted alcohol C10F15OH and very small amounts of [F–Al(OC10F15)3]−. 1H NMR (300.18 MHz, 298 K, o-DFB/THF): δ = 9.52 (br. s, 1H, C10F15OH) ppm. 19F NMR (282.45 MHz, 298 K, o-DFB/THF): δ = −223.7 (m, CF, 12F, Na[Al(OC10F15)4]), −223.2 (m, CF, 3F, C10F15OH), −191.3 (m, F, 1F, [F–Al(OC10F15)3]−), −122.2 (m, CF2, 12F, C10F15OH), −121.7 (m, CF2, 48F, Na[Al(OC10F15)4]) ppm. 23Na NMR (79.40 MHz, 298 K, o-DFB/THF): δ = −6.5 (s, 1Na, Na[Al(OC10F15)4]) ppm. 27Al NMR (78.22 MHz, 298 K, o-DFB/THF): δ = 34.9 (s, 1Al, Na[Al(OC10F15)4]), 42.5 (broad, 1Al, [F−Al(OC10F15)3]−) ppm.Preparation of Ag[Al(OC10F15)4] containing impurities by C10F15O−
Li[Al(OC10F15)4] (0.205 g, 0.152 mmol) containing impurities of the alcoholate C10F15O− (since this was one of the first attempts wherein the dried alcohol C10F15OH was not sublimed prior to synthesis of Li[Al(OC10F15)4] consequently containing NaOC10F15) and AgF (0.048 g, 0.378 mmol, 3.17 eq.) were suspended in o-DFB (5.0 mL) and sonicated for 14 h. After sedimentation of the solid an NMR was measured from the solution. The reaction mixture was filtered and crystallized by diffusion crystallization with n-pentane (5 mL) for purification. Two different sorts of crystals suitable for single crystal XRD were obtained as colorless needles ([Ag3(OC10F15)2(o-DFB)3][Al(OC10F15)4]) (3) and colorless blocks ([Ag(o-DFB)2][Al(OC10F15)4]) (2-oDFB). Since it was impossible to separate the two different sorts of crystals, the declaration of a yield is not reasonable at this point. For further investigation, an NMR spectrum of the isolated crystals containing both sorts of crystals was measured in CD2Cl2 containing undissolved solid. 1H NMR (400.17 MHz, 298 K, CD2Cl2): δ = 7.13 (m, 2H, o-DFB), 7.19 (m, 2H, o-DFB) ppm. 19F NMR (376.54 MHz, 298 K, CD2Cl2): δ = −223.3 (m, CF, 12F, Ag[Al(OC10F15)4]), −222.4 (m, CF, 3F, AgOC10F15), −139.4 (m, 2F, o-DFB), −122.6 (m, CF2, 12F, AgOC10F15), −121.6 (m, CF2, 48F, Ag[Al(OC10F15)4]) ppm. 27Al NMR (104.27 MHz, 298 K, CD2Cl2): δ = 35.0 (s, 1Al, Ag[Al(OC10F15)4]) ppm.Preparation of pure Ag(CH2Cl2)2[Al(OC10F15)4]
Li[Al(OC10F15)4] (0.855 g, 0.498 mmol) containing impurities of the alcohol C10F15OH and AgF (0.104 g, 0.820 mmol, 1.65 eq.) were suspended in o-DFB (3.0 mL) and sonicated for 10 h. The solvent o-DFB was removed under reduced pressure. CH2Cl2 (3.0 mL) was added, and the target compound was extracted 5 times by condensing the solvent back to the reaction mixture after filtration. n-pentane was added into the empty part of the H-cell (where the reaction took place) for diffusion crystallization. The target compound was obtained as colorless crystals (0.360 g, 0.200 mmol, 40%), washed with n-pentane (2× 5 mL), dried under reduced pressure, and analyzed by NMR spectroscopy. 19F NMR (282.45 MHz, 298 K, CD2Cl2): δ = −223.4 (m, CF, 12F, Ag[Al(OC10F15)4]), −122.0 (m, CF2, Ag[Al(OC10F15)4]), −121.6 (m, CF2, Ag[Al(OC10F15)4]) ppm. 27Al NMR (78.22 MHz, 298 K, CD2Cl2): δ = 34.7 (s, 1Al, Ag[Al(OC10F15)4]) ppm. FTIR (Diamant, ATR):![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 2966 (vw), 1351 (vw), 1268 (vs), 1106 (vw), 1038 (vw), 981 (m), 955 (vs), 852 (vw), 814 (vw), 758 (vw), 736 (vw), 679 (vw), 651 (vw), 533 (vw), 442 (vw), 402 cm−1.
= 2966 (vw), 1351 (vw), 1268 (vs), 1106 (vw), 1038 (vw), 981 (m), 955 (vs), 852 (vw), 814 (vw), 758 (vw), 736 (vw), 679 (vw), 651 (vw), 533 (vw), 442 (vw), 402 cm−1.
Preparation of [Ph3C][Al(OC10F15)4]
Li[Al(OC10F15)4] (0.260 g, 0.152 mmol) containing impurities of the alcohol C10F15OH and Ph3CCl (0.067 g, 0.240 mmol, 1.59 eq.) were suspended in SO2 (3.0 mL) and stirred at room temperature for six days. The reaction mixture was allowed to sediment and filtered afterwards. The sedimented solid was extracted five times by condensing SO2 back, stirring and filtration resulting in an intensive yellow solution on product side and a colorless solid on reactant side. SO2 was removed under reduced pressure. The yellow solid was washed with CH2Cl2 (5.0 mL) in order to remove impurities since the target compound is hardly soluble in CH2Cl2. The solid residue was dissolved in o-DFB (2.0 mL) and crystallized by diffusion crystallization with n-pentane (30 mL) for purification (Note: The progress of crystallization could be very well traced by the decolorization of the initially bright yellow solution). The yellow crystals (0.164 g, 0.084 mmol, 55%; Note: 65% yield considering that the used Li[Al(OC10F15)4] was contaminated by 0.73 eq. alcohol) were isolated after filtration and characterized by NMR spectroscopy. The discussed molecular structure was obtained from a previous attempt, where CH2Cl2 was used as solvent. Diffusion crystallization with n-pentane resulted in the formation of yellow crystals. 1H NMR (200.13 MHz, 298 K, CD3CN): δ = 7.76 (d, o-CH, 6H, [Ph3C]+), 7.92 (t, m-CH, 6H, [Ph3C]+), 8.33 (t, p-CH, 3H, [Ph3C]+) ppm. 7Li NMR (116.66 MHz, 298 K, CD3CN): δ = −2.7 (br. S, 1Li, Li+, only traces) ppm. 13C NMR (50.32 MHz, 298 K, CD3CN): δ = 130.2 (s, Ph, [Ph3C][Al(OC10F15)4], 140.2 (s, Ph, [Ph3C][Al(OC10F15)4], 143.1 (s, Ph, [Ph3C][Al(OC10F15)4], 143.2 (s, Ph, [Ph3C][Al(OC10F15)4], 212.1 (s, Ccarbenium, [Ph3C][Al(OC10F15)4] ppm. 19F NMR (282.45 MHz, 298 K, CD3CN): δ = −223.7 (m, CF, 12F, [Ph3C][Al(OC10F15)4]), −122.0 (m, CF2, 48F, [Ph3C][Al(OC10F15)4]) ppm. 27Al NMR (78.22 MHz, 298 K, CD3CN): δ = 34.8 (s, 1Al, [Ph3C][Al(OC10F15)4]) ppm. FTIR (Diamant, ATR): = 1585 (w), 1486 (vw), 1454 (vw), 1360 (w), 1293 (w), 1269 (vs), 1189 (vw), 1105 (vw), 978 (m), 955 (vs), 843 (vw), 807 (vw), 767 (vw), 756 (vw), 704 (vw), 678 (vw), 650 (vw), 624 (vw), 609 (vw), 533 (vw), 467 (vw), 441 (vw), 402 (vw) cm−1.
Preparation of [NO][Al(OC10F15)4] containing impurities by C10F15OH
Li[Al(OC10F15)4] (0.304 g, 0.177 mmol) containing impurities by alcohol C10F15OH and [NO][BF4] (0.025 g, 0.214 mmol, 1.21 eq.) were suspended in SO2 (3.0 mL) and stirred for 4 d at r.t. The reaction mixture was allowed to sediment and filtered. The solid residue was extracted once by condensing SO2 back followed by filtration. SO2 was removed under reduced pressure and the orange solid residue was washed with C6F6 (2.5 mL). The solvent was removed and the solid residue dried under reduced pressure and the target compound obtained as orange solid (0.143 g, 0.082 mmol, 46%) containing impurities of the alcohol. 1H NMR (400.17 MHz, 298 K, o-DFB): δ = 4.09 (m, 1H, C10F15OH) ppm. 19F NMR (376.54 MHz, 298 K, o-DFB): δ = −223.2 (m, CF, 12F, [NO][Al(OC10F15)4]), −222.8 (m, CF, 3F, C10F15OH), −163.6 (m, 6F, C6F6), −121.5 (m, CF2, 12F, C10F15OH), −121.3 (m, CF2, 48F, [NO][Al(OC10F15)4]) ppm. 27Al NMR (104.27 MHz, 298 K, o-DFB): δ = 35.4 (s, 1Al, [NO][Al(OC10F15)4]) ppm. FT-Raman: = 2332 (vw), 1292 (m), 1148 (vw), 727 (vw), 700 (vw), 667 (vw), 630 (vw), 573 (w), 500 (vw), 442 (w), 403 (vs), 372 (vs), 311 (s) cm−1.
Preparation of [P9][Al(OC10F15)4] containing impurities by C10F15OH
[NO][Al(OC10F15)4] (0.060 g, 0.034 mmol, containing impurities by alcohol C10F15OH) and P4 (0.011 g, 0.089 mmol, 2.60 eq.) were weighed into a J. Young valve NMR tube. After addition of CH2Cl2 (0.5 mL) the reaction mixture was sonicated for 20 h. A yellow solution containing also undissolved solid was obtained and analyzed by NMR spectroscopy. 1H NMR (200.13 MHz, 298 K, CH2Cl2): δ = 3.94 (br. s, 1H, C10F15OH) ppm. 19F NMR (188.31 MHz, 298 K, CH2Cl2): δ = −223.6 (m, CF, 12F, [P9][Al(OC10F15)4]), −222.5 (m, CF, 3F, C10F15OH), −121.9 (m, CF2, [P9][Al(OC10F15)4]), −121.5 (m, CF2, [P9][Al(OC10F15)4]), −121.1 (m, CF2, 12F, C10F15OH) ppm. 31P NMR (81.01 MHz, 298 K, CH2Cl2): δ = −522.1 (s, 4P, P4), −247.3 (m, 4P, [P9]+, PC, [P2P2PP2′P2′]+), 61.4 (m, 1P, [P9]+, PB, [P2P2PP2′P2′]+), 110.5 (m, 4P, [P9]+, PA, [P2P2PP2′P2′]+) ppm.Preparation of Tl[Al(OC10F15)4] containing impurities by C10F15OH/C10F15O−
Li[Al(OC10F15)4] (0.610 g, 0.355 mmol) containing impurities by alcohol C10F15OH and TlNO3 (0.209 g, 0.785 mmol, 2.21 eq.) were dissolved in o-DFB (6.0 mL), acetone (0.5 mL) and H2O (0.5 mL) and stirred at room temperature overnight. Additional H2O (20 mL) was added to dissolve the precipitated colorless solid. The reaction solution was sonicated for 4 h. The organic phase was separated, washed with H2O (3× 5 mL) and the solvents were removed under reduced pressure. The obtained colorless solid (0.442 g, 0.231 mmol, 65%) was analyzed by NMR spectroscopy showing impurities by alcoholate and only traces of Li+. The colorless solid was dissolved in o-DFB (1.5 mL) and crystallized with n-pentane. Initially, only amorphous solid precipitated, but after another few days, the formation of crystals could be observed, which were examined by NMR spectroscopy and single crystal X-ray diffraction. Since the obtained crystals are embedded in the amorphous solid, it was hard to separate larger amounts of crystals from the rest and therefore this is not a suitable purification method. NMR data before crystallization: 1H NMR (400.17 MHz, 298 K, o-DFB): δ = 2.31 (br. s, 6H, acetone) ppm. 7Li NMR (155.52 MHz, 298 K, o-DFB): δ = 2.1 (Li+, only traces, LiOC10F15 or Li[Al(OC10F15)4]) ppm. 19F NMR (282.45 MHz, 298 K, o-DFB): δ = −223.2 (m, CF, 12F, Tl[Al(OC10F15)4]), −222.8 (m, CF, 3F, C10F15OH/C10F15O−), −121.3 (m, CF2, 48F, Tl[Al(OC10F15)4]) ppm. 27Al NMR (78.22 MHz, 298 K, o-DFB): δ = 35.4 (s, 1Al, Tl[Al(OC10F15)4]) ppm. NMR Data after crystallization: 1H NMR (400.17 MHz, 298 K, CD2Cl2): δ = 2.15 (br. s, 6H, acetone) ppm. 7Li NMR (155.52 MHz, 298 K, CD2Cl2): no signal. 19F NMR (376.54 MHz, 298 K, CD2Cl2): δ = −223.4 (m, CF, 12F, Tl[Al(OC10F15)4]), −121.8 (m, CF2, 48F, Tl[Al(OC10F15)4]) ppm. 27Al NMR (104.27 MHz, 298 K, CD2Cl2): δ = 34.7 (s, 1Al, Tl[Al(OC10F15)4]) ppm. FTIR (Diamant, ATR): = 1697 (vw), 1686 (vw), 1350 (vw), 1292 (w), 1268 (vs), 1109 (vw), 979 (s), 954 (vs), 762 (vw), 678 (w), 651 (w), 643 (vw), 533 (vw), 441 (m), 402 (w) cm−1.
Preparation of [H(Et2O)2][Al(OC10F15)4]
Li[Al(OC10F15)4] (2.84 g, 1.65 mmol) containing impurities by alcohol C10F15OH and a solution of HCl in Et2O (2 M, 40 mL, 80.0 mmol) were stirred for five days at r.t. The solvent was removed under reduced pressure. The target compound was extracted with o-DFB (3× 25 mL) and the solvent was removed afterwards under reduced pressure. The solid residue (2.31 g) was washed with CH2Cl2 (4× 10 mL), dried under reduced pressure and the colorless solid (1.99 g, 1.07 mmol, 65%) analyzed by NMR spectroscopy. Additionally, crystals suitable for scXRD could be obtained by storage of [H(Et2O)2][Al(OC10F15)4] in o-DFB at −40 °C. 1H NMR (300.18 MHz, 298 K, o-DFB): δ = 1.63 (t, CH3, 12H, [H(Et2O)2][Al(OC10F15)4]), 4.27 (q, CH2, 8H, [H(Et2O)2][Al(OC10F15)4]), 16.20 (br. s, 1H, [H(Et2O)2][Al(OC10F15)4]) ppm. 7Li NMR (116.66 MHz, 298 K, o-DFB): δ = −0.1 (br. s, 1Li, Li+) ppm, only traces. 19F NMR (282.45 MHz, 298 K, o-DFB): δ = −223.2 (m, CF, 12F, [H(Et2O)2][Al(OC10F15)4]), −121.3 (m, CF2, 48F, [H(Et2O)2][Al(OC10F15)4]) ppm. 27Al NMR (78.22 MHz, 298 K, o-DFB): δ = 35.4 (s, 1Al, [H(Et2O)2][Al(OC10F15)4]) ppm. FTIR (Diamant, ATR): = 1392 (vw), 1353 (vw), 1295 (vw), 1269 (vs), 1255 (m), 1192 (vw), 1108 (vw), 1016 (vw), 981 (m), 955 (vs), 905 (w), 760 (vw), 679 (vw), 652 (w), 533 (vw), 443 (w), 402 (w) cm−1.
Preparation of Li[Al(OC10F15)4]·(Et2O)2
[H(Et2O)2][Al(OC10F15)4] (1.31 g, 0.759 mmol) and LiHMDS (0.127 g, 0.759 mmol, 1.0 eq.) were submitted into a Schlenk flask, suspended in o-DFB (7.0 mL) and stirred for 2.5 h at r.t. The solvent was removed under reduced pressure, the solid residue was washed with toluene (3× 5.0 mL) and dried under reduced pressure for several hours. The target compound was obtained as colorless solid (1.22 g, 0.681 mmol, 90%) and analyzed by NMR spectroscopy. 7Li NMR (116.66 MHz, 298 K, THF): δ = −0.3 (br. s, 1Li, Li+) ppm. 19F NMR (282.45 MHz, 298 K, THF): δ = −224.0 (m, CF, 12F, Li[Al(OC10F15)4]), −122.0 (m, CF2, 48F, Li[Al(OC10F15)4]) ppm. 27Al NMR (78.22 MHz, 298 K, THF): δ = 34.6 (s, 1Al, Li[Al(OC10F15)4]) ppm.Author contributions
A. Billion, M. Schorpp and I. Krossing conceptualized the study. A. Billion, R. Feser and L. Eisele performed syntheses and vibrational spectroscopic characterization. A. Billion and M. Schmitt performed quantum chemical calculations. M. Schorpp performed single crystal measurements. H. Scherer performed NMR spectroscopic analysis and data analysis. B. Butschke performed single crystal data analysis. T. Sonoda and H. Kawa provided the alcohol HO-C10F15. A. Billion wrote the article. I. Krossing supervised the work and co-wrote the article.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the Albert-Ludwigs-Universität Freiburg and by the DFG in the Sachbeihilfe. We would like to thank Fadime Bitgül for the measurement of the NMR spectra. The authors acknowledge support by the state of Baden-Württemberg through bwHPC and the German Research Foundation (DFG) through grant no INST 40/467-1 and 575-1 FUGG (JUSTUS1 and 2 cluster).References
- I. M. Riddlestone, A. Kraft, J. Schaefer and I. Krossing, Angew. Chem., Int. Ed., 2018, 57, 13982–14024 CrossRef CAS PubMed
.
- T. A. Engesser, M. R. Lichtenthaler, M. Schleep and I. Krossing, Chem. Soc. Rev., 2016, 45, 789–899 RSC
- H. F. T. Klare, L. Albers, L. Süsse, S. Keess, T. Müller and M. Oestreich, Chem. Rev., 2021, 121, 5889–5985 CrossRef CAS PubMed
- E. Y. Chen and T. J. Marks, Chem. Rev., 2000, 100, 1391–1434 CrossRef CAS PubMed
-
(a) S. P. Smidt, N. Zimmermann, M. Studer and A. Pfaltz, Chem. – Eur. J., 2004, 10, 4685–4693 CrossRef CAS PubMed
-
(a) M. C. Lipke, A. L. Liberman-Martin and T. D. Tilley, Angew. Chem., Int. Ed., 2017, 56, 2260–2294 CrossRef CAS PubMed
- A. B. A. Rupp and I. Krossing, Acc. Chem. Res., 2015, 48, 2537–2546 CrossRef CAS PubMed
- A. Shyamsunder, W. Beichel, P. Klose, Q. Pang, H. Scherer, A. Hoffmann, G. K. Murphy, I. Krossing and L. F. Nazar, Angew. Chem., Int. Ed., 2017, 56, 6192–6197 CrossRef CAS PubMed
-
(a) K.-C. Lau, T. J. Seguin, E. V. Carino, N. T. Hahn, J. G. Connell, B. J. Ingram, K. A. Persson, K. R. Zavadil and C. Liao, J. Electrochem. Soc., 2019, 166, A1510–A1519 CrossRef CAS
-
(a) Z. Li, O. Fuhr, M. Fichtner and Z. Zhao-Karger, Energy Environ. Sci., 2019, 12, 3496–3501 RSC
- G. C. Fish, J. M. Moreno-Naranjo, A. Billion, D. Kratzert, E. Hack, I. Krossing, F. Nüesch and J.-E. Moser, Phys. Chem. Chem. Phys., 2021, 23, 23886–23895 RSC
- D. Gesevičius, A. Neels, S. Yakunin, E. Hack, M. V. Kovalenko, F. Nüesch and J. Heier, ChemPhysChem, 2018, 19, 3356–3363 CrossRef PubMed
- A. G. Massey and A. J. Park, J. Organomet. Chem., 1964, 2, 245–250 CrossRef CAS
- J. Foropoulos and D. D. DesMarteau, Inorg. Chem., 1984, 23, 3720–3723 CrossRef CAS
- M. G. Fete, Z. Havlas and J. Michl, J. Am. Chem. Soc., 2011, 133, 4123–4131 CrossRef CAS PubMed
-
(a) I. Krossing and A. Reisinger, Coord. Chem. Rev., 2006, 250, 2721–2744 CrossRef CAS
-
(a) S. Bulut, P. Klose and I. Krossing, Dalton Trans., 2011, 40, 8114–8124 RSC
- I. Krossing, Chem. – Eur. J., 2001, 7, 490–502 CrossRef CAS PubMed
-
(a) S. M. Ivanova, B. G. Nolan, Y. Kobayashi, S. M. Miller, O. P. Anderson and S. H. Strauss, Chem. – Eur. J., 2001, 7, 503–510 CrossRef CAS PubMed
- S. Tsujioka, B. G. Nolan, H. Takase, B. P. Fauber and S. H. Strauss, J. Electrochem. Soc., 2004, 151, A1418 CrossRef CAS
- U. P. Preiss, G. Steinfeld, H. Scherer, A. M. T. Erle, B. Benkmil, A. Kraft and I. Krossing, Z. Anorg. Allg. Chem., 2013, 639, 714–721 CrossRef CAS
- J. J. Rockwell, G. M. Kloster, W. J. DuBay, P. A. Grieco, D. F. Shriver and S. H. Strauss, Inorg. Chim. Acta, 1997, 263, 195–200 CrossRef CAS
- I. M. Riddlestone, S. Keller, F. Kirschenmann, M. Schorpp and I. Krossing, Eur. J. Inorg. Chem., 2019, 2019, 59–67 CrossRef CAS
- Y. Sun, M. V. Metz, C. L. Stern and T. J. Marks, Organometallics, 2000, 19, 1625–1627 CrossRef CAS
-
(a) J. B. Lambert and Y. Zhao, Angew. Chem., Int. Ed. Engl., 1997, 36, 400–401 CrossRef CAS
- T. Köchner, T. A. Engesser, H. Scherer, D. A. Plattner, A. Steffani and I. Krossing, Angew. Chem., Int. Ed., 2012, 51, 6529–6531 CrossRef PubMed
-
(a) T. A. Engesser, C. Friedmann, A. Martens, D. Kratzert, P. J. Malinowski and I. Krossing, Chem. – Eur. J., 2016, 22, 15085–15094 CrossRef CAS PubMed
- M. Kuprat, M. Lehmann, A. Schulz and A. Villinger, Organometallics, 2010, 29, 1421–1427 CrossRef CAS
- S. B. Beil, S. Möhle, P. Enders and S. R. Waldvogel, Chem. Commun., 2018, 54, 6128–6131 RSC
- M. Niemann, B. Neumann, H.-G. Stammler and B. Hoge, Angew. Chem., Int. Ed., 2019, 58, 8938–8942 CrossRef CAS PubMed
- A. Wiesner, T. W. Gries, S. Steinhauer, H. Beckers and S. Riedel, Angew. Chem., 2017, 129, 8375–8378 CrossRef
- A. Reisinger, N. Trapp, I. Krossing, S. Altmannshofer, V. Herz, M. Presnitz and W. Scherer, Angew. Chem., Int. Ed., 2007, 46, 8295–8298 CrossRef CAS PubMed
-
(a) J. K. Kochi, Acc. Chem. Res., 1992, 25, 39–47 CrossRef CAS
- C. Armbruster, J. Hunger and I. Krossing, Unpublished data, 26.11.22.
- M. Rohde, L. O. Müller, D. Himmel, H. Scherer and I. Krossing, Chem. – Eur. J., 2014, 20, 1218–1222 CrossRef CAS PubMed
- D. Kratzert, J. J. Holstein and I. Krossing, J. Appl. Crystallogr., 2015, 48, 933–938 CrossRef CAS PubMed
- D. Kratzert and I. Krossing, J. Appl. Crystallogr., 2018, 51, 928–934 CrossRef CAS
- I. Krossing and I. Raabe, Chem. – Eur. J., 2004, 10, 5017–5030 CrossRef CAS PubMed
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed
- P. Erdmann, J. Leitner, J. Schwarz and L. Greb, ChemPhysChem, 2020, 21, 987–994 CrossRef CAS PubMed
- R. Herrero, J. Z. Dávalos, J.-L. M. Abboud, I. Alkorta and M. Mishima, Int. J. Mass Spectrom., 2007, 267, 302–307 CrossRef CAS
-
(a) M. Zhang, M. M. R. Badal, M. Pasikowska, T. Sonoda, M. Mishima, H. Fukaya, T. Ono, H.-U. Siehl, J.-L. M. Abboud and I. A. Koppel, Bull. Chem. Soc. Jpn., 2014, 87, 825–834 CrossRef CAS
- N. Trapp, H. Scherer, S. A. Hayes, R. J. F. Berger, A. Kütt, N. W. Mitzel, J. Saame and I. Krossing, Phys. Chem. Chem. Phys., 2011, 13, 6184–6191 RSC
- A. Kütt, I. Leito, I. Kaljurand, L. Sooväli, V. M. Vlasov, L. M. Yagupolskii and I. A. Koppel, J. Org. Chem., 2006, 71, 2829–2838 CrossRef PubMed
- R. W. Taft, I. A. Koppel, R. D. Topsom and F. Anvia, J. Am. Chem. Soc., 1990, 112, 2047–2052 CrossRef CAS
-
(a) K. O. Christe, D. A. Dixon, D. McLemore, W. W. Wilson, J. A. Sheehy and J. A. Boatz, J. Fluorine Chem., 2000, 101, 151–153 CrossRef CAS
- H. Böhrer, N. Trapp, D. Himmel, M. Schleep and I. Krossing, Dalton Trans., 2015, 44, 7489–7499 RSC
- P. Erdmann and L. Greb, ChemPhysChem, 2021, 22, 935–943 CrossRef CAS PubMed
- L. O. Müller, D. Himmel, J. Stauffer, G. Steinfeld, J. Slattery, G. Santiso-Quiñones, V. Brecht and I. Krossing, Angew. Chem., Int. Ed., 2008, 47, 7659–7663 CrossRef PubMed
- A. Wiesner, L. Fischer, S. Steinhauer, H. Beckers and S. Riedel, Chem. – Eur. J., 2019, 25, 10441–10449 CrossRef CAS PubMed
-
CRC Handbook of chemistry and physics, ed. W. M. Haynes, ed., CRC Press, Boca Raton, London, New York, 97th edn, 2017 Search PubMed
- P. Dabringhaus, A. Barthélemy and I. Krossing, Z. Anorg. Allg. Chem., 2021, 647, 1660–1673 CrossRef CAS
- I. Krossing, H. Brands, R. Feuerhake and S. Koenig, J. Fluorine Chem., 2001, 112, 83–90 CrossRef CAS
- A. Decken, H. D. B. Jenkins, G. B. Nikiforov and J. Passmore, Dalton Trans., 2004, 2496–2504 RSC
- A. Martens, P. Weis, M. C. Krummer, M. Kreuzer, A. Meierhöfer, S. C. Meier, J. Bohnenberger, H. Scherer, I. Riddlestone and I. Krossing, Chem. Sci., 2018, 9, 7058–7068 RSC
- J. Frötschel-Rittmeyer, M. Holthausen, C. Friedmann, D. Röhner, I. Krossing and J. J. Weigand, Sci. Adv., 2022, 8, eabq8613 CrossRef PubMed
- M. Gonsior, I. Krossing and N. Mitzel, Z. Anorg. Allg. Chem., 2002, 628, 1821 CrossRef CAS
- I. Krossing and A. Reisinger, Eur. J. Inorg. Chem., 2005, 2005, 1979–1989 CrossRef
- A. Schmidt, H. Koger, A. Barthélemy, G. Studer, B. Esser and I. Krossing, Batteries Supercaps, 2022, 5(12), e202200340, DOI:10.1002/batt.202200340
- A. Reisinger, N. Trapp and I. Krossing, Organometallics, 2007, 26, 2096–2105 CrossRef CAS
- L. Brammer, E. A. Bruton and P. Sherwood, Cryst. Growth Des., 2001, 1, 277–290 CrossRef CAS
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1006–1011 CrossRef CAS PubMed
- J. Cosier and A. M. Glazer, J. Appl. Crystallogr., 1986, 19, 105–107 CrossRef CAS
- SAINT V8.38A, Bruker AXS, Madison, Wisconsin, USA, 2015 Search PubMed.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3–10 CrossRef CAS PubMed
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2015, 71, 3–8 CrossRef PubMed
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef PubMed
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed
-
D. Kratzert, FinalCif, V104. Available at: https://dkratzert.de/finalcif.html Search PubMed
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS
-
(a) M. von Arnim and R. Ahlrichs, J. Comput. Chem., 1998, 19, 1746–1757 CrossRef CAS
-
(a) J. P. Perdew, Phys. Rev. B: Condens. Matter, 1986, 33, 8822–8824 CrossRef PubMed
- S. Grimme, J. G. Brandenburg, C. Bannwarth and A. Hansen, J. Chem. Phys., 2015, 143, 54107 CrossRef PubMed
-
(a) M. Sierka, A. Hogekamp and R. Ahlrichs, J. Chem. Phys., 2003, 118, 9136–9148 CrossRef CAS
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef
- F. Weigend, M. Häser, H. Patzelt and R. Ahlrichs, Chem. Phys. Lett., 1998, 294, 143–152 CrossRef CAS
- P. Deglmann, F. Furche and R. Ahlrichs, Chem. Phys. Lett., 2002, 362, 511–518 CrossRef CAS
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC
-
(a) C. Riplinger, B. Sandhoefer, A. Hansen and F. Neese, J. Chem. Phys., 2013, 139, 134101 CrossRef PubMed
-
(a) T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS
- S. Kozuch and J. M. L. Martin, Phys. Chem. Chem. Phys., 2011, 13, 20104–20107 RSC
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2224626, 2224627, 2224628 and 2224629. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt00199g |
| This journal is © The Royal Society of Chemistry 2023 |