DOI:
10.1039/D3DT00340J
(Paper)
Dalton Trans., 2023,
52, 5935-5942
Theoretical studies on the mechanism of molybdenum-catalysed deoxydehydration of diols†
Received
3rd February 2023
, Accepted 20th March 2023
First published on 28th March 2023
Abstract
Molybdenum-catalysed deoxydehydration (DODH) of vicinal diols to alkenes has been investigated using density functional theory (DFT). In particular, the mechanism of DODH of diols using a phosphane as a reductant and a [Mo(O)2(QR)2] complex (QR = acylpyrazolonate ligand) as a catalyst has been studied. This reaction was the first description of a Mo-catalysed DODH reaction. Two alternative routes, A and B, have been analysed in which the commonly recognised key steps in DODH processes have been considered: (i) activation of the diol by condensation and formation of a Mo-diolate intermediate, (ii) oxygen atom transfer to phosphane with reduction to a Mo(IV) species, and (iii) alkene extrusion from the Mo-diolate with regeneration of the starting dioxidomolybdenum(VI) complex. In pathway A, the activation of the diol by the molybdenum complex occurred before the oxygen atom transfer, while in pathway B, the oxygen atom transfer of the dioxidomolybdenum complex to the phosphane occurred before the diol activation. In both routes, the final step was alkene extrusion from the molybdenum-diolate species. Pathway B, in which the reduction by phosphane preceded the diol condensation, is energetically preferred to pathway A.
Introduction
The development of processes for the conversion of oxygenated biomass feedstocks into added value chemicals requires efficient and selective processes for oxygen removal and subsequent functionalization.1–5 The deoxydehydration process (DODH),6 in which diols are transformed into olefins (Scheme 1), is an example of such sustainable reactions and consequently is an area of research of enormous importance.7–12 Regarding transition-metal catalysed reactions, oxidorhenium systems are the most studied13 and, by using a variety of reductants14 (phosphanes, dihydrogen, sulphite, alcohols, etc.), good to excellent effectivities were reported.15 In the search for non-precious metals, catalysts based on vanadium16–20 and molybdenum21–30 were also investigated. Based on the observed selectivity, mechanistic probes and computational studies, it is generally accepted that the catalytic DODH reaction involves three basic processes: (i) condensation of a diol to the metal centre to yield a metal diolate species; (ii) reduction of the oxido-metal catalyst with the appropriate reductant;14 and (iii) extrusion of the alkene from the reduced metal diolate.
 |
| | Scheme 1 Transition-metal catalysed deoxydehydration of vicinal diols. | |
Our research group described years ago the DODH of vicinal diols catalysed by [Mo(O)2(QR)2] complexes, dioxidomolybdenum species with acylpyrazolonate ligands.21 This was the first reported example of a Mo-catalysed DODH reaction and, after this report, other research groups22–30 also investigated the Mo-catalysed DODH reaction in various scenarios (catalysts, reductants, solvents, reaction conditions, etc.) in homogeneous and heterogeneous systems.31–33 The DODH of diols catalysed by ammonium heptamolybdate was investigated by Fristrup and co-workers using density functional theory (DFT).25,26 In these studies, the Mo species was simplified as MoO3 to maintain the neutrality of the computational model system, and the reductants were the diol itself (1,2-propanediol or erythritol) or iPrOH, while phosphanes were not considered. In both cases, the reduction of Mo(VI) was found to be the rate-limiting step that occurred after diol activation. The absence of theoretical studies for the DODH reaction of diols catalysed by ligand-supported dioxido-Mo(VI) complexes prompted us to analyse the mechanism of our experimental results.21 For this reaction, two pathways may be considered (see Scheme 2). In pathway A, diol activation occurs before reduction by the phosphane through an oxygen atom transfer, while the opposite situation is observed in pathway B, namely reduction to Mo(IV) species by phosphane and then diol condensation with concomitant oxidation to Mo(VI). The catalytic cycle in both pathways has in common the final step corresponding to the ethylene extrusion from the Mo-diolate species. In particular, DFT calculations have been carried out to propose the reaction mechanism of the DODH of vicinal diols, catalysed by the [Mo(O)2(QR)2] species and using a phosphane as the reductant, and the results are presented and discussed here.
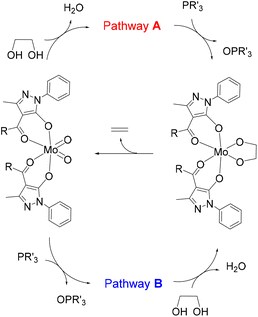 |
| | Scheme 2 Two pathways for the Mo-catalysed DODH reaction. | |
Computational details
The electronic structure and geometries of the model compounds were calculated using density functional theory at the B3LYP level.34,35 For optimisation, the Mo atom was described with the LANL2DZ basis set,36 while 6-311G** was used for the other atoms. This combination of the method and basis sets provided a good structural description of several dioxidomolybdenum [MoO2(QR)2] complexes, according to the comparison of the structural parameters of the optimised structures with those of their crystal structures.21 The molecular geometries of all model complexes were optimised without symmetry constraints. Frequency calculations were carried out at the same level of theory to identify all the stationary points as transition states (one imaginary frequency) or as minima (zero imaginary frequencies) and to provide thermal correction to free energies at 298.15 K and 1 atm. Solution-phase SCF energies of intermediates and transition states were calculated by a single point calculation on the in vacuo optimised structure using the CPCM solvation model in toluene.37 Gibbs free energies in toluene solution were estimated from the equation Gsolv = Esolv + (Ggas − Egas). The energy profiles are presented in terms of relative free energies derived from thermochemical analysis. The zero-energy reference was taken as the energy of the reagents before the reaction occurred (HOCH2CH2OH + [Mo(O)2(QMe)2] + PMe3). The DFT calculations were performed using the Gaussian 09 suite of programs.38 The coordinates of the optimised compounds are reported in Table S1 (ESI†).
Results and discussion
The proposed alternative pathways A and B (Scheme 2) were investigated by DFT calculations and their respective lowest energy profiles were found. First, a simplified model was considered using [Mo(O)2(Q)2], where the Q ligand is 4-formyl-1H-pyrazol-5-olate, to explore both catalytic cycles with less computational effort. Subsequently, a higher level of complexity was introduced by substituting the catalyst with [Mo(O)2(QMe)2], where QMe is the 4-acetyl-3-methyl-1-phenyl-1H-pyrazol-5-olate ligand. This model is rather similar to the catalysts [Mo(O)2(QR)2] (R = alkyl group) that experimentally showed activity in the DODH reaction.21 The selected diol was the simplest ethylene glycol, according to other related publications,17 while PMe3 was chosen as the reductant.39 The simplification of the actual reductant PPh3 by PMe3 has no significant effect according to related DFT studies of the DODH reaction.17 From an energetic point of view, the profiles of the two models are similar, although in the second model the energies of the intermediates are averagely higher, especially those of the transition states, even if certain intermediates are stabilised. This can be rationalised by considering the additional steric hindrance of the catalyst in the second model, particularly in the transition states, where the metal centre is even more crowded. Here, the discussion will be focused on the second model with the catalyst [Mo(O)2(QMe)2]. Several side reactions have been detected in the DODH process, such as oxidative cleavage of the diolate ligand, dehydration, and partial deoxygenation, which are responsible for the low selectivity of the Mo catalysts compared to that of Re systems. DFT analysis of MoO3-catalysed DODH included the oxidative cleavage of the diolate ligand in the mechanism.25,26 However, the consideration of such a step does not agree with our experimental results in which alkene yields were around 80%. For this reason, these side processes or alternative radical mechanisms were not investigated here. In the same way, the disproportionation reaction between Mo(IV) and Mo(VI) that yields the catalytically inactive species Mo(V) has not been taken into consideration. Although this reaction would cause a lower efficiency of the catalysis, it is not of interest for the purposes of the analysis of the DODH mechanism.
Theoretical characterisation of pathway A
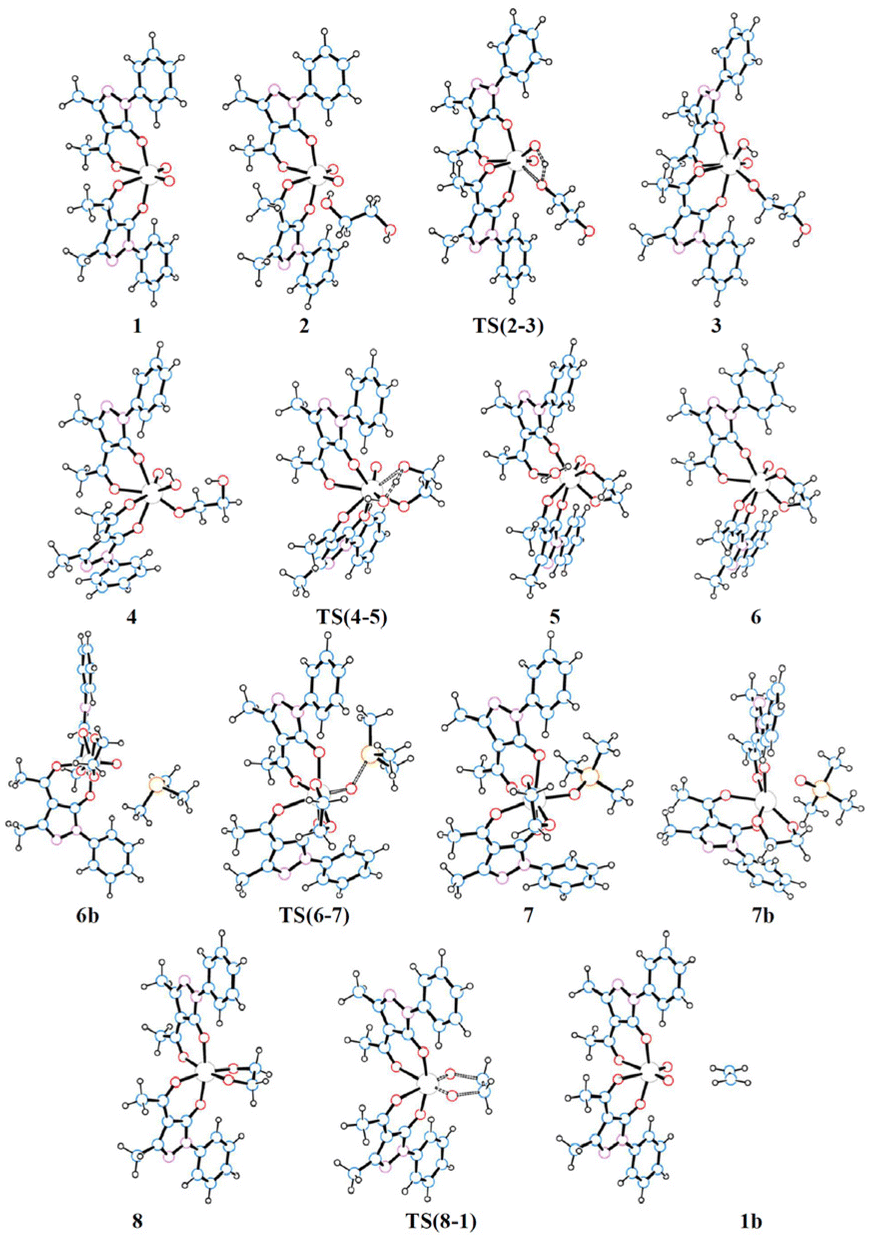
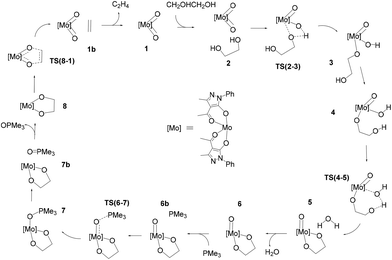
The schematic representation of pathway A is illustrated in Scheme 3, while the optimised structures of the intermediates and transition states involved are shown in Fig. 1. The relative Gibbs free energy profile in toluene is reported in Fig. 2 together with that of pathway B for an appropriate comparison. For the related gas phase profiles, see Fig. S1 (ESI).† The catalyst, 1, shows the typical cis-dioxido structure with a distorted octahedral geometry that is similar to those structurally or theoretically found for related complexes.21 The first step of the cycle, intermediate 2, consists of approaching of ethylene glycol towards 1 and its incipient interaction with the complex. The condensation occurs through the subsequent transition state TS(2–3) in which the H atom is transferred from the diol to one of the oxido groups of the Mo complex, while the same oxygen atom from the diol moves toward the metal centre. Therefore, the acceptor oxido group elongates to 1.836 Å (versus 1.707 Å of the other oxido group) and, at the same time, to accommodate the diol, it moves away increasing the OQ–Mo–Ooxo angle from 104.8° in 1 to 105.1° in TS(2–3). The H atom is placed halfway between the Ooxo and oxygen atoms of the diol with distances Ooxo–H of 1.178 Å and Odiol–H of 1.231 Å. This TS is 32.8 kcal mol−1 higher than 2, which is the highest ΔG difference of this cycle. The condensation terminates in 3 with the complex assuming a distorted pentagonal bipyramidal geometry. The newly formed bonds show Mo–O lengths of 1.988 and 1.940 Å to the diolate and OH ligands, respectively, with a Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond distance of 1.705 Å. The compound then rearranges to the lower energetic conformer 4 in which the diolate assumes the anti-conformation pointing the terminal OH group toward the hydroxido ligand. The stabilisation is due to the partial formation of an intramolecular hydrogen bond (–O–H⋯O length of 1.841 Å) between the terminal hydroxyl group of glycol (Odiol–H = 0.972 Å) and the hydroxido group (Ooxo–H = 0.968 Å). Also, the complex alters its geometry to a highly distorted capped octahedron, thus causing a shortening of the Mo–Odiol bond to 1.906 Å. This interaction drives the cycle through the next transition state TS(4–5) where the terminal oxygen of the glycol approaches the Mo centre (Mo–Odiol distance of 2.381 Å) and simultaneously transfers the H atom to the hydroxido ligand, also pushing it down and away from the molybdenum (Mo–OH length of 2.251 Å). The H atom is located halfway between the two oxygen atoms (1.204 and 1.193 Å). TS(4–5) resides 18.1 kcal mol−1 higher than intermediate 4. This hydrogen-transfer results in the complete removal of the water molecule from the coordination sphere of intermediate 5, restoring the pentagonal bipyramidal geometry. The coordination of the second oxygen of 1,2-ethanediol allows the formation of the diolate ligand with a Mo–O–C1–C2 dihedral angle of 29.3° and Mo–Odiol bond distances of 1.969 and 1.964 Å. The dissociation of water occurs, and the structure then rearranges making minor adjustments to further lower the energy, settling in intermediate 6. The cycle continues with the reductive oxygen atom transfer. First, the phosphane is placed around the reaction site, in the opposite direction of the diolate, where one acylpyrazolone leans downward to allow the interaction of PMe3 (6b). The actual oxygen atom transfer begins in the transition state TS(6–7) in which the phosphane moves toward the remaining oxido group (Mo–O–P angle of 130.1°) with phosphorus at 2.050 Å from oxygen and, at the same time, the Mo–Ooxo double bond elongates to 1.796 Å. This step fits well with similar oxygen atom transfer reactions in which the nucleophilic attack proceeds by the approaching of the phosphane to the π* MO orbital, with a M–O–P angle of about 130°, in an associative intermolecular process.17,39–41 The ΔG difference of 15.4 kcal mol−1 with respect to 6b compares well with related oxo-transfer processes.42,43 The formation of the PO bond is completed in 7, with a bond length of 1.534 Å. The phosphine oxide still remains coordinated to the metal through the oxygen atom with a Mo–O distance of 2.121 Å, and consequently the molybdenum reduces to Mo(IV). The structure then starts rearranging in 7b, with the dissociation of OPMe3, shifting from the pentagonal bipyramidal geometry to a partially capped trigonal prismatic geometry and relieving the steric pressure. Complete dissociation of the phosphine oxide occurs in 8 allowing the complex to achieve an octahedral geometry with a stabilisation of 14.6 kcal mol−1. The last step of the cycle involves the extrusion of ethylene through TS(8–1). In this TS, weakening of the C–O bonds of the diol (1.844 Å vs. 1.426 Å in 8) and the strengthening of Mo–O (1.792 vs. 1.937 Å) and C–C bonds (1.415 vs. 1.531 Å) are observed. These structural parameters are in agreement with an asynchronous concerted mechanism, the reverse of [3 + 2] cycloaddition.44 The barrier associated with TS(8–1), 22.7 kcal mol−1 with respect to 8, compares well with analogous barriers calculated for the alkene extrusion step in Re-diolate compounds,45,46 and is slightly higher than those found for related Mo-diolate complexes.25,26 Finally, the complete removal of the ethylene from 1b affords the regeneration of catalyst 1.
O bond distance of 1.705 Å. The compound then rearranges to the lower energetic conformer 4 in which the diolate assumes the anti-conformation pointing the terminal OH group toward the hydroxido ligand. The stabilisation is due to the partial formation of an intramolecular hydrogen bond (–O–H⋯O length of 1.841 Å) between the terminal hydroxyl group of glycol (Odiol–H = 0.972 Å) and the hydroxido group (Ooxo–H = 0.968 Å). Also, the complex alters its geometry to a highly distorted capped octahedron, thus causing a shortening of the Mo–Odiol bond to 1.906 Å. This interaction drives the cycle through the next transition state TS(4–5) where the terminal oxygen of the glycol approaches the Mo centre (Mo–Odiol distance of 2.381 Å) and simultaneously transfers the H atom to the hydroxido ligand, also pushing it down and away from the molybdenum (Mo–OH length of 2.251 Å). The H atom is located halfway between the two oxygen atoms (1.204 and 1.193 Å). TS(4–5) resides 18.1 kcal mol−1 higher than intermediate 4. This hydrogen-transfer results in the complete removal of the water molecule from the coordination sphere of intermediate 5, restoring the pentagonal bipyramidal geometry. The coordination of the second oxygen of 1,2-ethanediol allows the formation of the diolate ligand with a Mo–O–C1–C2 dihedral angle of 29.3° and Mo–Odiol bond distances of 1.969 and 1.964 Å. The dissociation of water occurs, and the structure then rearranges making minor adjustments to further lower the energy, settling in intermediate 6. The cycle continues with the reductive oxygen atom transfer. First, the phosphane is placed around the reaction site, in the opposite direction of the diolate, where one acylpyrazolone leans downward to allow the interaction of PMe3 (6b). The actual oxygen atom transfer begins in the transition state TS(6–7) in which the phosphane moves toward the remaining oxido group (Mo–O–P angle of 130.1°) with phosphorus at 2.050 Å from oxygen and, at the same time, the Mo–Ooxo double bond elongates to 1.796 Å. This step fits well with similar oxygen atom transfer reactions in which the nucleophilic attack proceeds by the approaching of the phosphane to the π* MO orbital, with a M–O–P angle of about 130°, in an associative intermolecular process.17,39–41 The ΔG difference of 15.4 kcal mol−1 with respect to 6b compares well with related oxo-transfer processes.42,43 The formation of the PO bond is completed in 7, with a bond length of 1.534 Å. The phosphine oxide still remains coordinated to the metal through the oxygen atom with a Mo–O distance of 2.121 Å, and consequently the molybdenum reduces to Mo(IV). The structure then starts rearranging in 7b, with the dissociation of OPMe3, shifting from the pentagonal bipyramidal geometry to a partially capped trigonal prismatic geometry and relieving the steric pressure. Complete dissociation of the phosphine oxide occurs in 8 allowing the complex to achieve an octahedral geometry with a stabilisation of 14.6 kcal mol−1. The last step of the cycle involves the extrusion of ethylene through TS(8–1). In this TS, weakening of the C–O bonds of the diol (1.844 Å vs. 1.426 Å in 8) and the strengthening of Mo–O (1.792 vs. 1.937 Å) and C–C bonds (1.415 vs. 1.531 Å) are observed. These structural parameters are in agreement with an asynchronous concerted mechanism, the reverse of [3 + 2] cycloaddition.44 The barrier associated with TS(8–1), 22.7 kcal mol−1 with respect to 8, compares well with analogous barriers calculated for the alkene extrusion step in Re-diolate compounds,45,46 and is slightly higher than those found for related Mo-diolate complexes.25,26 Finally, the complete removal of the ethylene from 1b affords the regeneration of catalyst 1.
 |
| | Scheme 3 Pathway A: catalytic cycle for the DODH of ethylene glycol with PMe3 catalysed by [Mo(O)2(QMe)2]. | |
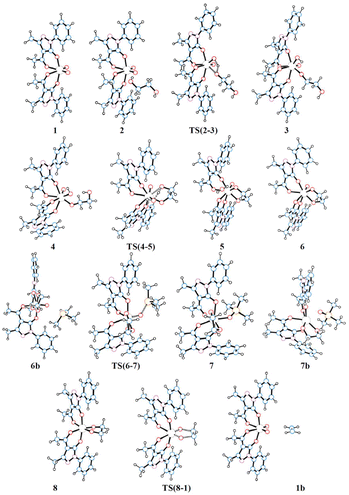 |
| | Fig. 1 Optimised structures of the intermediates and transition states of pathway A. | |
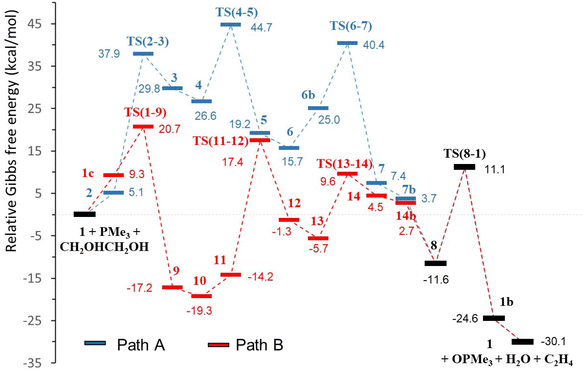 |
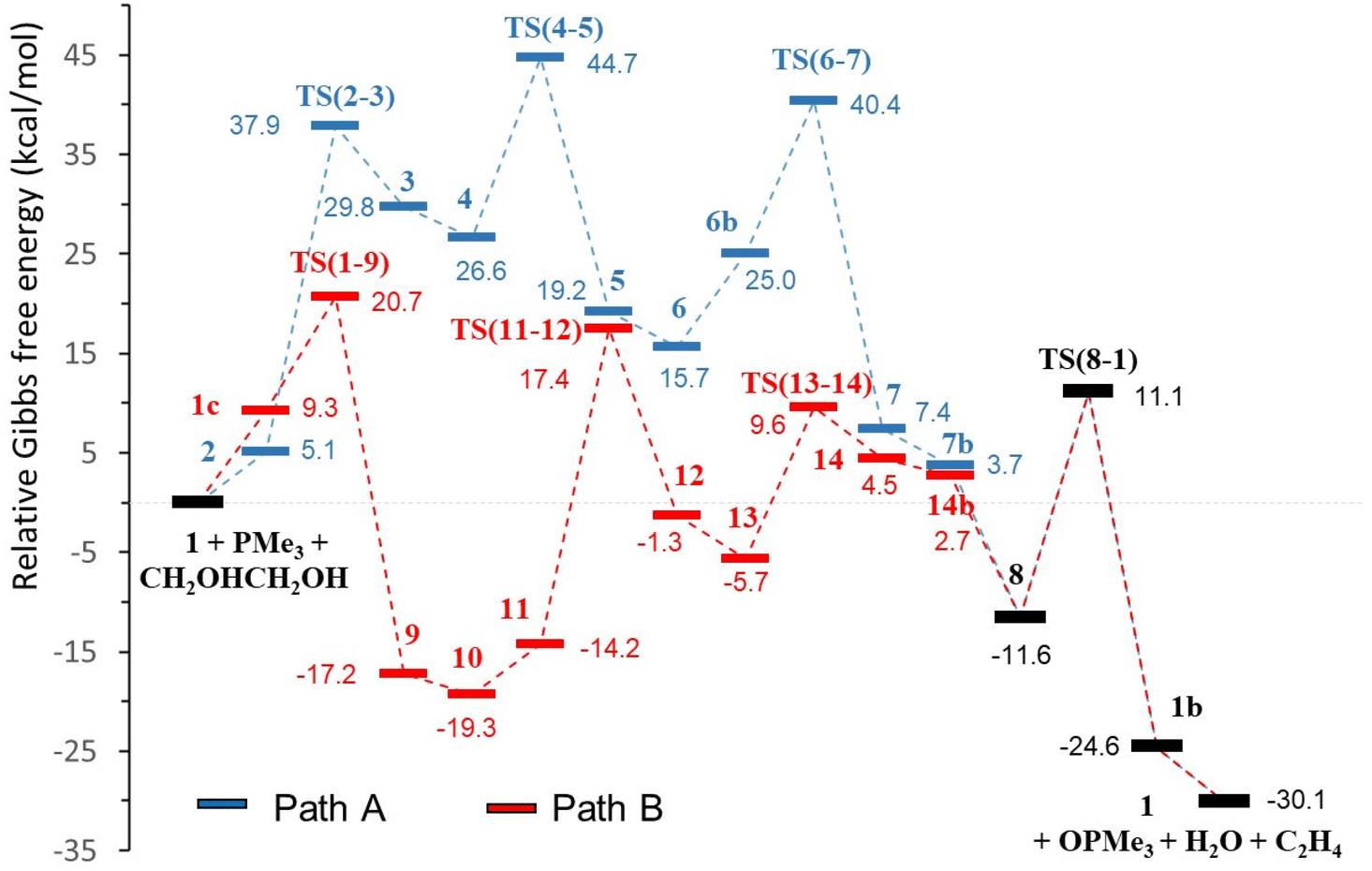
| | Fig. 2 Catalytic profiles, pathways A (blue) and B (red), for the DODH of ethylene glycol with PMe3 catalysed by [Mo(O)2(QMe)2] in toluene (relative ΔG in kcal mol−1, common steps in black). | |
Theoretical characterisation of pathway B
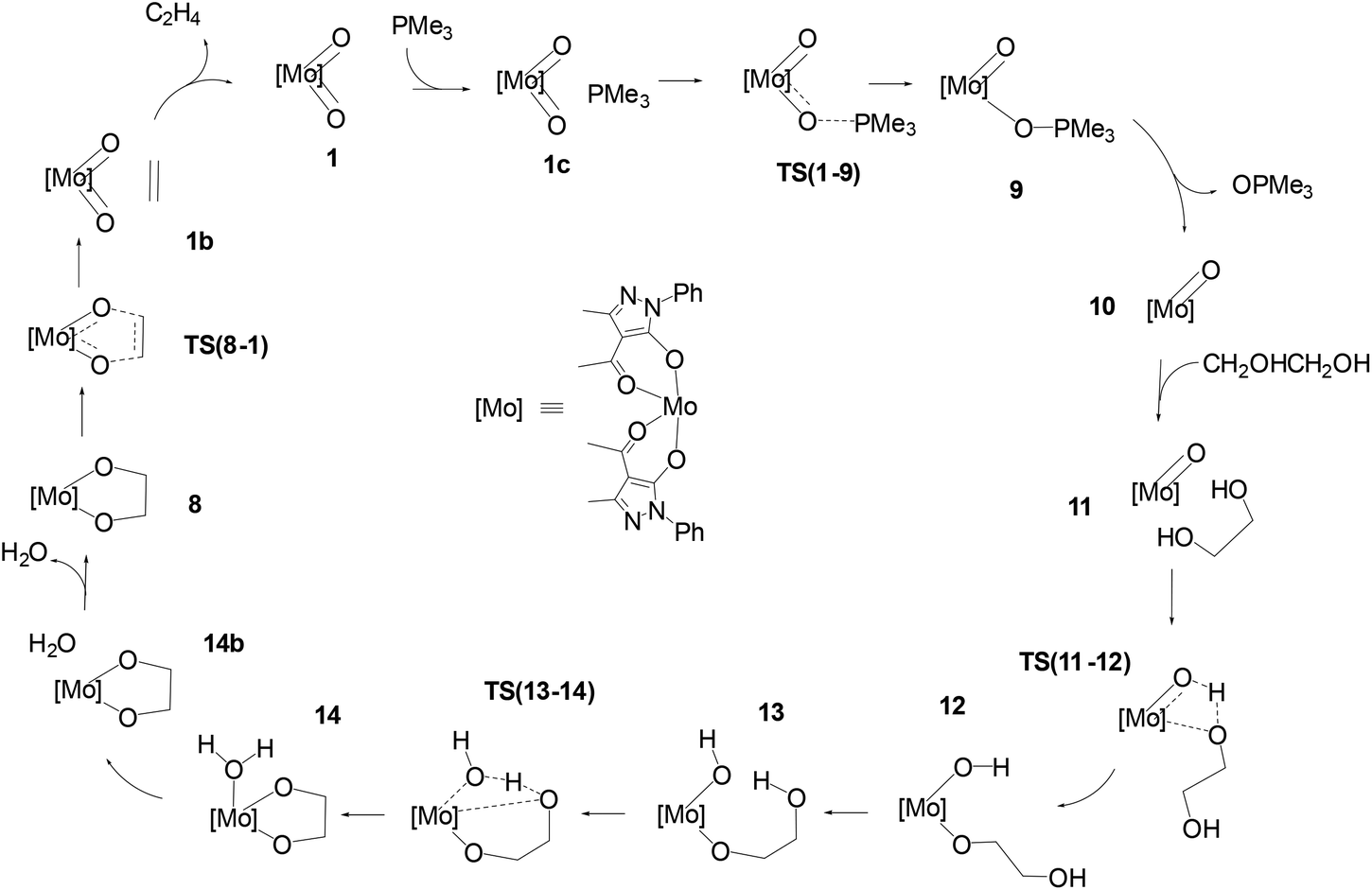
The schematic representation of path B is shown in Scheme 4, while the relative Gibbs free energy profile in toluene appears in Fig. 2. The optimised structures of the intermediates and transition states involved in this pathway are presented in Fig. 3.
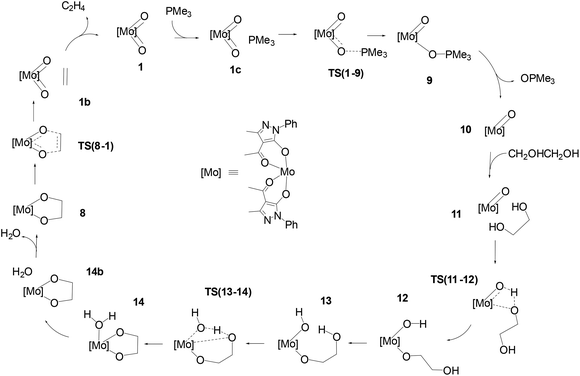 |
| | Scheme 4 Pathway B: catalytic cycle for the DODH of ethylene glycol with PMe3 catalysed by [Mo(O)2(QMe)2]. | |
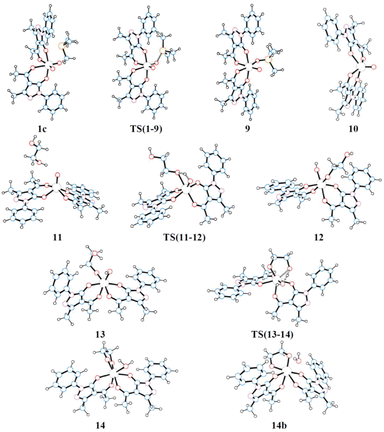 |
| | Fig. 3 Optimised structures of the intermediates and transition states of pathway B. | |
The first step of mechanism B involves the oxygen atom transfer from catalyst 1 to the PMe3 reductant. Initially, PMe3 approaches the catalyst passing through the local minima 1c to finally reach TS(1–9). In this TS, the P–O bond starts to form with a length of 2.049 Å and the typical Mo–O–P bond angle of 130.9° is found. TS(1–9) is also characterised by an elongated MoO bond (1.821 Å) compared to the other (1.702 Å). The energy required to overcome this barrier, in toluene, is equal to 11.4 kcal mol−1 with respect to 1c, which is less than the barrier present in pathway A for the same oxo-transfer step. The TS results in intermediate 9 where the P–O bond is formed (1.525 Å). The compound preserves the octahedral structure with an elongated Mo–O bond (2.222 Å) and PMe3 is placed at a P–O–Mo angle of 127.0°, almost above the remaining oxido group. Subsequent dissociation of phosphine oxide affords 10 which assumes a distorted square based pyramidal structure with the oxido group at the vertex position (MoO, 1.684 Å) and the Mo atom slightly elevated from the square plane. The two acylpyrazolone ligands are placed parallel in opposite directions and their oxygen atoms form the four points of the pyramid base. Intermediates 9 and 10 are the most stable species of profile B and this fact agrees with the experimental observation of related molybdenum(IV) complexes that can be prepared by the reduction of Mo(VI) derivatives with phosphanes.39,47 The process continues with the approach of the diol to the oxido group in intermediate 11 to reach the transition state TS(11–12). In this TS, one of the diol OH groups is placed closer to the MoO group (Ooxo–H, 1.061 and Odiol–H, 1.480 Å) and, at the same time, one of the two acylpyrazolone ligands is tilted, placing itself vertically with respect to the previous square planar plane. This rearrangement makes possible the simultaneous incipient coordination of the diol (Mo–O, 2.217 Å). This TS shows the greatest barrier involved in this pathway, 31.6 kcal mol−1 higher than intermediate 11. TS(11–12) ends in 12 in which the diol is fully coordinated to the metal centre (Mo–O, 1.947 Å) and the hydrogen atom is completely transferred to the oxido group (O–H = 0.968 Å). This intermediate shows a standard octahedral geometry that quickly shifts to the energetically favoured intermediate 13 where the diol rotates pointing the hydroxy group towards Mo–OH, positioning the hydrogen atom of the diol at 1.866 Å (Mo–OH⋯H–Odiol). This arrangement leads to the transition state TS(13–14). The hydrogen atom of the diol approaches the hydroxy group coordinated to Mo positioned halfway, while the second oxygen of the diol starts to advance closer to the metal centre, Mo–Odiol 2.234 Å, compared to the distance of 1.914 Å of the other oxygen atom already coordinated. One of the ligands rotates just to make room for the approaching oxygen atom, moving towards a pentagonal bipyramidal structure. The rearrangement gives intermediate 14, which assumes a capped octahedral geometry with the diol coordinated by both oxygen atoms (Mo–O lengths of 1.930 and 2.053 Å). Hydrogen transfer affords a coordinated water molecule with a distance from the metal centre Mo–OH2 of 2.290 Å. In intermediate 14b the water molecule moves further away, Mo–OH2 = 2.458 Å, and it finally dissociates in 8, where the compound recovers a distorted octahedral geometry. From 8 to 1 the mechanism is identical to that of pathway A.
Proposal of the DODH mechanism: discrimination between pathways
The profiles shown in Fig. 1 for paths A and B are relatively similar and to propose a valid mechanism for this Mo-catalysed DODH reaction it is necessary to carefully compare the calculated energies of both pathways. Table 1 shows the energies corresponding to the main processes of the reaction in toluene solution (see Table S2, ESI,† for data in the gas phase), namely, diol activation, oxo-transfer and water formation. Alkene extrusion is a common step in A and B and cannot be used for differentiation. In addition, according to previously defined criteria used for discrimination between mechanisms,48,49 other selected energies have been taken into account. These parameters are the highest barrier, the highest transition state encountered in the two energy profiles, the difference between the lowest and highest intermediate species, and the difference in the Gibbs energies of the TSs and the relative zero energy (in parentheses in Table 1). The comparison of the ΔG differences between a TS and its precedent intermediate shows slightly lower values for the steps of diol activation, oxo-transfer and water formation in profile B with respect to path A (Table 1). The higher ΔG difference for pathway A in the oxo-transfer step (15.4 kcal mol−1) is probably due to the heptacoordinated molybdenum environment caused by the presence of the diolate ligand in TS(6–7), while the molybdenum atom in TS(1–9) is located in a distorted octahedral geometry (11.4 kcal mol−1). In the same way, water formation requires overcoming a ΔG of 18.1 kcal mol−1 in A, TS(4–5), while the analogous parameter in B, TS(13–14), is lower, 15.3 kcal mol−1. Again, in this case, the reason may be the steric hindrance; the presence of the oxido group in TS(4–5) increases the steric pressure environment with respect to TS(13–14). Diol activation is the step with the highest ΔG difference in both pathways, but shows comparable energies (32.8 kcal mol−1 for A and 31.6 kcal mol−1 for B). Furthermore, considering the other energy parameters, we can see that the relative energy of the highest transition state (diol activation) is 44.7 kcal mol−1 for A, TS(6–7), while it is just 20.7 kcal mol−1 for B, TS(1–9). In fact, the difference in the Gibbs energies of the TSs and the relative zero energy are clearly higher in path A for all the steps. Except for the common alkene extrusion, all energies are lower in pathway B than in pathway A (Table 1). Additionally, the energetic span, which is defined as the difference in energy from the TDI (turnover determining intermediate) to the TDTS (turnover determining TS),50 is favourable for path B (36.7 kcal mol−1) with respect to path A (44.7 kcal mol−1). The TDI for path B is intermediate 10, which is in equilibrium with 9 and 11. For this path, the energetic span value of 36.7 kcal mol−1 is somewhat high, even at the experimental temperature of 110 °C. However, at this temperature, there will be a significant concentration of 11 for which the barrier decreases to 31.4 kcal mol−1, which is a more reasonable value. Therefore, we can support the hypothesis that the mechanism for [Mo(O)2(QMe)2]-catalysed DODH of ethylene glycol with a phosphane reductant proceeded through pathway B, that is, with the oxido-transfer process preceding diol condensation. This preferred mechanism B agrees very well with the experimental observation of a readily occurring reaction between [Mo(O)2(QCy)2] and PPh3 under nitrogen that gave OPPh3 and a paramagnetic molybdenum complex,21 and with the oxidation of PPh3 with dimethylsulphoxide by an oxido-transfer reaction catalysed by [Mo(O)2(QCy)2].21 The preference of pathway B was also observed in rhenium-catalysed DODH of vicinal diols both theoretically45 and experimentally.51 However, these results are in contrast to the DFT results reported by Fristrup and co-workers for the DODH catalysed by MoO3 in which the diol condensation was the first step.25,26 This is not surprising because there are several differences between both systems. The first factor is the reductant employed in the DODH reaction: our phosphane versus isopropanol or even the diol itself. More important is, in our opinion, the catalyst selected for each system. In the search for cheaper and commercially available Mo species, several types of molybdates were successfully used by Fristrup and co-workers.24 While they found good DODH results with ammonium heptamolybdate in neat diol, we observed that the use of this Mo catalyst and PPh3 as the reductant gave poorer results.21 The presence or absence of co-ligands in the oxido-molybdenum(VI) environment in conjunction with the selected reductant are certainly important factors that can govern the preferred mechanism in molybdenum-catalysed DODH reactions.
Table 1 Comparison of selected energies for pathways A and B, in toluene solution, in the [Mo(O)2(QMe)2]-catalysed DODH of ethylene glycol with the PMe3 reductant
| |
Path A |
Path B |
|
Difference in the calculated Gibbs energies of the transition states and their precedent intermediates. The difference in the calculated Gibbs energies of the TSs and the relative zero energy appears in parentheses.
|
| ΔGa (kcal mol−1) |
| Diol activation |
TS(2–3)
|
32.8 (37.9) |
TS(11–12)
|
31.6 (17.4) |
| Water formation |
TS(4–5)
|
18.1 (44.7) |
TS(13–14)
|
15.3 (9.6) |
| Oxo-transfer |
TS(6–7)
|
15.4 (40.4) |
TS(1–9)
|
11.4 (20.7) |
| Alkene extrusion |
TS(8–1)
|
22.7 (11.1) |
TS(8–1)
|
22.7 (11.1) |
| Energies (kcal mol−1) |
|
|
|
|
| Highest TS relative energy |
TS(6–7)
|
44.7 |
TS(1–9)
|
20.7 |
| Highest ΔG difference |
TS(2–3)
|
32.8 |
TS(11–12)
|
31.6 |
| Maximum relative ΔG between intermediates |
|
54.4 |
|
45.3 |
Conclusions
A DFT investigation of the mechanism of molybdenum catalysed DODH of vicinal diols to alkenes by phosphanes has been carried out using ethylene glycol, trimethylphosphane, and [Mo(O)2(QMe)2] as diol, phosphane, and catalyst models, respectively. According to previous proposals and studies,16,17 two pathways A and B have been investigated and a theoretical characterisation of the two catalytic DODH cycles along their lowest energy profiles has been carried out. In pathway A, condensation of the diol proceeds before oxygen atom transfer to the phosphane reductant, while the opposite situation is observed in pathway B, namely reduction by phosphane from Mo(VI) to Mo(IV) species by the oxido-transfer process prior to reoxidation to Mo(VI) by diol condensation. The catalytic cycle ends with a common final step, the ethylene extrusion from the molybdenum glycolate species, which regenerates the [Mo(O)2(QMe)2] catalyst. The ethylene glycol condensation shows a comparable ΔG difference between the TS and its precedent intermediate, but lower ΔG differences have been found in B for the oxido-transfer and water formation processes. These values, the energetic span and other energy data support pathway B as the preferred mechanism for the [Mo(O)2(QMe)2]-catalysed DODH of diols using a phosphane reductant. It is reasonable to assume that the present computational study will provide theoretical insights for future improvements in the Mo-catalysed DODH reaction.
Author contributions
The authors contributed equally to the investigation, data curation and writing – original draft preparation, review and editing, and they have read and agreed to the published version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The financial support from the Spanish Ministerio de Ciencia e Innovación (PGC2018-093443-B-I00) is gratefully acknowledged. The authors thank Centro de Servicios de Informática y Redes de Comunicaciones (CSIRC), Universidad de Granada, for providing the computing time. The reviewers’ comments in the first version of this article are also gratefully acknowledged.
References
- S. De, A. S. Burange and R. Luque, Green Chem., 2022, 24, 2267–2286 RSC.
- J. O. Metzger, ChemCatChem, 2013, 5, 680–682 CrossRef CAS.
- S. Dutta, ChemSusChem, 2012, 5, 2125–2127 CrossRef CAS PubMed.
- J. ten Dam and U. Hanefeld, ChemSusChem, 2011, 4, 1017–1034 CrossRef CAS PubMed.
- D. M. Alonso, J. Q. Bond and J. a. Dumesic, Green Chem., 2010, 12, 1493 RSC.
- F. C. Jentoft, Catal. Sci. Technol., 2022, 12, 6308–6358 RSC.
- C. Boucher-Jacobs and K. M. Nicholas, Top. Curr. Chem., 2014, 353, 163–184 CrossRef CAS PubMed.
-
S. Liu, J. Yi and M. M. Abu-Omar, in Reaction Pathways and Mechanisms in Thermocatalytic Biomass Conversion II, ed. Z. Schlaf and M. Zhang, Springer, Singapore, 2016, pp. 1–11 Search PubMed.
- K. A. DeNike and S. M. Kilyanek, R. Soc. Open Sci., 2019, 6, 191165 CrossRef PubMed.
- N. N. Tshibalonza and J.-C. M. Monbaliu, Green Chem., 2020, 22, 4801–4848 RSC.
- S. Suárez-Pantiga and R. Sanz, Org. Biomol. Chem., 2021, 19, 10472–10492 RSC.
- A. R. Petersen and P. Fristrup, Chem. – Eur. J., 2017, 23, 10235–10243 CrossRef CAS PubMed.
- G. K. Cook and M. A. Andrews, J. Am. Chem. Soc., 1996, 118, 9448–9449 CrossRef CAS.
- J. M. McClain II and K. M. Nicholas, ACS Catal., 2014, 4, 2109–2112 CrossRef.
- S. Raju, M.-E. Moret and R. J. M. Klein Gebbink, ACS Catal., 2015, 5, 281–300 CrossRef CAS.
- G. Chapman and K. M. Nicholas, Chem. Commun., 2013, 49, 8199–8201 RSC.
- A. Galindo, Inorg. Chem., 2016, 55, 2284–2289 CrossRef CAS PubMed.
- Y. Y. Jiang, J. L. Jiang and Y. Fu, Organometallics, 2016, 35, 3388–3396 CrossRef CAS.
- L. C. De Vicente Poutás, M. Castiñeira Reis, R. Sanz, C. S. López and O. N. Faza, Inorg. Chem., 2016, 55, 11372–11382 CrossRef PubMed.
- E. Aksanoglu, Y. H. Lim and R. A. Bryce, ChemSusChem, 2021, 14, 1545–1553 CrossRef CAS PubMed.
- L. Hills, R. Moyano, F. Montilla, A. Pastor, A. Galindo, E. Álvarez, F. Marchetti and C. Pettinari, Eur. J. Inorg. Chem., 2013, 2013, 3352–3361 CrossRef CAS.
- T. C. Siu, I. Silva, M. J. Lunn and A. John, New J. Chem., 2020, 44, 9933–9941 RSC.
- R. Tran and S. M. Kilyanek, Dalton Trans., 2019, 48, 16304–16311 RSC.
- J. R. Dethlefsen, D. Lupp, B. C. Oh and P. Fristrup, ChemSusChem, 2014, 7, 425–428 CrossRef CAS PubMed.
- J. R. Dethlefsen, D. Lupp, A. Teshome, L. B. Nielsen and P. Fristrup, ACS Catal., 2015, 5, 3638–3647 CrossRef CAS.
- D. Lupp, N. J. Christensen, J. R. Dethlefsen and P. Fristrup, Chem. – Eur. J., 2015, 21, 3435–3442 CrossRef CAS PubMed.
- K. Beckerle, A. Sauer, T. P. Spaniol and J. Okuda, Polyhedron, 2016, 116, 105–110 CrossRef CAS.
- M. Stalpaert and D. De Vos, ACS Sustainable Chem. Eng., 2018, 6, 12197–12204 CrossRef CAS.
- C. A. Navarro and A. John, Inorg. Chem. Commun., 2019, 99, 145–148 CrossRef CAS.
- J. Li, M. Lutz and R. J. M. Klein Gebbink, ChemCatChem, 2020, 12, 6356–6365 CrossRef CAS.
- D. Asada, T. Ikeda, K. Muraoka, Y. Nakagawa, K. Tomishige and A. Nakayama, J. Phys. Chem. C, 2022, 126, 20375–20387 CrossRef CAS.
- S. Albarracin-Suazo, L. Freitas de Lima e Freitas, B. MacQueen, A. Heyden, J. A. Lauterbach, E. Nikolla and Y. J. Pagán-Torres, ACS Sustainable Chem. Eng., 2022, 10, 5719–5727 CrossRef CAS.
- S. Hacatrjan, L. Liu, J. Gan, Y. Nakagawa, J. Cao, M. Yabushita, M. Tamura and K. Tomishige, Catal. Sci. Technol., 2022, 12, 2146–2161 RSC.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- A. Pastor, F. Montilla and A. Galindo, Polyhedron, 2015, 90, 233–238 CrossRef CAS.
- M. A. Pietsch and M. B. Hall, Inorg. Chem., 1996, 35, 1273–1278 CrossRef CAS PubMed.
- A. J. Millar, C. J. Doonan, P. D. Smith, V. N. Nemykin, P. Basu and C. G. Young, Chem. – Eur. J., 2005, 11, 3255–3267 CrossRef CAS PubMed.
- P. Basu, B. W. Kail, A. K. Adams and V. N. Nemykin, Dalton Trans., 2013, 42, 3071–3081 RSC.
- P. Basu, B. W. Kail and C. G. Young, Inorg. Chem., 2010, 49, 4895–4900 CrossRef CAS PubMed.
- K. P. Gable and F. A. Zhuravlev, J. Am. Chem. Soc., 2002, 124, 3970–3979 CrossRef CAS PubMed.
- S. Qu, Y. Dang, M. Wen and Z. X. Wang, Chem. – Eur. J., 2013, 19, 3827–3832 CrossRef CAS PubMed.
- P. Liu and K. M. Nicholas, Organometallics, 2013, 32, 1821–1831 CrossRef CAS.
- T. Robin, F. Montilla, A. Galindo, C. Ruiz and J. Hartmann, Polyhedron, 1999, 18, 1485–1490 CrossRef CAS.
- L. J. Goossen, D. Koley, H. L. Hermann and W. Thiel, Organometallics, 2006, 25, 54–67 CrossRef CAS.
- M. Drees, S. A. Hauser, M. Cokoja and F. E. Kühn, J. Organomet. Chem., 2013, 748, 36–45 CrossRef CAS.
- S. Kozuch and S. Shaik, Acc. Chem. Res., 2011, 44, 101–110 CrossRef CAS PubMed.
- J. R. Dethlefsen and P. Fristrup, ChemCatChem, 2015, 7, 1184–1196 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Catalytic profiles and comparison of computed barriers and selected energies for pathways A and B in gas phase. Coordinates of optimised compounds. See DOI: https://doi.org/10.1039/d3dt00340j |
| ‡ Present address: Chemistry Interdisciplinary Project (CHIP), School of Pharmacy, University of Camerino, 62032 Camerino MC, Italy. |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  and
Agustín
Galindo
and
Agustín
Galindo