
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCyclometalated platinum(II) complex as a selective light switch for G-quadruplex DNA†
Meenaxi
Saini
and
Tia E.
Keyes
 *
*
School of Chemical Science and National Centre for Sensor Research Dublin City University, Dublin 9, Ireland. E-mail: tia.keyes@dcu.ie
First published on 26th May 2023
Abstract
Cyclometalated 1,3-bis(8-quinolyl) phenyl chloroplatinum(II) (Pt1) shows selective luminescence transduction of G-quadruplex binding over duplex DNA. The effect is enhanced on association with parallel and hybrid G-quadruplex structures over other topologies. The kinetics of binding are studied for c-myc and the response is found to be partially reversible in a displacement assay.
G-quadruplexes (G4) are non-canonical nucleic acid structures formed in guanine-rich regions of both DNA and RNA in vivo, by self-assembly of four guanines via Hoogsteen hydrogen bonds.1 G4 structures form a number of topologies depending on the orientation of DNA strands including parallel, antiparallel, hybrid, and they may be intramolecular (formed through a single strand) or intermolecular (from multiple strands). They can also adopt various loop structures.1–3 The biological role of G4 structures is still emerging, but their importance in telomerase maintenance and regulation of gene expression, is well established and their role in cancer/oncogenes makes them particularly compelling both as therapeutic and diagnostic targets.4,5
Transition metal complexes are increasingly studied as small molecule ligands for G4.6–8 Amongst these, Pt(II) complexes, have been fairly widely explored as their square planar structures offer a flattened geometry that can promote binding to G4 structures through π-stacking/end-capping compared to the octahedra of Ruthenium and Rhenium which have been more widely studied in duplex DNA binding.9–13 Terpyridine complexes, particularly those with labile Cl ligands, such as 4′-tolyl-2,2′:6′,2′′-terpyridine Pt(II) chloride have been shown to have high binding affinity and specificity toward G-quadruplex14,15 and, end capping has been reported to be accompanied by platination of adenine loop sites on some G-quadruplex structures.16,17 Other non-covalent binding modes such as groove binding, electrostatic association and hydrogen bonding interactions can also occur and a number of Pt(II) complexes show more than one mode of interaction with G4s.17
Cyclometalated platinum(II) complexes bearing π-conjugated organic ligands have been much less widely studied in the context of G quadruplex binding than N-coordinated analogues.18–24 Although for tridentate ligand coordinated complexes they have advantageous photophysical properties that have been exploited across a range of domains,25–28 including as probes for biomolecules29 and bioimaging.30–33 Given their structural analogy to N,N,N Pt terpy complexes we rationalised they might be expected to show similar affinity but with superior addressability due to their photophysical properties in terms of both luminescent quantum yield and red emission wavelength.34,35 In particular, we are interested in G4 ligands that show molecular light switch properties, with weak/no emission from aqueous media that switches on, on target binding. Such behaviour has been widely observed in ruthenium complexes containing phenazine ligands.36,37 Rarer in Pt complexes,38 it has been noted in Pt(II) benzimidazole N,N,N complex on aggregation on binding to RNA39 and in Pt terpy complexes with an appended fluorophore.40 Kinetic control for Pt binding with G4 was reported very recently.41 Herein, we show that cyclometalated tridentate Pt complex 1,3-bis(8-quinolyl) phenyl chloroplatinum(II) (Pt1), under visible excitation, shows dramatic changes in emission amplitude and selective signal transduction of G quadruplex binding with sharp discrimination in behaviour between G4s and duplex DNA (Fig. 1).
 | ||
| Fig. 1 Chemical structure of Pt1. | ||
1,3-Bis(8-quinolyl) phenyl chloroplatinum(II) (Pt1) was prepared as reported by Williams et al. and structural analysis and photophysical properties conformed to their reported data.42 In aqueous PBS/KCl buffer at pH 7.0, Pt1 shows an absorbance feature at λmax ∼ 420 nm (Fig. S1†) attributed to d(Pt)/π(Cl) → π*(ligand) transition.42 Excitation at this wavelength results in intense luminescence in non-protic solvent (e.g. λem 600 nm, lifetime 865 ns in deaerated DMSO) (Fig. S2†) that is substantially and reversibly extinguished on titration with water (Fig S8†). In aerated aqueous PBS/KCl buffer at pH 7.0, emission is weak and short lived with an emission lifetime of 2 ns (∼99% amplitude) in aerated buffer (Fig. 2, Table S1†).
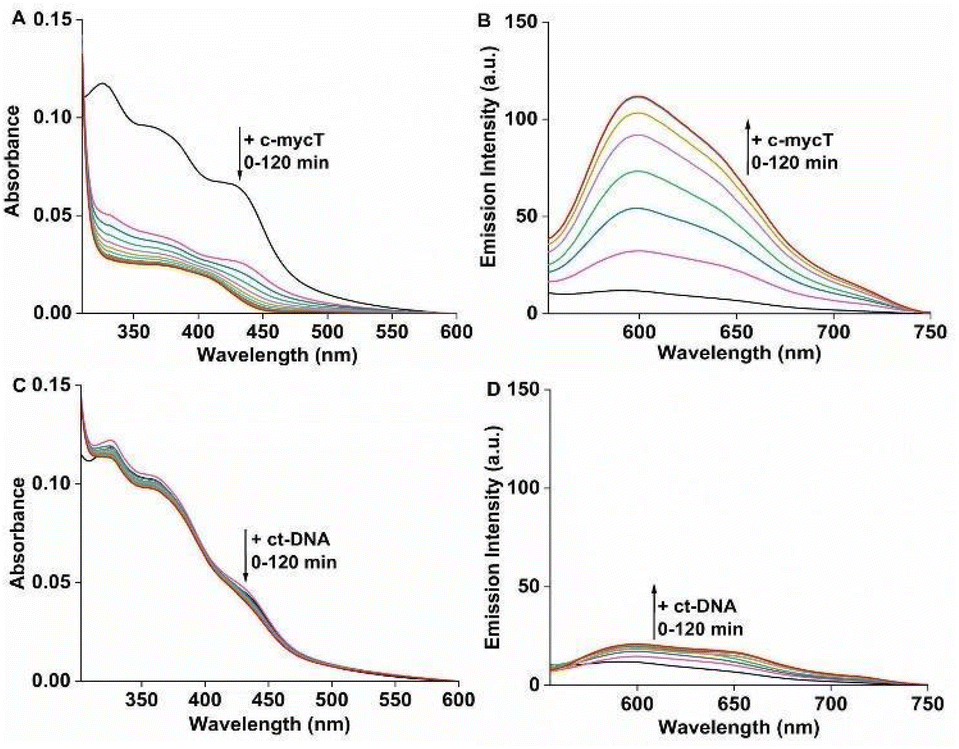 | ||
| Fig. 2 (A) Change in absorbance and (B) emission spectra of Pt1 complex (10 μM) in PBS/KCl buffer pH 7.0 with 30 μM c-mycT (c-mycT/Pt = 3) from 0 minute (black line) up to saturation (red, 120 minutes). (C and D) change in absorbance and emission of Pt1 complex in PBS/KCl buffer pH 7.0 with 100 μM ct-DNA (ct-DNA/Pt = 10) from 0 minute (black line) up to saturation (red, 120 minutes) respectively. λex = 420 nm. | ||
We compared Pt1 association across four G-rich oligonucleotides known to assemble into different representative G4 topologies: c-mycT and 21 KRAS form parallel G4, TBA forms antiparallel, 22 AG forms mix-hybrid and Inter G4 that forms an intermolecular G quadruplex structures. Binding was also compared with ct-DNA and short oligomer ds17A as representative duplex structures (full sequences are provided in ESI†). The impact of Pt1 incubation with c-mycT on its electronic spectroscopy is shown in Fig. 2A, where the d(Pt)/π(Cl) → π*(ligand) absorbance at 420 nm showed time-dependent hypochromicity in PBS/KCl buffer, shifting to approximately 406 nm with roughly 50% decrease in intensity. Such extensive change suggests Cl− loss on G4 association.15,23 Similar shifts in absorbance were seen on KRAS and 22AG association though without comparable loss in absorbance intensity (Fig. S3 and S4†). By contrast, absorbance changes for the other quadruplexes were modest (Fig. S5 and S6†), and with duplex (ct) DNA and duplex DNA oligomer ds17A (Fig. 2C and Fig. S7†) changes to absorbance were negligible.
Fig. 2B shows the time-dependent evolution of emission signal on incubation of Pt1 in aqueous media (PBS/KCl buffer, pH 7.0) with 3 molar equivalents of c-mycT. Both the magnitude of emission intensity change at saturation, and the rate (or indeed presence at all) of dynamic response were G4 topology dependent. The most dramatic response was observed on c-mycT association, where compared to aqueous buffer, emission increased approx. 14-fold on G4 association, yielding an emission quantum yield that exceeds that of the complex in non-aqueous media. A red shift of emission maxima by approx. 12 nm and alteration to the emission profile accompany the intensity change. Compensating for the large decrease in absorbance cross-section of the complex on c-mycT binding, the emission intensity increase is actually nearly 30-fold. KRAS and 22AG induced less dramatic, but nonetheless large increases in emission intensity (Fig. 3), of close to an order of magnitude on Pt1 binding compared to buffer.
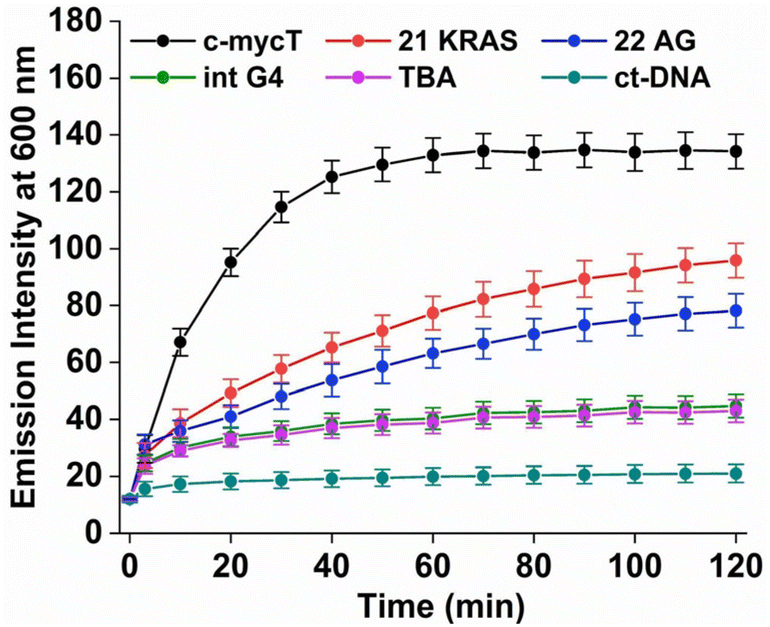 | ||
| Fig. 3 Temporal evolution of emission signal at λmax 600 nm of 10 μM Pt1 in PBS/KCl buffer pH 7.0 on addition of 30 μM G-quadruplexes [G-quadruplex/Pt1 = 3] and 100 μM ct-DNA. Emission intensities are not corrected for absorbance changes. λex = 420 nm. | ||
TBA and Int G4 association induced much weaker emission responses, where intensity increased approximately 4-fold compared to complex in buffer. For duplex DNA; ct-DNA and DNA oligomer ds17A, emission intensity changes were very weak (Fig. 2D and Fig. S7†). The final intensity increased less than 2-fold over complex in buffer solution. And, for TBA, Int G4 and duplex, intensity showed little time dependence. The addition of further concentrations of G4/duplex in each case did not alter the emission spectra further (Fig. S9†). The emission intensity changes were accompanied by increases to emission lifetime (Fig. S10 and Table S1†). Emission lifetime increased from predominantly 2 ns in aerated buffer to approximately 318 ns on association with duplex DNA, TBA or Int G4 whereas on binding to c-mycT emission lifetime was dramatically extended to 486 ns and to 424 ns and 351 ns for KRAS and 22AG respectively. Overall, there is clear distinction in signal transduction for both spectroscopic and photophysical data between Pt1 interaction with parallel and mixed hybrid quadruplexes showing enhanced emission compared to antiparallel, intermolecular quadruplex topology or to duplex DNA.
The binding constants for G4s and ctDNA were estimated from concentration-dependent emission intensity plots taken from time-equilibrated data (after 2 hours of equilibration). Kb for c-mycT was determined as (1.8 ± 0.2) × 105 M−1.43,44 The stoichiometry of binding for Pt1 – c-mycT G4 interaction was determined from Jobs plot as approximately 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Fig. S12†). The Kb of Pt1 were determined as (1.2 ± 0.1) × 105 M−1, (0.8 ± 0.06) × 105 M−1 for KRAS and 22 AG respectively, which are of the same order of magnitude as c-mycT. However, for duplex and the remaining quadruplex structures, binding was too weak to accurately determine (Table S1†). Therefore, Pt1 complex shows the greatest affinity for parallel and hybrid quadruplex with good selectivity over other topologies (Table S1†), including duplex DNA. The difference in binding affinities of the Pt1 complex for different quadruplex structures is presumably driven by differences in the loop structure and topology of quadruplex. A case in point is the variation in the binding affinity between c-mycT and KRAS. Both have parallel structures, confirmed from CD spectroscopy, with the same strand orientation and both comprise three quartets, but their loop structures are different. KRAS exhibits three regular propeller loops but c-mycT promoter P1, one of seven nuclease-hypersensitive elements (NHEs), NHE III1 of c-mycT gene, has been shown to provides unusual fold-back configuration and reversal loops which connect the G-tetrad layers, which are likely responsible for the variation in binding affinity.45,46
:1 (Fig. S12†). The Kb of Pt1 were determined as (1.2 ± 0.1) × 105 M−1, (0.8 ± 0.06) × 105 M−1 for KRAS and 22 AG respectively, which are of the same order of magnitude as c-mycT. However, for duplex and the remaining quadruplex structures, binding was too weak to accurately determine (Table S1†). Therefore, Pt1 complex shows the greatest affinity for parallel and hybrid quadruplex with good selectivity over other topologies (Table S1†), including duplex DNA. The difference in binding affinities of the Pt1 complex for different quadruplex structures is presumably driven by differences in the loop structure and topology of quadruplex. A case in point is the variation in the binding affinity between c-mycT and KRAS. Both have parallel structures, confirmed from CD spectroscopy, with the same strand orientation and both comprise three quartets, but their loop structures are different. KRAS exhibits three regular propeller loops but c-mycT promoter P1, one of seven nuclease-hypersensitive elements (NHEs), NHE III1 of c-mycT gene, has been shown to provides unusual fold-back configuration and reversal loops which connect the G-tetrad layers, which are likely responsible for the variation in binding affinity.45,46
However, the time dependence of the emission response for c-mycT, KRAS and 22AG, and the absence of such kinetic effects for the duplex, TBA and Int, are consistent with weak and non-covalent association in the latter three cases and may indicate covalent association in the former. By fitting the 600 nm intensity plots versus time to a pseudo first order rate equation (ΔI = A(1 − exp(−kt), where A is constant and k is observed reaction rate constant) for Pt1 interaction with c-mycT, KRAS and 22 AG, we obtained values of 0.066 ± 0.004 min−1, 0.021 ± 0.002 min−1, 0.019 ± 0.002 min−1 respectively for each G4 (Fig. S13†).
Again, it is notable that both affinity and observed rate constant is greatest for c-mycT. The analogous, N,N,N coordinated Pt TerpyCl complex has been reported to associate to the tetrad by terminus stacking as well as through coordination to adenine in G4 quadruplex. The clear distinction in behaviour between G4 topologies and duplex here, may be attributed to this same effect. The only site of adenine in the c-mycT is at the terminal of the sequence, so if adenine binding facilitates G4 interaction, one would expect replacement of adenine with thiamine to reduce the affinity and emission intensity of the associated complex. Therefore, to evaluate the potential role of adenine platination in emission enhancement, Pt1 solution was incubated with 3 molar equivalents of c-mycTT (where both adenines at 3′ terminus of c-mycT sequence are replaced by thymine). CD spectroscopy confirmed the fold was the same as c-mycT. However, the emission intensity increased only 6-fold compared to the complex in aqueous buffer (Fig. S14†) i.e. to only half the emission enhancement observed on c-mycT binding. These results strongly implicate the contribution of adenine platination to emission enhancement. Incubating the complex in buffer with adenine monophosphate alone, lead to only modest increases in emission enhancement (roughly 16% increase) so the effect is likely cooperative whereby adenine binding facilitates stronger non-specific interaction with the c-mycT G4 structure that maximizes its emission enhancement.
To further confirm the G4 structure is responsible for the emission enhancement, we carried out titrations of Pt1 against c-mycT in the presence of Li+ which is not expected to stabilise the G4.47,48 Correspondingly, the changes in emission intensity observed were similar to duplex DNA (Fig. S11†) indicating the Li+ is not stabilising the structure and that the response arises specifically from the G4 structure.
Finally, to evaluate the effect of conjugate binding on G4 DNA conformation, circular dichroism (CD) spectra were measured across different ratios of Pt1 to G4 DNA. The CD spectrum of c-mycT gave the expected negative band at 245 nm and a positive feature at 265 nm (Fig. S15†) characteristic of a predominantly parallel structure.49
With increasing concentration of Pt1 complex, Fig. 4A a positive induced CD (ICD) feature appeared at 420 nm, attributed to π-stacking at the tetrad terminal accompanied by adenine platination.16,17 The small change in molar ellipticity of the quadruplex (about 18% increase) at 245 nm and 265 nm indicates that Pt1 stabilises the quadruplex. Similar CD spectral changes were observed for 21 KRAS with increasing concentration of Pt1 (Fig. S16†).
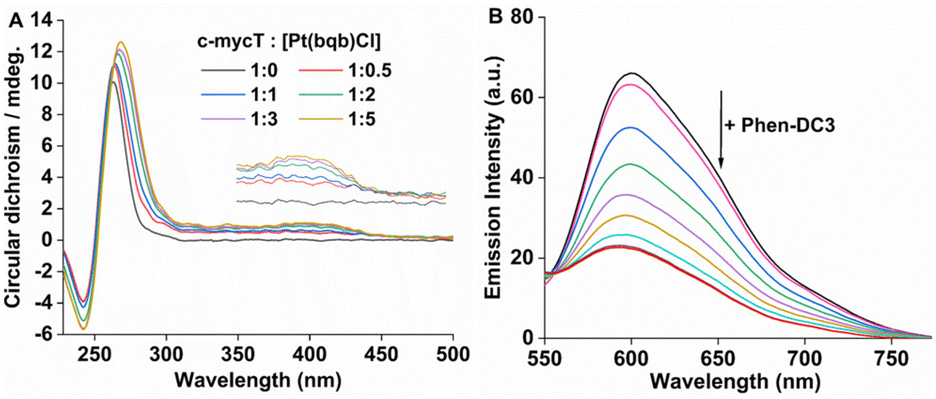 | ||
| Fig. 4 (A) Changes to the CD spectrum of c-mycT with increasing Pt1 concentration in PBS/KCl pH 7.0. (B) Changes to the emission spectra of Pt1 5 μM incubated with 15 μM c-mycT [c-mycT/Pt1 = 3] on addition of 5 μM to 100 μM Phen-DC3 (PBS/KCl pH.7.0). | ||
Finally, a fluorescence displacement study was performed by adding Phen-DC3 to the c-mycT-Pt1 complex after it had been allowed to assemble for 2 hours. Phen-DC3 is a selective G4 binder with high affinity for G4 quadruplex.50 As shown in Fig. 4B addition of Phen-DC3 results in a progressive reduction in the emission intensity of the c-mycT-Pt1 complex and the response saturates at 100 μM of Phen-DC3. Notably, the final emission intensity remains relatively higher than the Pt1 complex in buffer solution with a 3-fold decrease in intensity indicating the displacement of Pt1 with Phen-DC3 is incomplete. NMR spectroscopy studies suggested that Phen-DC3 associates with the guanine base of the top G-tetrad in G-quadruplex via extensive π-stacking.51 Reduction in the emission intensity of the c-mycT-Pt1 complex with the addition of Phen-DC3 also suggests both ligands (Pt1 and Phen-DC3) occupy similar binding sites on the quadruplex consistent with π-stacking interaction of Pt1 complex at terminal tetrad of G-quadruplex.
In summary, cyclometalated Pt complex; 1,3-bis(8-quinolyl) phenyl chloroplatinum(II) shows outstanding emission signal transduction on binding to adenine containing parallel or hybrid G-quadruplex DNA. Optical and photophysical properties show that association induced changes are much weaker with duplex DNA and antiparallel structures. The data indicate that cooperative non-covalent and covalent platination interactions may be responsible for the effect. CD spectroscopy shows stabilisation of the G4 structure for these three structures, with evidence of induced CD.
Overall, the photophysical data indicate that the quadruplex may be readily distinguished from DNA bound Pt complex with dramatic changes to emission lifetime that may facilitate lifetime imaging as a means to distinguish such structures in vivo.
Conflicts of interest
There are no conflicts of interest.Acknowledgements
This work is supported by Science Foundation Ireland under 19/FFP/6428, 12/RC/2276_P2 and EU.COFUND – Marie Skłodowska-Curie Actions.References
- M. L. Bochman, K. Paeschke and V. A. Zakian, Nat. Rev., 2012, 13, 770–780 CAS.
- T. Zok, N. Kraszewska, J. Miskiewicz, P. Pielacinska, M. Zurkowski and M. Szachniuk, Nucleic Acids Res., 2022, 50, D253–D258 CrossRef CAS PubMed.
- S. Ceschi, M. Berselli, M. Cozzaglio, M. Giantin, S. Toppo, B. Spolaore and C. Sissi, Nucleic Acids Res., 2022, 50, 1370–1381 CrossRef CAS PubMed.
- S. Pramanik, Y. Chen, H. Song, I. Khutsishvili, L. A. Marky, S. Ray, A. Natarajan, P. K. Singh and K. K. Bhakat, Nucleic Acids Res., 2022, 50, 3394–3412 CrossRef CAS PubMed.
- J. T. Grün, A. Blümler, I. Burkhart, J. W. Bartoschek, A. Heckel and H. Schwalbe, J. Am. Chem. Soc., 2021, 143, 6185–6193 CrossRef PubMed.
- A. Łęczkowska, J. G. Garcia, C. P. Arnaiz, B. Garcia, A. J. P. White and R. Vilar, Chem. – Eur. J., 2018, 24, 11785–11794 CrossRef PubMed.
- G. Farine, C. Migliore, A. Terenzi, F. L. Celso, A. Santoro, G. Bruno, R. Bonsignore and G. Barone, Eur. J. Inorg. Chem., 2021, 1332–1336 CrossRef CAS.
- O. Domarco, C. Kieler, C. Pirker, C. Dinhof, B. Englinger, J. M. Reisecker, G. Timelthaler, M. D. Garcia, C. Peinador, B. K. Keppler, W. Berger and A. Terenzi, Angew. Chem., Int. Ed., 2019, 58, 8007–8012 CrossRef CAS PubMed.
- F. E. Poynton, J. P. Hall, P. M. Keane, C. Schwarz, I. V. Sazanovich, M. Towrie, T. Gunnlaugsson, C. J. Cardin, D. J. Cardin, S. J. Quinn, C. Long and J. M. Kelly, Chem. Sci., 2016, 7, 3075–3084 RSC.
- J. D. Knoll and C. Turro, Coord. Chem. Rev., 2015, 282, 110–126 CrossRef PubMed.
- L. Holden, C. S. Burke, D. Cullinane and T. E. Keyes, RSC Chem. Biol., 2021, 2, 1021–1049 RSC.
- K. T. McQuaid, S. Takahashi, L. Baumgaertner, D. J. Cardin, N. G. Paterson, J. P. Hall, N. Sugimoto and C. J. Cardin, J. Am. Chem. Soc., 2022, 144, 5956–5964 CrossRef CAS PubMed.
- G. Li, L. Sun, L. Ji and H. Chao, Dalton Trans., 2016, 45, 13261–13276 RSC.
- E. Morel, C. Beauvineau, D. Naud-Martin, C. Landras-Guetta, D. Verga, D. Ghosh, S. Achelle, F. Mahuteau-Betzer, S. Bombard and M. P. Teulade-Fichou, Molecules, 2019, 24, 404 CrossRef PubMed.
- M. Chrzanowska, A. Katafias, A. Kozakiewicz and R. v. Eldik, Inorg. Chim. Acta, 2020, 504, 119449 CrossRef CAS.
- H. Bertrand, S. Bombard, D. Monchaud, E. Talbot, A. Guedin, J.-L. Mergny, R. Grunert, P. J. Bednarski and M. P. Teulade-Fichou, Org. Biomol. Chem., 2009, 7, 2864–2871 RSC.
- M. Trajkovski, E. Morel, F. Hamon, S. Bombard, M. P. Teulade-Fichou and J. Plavec, Chem. – Eur. J., 2015, 21, 7798–7807 CrossRef CAS PubMed.
- D.-L. Ma, C.-M. Che and S.-C. Yan, J. Am. Chem. Soc., 2009, 131(5), 1835–1846 CrossRef CAS PubMed.
- T. Kench, P. A. Summers, M. K. Kuimova, J. E. M. Lewis and R. Vilar, Angew. Chem., Int. Ed., 2021, 60, 10928–10934 CrossRef CAS PubMed.
- P. Wu, D.-L. Ma, C.-H. Leung, S.-C. Yan, N. Zhu, R. Abagyan and C.-M. Che, Chem. – Eur. J., 2009, 15, 13008–13021 CrossRef CAS PubMed.
- K. Suntharalingam, A. Łęczkowska, M. A. Furrer, Y. Wu, M. K. Kuimova, B. Therrien, A. J. P. White and R. Vilar, Chem. – Eur. J., 2012, 18, 16277–16282 CrossRef CAS PubMed.
- A. D. Richards and A. Rodger, Chem. Soc. Rev., 2007, 36, 471–483 RSC.
- A. I. Solomatina, P. S. Chelushkin, T. O. Abakumova, V. A. Zhemkov, M. Kim, I. Bezprozvanny, V. V. Gurzhiy, A. S. Melnikov, Y. A. Anufrikov, I. O. Koshevoy, S.-H. Su, P.-T. Chou and S. P. Tunik, Inorg. Chem., 2019, 58(1), 204–217 CrossRef CAS PubMed.
- B. S. McGhie, J. Sakoff, J. Gilbert, C. P. Gordon and J. R. Aldrich-Wright, Int. J. Mol. Sci., 2022, 23, 10469 CrossRef CAS PubMed.
- M. D. Aseman, M. Nikravesh, A. Abbasi and H. R. Shahsavari, Inorg. Chem., 2021, 60, 18822–18831 CrossRef PubMed.
- E. M. Bolitho, C. S. Cano, H. Shi, P. D. Quinn, M. Harkiolaki, C. Imberti and P. J. Sadler, J. Am. Chem. Soc., 2021, 143, 20224–20240 CrossRef CAS PubMed.
- D. M. Yufanyi, H. S. Abbo, S. J. J. Titinchi and T. Neville, Coord. Chem. Rev., 2020, 414, 213285 CrossRef CAS.
- G. Li, S. Liu, Y. Sun, W. Lou, Y.-F. Yang and Y. She, J. Mater. Chem. C, 2022, 10, 210–218 RSC.
- A. Zamora, E. Wachter, M. Vera, D. K. Heidary, V. Rodríguez, E. Ortega, V. Fernandez-Espín, C. Janiak, E. C. Glazer, G. Barone and J. Ruiz, Inorg. Chem., 2021, 60, 2178–2187 CrossRef CAS PubMed.
- M. Z. Shafikov, A. F. Suleymanova, R. J. Kutta, A. Gorski, A. Kowalczyk, M. Gapińska, K. Kowalski and R. Czerwieniec, J. Mater. Chem. C, 2022, 10, 5636–5647 RSC.
- W. A. Tarran, G. R. Freeman, L. Murphy, A. M. Benham, R. Kataky and J. A. G. Williams, Inorg. Chem., 2014, 53, 5738–5749 CrossRef CAS PubMed.
- K.-C. Tong, P.-K. Wan, C.-N. Lok and C.-M. Che, Chem. Sci., 2021, 12, 15229–15238 RSC.
- D. Septiadi, A. Aliprandi, M. Mauro and L. D. Cola, RSC Adv., 2014, 4, 25709–25718 RSC.
- D. A. Haque, L. Xu, R. A. Al-Balushi, M. K. Al-Suti, R. Ilmi, Z. Guo, M. S. Khan, W.-Y. Wong and P. R. Raithby, Chem. Soc. Rev., 2019, 48, 5547–5563 RSC.
- K. Li, G. S. M. Tong, Q. Wan, G. Cheng, W.-Y. Tong, W.-H. Ang, W.-L. Kwong and C.-M. Che, Chem. Sci., 2016, 7, 1653–1673 RSC.
- S. Shi, J. Zhao, X. Gao, C. Lv, L. Yang, J. Hao, H. Huang, J. Yao, W. Sun, T. Yao and L. Ji, Dalton Trans., 2012, 41, 5789–5793 RSC.
- C. Rajput, R. Rutkaite, L. Swanson, I. Haq and J. A. Thomas, Chem. – Eur. J., 2006, 12, 4611–4619 CrossRef CAS PubMed.
- D.-L. Ma, C.-M. Che and S.-C. Yan, J. Am. Chem. Soc., 2009, 131, 1835–1846 CrossRef CAS PubMed.
- A. S.-Y. Law, L. C.-C. Lee, K. K.-W. Lo and V. W.-W. Yam, J. Am. Chem. Soc., 2021, 143, 5396–5405 CrossRef CAS PubMed.
- S. Gama, I. Rodrigues, F. Mendes, I. C. Santos, E. Gabano, B. Klejevskaja, J. Gonzalez-Garcia, M. Ravera, R. Vilar and A. Paulo, J. Inorg. Biochem., 2016, 160, 275–286 CrossRef CAS PubMed.
- B.-C. Zhu, J. He, W. Liu, X.-Y. Xia, L.-Y. Liu, B.-B. Liang, H.-G. Yao, B. Liu, L.-N. Ji and Z.-W. Mao, Angew. Chem., Int. Ed., 2021, 60, 15340–15343 CrossRef CAS PubMed.
- K. L. Garner, L. F. Parkes, J. D. Piper and J. A. G. Williams, Inorg. Chem., 2010, 49, 476–487 CrossRef CAS PubMed.
- A. Mitra, S. Bhowmik and R. Ghosh, J. Photochem. Photobiol., 2021, 6, 100033 CrossRef.
- T.-P. Sheng, X.-X. Fan, G.-Z. Zheng, F.-R. Dai and Z.-N. Chen, Molecules, 2020, 25, 2656 CrossRef CAS PubMed.
- A. Ou, J. W. Schmidberger, K. A. Wilson, C. W. Evans, J. A. Hargreaves, M. Grigg, M. L. O'Mara, K. S. Iyer, C. S. Bond and N. M. Smith, Nucleic Acids Res., 2020, 48, 5766–5776 CrossRef CAS PubMed.
- D.-L. Ma, D. S.-H. Chan, W.-C. Fu, H.-Z. He, H. Yang, S.-C. Yan and C.-H. Leung, PLoS One, 2012, 7, e43278 CrossRef CAS PubMed.
- D. Bhattacharyya, G. M. Arachchilage and S. Basu, Front. Chem., 2016, 4, 38 CrossRef PubMed.
- J. You, H. Li, X.-M. Lu, W. Li, P.-Y. Wang, S.-X. Dou and X.-G. Xi, Biosci. Rep., 2017, 37, BSR20170771 CrossRef CAS PubMed.
- J. Carvalho, J. A. Queiroz and C. Cruz, J. Chem. Educ., 2017, 94, 1547–1551 CrossRef CAS.
- M. Deiana, J. Jamroskovic, I. Obi and N. Sabouri, Chem. Commun., 2020, 56, 14251–14254 RSC.
- W. J. Chung, B. Heddi, F. Hamon, M.-P. Teulade-Fichou and A. T. Phan, Angew. Chem., Int. Ed., 2014, 53, 999–1002 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3dt01188g |
| This journal is © The Royal Society of Chemistry 2023 |