
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
2-(Thienyl)quinoxaline derivatives and their application in Ir(III) complexes yielding tuneable deep red emitters†
Sophie A.
Fitzgerald
a,
Ellie N.
Payce
a,
Peter N.
Horton
b,
Simon J.
Coles
 b and
Simon J. A.
Pope
*a
b and
Simon J. A.
Pope
*a
aSchool of Chemistry, Main Building, Cardiff University, Cardiff CF10 3AT, UK. E-mail: popesj@cardiff.ac.uk
bUK National Crystallographic Service, Chemistry, University of Southampton, Highfield, Southampton, SO17 1BJ, UK
First published on 24th October 2023
Abstract
The synthesis and characterisation of eleven different 2-(thienyl)quinoxaline species that incorporate different points of functionality, including at the thiophene or quinoxaline rings, are described. These species display variable fluorescence properties in the visible region (λem = 401–491 nm) depending upon the molecular structures and extent of conjugation. The series of 2-(thienyl)quinoxaline species were then investigated as cyclometalating agents for Ir(III) to yield [Ir(C^N)2(bipy)]PF6 (where C^N = the cyclometalated ligand; bipy = 2,2′-bipyridine). Eight complexes were successfully isolated and fully characterised by an array of spectroscopic and analytical techniques. Two Ir(III) examples were structurally characterised in the solid state using single crystal X-ray diffraction; both structures confirmed the proposed formulations and coordination spheres in each case showing that the thiophene coordinates via a Ir–C bond. The photophysical properties of the complexes revealed that each complex is luminescent under ambient conditions with a range of emission wavelengths observed (665–751 nm) indicating that electronic tuning can be achieved via both the thienyl and quinoxaline moieties.
Introduction
Interest in transition metal-based deep red emitting luminophores1 has been driven by several areas of potential and realised application areas.2 In this context our own studies have investigated the development of quinoxaline containing ligands that can be utilised as cyclometalating agents for metal ions such as Ir(III) and Pt(II).3 For such applications, quinoxaline-based ligands present a useful alternative to more common 2-phenylpyridine or quinoline based systems. The electron-deficient quinoxaline ring typically results in longer wavelength absorption and emission features for Ir(III) complexes while often maintaining competitive luminescence efficiencies.4 These attributes have led, in particular, to the successful application of these types of complexes to energy upconversion studies5 and cellular bioimaging6 where efficient longer wavelength absorption is especially advantageous.We have sought to expand our repertoire of quinoxaline-containing ligands and considered the combination of the quinoxaline moiety with an electron rich thiophene-based donor component. There have been a small number of previous reports detailing the unique photophysical properties that result from the combination of thiophene and quinoxaline units in organic donor–acceptor (D–A) assemblies,7 including compounds bearing a 2,3-di(thiophen-2-yl)quinoxaline core,8 which displayed prominent intramolecular charge transfer (ICT) character. More recently, work on V-shaped D–A–D chromophores, where 1,5-thiophenylene acts as a π-conjugated spacer unit and either 6-cyano or 6,7-difluoroquinoxaline functioned as electron-acceptors have also been reported.9
While there are no reports on the coordination chemistry of thiophene–quinoxaline hybrid ligands, there are numerous examples, that date back to the 1970s, of organometallic complexes that contain a metalated thiophene unit. For example, 2-(2′-thienyl)pyridine was initially demonstrated as a ligand for Pt(II).10 In 1979, Nonoyama reported that Ir(III) complexes bearing 2-(2-thienyl)pyridine ligands were also attainable as cyclometalated species (Scheme 1, A).11
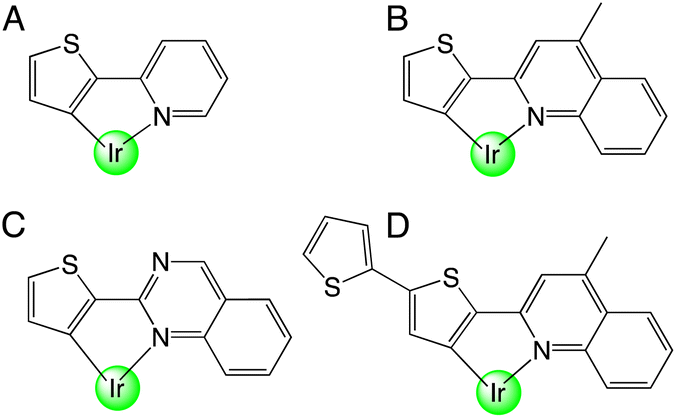 | ||
| Scheme 1 Examples of reported thiophene-containing cyclometalating ligands for Ir(III). | ||
In 2001, a series of heteroleptic [Ir(C^N)2(acac)] complexes allowed the photophysical comparison of species containing phenylpyridine (ppy), thienylpyridine (thp) and benzo-thienylpyridine (btp) cyclometalating ligands.12 The study showed that ligand-centred triplet states characterised the emission of thp and btp complexes (in contrast to 3MLCT for ppy13). Building on these reports, iterative ligand variants have been developed to achieve tuneable emission within the orange-red region of the visible range,14 including benzothiophene ligands for the deep red15 and near-IR region.16 In addition to their applications in OLEDs,17 recent developments have allowed cationic Ir(III) complexes based upon these ligand motifs to thrive in other areas such as photocatalytic H2 generation,18 electrochemiluminescent materials19 and biological fields.20 In addition thiophene-containing ligands combining quinoline or quinazoline (Scheme 1, B–D) units have also been reported and proven to display desirable emission properties for the development of OLED dopants.21
In the current work, a description of luminescent Ir(III) complexes that are formed from a range of new 2-(thienyl)quinoxaline type ligands is provided. The ligands can be functionalised at either the thiophene or quinoxaline units giving luminescent Ir(III) complexes that are tuneable within the deep red to near-IR regions.
Results and discussion
Synthesis and characterisation of the ligands
A series of eleven substituted 2-(thiophen-2-yl)quinoxaline species were synthesised, each incorporating different features and functionalities. Firstly, L1–5H, which vary in substitution at the 5-position of the thienyl component, were synthesised according to the route shown in Scheme 2. Ethanol was preferred as the solvent for the quinoxaline ring formation as in most cases the product precipitated upon cooling. However, our studies also revealed that an increase in yield of L1H was achievable using DMSO and NaHCO3 and heating at 120 °C.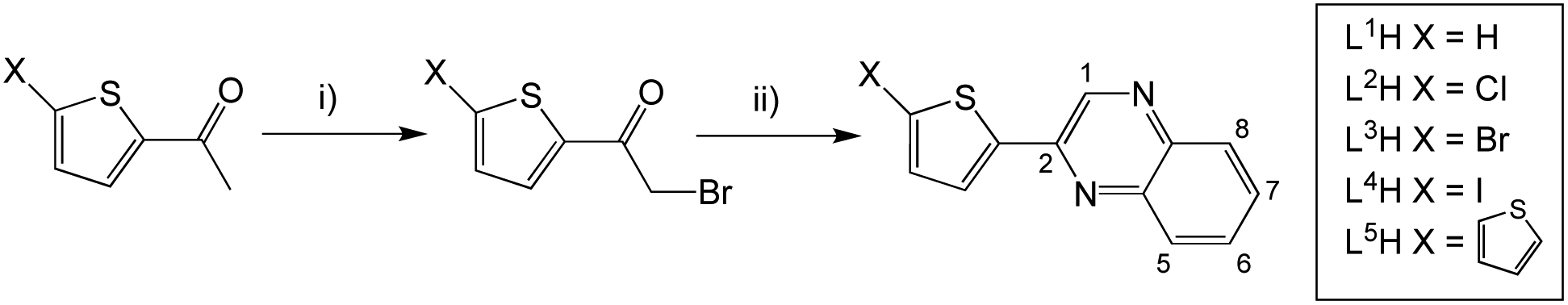 | ||
| Scheme 2 Synthesis of 2-(thiophen-2-yl)quinoxaline species with substitution at the thiophene ring. Reagents and conditions: (i) dioxane dibromide, 1,4-dioxane, rt; (ii) 1,2-phenylenediamine, EtOH, heat; glacial acetic acid can also be used for L4H. | ||
An exception to this general approach was noted in the synthesis of the iodo derivative, L4H. Various byproducts (observed in the 1H NMR spectrum) were noted following treatment of 5-iodo-2-acetylthiophene with dibromo-dioxane. Although the major product was the desired mono-brominated species (63%), di-bromination was also evident for this variant and was challenging to separate. Therefore, the synthetic methodology was altered wherein glacial acetic acid was used as the solvent and the mixture was stirred for a longer period in the absence of light. This adaption gave a much improved yield of the desired mono-brominated intermediate.
L6–9H were formed via an analogous synthetic procedure (Scheme 3). Here, substitution was introduced to the quinoxaline backbone by use of a disubstituted o-phenylenediamine or 2,3-diaminonaphthalene. L10H was synthesised from the β-diketone starting material, 2,2′-thenil (Scheme 4). Here, the reagents were once again dissolved in ethanol, but with a catalytic amount of glacial acetic acid added.
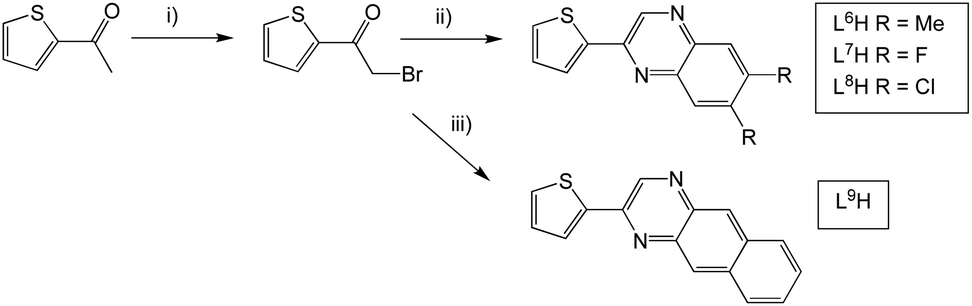 | ||
| Scheme 3 Synthesis of 2-(thiophen-2-yl)quinoxaline species with functionalisation at the quinoxaline ring. Reagents and conditions: (i) dioxane dibromide, 1,4-dioxane, rt; (ii) 1,2-phenylenediamine derivative, EtOH, heat; (iii) 2,3-diaminonaphthalene, EtOH, heat. | ||
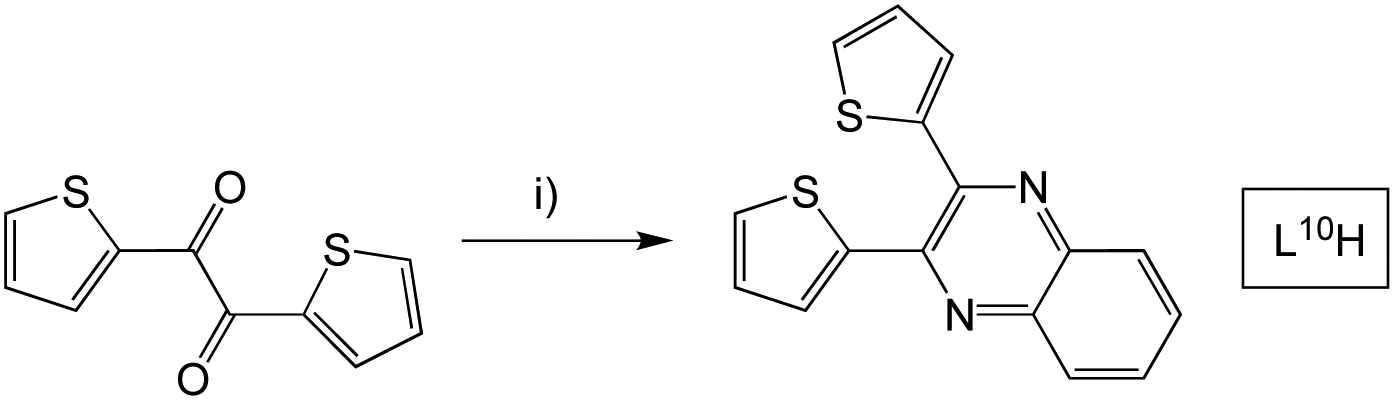 | ||
| Scheme 4 Synthesis of 2,3-di(thiophen-2-yl)quinoxaline species, L10H. Reagents and conditions: (i) 1,2-phenylenediamine, EtOH, AcOH, heat. | ||
Finally, 2-(thiophen-3-yl)quinoxaline (L11H) was synthesised to probe the influence of structural isomerism (cf.L1H), where the point of attachment was at the C3 position of the thiophene ring (Scheme 5). More forcing conditions were required for the initial bromination step with dibromo-dioxane added to the mixture at elevated temperatures and the reaction stirred for a longer period.
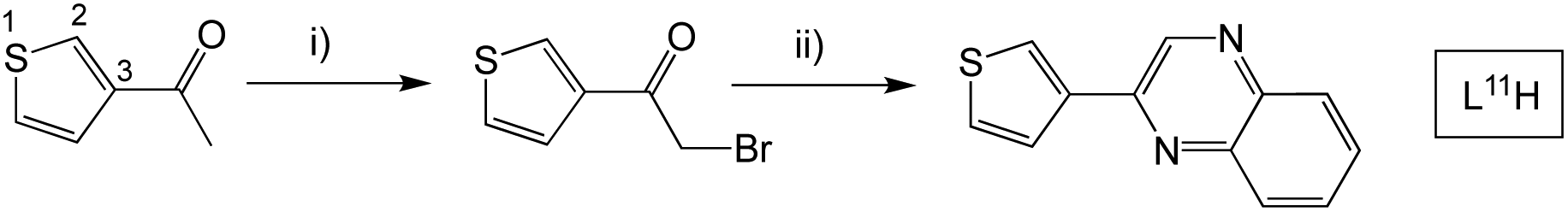 | ||
| Scheme 5 Synthesis of the isomeric 2-(thiophen-3-yl)quinoxaline species, L11H. Reagents and conditions: (i) dioxane dibromide, Et2O, 1,4-dioxane, heat; (ii) 1,2-phenylenediamine, EtOH, heat. | ||
Characterisation of the prospective ligands was achieved using the standard range of techniques. The 1H NMR spectra of L1–4H are presented in Fig. S1† and show an indicative singlet peak between 9.15–9.24 ppm corresponding to the uncoupled, deshielded proton at the 3-position of the quinoxaline ring. Similar features were observed for all these species except for L10H, as this proton is substituted for a secondary thiophene unit. Two multiplets between 8.10–7.99 ppm for L1–4H were related to the two pairs of protons on the quinoxaline moiety. The effect of electronegative substituents at the thiophene were observed in L1–4H with the chemical shift values for the two doublets on the thiophene ring differ more drastically as the halogen atom becomes more electronegative (Fig. S1, ESI†). Full details of the synthetic procedures and characterisation data for these species are presented in the Experimental section, with NMR spectra shown in the ESI.†
Synthesis and characterisation of the complexes
L1–11H were investigated as cyclometalating ligands for Ir(III) via the attempted formation of the corresponding chloro-bridged iridium dimers, [Ir(C^N)2(μ-Cl)2(C^N)2Ir]. Thus, the appropriate stoichiometric quantities of the free ligand, LnH, and IrCl3·xH2O were added to 2-methoxyethanol and heated at reflux for 48 hours. Subsequently, the putative dimeric species were cleaved into their monomeric, cationic counterparts by heating a solution of the dimer with 2,2′-bipyridine. For most of the target complexes, the dimer splitting reaction was carried out in chloroform due to the preferential solubility of the dimer species over alcoholic glycol ethers. Previous studies have shown that high yields can be obtained under relatively mild conditions, where >70% conversion can be achieved after a 19-hour reflux in DCM![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeOH (5:1) at 40 °C.22
:MeOH (5:1) at 40 °C.22
Interestingly, attempts to isolate the iridium complexes of the halogenated thiophene ligands (L2–4H) were unsuccessful with evidence for multiple species in the associated 1H NMR spectra. Thus, while the colour changes of the reaction mixture and absence of free ligand in the NMR spectra implied that a reaction had occurred with Ir(III), we were unable to isolate sufficient quantities of pure complex to confidently establish the coordination chemistry behaviour of L2–4H.
Pleasingly, the use of the remaining ligands in the series led to the successful preparation of the target complexes. These species were purified by column chromatography (silica gel; eluent was acetone followed by a solvent mixture of CH3CN:H2O:KNO3(sat.) (in a ratio of 14:2:1)); the desired products eluted as red or orange bands. Counterion exchange was performed to give the complexes as the hexafluorophosphate salts, [Ir(Ln)2(bipy)]PF6. Finally, recrystallisation from DCM and Et2O was also carried out to give the final complexes (Scheme 6). The complexes were obtained as highly coloured, air-stable solids, which were generally soluble in a wide range of organic solvents, including chloroform, acetonitrile, acetone and methanol.
 | ||
| Scheme 6 Structures of the Ir(III) complexes successfully isolated in this study. | ||
The complexes were initially characterised using NMR and IR spectroscopies and HRMS. The presence of the PF6− anion was confirmed by IR spectroscopy with a sharp band ca. 833 cm−1 relating to a v(P–F) stretching mode. Comparison of the 1H NMR spectra for L1H and [Ir(L1)2(bipy)]PF6 showed that all ligand-based proton resonances shift upon coordination to Ir(III). The downfield signature singlet of the 3-position of the quinoxaline ring was shifted upfield to 9.16 ppm. The most upfield resonance was attributed to the shielded thiophene proton adjacent to the point of cyclometalation. These spectra imply that cyclometalation occurs at the C3 position of the thiophene ring of L1: two doublets (3JHH values of 4.7 Hz) were noted for the remaining thiophene protons, which is consistent with previous studies.23
For [Ir(L7)2(bipy)]PF6, the 19F{1H} NMR spectrum showed two ligand-based doublets at −123.6 and −130.4 ppm (both 3JFF = 22 Hz) which were shifted from the corresponding free ligand (−129.3 and −130.9 ppm); the observed 3JFF values are consistent with values recorded for 1,2-difluorobenzene.24 Of course, the 19F ligand signals were in addition to that observed for the hexafluorophosphate which was present as a doublet (1JPF coupling in the range of 706–713 Hz) in all complexes between −73.1 and −71.6 ppm.
The 13C{1H} NMR spectra for the fluorinated compounds are displayed in Fig. S2,† where six doublets of doublets were seen in the spectrum of L7H, whilst five are visible in the corresponding complex (the peak at 118 ppm for [Ir(L7)2(bipy)]PF6 was coincident with the residual solvent peak from CD3CN); the C–F couplings were consistent with literature values.25 The carbon atom directly attached to the highly electronegative fluorine atoms are relatively deshielded (151.3–153.7 ppm) and possess a large coupling constant of 253.4–256.6 Hz in agreement with a 1JCF coupling. The resonances at 138.8–140.5 ppm were relatively downfield, owing to the electron-deficient nature of the quinoxaline ring with only subtle variations noted between the free ligand and the complex (see Table S1† for details and assignments). The NMR spectra for the ligand and complexes are shown in the ESI (Fig. S3–S37†).
HRMS data was obtained for all isolated complexes consistent with the cationic complex ion in each case. In the case of the chlorinated species, L2H, L8H and [Ir(L8)2(bipy)]PF6, the MS data revealed the isotopic 35/37Cl distributions expected for a mono-, di- and tetra-chlorinated species, respectively, in each case (see Fig. S38†).
Single crystal X-ray diffraction
Single crystals suitable for X-ray crystallographic studies were obtained for the methylated species, [Ir(L6)2(bipy)]PF6 (red, block-shaped crystals were grown from a solution of DCM and diethyl ether) and the dichloro analogue, [Ir(L8)2(bipy)]PF6 (red, rod-shaped crystals were grown from a solution of MeCN and diethyl ether); data collection parameters and ORTEP diagrams are presented in the ESI (Fig. S40†). Despite numerous attempts on different crystals the quality of the [Ir(L8)2(bipy)]PF6 structure was relatively poor. The diffraction produced diffuse spots (especially in one direction) and the intensities fell during each collection suggesting damage to the crystal. The crystal used was not single, and most probably some variant of a stacked plate with the distortion mainly about one axis; nonetheless attempts to integrate the data as multiple components produced much worse results. However, it is still sufficient to be confident about the skeletal structure, together with the identity of the complex and intrinsic details of the coordination sphere, especially when used in conjunction with other forms of characterisation. The data collection parameters are shown in Table S2.†The structures showed the expected formulation and ligand configurations within the complex (Fig. 1) and are, in essence, analogous. As expected, the complexes displayed a cis-C,C and trans-N,N configuration. The coordination sphere bond length values (Table 1) were consistent with related systems26 where cyclometalation was shown at the C3 position of the thiophene ring. The structural parameters were also comparable to previous examples that employ related 2-phenylquinoxaline cyclometalating ligands at Ir(III).27
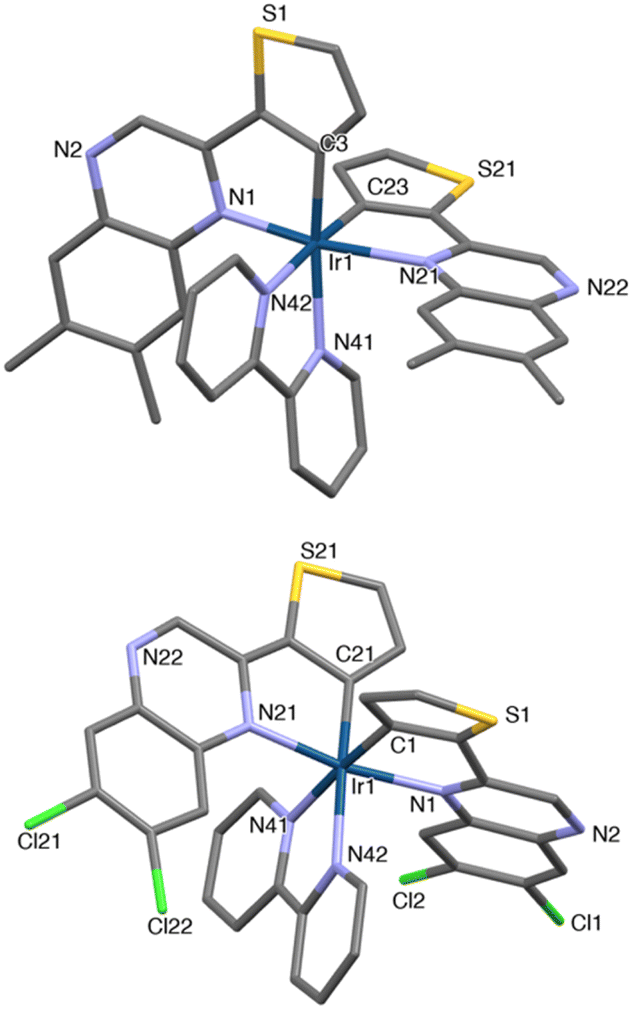 | ||
| Fig. 1 Structural representations of [Ir(L6)2(bipy)]PF6 (top) and [Ir(L8)2(bipy)]PF6 (bottom) obtained from X-ray diffraction. H-atoms and counter ions omitted for clarity. Only one of the three unique molecules of the asymmetric unit is shown for [Ir(L8)2(bipy)]PF6. | ||
| [Ir(L6)2(bipy)]PF6 | |||||
| Ir(1) | N(1) | 1.979(16) | Ir(1) | N(42) | 2.145(4) |
| Ir(1) | N(21) | 2.204(14) | Ir(1) | C(3) | 1.979(15) |
| Ir(1) | N(41) | 2.141(3) | Ir(1) | C(23) | 2.013(14) |
| [Ir(L8)2(bipy)]PF6 | |||||
| Ir(1) | N(1) | 2.110(14) | Ir(1) | N(42) | 2.143(9) |
| Ir(1) | N(21) | 2.063(15) | Ir(1) | C(1) | 1.97(2) |
| Ir(1) | N(41) | 2.059(13) | Ir(1) | C(21) | 1.951(19) |
Photophysical properties of 2-(thienyl)-quinoxaline species
The UV-vis absorption profiles of L1–11H in 10−5 M aerated acetonitrile solutions are shown in Fig. 2. All species displayed absorption features within the UV range, with some extending to the visible region. Despite the possibility for n → π* absorption bands to appear, all transitions shown can be attributed to allowed π → π* features due to their high molar absorptivities. Previous reports on 2-([2–2′,-bithiophen]-5-yl)quinoline suggests that low-energy π → π* singlet states can also possess some n → π* character, and depending on the energy difference between such states, efficient mixing can be achieved.28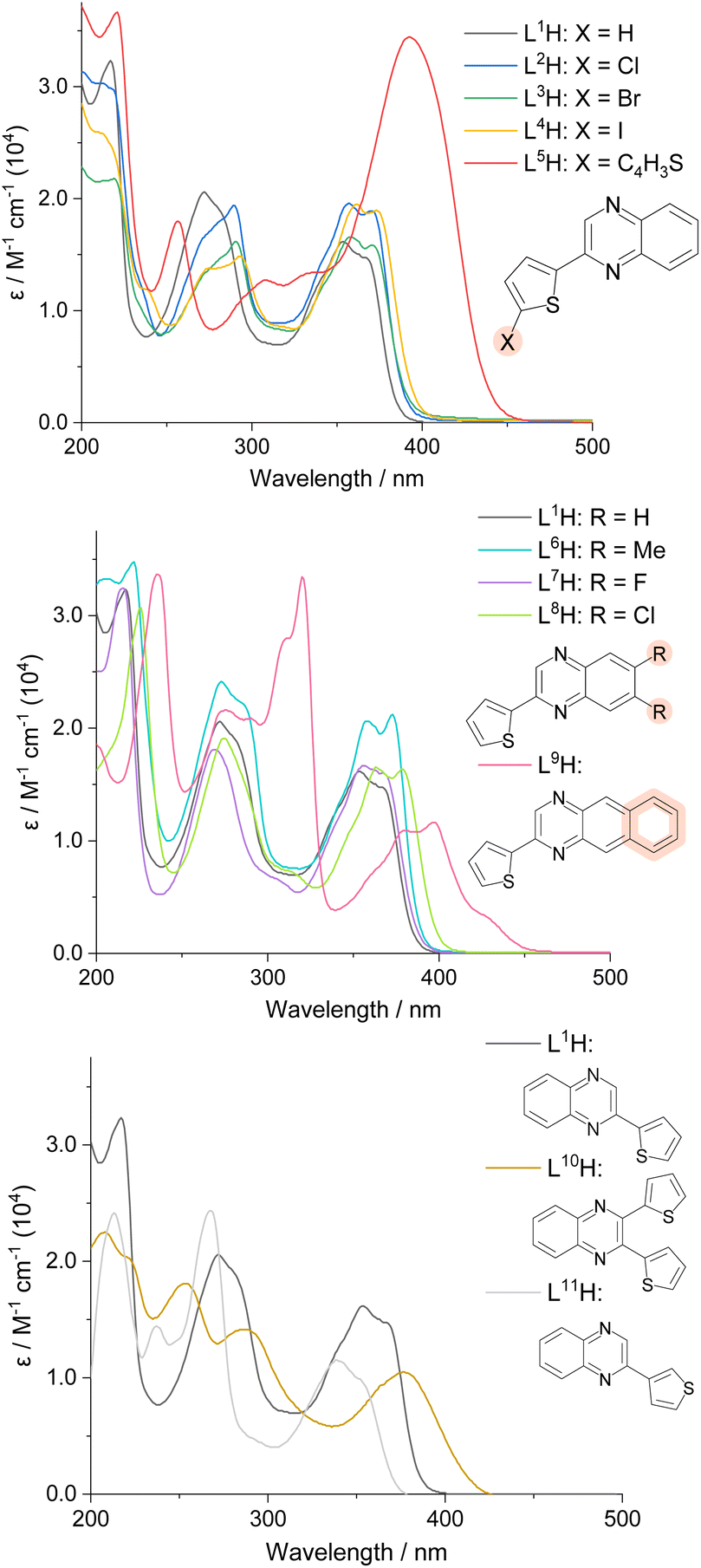 | ||
| Fig. 2 UV-vis spectra (MeCN) for the different thiophene-quinoxaline species. | ||
The spectra demonstrated that the position and nature of the substituent influenced the absorption wavelengths of the compounds. Substitution of the thiophene subunit with a halogen atom (L2–4H) or di-substitution of the quinoxaline backbone (L6–8H) produced subtle changes in peak appearance; addition of electronegative groups generally produced a subtle bathochromic shift. Comparison between the structural isomers L1H and L11H revealed a relative hypsochromic shift in λmax for the latter. Across the series, the lowest energy λmax value was observed for L9H (ca. 400 nm, with accompanying vibronic features extending to 450 nm), where the additional conjugation of the fused quinoxaline backbone (a benzo[g]quinoxaline) expectedly lowers the energy of the participating electronic transitions.
A high intensity peak (ε = 3.4 × 104 M−1 cm−1) at 392 nm was seen for L5H, where the π-conjugated system is extended by the additional thiophene ring. In comparison, when a second thiophene ring was added to the quinoxaline sub-unit (L10H), a lower ε value (1.0 × 104 M−1 cm−1) at λmax 378 nm was recorded. It should be noted that previous studies have shown that charge transfer can be observed from the thiophene ring donors to the quinoxaline acceptors,8 often explaining the observation of lower energy absorption bands.29
L1–11H were analysed by steady state luminescence spectroscopy (Table 2, Fig. 3). Following excitation at 350 nm, all compounds emit in the blue part of the visible range between 410–491 nm, with lifetimes typically < 3 ns which is indicative of a fluorescence in all cases. The species with the longest λem values, L5H and L9H, displayed the most bathochromically shifted emission bands at 491 and 488 nm, respectively. Subtle tunability in the emission properties was demonstrated by altering the substituents at the thiophene ring or the quinoxaline – for example, the iodine-substituted ligand was red-shifted by 8 nm compared to the chlorine analogue. Functionalisation at the quinoxaline ring appeared to promote more significant differences, where the dimethyl-substituted analogue showed a hypsochromic shift in emission (λem = 410 nm) when compared to the dichloro compound (λem = 431 nm). Interestingly, L11H possessed the most blue-shifted emission at 401 nm (cf. λem = 416 nm for L1H) suggesting that the point of attachment between thiophene and quinoxaline ring also impacts the electronic nature of these compounds.
| Compound | λ abs/nm |
λ
em
/nm |
τ
obs
/ns |
ϕ
F
/% |
|---|---|---|---|---|
| a Photophysical properties in aerated MeCN solutions at room temperature (10−5 M). λex = 350–400 nm. b Quinine sulfate in 0.1 M H2SO4 (Φ = 0.546)31 used as standard for quantum yield determination, λex = 350 nm. c Observed lifetimes, λex = 295 nm. | ||||
| L1H | 217, 272, 353, 368 | 416 | <1ns | 3.5 |
| L2H | 214, 289, 357, 369 | 422 | <1ns | 5.0 |
| L3H | 219, 290, 357, 370 | 423 | <1ns | 3.6 |
| L4H | 213, 275, 292, 361, 373 | 430 | <1ns | 1.7 |
| L5H | 221, 256, 307, 335, 392 | 491 | 2.7 | 26.4 |
| L6H | 222, 273, 357, 373 | 410 | <1ns | 4.7 |
| L7H | 215, 268, 355, 367 | 424 | <1ns | 5.8 |
| L8H | 226, 274, 363, 378 | 431 | <1ns | 6.0 |
| L9H | 235, 275, 311, 320, 379, 396, 426 | 488 | 1.3 | 3.6 |
| L10H | 208, 252, 286, 378 | 444 | <1ns | 19.0 |
| L11H | 213, 237, 267, 339, 351 | 401 | <1ns | 1.0 |
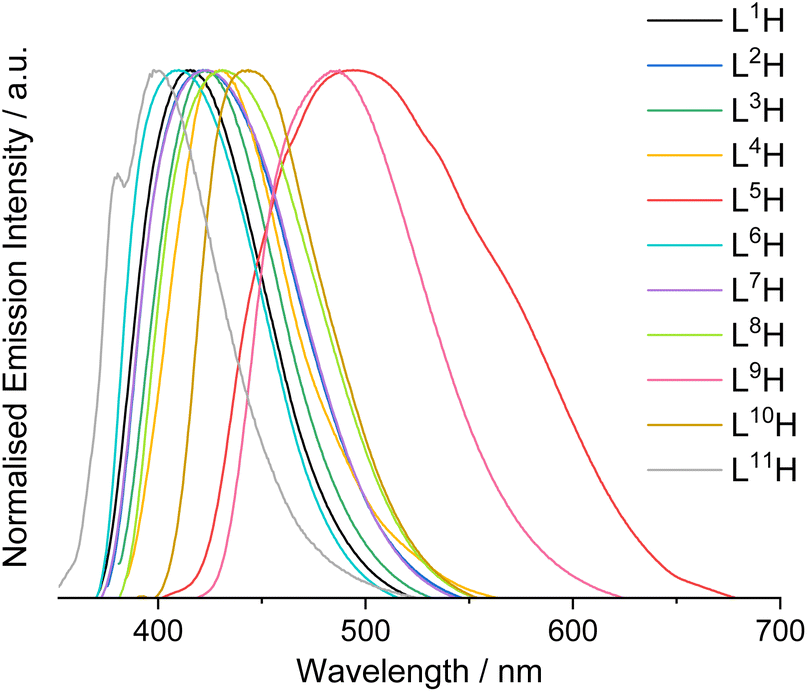 | ||
| Fig. 3 Normalised steady-state emission spectra for L1–11H in aerated MeCN (10−5 M, 293 K, λex = 350 nm). | ||
Photophysical properties of the complexes
UV-vis. absorption data for the Ir(III) complexes were also recorded in 10−5 M aerated acetonitrile solutions (Fig. 4). All complexes absorb throughout the UV range as well as into the visible region, with absorption bands spanning a large range of wavelengths. The peaks centred below 450 nm can be assigned to ligand-centred transitions, due to relatively high molar absorption coefficients, similar to those for the corresponding free ligands. Additional broad peaks were also observed at 475–600 nm with moderate magnitude ε values and were therefore attributed to spin-allowed CT absorptions. Previous studies on closely related 2-phenylquinoxaline complexes has shown that different types of CT transition are likely to contribute to these visible absorption bands, including MLCT, ILCT and LLCT (Table 3).30| λ abs/nm |
λ
em
/nm |
τ
obs
/ns |
ϕ
P
/% |
|
|---|---|---|---|---|
| a λ ex = 475–545 nm. b λ ex = 295 nm; deoxygenated value in parentheses. c [Ru(bipy)3][PF6]2 in MeCN (Φ = 0.018)34 used as standard for quantum yield determination, λex = 450 nm. | ||||
| [Ir(L1)2(bipy)]PF6 | 227, 297, 338, 371, 406, 485 | 668 | 192 (2499) | 4.0 |
| [Ir(L5)2(bipy)]PF6 | 233, 307, 358, 441, 532 | 751 | 141 (1258) | 3.1 |
| [Ir(L6)2(bipy)]PF6 | 228, 297, 337, 372, 406, 493 | 665 | 203 (2251) | 5.7 |
| [Ir(L7)2(bipy)]PF6 | 296, 337, 368, 406, 512 | 672 | 241 (3292) | 5.2 |
| [Ir(L8)2(bipy)]PF6 | 228, 303, 347, 375, 417, 525 | 680 | 209 (2899) | 5.1 |
| [Ir(L9)2(bipy)]PF6 | 244, 327, 312, 397, 421, 545 | 749 | 95 (209) | 0.3 |
| [Ir(L10)2(bipy)]PF6 | 238, 303, 355, 425, 503 | 685 | 151 (2225) | 2.6 |
| [Ir(L11)2(bipy)]PF6 | 205, 239, 284, 307, 348, 475 | 670 | 117 (1539) | 5.2 |
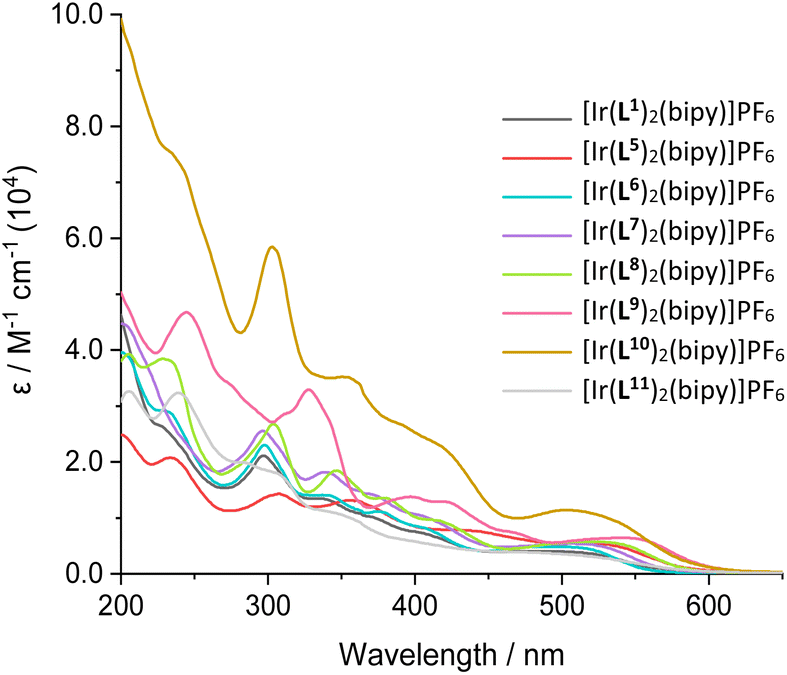 | ||
| Fig. 4 UV-vis. absorption spectra of the isolated cationic iridium complexes, [Ir(L)2(bipy)]PF6, measured in MeCN solutions (10−5 M, RT). | ||
In some cases, a broad tail was also evident which extended >600 nm; this weaker feature may be due to spin-forbidden CT bands due to enhanced intersystem crossing that is promoted by the heavy iridium atom. Comparison between the free ligands and their corresponding complexes provided clear insight into the effect of complexation. For example comparison of L9H with [Ir(L9)2(bipy)]PF6 shows (Fig. 5; see also Fig. S41–S47†) the presence of new CT bands at 450–600 nm that result from complexation with Ir(III). The wavelength maxima of the CT bands were strongly influenced by the nature of the cyclometalating ligands. For example, the most red-shifted CT band was observed for [Ir(L9)2(bipy)]PF6 (λmax = 545 nm), which is presumably due to the extended conjugation of the ligand. In contrast, [Ir(L11)2(bipy)]PF6 displayed the shortest wavelength CT peak (λmax = 475 nm), which was distinct from the value (485 nm) of its structural isomer, [Ir(L1)2(bipy)]PF6.
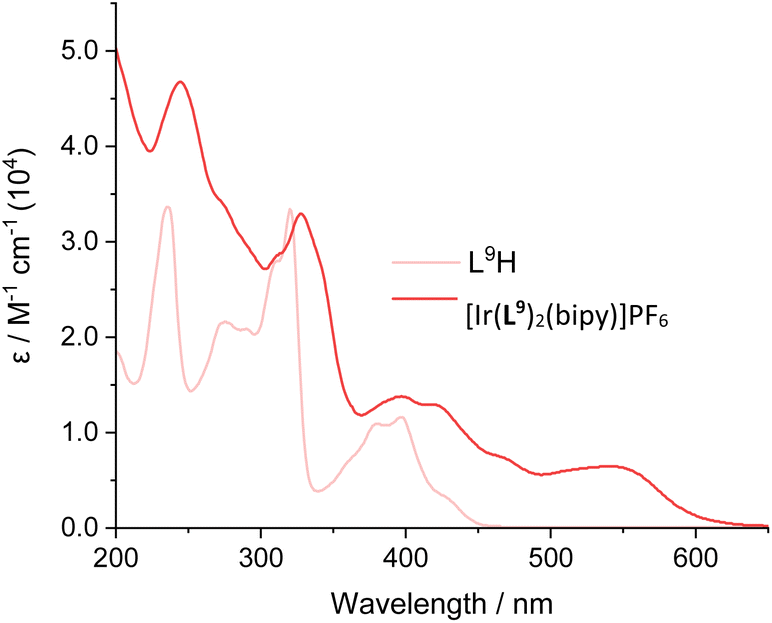 | ||
| Fig. 5 Comparison of the UV-vis absorption spectra of L9H and [Ir(L9)2(bipy)]PF6. | ||
Steady state emission data were collected for all Ir(III) complexes, wherein each complex was demonstrated to be a deep-red emitter, with two examples approaching the near-IR region. The complexes with the longest λem values were [Ir(L5)2(bipy)]PF6 (λem = 751 nm) and [Ir(L9)2(bipy)]PF6 (λem = 749 nm) showing that ligand conjugation at either the thiophene or quinoxaline moiety can lower the energy of the emitting state. It is noteworthy that the emission maximum of [Ir(L9)2(bipy)]PF6 is relatively blue-shifted compared to Ir(III) complexes formed from 2,3-diphenylbenzo[g]quinoxaline.32 Similarly, the bathochromic shift for [Ir(L5)2(bipy)]PF6vs. [Ir(L1)2(bipy)]PF6 may be due to the electron rich bithienyl moiety, which raises the HOMO level.33 Subtle tuning of the luminescence in the range of λem = 665–685 nm was also shown among the other examples where the methylated complex [Ir(L6)2(bipy)]PF6 was the most blue-shifted (λem = 665 nm), consistent with the destabilisation of the excited state due to electron donating groups at the quinoxaline ring (Fig. 6).30
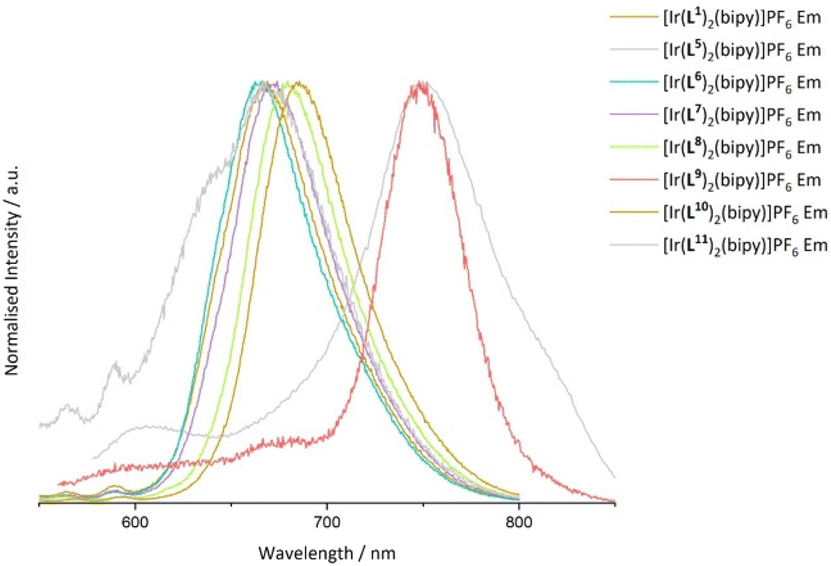 | ||
| Fig. 6 Comparison of the emission spectra for all complexes. Recorded in aerated MeCN. | ||
Time-resolved luminescence measurements revealed a single exponential decay profile in each case consistent with a single emissive process. All Ir(III) complexes displayed a significant enhancement in observed lifetimes compared to the free ligands with lifetimes in the range of 95–241 ns which are therefore consistent with a triplet emission in each case. Upon deoxygenation the lifetimes were typically further extended in to the microsecond domain (Table 3) confirming the inherent triplet nature of the emitting states (Fig. 48, ESI†). Of the near-IR emitting species, [Ir(L5)2(bipy)]PF6 demonstrated a longer triplet lifetime and higher quantum yield; [Ir(L9)2(bipy)]PF6 possessed a notably short lifetime even under deoxygenated conditions consistent with the weakly emissive character of this complex.
Low temperature (77 K) emission spectra were also collected for all complexes (Fig. S49, ESI†) using a EtOH/MeOH (3:1) frozen glass. A hypsochromic shift in emission wavelength maximum was noted in most cases (Fig. 7), which is commonly attributed to rigidochromism when emitting states are predominantly CT in nature. More pronounced features due to enhanced vibronic coupling were also clearly observed suggesting well defined coupling to ligand-based vibronic levels at low temperature.35
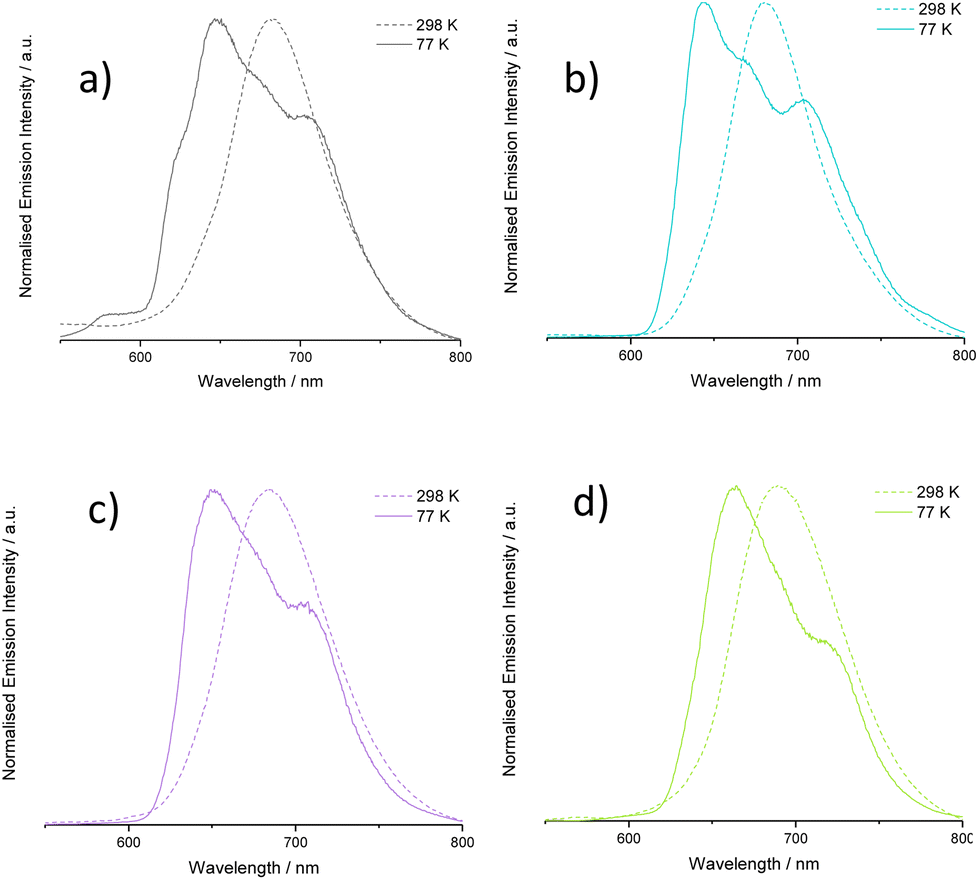 | ||
| Fig. 7 Comparison of emission spectra at 298 and 77 K for selected complexes: (a) [Ir(L1)2(bipy)]PF6, (b) [Ir(L6)2(bipy)]PF6, (c) [Ir(L7)2(bipy)]PF6, (d) [Ir(L8)2(bipy)]PF6. | ||
Conclusions
The current work has shown how both thiophene and quinoxaline rings can be integrated into ligand architectures that are suitable for organometallic coordination chemistry. 2-(Thienyl)quinoxaline species can be obtained with functionalisation across both ring systems to give a wide range of fluorescent variants. Coordination chemistry of these heterocyclic species with Ir(III) revealed reactivity that was generally predictable to give cyclometalated products. Resultant investigations showed that halogenation at the thiophene ring led to an apparent mixture of products which prevented isolation of the target complexes for L2–4H. Nonetheless, the successfully isolated Ir(III) complexes included a range of functionalities across the ligand architectures including bis-thiophene and benzo[g]quinoxaline moieties, both of which led to bathochromically shifted emission properties from the deep red to the near-IR region. Taken together these newly developed ligands provide opportunities for the development and application of deep red to NIR phosphorescent organometallic Ir(III) complexes.Experimental
General considerations
All reagents and solvents were commercially available and were used without further purification if not stated otherwise. 2-Bromo-1-(thiophen-3-yl)ethan-1-one,36 2-(thiophen-2-yl)quinoxaline (L1H),37 6,7-dimethyl-2-(thiophen-2-yl)quinoxaline (L6H),38 2,3-di(thiophen-2-yl)quinoxaline (L10H),39 and 2-(thiophen-3-yl)quinoxaline (L11H)40 have been reported previously, and synthetic procedures and characterisation data are presented in ESI.† For the measurement of 1H, 31P, and 13C NMR spectra a Bruker Fourier300 (300 MHz), Bruker AVANCE HD III equipped with a BFFO SmartProbe™ (400 MHz) or Bruker AVANCE III HD with BBO Prodigy CryoProbe (500 MHz) was used. The obtained chemical shifts δ are reported in ppm and are referenced to the residual solvent signal. Spin–spin coupling constants J are given in Hz. 31P and 13C(APT) spectra are proton decoupled unless otherwise stated.Low-resolution mass spectra were obtained by the staff at Cardiff University. High-resolution mass spectra were carried out at the EPSRC National Mass Spectrometry Facility at Swansea University. High resolution mass spectral (HRMS) data were obtained on a Waters MALDI-TOF mx at Cardiff University or on a Thermo Scientific LTQ Orbitrap XL. IR spectra were obtained from a Shimadzu IR-Affinity-1S FTIR. Reference to spectroscopic data are given for known compounds. UV-Vis studies were performed on a Shimadzu UV-1800 spectrophotometer as MeCN solutions (10−5 M). Photophysical data were obtained on a JobinYvon–Horiba Fluorolog spectrometer fitted with a JY TBX picosecond photodetection module as MeCN solutions. The pulsed source was a Nano-LED configured for 295 nm output operating at 1 or 500 MHz. Luminescence lifetime profiles were obtained using the JobinYvon–Horiba FluoroHub single photon counting module and the data fits yielded the lifetime values using the provided DAS6 deconvolution software. For the near-IR lifetime measurements on [Ir(L5)2(bipy)]PF6 and [Ir(L9)2(bipy)]PF6 the pulsed laser source was a Continuum Minilite Nd:YAG configured for 355 nm output in conjunction with a Hamamatsu R5509-73 detector (cooled to −80 °C using a C9940 housing).
X-ray crystallography
A suitable crystal for [Ir(L6)2(bipy)]PF6 was obtained and mounted on a MITIGEN holder in oil on a Rigaku FRE + diffractometer with Arc)Sec VHF Varimax confocal mirrors, a UG2 goniometer and HyPix 6000HE detector.41 For [Ir(L8)2(bipy)]PF6 a suitable crystal was selected and mounted on a MITIGEN holder in oil on Diamond Light Source Beamline I19-1. The crystals was kept at a steady T = 100 K during data collection. Using Olex242 the structures were solved with the ShelXT43 structure solution program, using the Intrinsic Phasing solution method. The models was refined with version 2018/3 of ShelXL44 using Least Squares minimisation. All non-H atoms were refined anisotropically and difference Fourier syntheses were employed in positioning idealized hydrogen atoms and were allowed to ride on their parent C-atoms.CCDC 2270408 and 2270409† contains supplementary X-ray crystallographic data for [Ir(L6)2(bipy)]PF6 and [Ir(L8)2(bipy)]PF6 respectively.
Synthesis of 2-(5-chlorothiophen-2-yl)quinoxaline (L2H)
Prepared similarly from 2-bromo-1-(5-chlorothiophen-2-yl)ethan-1-one (976 mg, 4.07 mmol) and 1,2-phenylenediamine (485 mg, 4.48 mmol) to give the product as an orange solid (164 mg, 16%). 1H NMR (400 MHz, CDCl3) δH = 9.17 (s, 1H), 8.10–7.99 (m, 2H), 7.79–7.67 (m, 2H), 7.63 (d, 3JHH = 4.0 Hz, 1H), 7.02 (d, 3JHH = 4.0 Hz, 1H) ppm. 13C{1H} NMR (101 MHz, CDCl3) δC = 146.7, 142.2, 141.6, 141.3, 141.2, 135.2, 130.8, 129.6, 129.3, 129.2, 127.8, 126.3 ppm. HRMS (EI): found m/z 246.0018, calc'd m/z 246.0018 for C12H7ClN2S. UV-vis (MeCN) λmax (ε × 104 L−1 mol−1 cm−1): 214 (3.0), 289 (1.9) 357 (2.0), 369 (1.9) nm. FTIR (solid, ATR) νmax/cm−1: 3098, 3077, 3007, 2369, 2345, 2322, 1751, 1611, 1547, 1487, 1466, 1431, 1364, 1348, 1323, 1312, 1300, 1225, 1213, 1198, 1123, 1082, 1022, 966, 935, 908, 856, 799, 768, 754, 675, 669, 615, 588, 567, 536, 482, 467, 455, 413.Synthesis of 2-(5-bromothiophen-2-yl)quinoxaline (L3H)
Prepared similarly from 2-bromo-1-(5-bromothiophen-2-yl)ethan-1-one (1.37 g, 4.81 mmol) and 1,2-phenylenediamine (0.57 g, 5.29 mmol) to give the product as a brown crystalline solid (0.52 g, 37%). 1H NMR (400 MHz, CDCl3) δH = 9.15 (s, 1H), 8.09–7.99 (m, 2H), 7.79–7.66 (m, 2H), 7.57 (d, 3JHH = 3.9 Hz, 1H), 7.15 (d, 3JHH = 3.9 Hz, 1H) ppm. 13C{1H} NMR (101 MHz, CDCl3) δC = 146.6, 144.1, 142.2, 141.6, 141.3, 131.5, 130.8, 129.6, 129.3, 129.2, 127.0, 117.9 ppm. HRMS (ESI): found m/z 290.9599, calc'd m/z 290.9592 for C12H8BrN2S. UV-vis (MeCN) λmax (ε × 104 L−1 mol−1 cm−1): 219 (2.2), 290 (1.6), 357 (1.7), 370 (1.6) nm. FTIR (solid, ATR) νmax/cm−1: 3310, 3084, 3073, 2997, 2949, 1661, 1510, 1389, 1312, 1287, 1215, 1190, 1146, 1067, 959, 924, 903, 870, 795, 762, 739, 662, 638, 573, 540.Synthesis of 2-(5-iodothiophen-2-yl)quinoxaline (L4H)
Prepared similarly from 2-bromo-1-(5-iodothiophen-2-yl)ethan-1-one (1.22 g, 3.69 mmol) and 1,2-phenylenediamine (399 mg, 3.69 mmol) to give the product as an off-white solid (540 mg, 43%). 1H NMR (400 MHz, CDCl3) δH = 9.16 (s, 1H), 8.10–8.00 (m, 2H), 7.79–7.67 (m, 2H), 7.49 (d, 3JHH = 3.9 Hz, 1H), 7.35 (d, 3JHH = 3.9 Hz, 1H) ppm. 13C{1H} NMR (126 MHz, CDCl3) δC = 148.4, 146.4, 142.2, 141.6, 141.5, 138.5, 130.8, 129.6, 129.3, 129.3, 128.0, 79.8 ppm. HRMS (ESI): found m/z 338.9456, calc'd m/z 338.9453 for C12H8127IN2S. UV-vis (MeCN) λmax (ε × 104 L−1 mol−1 cm−1): 213 (2.6), 275 (1.4), 292 (1.5), 361 (1.9), 373 (1.9) nm. FTIR (solid, ATR) νmax/cm−1: 3080, 3048, 3013, 1609, 1572, 1545, 1491, 1420, 1402, 1323, 1312, 1292, 1225, 1200, 1128, 1063, 1018, 999, 951, 932, 910, 881, 874, 856, 802, 797, 783, 768, 756, 739, 673, 613, 573, 538, 490, 469, 411.Synthesis of 2-([2,2′-bithiophen]-5-yl)quinoxaline (L5H)
Prepared similarly from 1-([2,2′-bithiophen]-5-yl)-2-bromoethan-1-one (112 mg, 0.388 mmol) and 1,2-phenylenediamine (50.3 mg, 0.465 mmol) to give the product as a yellow solid (83.2 mg, 73%). 1H NMR (500 MHz, CDCl3) δH = 9.22 (s, 1H), 8.11–8.05 (m, 2H), 7.78 (d, 3JHH = 3.8 Hz, 1H), 7.78–7.73 (m, 1H), 7.70 (ddd, 3JHH = 8.4, 6.9, 4JHH = 1.4 Hz, 1H), 7.34 (dd, 3JHH = 3.6, 4JHH = 1.1 Hz, 1H), 7.30 (dd, 3JHH = 5.1, 4JHH = 1.1 Hz, 1H), 7.27 (d, 3JHH = 3.9 Hz, 1H), 7.08 (dd, 3JHH = 5.1, 3.6 Hz, 1H) ppm. 13C{1H} NMR (126 MHz, CDCl3) δc = 147.3, 142.3, 142.1, 141.7, 141.1, 140.6, 137.1, 130.8, 129.4, 129.2, 129.1, 128.3, 128.0, 125.7, 125.0, 124.9 ppm. HRMS (ESI): found m/z 295.0358, calc'd m/z 295.0364 for C16H11N2S2. UV-vis (MeCN) λmax (ε × 104 L−1 mol−1 cm−1): 221 (3.7), 256 (1.8), 307 (1.3), 335 (1.3), 392 (3.4) nm. FTIR (solid, ATR) νmax/cm−1: 3100, 3082, 3063, 3046, 2918, 2849, 1609, 1571, 1553, 1537, 1514, 1489, 1468, 1445, 1423, 1341, 1317, 1227, 1202, 1132, 1119, 1070, 1053, 1013, 999, 949, 939, 930, 887, 858, 837, 812, 791, 783, 752, 681, 675, 611, 569, 550, 480, 455, 407.Synthesis of 6,7-difluoro-2-(thiophen-2-yl)quinoxaline (L7H)
Prepared similarly from 2-bromo-1-(thiophen-2-yl)ethan-1-one (250 mg, 1.22 mmol) and 4,5-difluoro-o-phenylenediamine (193 mg, 1.34 mmol) to give the product as an off-white solid (176 mg, 58%). 1H NMR (400 MHz, CDCl3) δH = 9.20 (s, 1H), 7.86 (dd, 3JHH = 3.7, 4JHH = 1.1 Hz, 1H), 7.82 (dd, 3JHF = 8.4, 4JHF = 1.9 Hz, 1H), 7.80 (dd, 3JHF = 8.3, 4JHF = 1.6 Hz, 1H), 7.57 (dd, 3JHH = 5.0, 4JHH = 1.1 Hz, 1H), 7.21 (dd, 3JHH = 5.0, 3.7 Hz, 1H) ppm. 13C{1H} NMR (101 MHz, CDCl3) δC = 152.9 (dd, 1JCF = 256.6, 2JCF = 16.1 Hz), 151.9 (dd, 1JCF = 256.0, 2JCF = 16.0 Hz), 147.7, 142.2, 141.7, 139.7 (dd, 3JCF = 11.1, 4JCF = 1.2 Hz), 138.6 (dd, 3JCF = 10.6, 4JCF = 1.1 Hz), 130.4, 128.7, 127.4, 115.1 (dd, 2JCF = 17.5, 3JCF = 1.9 Hz), 114.9 (dd, 2JCF = 17.5, 3JCF = 1.6 Hz) ppm. 19F{1H} NMR (376 MHz, CDCl3) δF = −129.3 (d, 3JFF = 21.1 Hz), −130.9 (d, 3JFF = 21.4 Hz) ppm. HRMS (ESI): found m/z 249.0293, calc'd m/z 249.0298 for C12H7N2F2S. UV-vis (MeCN) λmax (ε × 104 L−1 mol−1 cm−1): 215 (3.2), 268 (1.8), 355 (1.7), 367 (1.6) nm. FTIR (solid, ATR) νmax/cm−1: 3117, 3024, 1803, 1626, 1547, 1499, 1418, 1358, 1335, 1314, 1233, 1223, 1209, 1165, 1124, 1119, 1051, 999, 934, 920, 907, 864, 849, 833, 756, 731, 706, 646, 619, 611, 567, 486, 444.Synthesis of 6,7-dichloro-2-(thiophen-2-yl)quinoxaline (L8H)
Prepared similarly from 2-bromo-1-(thiophen-2-yl)ethan-1-one (250 mg, 1.22 mmol) and 4,5-dichloro-o-phenylenediamine (237 g, 1.34 mmol) to give the product as an off white solid (140 mg, 41%). 1H NMR (400 MHz, CDCl3) δH = 9.20 (s, 1H), 8.19 (s, 1H), 8.17 (s, 1H), 7.87 (dd, 3JHH = 3.8, 4JHH = 1.0 Hz, 1H), 7.59 (dd, 3JHH = 5.0, 4JHH = 1.1 Hz, 1H), 7.22 (dd, 3JHH = 5.0, 3.8 Hz, 1H) ppm. 13C{1H} NMR (101 MHz, CDCl3) δC = 148.4, 143.2, 141.7, 141.2, 140.1, 135.2, 133.7, 130.9, 129.9, 128.8, 128.8, 127.9 ppm. HRMS (ESI): found m/z 280.9695, calc'd m/z 280.9707 for C12H7N2SCl2. UV-vis (MeCN) λmax (ε × 104 L−1 mol−1 cm−1): 226 (3.1), 274 (1.9), 363 (1.7), 378 (1.6) nm.Synthesis of 2-(thiophen-2-yl)benzo[g]quinoxaline (L9H)
Prepared similarly from 2-bromo-1-(thiophen-2-yl)ethan-1-one (250 mg, 1.22 mmol) and 2,3-diaminonaphthalene (212 mg, 1.34 mmol), but instead of filtering the pure product, the yield was maximised by removing the solvent in vacuo to obtain the crude product. This was then purified by column chromatography (SiO2, CH2Cl2), where the product eluted as an orange band. The solvent was removed in vacuo to give the product as an orange solid (158 mg, 49%). 1H NMR (400 MHz, CDCl3) δH = 9.31 (s, 1H), 8.64 (s, 1H), 8.63 (s, 1H), 8.11 (dd, 3JHH = 4.6, 4JHH = 0.9 Hz, 1H), 8.09 (dd, 3JHH = 4.6, 4JHH = 0.9 Hz, 1H), 7.93 (dd, 3JHH = 3.7, 4JHH = 1.1 Hz, 1H), 7.60 (dd, 3JHH = 5.1, 4JHH = 1.1 Hz, 1H), 7.59–7.56 (m, 2H), 7.25 (dd, 3JHH = 5.0, 3.8 Hz, 1H) ppm. 13C{1H} NMR (101 MHz, CDCl3) δC = 147.5, 143.6, 142.8, 138.7, 138.2, 134.5, 133.6, 130.5, 128.7, 128.7, 128.5, 127.8, 127.7, 127.5, 127.1, 126.8 ppm. HRMS (ESI): found m/z 263.0632, calc'd m/z 263.0643 for C16H11N2S. UV-vis (MeCN) λmax (ε × 104 L−1 mol−1 cm−1): 235 (3.4), 275 (2.2), 311 (2.8), 320 (3.3), 379 (1.1), 396 (1.2), 426 (0.3) nm. FTIR (solid, ATR) νmax/cm−1: 3049, 2955, 2924, 2853, 1719, 1570, 1553, 1526, 1456, 1425, 1408, 1360, 1339, 1317, 1304, 1271, 1260, 1244, 1223, 1171, 1163, 1113, 1080, 1063, 995, 933, 912, 874, 849, 743, 700, 637, 608, 573, 561, 492, 469, 424.General procedure for the synthesis of [(Ir(Ln)2(μ-Cl))2]
The chloride-bridged dimer intermediates were synthesised according to the Nonoyama route.45 IrCl3·xH2O (1.0 eq.) and the free ligand, LnH (2.0 eq.) were dissolved in 2-methoxyethanol and distilled water (3:1, 10–20 mL) and the mixture was heated to reflux under an inert nitrogen atmosphere whilst stirring for 48 h. The reaction mixture was then cooled to room temperature, and precipitates formed upon the addition of distilled water (20–30 mL). The solids were collected by filtration under reduced pressure and washed with water (5–10 mL). The crude product was dissolved in dichloromethane and filtered under suction to remove any insoluble side-products, the solvent was removed in vacuo to give the products as brown/red solids (37–91%), which were used in subsequent steps without further purification or characterisation.
Synthesis of [Ir(L1)2(bipy)]PF6
[(Ir(L1)2(μ-Cl))2] (52.6 mg, 0.405 mmol) and 2,2′-bipyridine (14.5 mg, 0.930 mmol) were dissolved in chloroform (10 mL) and the solution was heated to reflux for 24 h under an inert nitrogen atmosphere. The reaction mixture was cooled to room temperature and the solvent was removed in vacuo. The crude solid was then purified by column chromatography (MeCN:H2O:KNO3 (aq. sat.) (14:2:1), SiO2), where the red band was collected. The solvent was removed in vacuo and the product was left to dry in a heated oven at ∼60 °C for 16 h. The product was dissolved in acetonitrile, and the insoluble salts were filtered off under suction. A saturated aqueous solution of NH4PF6 was added to the filtrate and stirred for 15 min to ensure the complex existed as a PF6 salt. The solvent was removed in vacuo, and then washed with distilled water to remove excess insoluble inorganic salts. The crude product was recrystallised from dichloromethane and diethyl ether to give the pure product as a red solid (10.2 mg, 14%). NMR (500 MHz, CDCl3) δH = 9.16 (s, 2H), 8.56 (d, 3JHH = 8.2 Hz, 2H), 8.15 (app. td, 3JHH = 8.0, 4JHH = 1.6 Hz, 2H), 8.02 (dd, 3JHH = 8.3, 4JHH = 1.4 Hz, 2H), 7.88 (dd, 3JHH = 5.5, 4JHH = 0.9 Hz, 2H), 7.58–7.54 (m, 4H), 7.54 (d, 3JHH = 4.7 Hz, 2H), 7.17 (ddd, 3JHH = 8.6, 7.0, 4JHH = 1.5 Hz, 2H), 6.81 (dd, 3JHH = 8.8, 4JHH = 0.8 Hz, 2H), 6.24 (dd, 3JHH = 11.2, 5.0 Hz, 2H) ppm. 13C{1H} NMR (126 MHz, CDCl3) δC = 160.2, 158.6, 156.2, 148.7, 142.2, 141.9, 141.2, 140.8, 137.9, 135.2, 132.9, 132.8, 131.2, 129.2, 128.7, 125.9, 121.8 ppm. 19F{1H} NMR (376 MHz, CDCl3) δF = −73.06 (d, 1JPF = 710.4 Hz) ppm. HRMS (ESI): found m/z 769.0957, calc'd m/z 769.0953 for C34H22N6S2191Ir. UV-vis (MeCN) λmax (ε × 104 L−1 mol−1 cm−1): 227 (2.6), 297 (2.1), 338 (1.3), 371 (1.0), 406 (0.7), 485 (0.4) nm. FTIR (solid, ATR) νmax/cm−1: 2959, 2922, 2853, 1537, 1499, 1447, 1429, 1389, 1260, 1061, 1018, 881, 839, 795, 756, 733, 633, 500.
Synthesis of [Ir(L5)2(bipy)]PF6
Prepared similarly from [(Ir(L5)2(μ-Cl))2] (20.7 mg, 0.013 mmol) and 2,2′-bipyridine (4.6 mg, 0.029 mmol) to give the product as a red solid (9.4 mg, 34%). 1H NMR (500 MHz, CDCl3) δH = 9.10 (s, 2H), 8.58 (d, 3JHH = 8.1 Hz, 2H), 8.17 (app. td, 3JHH = 8.1, 4JHH = 1.6 Hz, 2H), 8.07 (dd, 3JHH = 5.5, 4JHH = 0.9 Hz, 2H), 8.02 (dd, 3JHH = 8.2, 4JHH = 1.4 Hz, 2H), 7.60–7.50 (m, 4H), 7.25 (dd, 3JHH = 5.1 4JHH = 1.1 Hz, 2H), 7.21 (dd, 3JHH = 3.7, 4JHH = 1.1 Hz, 2H), 7.15 (ddd, 3JHH = 8.6, 7.1, 4JHH = 1.5 Hz, 2H), 6.99 (dd, 3JHH = 5.1, 3.7 Hz, 2H), 6.77 (d, 3JHH = 8.1 Hz, 2H), 6.32 (s, 2H) ppm. HRMS (ESI): found m/z 935.0718, calc'd m/z 935.0731 for C42H26N6S4193Ir. UV-vis (MeCN) λmax (ε × 104 L−1 mol−1 cm−1): 233 (2.1), 307 (1.4), 358 (1.3), 438 (0.8), 515 (0.6) nm. FTIR (solid, ATR) νmax/cm−1: 2961, 2849, 1701, 1533, 1499, 1439, 1387, 1260, 1084, 1015, 837, 797, 775, 700, 557, 474.Synthesis of [Ir(L6)2(bipy)]PF6
Prepared similarly from [(Ir(L6)2(μ-Cl))2] (67.0 mg, 0.052 mmol) and 2,2′-bipyridine (18.5 mg, 0.119 mmol) to give the product as a red solid (15.2 mg, 30%). 1H NMR (500 MHz, CDCl3) δH = 9.05 (s, 2H), 8.66 (d, 3JHH = 8.1 Hz, 2H), 8.17 (app. td, 3JHH = 8.0, 4JHH = 1.4 Hz, 2H), 7.92 (dd, 3JHH = 5.3, 4JHH = 0.7 Hz, 2H), 7.75 (s, 1H), 7.57–7.51 (m, 2H), 7.47 (d, 3JHH = 4.7 Hz, 2H), 6.54 (s, 2H), 6.23 (d, 3JHH = 4.7 Hz, 2H), 2.32 (s, 6H), 1.80 (s, 6H) ppm. 13C{1H} NMR (126 MHz, CDCl3) δC = 159.1, 157.1, 156.3, 148.9, 143.8, 141.1, 140.9, 140.5, 139.9, 139.7, 137.8, 134.0, 132.7, 130.2, 128.4, 125.9, 121.7, 20.6, 19.7 ppm. 19F{1H} NMR (376 MHz, CDCl3) δF = −73.03 (d, 1JPF = 712.0 Hz) ppm. HRMS (ESI): found m/z 825.1589, calc'd m/z 825.1579 for C38H30N6S2191Ir. UV-vis (MeCN) λmax (ε × 104 L−1 mol−1 cm−1): 229 (2.9), 298 (2.3), 342 (1.4), 375 (1.1), 406 (0.8), 496 (0.5) nm. FTIR (solid, ATR) νmax/cm−1: 3094, 2959, 2920, 2853, 1703, 1603, 1524, 1495, 1447, 1404, 1339, 1260, 1219, 1092, 1057, 1018, 833, 795, 767, 733, 662, 555, 465, 457, 419.Synthesis of [Ir(L7)2(bipy)]PF6
Prepared similarly from [(Ir(L7)2(μ-Cl))2] (70.0 mg, 0.048 mmol) and 2,2′-bipyridine (17.4 mg, 0.112 mmol) to give the product as a red solid (24.3 mg, 25%). 1H NMR (500 MHz, CD3CN) δH = 9.24 (s, 2H), 8.38 (d, 3JHH = 8.2 Hz, 2H), 8.12 (app. td, 3JHH = 7.9, 4JHH = 1.5 Hz, 2H), 7.96 (dd, 3JHH = 5.5, 4JHH = 0.9 Hz, 2H), 7.90 (3JHF = 10.5, 4JHF = 8.4 Hz, 2H), 7.72 (d, 3JHH = 4.8 Hz, 2H), 7.62 (ddd, 3JHH = 7.6, 5.6, 4JHH = 1.2 Hz, 2H), 6.62 (dd, 3JHF = 12.5, 4JHF = 7.8 Hz, 2H), 6.43 (d, 3JHH = 4.8 Hz, 2H) ppm. 19F{1H} NMR (376 MHz, CDCl3) δF −73.02 (d, 1JPF = 713.2 Hz), −123.59 (d, 3JFF-o = 22.2 Hz), −130.41 (d, 3JFF-o = 22.2 Hz) ppm. 13C NMR (126 MHz, CD3CN) δC = 161.6, 159.9, 156.7, 153.7 (dd, 1JCF = 255.0, 2JCF = 15.4 Hz), 151.3 (dd, 1JCF = 253.4, 2JCF = 15.4 Hz), 150.6, 144.5, 141.4, 140.5 (dd, 3JCF = 10.4, 4JCF = 1.2 Hz), 139.0, 138.8 (dd, 3JCF = 10.5, 4JCF = 1.4 Hz), 137.2, 133.8, 130.2, 126.1, 118.0 (dd, 2JCF = 17.9, 3JCF = 2.1 Hz) 110.5 (d, 2JCF = 22.8 Hz) ppm. HRMS (ESI): found m/z 843.0607, calc'd m/z 843.0600 for C34H18N6F4S2193Ir. UV-vis (MeCN) λmax (ε × 104 L−1 mol−1 cm−1): 297 (2.6), 340 (1.8), 369 (1.4), 404 (1.0), 509 (0.5) nm. FTIR (solid, ATR) νmax/cm−1: 3073, 2920, 2849, 1539, 1506, 1445, 1404, 1246, 1051, 835, 764, 733, 723, 613, 555, 509, 471.Synthesis of [Ir(L8)2(bipy)]PF6
Prepared similarly from [(Ir(L8)2(μ-Cl))2] (48.3 mg, 0.031 mmol) and 2,2′-bipyridine (11.0 mg, 0.070 mmol) to give the product as a red solid (25.2 mg, 78%). 1H NMR (500 MHz, CDCl3) δH = 9.14 (s, 2H), 8.66 (d, 3JHH = 8.2 Hz, 2H), 8.24 (app. td, 3JHH = 8.0, 4JHH = 1.6 Hz, 2H), 8.12 (s, 2H), 7.85 (dd, 3JHH = 5.6, 4JHH = 0.9 Hz, 2H), 7.68–7.64 (m, 2H), 7.65 (d, 3JHH = 4.8 Hz, 2H), 6.85 (s, 2H), 6.28 (d, 3JHH = 4.7 Hz, 2H) ppm. 13C{1H} NMR (126 MHz, CD3CN) δC = 162.2, 161.4, 156.7, 150.8, 145.5, 141.8, 141.6, 140.5, 139.0, 138.2, 136.5, 133.9, 132.8, 132.0, 130.4, 126.1, 124.4 ppm. 19F{1H} NMR (376 MHz, CDCl3) δF −73.11 (d, 1JPF = 713.0 Hz) ppm. HRMS (ESI): found m/z 904.9404, calc'd m/z 904.9394 for C34H18N6S235Cl4191Ir. UV-vis (MeCN) λmax (ε × 104 L−1 mol−1 cm−1): 205 (3.9), 228 (3.8), 303 (2.7), 347 (1.8), 379 (1.4), 416 (0.9), 523 (0.6) nm. FTIR (solid, ATR) νmax/cm−1: 3084, 2918, 2849, 1701, 1599, 1566, 1522, 1477, 1443, 1396, 1184, 1152, 1115, 1065, 833, 766, 691, 650, 555, 430, 420.Synthesis of [Ir(L9)2(bipy)]PF6
Prepared similarly from [(Ir(L9)2(μ-Cl))2] (77.4 mg, 0.052 mmol) and 2,2′-bipyridine (17.7 mg, 0.114 mmol) to give the product as a dark red solid (19.1 mg, 18%). 1H NMR (500 MHz, CD3CN) δH = 9.29 (s, 2H), 8.60 (s, 2H), 8.25 (d, 3JHH = 8.0 Hz, 2H), 8.18 (d, 3JHH = 4.7 Hz, 2H), 8.11 (app. t, 3JHH = 7.2 Hz, 2H), 8.04 (d, 3JHH = 8.4 Hz, 2H), 7.76–7.70 (m, 2H), 7.69 (d, 3JHH = 4.7 Hz, 2H), 7.56–7.46 (m, 2H), 7.46–7.38 (m, 2H), 7.37 (s, 2H), 6.96 (d, 3JHH = 8.4 Hz, 2H), 6.50 (d, 3JHH = 4.7 Hz, 2H) ppm. 13C{1H} NMR (126 MHz, CD3CN) δC = 162.5, 162.0, 156.7, 151.1, 145.1, 141.3, 139.9, 139.2, 138.5, 137.7, 135.0, 134.2, 133.2, 130.5, 130.1, 129.3, 129.1, 128.2, 128.0, 125.6, 121.0 ppm. 19F{1H} NMR (376 MHz, CD3CN) δF = −72.97 (d, 1JPF = 706.1 Hz) ppm. HRMS (ESI): found m/z 871.1296, calc'd m/z 871.1290 for C42H26N6S2193Ir. UV-vis (MeCN) λmax (ε × 104 L−1 mol−1 cm−1): 244 (4.7), 312 (2.9), 327 (3.3), 397 (1.4), 420 (1.3), 545 (0.6) nm. FTIR (solid, ATR) νmax/cm−1: 3096, 3055, 2916, 2849, 1705, 1601, 1557, 1479, 1456, 1443, 1427, 1375, 1344, 1323, 1314, 1184, 1163, 1152, 1138, 1119, 1053, 833, 766, 743, 733, 660, 555, 469, 444, 420.Synthesis of [Ir(L10)2(bipy)]PF6
Prepared similarly from [(Ir(L10)2(μ-Cl))2] (74.2 mg, 0.050 mmol) and 2,2′-bipyridine (19.0 mg, 0.105 mmol) to give the product as a dark red solid (55.8 mg, 57%). 1H NMR (500 MHz, CDCl3) δH = 8.55 (d, 3JHH = 7.6 Hz, 2H), 8.20 (app. t, 3JHH = 7.4 Hz, 2H), 8.16 (d, 3JHH = 5.2 Hz, 2H), 7.98 (dd, 3JHH = 8.3, 4JHH = 1.1 Hz, 2H), 7.72 (dd, 3JHH = 5.1, 4JHH = 1.1 Hz, 2H), 7.70–7.64 (m, 2H), 7.62 (dd, 3JHH = 3.6, 4JHH = 1.1 Hz, 2H), 7.54 (ddd, 3JHH = 8.1, 7.0, 4JHH = 1.0 Hz, 2H), 7.31 (dd, 3JHH = 5.1, 3.6 Hz, 2H), 7.30 (d, 3JHH = 4.9 Hz, 2H), 7.14 (ddd, 3JHH = 8.4, 7.0, 4JHH = 1.3 Hz, 2H), 6.97 (d, 3JHH = 8.5 Hz, 2H), 6.20 (d, 3JHH = 4.9 Hz, 2H) ppm. 13C{1H} NMR (126 MHz, CDCl3) δC = 160.7, 160.3, 156.1, 148.2, 146.7, 141.3, 141.2, 139.7, 138.5, 137.4, 135.8, 132.5, 132.3, 131.0, 130.9, 129.7, 129.5, 128.6, 127.7, 125.7, 122.2 ppm. 19F{1H} NMR (376 MHz, CDCl3) δF = −73.05 (d, 1JPF = 712.6 Hz) ppm. HRMS (ESI): found m/z 933.0696, calc'd m/z 933.0708 for C42H26N6S4191Ir. UV-vis (MeCN) λmax (ε × 104 L−1 mol−1 cm−1): 238 (7.4), 303 (5.8), 355 (3.5), 425 (2.1), 507 (1.1) nm. FTIR (solid, ATR) νmax/cm−1: 3094, 3061, 2918, 1603, 1506, 1483, 1472, 1441, 1429, 1416, 1300, 1323, 1231, 1144, 1130, 1076, 1051, 1032, 833, 762, 721, 637, 584, 555, 498, 457, 426, 420, 417.Synthesis of [Ir(L11)2(bipy)]PF6
The complex was prepared via an adapted method, where 2-ethoxyethanol (20 mL) was used as the solvent. [(Ir(L11)2(μ-Cl))2] (44.0 mg, 0.034 mmol) and 2,2′-bipyridine (11.2 mg, 0.071 mmol) were used, to give the product as a red solid (13.0 mg, 21%). 1H NMR (500 MHz, (CD3)2CO) δH = 9.60 (s, 2H), 8.73 (d, 3JHH = 8.2 Hz, 2H), 8.32 (dd, 3JHH = 8.0, 4JHH = 1.5 Hz, 2H), 8.21 (ddd, 3JHH = 5.7, 4JHH = 1.5, 0.5 Hz, 2H), 8.04 (dd, 3JHH = 8.3, 4JHH = 1.4 Hz, 2H), 8.03 (d, 3JHH = 5.2 Hz, 2H), 7.84 (m, 2H), 7.65 (m, 2H), 7.49 (d, 3JHH = 5.2 Hz, 2H), 7.31–7.15 (m, 2H), 7.04 (dd, 3JHH = 8.9, 4JHH = 0.9 Hz, 2H) ppm. 19F{1H} NMR (376 MHz, (CD3)2CO) δF −72.52 (d, 1JPF = 707.4 Hz). HRMS (ESI) [M – PF6]+: found m/z 771.0980, calc'd m/z 771.0977 for C34H22N6S2Ir. UV-vis (MeCN) λmax (ε × 104 L−1 mol−1 cm−1): 205 (3.3), 239 (3.2), 284 (2.0), 307 (1.8), 348 (1.1), 475 (0.4) nm. FTIR (solid, ATR) νmax/cm−1: 3353, 1605, 1534, 1507, 1471, 1447, 1428, 1383, 1314, 1295, 1208, 1070, 1023, 980, 914, 833, 756, 729, 630, 621, 555.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Cardiff University (Knowledge Economy Skills Scholarship via the Welsh Government's European Social Fund, to SAF) and STG Aerospace are thanked for financial support. We thank the staff of the Engineering and Physical Sciences Research Council (EPSRC) UK National Crystallographic Service at the University of Southampton. We thank Diamond Light Source for an award of beamtime on I19 (CY31778).References
- Recent examples include: P.-N. Lai, S. Yoon, Y. Wu and T. S. Teets, ACS Org. Inorg. Au, 2022, 2, 236 CrossRef CAS PubMed; S. Chen, H. Bi, W. Tian and Y. Liu, Molecules, 2022, 27, 286 CrossRef PubMed; P. Mandapati, J. D. Braun, I. B. Lozada, J. A. G. Williams and D. E. Herbert, Inorg. Chem., 2020, 59, 12504 CrossRef PubMed; P. Herr, A. Schwab, S. Kupfer and O. S. Wenger, ChemPhotoChem, 2022, e202200052 CrossRef; T Yu, D. P-K. Tsang, V. K.-M. Au, W. H. Lam, M.-Y. Chan and V. W.-W. Yam, Chem. – Eur. J., 2013, 19, 13418 CrossRef PubMed; E. Kabir, Y. Wu, S. Sittel, B.-L. Nguyen and T. S. Teets, Inorg. Chem. Front., 2020, 7, 1362 RSC; Y. Zhang, H.-C. Kao, C. Shi, C. Wu, M. Zhu, K. Li, C.-C. Wu and C. Yang, Chem. – Eur. J., 2022, 28, e202103543 CrossRef PubMed; C. E. Elgar, H. Y. Otaif, J. M. Beames, P. N. Horton, S. J. Coles, A. J. Hallett, S. P. O'Kell and S. J. A. Pope, Eur. J. Inorg. Chem., 2023, e202300102 CrossRef.
- J. Shum, P. K.-K. Leung and K. K.-W. Lo, Inorg. Chem., 2019, 58, 2231 CrossRef CAS PubMed; Y. Zhang and J. Qiao, iScience, 2021, 24, 102858 CrossRef PubMed; K. L. Smitten, P. A. Scattergood, C. Kiker, J. A. Thomas and P. I. P. Elliott, Chem. Sci., 2020, 11, 8928 RSC; O. J. Stacey and S. J. A. Pope, RSC Adv., 2013, 3, 25550 RSC; C. Caporale and M. Massi, Coord. Chem. Rev., 2018, 363, 71 CrossRef.
- For example: E. E. Langdon-Jones, A. J. Hallett, J. D. Routledge, D. A. Crole, B. D. Ward, J. A. Platts and S. J. A. Pope, Inorg. Chem., 2013, 52, 448 CrossRef CAS PubMed; S. A. Fitzgerald, X. Xiao, J. Zhao, P. N. Horton, S. J. Coles, R. C. Knighton, B. D. Ward and S. J. A. Pope, Chem. – Eur. J., 2023, 29, e202203241 CrossRef PubMed.
- F.-M. Hwang, H.-Y. Chen, P.-S. Chen, C.-S. Liu, Y. Chi, C.-F. Shu, F.-I. Wu, P.-T. Chou, S.-M. Peng and G.-H. Lee, Inorg. Chem., 2005, 44, 1344 CrossRef CAS PubMed.
- C. E. Elgar, H. Y. Otaif, X. Zhang, J. Zhao, P. N. Horton, S. J. Coles, J. M. Beames and S. J. A. Pope, Chem. – Eur. J., 2021, 27, 3427 CrossRef CAS PubMed; K. A. Phillips, T. M. Stonelake, K. Chen, Y. Hou, J. Zhao, S. J. Coles, P. N. Horton, S. J. Keane, E. C. Stokes, A. J. Hallett, S. P. O'Kell, J. M. Beames and S. J. A. Pope, Chem. – Eur. J., 2018, 24, 8577 CrossRef PubMed.
- A. H. Day, M. H. Ubler, H. L. Best, E. Lloyd-Evans, R. J. Mart, I. A. Fallis, R. K. Allemann, E. A. H. Al-Watter, N. I. Keymer, N. J. Buurma and S. J. A. Pope, Chem. Sci., 2020, 11, 1599 RSC.
- G. Angulo, J. Dobkowski, A. Kapturkiewicz and K. MacIolek, J. Photochem. Photobiol., A, 2010, 213, 101 CrossRef CAS.
- S. S. Mahadik, S. Chacko and R. M. Kamble, ChemistrySelect, 2019, 4, 10021 CrossRef CAS.
- T. N. Moshkina, E. V. Nosova, A. E. Kopotilova, B. Ośmiałowski, A. I. Reguant, P. A. Slepukhin, G. N. Lipunova, O. S. Taniya, A. A. Kalinichev and V. N. Charushin, Dyes Pigm., 2022, 110434 CrossRef CAS.
- T. J. Giordano and P. G. Rasmussen, Inorg. Chem., 1975, 14, 1628 CrossRef CAS.
- M. Nonoyama, Bull. Chem. Soc. Jpn., 1979, 52, 3749 CrossRef CAS.
- S. Lamansky, P. Djurovich, D. Murphy, F. Abdel-Razzaq, H.-E. Lee, C. Adachi, P. E. Burrows, S. R. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2001, 123, 4304 CrossRef CAS PubMed.
- S. Ladouceur and E. Zysman-Colman, Eur. J. Inorg. Chem., 2013, 2985 CrossRef CAS.
- M. L. Xu, G. Y. Wang, R. Zhou, Z. W. An, Q. Zhou and W. L. Li, Inorg. Chim. Acta, 2007, 360, 3149 CrossRef CAS.
- P.-N. Lai, C. H. Brysacz, K. Alam, N. A. Ayoub, T. G. Gray, J. Bao and T. S. Teets, J. Am. Chem. Soc., 2018, 140, 10198 CrossRef CAS PubMed.
- P. N. Lai, S. Yoon and T. S. Teets, Chem. Commun., 2020, 56, 8754 RSC.
- A. Liang, M. Luo, Y. Liu, H. Wang, Z. Wang, X. Zheng, T. Cao, D. Liu, Y. Zhang and F. Huang, Dyes Pigm., 2018, 159, 637 CrossRef CAS; W. Cho, G. Sarada, H. Lee, M. Song, Y. S. Gal, Y. Lee and S. H. Jin, Dyes Pigm., 2017, 136, 390 CrossRef.
- S. C. Yiu, P. Y. Ho, Y. Y. Kwok, X. He, Y. Wang, W. H. Yu, C. L. Ho and S. Huang, Chem. – Eur. J., 2022, e202104575 CrossRef CAS PubMed; S. Y. Takizawa, S. Katoh, A. Okazawa, N. Ikuta, S. Matsushima, F. Zeng and S. Murata, Inorg. Chem., 2021, 60, 4891 CrossRef PubMed.
- M. Bandini, M. Bianchi, G. Valenti, F. Piccinelli, F. Paolucci, M. Monari, A. Umani-Ronchi and M. Marcaccio, Inorg. Chem., 2010, 49, 1439 CrossRef CAS PubMed.
- Y. Park, G. S. Lee, H.-R. Choi, Y. Jeon, S. Y. Jeong, B. Noh, K.-C. Park, Y.-H. Kim and K.-C. Choi, Adv. Photonics Res., 2021, 2, 2100121 CrossRef CAS.
- C. H. Fan, P. Sun, T. H. Su and C. H. Cheng, Adv. Mater., 2011, 23, 2981 CrossRef CAS PubMed; Q. Mei, J. Weng, Z. Xu, B. Tong, Q. Hua, Y. Shi, J. Song and W. Huang, RSC Adv., 2015, 5, 97841 RSC; G. Szafraniec-Gorol, A. Słodek, M. Filapek, B. Boharewicz, A. Iwan, M. Jaworska, L. Zur, M. Sołtys, J. Pisarska, I. Grudzka-Flak, S. Czajkowska, M. Sojka, W. Danikiewicz and S. Krompiec, Mater. Chem. Phys., 2015, 162, 498 CrossRef.
- A. K. Pal, D. B. Cordes, A. M. Z. Slawin, C. Momblona, E. Ortĺ, I. D. W. Samuel, H. J. Bolink and E. Zysman-Colman, Inorg. Chem., 2016, 55, 10361 CrossRef CAS PubMed.
- A. M. Bünzli, H. J. Bolink, E. C. Constable, C. E. Housecroft, J. M. Junquera-Hernández, M. Neuburger, E. Ortí, A. Pertegás, J. J. Serrano-Pérez, D. Tordera and J. A. Zampese, Dalton Trans., 2013, 43, 738 RSC.
- N. Castillo, C. F. Matta and R. J. Boyd, J. Chem. Inf. Model., 2005, 45, 354 CrossRef CAS PubMed.
- F. J. Weigert and J. D. Roberts, J. Am. Chem. Soc., 1971, 93, 2361 CrossRef.
- For example: M. Bandini, M. Bianchi, G. Valenti, F. Piccinelli, F. Paolucci, M. Monari, A. Umani-Ronchi and M. Marcaccio, Inorg. Chem., 2010, 49, 1439 CrossRef CAS PubMed.
- T. M. Stonelake, K. A. Phillips, H. Y. Otaif, Z. C. Edwardson, P. N. Horton, S. J. Coles, J. M. Beames and S. J. A. Pope, Inorg. Chem., 2020, 59, 2266 CrossRef CAS PubMed.
- R. E. Atkinson and P. R. H. Speakman, J. Chem. Soc. B, 1971, 2077 RSC.
- J. Moussa, T. Cheminel, G. R. Freeman, L. M. Chamoreau, J. A. G. Williams and H. Amouri, Dalton Trans., 2014, 43, 8162 RSC.
- S. A. Fitzgerald, H. Y. Otaif, C. E. Elgar, N. Sawicka, P. N. Horton, S. J. Coles, J. M. Beames and S. J. A. Pope, Inorg. Chem., 2021, 60, 15467 CrossRef CAS PubMed.
- W. H. Melhuish, J. Phys. Chem., 1961, 65, 229 CrossRef CAS.
- H.-Y. Chen, C.-H. Yang, Y. Chi, Y.-M. Cheng, Y.-S. Yeh, P.-T. Chou, H.-Y. Hsieh, C.-S. Liu, S.-M. Peng and G.-H. Lee, Can. J. Chem., 2006, 84, 309 CrossRef CAS; C. Wang, L. Lystrom, H. Yin, M. Hetu, S. Kilina, S. A. McFarland and W. Sun, Dalton Trans., 2016, 45, 1636 Search PubMed; K. A. Phillips, T. M. Stonelake, P. N. Horton, S. J. Coles, A. J. Hallett, S. P. O'Kell, J. M. Beames and S. J. A. Pope, J. Organomet. Chem., 2019, 893, 11 CrossRef.
- C. E. Housecroft and E. C. Constable, Coord. Chem. Rev., 2017, 350, 155 CrossRef CAS.
- K. Suzuki, A. Kobayashi, S. Kaneko, K. Takehira, T. Yoshihara, H. Ishida, Y. Shiina, S. Oishi and S. Tobita, Phys. Chem. Chem. Phys., 2009, 11, 9850 RSC.
- A. Tsuboyama, H. Iwawaki, M. Furugori, T. Mukaide, J. Kamatani, S. Igawa, T. Moriyama, S. Miura, T. Takiguchi, S. Okada, M. Hoshino and K. Ueno, J. Am. Chem. Soc., 2003, 125, 12971 CrossRef CAS PubMed; Y. Sun, X. Yang, Z. Feng, B. Liu, D. Zhong, J. Zhang, G. Zhou and Z. Wu, ACS Appl. Mater. Interfaces, 2019, 11, 26152 CrossRef PubMed.
- M. Ye, Y. Wen, H. Li, Y. Fu and Q. Wang, Tetrahedron Lett., 2016, 57, 4983 CrossRef CAS.
- K. B. Harsha and K. S. Rangappa, RSC Adv., 2016, 6, 57154 RSC; K. B. Harsha, S. Rangappa, H. D. Preetham, T. R. Swaroop, M. Gilandoust, K. S. Rakesh and K. S. Rangappa, ChemistrySelect, 2018, 3, 5228 CrossRef CAS.
- S. Uçar and A. Daştan, J. Org. Chem., 2020, 85, 15502 CrossRef PubMed.
- F. Amaya-García, M. Caldera, A. Koren, S. Kubicek, J. Menche and M. M. Unterlass, ChemSusChem, 2021, 14, 1853 CrossRef PubMed.
- Y. Yang, F. Ni, W. M. Shu and A. X. Wu, Chem. – Eur. J., 2014, 20, 11776 CrossRef CAS PubMed.
- S. J. Coles and P. A. Gale, Chem. Sci., 2012, 3, 683 RSC.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 27, 3 Search PubMed.
- M. Nonoyama, Bull. Chem. Soc. Jpn., 1974, 47, 767 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures for ligand precursors, and additional spectra. CCDC 2270408 and 2270409. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt02193a |
| This journal is © The Royal Society of Chemistry 2023 |