
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Surface functionality of sub- to full-monolayer organic coverage of water aerosols determined by molecular dynamics simulations†
Aisling C.
Stewart‡
 ,
Martin J.
Paterson
and
Stuart J.
Greaves
*
,
Martin J.
Paterson
and
Stuart J.
Greaves
*
Institute of Chemical Sciences, Heriot-Watt University, Edinburgh EH14 4AS, UK. E-mail: S.J.Greaves@hw.ac.uk
First published on 17th July 2023
Abstract
The role that organic aqueous aerosols play within the atmosphere is highly dependent on their structures. This is influenced not only by the components that make up the organic layer of the aerosol, but also on the way that they pack together, as this affects the accessibility of key functional groups to incoming gaseous species. This is in turn impacted by the total coverage of organic molecules on the aerosol core. Here we present the results of a molecular dynamics study into how the surface coverage of three C18 fatty acids, stearic, oleic and linoleic acid, affects the structure of the resulting aerosol. Surface coverages ranging from significantly submonolayer to above monolayer coverages have been investigated. Acid chains are found to pack more tightly at higher coverages, and to be directed more along the surface normal. The surfaces are dominated by CH3 and CH2 groups at all coverages, however, changes in surface presence of HC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH and COOH can be used as a marker of when the monolayer coverage has been exceeded. The organic layers have been grown in a random and stepwise manner designed to mimic natural growth of aerosols in the atmosphere. Analysis focusses on the accessibility of different groups to an incoming ozone molecule and thus gives a measure of how the reactivity of aerosols may be affected by the organic surface coverage.
CH and COOH can be used as a marker of when the monolayer coverage has been exceeded. The organic layers have been grown in a random and stepwise manner designed to mimic natural growth of aerosols in the atmosphere. Analysis focusses on the accessibility of different groups to an incoming ozone molecule and thus gives a measure of how the reactivity of aerosols may be affected by the organic surface coverage.
Environmental significanceAerosols are key players in atmospheric processes including pollution cycles and radiative forcing. However, there are still many gaps in our understanding of their reactivities, due to the complexity of their structures. The structure of organic aqueous aerosol depends not only on the components that make up the organic layer but also on the combined surface coverage of these, as this influences the nature of their packing. Here we present a molecular dynamics study of different surface coverages of oleic acid, which is commonly used as an aerosol mimic, and two other C18 fatty acids, on water. The effect of surface coverage on the structure and functional group accessibility to incoming reactive species is discussed. |
Introduction
The significance of aerosols within the earth's atmosphere cannot be overstated; their roles include acting as catalysts, sources of pollution and key players in radiative forcing, to name a few.1–8 The origins and compositions of aerosols are as varied as their influences, with the task of determining their structures and the exact parts that they will play in atmospheric cycles one of the most complex problems that modern day atmospheric chemists face.1,9,10 Aerosols can contain components that are natural or anthropogenic and can be formed over marine or inland environments, with the exact environment of their formation playing a big role in determining their overall compositions. In addition to this, the make-up of an aerosol within the atmosphere is dynamic.11–17 When species in the atmosphere come into contact with an aerosol particle, they may either condense onto the surface of it or react with groups present on it, altering the chemical nature of these. Of particular interest are double bonds present on the surface of aerosols, as these are highly reactive towards atmospheric radicals, such as O3, OH and NO3.6,12,16,18–25 The oxidation of groups on the surface of aerosols alters their properties, both as participants in further chemical reactions and as cloud condensation nuclei (CCN).A commonly investigated type of aerosol consists of an aqueous core surrounded by a layer of organic matter.26 This organic matter can contain a variety of species, including alcohols, carboxylic acids and aromatic species.27–29 A class of molecules that have drawn particular attention are fatty acids. A variety of mono- and dicarboxylic acids of different chain lengths and degrees of unsaturation have been found within aerosols.27,28,30,31 One of the most well studied of these is oleic acid (cis-9-octadecenoic acid), which has often been used in laboratory studies as a mimic for aerosols, both on its own or with an aqueous core.9,19,20,32–34 The presence of the CC bond within oleic acid is of particular interest, as it is highly reactive towards ozone, and a range of studies have been carried out to investigate the kinetics and products of this reaction.17–20,35–37
The accessibility of double bonds at the aerosol–atmosphere interface and therefore the rate of reaction of this with atmospheric radicals is, however, highly dependent on the coverage and packing of organic molecules on the core. This coverage and packing can be affected by a number of factors including the concentrations of species in the atmosphere, humidity, pressure and factors such as salt concentration and pH of the aqueous core.38–42 Under different surface coverages, the aerosol may have different properties, for example in terms of its reactivity towards incoming species and in CCN activity.43–45 In order to be able to accurately model the behaviour of aerosols in different circumstances it is therefore critical to understand how the coverage of organic molecules at the water interface influences the presence of groups at the aerosol–atmosphere interface and the overall structure of the aerosol.
Field and laboratory studies of aerosols can be complimented by the use of computational methods, in particular molecular dynamics (MD) studies, which offer the chance to model systems with large numbers of atoms accurately, but in a manner that is not computationally prohibitive, as is the case for more quantum mechanical based treatments. A number of studies have probed systems with a water core and an organic outer layer, focusing in particular on the overall structure of these, and their ability to uptake water vapour and other gases and to coalesce to form larger particles.36–53 These studies often choose to look at a what is believed to be a monolayer of organic molecules on a water core, and to study the properties of such idealised aerosols in these saturated states.45,46,52,53 Monolayer coverage is defined, based on the work by Langmuir,54 as the point at which there is a layer of insoluble material exactly one molecule thick adsorbed onto the surface of the liquid or solid of interest and no more molecules can be accommodated in this single layer. However, in practice, it is not always obvious how many molecules to select when aiming for a monolayer, as this can depend on assumptions about the positions and packing densities at the water–organic interface, with studies often pre-defining the positions at which the organic molecules are expected to stick to the water core. An alternative approach has been to use the same number of organic molecules on different sizes of water surface to provide different monolayer compressions.25,55 Fatty acids are known to be able to cover a water surface without ordered packing at low concentrations/compressions, and will continue to provide ‘monolayer’ coverage as the concentration/compression increases until the point of monolayer collapse where the organic molecules start to form a bilayer.38 Because of this variation of what can be called a monolayer, for this work we define a full monolayer as: the point where addition of further molecules to a monolayer does not result in additional bonding with the water interface, and thus concentrations above this correspond to the start of formation of a bilayer.
Aerosols in the atmosphere are present with a range of surface coverages of organic material, depending on their age and the concentrations of water vapour and other gaseous species which they could potentially absorb. Some aging processes involving aerosols can also convert insoluble organic matter on the surface of an aerosol to species that may either dissolve within the bulk of the aerosol or be volatile and escape into the gaseous phase, again potentially changing the surface coverages of the organic matter. In the case of oleic acid, previous studies have noted that the ozonolysis of oleic acid on water produces, amongst other things, volatile nonanal and the highly soluble dicarboxylic acid, azelaic acid, thus in the absence of other species there is a net reduction in organic matter at the interface as the ozonolysis proceeds.32
It is known from the studies of self-assembled monolayers (SAMs) that the orientation and structure of molecules upon a substrate can vary significantly with surface coverage and molecule length.56–58 A logical extension is to examine whether this may be the case for aerosol systems, as well as to investigate how this change in orientation affects the groups that are present at the aerosol–atmosphere interface. The interface is known to be highly chemically active and key to CCN processes. Here we present to our knowledge the first study into how the fractional presence of different groups at the surface of an aerosol changes for different surface coverages and for different levels of unsaturation of fatty acid covering of an aerosol.
Most MD studies of carboxylic acid-on-water aerosols choose to focus on spherical water droplets surrounded by a set number of acid molecules.46–50,53 However, in practice, these simulated water droplets often tend to be significantly smaller than would be found in the majority of real-world applications, as the sizes of aerosols can range to multiple μm in diameter,1 which would involve huge computational costs. In contrast, the studies of SAMs often employ periodic boundary conditions, in order to expand a slab of given dimensions infinitely in the x and y directions (parallel to the surface).56,57 This allows for the mimicking of a much larger system as we discuss in ESI Section S3.†
In the present work we choose to look at the structures of different coverages of fatty acids on a water surface. We focus on samples of three different C18 fatty acids: stearic, oleic and linoleic acid (see Fig. 1). These are allowed to attach to a water sample that is infinite in the x and y directions, due to periodic boundary conditions, but which has an area of vacuum above and below it. The slabs are created by adding acid molecules in a random location and orientation one at a time to allow the overlayer to ‘grow’ in as natural a way as possible. For each of these acid-on-water ‘slabs’, nine different quantities of acid molecules, leading to nine different water surface coverages, are used. The resulting structures of these samples are investigated, to see how the conformations and positionings of the acids change with increasing surface concentration. This allows us to investigate how many acid molecules need to be added to a water sample of these dimensions in order to obtain full monolayer coverage. We focus in particular on the proportions of functional groups that would be accessible to an incoming reactive species at each of these coverages as this provides a method of investigating the likeliness of reactivity of aerosols of different coverages to incoming ozone molecules.59–62
 | ||
| Fig. 1 The structures of the three C18 fatty acids investigated this study. | ||
Methods
All simulations described in this paper were carried out using the GROMACS software package (version 2020.2)63,64 and run on the Cirrus EPCC cluster at Edinburgh University. The fatty acid molecules (stearic, oleic and linoleic acid, see Fig. 1) were described using the OPLS-AA force field,65–67 with parameters being obtained via the LigParGen online server.68–70 The water molecules were simulated using the extended simple point charge (SPC/E) model71 and constrained using the SETTLE algorithm.72 The choice of force field is discussed in our previous paper.73 The SPC/E water model was selected as it is known to accurately model surface properties of water, for example the surface tension, as discussed by Lovrić et al.74The procedure for generating the aerosol mimic was similar to that described in our previous work,73 with a few modifications. A box of dimensions 6.5 nm × 6.5 nm × 6.5 nm was filled with water molecules and equilibrated at 298.15 K before being expanded in the z direction to a total depth of 50 nm. This led to areas of vacuum forming above and below the water slab, to which acid molecules could be added. The total number of water molecules in each of the samples was 8925. Acid molecules were added one at a time at random locations and with random orientations within the area of vacuum both above and below the water slab. After the addition of each of the acid molecules, energy minimisation was carried out, followed by a short constant volume (NVT) equilibration step (50 ps), before a further molecule was added, up to a total of 70–150 acid molecules per interface (corresponding to 1.7–3.6 molecules per nm2 or 58–28 Å2 per molecule). Energy minimisation was carried out using the steepest-descent algorithm and was continued until the total energy of the simulation had converged. The NVT equilibration step employed the leap-frog algorithm,75 with a time step of 0.5 fs and a total length of 50 ps. Each sample contained water and a single acid species, and for each acid nine different quantities of acid were used – ranging from 70 to 150 molecules per interface in 10 molecule steps. The same number of acid molecules were added to the regions above and below the slabs, and a restraining potential was applied to the samples throughout all energy minimisation and NVT steps to ensure that there was no migration of molecules from the upper to the lower faces of the slab and vice versa. This ensured that the number of molecules on each face of the slab was known exactly. The restraining potential was a flat-bottomed potential, with no force exerted on molecules that were within the central 14 nm of the box (z = 0 ± 7 nm), and a harmonic potential with a force constant of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 kJ mol−1 nm−2 outside of this. This ensured that molecules cannot escape from the periodic box and re-enter it on the other side of the slab, thus giving exact control over how many acid molecules are present on each side of the slab, whilst not impacting on the energies of any of the molecules attached to the slab in the centre of the box.
000 kJ mol−1 nm−2 outside of this. This ensured that molecules cannot escape from the periodic box and re-enter it on the other side of the slab, thus giving exact control over how many acid molecules are present on each side of the slab, whilst not impacting on the energies of any of the molecules attached to the slab in the centre of the box.
After the designated number of acid molecules had been added to the simulation, a further NVT equilibration step (10 ns) was carried out, followed by a 10 ns production run. Both were run using a velocity Verlet algorithm76 and a time step of 0.5 fs. All the analysis described in this paper was carried out on the last 6 ns of these 10 ns production runs. All calculations were carried out at 298.15 K and employed both 3D periodic boundary conditions and particle-mesh Ewald long-range electrostatics. Further details about the simulation procedure can be found in Sections S1 and S2 of the ESI.† The use of periodic boundary conditions allowed for the slab to be used as a mimic for larger aerosol particles (>210 nm), as is discussed in the ESI Section S3.†
Results and discussion
During the simulation process acid molecules were added one by one to the simulation box and given a short period of time to equilibrate, before further molecules were added. In addition to this, both the position and orientation in which the acid molecules were added to the box were selected at random. Each of these criteria were chosen to mimic as closely as possible the growth of an aerosol in real world situations, as opposed to many previous studies in which all organic molecules were added simultaneously and/or at pre-determined locations and orientations.45,46,53,61 This makes prior assumptions about the way in which the molecules will attach to the aerosol and potentially biases the simulations towards a particular configuration.For each species and number concentration of acid studied, four separate samples were created, each one starting from a bare water sample and adding acid molecules at different random positions. This allowed for an investigation of whether samples of different starting points each achieved the same configurations and distributions of functional groups post-equilibration. The use of a slab of infinite dimensions in the x and y directions is a valid mimic of the surface of an aerosol, as these are generally large enough that their curvature does not have a significant effect on their properties on a molecular level (see discussion in our previous paper73 and Section S3 of the ESI†). The temperature used in this study was such that none of the acid molecules evaporated and fully escaped from the surface of the slabs over the course of the 10 ns production runs.
On addition of the acid molecules to the simulation box, they rapidly find the water surface and the carboxylic acid groups form hydrogen bonds to the water slab, with this taking place within the 50 ps equilibration stage that occurs between the addition of consecutive molecules. Snapshots of the final positions of the molecules at the end of some of the production runs are shown in Fig. 2. This shows a top-down view of the final configuration (last frame of the 10 ns production) for the two extreme coverages for each of the acid samples, presented using the Visual Molecular Dynamics (VMD) software.77 The acid molecules are shown in red and the water molecules in cyan. The water molecules are only visible for the slabs with lower numbers of acid molecules, showing that the monolayer is at least fully formed for each species by the time 150 molecules per interface has been reached. It can also be seen that for the partially covered slabs the acid molecules tend to clump together, presumably in order to increase chain–chain interactions between them. An extended version of this figure can be found in Section S4 of the ESI,† showing top-down VMD images for all coverages for all of the acids studied. Analogous images colour-coded by functional group can be found in ESI S5.†
 | ||
| Fig. 2 Top-down views of selected slabs for two different concentrations of each of the acids studied. Acid molecules are shown in red and water molecules in cyan. These images represent the final frame of a 10 ns trajectory, carried out at 298.15 K and under constant volume conditions (see the main text for details). The number of molecules per interface is given in white above the image. | ||
Fig. 3 shows side-on views of the upper parts of the same slabs, this time with the atoms colour-coded based on the functional groups to which they belong. Extended images of the full slabs are shown in the ESI (Section S6).† The water core is shown in white. Within the acid molecules the carboxylic acid groups are shown in blue, the methyl groups in red, the CH2 groups in green and the HCCH groups in black. From these side-on images, the preferential orientation of the chains with relation to the water core can clearly be seen – there is a marked excess of carboxylic acid groups close to the water and of methyl groups extending out into the vacuum. It can also be seen that for the stearic slabs there is a high degree of ordering of molecules with respect to each other, with chains almost fully extended and the angles that each molecule makes to the surface normal appearing similar between molecules. Such angles have been observed experimentally using X-ray diffraction39 and similar effects have been observed in MD simulations of methylamine on water.78
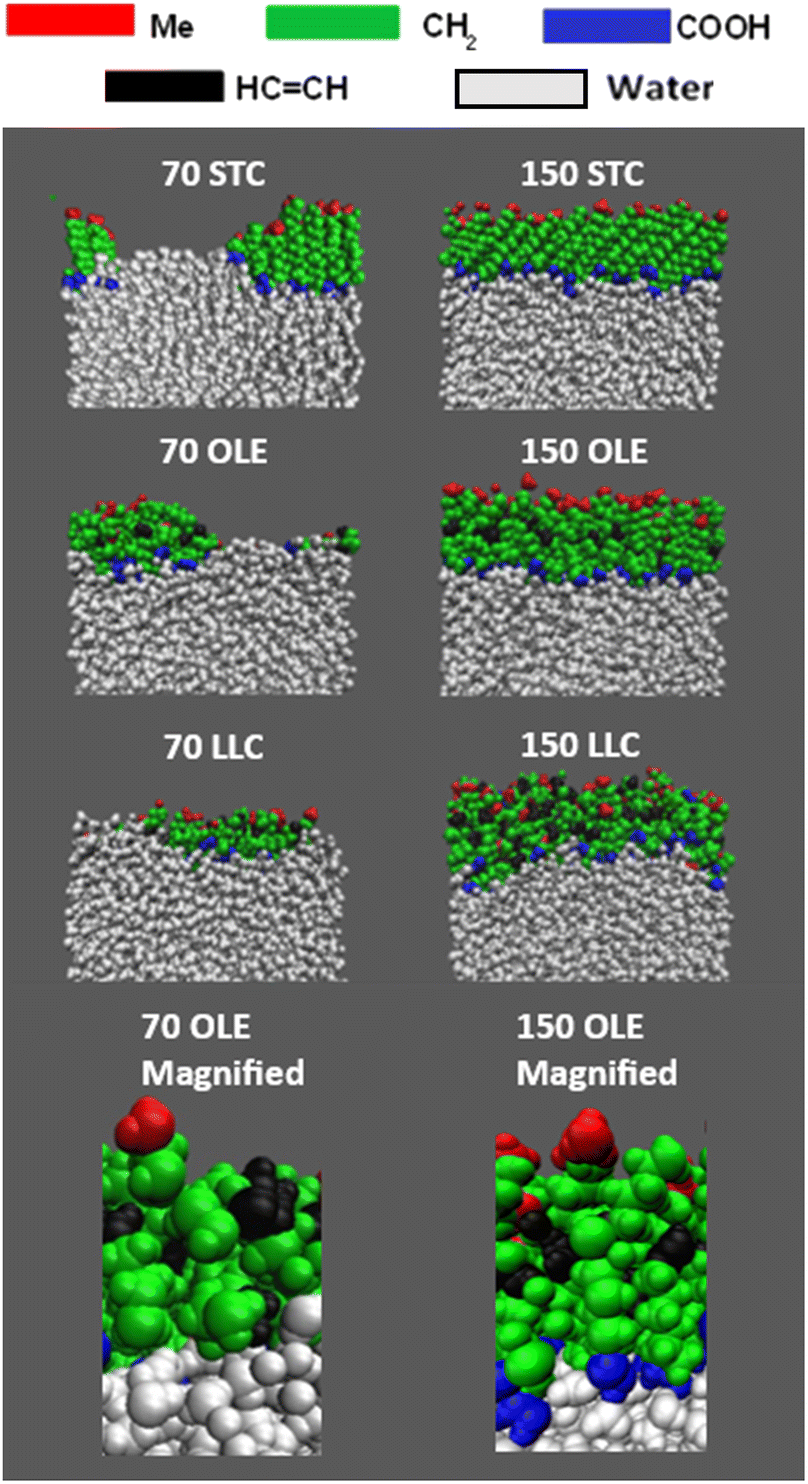 | ||
| Fig. 3 Side-on views of the upper sections of the slabs shown in Fig. 2, with the functional groups for each acid colour-coded, as per the key above the slab images. It can be seen that there is a much greater preference for COOH groups (blue) to be positioned close to the water molecules (white), with methyl groups (red) on the outermost parts of the samples. The bottom panels show zoomed in sections of the oleic acid slabs highlighting the atomic details provided by the all-atom forcefield. | ||
This ‘tilt angle’ distribution can be quantified using the gmx gangle function of GROMACS, as in Fig. 4. This figure displays the angles that long axes of the acid molecules make with the z axis of the simulation box, which is normal to the plane of the slab surface. The long axis of the acid molecules is defined by a vector running from C1 (the carbonyl carbon) to C18 (the methyl carbon) and the plots show the angles these vectors make with the z axis, binned into one degree bins. Fig. 4a–c show the full distributions of the molecules across the entire tilt angle range for each species studied. The population of each angular bin in these distributions has been divided by sin(θ) to take into account solid angle effects (discussed in Section S3 of the ESI† of our previous paper73). The main peaks of these distributions can be fitted with a Gaussian function, in order to obtain a measure of the most populated angle and the spread of the angular data. Fig. 4d shows the results of this fitting, giving the peak position of the Gaussian for each coverage. The corresponding standard deviations are shown in Section S7 of the ESI.† For all the plots, points shown are an average over the upper and lower interfaces of four slabs of the same number of acid molecules, with error bars representing one standard error of the mean. Analysis was only carried out at times >4 ns for each of the final production runs, to ensure that each of the slabs had first reached its equilibrium geometry. Averaging over the upper and lower faces was carried out in such a way that it was only the alignment of the C1–C18 vectors (parallel or perpendicular to the surface normal) that was taken into account and not the orientation (COOH group pointing towards or away from the water core). It was assumed that the overwhelming majority of the C1–C18 vectors would be directed with the COOH groups pointing towards the water core as this allows for maximum hydrogen bonding with the water. The validity of this assumption is demonstrated in Section S8 of the ESI.† For oleic and linoleic acid it can be seen that there is a strong relationship between the number of acid molecules and the peak of the angle fit: the more molecules that are present the smaller the angle between these molecules and the surface normal. This implies that when more molecules are present, these tend to pack together more tightly, and therefore there is a greater tendency for them to be aligned more vertically with respect to the slab. This allows the maximum number of hydrogen bonds to form between the acid molecules and the water sample. Such trends have been previously observed in the study of SAMs, where it has been noted that a decrease in the ‘tilt’ angle of molecules relative to the surface normal is associated with increasing surface coverage.56–58,79 Tighter more ordered packing at higher densities was also observed by Wadia et al. in their work on phospholipids on water.25 This pattern presents very similarly for both linoleic and oleic acids. For stearic acid, on the other hand, the trend is much less clear, with the average tilt angle, nearly independent of the number of molecules in the sample. This could reflect the greater flexibility of the stearic acid chains due to the lack of double bonds; stearic acid molecules may be able to more easily adopt a different conformation in order to fit further acid molecules in, instead of needing to tilt the entire molecule in order to accommodate this. In addition, this greater flexibility may allow stearic acid molecules to pack together more closely in partially filled monolayers, leading to islands of well-ordered fatty acids, which may have tilt angles closer to those of a full monolayer. We note that this island forming behaviour lasts to higher surface concentrations of stearic acid compared to oleic and linoleic acids, therefore the range of monolayer forming concentrations is smaller for stearic acid than the others. This tendency to take on different conformations in order to allow for the presence of further acid molecules may also be reflected in different preferences of the chains to be coiled or straightened when there are different numbers of acid molecules present. This can be investigated by looking at the distance between the two terminal carbons in a given chain – when the chain is fully extended this distance will be at its maximum value and when it is curled up this will be lower. Fig. 5 shows how the distribution of C1–C18 distances varies with the number of acid molecules present for each of the samples. As with the data in Fig. 4 each plot is an average of results from the upper and lower interfaces of four independent slabs of the same coverage of acid molecules, with the analysis being carried out at times >4 ns within the 10 ns production run. The C1–C18 distances were separated into 0.05 nm bins.
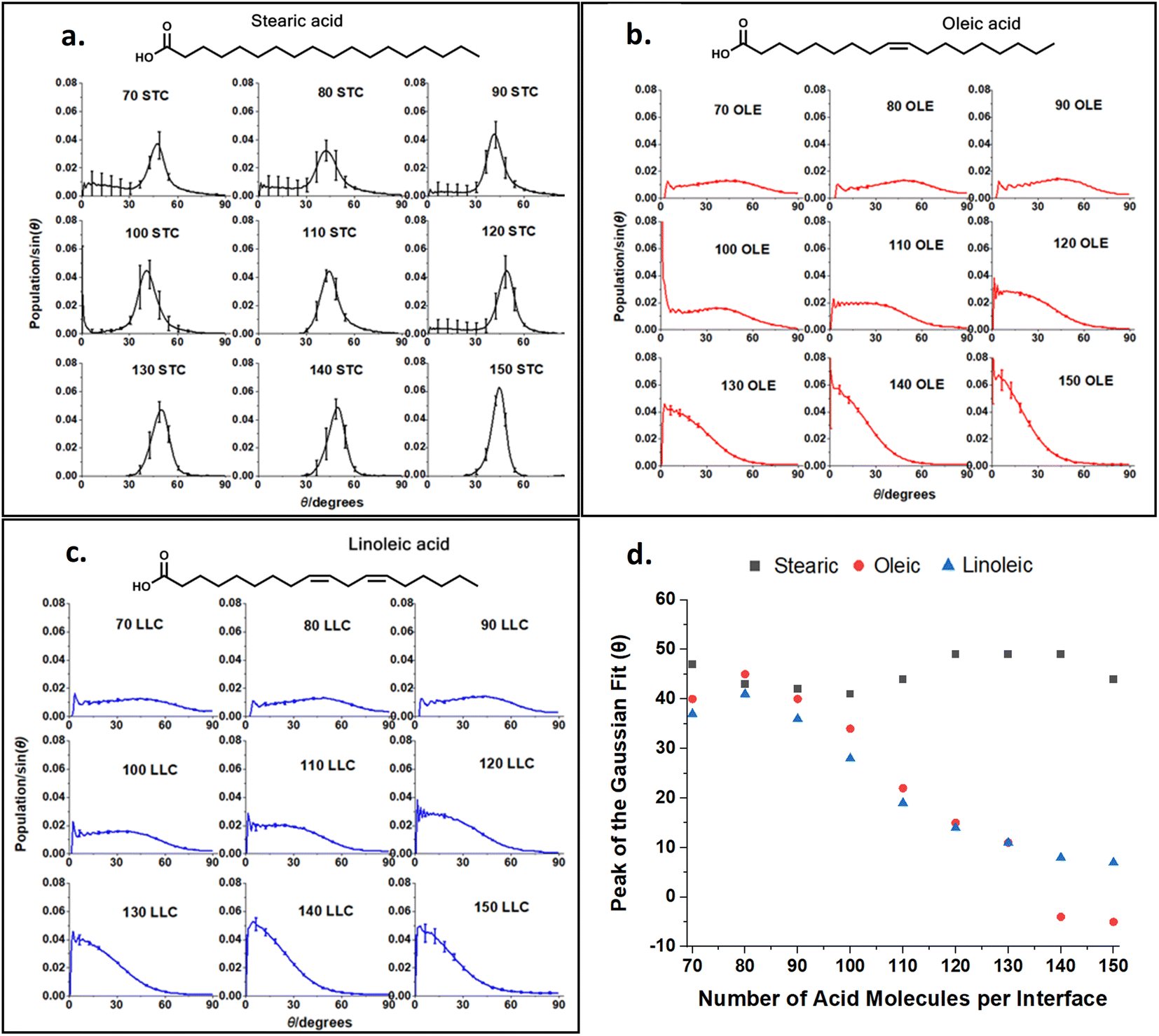 | ||
| Fig. 4 Distributions of the angles that the C1–C18 vector of the acids studied makes to the surface normal at different surface coverages of the acids, as denoted by the number of molecules per interface, taking into account solid angle effects. Plots (a–c) are colour-coded by species with the fatty acid that each set of plots represents shown above it for clarity. These graphs represent averages over times >4 ns for 10 ns production runs for four separately grown slabs. The error bars represent one standard error of the mean over these four runs. Data was binned into bins of one degree width. Plot (d) shows the peak of the Gaussian fits for each individual plot. All simulations were carried out at 298.15 K. | ||
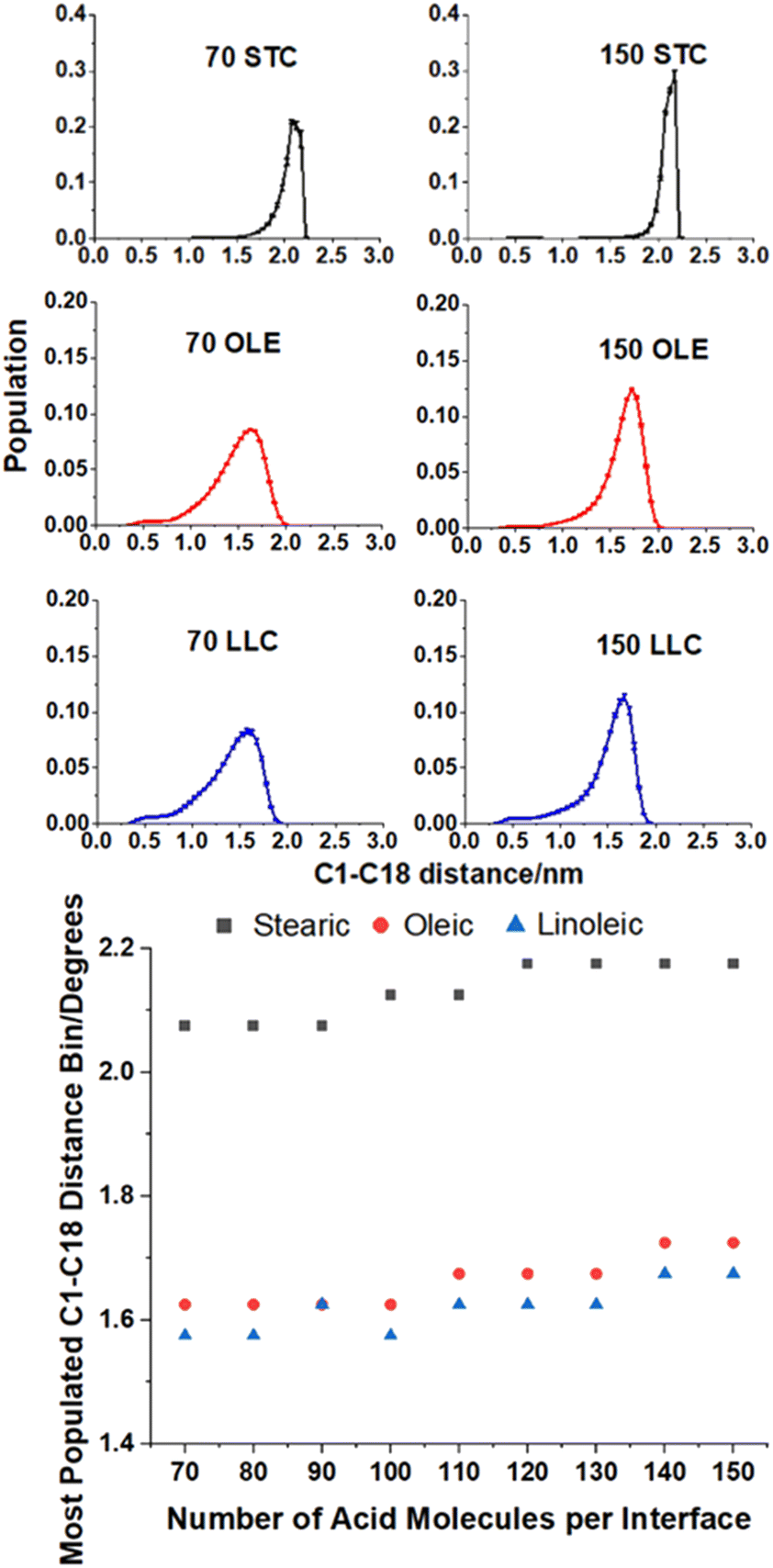 | ||
| Fig. 5 (Top) Distributions of C1–C18 distances for selected coverages of each of the acids. Populations are averages over four slabs, and at times >4 ns into a 10 ns production run. Distances histograms have bins of 0.05 nm. (Bottom) Plot showing how the most populated bin varies with surface coverage for each of the acids. All simulations were carried out at 298.15 K. | ||
The upper plots in Fig. 5 show comparisons of the distributions of the C1–C18 distances for the lowest and highest surface coverages of each of the acids studied, with the number and type of acid molecules per interface given above each plot. Analogous distributions for all of the intermediate surface coverages are given in Fig. S11–S13 of the ESI.† The bottom plot shows how the most populated C1–C18 distance varies with changing surface coverage for each of the acids. It can be seen from the plot that there is a slight increase in the C1–C18 distance for each of the acids as the surface coverage of increases. This suggests that there is a small tendency for molecules to become slightly straighter at higher surface area coverages, possibly as a way of packing more molecules into the slab and thus facilitating more COOH–H2O hydrogen bonding interactions and more alkene–alkene interactions between chains. This is in addition to the changes in the tilt angle demonstrated in Fig. 4, which result from the formation of a more tightly packed and better ordered monolayer. The stearic acid molecules have a larger end-to-end distance at all coverages, as the double bonds in oleic and linoleic acid enforce a kink in their structures and therefore prevent them from extending as fully. The fact that linoleic acid has two double bonds, whereas oleic acid only has one, means that this effect is slightly enhanced for linoleic acid, although this difference in average C1–C18 distance can be seen to be much smaller than that between that of stearic acid and the other two species.
The packing and ordering of organic molecules at the water–organic interface of an aerosol ultimately determines which groups will be exposed at the aerosol–air surface. This is of particular importance to atmospheric chemists as it is this what ultimately defines which groups will be present at the outermost edge of the aerosol, and therefore will be most available to undergo chemical processes involving incoming gaseous species. Whilst reactions do not take place exclusively at the interface, a high number of reactive groups, such as unsaturated moieties, at the surface increases the rate at which the aerosol can both act as a heterogenous catalysis as well as undergoing aging itself. This aging then has further implications for the composition and reactivity of the aerosol, as it can add a new range of functional groups that may not have been present in the primary material, or it may cause fragmentation of molecules, releasing volatile species into the air.
The accessible surface area (ASA) method80 is a well-established analytical method for determining which groups are present at the surface of a sample. ASA simulates an incoming atom or molecule as a probe sphere of a given radius. This probe approaches the slab from the vacuum side and samples those atoms that are present at the external side of the interface. Any atoms whose surface can come into contact with the probe's surface without the radius of the probe overlapping any other atoms are considered to be ‘accessible’ to incoming species of that size. The total surface area over which this is the case is the accessible surface area, however, this can be broken down by functional group type, in order to give a measure of the total surface area coverage of different groups at the interface of a sample that would be accessible to an incoming species of a particular size.
The ASA analysis in Fig. 6 also helps us to determine at which surface coverages water is no longer accessible to incoming radicals, suggesting that the aqueous core is fully covered by organic molecules. For oleic and linoleic acid this happens at relatively low numbers of acid molecules, with the water accessibility falling to virtually zero when there are 90+ acid molecules (1.7 molecules per nm2) on each interface. For stearic acid significantly higher numbers of organic molecules are needed for this to be the case, with ∼0.05 water coverage measured for slabs containing 130 stearic acid molecules (3.1 molecules per nm2). This suggests that the water cores of slabs containing stearic acid are accessible to incoming species up to higher number coverages of stearic acid, due to the closer packing of the stearic acid. This closer packing is a result of the absence of more rigid –HCCH– groups and leads to more ordered, tightly packed islands of acid molecules, with gaps exposing the water core between them.
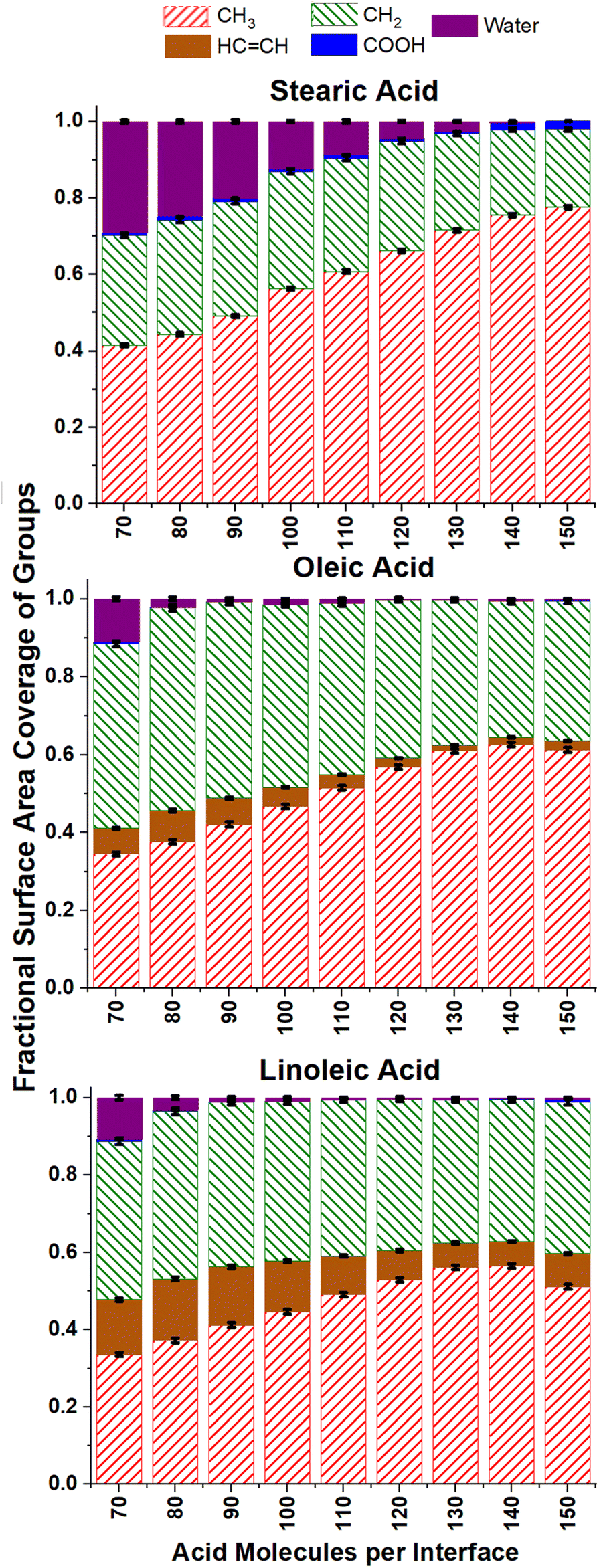 | ||
| Fig. 6 ASA results giving the fractional surface area coverage of different functional groups for different coverages of the three fatty acids, measured using a probe of radius 0.22 nm. Each bar represents the averaged results from four separate 10 ns production runs, with error bars representing one standard error of the mean. For each sample only data from times >4 ns was included. | ||
It can be seen from Fig. 6 that the ASA coverages are mainly made up of methyl and CH2 groups for all surface coverages, with very little COOH accessible at the surface. This is to be expected, as one would predict acid molecules to bond to the water core of the aerosol via hydrogen bonding using their COOH groups, thus leaving the hydrophobic hydrocarbon tails to point outwards. As the number of acid molecules within the samples increases, the fraction of the accessible surface that is made up of CH3 groups increases and that of CH2 decreases (seen more clearly in the ESI S10†). This is likely due to a closer packing of the chains, leaving less space for the probe to penetrate into the region between them, and thus making the CH2 groups less accessible. This effect is particularly pronounced for the stearic acid samples, possibly due to the tighter packing discussed above.
Of particular interest to atmospheric scientists is the accessibility of alkene double bonds to ozone molecules. It can be seen from Fig. 6 that the overall trend for this is that accessibility of HCCH groups primarily decreases with increasing surface coverage. This is again to be expected if one assumes that the vast majority of acid groups are bonded with the COOH groups inwards and the CH3 groups outwards and become increasingly tightly packed as monolayer coverage is approached. However, it can be seen that for all number concentrations of oleic and linoleic acid, there are non-negligible amounts of HCCH present at the surface, and even these small quantities will be significant with respect to the reactivity of the aerosols. It is therefore important to look at these results in more detail.
Fig. 7 shows how the accessible surface area coverage of selected groups varies with the number of acid molecules present. A more extensive version of this figure, showing all functional groups, can be found in the ESI S10.† It can be seen that the changes in the amounts of different groups present does not always vary linearly with the number of acid molecules, especially for oleic and linoleic acids. While HCCH presence at the surface seems to decrease with increasing coverage between 80 and 130 molecules, at higher numbers of acid molecules there appears to be an increase in this coverage. This is an indicator that full monolayer coverage has been reached, meaning that further molecules were not able to be packed so that they can access the water core of the aerosol, thus leaving HCCH groups exposed at the surface. Indeed, the slight increase in COOH groups present at the surface when >120 molecules are added may suggest that there are some molecules at these concentrations that are not bonded to the water core, but are present on an overlayer, thus allowing their COOH groups to be more accessible to incoming molecules.
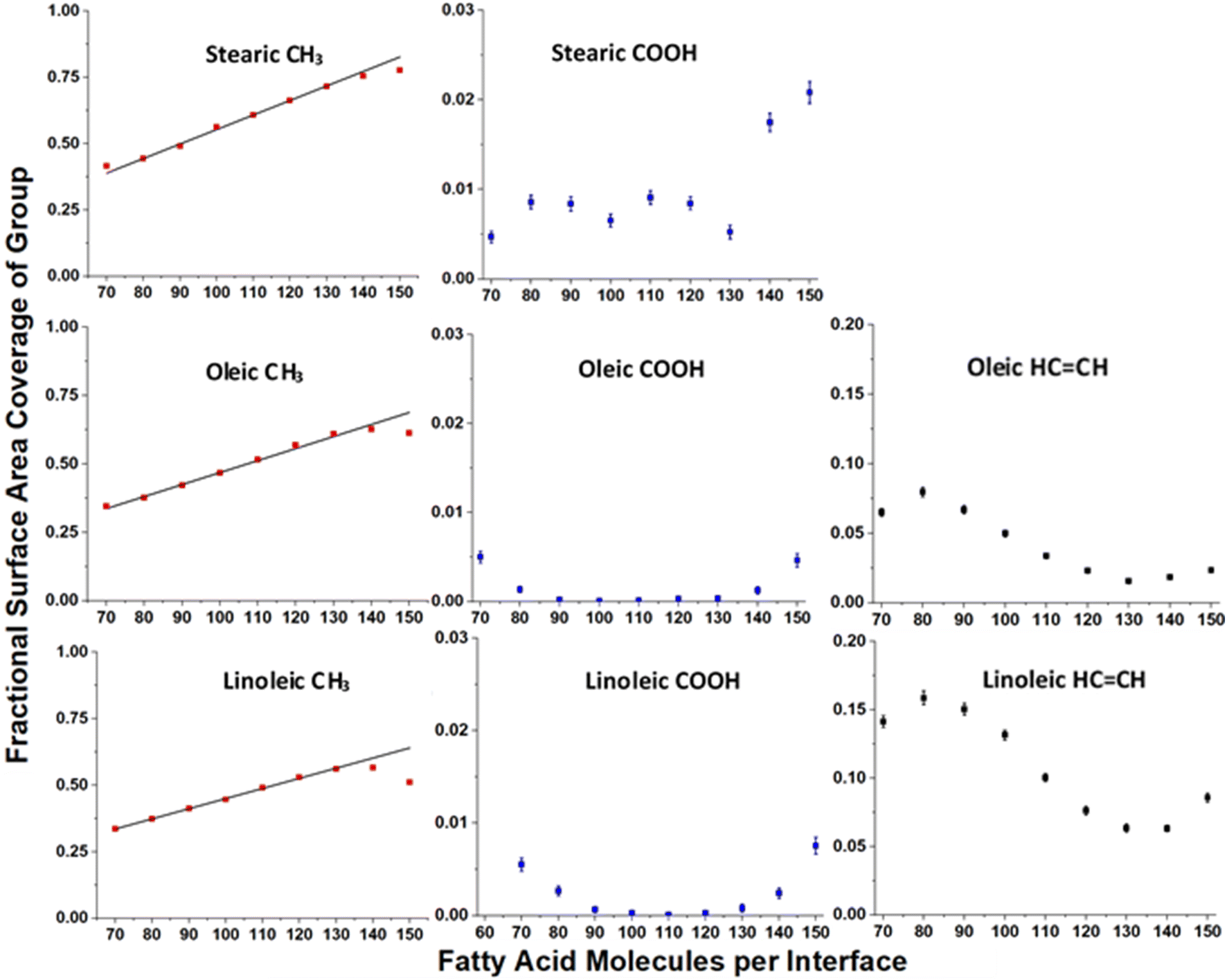 | ||
| Fig. 7 Variation of surface area coverages of different groups with numbers of fatty acid molecules per interface, based on the ASA results shown in Fig. 6. Each point represents the mean over four separately grown slabs, with the error bars representing one standard error of the mean. Only times >4 ns were included in the analysis. Note the difference in scales for each of the functional group types. The linear relationship between number of organic molecules and methyl coverage at lower fatty acid coverages is shown on the CH3 plots as a black trend line. It can be seen that for higher numbers of acid molecules the observed methyl coverage deviates from these trend lines. | ||
This is also reflected by the changes in methyl coverage with increasing number of organic molecules present. At number concentrations of <120 molecules per interface the methyl coverage is shown to increase in a near-linear fashion with increasing number of molecules, as demonstrated by the trend lines on the methyl plots in Fig. 7. This suggests that as molecules pack more closely together it becomes increasingly difficult for the probe species to sample atoms that are not on the very outermost ends of the fatty acid chains, and therefore the ASA results become dominated by the CH3 termini. At 130+ acid molecules per interface the proportion of methyl coverage starts to fall, presumably as a result of there being free acid molecules on top of the full monolayer, which would allow other groups for the probe molecule to interact with. The fact that 130 molecules per interface seems to mark a point at which there is a change in ASA trend for most groups (see also Fig. S14 of the ESI†), along with the fact that there is no water visible in the top-down images of the slabs at this coverage (Fig. S2–S4 of the ESI†) suggests that at 130 molecules per interface (corresponding to 3.1 molecules per nm2, 32 Å2 per molecule) monolayer coverage has been reached. This number of molecules corresponding to a monolayer coverage is also supported by changes in the number of fatty acid–fatty acid and fatty acid–water hydrogen bonds at increasing surface coverages, as discussed in Section S11 of the ESI.† The deviation of the methyl group coverage from the linear trend line is less noticeable for stearic acid than for the other species, which may be due to the stearic acid being able to form more tightly packed monolayers with larger numbers of molecules in them and thus fewer free acid molecules on top of them. When comparing the results for oleic and linoleic acid, the trends for all groups seem very similar, except for the fact that the presence of the additional HCCH group in linoleic acid leads to its larger surface coverage and a corresponding decreased surface coverage of the CH2 group. This, along with the results from the C1–C18 distance and tilt angle analysis suggests that the additional HCCH group in linoleic acid has very little effect on the packing of the chains and primarily changes what is present at the surface by statistical effects only.
Conclusions
The growth of aerosols around an aqueous core is a highly complex and dynamic problem, with the exact nature and number of different species present at the aqueous–organic interface dependent on a large number of factors involving both the molecules that are present in the environment in which it is formed and the reactions that take place at its surface to age it. These reactions are in turn affected by the accessibility of different groups at the aerosol–air interface.The work here uses molecular dynamics simulations to investigate how the structure of the organic layer of aqueous aerosol with radius >210 nm varies as the surface coverage of organic molecules increases. The chosen aerosol mimics were slabs of water with stearic, oleic or linoleic acid molecules added one by one, in order to mimic stepwise aerosol growth. Changes in the orientations of individual molecules at different acid coverages were discussed. It is found that at higher acid concentrations linoleic and oleic acids tend to be orientated with their chains more aligned to the surface normal, in order to allow for the better packing of acid molecules into the surface layer. No strong trend was found for stearic acid, with this attributed to the more flexible nature of the chains allowing for other methods of efficient packing even at low coverages. There was also found to be a slight tendency for chains to be more uncurled at higher surface coverages, for all acid species.
The accessibility of different groups to an incoming species was then investigated using the accessible surface area analysis method. It was found that at all surface coverages the surface largely consisted of CH3 and CH2 groups with only small HCCH and COOH presence. The surface area presence of these HCCH and COOH groups did, however, vary with the number of acid molecules present, first decreasing, as tighter packing prevented the probe from accessing groups further into the slab, before increasing as the monolayer coverage was exceeded (monolayer collapse). Based on these changes, and on visual inspection of the slabs it was determined that for a periodic slab with surface dimensions of 6.5 × 6.5 nm full monolayer coverage is reached on addition of around 130 molecules, which corresponds to coverages of 3.1 molecules per nm2 or 32 Å2 per molecule. The trends were, however, less clear for stearic acid than for the other two organic species.
It has been demonstrated that the structure of the organic layer of model aerosols changes significantly with surface coverage, even in the range that could be considered a monolayer, as does the accessibility of different groups to an incoming species. It is this accessibility that defines the reactivity of an aerosol particle towards species present in the atmosphere, both altering its ability to catalyse atmospheric reactions and determining its own evolution due to aging processes. The work here gives a means of directly examining how surface coverage is linked to functional group presence at the aerosol–atmospheric interface and therefore gives a unique insight into how this coverage may affect the reactivity of the aerosol. A deeper understanding of how the structure of aerosols affects their reactivities will inform better modelling of the formation and evolution of these and thus ultimately lead to a greater understanding of the roles that they play in key atmospheric processes.
Conflicts of interest
There are no conflicts to declare between the authors.Acknowledgements
The calculations in this work were carried out on the Cirrus UK National Tier-2 HPC Service at EPCC (https://www.cirrus.ac.uk) funded by the University of Edinburgh and EPSRC (EP/P020267/1), with access via the EPSRC Tier 2 Open Access RAP. S. J. G. thanks the EPSRC for funding via the EP/T021675/1 Program and EP/P001459/1 Platform grants.References
- C. E. Kolb and D. R. Worsnop, Chemistry and Composition of Atmospheric Aerosol Particles, Annu. Rev. Phys. Chem., 2012, 63, 471–491 CrossRef CAS PubMed.
- T. Novakov and J. E. Penner, Large Contribution of Organic Aerosols to Cloud-Condensation-Nuclei Concentrations, Nature, 1993, 365, 823–826 CrossRef CAS.
- V. Ramanathan, P. J. Crutzen, J. T. Kiehl and D. Rosenfeld, Aerosols, Climate, and the Hydrological Cycle, Science, 2001, 294, 2119–2124 CrossRef CAS PubMed.
- R. J. Charlson, J. H. Seinfeld, A. Nenes, M. Kulmala, A. Laaksonen and M. C. Facchini, Reshaping the Theory of Cloud Formation, Science, 2001, 292, 2025–2026 CrossRef CAS PubMed.
- J. Merikanto, D. V. Spracklen, G. W. Mann, S. J. Pickering and K. S. Carslaw, Impact of Nucleation on Global CCN, Atmos. Chem. Phys., 2009, 9, 8601–8616 CrossRef CAS.
- A. D. Estillore, J. V. Trueblood and V. H. Grassian, Atmospheric Chemistry of Bioaerosols: Heterogeneous and Multiphase Reactions with Atmospheric Oxidants and Other Trace Gases, Chem. Sci., 2016, 7, 6604–6616 RSC.
- J. Zhong, M. Kumar, J. S. Francisco and X. C. Zeng, Insight into Chemistry on Cloud/Aerosol Water Surfaces, Acc. Chem. Res., 2018, 51, 1229–1237 CrossRef CAS PubMed.
- U. Pöschl and M. Shiraiwa, Multiphase Chemistry at the Atmosphere-Biosphere Interface Influencing Climate and Public Health in the Anthropocene, Chem. Rev., 2015, 115, 4440–4475 CrossRef PubMed.
- U. K. Krieger, C. Marcolli and J. P. Reid, Exploring the Complexity of Aerosol Particle Properties and Processes using Single Particle Techniques, Chem. Soc. Rev., 2012, 41, 6631–6662 RSC.
- J. H. Kroll and J. H. Seinfeld, Chemistry of Secondary Organic Aerosol: Formation and Evolution of Low-Volatility Organics in the Atmosphere, Atmos. Environ., 2008, 42, 3593–3624 CrossRef CAS.
- L. Wang, A. F. Khalizov, J. Zheng, W. Xu, Y. Ma, V. Lal and R. Zhang, Atmospheric Nanoparticles Formed from Heterogeneous Reactions of Organics, Nat. Geosci., 2010, 3, 238–242 CrossRef CAS.
- T. Moise and Y. Rudich, Reactive Uptake of Ozone by Aerosol-Associated Unsaturated Fatty Acids: Kinetics, Mechanism, and Products, J. Phys. Chem. A, 2002, 106, 6469–6476 CrossRef CAS.
- J. V. Trueblood, X. Wang, V. W. Or, M. R. Alves, M. V. Santander, K. A. Prather and V. H. Grassian, The Old and the New: Aging of Sea Spray Aerosol and Formation of Secondary Marine Aerosol through OH Oxidation Reactions, ACS Earth Space Chem., 2019, 3, 2307–2314 CrossRef CAS.
- O. Laskina, H. S. Morris, J. R. Grandquist, Z. Qin, E. A. Stone, A. V. Tivanski and V. H. Grassian, Size Matters in the Water Uptake and Hygroscopic Growth of Atmospherically Relevant Multicomponent Aerosol Particles, J. Phys. Chem. A, 2015, 119, 4489–4497 CrossRef CAS PubMed.
- M. D. King, K. C. Thompson and A. D. Ward, Laser Tweezers Raman Study of Optically Trapped Aerosol Droplets of Seawater and Oleic Acid Reacting with Ozone: Implications for Cloud-Droplet Properties, J. Am. Chem. Soc., 2004, 126, 16710–16711 CrossRef CAS PubMed.
- J. Zahardis and G. A. Petrucci, The Oleic Acid-Ozone Heterogeneous Reaction System: Products, Kinetics, Secondary Chemistry, and Atmospheric Implications of a Model System - A Review, Atmos. Chem. Phys., 2007, 7, 1237–1274 CrossRef CAS.
- T. Thornberry and J. P. D. Abbatt, Heterogeneous Reaction of Ozone with Liquid Unsaturated Fatty Acids: Detailed Kinetics and Gas-Phase Product Studies, Phys. Chem. Chem. Phys., 2004, 6, 84–93 RSC.
- J. W. Morris, P. Davidovits, J. T. Jayne, J. L. Jimenez, Q. Shi, C. E. Kolb, D. R. Worsnop, W. S. Barney and G. Cass, Kinetics of Submicron Oleic Acid Aerosols with Ozone: A Novel Aerosol Mass Spectrometric Technique, Geophys. Res. Lett., 2002, 29, 71 Search PubMed.
- H. M. Hung and P. Ariya, Oxidation of Oleic Acid and Oleic Acid/Sodium Chloride(aq) Mixture Droplets with Ozone: Changes of Hygroscopicity and Role of Secondary Reactions, J. Phys. Chem. A, 2007, 111, 620–632 CrossRef CAS PubMed.
- Y. Katrib, S. T. Martin, H. M. Hung, Y. Rudich, H. Zhang, J. G. Slowik, P. Davidovits, J. T. Jayne and D. R. Worsnop, Products and Mechanisms of Ozone Reactions with Oleic Acid for Aerosol Particles having Core-Shell Morphologies, J. Phys. Chem. A, 2004, 108, 6686–6695 CrossRef CAS.
- G. D. Smith, E. Woods, C. L. DeForest, T. Baer and R. E. Miller, Reactive Uptake of Ozone by Oleic Acid Aerosol Particles: Application of Single-Particle Mass Spectrometry to Heterogeneous Reaction Kinetics, J. Phys. Chem. A, 2002, 106, 8085–8095 CrossRef CAS.
- J. D. Hearn, A. J. Lovett and G. D. Smith, Ozonolysis of Oleic Acid Particles: Evidence for a Surface Reaction and Secondary Reactions Involving Criegee Intermediates, Phys. Chem. Chem. Phys., 2005, 7, 501–511 RSC.
- T. Nah, S. H. Kessler, K. E. Daumit, J. H. Kroll, S. R. Leone and K. R. Wilson, OH-Initiated Oxidation of Sub-Micron Unsaturated Fatty Acid Particles, Phys. Chem. Chem. Phys., 2013, 15, 18649–18663 RSC.
- F. Sebastiani, R. A. Campbell and C. Pfrang, Night-Time Oxidation at the Air–Water Interface: Co-Surfactant Effects in Binary Mixtures, Environ. Sci.: Atmos., 2022, 2, 1324–1337 CAS.
- Y. Wadia, D. J. Tobias, R. Stafford and B. J. Finlayson-Pitts, Real-Time Monitoring of the Kinetics and Gas-Phase Products of the Reaction of Ozone with Unsaturated Phospholipid at the Air-Water Interface, Langmuir, 2000, 16, 321–9330 CrossRef.
- D. J. Donaldson and V. Vaida, Organic Films at the Air–Aqueous Boundary, Chem. Rev., 2006, 106, 1445–1461 CrossRef CAS PubMed.
- A. Chebbi and P. Carlier, Carboxylic Acids in the Troposphere, Occurrence, Sources, and Sinks: A Review, Atmos. Environ., 1996, 30, 4233–4249 CrossRef CAS.
- U. Pöschl, Atmospheric Aerosols: Composition, Transformation, Climate and Health Effects, Angew. Chem., Int. Ed., 2005, 44, 7520–7540 CrossRef PubMed.
- V. McNeill, N. Sareen and A. N. Schwier, Surface-Active Organics in Atmospheric Aerosols, in Atmospheric and Aerosol Chemistry, ed. V. McNeill and P. Ariya, Springer, Berlin, 2013, vol. 339, pp. 201–259 Search PubMed.
- J. C. Marty, A. Saliot, P. Buat-Ménard, R. Chesselet and K. A. Hunter, Relationship Between the Lipid Compositions of Marine Aerosols, the Sea Surface Microlayer, and Subsurface Water, J. Geophys. Res., 1979, 84, 5707–5716 CrossRef CAS.
- Y. Cheng, S.-M. Li, A. Leithead, P. C. Brickell and W. R. Leaitch, Characterizations of cis-Pinonic Acid and n-Fatty Acids on Fine Aerosols in the Lower Fraser Valley during Pacific 2001 Air Quality Study, Atmos. Environ., 2004, 38, 5789–5800 CrossRef CAS.
- M. D. King, A. R. Rennie, K. C. Thompson, F. N. Fisher, C. C. Dong, R. K. Thomas, C. Pfrang and A. V. Hughes, Oxidation of Oleic Acid at the Air-Water Interface and its Potential Effects on Cloud Critical Supersaturations, Phys. Chem. Chem. Phys., 2009, 11, 7699–7707 RSC.
- M. D. King, K. C. Thompson, A. D. Ward, C. Pfrang and B. R. Hughes, Oxidation of Biogenic and Water-Soluble Compounds in Aqueous and Organic Aerosol Droplets by Ozone: A Kinetic and Product Analysis Approach using Laser Raman Tweezers, Faraday Discuss., 2007, 137, 173–192 RSC.
- C. Pfrang, K. Rastogi, E. R. Cabrera-Martinez, A. M. Seddon, C. Dicko, A. Labrador, T. S. Plivelic, N. Cowieson and A. M. Squires, Complex Three-Dimensional Self-Assembly in Proxies for Atmospheric Aerosols, Nat. Commun., 2017, 8, 1724 CrossRef CAS PubMed.
- A. Milsom, A. M. Squires, A. D. Ward and C. Pfrang, The Impact of Molecular Self-Organisation on the Atmospheric Fate of Cooking Aerosol Proxy, Atmos. Chem. Phys., 2022, 22, 4895–4907 CrossRef CAS.
- E. González-Labrada, R. Schmidt and C. E. DeWolf, Kinetic Analysis of the Ozone Processing of an Unsaturated Organic Monolayer as a Model of an Aerosol Surface, Phys. Chem. Chem. Phys., 2007, 9, 5814–5821 RSC.
- P. Ziemann, Aerosol Products, Mechanisms, and Kinetics of Heterogeneous Reactions of Ozone with Oleic Acid in Pure and Mixed Particles, Faraday Discuss., 2005, 130, 469–490 RSC.
- S. Seok, T. J. Kim, S. Y. Hwang, Y. D. Kim, D. Vaknin and D. Kim, Imaging of Collapsed Fatty Acid Films at Air-Water Interfaces, Langmuir, 2009, 25, 9262–9269 CrossRef CAS PubMed.
- V. M. Kaganer, I. R. Peterson, R. M. Kenn, M. C. Shih, M. Durbin and P. Dutta, Tilted Phases of Fatty Acid Monolayers, J. Chem. Phys., 1995, 102, 9412–9422 CrossRef CAS.
- S. Sam, S. Krem, J. Lee and D. Kim, Recovery of Fatty Acid Monolayers by Salts Investigated by Sum Frequency Generation Spectroscopy, J. Phys. Chem. B, 2022, 126, 643–649 CrossRef CAS PubMed.
- U. K. Krieger, C. Marcollia and J. P. Reid, Exploring the Complexity of Aerosol Particle Properties and Processes using Single Particle Techniques, Chem. Soc. Rev., 2012, 41, 6631–6662 RSC.
- B. Ervens, G. Feingold and S. M. Kreidenweis, Influence of Water-Soluble Organic Carbon on Cloud Drop Number Concentration, J. Geophys. Res.: Atmos., 2005, 110, D18211 CrossRef.
- M. D. King, A. R. Rennie, K. C. Thompson, F. N. Fisher, C. C. Dong, R. K. Thomas, C. Pfrang and A. V. Hughes, Oxidation of Oleic Acid at the Air–Water Interface and its Potential Effects on Cloud Critical Supersaturations, Phys. Chem. Chem. Phys., 2009, 11, 7699–7707 RSC.
- H.-M. Hung and P. Ariya, Oxidation of Oleic Acid and Oleic Acid/Sodium Chloride(aq) Mixture Droplets with Ozone: Changes of Hygroscopicity and Role of Secondary Reactions, J. Phys. Chem. A, 2007, 111, 620–632 CrossRef CAS PubMed.
- C. Y. Pak, W. Li and Y. L. S. Tse, Free Energy and Dynamics of Organic-Coated Water Droplet Coalescence, J. Phys. Chem. C, 2020, 124, 8749–8757 CrossRef CAS.
- X. Ma, P. Chakraborty, B. J. Henz and M. R. Zachariah, Molecular Dynamic Simulation of Dicarboxylic Acid Coated Aqueous Aerosol: Structure and Processing of Water Vapor, Phys. Chem. Chem. Phys., 2011, 13, 9374–9384 RSC.
- X. Li, T. Hede, Y. Tu, C. Leck and H. Ågren, Surface-Active cis-Pinonic Acid in Atmospheric Droplets: A Molecular Dynamics Study, J. Phys. Chem. Lett., 2010, 1, 769–773 CrossRef CAS.
- M. H. H. Pomata, D. Laria, M. S. Skaf and M. D. Elola, Molecular Dynamics Simulations of AOT-Water/Formamide Reverse Micelles: Structural and Dynamical Properties, J. Chem. Phys., 2008, 129, 244503 CrossRef PubMed.
- J. Chowdhary and B. M. Ladanyi, Molecular Simulation Study of Water Mobility in Aerosol-AOT Reverse Micelles, J. Phys. Chem. A, 2011, 115, 6306–6316 CrossRef CAS PubMed.
- J. Chowdhary and B. M. Ladanyi, Molecular Dynamics Simulation of Aerosol-AOT Reverse Micelles, J. Phys. Chem. B, 2009, 113, 15029–15039 CrossRef CAS PubMed.
- G. Ergin and S. Takahama, Carbon Density is an Indicator of Mass Accommodation Coefficient of Water on Organic-Coated Water Surface, J. Phys. Chem. A, 2016, 120, 2885–2893 CrossRef CAS PubMed.
- S. Takahama and L. M. Russell, A Molecular Dynamics Study of Water Mass Accommodation on Condensed Phase Water Coated by Fatty Acid Monolayers, J. Geophys. Res.: Atmos., 2011, 116, D02203 CrossRef.
- P. Chakraborty and M. R. Zachariah, On the Structure of Organic-Coated Water Droplets: From “Net Water Attractors” to “Oily” Drops, J. Geophys. Res.: Atmos., 2011, 116, D21205 CrossRef.
- I. Langmuir, The Adsorption of Gases on Plane Surfaces of Glass, Mica and Platinum, J. Am. Chem. Soc., 1918, 40, 1361–1403 CrossRef CAS.
- J. Vieceli, O. L. Ma and D. J. Tobias, Uptake and Collision Dynamics of Gas Phase Ozone at Unsaturated Organic Interfaces, J. Phys. Chem. A, 2004, 108, 5806–5814 CrossRef CAS.
- O. M. Roscioni, L. Muccioli, A. Mityashin, J. Cornil and C. Zannoni, Structural Characterization of Alkylsilane and Fluoroalkylsilane Self-Assembled Monolayers on SiO2 by Molecular Dynamics Simulations, J. Phys. Chem. C, 2016, 120, 14652–14662 CrossRef CAS.
- J. M. Castillo, M. Klos, K. Jacobs, M. Horsch and H. Hasse, Characterization of Alkylsilane Self-Assembled Monolayers by Molecular Simulation, Langmuir, 2015, 31, 2630–2638 CrossRef CAS PubMed.
- D. M. Duffy and J. H. Harding, Modeling the Properties of Self-Assembled Monolayers Terminated by Carboxylic Acids, Langmuir, 2005, 21, 3850–3857 CrossRef CAS PubMed.
- R. Vácha, L. Cwiklik, J. Řezáč, P. Hobza, P. Jungwirth, K. Valsaraj, S. Bahr and V. Kempter, Adsorption of Aromatic Hydrocarbons and Ozone at Environmental Aqueous Surfaces, J. Phys. Chem. A, 2008, 112, 4942–4950 CrossRef PubMed.
- T. P. Liyana-Arachchi, K. T. Valsaraj and F. R. Hung, Molecular Simulation Study of the Adsorption of Naphthalene and Ozone on Atmospheric Air/Ice Interfaces, J. Phys. Chem. A, 2011, 115, 9226–9236 CrossRef CAS PubMed.
- P. Chakraborty and M. R. Zachariah, Sticking Coefficient and Processing of Water Vapor on Organic-Coated Nanoaerosols, J. Phys. Chem. A, 2008, 112, 966–972 CrossRef CAS PubMed.
- A. C. Stewart, M. J. Paterson and S. J. Greaves, Chemical Functionality at the Liquid Surface of Pure Unsaturated Fatty Acids, Environ. Sci.: Atmos., 2021, 1, 498–507 CAS.
- H. J. C. Berendsen, D. Van der Spoel and R. Van Drunen, GROMACS: A Message-Passing Parallel Molecular Dynamics Implementation, Comput. Phys. Commun., 1995, 91, 43–56 CrossRef CAS.
- D. Van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. C. Berendsen, GROMACS: Fast, Flexible, and Free, J. Comput. Chem., 2005, 26, 1701–1718 CrossRef CAS PubMed.
- M. J. Robertson, W. L. Tirado-Rives and J. Jorgensen, Improved Peptide and Protein Torsional Energetics with the OPLS-AA Force Field, J. Chem. Theory Comput., 2015, 11, 3499–3509 CrossRef CAS PubMed.
- D. L. Beveridge and W. L. Jorgensen, The OPLS (Optimized Potentials for Liquid Simulations) Potential Functions for Proteins, Energy Minimizations for Crystals of Cyclic Peptides and Crambin, Annu. Rev. Biophys. Bioeng., 1988, 110, 39 Search PubMed.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS.
- L. S. Dodda, J. Z. Vilseck, J. Tirado-Rives and W. L. Jorgensen, 1.14*CM1A-LBCC: Localized Bond-Charge Corrected CM1A Charges for Condensed-Phase Simulations, J. Phys. Chem. B, 2017, 121, 3864–3870 CrossRef CAS PubMed.
- L. S. Dodda, I. C. De Vaca, J. Tirado-Rives and W. L. Jorgensen, LigParGen Web Server: An Automatic OPLS-AA Parameter Generator for Organic Ligands, Nucleic Acids Res., 2017, 45, W331–W336 CrossRef CAS PubMed.
- W. L. Jorgensen and J. Tirado-Rives, Potential Energy Functions for Atomic-Level Simulations of Water and Organic and Biomolecular systems, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 6665–6670 CrossRef CAS PubMed.
- H. J. C. Berendsen, J. R. Grigera and T. P. Straatsma, The Missing Term in Effective Pair Potentials, J. Phys. Chem., 1987, 91, 6269–6271 CrossRef CAS.
- S. Miyamoto and P. A. Kollman, Settle: An Analytical Version of the SHAKE and RATTLE Algorithm for Rigid Water Models, J. Comput. Chem., 1992, 13, 952–962 CrossRef CAS.
- A. C. Stewart, M. J. Paterson and S. J. Greaves, The Influence of Saturation on the Surface Structure of Mixed Fatty Acid-on-Water Aerosol: A Molecular Dynamics Study, Environ. Sci.: Atmos., 2022, 2, 1516–1525 CAS.
- J. Lovrić, D. Duflot, M. Monnerville, C. Toubin and S. Briquez, Water-Induced Organization of Palmitic Acid at the Surface of a Model Sea Salt Particle: A Molecular Dynamics Study, J. Phys. Chem. A, 2016, 120, 10141–10149 CrossRef PubMed.
- R. W. Hockney, The Potential Calculation and Some Applications, Methods Comput. Phys., 1970, 9, 136 Search PubMed.
- W. C. Swope, H. C. Andersen, P. H. Berens and K. R. Wilson, A Computer-Simulation Method for the Calculation of Equilibrium-Constants for the Formation of Physical Clusters of Molecules: Application to Small Water Clusters, J. Chem. Phys., 1982, 76, 637–649 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, VMD - Visual Molecular Dynamics, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- R. D. Hoehn, M. A. Carignano, S. Kais, C. Zhu, J. Zhong, X. C. Zeng, J. S. Francisco and I. Gladich, Hydrogen bonding and orientation effects on the accommodation of methylamine at the air-water interface, J. Chem. Phys., 2016, 144, 214701 CrossRef PubMed.
- T. Vallant, J. Kattner, H. Brunner, U. Mayer and H. Hoffmann, Investigation of the Formation and Structure of Self-Assembled Alkylsiloxane Monolayers on Silicon using in situ Attenuated Total Reflection Infrared Spectroscopy, Langmuir, 1999, 15, 5339–5346 CrossRef CAS.
- F. Eisenhaber, P. Lijnzaad, P. Argos, C. Sander and M. Scharf, The Double Cubic Lattice Method: Integration of Surface Area and Volume Efficient Approaches to Numerical Molecular Assemblies and to Dot Surface Contouring of Molecular Assemblies, J. Comput. Chem., 1995, 16, 273–284 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ea00148a |
| ‡ Current address: Department of Chemistry, College of Science, Swansea University, Swansea, SA2 8PP, United Kingdom. |
| This journal is © The Royal Society of Chemistry 2023 |