
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanistic study of the complex photooxidation of allyl methyl sulfide (AMS): reaction paths and products of addition under different atmospheric conditions†
Alejandro L.
Cardona
 a,
María B.
Blanco
a,
Mariano A.
Teruel
*a and
Oscar N.
Ventura
*b
a,
María B.
Blanco
a,
Mariano A.
Teruel
*a and
Oscar N.
Ventura
*b
a(L.U.Q.C.A), Laboratorio Universitario de Química y Contaminación del Aire, Instituto de Investigaciones en Fisicoquímica de Córdoba (INFIQC), CONICET, Dpto. de Fisicoquímica, Facultad de Ciencias Químicas, Universidad Nacional de Córdoba, Ciudad Universitaria, 5000 Córdoba, Argentina. E-mail: Mariano.Teruel@unc.edu.ar
bComputational Chemistry and Biology Group (CCBG), Detema, Facultad de Química, Universidad de la República, Montevideo, Uruguay. E-mail: Oscar.N.Ventura@gmail.com
First published on 5th May 2023
Abstract
The addition mechanism of the OH-initiated oxidation of allyl methyl sulfide (AMS) under atmospheric conditions was studied theoretically using both density functional theory (DFT) and the SVECV-f12 composite method. We found that the addition does not occur directly but rather through a pre-reactive complex that serves as a previous stage for addition at both the C1 and C2 positions. However, based on thermodynamics the addition at C2 possibly occurs by branching of the C1 addition mechanism. Once the addition proceeds, the reaction with O2 under atmospheric conditions produces RO2 radicals that can decompose in multiple ways. Complex mechanisms of intramolecular rearrangement and decomposition both in the absence and presence of NOx have been examined. The thermodynamically most favourable decomposition paths produce 2-hydroxy-acetaldehyde, 2-methyl-thio-acetaldehyde, formaldehyde, and the methyl thiomethyl peroxy (MSP) intermediate. This latter species is proposed as the main source of sulfur dioxide (SO2), which is the product found in the highest yield during the experimental determinations.
Environmental significanceSulfur-containing volatile organic compounds (VOSCs), which can be emitted in significant quantities during the decomposition of organic matter, produce mainly SO2 during their atmospheric oxidation, a species with the ability to acidify the atmosphere and to form aerosols. Furthermore, in the case of unsaturated sulfides, the degradation mechanism could be extremely complex and gives rise to multiple possible products, some of which are expected and observed, while others are unusual and have not been observed yet. The theoretical study of the reaction mechanisms presented in this paper using high accuracy methods allows for an understanding of how these reactions take place. Moreover, this study opens the exciting possibility of new experimental studies that can lead to identification of new products predicted theoretically. |
1 Introduction
Allyl methyl sulfide (AMS) is one of the simplest allyl sulfides, a member of the family of volatile organosulfur compounds (VOSCs), with just a methyl group attached to the sulfur atom. This sulfur containing VOC is emitted into the atmosphere mainly by anaerobic decomposition of organic matter1,2 and food wastes.3,4 The gas phase decomposition of VOSCs has important environmental consequences; for instance, they contribute to the acidification of the atmosphere and to aerosol formation.5 Once in the atmosphere, their main path is the gas phase degradation initiated by OH radicals,6 as in other unsaturated compounds. This reaction usually initiates via addition to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond,7,8 leading to the formation of simpler species.
C double bond,7,8 leading to the formation of simpler species.
As in the case of dimethyl sulfide (DMS) degradation, the formation of the methyl thiomethyl radical 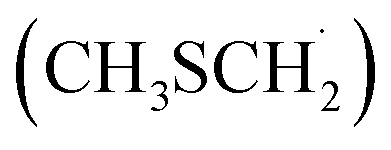 is observed in the oxidation of AMS.9–11 This radical reacts rapidly with atmospheric O2 to form methyl thiomethyl peroxy radical (MSP, H3CSCH2OO˙).12 Using electronic structure calculations and RRKM simulations, previous studies showed that an intramolecular H-shift is fast enough to compete with bimolecular reactions under atmospheric conditions.12,13 Besides, experimental findings reveal a two-step isomerization process of MSP radicals, finally forming HOOCH2SCHO accompanied by OH recycling.12 In this context, Veres et al. recently reported evidence of previously unquantified DMS oxidation product, hydroperoxymethyl thioformate, from global-scale, airborne observations.14
is observed in the oxidation of AMS.9–11 This radical reacts rapidly with atmospheric O2 to form methyl thiomethyl peroxy radical (MSP, H3CSCH2OO˙).12 Using electronic structure calculations and RRKM simulations, previous studies showed that an intramolecular H-shift is fast enough to compete with bimolecular reactions under atmospheric conditions.12,13 Besides, experimental findings reveal a two-step isomerization process of MSP radicals, finally forming HOOCH2SCHO accompanied by OH recycling.12 In this context, Veres et al. recently reported evidence of previously unquantified DMS oxidation product, hydroperoxymethyl thioformate, from global-scale, airborne observations.14
In our recently published experimental work, we determined the rate coefficient of the gas phase degradation of AMS initiated by OH radicals where the main reaction products were detected and quantified.15 Our findings led us to assume that the degradation mechanism could be more complex than for other unsaturated compounds, due to the presence of the sulfur atom in the molecule. In the case of alkyl sulfides, it has been proposed that in the reaction with OH radicals, those can weakly bind to the sulfur atom, leading to a prereactive complex which has been related to a negative temperature dependence.16,17 Notwithstanding that high yields of SO2 were determined, as expected, the formation mechanism of this and other products is not clear enough and the distribution between the mechanisms of addition and abstraction of hydrogen is still uncertain.
Although there are a few theoretical studies on AMS, they mostly aimed to understand gas phase thermal unimolecular decomposition.18,19 To the best of our knowledge, there are no computational studies on gas-phase degradation initiated by atmospheric radicals. Therefore, in this work we explore in depth the energetics of the addition reaction mechanism involved in the gas phase degradation of AMS initiated by OH radicals using computational methods, to better understand and complement our previous experimental finding o allyl sulfides.15
2 Computational details
Geometry optimizations and vibrational analysis of stationary points involved in the gas phase degradation of AMS initiated by OH radicals via addition to the C–C double bond were performed using two hybrid exchange–correlation density functional theory (DFT) methods; M06-2X with empirical Grimme dispersion (GD3) and MN15.20–22 Dunning's cc-pVTZ and aug-cc-pVTZ basis sets were employed for calculations.23 This is not a random choice. An extensive study of DFT functionals applied to the GMTKN55 database by Goerigk et al.24 showed that dispersion corrected Minnesota functionals are among the top performers, unless one resorts to the much more expensive double hybrid functionals. Since no multiconfigurational species are expected to have relevance in this study, the M06-2X method is appropriate, and the D3 dispersion is necessary, as noted by Goerik et al., even if the original method included part of the dispersion in the determination of the parameters.For local minima, reactants (R), intermediates (IM), product complexes (PW) and final products (P), the absence of imaginary frequencies was verified. For transition states (TSs), the presence of a single imaginary frequency confirmed that the structures have a translational mode and are maximum w.r.t. the reaction coordinates. Furthermore, intrinsic reaction coordinate (IRC) analyses were performed to confirm that the structure is the correct TS for the path, i.e., that it connects appropriate reactants and products.
In order to get more accurate energies, the recently developed SVECV-f12 composite method was applied.25 This protocol starts by performing M06-2X-D3 geometry optimization and frequency evaluation. The total energy is then obtained at the optimum geometries by the explicitly correlated CCSDS(T)-F12 method26,27 extrapolated to the complete basis set limit using the cc-pVDZ-F12 and cc-pVTZ-F12 basis sets.28 Finally, the core valence correlation energy is calculated by employing the MP2 method with the cc-pVCTZ basis set.23 The protocol has been tested specifically for the prediction of barriers of reaction for several archetypical systems25 and found to produce results with an accuracy of 0.5 kcal mol−1 or better, in comparison with experiments and/or more sophisticated and expensive methods of calculation.
T1 diagnostics for all the species were checked and found to be ≤0.03, indicating that the use of the single-reference method to describe the wave function is appropriate. DFT calculations were performed using the Gaussian 16 software package,29 while the SVECV-f12 calculations were performed using the Molpro 2020 30 and ORCA V5.0 31–33 programs.
3 Results and discussion
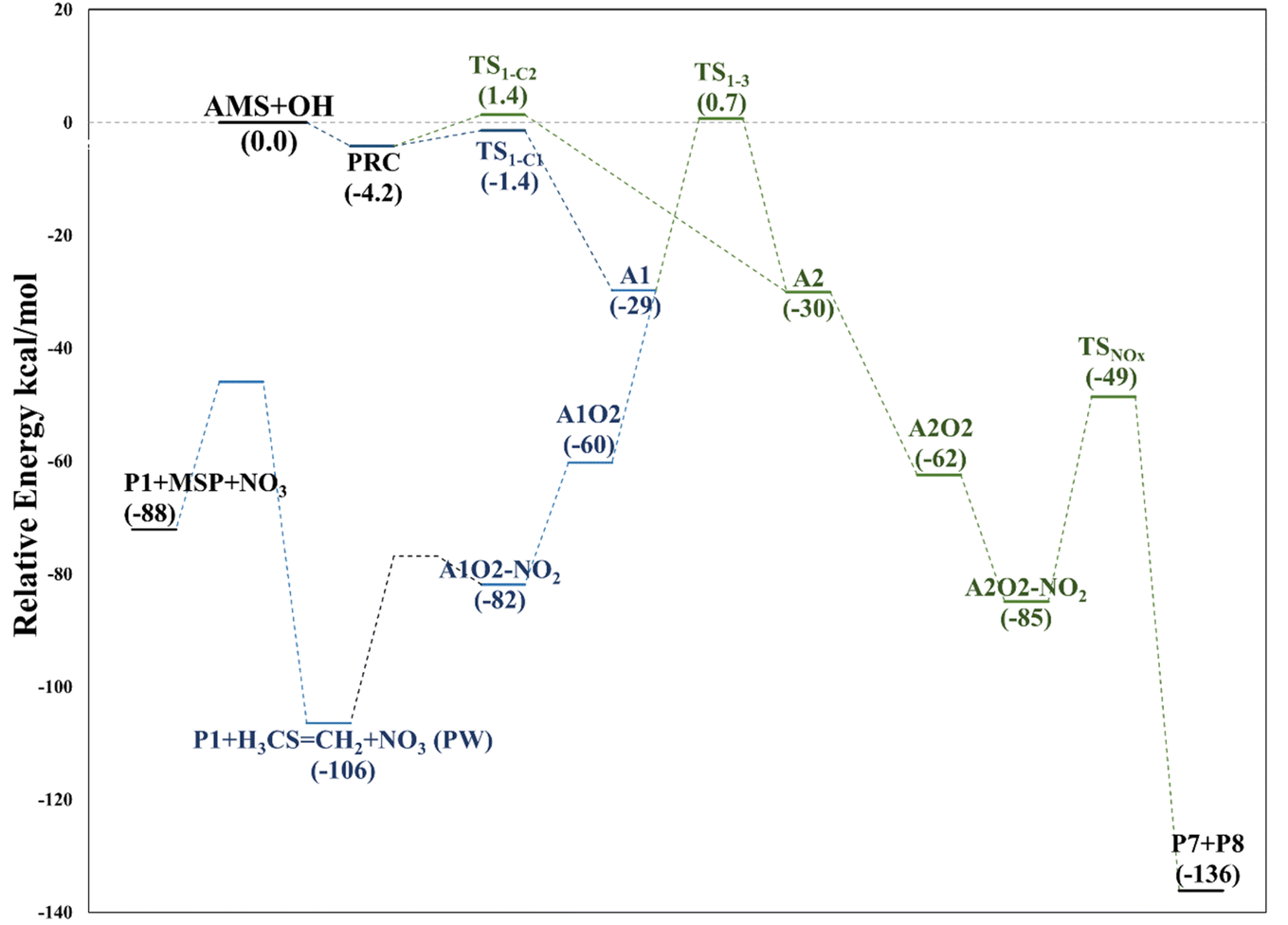
We have computationally explored the complete oxidation mechanism of AMS initiated by the OH radical addition to both the α and β positions of the vinyl moiety (H2Cα = CβHR) labelled as C1 and C2 from now on. Accordingly, C3 is the carbon bonded to C2 and sulfur and C4 is the carbon atom in the methyl group.Performing an optimized scan using the C2–C3–S–C4 dihedral angle as the dependent variable (while optimizing all the rest of the geometrical parameters, including the C1–C2–C3–S dihedral angle) we found three local minima, as shown in Fig. 1. From these results the difference between the three conformers AMS_1, AMS_2 and AMS_3 is about 2 kcal mol−1 with all the methods used. Since the corresponding isomerization barriers are easily overcome at room temperature, it is reasonable to assume that the three isomers are in equilibrium and ruled by a Boltzmann distribution. Using the best free energies (at the SVECV-f12 level) for the optimized geometries of the three isomers, we obtained relative concentrations of 83%, 5% and 11% respectively (using optimum geometries and the best method of calculation, it was found that AMS_2 is less stable than AMS_3 and both are less stable than AMS_1). Thus, taking this equilibrium into account, the reaction will proceed from the AMS_1 isomer preferentially. Therefore, all energies in the following are displayed relative to that of the AMS_1 isomer. Initially, the results for OH addition and further oxidation leading to products are described assuming that ROO˙ radicals produce RO˙ + O2via peroxy radical recombination. Then, two alternative routes for the mechanism of decomposition of ROO˙ radicals were explored: the reaction with NO2 and intramolecular hydrogen migration leading to the formation of OH radicals.
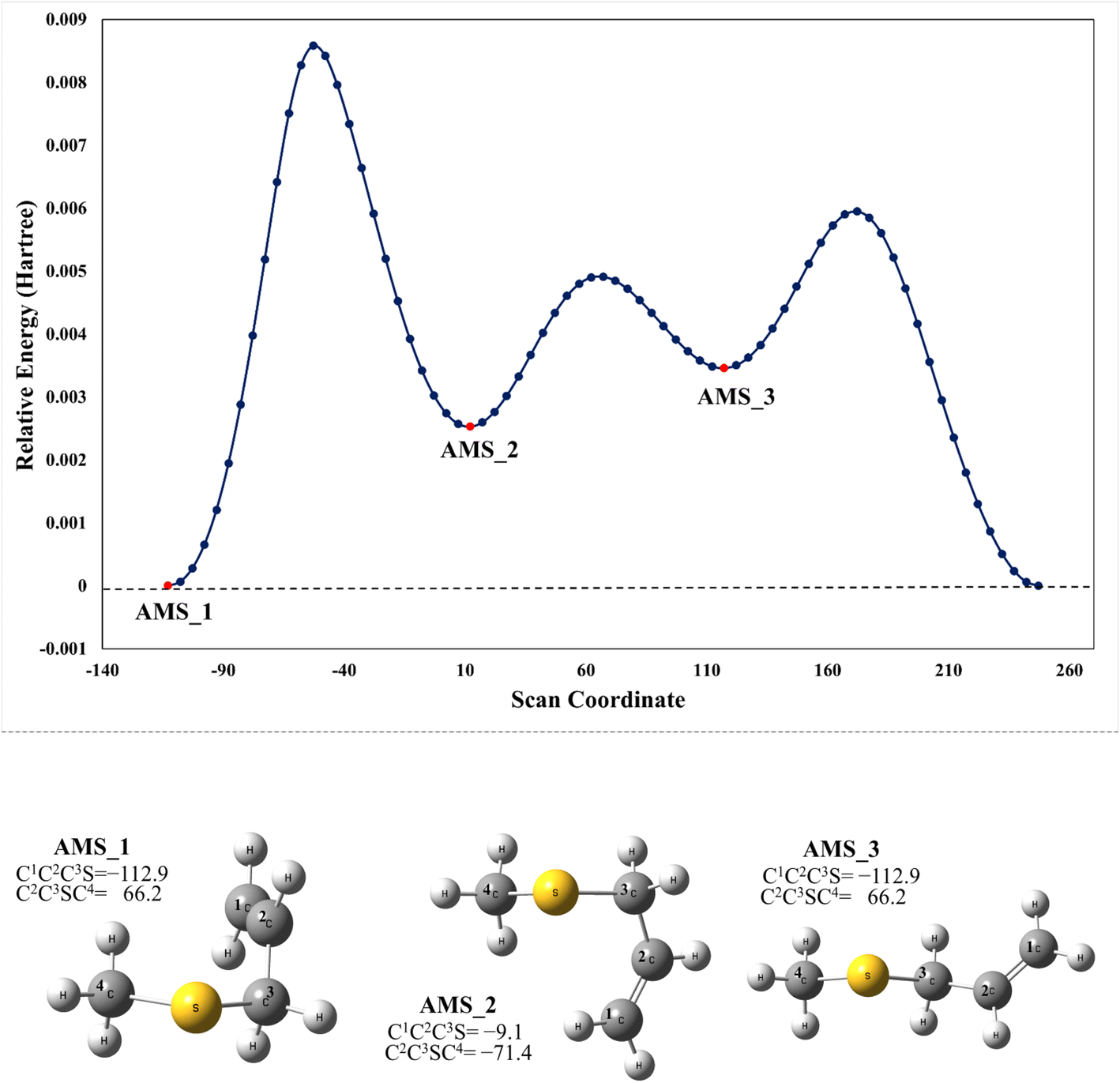 | ||
| Fig. 1 Upper panel: M06-2X/GD3/aug-cc-pVTZ energy at the optimized geometries on a scan along the C2–C3–S–C4 dihedral angle in AMS. Lower panel: optimized structures of the local minimum energy conformations (XYZ coordinates of these and the other structures considered in this paper are compiled in the ESI†). | ||
3.1 Addition of OH at C1
Fig. 2 shows the suggested mechanism for the OH radical addition to AMS. Our findings suggest that the addition at C1 occurs through a well-defined (but submerged) transition state that is preceded by the formation of a pre-reactive complex (PRC) in which the hydroxyl hydrogen interacts with the sulfur atom of the AMS, while the oxygen is positioned towards the double bond and there is no interaction between sulfur and oxygen, forming a dipole–dipole complex as previously reported for DMS.34 The PRC is 4.2, 4.0 and 5.6 kcal mol−1 below reactants at the M06-2X-D3/aug-cc-pVTZ, MN15/aug-cc-pVTZ and SVECV-f12 levels of theory, respectively. It is worth mentioning that the PRC is an addition complex for both the C1 and C2 positions.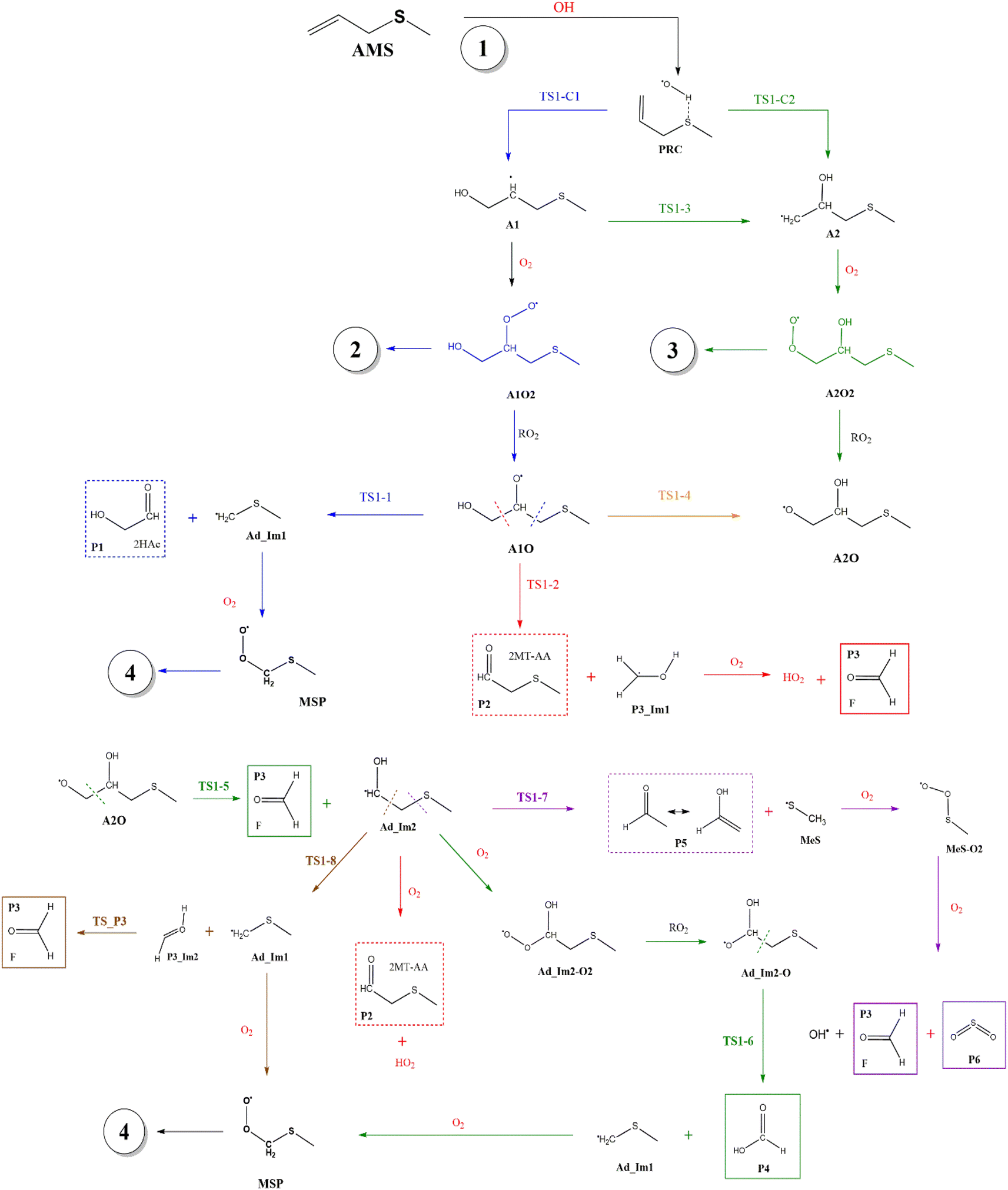 | ||
| Fig. 2 General mechanism of the OH radical addition to AMS in the absence of NOx radicals. Products are shown enclosed in boxes: identified (solid lines) and newly proposed (dashed lines). | ||
This addition proceeds through TS1-C1 which is 2.8 kcal mol−1 above the PRC but still submerged by 2.8 kcal mol−1 relative to reactants forming the intermediate radical A1 and all the subsequent oxidation steps are submerged relative to the reactants (Fig. 3, the data used to build this graph and the next ones are contained in Tables S2–S5 in the ESI†).
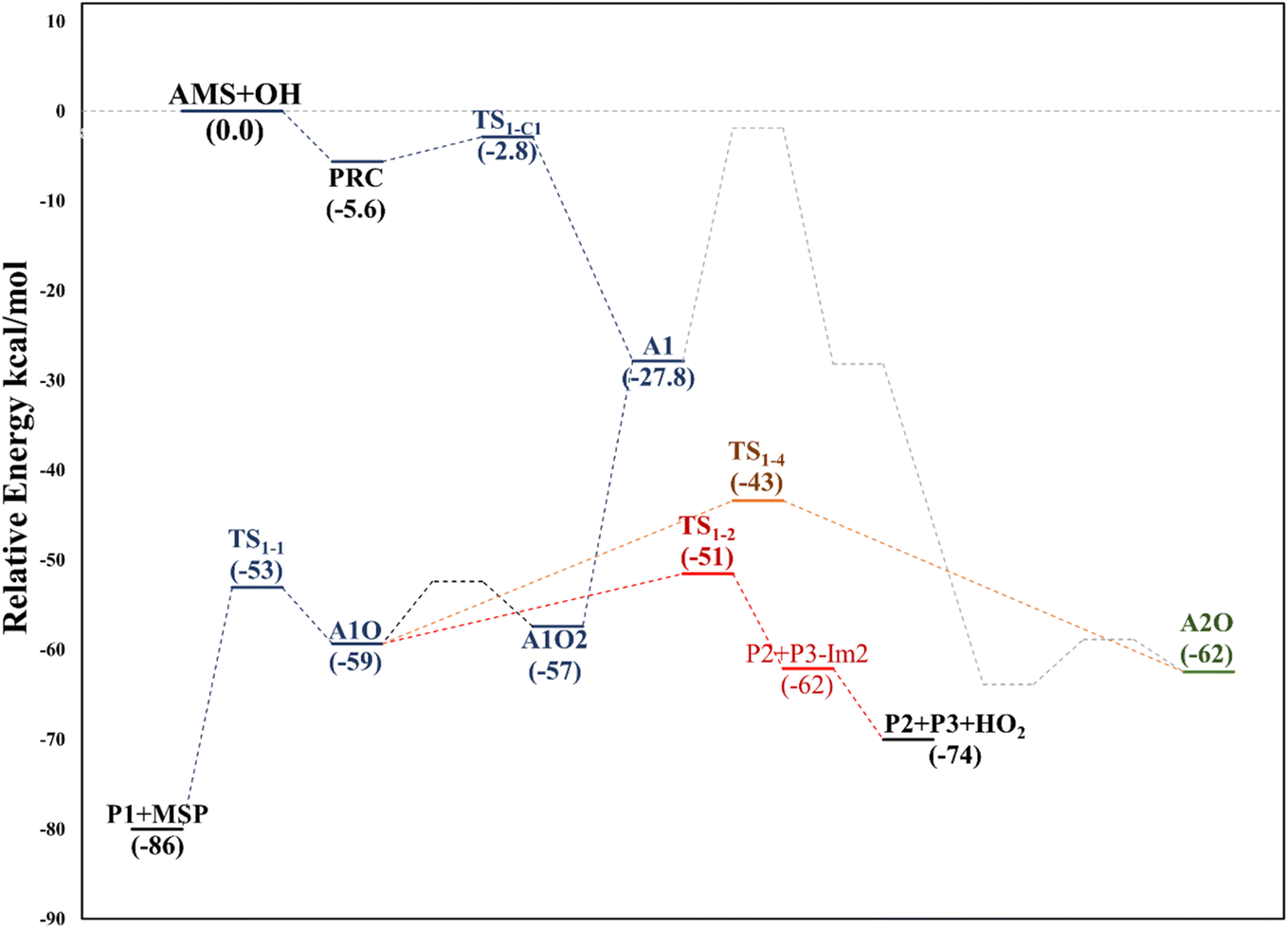 | ||
| Fig. 3 Partial relative energy diagram for the OH addition to the allyl group in the AMS oxidation initiated by OH radicals at the SVECV-f12 level. Only the addition through A1 (i.e., the addition of OH at C1 in the absence of NOx) is shown explicitly. | ||
Under atmospheric conditions, the A1 intermediate can further react with O2 to produce the peroxy radical A1O2 (ROO˙). The latter can undergo an isomerization process involving intramolecular hydrogen transfer, leading to products which will be discussed later on (reaction path 2 in Fig. 2). Alternatively, it can be transformed into alkoxy radical A1O (RO˙) through three possible routes: (1) a not completely established recombination mechanism involving two molecules of the peroxy radical and further reaction liberating an oxygen molecule (dashed line in Fig. 3), (2) reaction with NOx (see Section 3.3), or (3) further reaction with OH to produce hydroperoxyl radicals.35–38 The so formed A1O intermediate radical can decompose through the transition state TS1-1, breaking the C1–C2 bond, leading to 2-hydroxy-acetaldehyde (P1) and the methyl thiomethyl radical (H3CSC·H2, Ad_Im1). Subsequently, Ad_Im1 reacts further with O2 to give the MSP radical whose decomposition mechanism is well known since it has been studied as part of the DMS oxidation process.10,12,13,39 We performed anyway a study of the oxidation of this radical at the same level of calculation we used for the rest of the work. The results are described in Section 3.5.
Alternatively, the A1O intermediate can decompose by cleavage of the C2–C3 bond through the TS1-2 transition state. This reaction leads to 2-methyl-thio-acetaldehyde (P2), formaldehyde (P3), and HO2 (see Fig. 2). P3 is produced by both C1 and C2 addition paths, so it is not useful to characterize this one. P2 instead would be characteristic of this path, as P1 was from the reaction through TS1-1. However, it should be noted that the formation of P1 was not experimentally verified by comparing the reference spectrum of P1 with the post-reaction spectrum of AMS + OH (see ref. 15). This could mean that it does not form, forms in proportions that are not detectable experimentally, or suffers from secondary processes such as deposition on the walls of the reactor, photolysis, or a reaction with HO2 radicals which will lead mainly to formaldehyde, formic acid, methanol, CO and OH radicals.40,41 It can also undergo photoinduced keto–enol isomerization to 1,2-ethenediol via the double-hydrogen shift mechanism, which, as has been reported, can be thermodynamically more favorable than photodissociation channels.42 And, last but not least, the calculated height of the barrier may be too low, although we don't expect the SVECV-f12 protocol to exhibit such an error in the relative energies of TS1-1 and TS1-2. P2 was not identified experimentally among the products, mainly because of the lack of pure species to measure the IR spectra.
3.2 Addition of OH at C2
As mentioned above, the PRC is also a complex for addition to the C2 position through TS1-C2 which is slightly submerged at 5.5 kcal mol−1 above the PRC. However, as part of our search for possible reaction paths we have found TS1-3, a transition state connecting A2 to A1 (Fig. 4) which is slightly less stable than the former, similarly to what was reported previously for the mechanism of addition of OH to 2-fluoropropene.43 This finding is consistent with our supposition that an addition mechanism to C2 does not occur directly, but is a branch of the addition path to C1. TS1-3 is 0.3 kcal mol−1 above reactants at the M06-2X-D3/aug-cc-pVTZ level but 1.4 and 1.9 kcal mol−1 below them at the MN15/aug-cc-pVTZ and SVECV-f12 levels of theory, respectively. As can be seen in Fig. 4, the IRC in the region of TS1-3 is considerably flat and broad, indicating the presence of nearly zero eigenvalues, a degenerate condition that explains why A2 could not be directly reachable from the reactants but nonetheless representing a feasible reaction path.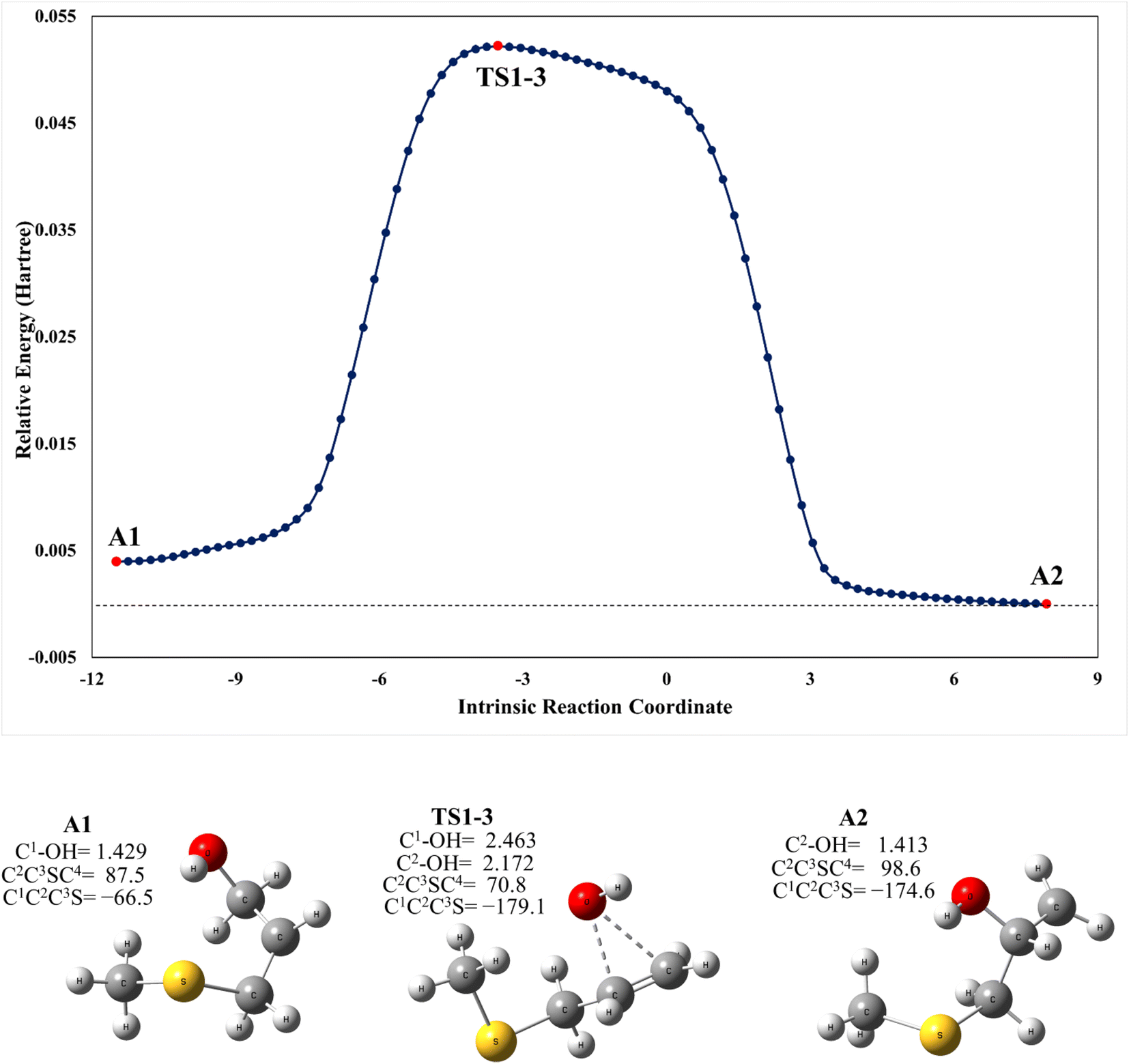 | ||
| Fig. 4 Upper panel: a plot of intrinsic reaction coordinates for the transition state between the A1 and A2 intermediaries, TS1-3. Lower panel: optimized structures at the M06-2X/GD3/aug-cc-pVTZ level (XYZ coordinates of these species are collected in the ESI†). | ||
O2 addition produces A2O2 that would eventually generate the A2O alkoxy intermediate. In accordance with our assumption, we found another TS that connects the C2 addition path with the addition on C1. TS1-4 is an H-shift five-member cyclic transition state that goes from A1O to A2O which has approximately 6 kcal mol−1 lower energy. TS1-4 is below reactants by 45.0, 50.6 and 43.4 kcal mol−1 at the M06-2X-D3/aug-cc-pVTZ, MN15/aug-cc-pVTZ and SVECV-f12 levels of theory. Although it seems more likely that the exchange between mechanisms occurs through this TS, the TS1-4 barrier is approximately 10 kcal mol−1 above the TS1-1 and TS1-2 transition states that reach products from A1O (Fig. 5). A2O will release formaldehyde (P3) via C1–C2 bond cleavage to form the 1-hydroxy-2-(methylthio)-ethyl radical intermediate (CH3SCH2CH˙(OH), Ad_Im2), which can react with O2 or decompose via bond cleavage. The reaction with oxygen can take two paths, H-abstraction to form P2 and HO2 or O2 addition and recombination to get the alkoxy intermediate, which can in turn decompose to formic acid (P4) and MSP (prior O2 addition) viaTS1-6. Bond cleavage between C3 and S through TS1-7 produces acetaldehyde (or vinyl alcohol, P5); further oxidation of the H3CS intermediate generates sulfur dioxide (P6), P3 and regenerates the OH radical by H-shifting on the H3CO2 intermediate. Decomposition through the breaking of the C2–C3 bond produces MSP and hydroxy methylene (HC·OH, P3_Im2) that produce P3viaTS_P3. This has not been considered in Fig. 5 since the height of the TS1-8 barrier is about 10 kcal mol−1 above reactants.
 | ||
| Fig. 5 Relative energy diagram for the OH allyl addition in the AMS oxidation initiated by the OH radical at the SVECV-f12 level. A2: addition at C2 in the absence of NOx. | ||
Based again on the values collected in Tables S2–S5 in the ESI,† we have collected in Fig. 5 the schematics of the relative energies in the path for the addition of OH at C2, and both paths have been merged and shown in Fig. 6 to have an easier comparison. It is noticeable in Fig. 6 that both paths through intermediaries A1 and A2 are in equilibrium. Even if A2 cannot be obtained directly from the reactants, which prefer to evolve toward A1, the transition state for the isomerization is under the reactants (at the best level of calculation) and actually A2 is slightly more stable than A1. The reaction of both intermediaries with O2 finally produces the radicals A1O and A2O, with the latter being more stable, and again there is an equilibrium between them with a transition state which is very submerged with respect to reactants.
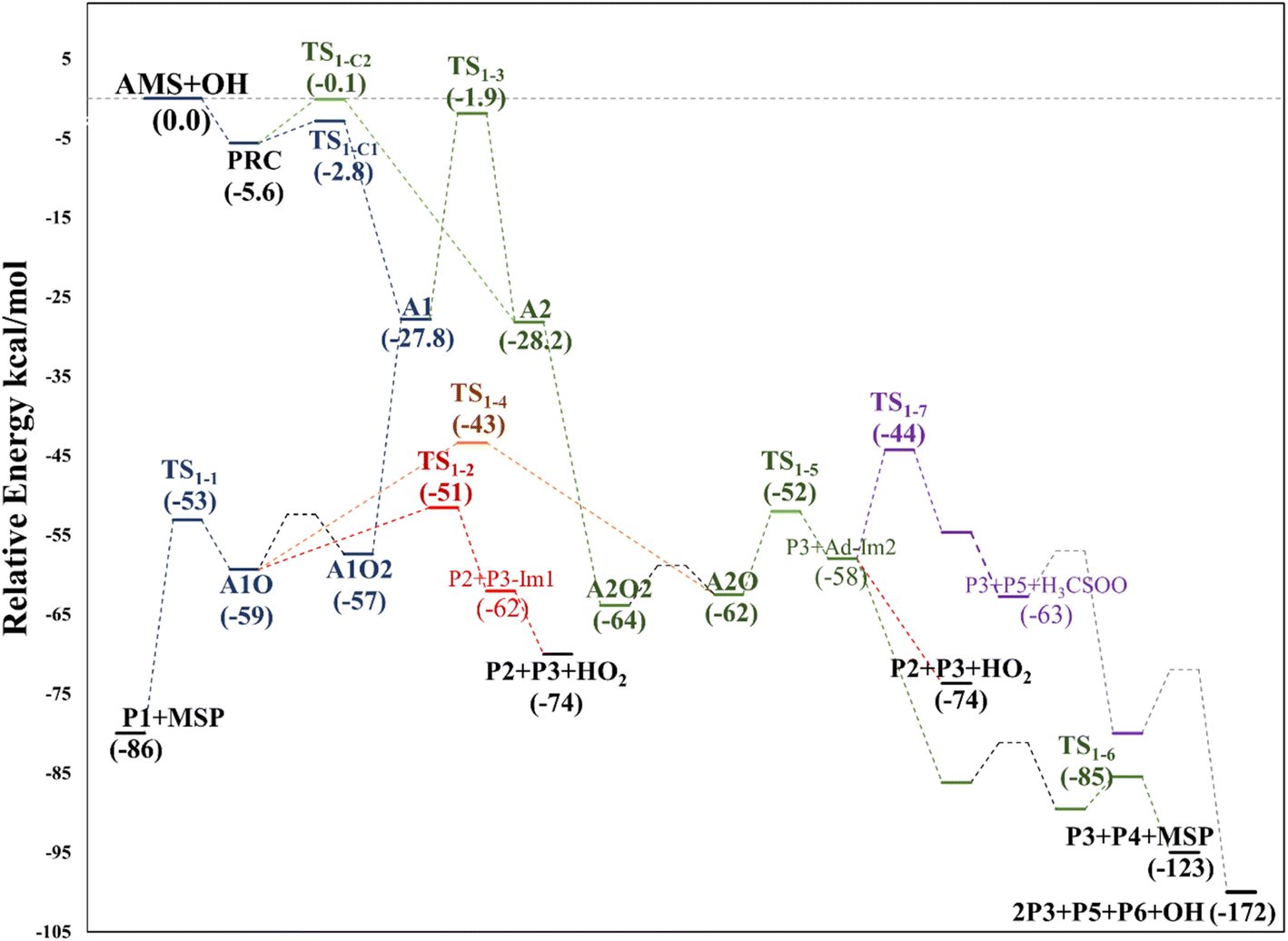 | ||
| Fig. 6 Relative energy diagram for the OH allyl addition in the AMS oxidation initiated by OH radicals at the SVECV-f12 level. Addition paths in the absence of NOx. | ||
Both can surmount a small barrier (7–8 kcal mol−1) to give exactly the same products, P2, P3 and HO2. The only fact that makes the real difference is that A1O can react further with P1 and MSP, while A2O cannot produce P1. On the basis of purely thermochemical reasons, it is to be expected that the mechanism of addition to C1 is preferred with the concomitant appearance of P1 (2-hydroxy acetaldehyde). However, if the barrier of TS1-4 (which is 9.7 kcal mol−1 above TS1-1 but still submerged) is exceeded to get to A2O, the latter would produce P3, P4 and MSP (−123.3 kcal mol−1) viaTS1-5 and TS1-6 (2.3 and 32.4 kcal mol−1 below TS1-1) making it a possible and competitive route, supporting the fact that both P3 and P4 have been identified experimentally. The reaction of MSP ends up producing several reaction products as will be described later.
3.3 Reactions of the peroxy radicals with NO2
Knowledge of the pathways for the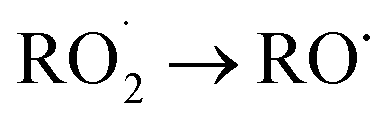 transformation is important for radical recycling in the atmosphere. In this section, we explore the formation of alkoxy radicals by the reaction of the peroxy radicals A1O2 and A2O2 with NO2. In our previous experimental work, NO2 formation was expected when methyl nitrite was used as the precursor of OH radicals (without extra addition of NO). The first step in the reaction with NO2 of both peroxy radicals is the formation of an organonitrate intermediate (ROONO2). It is actually known that organonitrates can serve as NOx reservoirs and can make up a significant fraction of secondary organic aerosols (SOAs).44,45
transformation is important for radical recycling in the atmosphere. In this section, we explore the formation of alkoxy radicals by the reaction of the peroxy radicals A1O2 and A2O2 with NO2. In our previous experimental work, NO2 formation was expected when methyl nitrite was used as the precursor of OH radicals (without extra addition of NO). The first step in the reaction with NO2 of both peroxy radicals is the formation of an organonitrate intermediate (ROONO2). It is actually known that organonitrates can serve as NOx reservoirs and can make up a significant fraction of secondary organic aerosols (SOAs).44,45
The intermediate A1O2–NO2 decomposes directly, forming the alkoxy radical A1O. Once NO3 is released, there is a C2–C3 bond cleavage leading to P1 and the Ad_Im1 radical; the latter eventually produces MSP by O2 addition (Fig. 7).
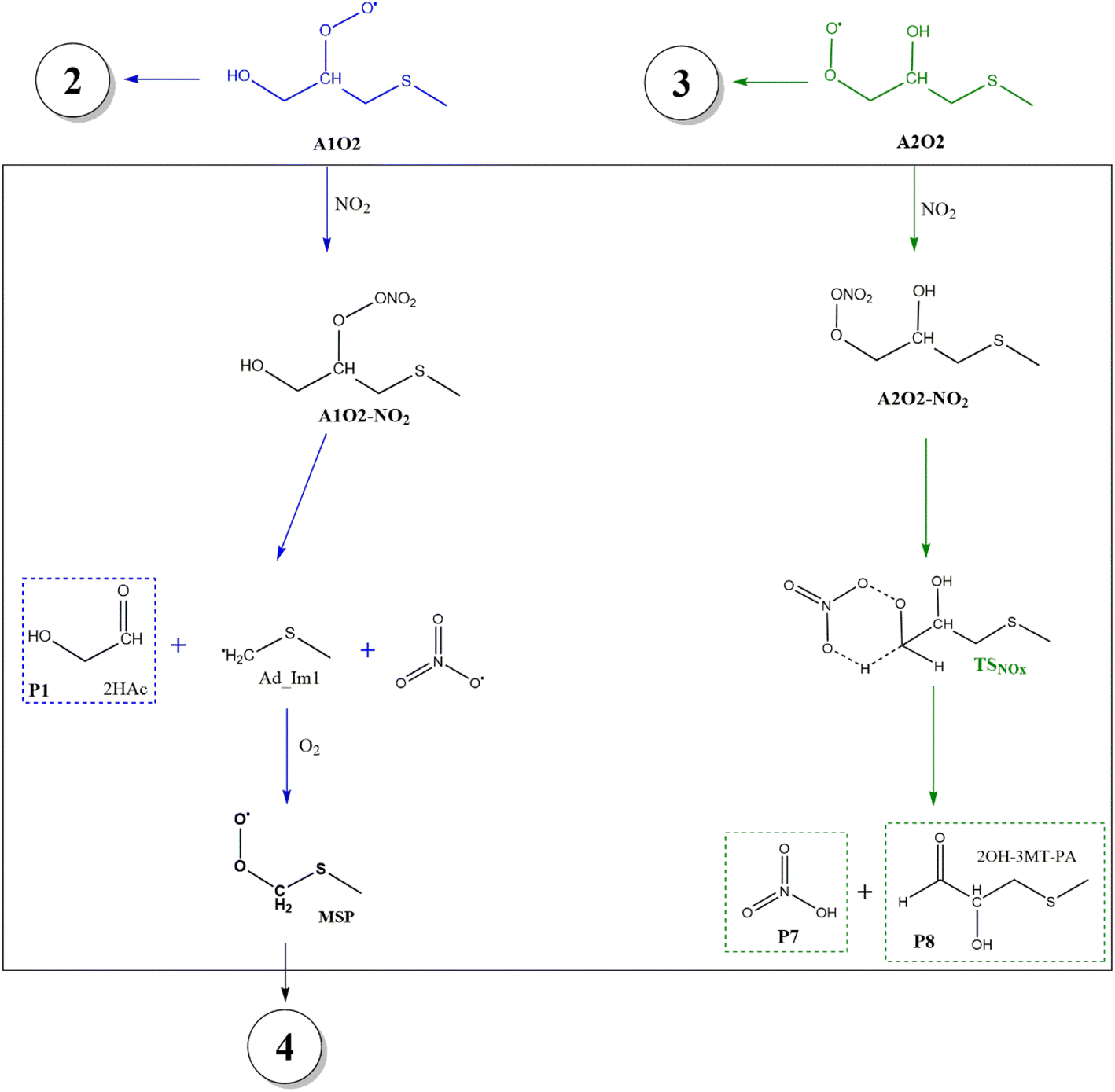 | ||
| Fig. 7 General mechanism for decomposition of RO2 radicals formed in the AMS oxidation initiated by OH radicals in the presence of NOx. Proposed products are shown enclosed in dashed line boxes. | ||
The decomposition of the A2O2–NO2 intermediate occurs through a six-member intermolecular H-abstraction TS, which is 48.6 and 57.0 kcal mol−1 below reactants and 36.3 and 33.5 kcal mol−1 above A2O2–NO2 at the M06-2X-D3/aug-cc-pVTZ and MN15/aug-cc-pVTZ levels of theory, respectively. We calculated in this study that TSNOx leads to HNO3 (P7) and 2-hydroxy-3-methylthio-propanaldehyde (P8). However, these products have not been observed experimentally yet. The relative energies in this process are shown in Fig. 8.
 | ||
| Fig. 8 Relative energy diagram for decomposition of RO2 radicals formed in the AMS oxidation initiated by OH radicals in the presence of NOx at the M06-2X/GD3/aug-cc-pVTZ level. | ||
3.4 Intramolecular rearrangement mechanism in RO2 intermediates
Based on the experimental and theoretical studies for the MSP radical.12–14 we have investigated the similar internal rearrangement mechanism for the RO2 radicals formed in the oxidation of AMS, since there are five possible positions for internal H-abstraction, which may reveal new pathways and reaction products. The scheme with the possible paths is represented in Fig. 9.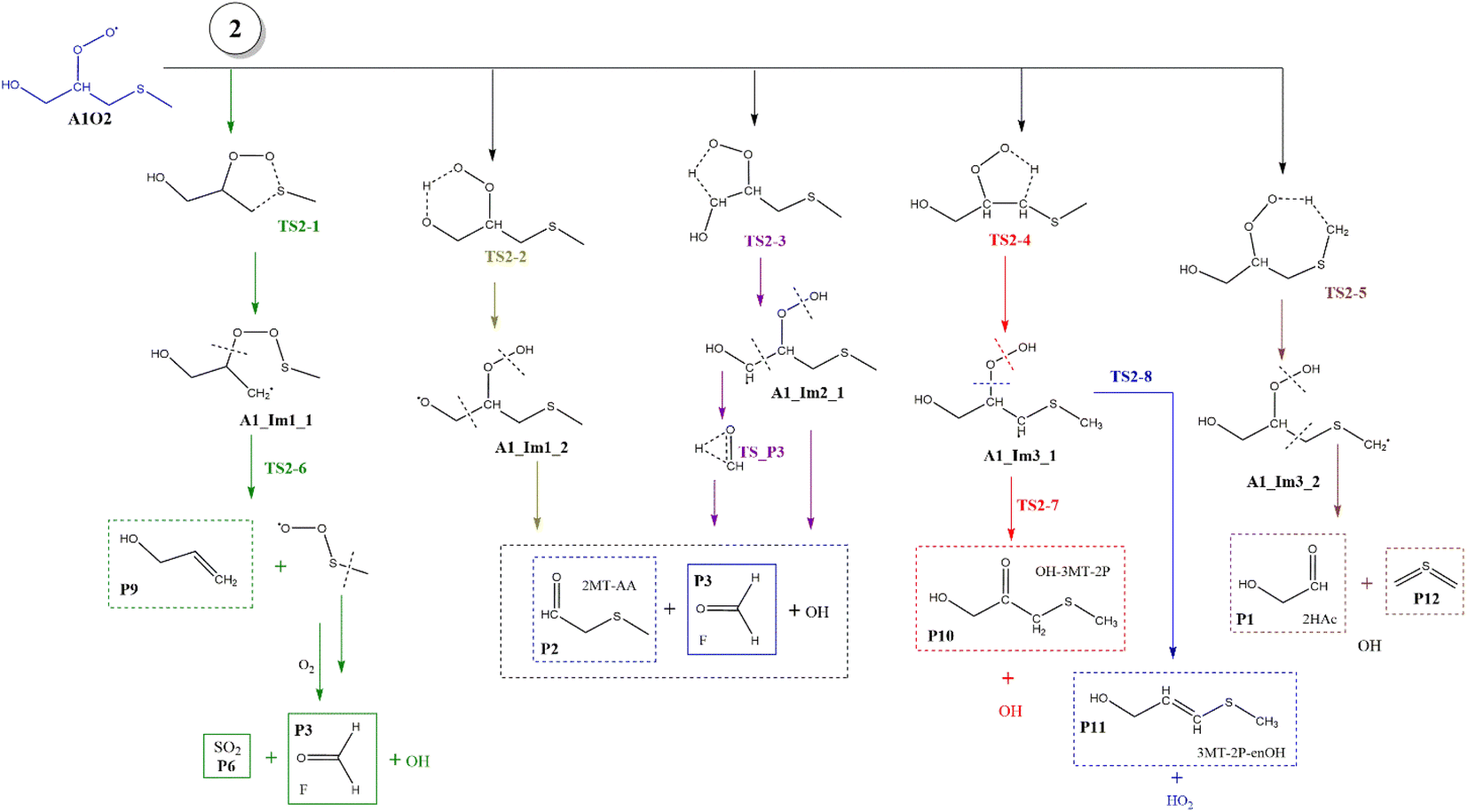 | ||
| Fig. 9 General mechanism of A1O2 intermediate (addition to C1) decomposition in the absence of NOx. Products are shown enclosed in boxes: identified (solid line) and proposed (dashed line). | ||
The internal H-migration from C1 to ROO˙ occurs from the A1_Im2 intermediate. In this structure the value of the HO–C1–C2–OO˙ dihedral angle is 175.0°, leaving the HO and ROO˙ groups in opposite directions. The ROO˙–HC1 distance in the A1_Im2 intermediate is 2.550 Å and the abstraction proceeds through a five-member cyclic TS (TS2-3) in which that distance is reduced to 1.290 Å to obtain the A1_Im2_1 intermediate. The barrier for this process is 26.5 kcal mol−1 from A1_Im2 and 33.1 kcal mol−1 below reactants (Table S2†). Once the A1_Im2_1 intermediate decomposes, it generates P2, the OH radical and the HCOH species that eventually produces P3 (see Fig. 9).
Finally, from the A1_Im3 intermediate, the internal H-abstraction could occur at the C3 and C4 positions. Abstraction at C3 produces A1_Im3_1 through a five-member cyclic TS (TS2-4) which is 27.0 kcal mol−1 above A1_Im3. The A1_Im3_1 intermediate could decompose into two sets of products, OH radicals and hydroxy-3-methylthio-2-propanone (P10) by O–O bond cleavage or HO2 radicals and 3-methylthio-2-propenol (P11) by C–O bond cleavage. Instead, the abstraction at the C4 position (methyl group) goes through a cyclic TS (TS2-5) with a barrier of 19.2 kcal mol−1 which is the lowest in the A1O2 mechanism (Table S2†). Intermediate A1_Im3_2 decomposes to P1 and thioformaldehyde-S-methylide (P12).34,46–48 The energies of all these processes are schematized in Fig. 10.
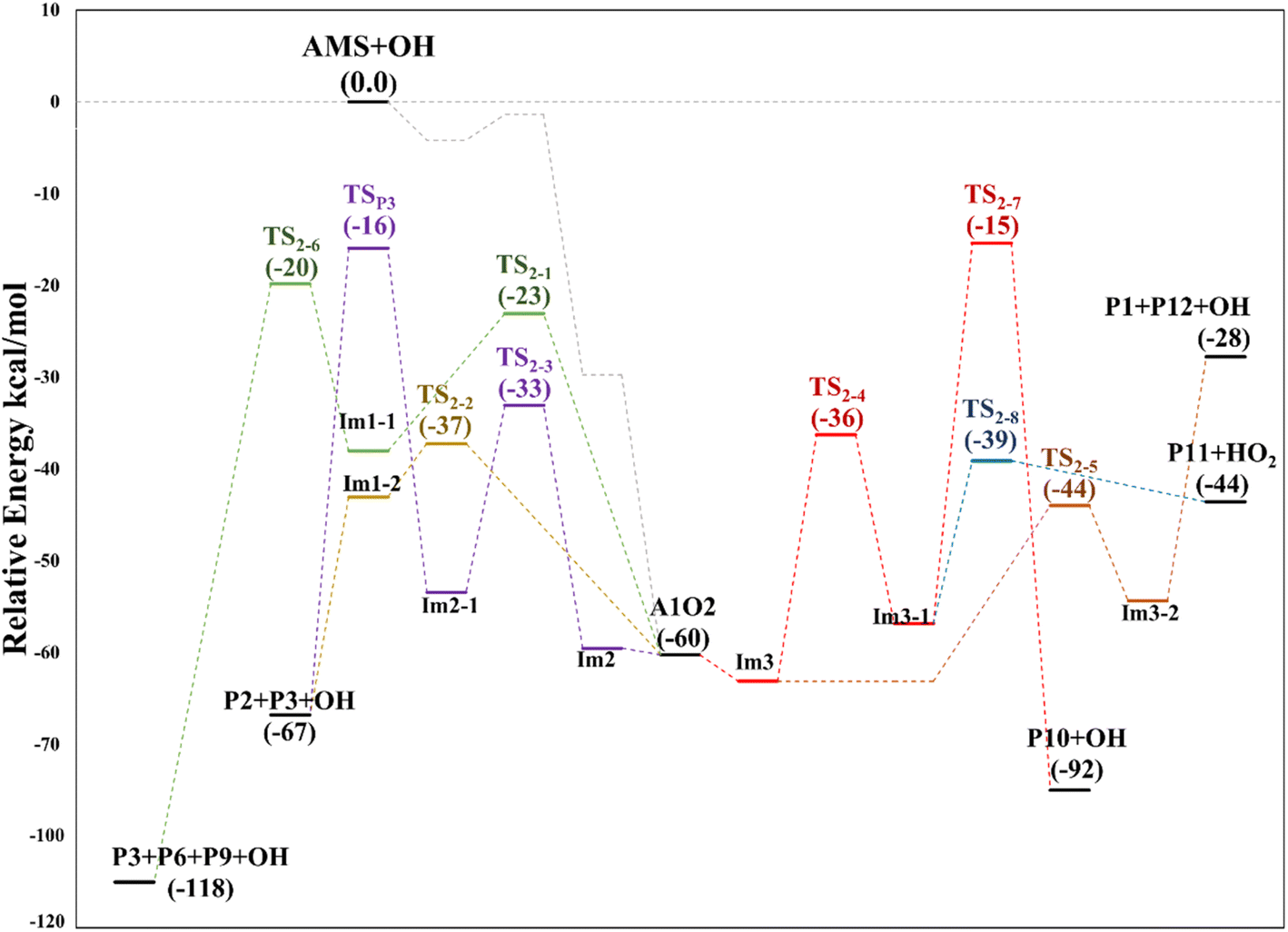 | ||
| Fig. 10 Relative energy diagram for decomposition of the A1O2 intermediate (addition to C1) formed in the AMS oxidation initiated by OH radicals in the absence of NOx at the M06-2X/GD3/aug-cc-pVTZ level. | ||
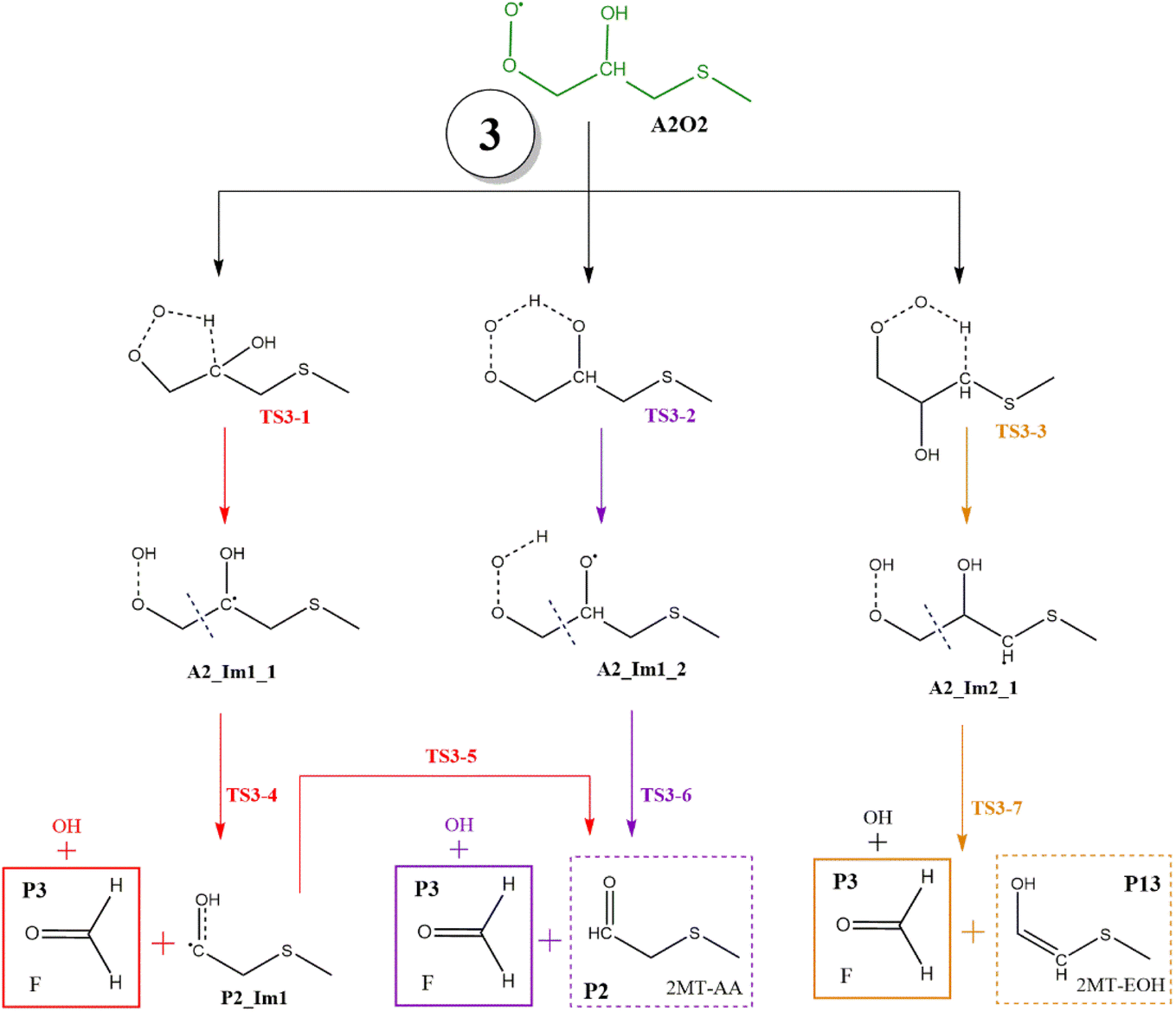 | ||
| Fig. 11 General mechanism of A2O2 intermediate (addition to C2) decomposition in the absence of NOx. Products are shown enclosed in boxes: identified (solid line) and proposed (dashed line). | ||
In all paths, the internal H-abstraction produces an intermediate with the HOOCH2R form, and eventually the HOOCH2- moiety ends up giving OH radicals and P1. Both internal H-abstractions at C2 will also produce P2 through the transition states TS3-1 and TS3-2, whose barriers are 29.3 and 19.3 kcal mol−1 relative to A2_Im1, respectively (Table S3†). However, to obtain P2 from the ˙C(HO)CH2SCH3 radical formed in R3-1 (P2_Im1), it is necessary to reach a high energy barrier of 10.9 kcal mol−1 above the reactants (TS3-5), not a plausible process. Internal H-abstraction from C3 goes through the transition state TS3-3, which has a barrier of 20.6 kcal mol−1 relative to A2_Im2. Final products of this path are P1, OH, and 2-methylthioethanol (P13). The energy schematics are shown in Fig. 12.
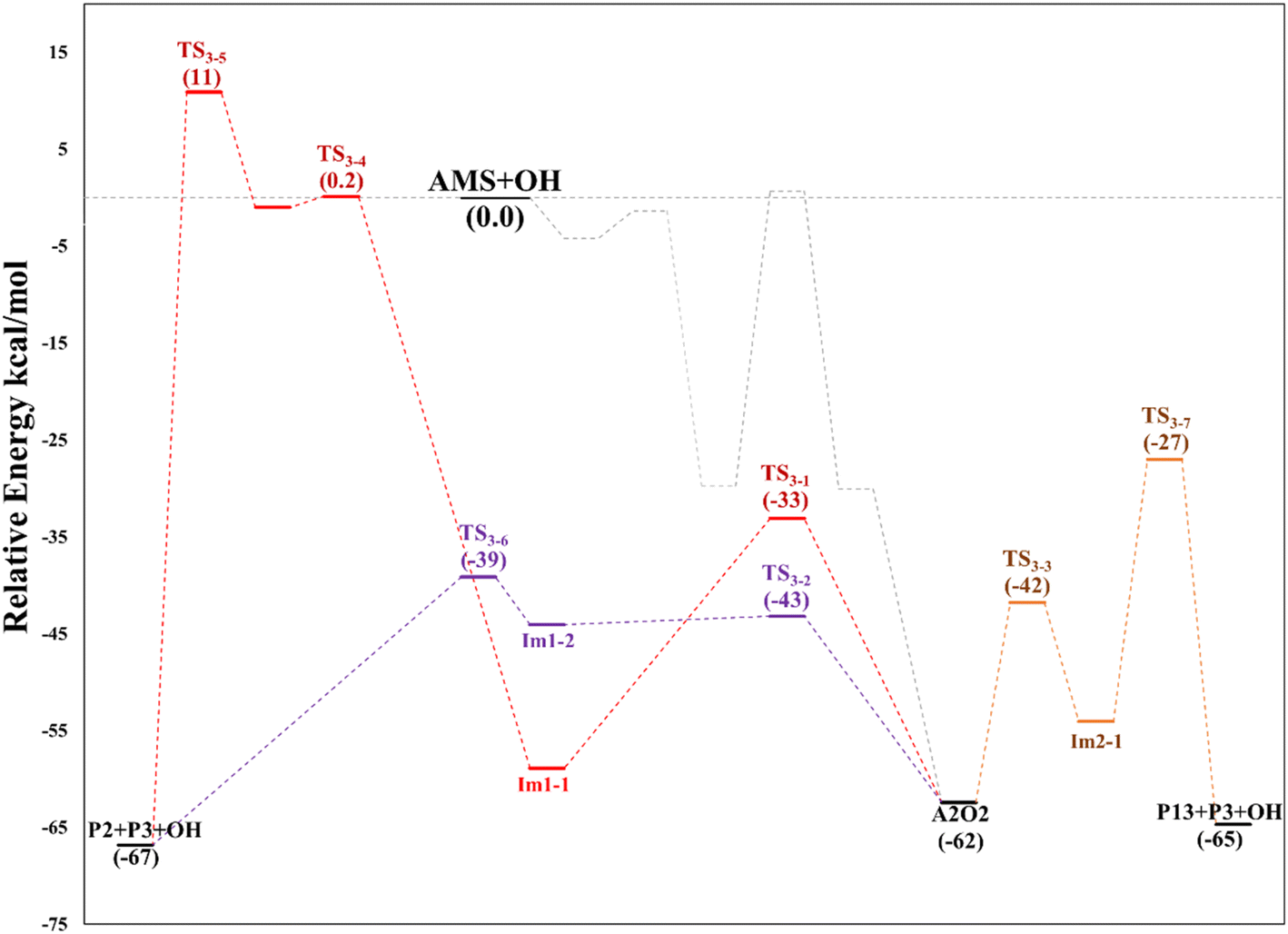 | ||
| Fig. 12 Relative energy diagram for decomposition of the A2O2 intermediate (addition to C2) formed in the AMS oxidation initiated by OH radicals in the absence of NOx at the M06-2X/GD3/aug-cc-pVTZ level. | ||
3.5 MSP decomposition
Although the radical decomposition mechanism of MSP is generally well explored, in this section we report an in depth study of all possible degradation pathways at the same level of calculation as we treated the rest of the mechanism, including intramolecular rearrangement and H-shift paths (Fig. 13). The best-known path for MSP decomposition is recombination to the corresponding alkoxy radical MSO and C–S bond cleavage to form P3 and the methylthiyl radical (CH3S˙). The CH3S˙ radical can either dimerize to dimethyl disulfide (DMDS) or form the methylthiyl peroxy radical (CH3SOO˙) by adding oxygen; the latter then produces another P3 molecule and P6 by S–C bond cleavage. Both the formation of DMDS and P3 + P6 paths occur with low barriers once recombination of RO2 radicals occurs (Fig. 14).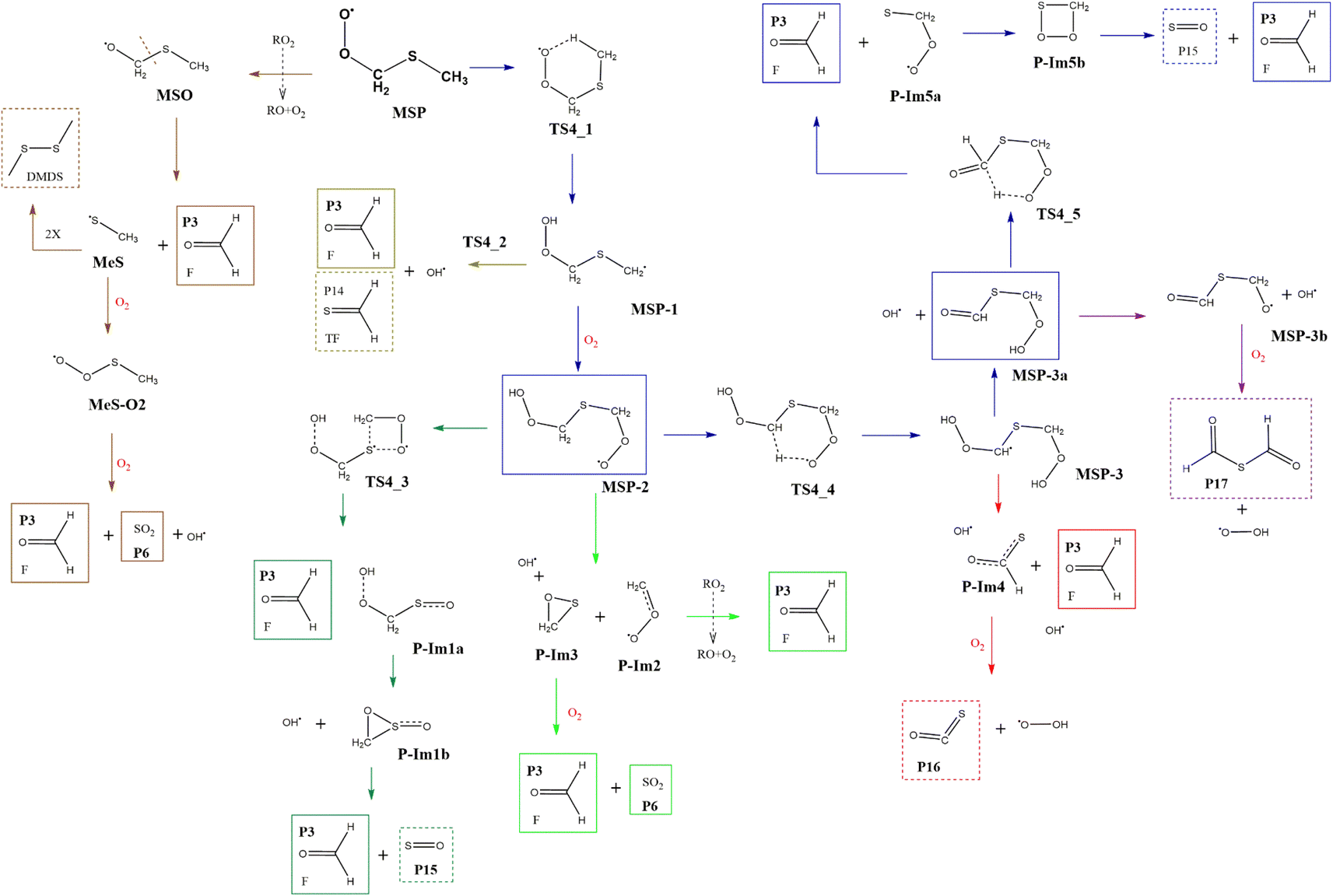 | ||
| Fig. 13 General mechanism of MSP intermediate decomposition in the absence of NOx. Products are shown enclosed in boxes: identified (solid line) and proposed (dashed line). | ||
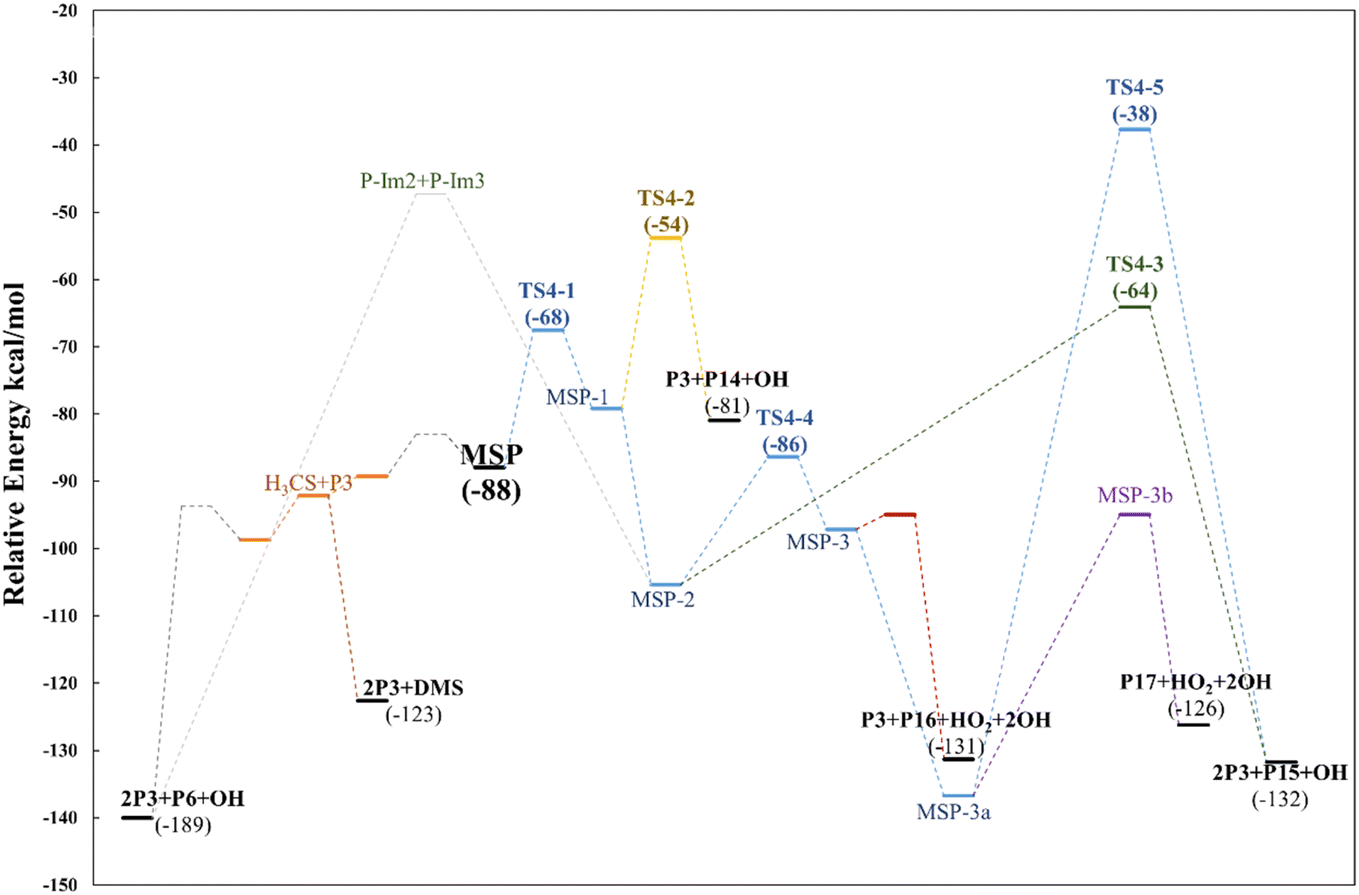 | ||
| Fig. 14 Relative energy diagram for decomposition of the MSP intermediate formed in the AMS oxidation initiated by OH radicals in the absence of NOx at the M06-2X/GD3/aug-cc-pVTZ level. | ||
However, as previously addressed both theoretically and experimentally,12,13,49 in the absence of NOx and at low concentrations of HO2 or RO2 radicals, the MSP isomerization path could be competitive with bimolecular reactions. The first isomerization step via internal H-abstraction has a barrier of approximately 20 kcal mol−1 (energies relative to MSP in this section) and proceeds through a cyclic TS (TS4-1) in which the hydrogen moves from the methyl group to the ROO˙ moiety to generate the MSP-1 intermediate. MSP-1 produces P3, thioformaldehyde (P14) and the OH radical if the reaction proceeds via S–C bond cleavage (TS4-2), with a barrier of 25 kcal mol−1 relative to MSP-1. However, although the MSP-1 intermediate is 9 kcal mol−1 above MSP, the reaction with O2 is barrierless and forms MSP-2, which was already detected experimentally by Berndt et al.12 and is 17.4 kcal mol−1 below MSP (Fig. 14).
Both energies calculated using the SVECV-f12 protocol for TS4-1 and MSP-1, −5.1 and −17.3 kcal mol−1 respectively, (relative to CH3SOO˙ + O2), are in good agreement with those obtained at the UCBS-QB3, ROCBS-QB3, and M06-2X levels by Wu et al.13 However, their energies at the UCBS-QB3 (1.5 kcal mol−1) and ROCBS-QB3 (2.3 kcal mol−1) levels for TS4-2 disagree with that predicted by themselves at the M06-2X level (9.1 kcal mol−1) and with what we obtained using the SVECV-f12 protocol (8.0 kcal mol−1) relative to CH3SOO˙ + O2.
MSP-2 can produce two molecules of P3, OH radical and sulfur monoxide (P15) through the transition state TS4-3 (41.2 kcal mol−1), decompose by breaking the S–C bond, forming two molecules of P3 and one of P6, or can undergo an internal hydrogen migration (through TS4-4, at 19.0 kcal mol−1) generating the MSP-3 intermediary which decomposes to P3, carbonyl sulfide (P16), and HO2 releasing two OH radicals. OH loss from MSP-3 is highly exothermic and produces the MSP-3a intermediate, which was also detected experimentally by Berndt et al.12MSP-3a can then decompose by two different paths in which one OH radical is released for each molecule of the intermediate: (i) decomposition to P17 and HO2 or (ii) formation of two molecules of P3 and one of P15 by intermolecular migration of H (TS4-5, 99.1 kcal mol−1). Wu et al.13 also proposed the reaction of MSP-3a with OH radicals.
Since all transition states are submerged by many kcal mol−1, all paths are kinetically open, but from a thermodynamic point of view, the route leading to 2P3 + P6 is clearly favoured, which agrees with the products observed experimentally. However, it is not implausible that variation in the experimental conditions could lead to other products, of which the one that looks more interesting is formic thioanhydride (P17).
4 Conclusions
As usual for unsaturated compounds, addition to the double bond in the OH-initiated oxidation of AMS is an important decomposition pathway. Although AMS has two possible OH addition positions, the results obtained in this work lead us to assume that the addition does not occur directly but rather through a pre-reactive complex that serves as a previous stage for addition at both the C1 and C2 positions. Based on the energetics of the TSs for the addition pathways, it is also possible to suggest that the addition at C2 could be a branch of the C1 addition mechanism. The addition path to C1 generates 2-hydroxy-acetaldehyde (P1), 2-methyl-thio-acetaldehyde (P2), formaldehyde (P3), and the intermediate methyl thiomethyl peroxy (MSP) radical. If the reaction follows the path in which the OH is attached to C2 instead, the mechanism is more complex. P2, P3, formic acid (P4), acetaldehyde (P5), sulfur dioxide (P6) and the intermediate MSP are generated.In the oxidation initiated by OH, independent of whether at C1 or C2, secondary RO2 radicals are formed by a reaction with oxygen. Further reactions of these radicals with NO2 molecules (simulating polluted atmospheres) and the intramolecular rearrangement implying internal H-shift and subsequent oxidations (pristine atmospheres) were proposed. The energetics and degradation mechanisms of these pathways suggest that it is possible that, in addition to the experimentally identified products, more complex sulfide compounds may be formed, but have not been detected yet. The MSP intermediate decomposition is the main source of sulfur dioxide (P6), which is the product found with the highest yield during the AMS experimental determinations. Again, intramolecular rearrangement and successive oxidations open up a range of new pathways that are thermodynamically accessible and generate products that have not been previously proposed.
In this work, two different levels of theory have been used: DFT and the recently developed SVECV-f12 composite method. Although the results in general show good agreement, thus validating in general the use of DFT methods, some differences were observed in the reaction barrier values, whose accuracy is essential for the calculation of theoretical rate coefficients. Additional calculations for the H abstraction mechanism are currently being carried out to determine rate constants for both oxidation paths (double bond addition and H abstraction) in order to obtain an overall rate constant that allows a full comparison between the theoretical and experimental results.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. L. C. acknowledges CONICET (Argentina) for a doctoral fellowship and support. M. B. B. wishes to acknowledge the Alexander von Humboldt Foundation for the support. M. T. acknowledges FONCYT (Argentina), SECYT (Argentina), and CONICET (Argentina) for financial support of this research. O. N. V. thanks Pedeciba (Uruguay), CSIC (Uruguay) and ANII (Uruguay) for continuing support to the research in their group. Calculations reported in this paper were carried out using ClusterUY (site: https://www.cluster.uy), the cluster at the CCBG (Uruguay) and at the CCAD-UNC, which is part of SNCAD-MinCyT, Argentina. The authors want to thank the reviewers for the careful reading that allowed us to improve the quality and clarity of the manuscript.References
- T. Wu, X. Wang, D. Li and Z. Yi, Emission of volatile organic sulfur compounds (VOSCs) during aerobic decomposition of food wastes, Atmos. Environ., 2010, 44, 5065–5071 CrossRef CAS.
- A. Muezzinoglu, A study of volatile organic sulfur emissions causing urban odors, Chemosphere, 2003, 51, 245–252 CrossRef CAS PubMed.
- H.-X. Cao, K.-X. Zhu, J.-G. Fan and L. Qiao, Garlic-Derived Allyl Sulfides in Cancer Therapy, Anti-Cancer Agents Med. Chem., 2014, 14, 793–799 CrossRef CAS PubMed.
- A. K. Sinha, S. A. Farooqui, A. Sharma, A. Mishra and V. Verma, Reactivity of allyl methyl sulphide, the in vitro metabolite of garlic, with some amino acids and with phospholipid involved in viral infections, J. Biomol. Struct. Dyn., 2022, 40, 565–571 CrossRef CAS PubMed.
- R. J. Charlson, J. E. Lovelock, M. O. Andreae and S. G. Warren, Oceanic phytoplankton, atmospheric sulphur, cloud albedo and climate, Nature, 1987, 326, 655–661 CrossRef CAS.
- B. J. Finlayson-Pitts and J. N. Pitts Jr, Chemistry of the Upper and Lower Atmosphere: Theory, Experiments, and Applications, Elsevier, 1999 Search PubMed.
- M. P. Walavalkar, S. Vijayakumar, A. Sharma, B. Rajakumar and S. Dhanya, Is H Atom Abstraction Important in the Reaction of Cl with 1-Alkenes?, J. Phys. Chem. A, 2016, 120, 4096–4107 CrossRef CAS PubMed.
- C. Pfrang, M. D. King, C. E. Canosa-Mas and R. P. Wayne, Structure-activity relations (SARs) for gas-phase reactions of NO3, OH and O3 with alkenes: an update, Atmos. Environ., 2006, 40, 1180–1186 CrossRef CAS.
- I. Barnes, K. H. Becker and I. Patroescu, FTIR product study of the OH initiated oxidation of dimethyl sulphide: observation of carbonyl sulphide and dimethyl sulphoxide, Atmos. Environ., 1996, 30, 1805–1814 CrossRef CAS.
- M. Albu, I. Barnes, K. H. Becker, I. Patroescu-Klotz, R. Mocanu and T. Benter, Rate coefficients for the gas-phase reaction of OH radicals with dimethyl sulfide: temperature and O2 partial pressure dependence, Phys. Chem. Chem. Phys., 2006, 8, 728–736 RSC.
- I. Barnes, V. Bastian and K. H. Becker, Kinetics and mechanisms of the reaction of OH radicals with dimethyl sulfide, Int. J. Chem. Kinet., 1988, 20, 415–431 CrossRef CAS.
- T. Berndt, W. Scholz, B. Mentler, L. Fischer, E. H. Hoffmann, A. Tilgner, N. Hyttinen, N. L. Prisle, A. Hansel and H. Herrmann, Fast Peroxy Radical Isomerization and OH Recycling in the Reaction of OH Radicals with Dimethyl Sulfide, J. Phys. Chem. Lett., 2019, 10, 6478–6483 CrossRef CAS PubMed.
- R. Wu, S. Wang and L. Wang, New mechanism for the atmospheric oxidation of dimethyl sulfide. The importance of intramolecular hydrogen shift in a CH3SCH2OO radical, J. Phys. Chem. A, 2015, 119, 112–117 CrossRef CAS PubMed.
- P. R. Veres, J. Andrew Neuman, T. H. Bertram, E. Assaf, G. M. Wolfe, C. J. Williamson, B. Weinzierl, S. Tilmes, C. R. Thompson, A. B. Thames, J. C. Schroder, A. Saiz-Lopez, A. W. Rollins, J. M. Roberts, D. Price, J. Peischl, B. A. Nault, K. H. Møller, D. O. Miller, S. Meinardi, Q. Li, J. F. Lamarque, A. Kupc, H. G. Kjaergaard, D. Kinnison, J. L. Jimenez, C. M. Jernigan, R. S. Hornbrook, A. Hills, M. Dollner, D. A. Day, C. A. Cuevas, P. Campuzano-Jost, J. Burkholder, T. Paul Bui, W. H. Brune, S. S. Brown, C. A. Brock, I. Bourgeois, D. R. Blake, E. C. Apel and T. B. Ryerson, Global airborne sampling reveals a previously unobserved dimethyl sulfide oxidation mechanism in the marine atmosphere, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 4505–4510 CrossRef CAS PubMed.
- A. L. Cardona, R. G. Gibilisco, C. B. Rivela, M. B. Blanco, I. Patroescu-Klotz, N. Illmann, P. Wiesen and M. A. Teruel, Kinetics, product distribution and atmospheric implications of the gas-phase oxidation of allyl sulfides by OH radicals, Chemosphere, 2022, 288, 132546 CrossRef CAS PubMed.
- I. Barnes, J. Hjorth, N. Mihalapoulos and N. Mihalopoulos, Dimethyl sulfide and dimethyl sulfoxide and their oxidation in the atmosphere, Chem. Rev., 2006, 106, 940–975 CrossRef CAS PubMed.
- P. H. Wine, N. M. K.reutter, C. A. Gump and A. R. Ravishankara, Kinetics of OH reactions with the atmospheric sulfur compounds H2S, CH3SH, CH3SCH3, and CH3SSCH3, J. Phys. Chem., 1981, 85, 2660–2665 CrossRef CAS.
- L. Aristizabal, M. Ángel, C. Orozco, P. Ruiz, J. Quijano and R. Notario, Computational study of the thermal decomposition and the thermochemistry of allyl ethers and allyl sulfides, Struct. Chem., 2018, 29, 897–907 CrossRef CAS.
- M. Izadyar and M. R. Gholami, Substituent effects on the gas phase reactivity of alkyl allyl sulfides, a theoretical study, J. Mol. Struct.: THEOCHEM, 2006, 759, 11–15 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other function, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132(15), 154104 CrossRef PubMed.
- H. S. Yu, X. He, S. L. Li and D. G. Truhlar, MN15: a Kohn-Sham global-hybrid exchange-correlation density functional with broad accuracy for multi-reference and single-reference systems and noncovalent interactions, Chem. Sci., 2016, 7, 5032–5051 RSC.
- D. E. Woon and T. H. Dunning, Gaussian basis sets for use in correlated molecular calculations. V. Core-valence basis sets for boron through neon, J. Chem. Phys., 1995, 103, 4572–4585 CrossRef CAS.
- L. Goerigk, A. Hansen, C. Bauer, S. Ehrlich, A. Najibi and S. Grimme, A look at the density functional theory zoo with the advanced GMTKN55 database for general main group thermochemistry, kinetics and noncovalent interactions, Phys. Chem. Chem. Phys., 2017, 19, 32184–32215 RSC.
- O. N. Ventura, M. Kieninger, A. Katz, M. Vega-Teijido, M. Segovia and K. Irving, SVECV-f12: benchmark of a composite scheme for accurate and cost effective evaluation of reaction barriers, Int. J. Quantum Chem., 2021, 121, 1–19 CrossRef.
- T. B. Adler, G. Knizia and H.-J. Werner, A simple and efficient CCSD(T)-F12 approximation, J. Chem. Phys., 2007, 127, 221106 CrossRef PubMed.
- G. Knizia, T. B. Adler and H.-J. Werner, Simplified CCSD(T)-F12 methods: theory and benchmarks, J. Chem. Phys., 2009, 130, 054104 CrossRef PubMed.
- K. A. Peterson, T. B. Adler and H.-J. Werner, Systematically convergent basis sets for explicitly correlated wavefunctions: the atoms H, He, B–Ne, and Al–Ar, J. Chem. Phys., 2008, 128, 084102 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, 2019.
- H.-J. Werner, P. J. Knowles, F. R. Manby, J. A. Black, K. Doll, A. Heßelmann, D. Kats, A. Köhn, T. Korona, D. A. Kreplin, Q. Ma, T. F. Miller, A. Mitrushchenkov, K. A. Peterson, I. Polyak, G. Rauhut and M. Sibaev, The Molpro quantum chemistry package, J. Chem. Phys., 2020, 152, 144107 CrossRef CAS PubMed.
- F. Neese, The ORCA program system, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, The ORCA quantum chemistry program package, J. Chem. Phys., 2020, 152, 224108 CrossRef CAS PubMed.
- F. Neese, Software update: the ORCA program system—Version 5.0, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 1–15 Search PubMed.
- Z. Salta, J. Lupi, V. Barone and O. N. Ventura, H-Abstraction from Dimethyl Sulfide in the Presence of an Excess of Hydroxyl Radicals. A Quantum Chemical Evaluation of Thermochemical and Kinetic Parameters Unveils an Alternative Pathway to Dimethyl Sulfoxide, ACS Earth Space Chem., 2020, 4, 403–419 CrossRef CAS.
- R. Atkinson, Rate constants for the atmospheric reactions of alkoxy radicals: an updated estimation method, Atmos. Environ., 2007, 41, 8468–8485 CrossRef CAS.
- A. Bossolasco, E. P. Faragó, C. Schoemaecker and C. Fittschen, Rate constant of the reaction between CH3O2 and OH radicals, Chem. Phys. Lett., 2014, 593, 7–13 CrossRef CAS.
- E. P. Faragó, C. Schoemaecker, B. Viskolcz and C. Fittschen, Experimental determination of the rate constant of the reaction between C2H5O2 and OH radicals, Chem. Phys. Lett., 2015, 619, 196–200 CrossRef.
- C. Fittschen, L. K. Whalley and D. E. Heard, The Reaction of CH3O2 Radicals with OH Radicals: a Neglected Sink for CH3O2 in the Remote Atmosphere, Environ. Sci. Technol., 2014, 48, 7700–7701 CrossRef CAS PubMed.
- I. Barnes, K. H. Becker and N. Mihalopoulos, An FTIR product study of the photooxidation of dimethyl disulfide, J. Atmos. Chem., 1994, 18, 267–289 CrossRef CAS.
- C. Bacher, G. S. Tyndall and J. J. Orlando, The Atmospheric Chemistry of Glycolaldehyde, J. Atmos. Chem., 2001, 39, 171–189 CrossRef CAS.
- I. Magneron, A. Mellouki, G. Le Bras, G. K. Moortgat, A. Horowitz and K. Wirtz, Photolysis and OH-Initiated Oxidation of Glycolaldehyde under Atmospheric Conditions, J. Phys. Chem. A, 2005, 109, 4552–4561 CrossRef CAS PubMed.
- S. So, U. Wille and G. da Silva, A Theoretical Study of the Photoisomerization of Glycolaldehyde and Subsequent OH Radical-Initiated Oxidation of 1,2-Ethenediol, J. Phys. Chem. A, 2015, 119, 9812–9820 CrossRef CAS PubMed.
- C. B. Rivela, A. L. Cardona, M. B. Blanco, I. Barnes, M. Kieninger, O. N. Ventura and M. A. Teruel, Degradation mechanism of 2-fluoropropene by Cl atoms: experimental and theoretical products distribution studies, Phys. Chem. Chem. Phys., 2022, 24, 5094–5108 RSC.
- N. L. Ng, S. S. Brown, A. T. Archibald, E. Atlas, R. C. Cohen, J. N. Crowley, D. A. Day, N. M. Donahue, J. L. Fry, H. Fuchs, R. J. Griffin, M. I. Guzman, H. Herrmann, A. Hodzic, Y. Iinuma, J. L. Jimenez, A. Kiendler-Scharr, B. H. Lee, D. J. Luecken, J. Mao, R. McLaren, A. Mutzel, H. D. Osthoff, B. Ouyang, B. Picquet-Varrault, U. Platt, H. O. T. Pye, Y. Rudich, R. H. Schwantes, M. Shiraiwa, J. Stutz, J. A. Thornton, A. Tilgner, B. J. Williams and R. A. Zaveri, Nitrate radicals and biogenic volatile organic compounds: oxidation, mechanisms, and organic aerosol, Atmos. Chem. Phys., 2017, 17, 2103–2162 CrossRef CAS PubMed.
- B. H. Lee, C. Mohr, F. D. Lopez-Hilfiker, A. Lutz, M. Hallquist, L. Lee, P. Romer, R. C. Cohen, S. Iyer, T. Kurtén, W. Hu, D. A. Day, P. Campuzano-Jost, J. L. Jimenez, L. Xu, N. L. Ng, H. Guo, R. J. Weber, R. J. Wild, S. S. Brown, A. Koss, J. de Gouw, K. Olson, A. H. Goldstein, R. Seco, S. Kim, K. McAvey, P. B. Shepson, T. Starn, K. Baumann, E. S. Edgerton, J. Liu, J. E. Shilling, D. O. Miller, W. Brune, S. Schobesberger, E. L. D'Ambro and J. A. Thornton, Highly functionalized organic nitrates in the southeast United States: contribution to secondary organic aerosol and reactive nitrogen budgets, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 1516–1521 CrossRef CAS PubMed.
- Z. Salta, J. Lupi, N. Tasinato, V. Barone and O. N. Ventura, Unraveling the role of additional OH-radicals in the H-abstraction from dimethyl sulfide using quantum chemical computations, Chem. Phys. Lett., 2020, 739, 136963 CrossRef CAS.
- Z. Salta, M. E. Segovia, A. Katz, N. Tasinato, V. Barone and O. N. Ventura, Isomerization and Fragmentation Reactions on the [C2SH4] Potential Energy Surface: The Metastable Thione S -Methylide Isomer, J. Org. Chem., 2021, 86, 2941–2956 CrossRef CAS PubMed.
- Z. Salta, M. Vega-Teijido, A. Katz, N. Tasinato, V. Barone and O. N. Ventura, Dipolar 1,3-cycloaddition of thioformaldehyde S-methylide (CH2SCH2) to ethylene and acetylene. A comparison with (valence) isoelectronic O3, SO2, CH2OO and CH2SO, J. Comput. Chem., 2022, 43(21), 1420–1433 CrossRef CAS PubMed.
- T. Berndt, J. Chen, K. H. Møller, N. Hyttinen, N. L. Prisle, A. Tilgner, E. H. Hoffmann, H. Herrmann and H. G. Kjaergaard, SO2 formation and peroxy radical isomerization in the atmospheric reaction of OH radicals with dimethyl disulfide, Chem. Commun., 2020, 56, 13634–13637 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Optimized geometries and vibrational frequencies of species considered in the AMS oxidation initiated by OH radicals and thermochemistry tables. See DOI: https://doi.org/10.1039/d3ea00010a |
| This journal is © The Royal Society of Chemistry 2023 |