
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Co-photolysis of mixed chromophores affects atmospheric lifetimes of brown carbon†
Yalin
Wang
a,
Tian
Qiu
a,
Cong
Zhang
a,
Tianwei
Hao
a,
Brix Raphael
Go
b,
Ruifeng
Zhang
b,
Masao
Gen
c,
Man Nin
Chan
 d,
Dan Dan
Huang
e,
Xinlei
Ge
f,
Junfeng
Wang
f,
Lin
Du
g,
Ru-Jin
Huang
h,
Qi
Chen
i,
Ka In
Hoi
a,
Kai Meng
Mok
a,
Chak K.
Chan
bj and
Yong Jie
Li
*a
d,
Dan Dan
Huang
e,
Xinlei
Ge
f,
Junfeng
Wang
f,
Lin
Du
g,
Ru-Jin
Huang
h,
Qi
Chen
i,
Ka In
Hoi
a,
Kai Meng
Mok
a,
Chak K.
Chan
bj and
Yong Jie
Li
*a
aDepartment of Civil and Environmental Engineering, Department of Ocean Science and Technology, Centre for Regional Oceans, Faculty of Science and Technology, University of Macau, Macau 999078, China. E-mail: yongjieli@um.edu.mo
bSchool of Energy and Environment, City University of Hong Kong, Hong Kong 999077, China
cInstitute of Multidisciplinary Research for Advanced Materials, Tohoku University, 2-1-1, Katahira, Sendai, Miyagi 980-8577, Japan
dEarth System Science Programme, Faculty of Science, Institute of Environment, Energy, and Sustainability, The Chinese University of Hong Kong, Hong Kong 999077, China
eShanghai Academy of Environmental Sciences, Shanghai 200233, China
fJiangsu Key Laboratory of Atmospheric Environment Monitoring and Pollution Control (AEMPC), School of Environmental Science and Engineering, Nanjing University of Information Science & Technology, Nanjing 210044, China
gEnvironment Research Institute, Shandong University, Shanda South Road 27, Shandong 250100, China
hState Key Laboratory of Loess and Quaternary Geology, Institute of Earth Environment, Chinese Academy of Sciences, Xi'an 710061, China
iState Key Joint Laboratory of Environmental Simulation and Pollution Control, BIC-ESAT, IJRC, College of Environmental Sciences and Engineering, Peking University, Beijing 100871, China
jPhysical Sciences and Engineering Division, King Abdullah University of Science and Technology, Kingdom of Saudi Arabia
First published on 26th June 2023
Abstract
Brown carbon (BrC) affects radiation budget and thus global climate by absorbing light, during which photolysis can also occur and serve as an important sink of BrC. Yet, the interactive roles of mixed chromophores during BrC co-photolysis, which is anticipated in ambient aerosol particles, is seldom explored, making a model representation of atmospheric lifetimes of BrC highly uncertain. Herein, we investigate the photolysis of four typical atmospheric BrC chromophores (two methoxyphenols, MPs, and two nitrophenols, NPs), alone or mixed, with a wide range of concentrations as anticipated in atmospheric cloud/fog and aerosol droplets. The photo-decay rate constants (k) for the photolysis of single chromophores generally decrease as BrC concentration increases, but the effective quantum yields (Φe) are relatively constant in optically thin solutions; the Φe values increase by one order of magnitude in optically thick solutions for most BrC chromophores studied and even exceed 1 when BrC concentrations are high (e.g., 500 mM in isopropanol solutions), presumably due to secondary reactions involving their triplet states. During co-photolysis of two mixed chromophores, MPs increase the Φe values of NPs by factors of 4–26, but NPs affect the Φe values of MPs insignificantly, indicating a stronger sensitizing effect of MPs than NPs. In contrast, NPs mainly exert a shadowing effect on MP degradation, which only affects photo-decay rate constant but not effective quantum yield and is negligible for fine particles. Estimation of atmospheric lifetimes suggests that sensitizing by MPs can accelerate NP degradation by a factor of up to 30 while shadowing between them plays minor roles in fine particles but non-negligibly in coarse particles.
Environmental significanceAtmospheric brown carbon (BrC) absorbs light and undergoes photolysis simultaneously, thereby affecting global climate and atmospheric chemistry. Co-photolysis is expected for complex BrC chromophore mixtures in ambient aerosol particles. The interactive roles between different chromophores during BrC co-photolysis were equivocally explained as competition for light or for reactive species previously, with little consideration of the potential cross-sensitizing effects. We herein study the co-photolysis of methoxyphenols and nitrophenols from a perspective of shadowing and sensitizing. The shadowing effect does not affect atmospheric lifetimes in small particles but might do so in coarse particles. The sensitizing effect of methoxyphenols can significantly accelerate the degradation of nitrophenols, but not vice versa. This work elucidates the effects of BrC type, BrC concentration, and particle size on atmospheric lifetimes due to co-photolysis from the perspective of shadowing and sensitizing, which should be considered in chemical transport models for complex aerosol particles with mixed BrC chromophores. |
1 Introduction
Atmospheric brown carbon (BrC) can efficiently absorb radiation in the near-ultraviolet (UV) and visible ranges, affecting air quality, atmospheric chemistry, and climate forcing.1–4 There are two major sources of BrC: (1) direct emissions such as the combustion of biomass5–7 or fossil fuels8,9 and (2) secondary formation of electron-rich products such as carbonyls, aromatics, as well as nitrophenols.10–13 The positive radiative forcing of BrC is attributable to the photophysical process of energy dissipation from their electronically excited states upon light absorption.4,14 Photochemical processes of the excited BrC, on the other hand, alter their chemical identities,15–17 forming either less-absorbing (bleaching)18,19 or more-absorbing (further browning) products.20–22 Photosensitized reactions of BrC through their triplet state (3BrC*) also lead to the formation of reactive oxygen species (ROS) such as hydroxyl radical (OH), superoxide (O2−), and singlet oxygen (1O2).20,23–25 These species participate in atmospheric chemistry by acting as oxidants and accelerate the degradation of BrC via secondary reactions,26,27 thereby affecting atmospheric lifetimes of BrC. The atmospheric lifetimes of the chromophores during various bleaching processes28,29 dictate to a great extent the warming potency of BrC.30,31Experimental studies of BrC photodegradation have been conducted with direct photolysis,32,33 as well as those mediated by H2O2 (as a precursor of OH radicals),33,34 photosensitizers,23,35 and nitrate.20,21 The photochemical lifetime (τ, s), as the reciprocal of the photolysis rate constant (j, s−1) under solar irradiation, is linked to the experimentally determined pseudo first-order photo-decay rate constant (k, s−1) through intrinsic quantum yield of BrC degradation (Φ, dimensionless). All these parameters are wavelength-dependent and can be obtained from experiments using lights with wavelengths as much atmospherically relevant as possible. To obtain Φ from k, experiments were also generally conducted in optically thin (dilute) solutions, as in cloud and fog waters, to avoid light competition and secondary reactions.35,36 The BrC concentration in aerosol liquid water, however, can be orders of magnitude higher than those in cloud or fog waters.36–39 Photolysis experiments of BrC are still widely performed in bulk photochemical reactors, in need of comprehensive optical and chemical characterizations. To simulate BrC photolysis at high concentrations as in aerosol particles, light competition is highly probable in those reactors because light attenuation by a long optical path of one or a few centimeters (cm) would be much higher than that in fine aerosol particles of less than 1 micrometer (μm). This effect might lead to concentration dependence of measured photodegradation rates. Indeed, concentration-dependent photodegradation rates of BrC were observed,23,35 which was attributed to the decrease in volume-averaged photon flux. Such dependence was quantified using an internal light screening factor that characterized the self-shadowing effect.35 On the other hand, at high concentrations for BrC photolysis, secondary reactions are deemed inevitable if the chromophores are capable of photosensitizing. Self-sensitizing normally accelerates the photodegradation of BrC via their triplet states or the reactive intermediates (e.g., ROS) generated.23,40,41
A light-absorbing subset of compositionally highly complex organic aerosol (OA),42,43 BrC is also a mixture44 with chromophores originating from both primary emission and secondary formation.15 The photophysical process of light absorption by mixed BrC chromophores is not simply additive, with featureless tailing absorption extending to longer wavelengths, due to the formation of charge-transfer complexes45 and/or almost a continuum of electronic states.46 It is anticipated that the photochemical processes of mixed BrC chromophores are not additive either because the chemistry involved is even more intertwined and non-linear than light absorption. In ambient aerosol particles, inorganics such as sulfate and nitrate are normally mixed with BrC chromophores. Therefore, degradation of BrC in mixtures with those inorganic components has been studied widely,21,47–50 including our previous study51 on interactive photolysis of BrC and nitrate. There are, however, few investigations on the interactions between mixed BrC chromophores during (co)-photolysis,23,35,52 which are also generally expected in ambient aerosol particles.53,54 The cross-shadowing (light screening) and cross-sensitizing (secondary reaction) effects during co-photolysis of BrC chromophores are even less explored than the concentration dependence, hindering accurate estimation of their atmospheric lifetimes to assess their effects on global climate and atmospheric chemistry.
We investigate herein the shadowing and sensitizing effects in the co-photolysis of different BrC chromophores using a bulk photochemical reactor, especially in elevated concentrations approaching those in ambient aerosol particles. The shadowing effect in this work refers to the light competition between BrC chromophores and is estimated with relative light absorption fraction fi(λ) from their absorptivity and concentration. The sensitizing effect refers to the oxidation reaction initiated by the excited triplet state BrC and contributes to BrC degradation, including direct H-abstraction, electron transfer, energy transfer, and ROS generation. We conjecture that the shadowing and sensitizing effects depend on the structural and absorptive features of the chromophores, i.e., BrC types. We test this hypothesis by investigating the photolysis of two methoxyphenols (MPs: vanillic acid, VA, and vanillin, VL) and two nitrophenols (NPs: 4-nitrocatechol, 4NC, and 4-nitrophenol, 4NP) under different concentrations, alone or mixed. These phenolic compounds are typical BrC chromophores from primary emission (e.g., MPs from biomass burning)55,56 and/or secondary formation (e.g., NPs from NO2-mediated oxidation of aromatics).57 The structures of the four model BrC chromophores are shown in Fig. S1 in the ESI,† and their absorption spectra in Fig. S2,† together with the emission spectra of the lamp used and typical solar radiation. Experiments were mainly conducted in air-saturated aqueous solutions, but selected experiments were also performed in an organic solvent or under N2-saturated conditions to investigate the matrix dependence and the roles of ROS, respectively. Mass spectrometric analysis was also performed for some experiments to probe the product formation during (co-)photolysis of selected chromophores. Shadowing and sensitizing effects induced by these BrC model compounds are discussed with experimentally determined photo-decay rate constants (k), calculated effective quantum yields of BrC degradation (Φe), and estimated atmospheric lifetimes (τ) via transformed photolysis rate constants (j) as in aerosol particles.
2 Materials and methods
2.1 Chemicals
VA (≥97%), VL (99%), 4NC (97%), and 4NP (99%) were obtained from Sigma-Aldrich. Their structures and absorption spectra are shown in Fig. S1 and S2, respectively, in the ESI.† Isopropanol (IPA, ≥99.7%) was from Xilong Scientific, sulfuric acid (H2SO4, 95% wt) was from Acros Organics, and sodium hydroxide (NaOH, 98%) was from Fluka. Solutions were prepared with deionized (DI) water from a Milli-Q water purification system (Direct-Q 3UV system). All chemicals were used as received without further purification.2.2 Photolysis and measurements
We used a photochemical reactor equipped with a xenon lamp (NBeT Co. Ltd., 300 W), a 100 mL Pyrex glass reactor with a magnetic stirrer, a quartz window (GGS1, NBeT-PH100), and a circulating water-cooled system (298 ± 1 K) to perform the BrC photolysis experiments, as in previous work.51 The output light of the lamp in this study was filtered by an optical filter (Newport, FSQ-WG305) to screen off the light below 309 nm to simulate solar radiation. The photon flux was measured by a spectrometer (Brolight, BIM-6002), with the spectrum shown in Fig. S2.† We performed photolysis under different conditions as shown in Tables S1–S3.† The concentrations of these BrC compounds were set as 0.02, 0.05, 0.5, 2, and 5 mM for direct photolysis in aqueous air-saturated solutions. Each BrC chromophore of 0.05 or 0.5 mM was mixed with another BrC chromophore to study the co-photolysis of BrC. It should be noted that the light absorption of BrC containing carboxylic or phenolic moieties exhibits pH dependence.58 Therefore, all aqueous solutions in this study were adjusted to a fixed pH of 5 by H2SO4 or NaOH before photolysis. We also used IPA instead of water as the solvent for VL and 4NC (Table S2†) in some selected experiments to study the matrix effect or to obtain VL solution with high enough concentration (500 mM) in some cases. To investigate the roles of molecular oxygen (ROS generation) during the photolysis, some experiments were conducted under N2-saturated conditions, in which nitrogen (N2, 99.999%, 0.5 L min−1) was introduced into solutions for 30 min before irradiation and continued throughout the photolysis experiments.20 Unless otherwise specified, the data are from air-saturated conditions. Each experiment was repeated independently at least 3 times, and measurements were conducted in triplicate. The error bar in the figures represents the standard deviations of three experimental results.Aliquots of illuminated solutions were collected for precursor concentration measurements with liquid chromatography, as in our previous study.21 The pseudo-first-order decay rate constant, k, was estimated by fitting the exponential decay of the BrC compounds.21,23 Products were analyzed from some experiments with a high-performance liquid chromatograph-mass spectrometer equipped with an Orbitrap mass analyzer (HPLC-Orbitrap-MS, Thermo) as in our previous study.21 The mass spectra were obtained in the negative mode (the positive ion mode was tested, but signals were relatively low). The m/z scan range was set from 50 to 500 dalton (Da). The mass spectral data were analyzed with a free toolbox MZmine (https://mzmine.github.io/).
2.3 Calculations
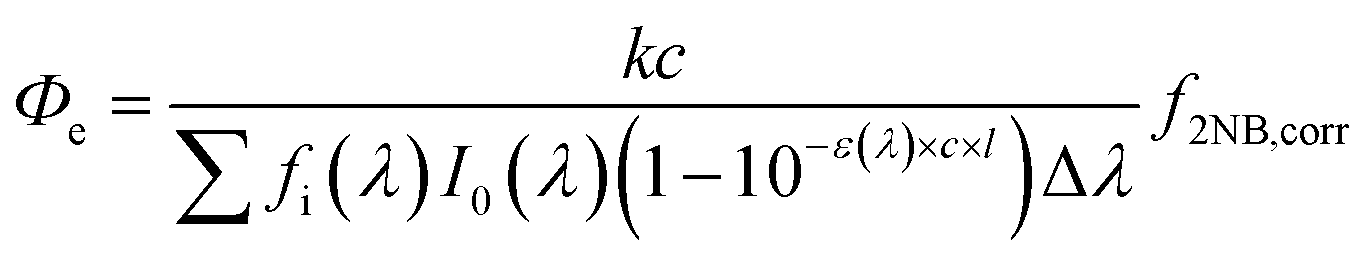 | (1) |
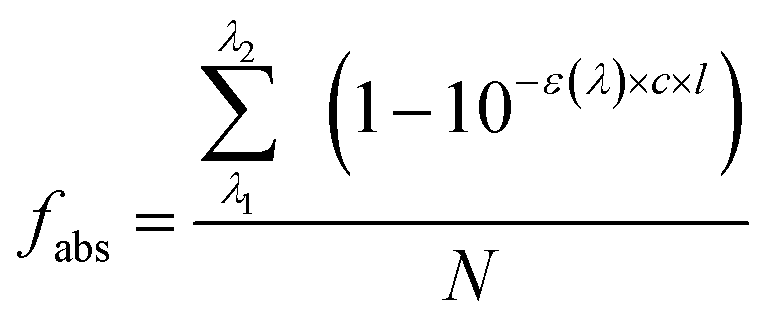 | (2) |
| j1 =3.82 × 10−21Φ∑F(λ)ε(λ)Δλ | (3) |
In concentrated aerosol particles, however, the light might not reach every single BrC molecule, and the effect is more prominent if the light path is longer (e.g., for larger particles). Therefore, another equation as below,59 was used to take concentration and droplet size into account.
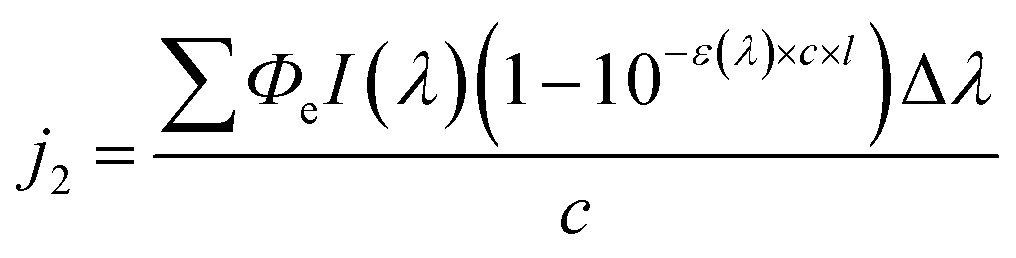 | (4) |
We refer to this method as Method 2, and j2 is applicable to all cases regarding the BrC concentration range. The reciprocal of j1 or j2 is taken as the atmospheric lifetime (τ) of the BrC chromophore due to photolysis:
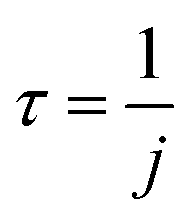 | (5) |
More details about the estimation of atmospheric lifetimes are provided in Section S3 in the ESI.†
3 Results and discussion
3.1 Decay rate constant
The pseudo first-order decay rate constants (k, Fig. 1) during aqueous-phase photolysis generally decrease as BrC concentrations increase, consistent with previous studies that attributed this trend to competition for light (self-shadowing).23,35,61,62 At low concentrations (≤0.05 mM), the k values of MPs (VA and VL, ∼5 × 10−4 s−1) are approximately 10 times higher than those of NPs (4NC and 4NP, ∼5 × 10−5 s−1), consistent with our previous study using 254 nm and 313 nm lamps.21 Above 0.5 mM, the k values of VA rapidly decrease to ∼5 × 10−5 s−1 (Fig. 1A), while those downward trends of VL, 4NC, and 4NP slow down or even become stable (Fig. 1B–D). The latter three cases indicate accelerating effects, most likely self-sensitizing, that dampen further decrease of k values at high concentrations, which is discussed later with estimated Φe (light-normalized, see eqn (1) in Materials and methods).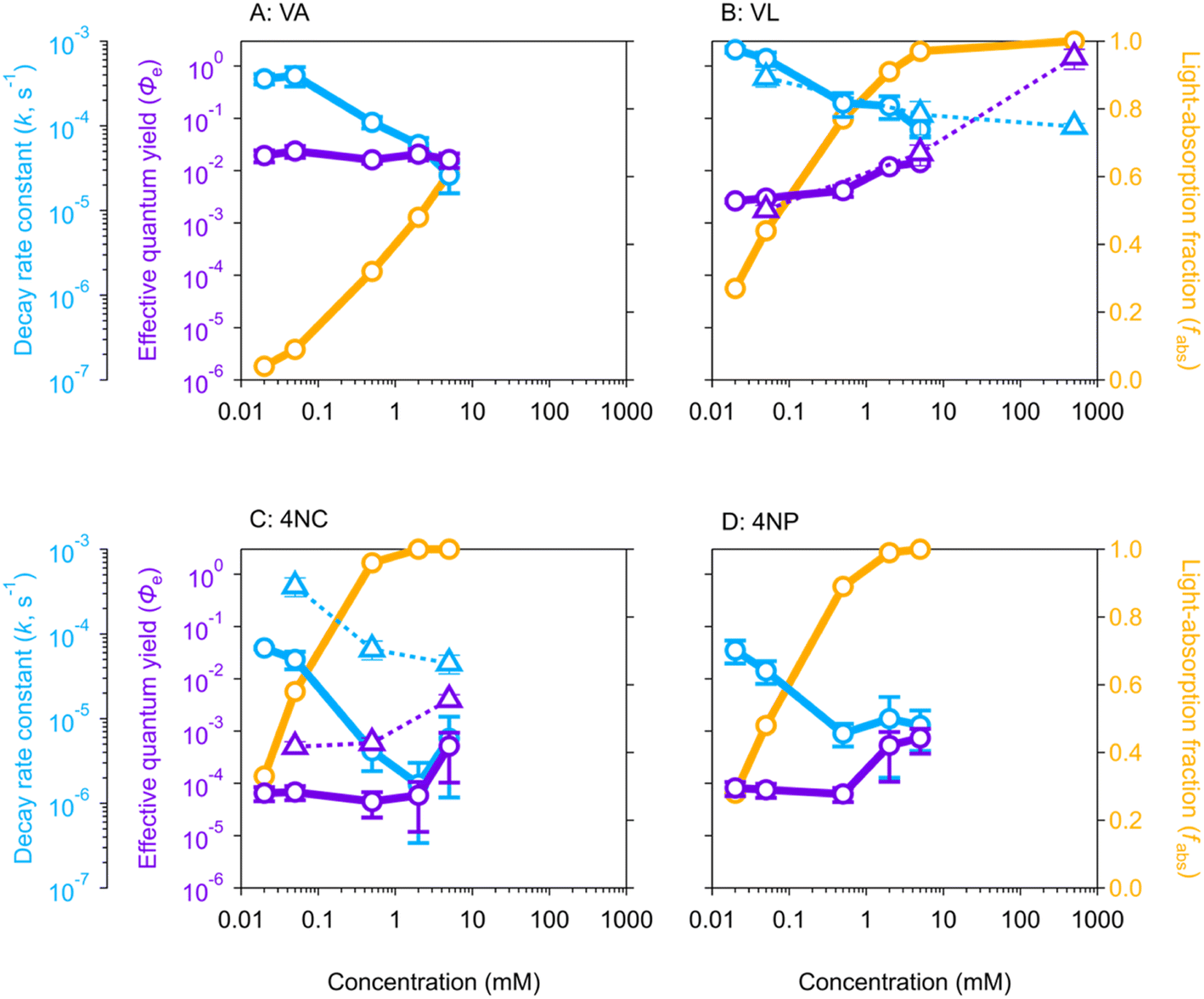 | ||
| Fig. 1 Pseudo-first-order decay rate constant (k) (cyan symbols), effective quantum yield (Φe) (violet symbols), and light-absorption fraction (fabs) (orange symbols) of (A) vanillic acid (VA), (B) vanillin (VL), (C) 4-nitrocatechol (4NC), and (D) 4-nitrophenol (4NP) as a function of concentration. The hollow circles connected with solid lines are data from experiments in aqueous solutions, while the hollow triangles connected with dashed lines are data from experiments in isopropanol (IPA) solutions. | ||
Similar gradually decreasing trends of k values are also observed in isopropanol (IPA) solutions for VL and 4NC (Fig. 1B and C, respectively), again attributable to self-shadowing. A notable difference between VL and 4NC is that the k values of VL in IPA and in H2O are comparable, while those of 4NC are approximately one order of magnitude higher in IPA than in H2O. The former can be explained by the previous finding that VL mainly degrades via self-quenching of its triplet state (3VL*)63 and thus is less prone to matrix effect, while the latter suggests a matrix dependence attributable to the disparity in the quenching efficiency by aqueous and organic solvents.64,65 Photo-excited nitroaromatics such as 34NC* preferably react by abstracting hydrogen,66,67 which is more readily available from the C–H bonds in IPA than from the O–H bonds in H2O.64,66 This observation indicates that the commonly present organic matrix in atmospheric aerosols might enhance the nitrophenol degradation. Notably, organic matrix might scavenge reactive radicals such as OH and then suppress the BrC oxidation. The comparable Φe values of VL in IPA and H2O, however, indicate that reactive radicals quenched by IPA have a minor effect on BrC degradation or that such reduction in VL degradation is replenished by other unknown mechanisms.
The k values for co-photolysis of BrC are shown in Fig. S3.† There are three mixing cases: (1) MP + MP, (2) NP + NP, and (3) MP + NP. The k values of the first BrC chromophore in the mixture mostly decrease in the presence of the second, and the higher the concentration (from 0.05 to 0.5 mM) of the second chromophore, the lower the k values of the first. Therefore, the self-shadowing effect of BrC (as its own concentration increases, as discussed in the section above) or cross-shadowing by another chromophore can decrease the k values via light competition. However, there are exceptions. The k values of 4NC and 4NP in the presence of VA or VL increase by factors of 1.4–38.4 (Fig. S3C and D†), suggesting that cross-sensitizing by MPs on NPs outweighs cross-shadowing, thereby accelerating NP degradation. Similarly, k values of MPs (i.e., VL) decrease by 33–90% (Fig. S3B†) in the presence of NPs (i.e., 4NC), but increase by 38–66% for k values of 4NC in the presence of VL (Fig. S3C†), are also observed in IPA solutions. These observations demonstrate that the cross-sensitizing ability of MPs is superior to that of NPs, and such ability shows little matrix dependence.
3.2 Effective quantum yield
The violet symbols in Fig. 1 are the estimated Φe values (by eqn (1) in Materials and methods) as a function of BrC concentration. The Φe value of VA (Fig. 1A) is relatively constant at (1.9 ± 0.3) × 10−2 in the whole concentration range studied (0.02–5 mM), and those of VL [(3.2 ± 0.8) × 10−3], 4NC [(5.9 ± 0.4) × 10−5], and 4NP [(7.3 ± 0.9) × 10−5] are also quite stable in the low-concentration range of 0.02–0.5 mM (Fig. 1B–D, respectively). The average Φe values of VL and 4NP in optically thin (dilute) solutions are comparable to results reported in previous studies,35,68 while those of VA and 4NC are two orders of magnitude higher than those from previous studies65,69 that might differ from ours in the light intensity of the lamp or the calculation method for quantum yield. The Φe values of VL, 4NC, and 4NP increase substantially above 1 mM (Fig. 1B–D), which is different from those of VA (Fig. 1A), a distinction also observed for k values as discussed in the section above. The steady Φe values translated from the rapid decrease in k values of VA as concentration increases suggest that concentration dependence is eliminated by considering the self-shadowing effect with light normalization (eqn (1)). The increases in Φe values of the other three BrC chromophores, however, emphasize that self-sensitizing dominates over self-shadowing at high concentrations.In IPA solutions for VL and 4NC, the greatly enhanced Φe values under much higher concentrations (Fig. 1B and C) again suggest self-sensitizing during the photolysis of these two BrC chromophores as concentration increases. In IPA solutions from 0.05–5 mM, 4NC has much higher Φe values in IPA [(4 ± 1) × 10−3 at 5 mM] than in H2O [(5 ± 4) × 10−4 at 5 mM] (Fig. 1C), while VL has comparable Φe values in H2O and IPA (Fig. 1B), with reasons discussed in the previous section for k values. The Φe value of VL at 500 mM in IPA is even higher than unity (1.48 ± 0.61), suggesting that self-sensitizing in an organic matrix at such a high concentration leads to cascade degradation of VL beyond the assumption of one reaction per photon absorbed. Although organic concentrations in aerosols are not well constrained due to some factors such as solubility, volatility, and phase,39 the BrC concentration can reach tens of mM37 or even higher in the interfacial region of the particles because of surface enhancement of organics.70 Therefore, this observation implies possible enhancement on BrC photodegradation in the aerosol organic matrix where BrC is concentrated.
Fig. 2 shows the Φe values for co-photolysis (color symbols) as compared to those in the photolysis of a single chromophore (grey symbols). The Φe values of VA change insignificantly in the presence of 4NC, but they increase by approximately one order of magnitude (from 0.019 ± 0.003 to 0.165 ± 0.085) with 0.5 mM of VL (Fig. 2A). Similarly, adding 4NC does not change the Φe value of 4NP, but 0.5 mM of VL increases it by a factor of 4 (Fig. 2D). For VL (Fig. 2B), the presence of either MP (VA) or NP (4NC) does not increase its Φe values in H2O or in IPA. The Φe values of 4NC in H2O (Fig. 2C) are insignificantly affected by 4NP but increased by factors of 5 and 26 in the presence of 0.5 mM of VA and VL, from (5.94 ± 1.26) × 10−5 to (2.85 ± 1.22) × 10−4 and to (1.55 ± 0.07) × 10−3, respectively. A similar increase in Φe values [from (5.5 ± 0.06) × 10−4 to (1.45 ± 0.43) × 10−3] are also observed in IPA for 4NC in the presence of VL (Fig. 2C). These observations for Φe values during co-photolysis further illustrate the better cross-sensitizing ability of MPs than NPs.
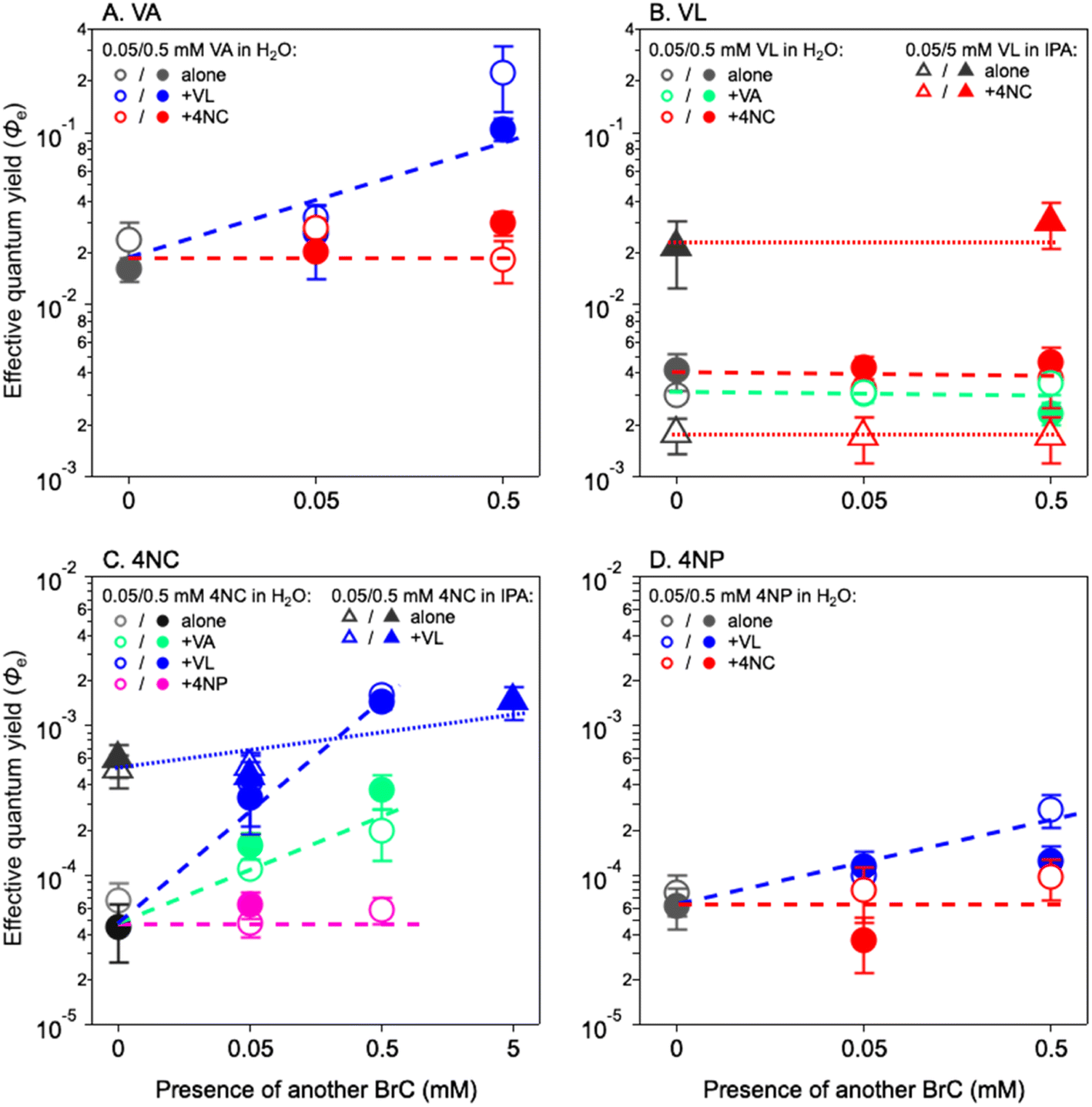 | ||
| Fig. 2 Effective quantum yield of (A) vanillic acid (VA), (B) vanillin (VL), (C) 4-nitrocatechol (4NC), and (D) 4-nitrophenol (4NP) under three mixing conditions: case 1 of MP + MP, case 2 of NP + NP, and case 3 of MP + NP. The circles are data from experiments in aqueous solution, while the triangles are data from experiments in isopropanol (IPA) solutions. The hollow and solid symbols represent the data for lower concentrations (0.05 mM) and higher concentrations (0.5 or 5 mM), respectively. The dashed and dotted lines are not fitting results and are merely to guide the eyes. | ||
The above interpretation can be understood from another perspective. In optically thin solutions, almost every BrC chromophore molecule can absorb light, upon which photodegradation may occur. Therefore, the quantum yield for direct photolysis should be concentration independent, given the assumption that the photolysis event proportionally scales with BrC concentration when there are sufficient photons. This can explain the relatively stable Φe values of VA in the 0–5 mM range and of VL/4NC/4NP in the 0–0.5 mM range (Fig. 1). In optically thick solutions, however, due to light depletion, some BrC chromophore molecules cannot absorb light and remain in their ground states. As BrC concentration increases, the ground-state BrC tends to react with reactive intermediates generated from photosensitized reactions and results in higher Φe values, as those of VL/4NC/4NP with concentration from 0.5 to 5 mM (Fig. 1B–D). In our experiments, it seems that partial shadowing (light not depleted, Fig. 1A) occurred for VA at concentrations up to 5 mM and for VL/4NC/4NP at concentrations up to 0.5 mM, which only reduced the k values but did not affect Φe values. For VL/4NC/4NP at concentrations higher than 0.5 mM, however, almost complete shadowing (light depleted, Fig. 1B–D) occurred, and sensitizing started to play a more important role in their photodegradation, leading to enhanced Φe values at higher concentrations.
The BrC chromophores in their triplet excited state can be a precursor of ROS upon the reactions with dissolved O2 and/or other substrates (e.g., phenols themselves) during the photolysis.20,25 To further investigate in the cases of light depletion whether the reactive intermediates from sensitizing are mainly ROS, we conducted aqueous-phase photolysis for VA and VL with limited dissolved O2 (N2-saturated solutions, Table S3†). The Φe values of these two MPs under N2-saturated conditions (Fig. S4†) were 5–10 times lower than those under air-saturated conditions, which is consistent with previous studies.20,71 This result indicates that ROS plays a role in enhancing the degradation of these BrC chromophores. In addition, VA does not show an obvious change in Φe values as its concentration increases under N2-saturated conditions as under air-saturated conditions (Fig. S4A†); neither does VL under N2-saturated conditions, which is different from the increasing trend from 0.5 mM to 5 mM (with high fabs values) under air-saturated conditions for VL (Fig. S4B†). Therefore, our result indicates that ROS does play a role in the increasing Φe values of MPs at high concentrations by reacting with ground-state BrC (i.e., those BrC molecules that do not receive light). This effect is mainly caused by sensitizing but is also indirectly manifested by shadowing that leaves more BrC chromophores in the ground state.
To further investigate which one of the first two mechanisms holds, co-photolysis of VL and 4NC was conducted under N2-saturated solutions (Table S3†). If the first mechanism holds, VL will not enhance the Φe values of 4NC in N2-saturated solutions because of the limited availability of ROS; if the second mechanism holds, VL will increase the Φe values of 4NC in N2-saturated solutions, while the Φe values of VL will be reduced by 4NC, because of limited reactions between VL and ROS to offset the consumption of 3VL* by 4NC. In N2-saturated solutions, the Φe values of VL decreased by factors of 6–7 and 35–65 in the presence of 0.05 mM and 0.5 mM of 4NC, respectively, which are different from the relatively constant Φe values of VL in air-saturated solutions (Fig. 3A). The Φe values of 4NC, in contrast, increased by factors of 4–44 and 7–121 with VL of 0.05 mM to 0.5 mM, respectively, similar to the increase in air-saturated conditions (Fig. 3B). The results thus support the second mechanism that NP degradation is mainly driven by reaction with 3MP*, albeit reactions with ROS might have also played a role. The proposed pathways for this mechanism are shown in Scheme S1 in the ESI.†
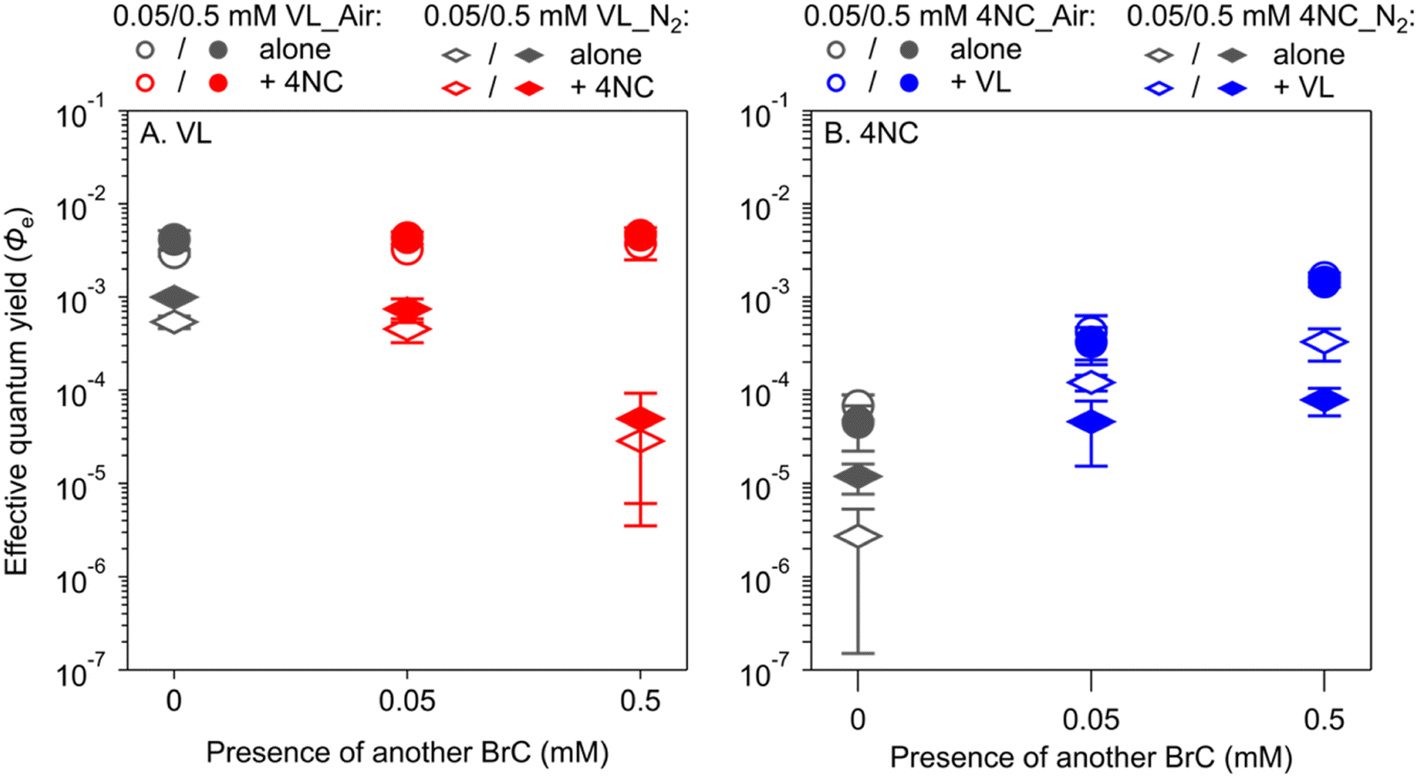 | ||
| Fig. 3 Comparison of effective quantum yields of (A) vanillin (VL) and (B) 4-nitrocatechol (4NC) alone or mixed under air-saturated and N2-saturated conditions. The hollow and solid symbols represent the data for a lower concentration (0.05 mM) and a higher concentration (0.5 mM), respectively. | ||
The product formation from co-photolysis of VL and 4NC also supports the second mechanism (reaction between 4NC and 3VL*). Table S4† shows the main identified products of VL by hydroxylation, dimerization, and cross-reaction from the mass spectrometric analysis. In the VL (0.5 mM) + 4NC (0.05 mM) solutions, the cross-reaction product (C14H11NO7) between 4NC and VL was generated under both air- and N2-saturated conditions (Fig. 4), although its efficient decomposition after 15 min was observed under air-saturated conditions. The production of VL dimer (C16H14O6) by self-quenching (VL + 3VL*),63 however, was 1.5–1.7 times less during co-photolysis than during photolysis alone (Fig. 4A) under air-saturated conditions at 30 and 40 min; an even greater reduction of its formation (2.7–3.9 times) under N2-saturated conditions from 15–60 min was observed. These observations suggest that the presence of 4NC suppresses the reaction between VL and 3VL* (less formation of VL dimer C16H14O6), which was replaced by the reaction between 4NC and 3VL* (forming cross-reaction product C14H11NO7).
 | ||
| Fig. 4 Peak areas of main products of VL (0.5 mM) photolysis alone or co-photolysis with 4NC (0.05 mM) under (A) air-saturated and (B) N2-saturated conditions, respectively. The formula of C16H14O6 is dimerization product(s), C8H8O4 hydroxylation product(s), and C14H11NO7 cross-reaction product(s). The formulae for these products were derived from the corresponding quasi-molecular ions ([M − H]−). | ||
On the other hand, the consumption of 3VL* by 4NC was indeed offset by the ROS oxidation, which leads to significant increase of hydroxylation products (C8H8O4) under air-saturated conditions (Fig. 4A) but not so under N2-saturated conditions (Fig. 4B). Therefore, oxidation by ROS in air-saturated solutions is of importance in compensating the decrease of reaction 3MP* + MP, thus maintaining their Φe values (Fig. 3A). In anoxic conditions as in N2-saturated solutions, however, oxidation of MPs (e.g., VL) is suppressed in the presence of 4NC, because the reaction between 3VL* and 4NC consumes substantial amounts of 3VL*. Such suppression of reaction between ground-state VL and 3VL*, which is the main driving force for VL photodegradation when it photolyzes alone, leads to one to two orders of magnitude lower Φe values for VL under N2-saturated conditions (Fig. 3A) at higher concentrations.
3.3 Estimated atmospheric lifetime
The concentration-dependent Φe values of BrC chromophores during their (co-)photolysis were used to estimate their photolysis rate constants (j, s−1) using Method 1 (eqn (3)) and Method 2 (eqn (4)) as shown in Materials and methods. These two methods differ in that Method 1 assumes optically thin solutions, and therefore optical path (l) is nonessential and ignored, while Method 2 considers the shadowing effect and keeps l, which is particle radius in ambient aerosol particles or solution depth in our experiments. The ratio between the j values obtained by these two methods, termed as the j1-to-j2 ratio (Fig. S5†), is close to unity at low concentrations (<5 mM) but increases substantially at higher ones (50–500 mM) for chromophores with a molar absorption coefficient (ε) larger than 1000 mol−1 L cm−1. This analysis reveals the potential effect of particle size on the estimated atmospheric lifetimes for highly light-absorbing BrC chromophores. Shadowing effect is largely negligible for small particles and could only be significant for large particles (>1 μm) with highly absorbing (ε > 1000 mol−1 L cm−1) and highly concentrated chromophores (possible in aerosol particles).To be compatible with previous studies, we used Method 1 to calculate the j values for 100 nm particles, which are comparable with those calculated by Method 2 at this small particle size, and consideration of size effect is not necessary even for concentrated conditions. The j values of these BrC chromophores in H2O and IPA are shown in Fig. S6,† from which their atmospheric lifetimes (τ) were also calculated with eqn (5) (see Materials and methods) and are shown in Fig. 5. The τ values of VA, VL, 4NC, and 4NP via aqueous-phase photolysis in small particles (100 nm) at optically thin solutions (0.02–5 mM for VA; 0.02–0.5 mM for VL/4NC/4NP) are 10.7 ± 1.7 h, 1.2 ± 0.3 h, 12.4 ± 2.6 h, and 14.5 ± 1.9 h, respectively (Fig. 5 and Table 1). In IPA solutions at a low concentration (0.05 mM), the τ value of VL (2.2 ± 0.5 h) is comparable with that in H2O, while that of 4NC is about 10 times shorter (1.3 ± 0.2 h) than that in H2O, indicating faster degradation of 4NC in organics-dominated particles.
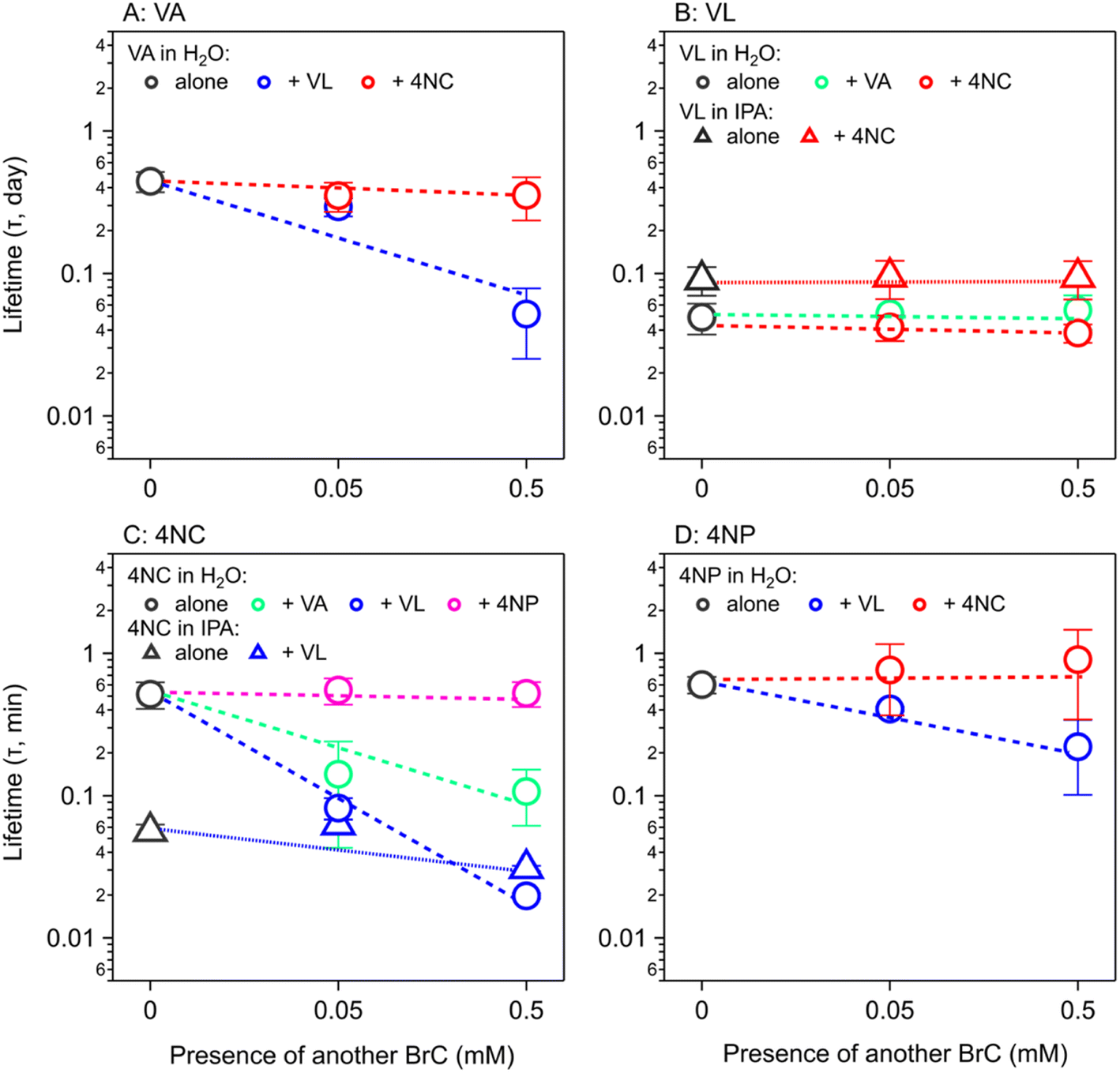 | ||
| Fig. 5 Estimation of atmospheric lifetimes (τ, h) of (A) vanillic acid (VA), (B) vanillin (VL), (C) 4-nitrocatechol (4NC), and (D) 4-nitrophenol (4NP) alone (in black) and under three mixing conditions: MP + MP, NP + NP, and MP + NP. The estimation assumes particles of 100 nm radius and concentration of 0.02–0.5 mM. The circles used data from experiments in aqueous solutions, while the triangles are data from experiments in isopropanol (IPA) solutions. The dashed and dotted lines are not fitting results and are merely to guide the eyes. | ||
| Gas-phase OH oxidation | Aq. OH oxidation | Aq. photolysis | ||||||
|---|---|---|---|---|---|---|---|---|
| k OH,g | [OH]g | τ OH,g (h) | k OH,aq. | [OH]aq. | τ OH,aq. (h) | τ hv,aq. (h) | τ hv,aq.,min (h) | |
| a k OH,g: ×10−12 cm3 per molecule per s; [OH]g: ×106 molecule per cm3; kOH,aq.: ×109 M s−1; [OH]aq.: ×10 M. b Using 2NP data due to the lack of data for 4NP. c Mean value at urban case in cloud water. d τ hv,aq.: estimated from optically thin (<0.5 mM) and small particles (100 nm) in this study. e τ hv,aq.,min: minimum τ values due to sensitizing in this study. | ||||||||
| VA | 4.72 (ref. 74) | 1.5 (ref. 75) | 39.2 | 9.8 (ref. 76) | 0.35 (ref. 77)c | 8.1 | 10.7 ± 1.7 | 1.2 ± 0.6 |
| VL | 5.72 (ref. 78) | 32.4 | 3.3 (ref. 79) | 14.1 | 1.2 ± 0.3 | 1.2 ± 0.3 | ||
| 4NC | 1.27 (ref. 80) | 145.8 | 5.0 (ref. 19) | 15.9 | 12.4 ± 2.6 | 0.5 ± 0.02 | ||
| 4NP | 0.9 (ref. 81)b | 205.8 | 4.7 (ref. 82) | 16.9 | 14.5 ± 1.9 | 5.3 ± 2.9 | ||
For the co-photolysis of MP + MP (VA + VL), the τ values of VA are shortened by 30% and 90% (to approximately 1 h) in the presence of VL of 0.05 mM and 0.5 mM, respectively (Fig. 5A), while those of VL is insignificantly affected by VA (Fig. 5B). For NP + NP (4NC + 4NP) (Fig. 5C and D), their τ values are also insignificantly affected by each other. For MP + NP (VA/VL + 4NC/4NP), the τ values of MPs are affected insignificantly by NPs (Fig. 5A and B), while those of NPs are greatly shortened by MPs to less than a half or even less than one-tenth (Fig. 5C and D). In IPA solutions, the τ values of VL are insignificantly affected by 4NC (Fig. 5B), while those of 4NC are shortened by 40% in the presence of VL (Fig. 5C). This acceleration effect of VL on 4NC in IPA is not as strong as that in H2O, but still reduces the τ value of 4NC to <1 h. In general, BrC chromophores (especially NPs) tend to photolyze faster in the organic matrix that is commonly found in atmospheric aerosol particles. As a comparison, a previous study also showed a decreased lifetime of 4NP from 11 h to 8 h in the presence of α-pinene-derived secondary organic aerosol (SOA),68 which was attributed to the oxidation by OH radicals from photolysis of fresh SOA as a photochemically labile organic matrix.
Field measurements indicated that BrC had short lifetimes (or half-life times) of 9–15 h.13,28 Our estimated τ values for aqueous-phase photolysis in optically thin solutions (10–15 h for VA/4NC/4NP and 1.2 h for VL, Table 1) are similar to or shorter than those due to aqueous-phase OH oxidation (8–17 h, Table 1); they are even much shorter than those due to gas-phase OH oxidation (30–200 h, Table 1), although such comparison is not straightforward without considering gas-particle partitioning. For different organic compounds, their partitioning between the gas and particle phases is governed by the vapor pressure, octanol–air partition coefficient, and/or solubility, etc.72 and affects their lifetime estimation, which is beyond the scope of the current study. Despite that photolysis rates of BrC chromophores are highly molecule-specific and should therefore not be extrapolated to structurally similar compounds,32 our results highlight that aqueous-phase photolysis can be a significant sink of BrC chromophores. In addition, we show that the atmospheric lifetimes of NPs are shortened by a factor of up to 30 in the presence of MPs (Fig. 5C), potentially making co-photolysis with sensitizing effect a dominating sink for some BrC chromophores.
We herein used experimental data of VL in IPA to illustrate the combined shadowing and sensitizing effects in the estimation of its atmospheric lifetime with different concentrations and particle sizes, as shown in Section S3 in the ESI.† For a 100 nm particle containing 0.05 mM or 500 mM VL, there is no significant difference in the τ values of VL (Fig. S7B†) because of little shadowing effect caused by reduced volume-normalized photon flux. As particle radius increases, however, the τ value of VL of 500 mM increases by 32% in a 1000 nm particle (Fig. S7B†), where the Φe value is similar, but shadowing effect (fabs = 0.28) plays a role compared to the 0.05 mM scenario; an even more drastic difference of a 100 time decrease is observed for a 10 μm particle from the 0.05 mM to the 500 mM scenarios (Fig. S7B†). The drastic difference in τ value for larger particles is due mainly to the greatly enhanced Φe value (1.48 ± 0.61) caused by sensitizing for the 500 mM scenario. In addition, such sensitizing effect is indirectly amplified by the shadowing effect (light depletion, fabs = 0.65) that leaves more VL in the ground state to react with reactive intermediates (e.g., ROS) instead of undergoing direct photolysis. Therefore, the combined effect of shadowing and sensitizing, together with that from particle size, might either shorten or lengthen BrC lifetimes.
4 Conclusions and atmospheric implications
The shadowing effect plays a minor role in BrC photolysis in fine particles in which BrC is commonly found; it might indirectly manifest more efficient sensitizing in coarse particles containing highly absorbing chromophores, which is also possible, as found in field studies. It has been shown that the absorption-size distributions of BrC mainly peaked in the accumulation mode at ∼500 nm,6 where the shadowing effect should be negligible. The same study, however, also revealed a certain contribution of BrC absorption from particles larger than 5 μm.6 Indeed, VL showed a bimodal pattern with equivalent peaks in the fine (0.4–1 μm) and coarse (3–10 μm) modes,73 which might be due to its evaporation (relatively high volatility) from fine particles and then dissolution (slight water solubility) in the coarse particles containing a large amount of liquid water.33,73 Therefore, although shadowing affects the atmospheric lifetimes of BrC in fine particles negligibly, it might be important in cases where BrC is found in coarse particles.The sensitizing effect can shorten the atmospheric lifetimes of BrC chromophores during their co-photolysis, regardless of particle size. The photosensitization ability of the four BrC chromophores studied here has the sequence of VL > VA > 4NC ≈ 4NP. The atmospheric lifetime of 4NC was shortened from 12 h to 0.5 h (Table 1) in the presence of the most capable photosensitizer in this study (VL). Based on our analysis, we arrived at two major conclusions regarding the sensitizing ability during co-photolysis: (1) MPs can greatly shorten the atmospheric lifetimes of NPs but not vice versa (Fig. 5), which is consistent with results in our previous study using lamps of different wavelengths;21 and (2) among MPs, VL can shorten the atmospheric lifetimes of VA but not vice versa (Fig. 5A and B), which might be due to the presence of a conjugated carbonyl group that makes VL a better photosensitizer.41 In fact, the atmospheric lifetime of VL is overwhelmingly affected by self-sensitization, as suggested by the strong concentration dependence of Φe value (Fig. 1B). Yet, VL was considered a relatively mild photosensitizer41 with lower quantum yields of excited triplet state among many other BrC chromophores. If that is the case, the sensitizing effect of those more capable photosensitizers in aerosol particles may shorten the atmospheric lifetimes of other BrC chromophores (or particulate components in general) to the extent that is greater than those observed here (up to a factor of 30, Fig. 5).
The overall effect of shadowing and sensitizing during co-photolysis depends on particle size, concentration, and the sensitization ability of the chromophores. Therefore, the estimation of atmospheric lifetimes for BrC chromophores might suffer from high uncertainties because of our limited knowledge of their chemical identities, not to mention their sensitization abilities. Such uncertainties in atmospheric lifetimes may affect the modelling accuracy of the impacts on global climate and atmospheric chemistry induced by complex mixed BrC chromophores in aerosol particles. Chemical transport models should also consider such accelerated degradation of BrC chromophores or particulate species in general.
Author contributions
YJL and YW designed the study. YW, TQ, and CZ performed the experiments. YW and CZ analyzed the data. YW, TH, BG, RZ, MG, MNC, DDH, XG, JW, LD, RJH, QC, CKC, and YJL performed data interpretation and presentation of figures. YW, KIH, KMM, CKC, and YJL wrote the first draft of the manuscript, and all authors contributed to the editing of the article.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Science and Technology Development Fund, Macau SAR (file no. 0019/2020/A1), and a multiyear research grant (no. MYRG2022-00027-FST) from the University of Macau.References
- V. Ramanathan, F. Li, M. V. Ramana, P. S. Praveen, D. Kim, C. E. Corrigan, H. Nguyen, E. A. Stone, J. J. Schauer, G. R. Carmichael, B. Adhikary and S. C. Yoon, Atmospheric brown clouds: hemispherical and regional variations in long-range transport, absorption, and radiative forcing, J. Geophys. Res.: Atmos., 2007, 112, D22S21 CrossRef.
- M. O. Andreae and A. Gelencsér, Black carbon or brown carbon? The nature of light-absorbing carbonaceous aerosols, Atmos. Chem. Phys., 2006, 6(10), 3131–3148 CrossRef CAS.
- T. C. Bond and R. W. Bergstrom, Light Absorption by Carbonaceous Particles: An Investigative Review, Aerosol Sci. Technol., 2006, 40(1), 27–67 CrossRef CAS.
- Y. Feng, V. Ramanathan and V. R. Kotamarthi, Brown carbon: a significant atmospheric absorber of solar radiation?, Atmos. Chem. Phys., 2013, 13(17), 8607–8621 CrossRef.
- Y. Chen and T. C. Bond, Light absorption by organic carbon from wood combustion, Atmos. Chem. Phys., 2010, 10(4), 1773–1787 CrossRef CAS.
- J. Liu, M. Bergin, H. Guo, L. King, N. Kotra, E. Edgerton and R. J. Weber, Size-resolved measurements of brown carbon in water and methanol extracts and estimates of their contribution to ambient fine-particle light absorption, Atmos. Chem. Phys., 2013, 13(24), 12389–12404 CrossRef.
- R. Saleh, E. S. Robinson, D. S. Tkacik, A. T. Ahern, S. Liu, A. C. Aiken, R. C. Sullivan, A. A. Presto, M. K. Dubey, R. J. Yokelson, N. M. Donahue and A. L. Robinson, Brownness of organics in aerosols from biomass burning linked to their black carbon content, Nat. Geosci., 2014, 7(9), 647–650 CrossRef CAS.
- T. C. Bond, Spectral dependence of visible light absorption by carbonaceous particles emitted from coal combustion, Geophys. Res. Lett., 2001, 28(21), 4075–4078 CrossRef.
- M. Yang, S. G. Howell, J. Zhuang and B. J. Huebert, Attribution of aerosol light absorption to black carbon, brown carbon, and dust in China – interpretations of atmospheric measurements during EAST-AIRE, Atmos. Chem. Phys., 2009, 9(6), 2035–2050 CrossRef CAS.
- J. Laskin, A. Laskin, S. A. Nizkorodov, P. Roach, P. Eckert, M. K. Gilles, B. Wang, H. J. Lee and Q. Hu, Molecular Selectivity of Brown Carbon Chromophores, Environ. Sci. Technol., 2014, 48(20), 12047–12055 CrossRef CAS PubMed.
- L. Yu, J. Smith, A. Laskin, C. Anastasio, J. Laskin and Q. Zhang, Chemical characterization of SOA formed from aqueous-phase reactions of phenols with the triplet excited state of carbonyl and hydroxyl radical, Atmos. Chem. Phys., 2014, 14(24), 13801–13816 CrossRef.
- E. L. Shapiro, J. Szprengiel, N. Sareen, C. N. Jen, M. R. Giordano and V. F. McNeill, Light-absorbing secondary organic material formed by glyoxal in aqueous aerosol mimics, Atmos. Chem. Phys., 2009, 9(7), 2289–2300 CrossRef CAS.
- H. Forrister, J. Liu, E. Scheuer, J. Dibb, L. Ziemba, K. L. Thornhill, B. Anderson, G. Diskin, A. E. Perring, J. P. Schwarz, P. Campuzano-Jost, D. A. Day, B. B. Palm, J. L. Jimenez, A. Nenes and R. J. Weber, Evolution of brown carbon in wildfire plumes, Geophys. Res. Lett., 2015, 42(11), 4623–4630 CrossRef CAS.
- A. Zhang, Y. Wang, Y. Zhang, R. J. Weber, Y. Song, Z. Ke and Y. Zou, Modeling the global radiative effect of brown carbon: a potentially larger heating source in the tropical free troposphere than black carbon, Atmos. Chem. Phys., 2020, 20(4), 1901–1920 CrossRef CAS.
- A. Laskin, J. Laskin and S. A. Nizkorodov, Chemistry of Atmospheric Brown Carbon, Chem. Rev., 2015, 115(10), 4335–4382 CrossRef CAS PubMed.
- H. Herrmann, T. Schaefer, A. Tilgner, S. A. Styler, C. Weller, M. Teich and T. Otto, Tropospheric Aqueous-Phase Chemistry: Kinetics, Mechanisms, and Its Coupling to a Changing Gas Phase, Chem. Rev., 2015, 115(10), 4259–4334 CrossRef CAS PubMed.
- R. F. Hems, E. G. Schnitzler, C. Liu-Kang, C. D. Cappa and J. P. D. Abbatt, Aging of Atmospheric Brown Carbon Aerosol, ACS Earth Space Chem., 2021, 5(4), 722–748 CrossRef CAS.
- R. Zhao, A. K. Y. Lee, L. Huang, X. Li, F. Yang and J. P. D. Abbatt, Photochemical processing of aqueous atmospheric brown carbon, Atmos. Chem. Phys., 2015, 15(11), 6087–6100 CrossRef CAS.
- R. F. Hems and J. P. D. Abbatt, Aqueous Phase Photo-oxidation of Brown Carbon Nitrophenols: Reaction Kinetics, Mechanism, and Evolution of Light Absorption, ACS Earth Space Chem., 2018, 2(3), 225–234 CrossRef CAS.
- B. R. G. Mabato, Y. Lyu, Y. Ji, Y. J. Li, D. D. Huang, X. Li, T. Nah, C. H. Lam and C. K. Chan, Aqueous secondary organic aerosol formation from the direct photosensitized oxidation of vanillin in the absence and presence of ammonium nitrate, Atmos. Chem. Phys., 2022, 22(1), 273–293 CrossRef CAS.
- Y. Wang, W. Huang, L. Tian, Y. Wang, F. Li, D. D. Huang, R. Zhang, B. R. Go Mabato, R.-J. Huang, Q. Chen, X. Ge, L. Du, Y. G. Ma, M. Gen, K. I. Hoi, K. M. Mok, J. Z. Yu, C. K. Chan, X. Li and Y. J. Li, Decay Kinetics and Absorption Changes of Methoxyphenols and Nitrophenols during Nitrate-Mediated Aqueous Photochemical Oxidation at 254 and 313 nm, ACS Earth Space Chem., 2022, 6(4), 1115–1125 CrossRef CAS.
- W. Jiang, M. V. Misovich, A. P. S. Hettiyadura, A. Laskin, A. S. McFall, C. Anastasio and Q. Zhang, Photosensitized Reactions of a Phenolic Carbonyl from Wood Combustion in the Aqueous Phase-Chemical Evolution and Light Absorption Properties of AqSOA, Environ. Sci. Technol., 2021, 55(8), 5199–5211 CrossRef CAS.
- J. D. Smith, V. Sio, L. Yu, Q. Zhang and C. Anastasio, Secondary Organic Aerosol Production from Aqueous Reactions of Atmospheric Phenols with an Organic Triplet Excited State, Environ. Sci. Technol., 2014, 48(2), 1049–1057 CrossRef CAS PubMed.
- D. Vione, V. Maurino, C. Minero, E. Pelizzetti, M. A. J. Harrison, R.-I. Olariu and C. Arsene, Photochemical reactions in the tropospheric aqueous phase and on particulate matter, Chem. Soc. Rev., 2006, 35(5), 441–453 CAS.
- C. George, M. Brüggemann, N. Hayeck, L. Tinel and J. Donaldson, Interfacial Photochemistry, in Physical Chemistry of Gas-Liquid Interfaces, ed. J. A. Faust and J. E. House, Elsevier, 2018, ch. 14, pp. 435–457 Search PubMed.
- E. Gómez Alvarez, H. Wortham, R. Strekowski, C. Zetzsch and S. Gligorovski, Atmospheric Photosensitized Heterogeneous and Multiphase Reactions: From Outdoors to Indoors, Environ. Sci. Technol., 2012, 46(4), 1955–1963 CrossRef.
- M. Le Bechec, T. Pigot and S. Lacombe, Chemical Quenching of Singlet Oxygen and Other Reactive Oxygen Species in Water: A Reliable Method for the Determination of Quantum Yields in Photochemical Processes?, ChemPhotoChem, 2018, 2(7), 622–631 CrossRef CAS.
- L. T. Fleming, P. Lin, J. M. Roberts, V. Selimovic, R. Yokelson, J. Laskin, A. Laskin and S. A. Nizkorodov, Molecular composition and photochemical lifetimes of brown carbon chromophores in biomass burning organic aerosol, Atmos. Chem. Phys., 2020, 20(2), 1105–1129 CrossRef CAS.
- E. G. Schnitzler, N. G. A. Gerrebos, T. S. Carter, Y. Huang, C. L. Heald, A. K. Bertram and J. P. D. Abbatt, Rate of atmospheric brown carbon whitening governed by environmental conditions, Proc. Natl. Acad. Sci. U. S. A., 2022, 119(38), e2205610119 CrossRef CAS.
- N. A. June, X. Wang, L.-W. A. Chen, J. C. Chow, J. G. Watson, X. Wang, B. H. Henderson, Y. Zheng and J. Mao, Spatial and Temporal Variability of Brown Carbon in the United States: Implications for Direct Radiative Effects, Geophys. Res. Lett., 2020, 47(23), e2020GL090332 CrossRef CAS.
- H. Brown, X. Liu, R. Pokhrel, S. Murphy, Z. Lu, R. Saleh, T. Mielonen, H. Kokkola, T. Bergman, G. Myhre, R. B. Skeie, D. Watson-Paris, P. Stier, B. Johnson, N. Bellouin, M. Schulz, V. Vakkari, J. P. Beukes, P. G. van Zyl, S. Liu and D. Chand, Biomass burning aerosols in most climate models are too absorbing, Nat. Commun., 2021, 12(1), 277 CrossRef CAS PubMed.
- S. A. Epstein, E. Tapavicza, F. Furche and S. A. Nizkorodov, Direct photolysis of carbonyl compounds dissolved in cloud and fog∼droplets, Atmos. Chem. Phys., 2013, 13(18), 9461–9477 CrossRef.
- Y. J. Li, D. D. Huang, H. Y. Cheung, A. K. Y. Lee and C. K. Chan, Aqueous-phase photochemical oxidation and direct photolysis of vanillin – a model compound of methoxy phenols from biomass burning, Atmos. Chem. Phys., 2014, 14(6), 2871–2885 CrossRef.
- Y. L. Sun, Q. Zhang, C. Anastasio and J. Sun, Insights into secondary organic aerosol formed via aqueous-phase reactions of phenolic compounds based on high resolution mass spectrometry, Atmos. Chem. Phys., 2010, 10(10), 4809–4822 CrossRef CAS.
- J. D. Smith, H. Kinney and C. Anastasio, Phenolic carbonyls undergo rapid aqueous photodegradation to form low-volatility, light-absorbing products, Atmos. Environ., 2016, 126, 36 CrossRef CAS.
- D. Vione, A. Albinet, F. Barsotti, M. Mekic, B. Jiang, C. Minero, M. Brigante and S. Gligorovski, Formation of substances with humic-like fluorescence properties, upon photoinduced oligomerization of typical phenolic compounds emitted by biomass burning, Atmos. Environ., 2019, 206, 197–207 CrossRef CAS.
- H. A. Al-Abadleh, Aging of atmospheric aerosols and the role of iron in catalyzing brown carbon formation, Environ. Sci.: Atmos., 2021, 1(6), 297–345 CAS.
- H. Lamkaddam, J. Dommen, A. Ranjithkumar, H. Gordon, G. Wehrle, J. Krechmer, F. Majluf, D. Salionov, J. Schmale, S. Bjelić, K. S. Carslaw, I. El Haddad and U. Baltensperger, Large contribution to secondary organic aerosol from isoprene cloud chemistry, Sci. Adv., 2021, 7(13), eabe2952 CrossRef CAS.
- J. D. Cope, K. H. Bates, L. N. Tran, K. A. Abellar and T. B. Nguyen, Sulfur radical formation from the tropospheric irradiation of aqueous sulfate aerosols, Proc. Natl. Acad. Sci. U. S. A., 2022, 119(36), e2202857119 CrossRef CAS.
- K. Z. Aregahegn, B. Nozière and C. George, Organic aerosol formation photo-enhanced by the formation of secondary photosensitizers in aerosols, Faraday Discuss., 2013, 165, 123–134 RSC.
- T. Felber, T. Schaefer, L. He and H. Herrmann, Aromatic Carbonyl and Nitro Compounds as Photosensitizers and Their Photophysical Properties in the Tropospheric Aqueous Phase, J. Phys. Chem. A, 2021, 125(23), 5078–5095 CrossRef CAS PubMed.
- A. H. Goldstein and I. E. Galbally, Known and unexplored organic constituents in the earth's atmosphere, Environ. Sci. Technol., 2007, 41(5), 1514–1521 CrossRef CAS PubMed.
- M. Glasius and A. H. Goldstein, Recent Discoveries and Future Challenges in Atmospheric Organic Chemistry, Environ. Sci. Technol., 2016, 50(6), 2754–2764 CrossRef CAS.
- R. J. Huang, L. Yang, J. C. Shen, W. Yuan, Y. Q. Gong, H. Y. Ni, J. Duan, J. Yan, H. B. Huang, Q. H. You and Y. J. Li, Chromophoric Fingerprinting of Brown Carbon from Residential Biomass Burning, Environ. Sci. Technol. Lett., 2021, 9(2), 102–111 CrossRef.
- S. M. Phillips and G. D. Smith, Light Absorption by Charge Transfer Complexes in Brown Carbon Aerosols, Environ. Sci. Technol. Lett., 2014, 1(10), 382–386 CrossRef CAS.
- H. Sun, L. Biedermann and T. C. Bond, Color of brown carbon: a model for ultraviolet and visible light absorption by organic carbon aerosol, Geophys. Res. Lett., 2007, 34(17), L17813 CrossRef.
- W. Zhou, M. Mekic, J. Liu, G. Loisel, B. Jin, D. Vione and S. Gligorovski, Ionic strength effects on the photochemical degradation of acetosyringone in atmospheric deliquescent aerosol particles, Atmos. Environ., 2019, 198, 83–88 CrossRef CAS.
- G. Loisel, M. Mekic, S. Liu, W. Song, B. Jiang, Y. Wang, H. Deng and S. Gligorovski, Ionic strength effect on the formation of organonitrate compounds through photochemical degradation of vanillin in liquid water of aerosols, Atmos. Environ., 2021, 246, 118140 CrossRef CAS.
- M. Mekic, M. Brigante, D. Vione and S. Gligorovski, Exploring the ionic strength effects on the photochemical degradation of pyruvic acid in atmospheric deliquescent aerosol particles, Atmos. Environ., 2018, 185, 237–242 CrossRef CAS.
- J. Yang, W. C. Au, H. Law, C. H. Lam and T. Nah, Formation and evolution of brown carbon during aqueous-phase nitrate-mediated photooxidation of guaiacol and 5-nitroguaiacol, Atmos. Environ., 2021, 254, 118401 CrossRef CAS.
- Y. Wang, D. D. Huang, W. Huang, B. Liu, Q. Chen, R. Huang, M. Gen, B. R. G. Mabato, C. K. Chan, X. Li, T. Hao, Y. Tan, K. I. Hoi, K. M. Mok and Y. J. Li, Enhanced Nitrite Production from the Aqueous Photolysis of Nitrate in the Presence of Vanillic Acid and Implications for the Roles of Light-Absorbing Organics, Environ. Sci. Technol., 2021, 55(23), 15694–15704 CrossRef CAS PubMed.
- C. Anastasio, B. C. Faust and C. J. Rao, Aromatic Carbonyl Compounds as Aqueous-Phase Photochemical Sources of Hydrogen Peroxide in Acidic Sulfate Aerosols, Fogs, and Clouds. 1. Non-Phenolic Methoxybenzaldehydes and Methoxyacetophenones with Reductants (Phenols), Environ. Sci. Technol., 1997, 31(1), 218–232 CrossRef CAS.
- J. P. S. Wong, M. Tsagkaraki, I. Tsiodra, N. Mihalopoulos, K. Violaki, M. Kanakidou, J. Sciare, A. Nenes and R. J. Weber, Atmospheric evolution of molecular-weight-separated brown carbon from biomass burning, Atmos. Chem. Phys., 2019, 19(11), 7319–7334 CrossRef CAS.
- C. Liu-Kang, P. J. Gallimore, T. Liu and J. P. D. Abbatt, Photoreaction of biomass burning brown carbon aerosol particles, Environ. Sci.: Atmos., 2022, 2(2), 270–278 CAS.
- C. D. Simpson, M. Paulsen, R. L. Dills, L. J. S. Liu and D. A. Kalman, Determination of Methoxyphenols in Ambient Atmospheric Particulate Matter: Tracers for Wood Combustion, Environ. Sci. Technol., 2005, 39(2), 631–637 CrossRef CAS.
- Y. Sun, F. Xu, X. Li, Q. Zhang and Y. Gu, Mechanisms and kinetic studies of OH-initiated atmospheric oxidation of methoxyphenols in the presence of O2 and NOx, Phys. Chem. Chem. Phys., 2019, 21(39), 21856–21866 RSC.
- M. A. J. Harrison, S. Barra, D. Borghesi, D. Vione, C. Arsene and R. Iulian Olariu, Nitrated phenols in the atmosphere: a review, Atmos. Environ., 2005, 39(2), 231–248 CrossRef CAS.
- S. M. Phillips, A. D. Bellcross and G. D. Smith, Light Absorption by Brown Carbon in the Southeastern United States is pH-dependent, Environ. Sci. Technol., 2017, 51(12), 6782–6790 CrossRef CAS.
- H. Lignell, S. A. Epstein, M. R. Marvin, D. Shemesh, B. Gerber and S. Nizkorodov, Experimental and Theoretical Study of Aqueous cis-Pinonic Acid Photolysis, J. Phys. Chem. A, 2013, 117(48), 12930–12945 CrossRef CAS PubMed.
- B. J. Finlayson-Pitts and J. N. Pitts, Spectroscopy and Photochemistry: Fundamentals, in Chemistry of the Upper and Lower Atmosphere, ed. B. J. Finlayson-Pitts and J. N. Pitts, Academic Press, San Diego, 2000, ch. 3, pp. 43–85 Search PubMed.
- K. Li, P. Zhang, L. Ge, H. Ren, C. Yu, X. Chen and Y. Zhao, Concentration-dependent photodegradation kinetics and hydroxyl-radical oxidation of phenicol antibiotics, Chemosphere, 2014, 111, 278–282 CrossRef CAS PubMed.
- J. Niu, Y. Li and W. Wang, Light-source-dependent role of nitrate and humic acid in tetracycline photolysis: kinetics and mechanism, Chemosphere, 2013, 92(11), 1423–1429 CrossRef CAS PubMed.
- T. A. Gawargy, B. Wang and J. C. Scaiano, Unveiling the Mechanism for the Photochemistry and Photodegradation of Vanillin, Photochem. Photobiol., 2022, 98(2), 429–433 CrossRef CAS.
- H. Lignell, M. L. Hinks and S. A. Nizkorodov, Exploring matrix effects on photochemistry of organic aerosols, Proc. Natl. Acad. Sci. U. S. A., 2014, 111(38), 13780 CrossRef CAS.
- A. B. Dalton and S. A. Nizkorodov, Photochemical Degradation of 4-Nitrocatechol and 2,4-Dinitrophenol in a Sugar-Glass Secondary Organic Aerosol Surrogate, Environ. Sci. Technol., 2021, 55(21), 14586–14594 CrossRef CAS.
- J. A. Barltrop and N. J. Bunce, Organic photochemistry. Part VIII. The photochemical reduction of nitro-compounds, J. Chem. Soc. C, 1968, 1467–1474 RSC.
- H. Shizunobu and K. Koji, The Photochemical Reduction of Nitrobenzene and Its Reduction Intermediates. X. The Photochemical Reduction of the Monosubstituted Nitrobenzenes in 2-Propanol, Bull. Chem. Soc. Jpn., 1972, 45(2), 549–553 CrossRef.
- T. Braman, L. Dolvin, C. Thrasher, H. Yu, E. Q. Walhout and R. E. O'Brien, Fresh versus Photo-recalcitrant Secondary Organic Aerosol: Effects of Organic Mixtures on Aqueous Photodegradation of 4-Nitrophenol, Environ. Sci. Technol. Lett., 2020, 7(4), 248–253 CrossRef CAS.
- W. Shuai, C. Liu, Y. Wang, F. Zhu, D. Zhou and J. Gao, (Fe3+)-UVC-(aliphatic/phenolic carboxyl acids) systems for diethyl phthalate ester degradation: a density functional theory (DFT) and experimental study, Appl. Catal., A, 2018, 567, 20–27 CrossRef CAS.
- B. Minofar, P. Jungwirth, M. R. Das, W. Kunz and S. Mahiuddin, Propensity of Formate, Acetate, Benzoate, and Phenolate for the Aqueous Solution/Vapor Interface: Surface Tension Measurements and Molecular Dynamics Simulations, J. Phys. Chem. C, 2007, 111(23), 8242–8247 CrossRef CAS.
- Y. Chen, N. Li, X. Li, Y. Tao, S. Luo, Z. Zhao, S. Ma, H. Huang, Y. Chen, Z. Ye and X. Ge, Secondary organic aerosol formation from 3C*-initiated oxidation of 4-ethylguaiacol in atmospheric aqueous-phase, Sci. Total Environ., 2020, 723, 137953 CrossRef CAS PubMed.
- I. T. Cousins and D. Mackay, Gas–Particle Partitioning of Organic Compounds and Its Interpretation Using Relative Solubilities, Environ. Sci. Technol., 2001, 35(4), 643–647 CrossRef CAS PubMed.
- G. Wang, C. Chen, J. Li, B. Zhou, M. Xie, S. Hu, K. Kawamura and Y. Chen, Molecular composition and size distribution of sugars, sugar-alcohols and carboxylic acids in airborne particles during a severe urban haze event caused by wheat straw burning, Atmos. Environ., 2011, 45(15), 2473–2479 CrossRef CAS.
- C. Liu and C. Zeng, Heterogeneous kinetics of methoxyphenols in the OH-initiated reactions under different experimental conditions, Chemosphere, 2018, 209, 560–567 CrossRef CAS PubMed.
- J. Mao, X. Ren, W. H. Brune, J. R. Olson, J. H. Crawford, A. Fried, L. G. Huey, R. C. Cohen, B. Heikes, H. B. Singh, D. R. Blake, G. W. Sachse, G. S. Diskin, S. R. Hall and R. E. Shetter, Airborne measurement of OH reactivity during INTEX-B, Atmos. Chem. Phys., 2009, 9(1), 163–173 CrossRef CAS.
- S. Tang, F. Li, N. T. Tsona, C. Lu, X. Wang and L. Du, Aqueous-Phase Photooxidation of Vanillic Acid: A Potential Source of Humic-Like Substances (HULIS), ACS Earth Space Chem., 2020, 4(6), 862–872 CrossRef CAS.
- H. Herrmann, D. Hoffmann, T. Schaefer, P. Bräuer and A. Tilgner, Tropospheric Aqueous-Phase Free-Radical Chemistry: Radical Sources, Spectra, Reaction Kinetics and Prediction Tools, ChemPhysChem, 2010, 11(18), 3796–3822 CrossRef CAS PubMed.
- Y. H. Sun, L. Liu, M. Li, X. X. Chen and F. Xu, Theoretical investigation on the mechanisms and kinetics of OH/NO3− initiated atmospheric oxidation of vanillin and vanillic acid, Chemosphere, 2022, 288, 132544 CrossRef CAS PubMed.
- H. S. Mahal, L. P. Badheka and T. Mukherjee, Radical scavenging properties of a flavouring agent–Vanillin, Res. Chem. Intermed., 2001, 27(6), 595–604 CrossRef CAS.
- C. Roman, C. Arsene, I. G. Bejan and R. I. Olariu, Investigations into the gas-phase photolysis and OH radical kinetics of nitrocatechols: implications of intramolecular interactions on their atmospheric behaviour, Atmos. Chem. Phys., 2022, 22(4), 2203–2219 CrossRef CAS.
- E. S. C. Kwok and R. Atkinson, Estimation of hydroxyl radical reaction rate constants for gas-phase organic compounds using a structure-reactivity relationship: an update, Atmos. Environ., 1995, 29(14), 1685–1695 CrossRef CAS.
- B. Witkowski, P. Jain and T. Gierczak, Aqueous chemical bleaching of 4-nitrophenol brown carbon by hydroxyl radicals; products, mechanism, and light absorption, Atmos. Chem. Phys., 2022, 22(8), 5651–5663 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ea00073g |
| This journal is © The Royal Society of Chemistry 2023 |