
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
NO3 reactivity measurements in an indoor environment: a pilot study†
Patrick
Dewald
 ,
Jos
Lelieveld
and
John N.
Crowley
*
,
Jos
Lelieveld
and
John N.
Crowley
*
Department of Atmospheric Chemistry, Max-Planck-Institute for Chemistry, 55128 Mainz, Germany. E-mail: john.crowley@mpic.de
First published on 3rd November 2023
Abstract
We present the first direct indoor measurements of VOC-induced nitrate radical (NO3) reactivity (kNO3) together with measurements of nitric oxide (NO), nitrogen dioxide (NO2), ozone (O3) and dinitrogen pentoxide (N2O5) inside a laboratory during a four-day period in October 2021 in a suburban area (Mainz, Germany). Indoor mixing ratios of O3 ranged from <2–28 ppbv and those of NO2 from 4.5–27 ppbv. The rapid ventilation of the room (air change rates of ∼4 h−1) meant that indoor mixing ratios mirrored the variability in NO2 and O3 outdoors. NO3 production rates were between <0.02 and 0.12 pptv s−1 with indoor N2O5 mixing ratios increasing to 4–29 pptv during five NO-depleted day- or nighttime periods when kNO3 was between 0.04 and 0.2 s−1. Steady-state calculations resulted in a peak NO3 mixing ratio of 6 pptv. A comparison of measured N2O5 mixing ratios to those derived from steady-state calculations and the equilibrium coefficient for the NO2, NO3, N2O5 system showed very good agreement, indicating that heterogeneous reactions do not contribute significantly to the overall NO3 loss rate (LNO3). During these five periods, NO3 was mostly lost to NO and VOCs, the latter contributing on average 65% to LNO3. This pilot study underlines the necessity of further indoor NO3 reactivity measurements and that the nitrate radical can be a significant indoor oxidizing agent when the room is sufficiently ventilated during episodes of moderate outdoor air pollution.
Environmental significanceThe nitrate radical (NO3), formed by the reaction between ozone (O3) and nitrogen oxide (NO2), is an important nocturnal oxidant of unsaturated volatile organic compounds in the atmosphere. While its effect on the atmospheric lifetime of NOx has been extensively studied outdoors, indoor studies of NO3 are very limited. The short atmospheric lifetime of NO3 makes its detection challenging. We demonstrate the first direct measurement of NO3 reactivity indoors, providing an alternative way to assess NO3 concentrations under polluted conditions. Our measurements suggest that NO3 can be the dominant indoor oxidant of limonene, which is often released indoors owing to its presence in cleaning agents. This study emphasizes that NO3 chemistry can significantly impact indoor air quality. |
1 Introduction
Since a great fraction of human lifetime is spent indoors, the composition of indoor air can have a significant impact on human health.1 Analogous to outdoor environments, the major indoor oxidants are ozone (O3), the hydroxyl radical (OH) and the nitrate radical (NO3).2–5 NO3 is produced by the oxidation of nitrogen dioxide (NO2) by ozone (O3), both of which are usually present in indoor air from ventilation of outside air:4| NO2 + O3 → NO3 + O2 | (R1) |
Dinitrogen pentoxide (N2O5) is formed via reaction of NO3 and NO2 and is in thermal equilibrium with both:6
| NO3 + NO2 + M → N2O5 + M | (R2) |
| N2O5 + M → NO3 + NO2 + M | (R3) |
In outdoor environments during the day, NO3 is removed rapidly by reaction with NO (R4) and via photolysis by sunlight ((R5a) and (R5b)).7
| NO3 + NO → 2 NO2 | (R4) |
| NO3 + hv (λ ≈ 400 – 640 nm) → NO2 + O | (R5a) |
| NO3 + hv (λ ≈ 585 – 640 nm) → NO + O2 | (R5b) |
NO3 photolysis rates indoors are sufficiently diminished compared to outside so that (R5a) and (R5b) can be neglected and NO3 gains in importance, relative to O3 and OH, as an oxidizing agent.4,10,11 As evident from (R1), NO3 production (and its subsequent chemistry) relies on the presence of O3 and NO2. Elevated indoor ozone (and NOx) levels are particularly common in urban buildings equipped with ventilation systems or when windows are open,12,13 so that indoor NO3 production rates become significant. Indoor ozone can be consumed by NO (R6) in poorly ventilated, residential environments,14 where a major (indoor) source of NO is gas cooking.4
| NO + O3 → NO2 + O2 | (R6) |
Unsaturated volatile organic compounds (VOCs) such as terpenes, which are highly reactive towards NO3,15 are often abundant in indoor environments as they are present in detergents and cleaning agents.16 In some environments (e.g. forested regions), reactions with unsaturated hydrocarbons not only become the dominant NO3 removal process at night, but even compete with (R4), (R5a) and (R5b) during the day.8,9 In addition, the reaction of NO3 with VOCs (R7) leads (among other products) to the formation of alkyl nitrates (RONO2) or nitric acid (HNO3), which, in outdoor environments can transfer to the particle phase leading to secondary organic aerosols (SOA) and/or particulate nitrate.17,18
| NO3 + VOCs (+O2) →→ products (e.g. RONO2, HNO3) | (R7) |
While the impact of O3 and OH on indoor air quality has been extensively investigated,3,14,19,20 the number of studies examining the role of the nitrate radical in indoor environments is very limited. Along with some model calculations and steady-state calculations which suggest that indoor NO3 levels are below 0.04 parts per trillion by volume (pptv),13,21–24 only a few direct measurements of NO3 or its equilibrium partner N2O5 are available.25–27 Arata et al.26 detected several pptv of NO3 in a poorly-ventilated residential kitchen when the NO3 production rate was artificially enhanced by continuous addition of “synthetic” O3 (up to 40 parts per billion by volume, ppbv). Detectable mixing ratios of N2O5 and NO3 in the lower pptv range have been reported for ventilated rooms (exchange rates of 3.8 h−1 and 7 h−1) in an office building and an athletic facility.25,27
This limited number of studies indicates that, in poorly ventilated rooms, high NO3 loss rates make it difficult to assess the impact of the nitrate radical yet no direct indoor NO3 reactivity measurements have been reported. In addition, with the outbreak of the COVID-19 pandemic, rapid ventilation of indoor environments has become increasingly important to reduce viral loads.28,29 Rapid ventilation of outside air results in the transport of photochemically generated O3 and NOx into the indoor environment. Clearly, indoor measurement of NO3 mixing ratios, NO3 production rates and NO3 reactivity would thus help to assess the fate of NO3 radicals in such an environment. In this study, we report the first measurements of VOC-induced NO3-reactivity along with mixing ratios of the nitrogen oxides NO, NO2, NO3 and N2O5 together with O3 from a well-ventilated laboratory over a weekend period in October 2021 in Mainz (Germany). Quantifying VOC-induced NO3 reactivity together with the above-mentioned set of measurements enables identification of the dominant NO3 loss processes which may allow qualitative conclusions about the formation of organic nitrates indoors to be drawn. The purpose of this study is thus to evaluate whether NO3 reactivity measurements provide us with new insights into indoor oxidation processes.
2 Experimental
The laboratory used in this study has a volume of ∼220 m3 (floor area = 61 m2) and was mostly unoccupied during the measurement period in order to reduce the impact of the human emissions on the observations. The room itself is located at the Max-Planck-Institute for Chemistry (MPIC) that is situated in direct vicinity to commercial, residential and university buildings. Busy two- and four-lane roads leading to the city center of Mainz (5 km, 217![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 inhabitants) are adjacent to the institute. Mainz is part of the densely populated and industrialized Rhine-Main area close to Frankfurt and Wiesbaden. A ventilation system, that was continuously operated in “night-mode” (i.e. reduced air change rate, see Supplement S1†) during the study period constantly replenished the room with urban (polluted) air from outside. The air change rate (kchange) was estimated using a tracer-gas approach30 in which limonene or 2,3-dimethyl-2-butene was released into the laboratory and its decay in concentration was monitored. The concentration of limonene or 2,3-dimethyl-2-butene was not measured directly but by the change (reduction) in NO3 reactivity as their mixing ratios decreased mainly due to exchange with outdoor air. Following a phase of mixing (<1 min) initial concentrations of ≈ 200 pptv (for limonene) decrease to roughly zero in ≈ 30 min. The decay of limonene and of 2,3-dimethyl-2-butene is exponential, enabling decay constants (or air change rate constants) of 5.76 h−1 (limonene) and 4.3 h−1 (2,3-dimethyl-2-butene) to be derived. The faster decay term for limonene is likely related to its indoor oxidation and wall loss, which are both expected to be more rapid than for 2,3-dimethyl-2-butene. Due to the bias caused by deposition and chemical loss processes, the values derived by our approach serve as an upper limit of the true air change rate. In the case of limonene, gas-phase losses contribute ≈ 30% to the overall decay rate. The air change rate of our laboratory during the measurement period was less than 4 h−1. The procedure and results of the air change rate determination are found in more detail in the Supplement (S1†). Note that a more volatile and less reactive tracer such as carbon dioxide (CO2), which is readily available through respiration and can be detected by inexpensive sensors, represents an alternative tracer.
000 inhabitants) are adjacent to the institute. Mainz is part of the densely populated and industrialized Rhine-Main area close to Frankfurt and Wiesbaden. A ventilation system, that was continuously operated in “night-mode” (i.e. reduced air change rate, see Supplement S1†) during the study period constantly replenished the room with urban (polluted) air from outside. The air change rate (kchange) was estimated using a tracer-gas approach30 in which limonene or 2,3-dimethyl-2-butene was released into the laboratory and its decay in concentration was monitored. The concentration of limonene or 2,3-dimethyl-2-butene was not measured directly but by the change (reduction) in NO3 reactivity as their mixing ratios decreased mainly due to exchange with outdoor air. Following a phase of mixing (<1 min) initial concentrations of ≈ 200 pptv (for limonene) decrease to roughly zero in ≈ 30 min. The decay of limonene and of 2,3-dimethyl-2-butene is exponential, enabling decay constants (or air change rate constants) of 5.76 h−1 (limonene) and 4.3 h−1 (2,3-dimethyl-2-butene) to be derived. The faster decay term for limonene is likely related to its indoor oxidation and wall loss, which are both expected to be more rapid than for 2,3-dimethyl-2-butene. Due to the bias caused by deposition and chemical loss processes, the values derived by our approach serve as an upper limit of the true air change rate. In the case of limonene, gas-phase losses contribute ≈ 30% to the overall decay rate. The air change rate of our laboratory during the measurement period was less than 4 h−1. The procedure and results of the air change rate determination are found in more detail in the Supplement (S1†). Note that a more volatile and less reactive tracer such as carbon dioxide (CO2), which is readily available through respiration and can be detected by inexpensive sensors, represents an alternative tracer.
The east side of the room featured windows to an inner courtyard. Light entering the room through the permanently closed windows was attenuated by a fine-meshed sunscreen. The room lights (fluorescent strip-lamps) were turned off during the entire period. Spectral-radiometric measurements verified that NO3 photolysis rates (JNO3) resulting from the attenuated daylight (<10−6 s−1) were insignificant (see Supplement, Fig. S2†).
The laboratory was equipped with four instruments described below, each one connected to a central exhaust system. The NO bottles used to run the cavity ring-down spectrometers (CRDS) were stored in a separate, ventilated safety cabinet. All gas lines were thoroughly checked for leakages. By avoiding potential laboratory (chemical) emissions, the composition of the air should thus be comparable to other ventilated rooms. The measurements were carried out at the institute during a four-day period from Friday to Tuesday in October 2021 during which the maximum outdoor daytime and nighttime temperatures were 14 °C and 5 °C, respectively. The weather during the weekend was dominated by clouds and fog, the only extended sunny period was on October 16 between 07:00 and 15:00 UTC.
2.1 NO3 reactivity
The NO3 reactivity (kNO3) was directly measured with a cavity ring-down spectrometer that was coupled to a flowtube (FT-CRDS) as detailed in Liebmann et al.31 After accounting for the impact of NOx (see below), this instrument quantifies the total gas-phase NO3 reactivity (i.e. the inverse of the NO3 lifetime) towards VOCs, so that kNO3 is equal to the summed first-order loss rate Σki[VOC]i with the concentration of a VOC [VOC]i and the corresponding rate coefficient ki for its reaction with NO3(R7). A commercial zero-air generator (CAP 180, Fuhr GmbH) provides 400 standard (STP) cubic centimeters per minute (sccm) of zero-air which is passed over a mercury lamp (Penray) to generate O3 at ca. 400 ppbv. This flow is mixed with a flow of NO (3 sccm of 1 parts per million by volume (ppmv) in N2, Air Liquide) and directed through a thermostated Teflon-coated (FEPD 121, Chemours) reactor (30 °C, 1.3 bar). During the residence time of ca. 5 min, O3 sequentially oxidizes NO to NO2(R5) and then to NO3(R1), which reacts with NO2 to form N2O5(R2). Passing the gas through a 15 cm long piece of PFA (perfluoroalkoxy) tubing (outer diameter (OD) of 1/4 in. = 0.635 cm) heated to 140 °C converts N2O5 to NO3 and NO2(R3). The flow containing NO3 is then mixed with 2800 sccm synthetic or ambient air and directed through a Teflon-coated (FEPD 121, Chemours) flowtube, where the gas-mixture has a time of 11 s to react. The NO3 that survives the flowtube is quantified using cavity ring-down spectroscopy (CRDS) at a wavelength of 662 nm. The basic principle of the instrument thus relies on comparing the level of synthetic NO3 in zero-air (i.e. total NO3 reactivity = 0 s−1), to those in indoor air. The ring-down time in the absence of NO3 was determined by the periodic addition of sufficient NO (3 sccm of 100 ppmv NO in N2, Air Liquide) to titrate the NO3 completely. Zero-air was humidified to match indoor-air humidity using a permeation-tube based humidification system (PermaPure, MH-070-24F-4) filled with deionized water (LiChrosolv, Merck). Indoor air was sampled through 1 m 1/4 in. (OD) PFA tubing equipped with a Teflon membrane filter (Pall Corp., 47 mm, 0.2 μm pore) to protect the mirrors and through a 2 L (uncoated) borosilicate glass flask heated to 45 °C to remove both ambient NO3 and N2O5, which would bias the measurement.Indoor air was dynamically diluted with zero-air during periods with highly reactive ambient air, which extends the upper limit of measurable reactivities to 1.7 s−1. The variability of the NO3 source and cavity instabilities results in a lower limit of detection (LOD) of 0.006 s−1 and contribute ∼18% to the measurement uncertainty.
As described by Liebmann et al.,31 the presence of NO, NO2, O3 (and thus reactions ((R1–R4) and (R6) and (R7)) and NO3 losses on the walls of the flowtube (NO3 wall loss rate = 0.001 s−1) require numerical simulations in order to extract the NO3 reactivity (kNO3) that can be attributed to VOCs (R7). Due to the numerical correction procedure, the total uncertainty of the measurement is dependent on the ratio of ambient NO2 and kNO3.31 Note that conversion of NO to additional NO2via(R6) in the flowtube affects NO2/kNO3 which is why accounting for the impact of NO becomes significant in polluted conditions not only because of (R4). If, for example, a reactivity of 0.15 s−1 is measured in the presence of 12 ppbv NO2, the additional uncertainty introduced by the numerical simulations would be ∼24%, leading to a total measurement uncertainty (TMU) of 30% which corresponds to the median TMU of this instrument during the study.
2.2 NO3 and N2O5
Ambient NO3 and N2O5 mixing ratios were monitored using the two 662 nm cavities of the five-channel cavity ring-down spectrometer (5Ch-CRDS), described by Sobanski et al.32 In this instrument, N2O5 is quantitatively dissociated to NO3(R3) by passing the sample air through a Teflon-coated glass tube heated to 95 °C prior to entering the cavity at the same temperature. The heated cavity consequently detects the sum of NO3 + N2O5. Similar to the FT-CRDS, the “baseline” (i.e. the ring-down time in the absence of the absorbing species) was determined by adding NO to the cavity (6 sccm of 100 ppmv NO in N2, Air Liquide). Air was sampled at 15 standard (STP) liters per minute (SLPM) through 10 cm of 1/4 in. (OD) PFA tubing and a Teflon membrane filter (Pall Corp., 47 mm, 0.2 μm pore) with the heated cavity sampling 7 SLPM and the unheated one 8 SLPM. To reduce NO3 loss through the inlet, an automatic filter changer normally replaces the inlet filter every hour. Unfortunately, the filter changer was not available during the measurement period. The data was corrected for cavity losses of NO3, the effective NO3 cross-section at both cavity temperatures and the impact of the mirror purge gas flow as described in Sobanski et al.32 Taking the standard deviation (2σ) from the baseline variability of the whole 10 min-averaged data set results in LODs of 0.9 pptv and 1.5 pptv for NO3 and N2O5, respectively. The total uncertainty associated to the NO3 and N2O5 measurements is 25% and 28%, respectively.2.3 NO and NO2
NO and NO2 were measured with a two-channel, cavity ring-down spectrometer33 operated at 405 nm. One cavity directly samples ambient air to detect NO2, while the other samples via additional tubing (1 m 1/2 inch (OD) PFA) that forms a reaction volume in which NO is oxidized to NO2via the addition of ∼3 ppmv O3(R6). The second cavity thus detects the sum of NO and NO2 so that NO mixing ratios were derived by the difference signal. Each cavity sampled indoor air at a flow of 2 SLPM through ∼2 m 1/2 in. (OD) PFA tubing and a Teflon membrane filter (Pall Corp., 47 mm, 0.2 μm pore). Based on the noise level and the variability in the baseline, the LODs of the NO and NO2 measurements are 132 pptv and 57 pptv with associated uncertainties of 11% and 9%, respectively.Since the correction of the NO3 reactivity measurements requires accurate NO2 measurements, NO2 mixing ratios were additionally monitored with a cavity of the 5Ch-CRDS operated at 405 nm. A comparison of both NO2 measurements is presented in the Supplement (Fig. S3†) and reveals excellent agreement between the two-channel and five-channel instruments. As both instruments were placed in opposed corners of the laboratory, this comparison also confirms that the air in the room is well mixed.
2.4 O3 and other auxiliary measurements
Ozone mixing ratios were measured with a commercial ozone monitor (2B Technologies, model 205) based on UV absorption. The instruments detects O3 mixing ratios > 2 ppbv with an uncertainty of 5%.Relative humidity (RH) and temperature (T) measurements were monitored with the NO3 reactivity setup (see 2.1) by separately sampling 500 sccm ambient air over a commercial hygrometer (Innovative Sensor Technology, HYT939) with an accuracy of ±1.8% (RH) and ±0.2 °C (T). Total (i.e. non-size-segregated) particle number densities were sporadically measured after the pilot study using a condensation particle counter (CPC, TSI, model 3025a).
Indoor photolysis frequencies were determined using a spectral-radiometer (Metcon GmbH) with a single monochromator and 512 pixel CCD array as a detector (275–640 nm). The thermostated monochromator-detector unit was attached via a 10 m optical fiber to an integrating hemispheric quartz dome that was placed at a height of ca. 2 m in the center of the room. Light fluxes were converted to photolysis rate constants for NO3 ((R5a) and (R5b), JNO3) and NO2 (JNO2) using molecular parameters recommended by the IUPAC and NASA evaluation panels.34,35
3 Results and discussion
A time-series of the mixing ratios of NO, NO2, O3, NO3 and N2O5 as well as RH, T and kNO3 is given in Fig. 1 together with calculated quantities (e.g. production and loss rates of NO3) that are discussed in section 3.2. Within the measurement period, the relative humidity varied between 24% and 36% at a fairly constant temperature of 297–298 K (panel a). NO (<132 pptv to 41 ppbv, panel b), NO2 (4.5 to 27 ppbv, panel b) and O3 (<2 ppbv to 28 ppbv, panel c) mixing ratios were quite variable. During the periods in which NO was high, likely a result of nearby automobile emissions, the NO3 reactivity exceeded the instrument's upper LOD of 1.7 s−1 (panel d), which was reached when the NO mixing ratio exceeded 2.5 ppbv.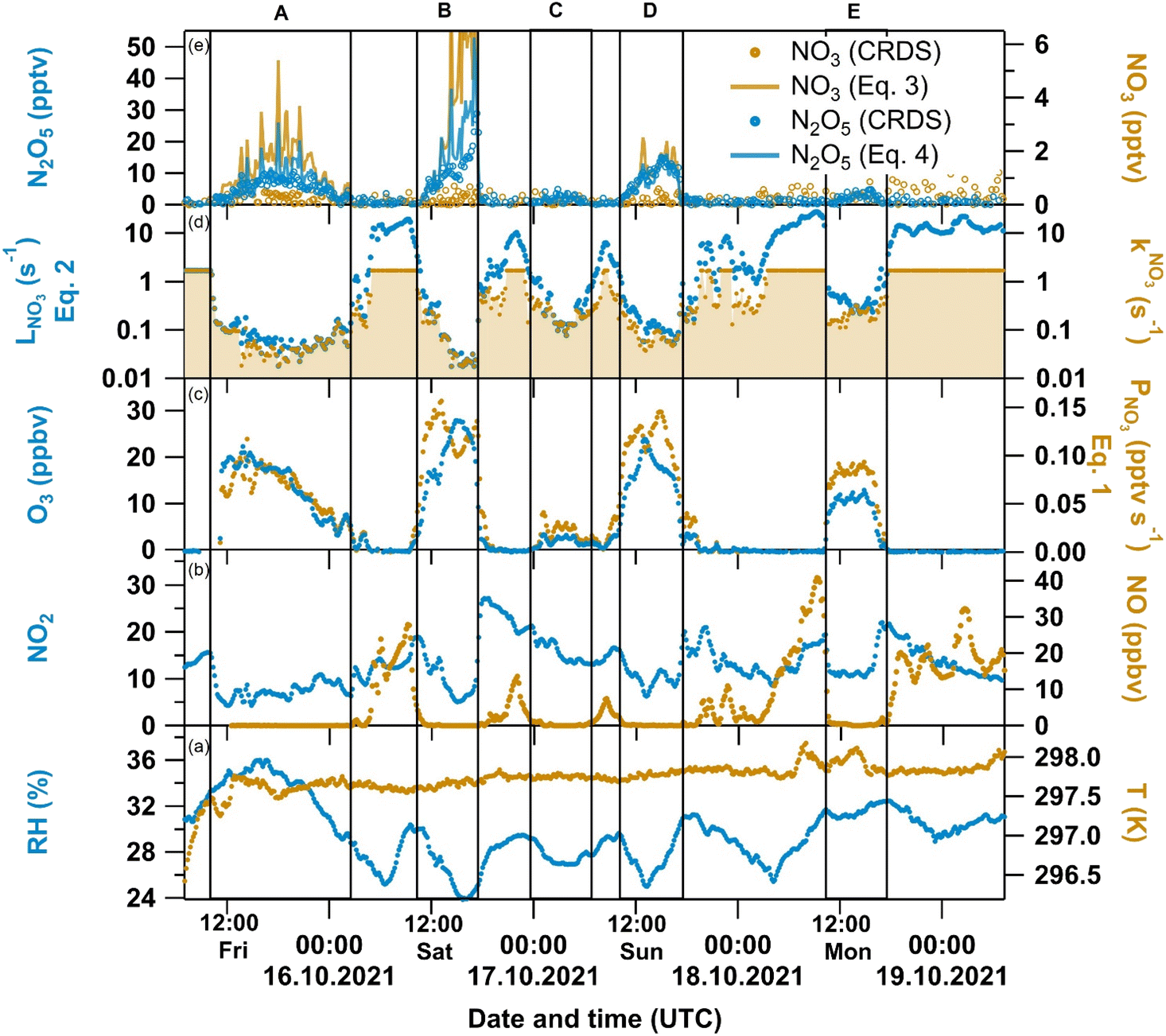 | ||
| Fig. 1 Overview of directly measured and derived quantities during a weekend period inside a ventilated laboratory: (a) relative humidity RH (blue, left axis) and temperature T (orange, right axis); (b) NO2 (blue, left axis) and NO (orange, right axis) (c) O3 (blue, left axis) and NO3 production rate PNO3from (Eq. 1) (blue, right axis); (d) Total NO3 loss rate LNO3 from (Eq. 2) (blue, left axis) and NO3 reactivity kNO3 (LOD of 1.7 s−1, orange with same-coloured shaded area to mark its contribution to LNO3); (e) N2O5 (blue circles, left axis), calculated N2O5 from (Eq. 4) (blue line, left axis), NO3 (orange circles, right axis) and calculated NO3 from (Eq. 3) (orange line, right axis). | ||
We have divided the measurements into five periods (A–E in Fig. 1), during which N2O5 mixing ratios (panel e) were >2 pptv and NO mixing ratios were close to or below the LOD, whereas O3 mixing ratios were large. The anti-correlation between NO and O3 is readily understood considering the efficient conversion of NO to NO2via(R6), which has the following consequences: (1) The presence of O3 (together with NO2) enables the production of both NO3 and N2O5. (2) The lack of NO leads to lower NO3 reactivities (i.e. higher NO3 lifetimes) of between 0.04 to 0.2 s−1. This is reflected in measurable amounts of N2O5 (up to 29 pptv) during period B.
In this study, NO3 remained undetected. Considering the thermal equilibrium between NO2, NO3 and N2O5 ((R2) and (R3)) and the measured NO2 and N2O5 mixing ratios, this is contradictory to calculations using evaluated rate coefficients for k2 and k3,35 which indicated the presence of 2–4 pptv NO3. The most likely explanation is that NO3 (a reactive radical) was lost during sampling through the inlet tubing and filter, while both N2O5 and NO2 pass almost loss-free through the system. After use over the 4 days period, the inlet filter was clearly contaminated with dark-coloured particles, presumably black-carbon. A picture of this filter in comparison to an unused filter is shown in the Supplement (Fig. S4†). A previous study36 on the interaction of NO3 and N2O5 with urban aerosols collected on filter samples at the same location showed no measurable uptake for N2O5, whereas NO3 was lost efficiently with γ(NO3)/γ(N2O5) > 15, where γ is the net-uptake coefficient for heterogeneous loss.
Outdoors, NO3 and N2O5 are usually only observed during nighttime due to NO3 photolysis rates of typically up to 0.17 s−1 at noon,7 whereas in this study N2O5 was detected independently of the diel cycle. In Fig. 1, period A covers a daytime–nighttime transition, the N2O5 peaks in periods B, D and E are around noontime, while period C is a nighttime period. However, an air change rate of 4 h−1 is sufficient to entrain N2O5 originating from outside into the laboratory especially at night. To assess the potential impact of outdoor N2O5 on our measurement, we compare the calculated in situ indoor N2O5 production rate Pindoor(N2O5) with the production rate Poutdoor(N2O5) caused by infiltration of N2O5 originating from outside. According to (R2) and (Eq. 4), Pindoor(N2O5) is equal to k2[NO2][NO3] ≈ k1k2[NO2]2[O3]/LNO3. Calculation of Pindoor(N2O5) from our measurements of NO2, O3, NO and kNO3 results in values of 0.1–2.1 pptv s−1 during periods A–E. Schuster et al.37 reported nighttime N2O5 mixing ratios outside the institute in October 2007 of up to 80 pptv which would result in an production rate Poutdoor(N2O5) = [N2O5]outdoor × kchange of ≈ 0.01 pptv s−1. Unless several hundreds of pptv of N2O5 were constantly abundant outside, Poutdoor(N2O5) is very unlikely to affect our N2O5 levels measured inside.
3.1 Comparison of indoor and outside air
With an air change rate of up to 4 h−1, the composition of the air in the laboratory is strongly influenced by that outside. Fig. 2 compares the indoor mixing ratios of O3 and NO2 to those from a local air-quality measurement station38 located in Mainz-Mombach, ca. 4 km north of the Max-Planck-Institute. For both trace gases, the indoor/outdoor diel profiles are very similar. Outdoor O3 is close to zero at nighttime, which is the result of deposition to surfaces and also reaction with NO forming NO2(R6). Outdoors, NO2 is rapidly photolysed during the day (R8) to re-generate O3(R9), whereas indoor photolysis rates for NO2 (JNO2) are very low (see Fig. S2 in the Supplement†) and there are no significant in situ sources of O3 in the laboratory. The availability of O3 indoors thus depends entirely on exchange with outdoor air and the strong co-variance is expected. Outdoors, the nighttime increase in NO2 (resulting from the oxidation of locally emitted NO) results in rapid formation of O3 at the start of the day as NO2 is photolysed (to O(3P) and thus O3).| NO2 + hν → NO + O(3P) | (R8) |
| O(3P) + O2 + M → O3 + M | (R9) |
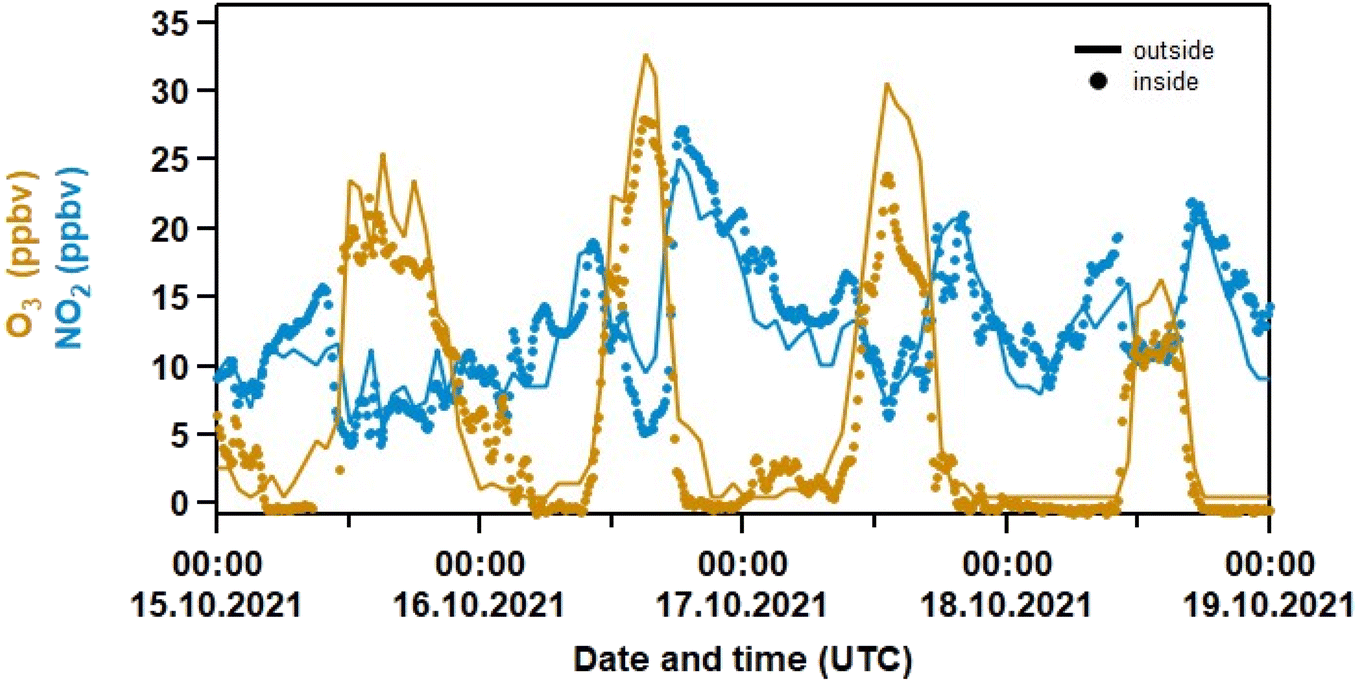 | ||
| Fig. 2 Measurements of O3 and NO2 inside the laboratory (dots, 10 min data) and outside at a station in Mainz–Mombach (solid line, 1 h data). The outdoor data was taken from the German Environment Agency38 and converted from μg cm−3 to ppbv using a pressure of 760 Torr and a temperature of 298 K. | ||
There is no obvious lag in the indoor O3 data at sunrise compared to outdoors, which implies that the air change rate is more rapid than the production and loss terms for O3. The indoor-to-outdoor ratio (I/O) of O3 and NO2 was obtained by measuring both NO2 and O3 sequentially inside the laboratory and directly outside by passing the inlet through a port in the wall (Fig. 3). In this case, I/O(O3) was determined to be 0.63 ± 0.03 (1σ). Note that windows are shut at all times in the laboratory, and that outdoor air passes through a compressor, metal piping and filters before entering the labs so O3 is expected to be lost, which explains the lower than unity indoor/outdoor ratio. An average I/O of 0.25 was recently reported in a review summarizing measurements in ca. 2000 indoor environments.14
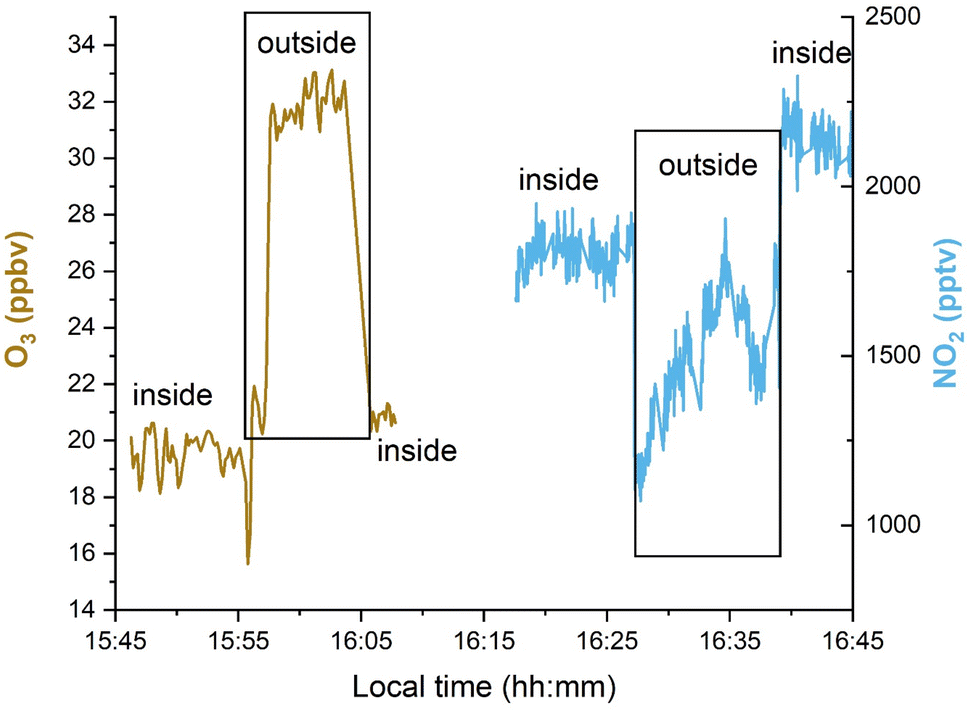 | ||
| Fig. 3 Sequential measurements of O3 and NO2 inside and outside the laboratory on the 21st October 2021. | ||
I/O(O3) can be assessed by assuming that the air change rate kchange and surface removal rate ksurface of O3 are much faster than its outdoor variability so that I/O(O3) = kchange/(kchange + ksurface).12 Using an air change rate of 4 h−1 and a surface removal rate for O3 as reported for a laboratory environment of 2.5 h−1,12 I/O(O3) = 0.62 is derived, which is close to the observed value. However, I/O(O3) is affected by several parameters such as the air change rate, abundance of NO, loss rate on indoor surfaces (highly dependent on the material), transmission loss through ventilation systems and the presence of humans.14 The values of ksurface may consequently be very different for two non-identical laboratories. Since indoor O3 only reached elevated levels when NO ≤ 150 pptv resulting in a maximum O3 loss rate of 0.25 h−1, the contribution of (R6) was neglected.
As mentioned above, the oxidation of NO by O3 not only leads to O3 loss, but also to formation of NO2. As shown in Fig. 3, the indoor mixing ratios of NO2 (1800–2100 pptv) were indeed higher than outside (1100 to 1700 pptv). The high variability in NO2 outside makes it impossible to accurately quantify a value for I/O(NO2), which is highly dependent on the availability of indoor NO2 sources, the ratio between kchange + ksurface (similarly to O3), season (photochemistry) and location. Models and long-term experiments show that I/O(NO2) is often above or close to unity in non-domestic, strongly ventilated rooms in urban areas similar to our laboratory.39,40
In summary, Fig. 2 and 3 underline (1) that (for a given ventilation rate) the indoor abundance of the NO3 precursors (NO2 and O3) are mainly determined by the air composition outside and (2) that (R6), similarly to outdoors, explains the distinct anti-correlation between O3 and NOx measured indoors as observed in Fig. 1.
In this laboratory environment, in which the indoor air is supplied by a compressor/filter system, the particle concentration is greatly reduced compared to outdoors. A few checks (after our pilot study) showed that the typical number density indoor-to-outdoor ratio, I/O(Npart), was close to 0.1. As shown below, the low particle number density results in low aerosol surface areas, hence leading to insignificant losses of e.g. N2O5 or NO3 to particles in ambient air compared to other surfaces and/or reactants. Nevertheless, accumulation of ambient particles on our inlet filter with time results in quantitative NO3 removal prior to entering our cavity.36 The PTFE membrane filters are usually changed (automatically) on an hourly basis during measurements to avoid this issue.32,41
3.2 Measured versus calculated NO3 and N2O5 mixing ratios
NO3 and N2O5 mixing ratios can be calculated from a stationary-state approximation if measurements of NO2 and O3 mixing ratios as well as NO3 reactivity are available:41–47 By using the rate coefficient k1 for the reaction between NO2 and O3(R1) and the mixing ratios of O3 and NO2, the NO3 production rate (PNO3) can be assessed:| PNO3 = k1[NO2][O3] | (Eq. 1) |
In our analysis, the NO3 reactivity kNO3 is corrected for the impact of NOx. Thus, in order to derive the overall NO3 loss rate LNO3 (assuming only the overall gas-phase loss rate kgas is relevant compared to heterogeneous loss rate khet), the pseudo-first-order loss rate constant for reaction with NO has to be added:
| LNO3 = kgas + khet ≈ kNO3 + k4[NO], | (Eq. 2) |
With k4 being the rate coefficient for the reaction between NO and NO3(R4). In stationary state, i.e. d[NO3]/dt ≈ 0 the NO3 loss rate is in balance with its production rate (Eq. 3) and its steady-state concentration [NO3]ss can be calculated:
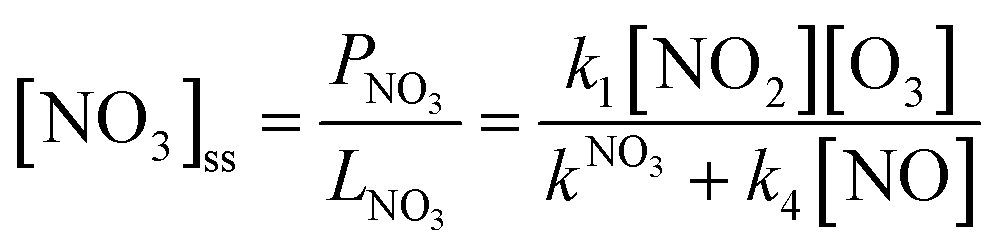 | (Eq. 3) |
This approximation is valid as long as the system is in equilibrium ((R2) and (R3)), NO3 loss rates are sufficiently large and NO2 does not vary too much on short time-scales.48,49 The thermal equilibrium constant Keq = k2/k3 (ref. 35) enables derivation of N2O5 mixing ratios:
| [N2O5]eq = Keq[NO2][NO3]ss | (Eq. 4) |
The NO3 production and loss rates, and steady-state mixing ratios of NO3 and N2O5 calculated using (Eq. 1) to (Eq. 4) are displayed in Fig. 1. When both O3 and NO2 were present (i.e. when O3 was not removed by NO) the NO3 production rates were between 0.02 and 0.12 pptv s−1. Since the abundance of O3 was the limiting factor in NO3 production in this study, PNO3 mostly follows the diel pattern of O3 (Fig. 1, panel c). During NO-rich periods, O3 is not only entirely depleted (so that PNO3 ≈ 0 pptv s−1), but LNO3 also increases to 10 s−1 (panel d), which is why neither NO3 nor N2O5 was expected. Furthermore, Fig. 1 (panel e) shows good agreement between measured and calculated N2O5 mixing ratios. At higher N2O5 mixing ratios, up to 6 pptv of NO3 would have been present at equilibrium (with NO2 and N2O5), which would have been detectable by our instrument. The lack of a NO3 signal is attributed to its loss on a contaminated filter (see above).
In order to analyze the conditions under which indoor N2O5 is observable, we focus on period D (Fig. 4) which we consider representative of all periods in which N2O5 was detected. The uncertainties in PNO3, LNO3, [NO3]ss and [N2O5]eq were derived from propagation of the uncertainties associated with the measurements (see section 2) as well as with the rate coefficients k1 (15%) and Keq (20%).34,35 In Fig. 4, N2O5 (panel e) starts to increase around 10:00 UTC and O3 mixing ratios (panel c) increase from < 2 ppbv to 20 ppbv (at 14:00). At the same time, NO (panel b) decreases from 3 ppbv to close to, or below the LOD so that NO3 loss rates (panel d) are around 0.04–0.1 s−1. The NO3 production rate (panel c) of ca. 0.06 pptv s−1 (circa 20 ppb O3 and 10 ppbv NO2) is sufficient to generate detectable amounts of NO3/N2O5 (panel e). These observations emphasize that the presence of N2O5 (and NO3) goes hand-in-hand with O3 mixing ratios that are sufficient for removal of NO (and its subsequent conversion to NO2). As indicated in section 3.1, the abundance of indoor NO2 and O3 is mostly determined by their outdoor mixing ratios and the air change rate. Between 14:00 and 16:00 UTC in Fig. 4, we have 2 pptv NO3 and 20 ppbv O3. If we assume an OH concentration of 7 × 105 molecules cm−3,20,50 the loss rate constants for an indoor VOC such as limonene would be 5.91 × 10−4 s−1 (NO3 loss), 1.08 × 10−4 s−1 (O3 loss) and 1.15 × 10−4 s−1 (OH loss). During period D, NO3 is thus the major oxidant of limonene and presumably other unsaturated compounds that display similar reactivity to NO3. In contrast to the reaction with O3 and OH which form carbonyl compounds (e.g. limona ketone, 4-acetyl-1-methyl-1-cyclohexene) and peroxy acetyl nitrate (CH3C(O)O2NO2, PAN) indoors,51 the NO3-initiated oxidation of limonene results in the formation of alkyl nitrates (RONO2) in high yields (30–67%).35 Note that the reaction with NO3 would only contribute ca. 30% to the total VOC loss rate when the maximum air change rate of 4 h−1 is taken into account.
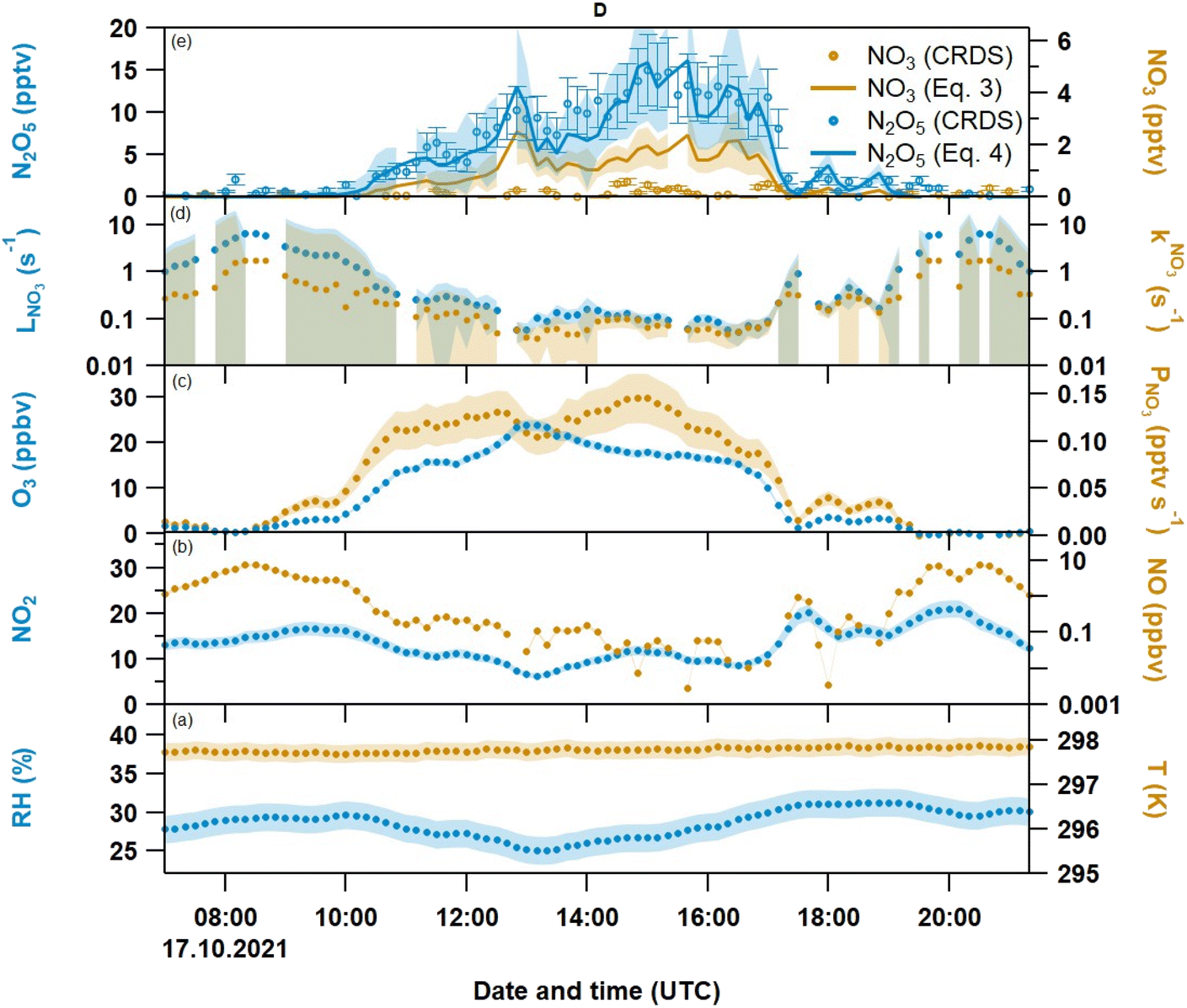 | ||
| Fig. 4 Same as Fig. 1, but expanded to display period D. Total uncertainties are shown by shaded areas in the same color as the corresponding traces. In the case of directly measured NO3 and N2O5, the uncertainties are shown as error bars. | ||
The good agreement between the measured and calculated N2O5 mixing ratios (Fig. 4) is further examined in Fig. 5 in which these quantities are plotted against each other. For this, mixing ratios at or below the LOD have been removed. A bivariate, linear regression yields an intercept of (−0.16 ± 0.34) pptv and a slope of 1.00 ± 0.09, indicating very good agreement within associated uncertainties. As heterogeneous loss processes of both N2O5 and NO3 are neglected in the calculation of N2O5 mixing ratios, and reactions of NO3 with RO2 (or other radicals) do not contribute to the measured loss term, we conclude that (within the associated uncertainty of this analysis), the loss of NO3 (and thus indirectly N2O5) is dominated by reactions with VOCs and NO. The uptake of N2O5 to aqueous particles can be an important term outdoors, but in an indoor environments which is supplied with fresh-air via filters, particle concentrations indoors are too low for this to contribute significantly (see section 3.1). In addition, the RH was relatively low so that the water content of aerosol particles is expected to be minor, which lowers the N2O5 uptake coefficient.52
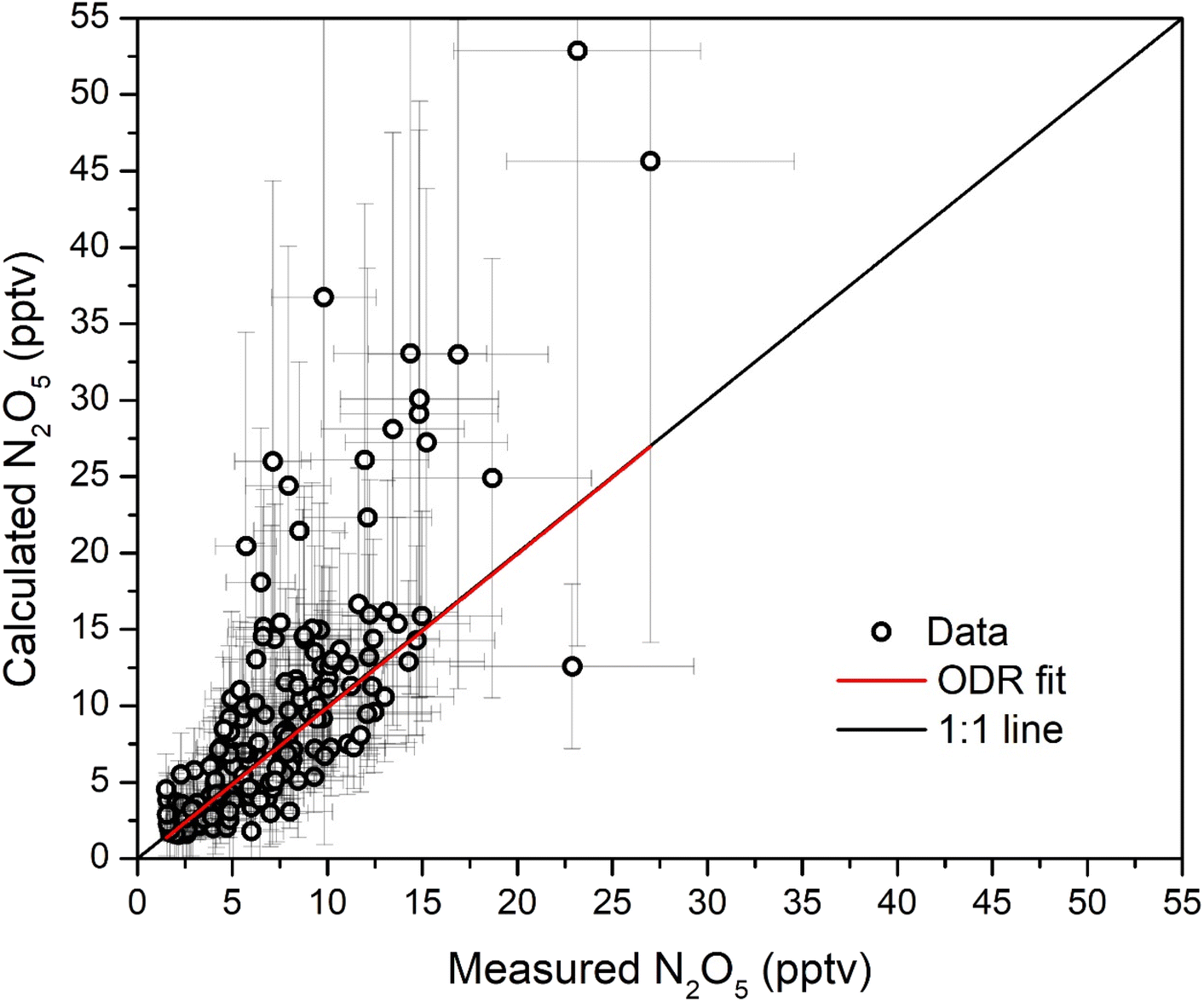 | ||
| Fig. 5 N2O5 mixing ratios calculated from (Eq. 4)versus those directly measured. The red solid line shows an orthogonal distance regression (ODR,57 slope: 1.00 ± 0.09, intercept: (−0.16 ± 0.34) pptv, Pearson correlation coefficient r = 0.8) while the black line denotes ideal 1:1 agreement. | ||
During period B (see Fig. 1, upper panel) a greater deviation between measured and calculated N2O5 mixing ratios is observed, which results in some of the scatter in Fig. 5. Period B is the only sunny period of the measurement period and an increase of 0.016 s−1 in the NO3 loss rate constant would be necessary to bring measured and calculated values into agreement. Such a reactivity would be caused by just 25 pptv of NO, which is below the LOD of the NO instrument.
3.3 Indoor fate of the nitrate radical
As indicated in section 1, reactions of NO3 with VOCs may lead to NOx removal from the gas-phase, while reaction with NO leads to reformation of NO2. Fig. 1 (panel d) suggests that the contribution of both reaction paths to LNO3 varies. We thus define the fractional contribution F of NO3 reactions with VOCs (represented by kNO3) to the overall NO3 loss rate LNO3: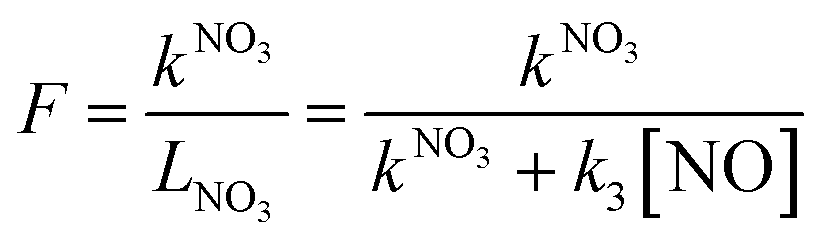 | (Eq. 5) |
The resulting fractional contributions are plotted together with measured (VOC-induced) NO3 reactivities in Fig. 6. The mean fractional contribution of VOCs is 0.46 ± 0.31 (1σ). This implies that on average NO3 was consumed roughly equally by NO and VOCs during the measurement period. It should be kept in mind that both the upper LOD of the kNO3 measurement and the lower LOD of the NO measurement bias the calculated fractional contributions to higher values (potential overestimation of kNO3 during NO-dominated periods and potential underestimation of NO-induced reactivity during VOC-dominated periods). During periods A–E, when N2O5 was above its LOD, the mean fractional contribution occasionally increased to 1, with a mean of 0.65 ± 0.32 (1σ). Both values are comparable to the nighttime outdoor values of 0.5–0.6 as observed at the summit of a semi-rural mountain site (impacted by both NO soil emissions and biogenic VOCs) ca. 30 km from the laboratory,53 but lower than the values close to 1 as observed in forested regions where NO3 reactivity is dominated by terpenes at night.8,9 The closer agreement with nighttime outdoor conditions in urban environments is easily understood considering the lack of NO3 photolysis and the abundance of NOx throughout the diel cycle. Moravek et al.27 identified reaction with NO to be the dominant loss process in a highly-ventilated athletics facility during daytime, whereas the thermal equilibrium to N2O5 gained importance in the afternoon. Such a distinct diurnal variation is not observed in our short study.
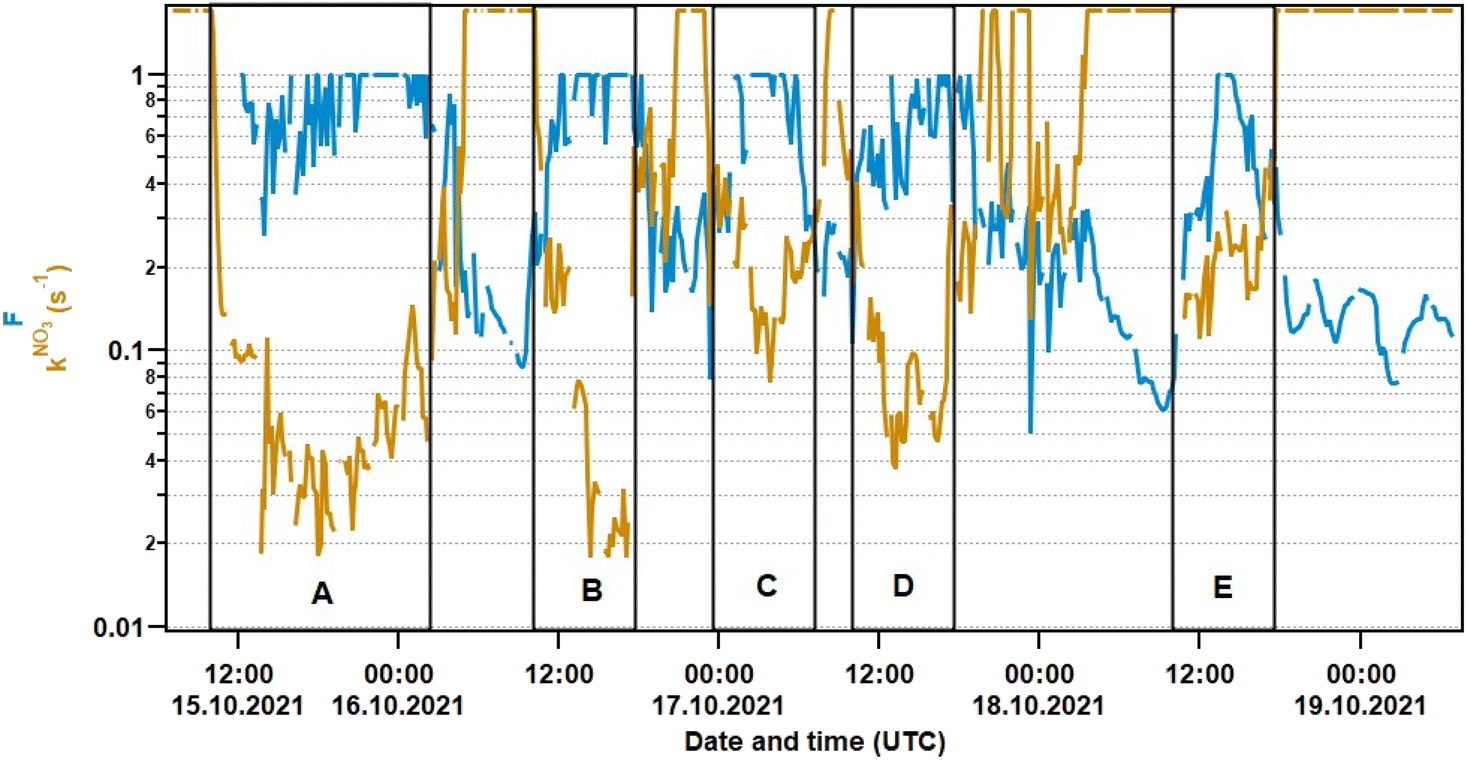 | ||
| Fig. 6 NO3 reactivity (kNO3) and fractional contribution F (Eq. 5) of kNO3 to the overall NO3 loss rate LNO3. | ||
Fig. 6 also shows that reaction with VOCs is a relevant NO3 loss process most of the time in this particular environment. Unfortunately, there are no simultaneous VOC measurements available for this pilot study. Monoterpenes such as limonene are a class of organic compounds that are abundant in indoor environments due to their presence in detergents and cleaning agents16,19,27 and which are highly reactive towards NO3.15 Terpenes or other unsaturated compounds released from indoor sources are thus the most likely organic reactants for NO3. Reactive VOCs may however also originate from outside: the laboratory used in this study is located in the direct vicinity of deciduous trees, which at some times of the year can represent a significant source of isoprene as well as monoterpenes.54–56 However, as our measurements were carried out in mid October with weak insolation and low temperatures, both of which result in low levels of biogenic activity, this is unlikely to represent the major source of indoor reactivity in this study.
4 Comparison to other indoor studies
In contrast to O3 and OH, the nitrate radical has not been studied in indoor environments to a similar extent. Table 1 gives an overview of maximum mixing ratios of NO3 (and N2O5) that were detected or modelled in different indoor environments.| Reference | Environment | Method | Max. NO3/N2O5 (pptv) | Air change rate (h−1) |
|---|---|---|---|---|
| a CRDS: Cavity ring-down spectroscopy, CIMS: chemical ionization mass spectrometry. | ||||
| Carslaw21 | Residential | Model calculation | 0.03 (NO3) | 2 |
| Nøjgaard25 | Office (Copenhagen) | Indirect measurement | 58 (NO3 + N2O5) | 3.76 |
| Waring et al.22 | Residential | Model calculation | 0.07 (NO3) | 0.5 |
| Zhou et al.13 | Residential (New York) | Steady-state calculation | 6.7 × 10−4 (NO3) | 0.65 |
| Arata et al.26 | Residential (Oakland) | CRDS | 4 (NO3) | 1–1.4 |
| Price et al.23 | Museum (Boulder) | Steady-state calculation | 0.04 (NO3) | 0.8 |
| Moravek et al.27 | Athletic facility (Boulder) | CIMS | 4 (N2O5) | 7 |
| Link et al.24 | Residential (Gaithersburg) | Model calculation | 0.3 (NO3) | 0.24 |
| This study | Laboratory (Mainz) | CRDS (N2O5) | 29 (N2O5) | <4 |
| Steady-state calculation (NO3) | 6 (NO3) | |||
As indicated in Fig. 1, up to 29 pptv of N2O5 were detected in our laboratory. The observation is comparable to 58 pptv of N2O5 + NO3 (with N2O5 as the dominant fraction) that was observed inside an office building in Copenhagen, Denmark.25 Note that both indoor environments were unoccupied, feature a comparable air change rate and are situated in urban areas. Arata et al.26 reported 190 pptv of N2O5, if 40 ppbv O3 was artificially added by a commercial ozone generator in order to force (together with 50–100 ppbv of NO2) a very high NO3 production rate of 7 ppbv h−1. This is at least a factor of 10 higher than our values of 0.04 to 0.2 pptv s−1 for PNO3 and accounts for the lower N2O5 mixing ratios measured in our laboratory.
Recently, up to 4 pptv N2O5 (similar to our measurements in period C and E) were observed inside an athletics facility, which was not only more strongly ventilated (with an air change rate of 7 h−1), but also occupied by humans.27 In this case, NO3 production rates between 0.025 and 0.3 pptv s−1 are similar to ours, so the lower maximum N2O5 mixing ratio is a reflection of higher NO3 loss rates of up to 8 s−1 (at high-NO) compared to our median LNO3 of 0.15 s−1.
In a study by Price et al.,23 oxidation via NO3 contributes ∼10% to the total VOC loss in a museum that is ventilated with an air change rate of only 0.8 h−1. The dominant loss process in this museum is ventilation with the residual contribution of 90%. Despite the fact that our air change rate is higher by a factor of 5, we estimated a higher NO3 contribution of ∼30% (see above). This discrepancy is explained by different indoor NO3 levels: According to steady-state calculations, only 0.04 pptv NO3 were present in the museum which is two orders of magnitudes lower compared to our values.
Link et al.24 identified ventilation to be the dominant VOC sink (88%) in a residential building featuring an even lower air change rate and modelled NO3 level of 0.2 h−1 and 0.02 pptv, respectively. When 70 ppbv of O3 were added artificially, oxidative processes competed with ventilation. In this case, ozonolysis (and OH production) drastically reduced the fractional contribution of (R7) to ∼10%. Again, our higher fractional contribution of (R7) to the overall VOC loss is consequently reflected in our higher indoor NO3 levels compared to those in Link et al.24
The NO3 mixing ratios calculated from NO2 and N2O5 mixing ratios (Fig. 1, panel e) are between 2 and 6 pptv and thus similar to the 3–4 pptv NO3 that were directly measured in a residential kitchen26 despite the great difference in PNO3. The comparable NO3 levels are accordingly caused by the higher NO3 reactivity of 0.8 s−1 (calculated from VOC measurements) in the residential kitchen. In any case, mixing ratios of a few pptv clearly exceed indoor NO3 levels predicted in model or steady-state calculations for residential environments by a factor of ∼100.13,21–24 This underlines the necessity of more direct measurements of NO3 mixing ratios, NO3 reactivity, the traces gases responsible for the loss of NO3 and also the products (e.g. organic nitrates) of indoor NO3–VOC interactions.
5 Conclusions
We present the first direct NO3 reactivity measurement in a ventilated indoor environment. Our pilot study suggests that the nitrate radical concentration increases, when (1) indoor air is continuously exchanged with outdoor air so that both O3 and NO2 are available, (2) NO is (almost) entirely depleted by O3 and (3) the room is not directly exposed to sunlight. A high ventilation rate (∼4 h−1) resulted in a high correlation between indoor and outdoor mixing ratios of NO2 and O3. Measured N2O5 and calculated NO3 mixing ratios peaked at 29 and 6 pptv, which are significantly higher than reported in model calculations21,22 but which agree with observations made in other ventilated (non-residential) rooms.25,27 We demonstrate that, in indoor environments when highly polluted conditions impede the formation of detectable amounts of NO3, measuring the NO3 reactivity simultaneously with NO2 and O3 represents an alternative way to assess NO3 mixing ratios. Furthermore, our NO3 measurements emphasized the necessity of frequent inlet filter changes as common in outdoor field measurements. By comparing calculated with directly measured NO3 reactivities, we find that the most important loss processes for NO3 are reactions with NO and VOCs (such as monoterpenes), the latter thus providing an indoor source of organic nitrates. Direct NO3 reactivity measurements can therefore contribute to identify the indoor fate of the nitrate radical.Author contributions
Patrick Dewald: conceptualization; data curation; formal analysis; investigation; methodology; visualization; writing – original draft; writing – review & editing. John N. Crowley: conceptualization; supervision; validation; writing – review & editing. Jos Lelieveld: resources; supervision; validation; writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Chemours for provision of a FEP sample used to coat the flowtube reactor.References
- N. E. Klepeis, W. C. Nelson, W. R. Ott, J. P. Robinson, A. M. Tsang, P. Switzer, J. V. Behar, S. C. Hern and W. H. Engelmann, The National Human Activity Pattern Survey (NHAPS): a resource for assessing exposure to environmental pollutants, J. Exposure Anal. Environ. Epidemiol., 2001, 11, 231–252 CrossRef CAS PubMed.
- C. J. Weschler, M. Brauer and P. Koutrakis, Indoor Ozone and Nitrogen-Dioxide - a Potential Pathway to the Generation of Nitrate Radicals, Dinitrogen Pentaoxide, and Nitric-Acid Indoors, Environ. Sci. Technol., 1992, 26, 179–184 CrossRef CAS.
- C. J. Weschler and H. C. Shields, Production of the hydroxyl radical in indoor air, Environ. Sci. Technol., 1996, 30, 3250–3258 CrossRef CAS.
- C. J. Young, S. Zhou, J. A. Siegel and T. F. Kahan, Illuminating the dark side of indoor oxidants, Environ. Sci.: Processes Impacts, 2019, 21, 1229–1239 RSC.
- N. Zannoni, P. S. J. Lakey, Y. Won, M. Shiraiwa, D. Rim, C. J. Weschler, N. J. Wang, L. Ernle, M. Z. Li, G. Bekö, P. Wargocki and J. Williams, The human oxidation field, Science, 2022, 377, 1071–1076 CrossRef CAS PubMed.
- R. P. Wayne, I. Barnes, P. Biggs, J. P. Burrows, C. E. Canosamas, J. Hjorth, G. Lebras, G. K. Moortgat, D. Perner, G. Poulet, G. Restelli and H. Sidebottom, The Nitrate Radical - Physics, Chemistry, and the Atmosphere, Atmos. Environ., 1991, 25, 1–203 CrossRef.
- S. S. Brown and J. Stutz, Nighttime radical observations and chemistry, Chem. Soc. Rev., 2012, 41, 6405–6447 RSC.
- J. Liebmann, E. Karu, N. Sobanski, J. Schuladen, M. Ehn, S. Schallhart, L. Quéléver, H. Hellen, H. Hakola, T. Hoffmann, J. Williams, H. Fischer, J. Lelieveld and J. N. Crowley, Direct measurement of NO3 radical reactivity in a boreal forest, Atmos. Chem. Phys., 2018, 18, 3799–3815 CrossRef CAS.
- J. M. Liebmann, J. B. A. Muller, D. Kubistin, A. Claude, R. Holla, C. Plaß-Dülmer, J. Lelieveld and J. N. Crowley, Direct measurements of NO3-reactivity in and above the boundary layer of a mountain-top site: Identification of reactive trace gases and comparison with OH-reactivity, Atmos. Chem. Phys., 2018, 18, 12045–12059 CrossRef CAS.
- A. Gandolfo, V. Gligorovski, V. Bartolomei, S. Tlili, E. G. Alvarez, H. Wortham, J. Kleffmann and S. Gligorovski, Spectrally resolved actinic flux and photolysis frequencies of key species within an indoor environment, Build. Environ., 2016, 109, 50–57 CrossRef.
- S. Zhou and T. F. Kahan, Spatiotemporal characterization of irradiance and photolysis rate constants of indoor gas-phase species in the UTest house during HOMEChem, Indoor Air, 2022, 32, e12964 CAS.
- C. J. Weschler, Ozone in indoor environments: Concentration and chemistry, Indoor Air, 2000, 10, 269–288 CrossRef CAS PubMed.
- S. Zhou, C. J. Young, T. C. VandenBoer, S. F. Kowal and T. F. Kahan, Time-Resolved Measurements of Nitric Oxide, Nitrogen Dioxide, and Nitrous Acid in an Occupied New York Home, Environ. Sci. Technol., 2018, 52, 8355–8364 CrossRef CAS PubMed.
- W. W. Nazaroff and C. J. Weschler, Indoor ozone: Concentrations and influencing factors, Indoor Air, 2022, 32, e12942 CAS.
- N. L. Ng, S. S. Brown, A. T. Archibald, E. Atlas, R. C. Cohen, J. N. Crowley, D. A. Day, N. M. Donahue, J. L. Fry, H. Fuchs, R. J. Griffin, M. I. Guzman, H. Herrmann, A. Hodzic, Y. Iinuma, J. L. Jimenez, A. Kiendler-Scharr, B. H. Lee, D. J. Luecken, J. Mao, R. McLaren, A. Mutzel, H. D. Osthoff, B. Ouyang, B. Picquet-Varrault, U. Platt, H. O. T. Pye, Y. Rudich, R. H. Schwantes, M. Shiraiwa, J. Stutz, J. A. Thornton, A. Tilgner, B. J. Williams and R. A. Zaveri, Nitrate radicals and biogenic volatile organic compounds: oxidation, mechanisms, and organic aerosol, Atmos. Chem. Phys., 2017, 17, 2103–2162 CrossRef CAS PubMed.
- W. W. Nazaroff and C. J. Weschler, Cleaning products and air fresheners: exposure to primary and secondary air pollutants, Atmos. Environ., 2004, 38, 2841–2865 CrossRef CAS.
- N. L. Ng, A. J. Kwan, J. D. Surratt, A. W. H. Chan, P. S. Chhabra, A. Sorooshian, H. O. T. Pye, J. D. Crounse, P. O. Wennberg, R. C. Flagan and J. H. Seinfeld, Secondary organic aerosol (SOA) formation from reaction of isoprene with nitrate radicals (NO3), Atmos. Chem. Phys., 2008, 8, 4117–4140 CrossRef CAS.
- J. L. Fry, A. Kiendler-Scharr, A. W. Rollins, T. Brauers, S. S. Brown, H. P. Dorn, W. P. Dube, H. Fuchs, A. Mensah, F. Rohrer, R. Tillmann, A. Wahner, P. J. Wooldridge and R. C. Cohen, SOA from limonene: role of NO3 in its generation and degradation, Atmos. Chem. Phys., 2011, 11, 3879–3894 CrossRef CAS.
- G. Bekö, P. Wargocki, N. J. Wang, M. Z. Li, C. J. Weschler, G. Morrison, S. Langer, L. Ernle, D. Licina, S. Yang, N. Zannoni and J. Williams, The Indoor Chemical Human Emissions and Reactivity (ICHEAR) project: Overview of experimental methodology and preliminary results, Indoor Air, 2020, 30, 1213–1228 CrossRef PubMed.
- N. Carslaw, L. Fletcher, D. Heard, T. Ingham and H. Walker, Significant OH production under surface cleaning and air cleaning conditions: Impact on indoor air quality, Indoor Air, 2017, 27, 1091–1100 CrossRef CAS PubMed.
- N. Carslaw, A new detailed chemical model for indoor air pollution, Atmos. Environ., 2007, 41, 1164–1179 CrossRef CAS.
- M. S. Waring and J. R. Wells, Volatile organic compound conversion by ozone, hydroxyl radicals, and nitrate radicals in residential indoor air: Magnitudes and impacts of oxidant sources, Atmos. Environ., 2015, 106, 382–391 CrossRef CAS PubMed.
- D. J. Price, D. A. Day, D. Pagonis, H. Stark, L. B. Algrim, A. V. Handschy, S. Liu, J. E. Krechmer, S. L. Miller, J. F. Hunter, J. A. de Gouw, P. J. Ziemann and J. L. Jimenez, Budgets of Organic Carbon Composition and Oxidation in Indoor Air, Environ. Sci. Technol., 2019, 53, 13053–13063 CrossRef CAS PubMed.
- M. F. Link, J. Li, J. C. Ditto, H. Huynh, J. Yu, S. M. Zimmerman, K. L. Rediger, A. Shore, J. P. D. Abbatt, L. A. Garofalo, D. K. Farmer and D. Poppendieck, Ventilation in a Residential Building Brings Outdoor NOx Indoors with Limited Implications for VOC Oxidation from NO3 Radicals, Environ. Sci. Technol., 2023, 57(43), 16446–16455 CrossRef PubMed.
- J. K. Nøjgaard, Indoor measurements of the sum of the nitrate radical, NO3, and nitrogen pentoxide, N2O5 in Denmark, Chemosphere, 2010, 79, 898–904 CrossRef PubMed.
- C. Arata, K. J. Zarzana, P. K. Misztal, Y. J. Liu, S. S. Brown, W. W. Nazaroff and A. H. Goldstein, Measurement of NO3 and N2O5 in a Residential Kitchen, Environ. Sci. Technol. Lett., 2018, 5, 595–599 CrossRef CAS.
- A. Moravek, T. C. VandenBoer, Z. Finewax, D. Pagonis, B. A. Nault, W. L. Brown, D. A. Day, A. V. Handschy, H. Stark, P. Ziemann, J. L. Jimenez, J. A. de Gouw and C. J. Young, Reactive Chlorine Emissions from Cleaning and Reactive Nitrogen Chemistry in an Indoor Athletic Facility, Environ. Sci. Technol., 2022, 56, 15408–15416 CrossRef CAS PubMed.
- Y. Shah, J. W. Kurelek, S. D. Peterson and S. Yarusevych, Experimental investigation of indoor aerosol dispersion and accumulation in the context of COVID-19: Effects of masks and ventilation, Phys. Fluids, 2021, 33, 073315 CrossRef CAS PubMed.
- R. A. Cox, M. Ammann, J. N. Crowley, P. T. Griffiths, H. Herrmann, E. H. Hoffmann, M. E. Jenkin, V. F. McNeill, A. Mellouki, C. J. Penkett, A. Tilgner and T. J. Wallington, Opinion: The germicidal effect of ambient air (open-air factor) revisited, Atmos. Chem. Phys., 2021, 21, 13011–13018 CrossRef CAS.
- M. H. Sherman, Tracer-Gas Techniques for Measuring Ventilation in a Single Zone, Build. Environ., 1990, 25, 365–374 CrossRef.
- J. M. Liebmann, G. Schuster, J. B. Schuladen, N. Sobanski, J. Lelieveld and J. N. Crowley, Measurement of ambient NO3 reactivity: Design, characterization and first deployment of a new instrument, Atmos. Meas. Tech., 2017, 10, 1241–1258 CrossRef CAS.
- N. Sobanski, J. Schuladen, G. Schuster, J. Lelieveld and J. N. Crowley, A five-channel cavity ring-down spectrometer for the detection of NO2, NO3, N2O5, total peroxy nitrates and total alkyl nitrates, Atmos. Meas. Tech., 2016, 9, 5103–5118 CrossRef CAS.
- N. Friedrich, I. Tadic, J. Schuladen, J. Brooks, E. Darbyshire, F. Drewnick, H. Fischer, J. Lelieveld and J. N. Crowley, Measurement of NOx and NOy with a thermal dissociation cavity ring-down spectrometer (TD-CRDS): instrument characterisation and first deployment, Atmos. Meas. Tech., 2020, 13, 5739–5761 CrossRef CAS.
- J. B. Burkholder, S. P. Sander, J. Abbatt, J. R. Barker, R. E. Huie, C. E. Kolb, M. J. Kurylo, V. L. Orkin, D. M. Wilmouth and P. H. Wine, Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies, Evaluation No. 18, JPL Publication 15-10, Jet Propulsion Laboratory, Pasadena, https://jpldataeval.jpl.nasa.gov, accessed 23 July 2023 Search PubMed.
- IUPAC, Task Group on Atmospheric Chemical Kinetic Data Evaluation, ed. M. Ammann, R. A. Cox, J. N. Crowley, H. Herrmann, M. E. Jenkin, V. F. McNeill, A. Mellouki, M. J. Rossi, J. Troe and T. J. Wallington, https://iupac.aeris-data.fr/en/home-english/, accessed 23 July 2023 Search PubMed.
- M. J. Tang, J. Thieser, G. Schuster and J. N. Crowley, Uptake of NO3 and N2O5 to Saharan dust, ambient urban aerosol and soot: a relative rate study, Atmos. Chem. Phys., 2010, 10, 2965–2974 CrossRef CAS.
- G. Schuster, I. Labazan and J. N. Crowley, A cavity ring down/cavity enhanced absorption device for measurement of ambient NO3 and N2O5, Atmos. Meas. Tech., 2009, 2, 1–13 CrossRef CAS.
- Umweltbundesamt, https://www.umweltbundesamt.de/en/data/air/air-data/, accessed 12 June 2023 Search PubMed.
- Y. Hu and B. Zhao, Relationship between indoor and outdoor NO2: A review, Build. Environ., 2020, 180, 106909 CrossRef.
- T. Grontoft, The influence of photochemistry on outdoor to indoor NO2 in some European museums, Indoor Air, 2022, 32, e12999 CrossRef PubMed.
- J. N. Crowley, G. Schuster, N. Pouvesle, U. Parchatka, H. Fischer, B. Bonn, H. Bingemer and J. Lelieveld, Nocturnal nitrogen oxides at a rural mountain site in south-western Germany, Atmos. Chem. Phys., 2010, 10, 2795–2812 CrossRef CAS.
- U. F. Platt, A. M. Winer, H. W. Biermann, R. Atkinson and J. N. Pitts, Measurement of Nitrate Radical Concentrations in Continental Air, Environ. Sci. Technol., 1984, 18, 365–369 CrossRef CAS PubMed.
- F. Heintz, U. Platt, H. Flentje and R. Dubois, Long-term observation of nitrate radicals at the tor station, Kap Arkona (Rügen), J. Geophys. Res.: Atmos., 1996, 101, 22891–22910 CrossRef CAS.
- M. Martinez, D. Perner, E. M. Hackenthal, S. Kulzer and L. Schutz, NO3 at Helgoland during the NORDEX campaign in October 1996, J. Geophys. Res.: Atmos., 2000, 105, 22685–22695 CrossRef CAS.
- S. S. Brown, H. Stark, T. B. Ryerson, E. J. Williams, D. K. Nicks, M. Trainer, F. C. Fehsenfeld and A. R. Ravishankara, Nitrogen oxides in the nocturnal boundary layer: Simultaneous in situ measurements of NO3, N2O5, NO2, NO, and O3, J. Geophys. Res.: Atmos., 2003, 108, 4299 Search PubMed.
- S. S. Brown, J. A. Degouw, C. Warneke, T. B. Ryerson, W. P. Dube, E. Atlas, R. J. Weber, R. E. Peltier, J. A. Neuman, J. M. Roberts, A. Swanson, F. Flocke, S. A. McKeen, J. Brioude, R. Sommariva, M. Trainer, F. C. Fehsenfeld and A. R. Ravishankara, Nocturnal isoprene oxidation over the Northeast United States in summer and its impact on reactive nitrogen partitioning and secondary organic aerosol, Atmos. Chem. Phys., 2009, 9, 3027–3042 CrossRef CAS.
- N. Sobanski, M. J. Tang, J. Thieser, G. Schuster, D. Pöhler, H. Fischer, W. Song, C. Sauvage, J. Williams, J. Fachinger, F. Berkes, P. Hoor, U. Platt, J. Lelieveld and J. N. Crowley, Chemical and meteorological influences on the lifetime of NO3 at a semi-rural mountain site during PARADE, Atmos. Chem. Phys., 2016, 16, 4867–4883 CrossRef CAS.
- S. S. Brown, H. Stark and A. R. Ravishankara, Applicability of the steady state approximation to the interpretation of atmospheric observations of NO3 and N2O5, J. Geophys. Res.: Atmos., 2003, 108, 4539 Search PubMed.
- P. Dewald, J. M. Liebmann, N. Friedrich, J. Shenolikar, J. Schuladen, F. Rohrer, D. Reimer, R. Tillmann, A. Novelli, C. M. Cho, K. M. Xu, R. Holzinger, F. Bernard, L. Zhou, W. Mellouki, S. S. Brown, H. Fuchs, J. Lelieveld and J. N. Crowley, Evolution of NO3 reactivity during the oxidation of isoprene, Atmos. Chem. Phys., 2020, 20, 10459–10475 CrossRef CAS.
- C. J. Weschler and H. C. Shields, Measurements of the hydroxyl radical in a manipulated but realistic indoor environment, Environ. Sci. Technol., 1997, 31, 3719–3722 CrossRef CAS.
- N. Carslaw, A mechanistic study of limonene oxidation products and pathways following cleaning activities, Atmos. Environ., 2013, 80, 507–513 CrossRef CAS.
- T. H. Bertram and J. A. Thornton, Toward a general parameterization of N2O5 reactivity on aqueous particles: the competing effects of particle liquid water, nitrate and chloride, Atmos. Chem. Phys., 2009, 9, 8351–8363 CrossRef CAS.
- P. Dewald, C. M. Nussbaumer, J. Schuladen, A. Ringsdorf, A. Edtbauer, H. Fischer, J. Williams, J. Lelieveld and J. N. Crowley, Fate of the nitrate radical at the summit of a semi-rural mountain site in Germany assessed with direct reactivity measurements, Atmos. Chem. Phys., 2022, 22, 7051–7069 CrossRef CAS.
- C. Calfapietra, S. Fares, F. Manes, A. Morani, G. Sgrigna and F. Loreto, Role of Biogenic Volatile Organic Compounds (BVOC) emitted by urban trees on ozone concentration in cities: A review, Environ. Pollut., 2013, 183, 71–80 CrossRef CAS PubMed.
- E. C. Lahr, G. W. Schade, C. C. Crossett and M. R. Watson, Photosynthesis and isoprene emission from trees along an urban–rural gradient in Texas, Global Change Biol., 2015, 21, 4221–4236 CrossRef PubMed.
- Y. Gao, M. Ma, F. Yan, H. Su, S. Wang, H. Liao, B. Zhao, X. Wang, Y. Sun, J. R. Hopkins, Q. Chen, P. Fu, A. C. Lewis, Q. Qiu, X. Yao and H. Gao, Impacts of biogenic emissions from urban landscapes on summer ozone and secondary organic aerosol formation in megacities, Sci. Total Environ., 2022, 814, 152654 CrossRef CAS PubMed.
- D. York, Least-Squares Fitting of a Straight Line, Can. J. Phys., 1966, 44, 1079–1086 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ea00137g |
| This journal is © The Royal Society of Chemistry 2023 |