
Efficient and stable formamidinium–caesium perovskite solar cells and modules from lead acetate-based precursors†
Jie
Zhao
 ab,
Sebastian O.
Fürer
ab,
David P.
McMeekin
ab,
Qingdong
Lin
ab,
Pin
Lv
c,
Jisheng
Ma
de,
Wen Liang
Tan
d,
Chao
Wang
d,
Boer
Tan
ab,
Anthony S. R.
Chesman
fg,
Huiyu
Yin
h,
Andrew D.
Scully
f,
Christopher R.
McNeill
d,
Wenxin
Mao
*ab,
Jianfeng
Lu
*ch,
Yi-Bing
Cheng
hi and
Udo
Bach
*ab
ab,
Sebastian O.
Fürer
ab,
David P.
McMeekin
ab,
Qingdong
Lin
ab,
Pin
Lv
c,
Jisheng
Ma
de,
Wen Liang
Tan
d,
Chao
Wang
d,
Boer
Tan
ab,
Anthony S. R.
Chesman
fg,
Huiyu
Yin
h,
Andrew D.
Scully
f,
Christopher R.
McNeill
d,
Wenxin
Mao
*ab,
Jianfeng
Lu
*ch,
Yi-Bing
Cheng
hi and
Udo
Bach
*ab
aDepartment of Chemical and Biological Engineering, Monash University, Victoria 3800, Australia. E-mail: Wenxin.Mao@monash.edu; Udo.Bach@monash.edu
bARC Center of Excellence in Exciton Science, Monash University, Victoria 3800, Australia
cState Key Laboratory of Silicate Materials for Architectures, Wuhan University of Technology, Wuhan 430070, China. E-mail: Jianfeng.lu@whut.edu.cn
dDepartment of Materials Science and Engineering, Monash University, Victoria 3800, Australia
eMonash X-ray Platform, Monash University, Victoria 3800, Australia
fCSIRO Manufacturing, Clayton, Victoria 3168, Australia
gThe Melbourne Centre for Nanofabrication, Victorian Node of the Australian National Fabrication Facility, Clayton, Victoria 3168, Australia
hFoshan Xianhu Laboratory of the Advanced Energy Science and Technology Guangdong Laboratory, Foshan 528216, China
iState Key Laboratory of Advanced Technology for Materials Synthesis and Processing, Wuhan University of Technology, Wuhan 430070, China
First published on 1st December 2022
Abstract
Controlling the crystallization process of perovskite thin films to obtain a high-quality material is one of the most challenging aspects for upscaling perovskite solar cell (PSC) technology. The use of non-halide lead sources, such as lead acetate, is a potential solution to this issue due to the fast perovskite crystallization process triggered by the facile removal of acetate during post-annealing. However, to date, lead acetate has been used exclusively as a precursor for the synthesis of methylammonium (MA) or caesium (Cs) based perovskites, which are unstable and less efficient. Here, we expand the lead acetate precursor route to form mixed A-cation perovskites, namely, formamidinium–caesium lead perovskite. High-quality large-area formamidinium–caesium mixed-cation perovskite films were produced by blade-coating a lead acetate-based precursor formulation in an ambient laboratory environment, with the use of NH4+ as a volatile cation to drive off acetate during annealing, leading to formation of PSCs with a power conversion efficiency (PCE) of up to 21.0%. Blade coated mini-modules with an aperture area of 10 cm2 displayed PCEs of up to 18.8%. The encapsulated PSCs showed excellent thermal stability, with no evidence of efficiency loss after 3300 hours at 65 °C.
Broader contextPerovskite solar cells (PSCs) have attracted widespread attention as a promising photovoltaic technology due to the outstanding photovoltaic properties of perovskite active layers. The power conversion efficiency (PCE) of PSCs is rapidly approaching their theoretical limit. However, scalability and stability remain the main obstacles restricting their commercialization. Lead acetate-based precursors have produced ultrasmooth perovskite thin films with only short annealing times and without the need for an antisolvent. The accelerated perovskite crystallization process and facile processability make lead acetate a promising lead source for large-scale fabrication of PSCs. However, to date, it has only been used as a precursor for the synthesis of methylammonium (MA) or caesium (Cs) based perovskites due to a limited understanding of side-reactions occurring in the precursor solution, which limits its application in the field. For the lead acetate route to yield high purity perovskite thin films, a compatible cation is required for effective thermalisation of the volatile components. Here we outline why the conventionally used methylammonium cation cannot be used in conjunction with formamidinium (FA) and show that replacing it with ammonium (NH4+) addresses this issue. This expands the lead acetate route to all formamidinium–caesium (FACs) perovskites, and hence makes it viable for all high-performing and industrially relevant perovskite compositions. Furthermore, efficient and stable PSCs and perovskite solar modules (PSMs) can be produced from our precursor solution using industrially scalable blade coating techniques in an ambient air environment. These results and insights will stimulate further work on the use of lead acetate to facilitate stable and efficient large area PSCs and thereby contribute to the commercialization of this technology. |
Introduction
Over the past decade, metal halide perovskite solar cells (PSCs) have become one of the most promising photovoltaic technologies due to the outstanding photovoltaic properties of perovskite active layers, such as tuneable band gaps, high absorption coefficients and long carrier diffusion lengths.1–5 Recently, the power conversion efficiency (PCE) of PSCs has reached a certified value of 25.7%.6 Although the PCE of PSCs is now approaching close to that of more traditional solar cell technologies, scalability and stability remain primary technical challenges for successful commercialization of PSC technology. One of the most important considerations for achieving highly efficient and stable large-area PSCs resides in the fabrication of perovskite active layers.Many factors affect the perovskite film quality, such as the fabrication protocols,7–9 perovskite compositions,2,10–12 and the defect passivation method.13–15 Among others, the choice of precursors is also very important. Most commonly, perovskites are formed from lead halide and A-cation halides as precursors: AX + PbX2 → APbX3 (A is methylammonium MA+, formamidinium FA+, and/or caesium Cs+, X is halide iodide I− and/or bromide Br−).16,17 Precursor solutions can also be produced directly from pre-synthesised perovskites or solvent-adducts, such as MAPbI3·DMF.18–20 Alternatively, strategies exploit the volatile nature of side-products from the perovskite synthesis to produce lead halide perovskite films. For example, Snaith et al. utilised the volatility of MACl to produce MAPbI3 from MAI and PbCl2, 3 MAI + PbCl2 → MAPbI3 + 2 MACl, in their seminal PSC paper in 2012.21 Also, lead acetate (PbAc2) can be used as a lead source to form ultrasmooth and well-crystallized MAPbI3 thin films, 3 MAI + PbAc2 → MAPbI3 + 2 MAAc, due to the accelerated crystallization facilitated by the facile removal of volatile MAAc during the post-annealing process.22
Lead iodide-based fabrication methods generally require stringent control of the crystallization conditions. To date, antisolvent quenching is the most common method to control the initial nucleation processes during perovskite film formation.7,23 However, scaling this method in a manufacturing environment is challenging. Although, it was reported that the use of nitrogen blades can help produce high quality perovskite thin films from slot-die coating or blade coating without the use of an antisolvent,24,25 the addition of strongly coordinating solvents such as DMSO or NMP is widely carried out to form perovskite–solvent intermediates and control the perovskite crystallization.26–29 The addition of these solvents has been found to be detrimental to the device stability as they can easily be trapped in the films and lead to the formation of voids at the perovskite substrate interface due to their high boiling points and strong coordination ability.30–34 Although, there is still an ongoing debate in the literature on the impact of these voids with a recent study suggesting that they are not necessarily detrimental to the device stability.35 By using PbAc2 as the lead source these problems can be avoided as neither an antisolvent nor the addition of a high-boiling point solvent, such as DMSO or NMP, is required to control the crystallization. The volatility of the side products drives rapid crystallisation, which in turn leads to ultrasmooth, pinhole free thin films. More importantly, it has been shown that the use of PbAc2 in the precursor solution can help produce efficient MAPbI3 PSCs in ambient air (relative humidity >50%),36 so lead acetate is a potenital lead source for the scalable fabrication of PSCs.
Surprisingly, however, pure PbAc2 has so far only been used as a precursor for the synthesis of MAPbI3 or CsPbI3 perovskites.22,37 With the rise of mixed cation (FACs) perovskites, which have more suitable band gaps for high efficiency devices and improved stability,2,38,39 there is a strong incentive to expand the PbAc2 route to make it applicable to this new class of perovskites. In fact, since the first successful synthesis of methylammonium-based perovskites using a lead acetate precursor in 2015, no FACs mixed-cation perovskite has been produced from lead acetate precursors. When adopting the PbAc2 route for the synthesis of FA or FACs perovskites, one could envisage a synthetic route developed for MAPbI3 that replaces one or all 3 equivalents of MA+ with FA+ (see Fig. 1(A)). Such synthetic avenues have previously been explored by Luo et al. who reported that the synthesis of FAPbI3 from pure PbAc2 as the lead source and either pure FAI or a FAI/MAI mixture was not possible.40
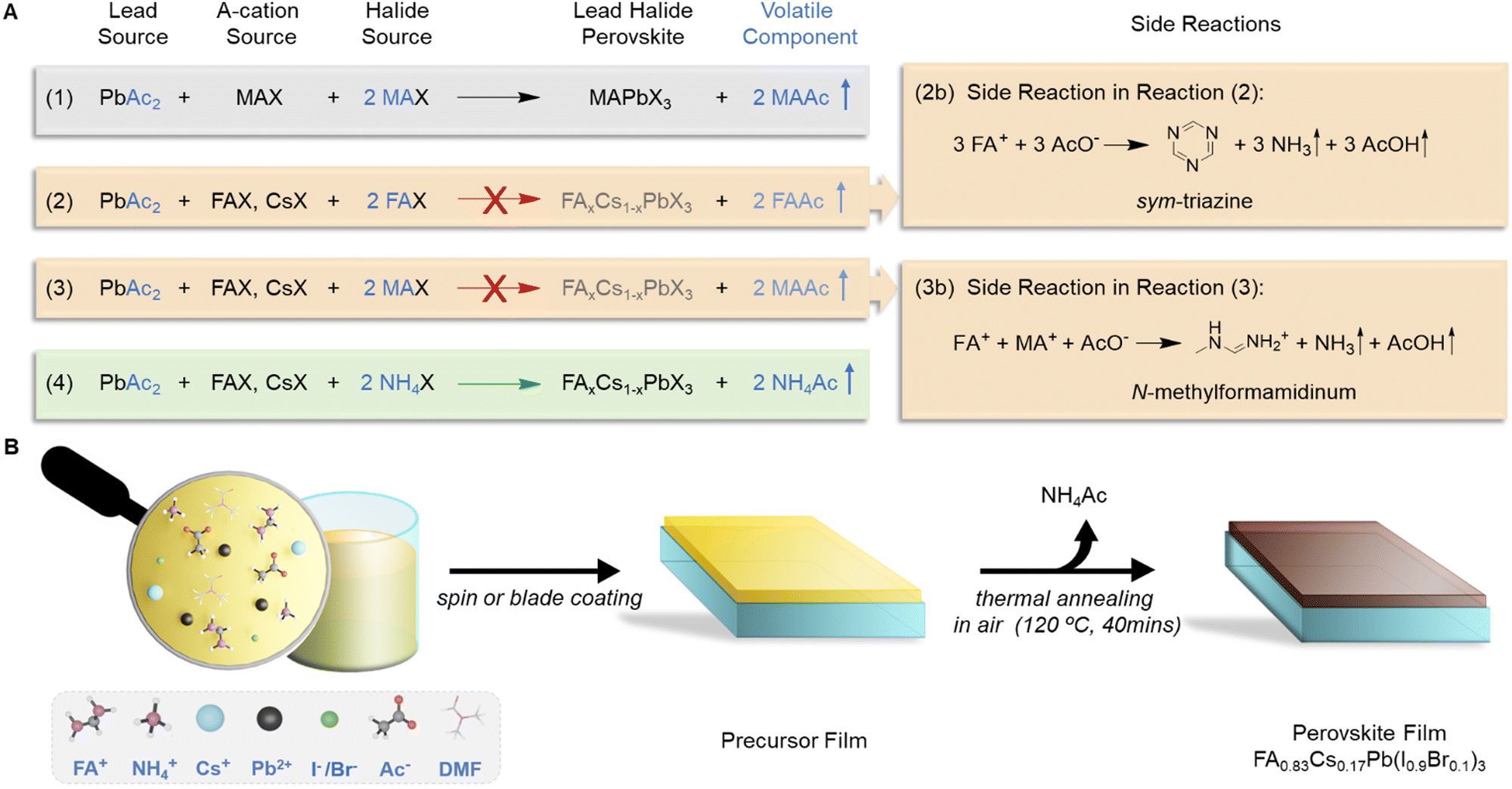 | ||
| Fig. 1 Effect of different volatile cations in PbAc2-based precursors on the formation of FACs perovskite. (A) Different synthetic avenues for the synthesis of MA- and FACs-perovskites using the PbAc2 route and proposed side reactions. (B) Schematic diagram of the fabrication process of the FACs perovskite from a PbAc2-based precursor solution with NH4+ as the volatile cation (abbreviated to A-Ac-FACs). | ||
In this work, we explore the molecular mechanisms that impede the synthesis of FA-based perovskites from PbAc2-based precursors and provide a simple solution in which ammonium (NH4+) is used as the volatile cation to successfully expand the PbAc2 route to achieve FACs mixed-cation perovskites. We used X-ray diffraction (XRD) measurements and nuclear magnetic resonance (NMR) spectroscopy to systematically study the chemical reactions in FA-based precursor solutions containing various different volatile cations [FA+, MA+ and ammonium (NH4+)]. We found that acetate catalyses the detrimental trimerization of FA+ to form sym-triazine in the precursor solution. If MA+ is added as a volatile cation, acetate triggers a nucleophilic reaction between MA+ and FA+, forming N-methyl formamidinium iodide (MFAI) and N,N′-dimethyl formamidinium iodide (DMFAI), both of which hinder the formation of perovskites. In contrast, no side-product is formed if NH4+ is added into the precursor solution. Based on these insights into the precursor chemistry we were able to produce high-quality FA0.83Cs0.17Pb(I0.9Br0.1)3 perovskite thin films via blade coating in ambient air by using NH4+ as the volatile cation. PSCs printed via the PbAc2 route exhibit a high PCE of up to 21.0% and excellent long-term thermal stabilities. To the best of our knowledge, this is the first time that FACs PSCs have been fabricated successfully from PbAc2-based precursors. These results represent the highest PCE achieved to date for a PSC fabricated from a non-halide lead source (Table S1, ESI†). A perovskite mini-module displaying a PCE of 18.8% (aperture area 10.0 cm2) was also fabricated in air using our PbAc2-based precursor approach.
Results and discussion
Lead acetate-based precursor solution chemistry
Perovskites form readily when equimolar ratios of lead halide and A-cation halides are reacted, with no side-products being formed during the reaction. Lead acetate itself is not a source of halides. Accordingly, in order to form 1 mole of a perovskite, lead acetate needs to be reacted with 3 mole equivalents of halide salts. One mole equivalent is typically provided in the form of A-cation halides. The additional 2 molar equivalents of halides (RX) are needed to form a volatile acetate compound RAc during the reaction, which enters the vapour phase during the drying and thermal annealing of the film. In the case of common MAPbI3 synthesis, volatile methylammonium acetate is initially formed and then removed from the film (equation 1 in Fig. 1(A)).For the formation of formamidinium caesium mixed cation perovskites (FACs), the reaction is generally described using the following equation:
| x FAX + (1 − x) CsX + PbAc2 + 2 RX → FAxCs1−xPbX3 + 2 RAc (X = Br and I and R = organic volatile cations). |
Fig. 1(A) shows three possible synthetic routes for the PbAc2-based formation of FACs perovskites that differ in the choice of the volatile cation. The first approach uses FA+ as the volatile cation and is based on an earlier report by Luo et al., who attempted to synthesise FAPbI3 perovskites directly from FAI and PbAc2.40 They observed the formation of a non-perovskite phase in the films that remained even after annealing at 170 °C, above the phase transition temperature of δ-FAPbI3.41 Our own experiments confirm these findings and prompted us to study the origin of this non-perovskite phase. We prepared FACs perovskite films from a PbAc2-based precursor with FA+ as the volatile cation (abbreviated as FA-Ac-FACs) and dissolved the films in DMSO-d6 to record the 1H NMR spectrum. Fig. S1 (ESI†) shows the NMR spectrum of films made from a fresh FA-Ac-FACs solution. In addition to the expected peak from FA at 7.85 ppm we found a peak at 9.31 ppm that can be assigned to sym-triazine.42 The formation of sym-triazine has previously been reported in aged precursor solutions prepared from PbI2 and as a degradation product of FA-based perovskites.42,43 Replacing PbI2 with PbAc2 appears to accelerate the formation of sym-triazine in the precursor solution, leading to the formation of non-perovskite films in the perovskite films.
To avoid this detrimental side-reaction from happening, we adopted the PbAc2 synthesis route for MAPbI3 by replacing one molar equivalent of MAI with the desired A cations (FACs), such as 0.83 FAX + 0.17 CsX + PbAc2 + 2 MAI → FA0.83Cs0.17PbX3 + 2 MAAc (precursor solution of this route is abbreviated as MA-Ac-FAAc). As expected, by using MA+ as the volatile cation the formation of sym-triazine was avoided. However, the XRD pattern (Fig. S2, ESI†) of the prepared film shows an additional peak at 11.51°, which is assigned to the formation of N-methyl formamidinium iodide (MFAI) based on previous reports.42 To confirm this, we recorded the 1H NMR spectra of solutions from these thin films and compared them those of the films made via the PbI2 route (Fig. S3, ESI†). In order to avoid broadening of the NMR signals due to resonance structures the solutions were spiked with HI following a previously reported protocol.44 The reference spectrum shows three characteristic peaks of FAI at 8.99 ppm, 8.66 ppm and 7.84 ppm originating from the NH2- and CH-protons, respectively.44 In the spectrum of the dissolved MA-Ac-FACs films these three peaks are shifted to lower field. Additional peaks at 9.46 ppm and 2.80–3.02 ppm appear, with a relative integration of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, which is consistent with the formation of MFA+ (Fig. S4, ESI†). Moreover, the 2D 1H–1H COSY spectrum (Fig. S5, ESI†) confirms the formation of MFAI. We also measured the mass spectrum of dissolved MA-Ac-FACs films (Fig. S6, ESI†). The peak at m/z of 59 confirms the presence of MFA as well.
:3, which is consistent with the formation of MFA+ (Fig. S4, ESI†). Moreover, the 2D 1H–1H COSY spectrum (Fig. S5, ESI†) confirms the formation of MFAI. We also measured the mass spectrum of dissolved MA-Ac-FACs films (Fig. S6, ESI†). The peak at m/z of 59 confirms the presence of MFA as well.
In contrast, when NH4+ was used as the volatile cation in the precursor solution (abbreviated as A-Ac-FAAc), the 1H NMR spectrum showed the same peaks at 8.99 ppm, 8.66 ppm and 7.84 ppm as in the reference spectra with no impurities in the spectrum (Fig. S3, ESI†). This suggests a clean synthesis of the FACs-perovskite with no detrimental side reactions and agrees well with the diffraction peaks shown in Fig. S2 (ESI†) where we did not find any impurity phases for the films based on the A-Ac-FACs precursor solution.
From this we conclude that there are two different side-reactions occurring in the PbAc2-based precursor solutions depending on the type of volatile cation used (Fig. 1(A)). In the case of FA-Ac-FACs, the decomposition of FA leads to the formation of sym-triazine (eqn (2b) in Fig. 1, and the detailed mechanism in Fig. S7, ESI†), which hinders the formation of the perovskite phase. When MA is used as the volatile cation, nucleophilic attack of methylamine at the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond of FA followed by elimination of ammonia leads to the formation of MFAI (eqn (3b) in Fig. 1). Moreover, a second methylamine can further react with MFAI and forms N,N′-dimethyl formamidinium iodide (DMFAI), which irreversibly consumes FA+ (detailed mechanism is shown in Fig. S8, ESI†). Key to both of these reactions is the high pKa (13.5) of acetic acid in DMF.45 While both reactions happen over longer time periods and at elevated temperatures in PbI2-based precursor solutions, the high basicity of acetate in DMF significantly accelerates these side-reactions.43 Consequently, we observed the presence of MFAI even in fresh precursor solutions (Fig. S9 and S10, ESI†), confirming the accelerated reaction kinetics. After 24 hours of precursor aging, the concentration of MFAI and DMFAI increased while the concentration of MAI decreased (Fig. S11, ESI†), further supporting our reaction mechanism.
N bond of FA followed by elimination of ammonia leads to the formation of MFAI (eqn (3b) in Fig. 1). Moreover, a second methylamine can further react with MFAI and forms N,N′-dimethyl formamidinium iodide (DMFAI), which irreversibly consumes FA+ (detailed mechanism is shown in Fig. S8, ESI†). Key to both of these reactions is the high pKa (13.5) of acetic acid in DMF.45 While both reactions happen over longer time periods and at elevated temperatures in PbI2-based precursor solutions, the high basicity of acetate in DMF significantly accelerates these side-reactions.43 Consequently, we observed the presence of MFAI even in fresh precursor solutions (Fig. S9 and S10, ESI†), confirming the accelerated reaction kinetics. After 24 hours of precursor aging, the concentration of MFAI and DMFAI increased while the concentration of MAI decreased (Fig. S11, ESI†), further supporting our reaction mechanism.
As in the case of FA+ and MA+, acetate can deprotonate NH4+ leading to the formation of ammonia, NH3. The key difference to FA+ and MA+ is that after nucleophilic attack with NH3 followed by elimination of NH3, FA is formed again (Fig. S12, ESI†). Hence no detrimental side-product is formed, which allows all FA+ cations to be utilised in the perovskite crystal lattice. We also noticed that there is no sym-triazine formed in our A-Ac-FACs solution. This is possibly because FA+ tends to react faster with NH4+ rather than with itself due to the stronger nucleophilicity of NH4+. In summary, pure FACs perovskites can only be formed when NH4+ is used as the volatile cation, while FA+ and MA+ lead to formation of side products and the formation of non-perovskite phases. Our results demonstrate the crucial role of volatile cations in PbAc2-based precursors for the successful formation of perovskites.
Fig. 1(B) shows a schematic diagram of the FACs perovskite fabrication process from our A-Ac-FACs precursor solution. The FACs perovskite thin film can be produced by either spin coating without the use of an antisolvent or blade coating. For blade coating, a nitrogen flow is applied to mimic the drying condition of spin coating and help remove the solvent and NH4Ac. The fast crystallization triggered by the evaporation of NH4Ac during the annealing process contributes to the formation of high-quality large-area FACs perovskite thin films. Fig. S13 (ESI†) shows the photograph of a blade coated FACs perovskite thin film on a 10 cm-by-10 cm substrate.
Characterization of perovskite thin films
To study the effect of the two lead sources on the perovskite film quality, we compare FA0.83Cs0.17Pb(I0.9Br0.1)3 perovskite thin films prepared from our newly developed PbAc2-based precursor solutions A-Ac-FAAc (target) to films with the same stoichiometry produced following the conventional PbI2 route (reference).46 For reference films, we used the optimised antisolvent-assisted crystallization method established in the literature.7,8,46 For our target films, FACs perovskite thin films were spin coated from our A-Ac-FACs precursor solution without the need for an antisolvent quenching step due to the described accelerated crystallisation. Hypophosphorous acid (HPA) was added to the A-Ac-FACs precursor solution to improve the grain size and optoelectronic properties as reported for MAPbI3 films prepared via the PbAc2 route.47 In our experiments we found that HPA significantly reduces the perovskite film quality following the PbI2 route (Fig. S14 and S15, ESI†); hence we omitted this additive in the PbI2-based precursor solutions for the following experiments. Additional details regarding the fabrication procedure are provided in the experimental section. We employed a FA0.83Cs0.17Pb(I0.9Br0.1)3 composition for single-junction solar cell application to maximize efficiency and stability.46 Moreover, our NH4Ac approach allows for introduction of different Br− contents (Fig. S16 and S17, ESI†) to fine-tune the band gap, as required for tandem solar cell applications, emphasising the versatility of our method.Fig. 2(A) shows the X-ray diffraction (XRD) patterns of the reference and target FA0.83Cs0.17Pb(I0.9Br0.1)3 films, confirming that the desired perovskite composition can be formed using the PbAc2 route without the formation of non-perovskite phases. 1H-NMR spectra of the perovskite films dissolved in DMSO-d6 further demonstrate that NH4+ was removed completely after annealing at 120 °C for 40 min (Fig. S18, ESI†). Synchrotron-based two-dimensional grazing-incidence X-ray scattering (GIWAXS) measurements were also performed for the films (Fig. S19, ESI†). Both films show similar perovskite crystallite orientation with the reference film being slightly more oriented. The random perovskite orientation of the target film might be attributed to the fast crystallization driven by the volatile side product NH4Ac. Zhang et al. recently demonstrated that orientational diversity can decrease the degree of lattice distortion and help to relax the microstrain in the crystal.48 As such, we performed the refinement of our XRD pattern to calculate the microstrain of the perovskite films. Indeed, our target film shows a smaller degree of microstrain 5.39 × 10−4 (±5 × 10−5) vs. 6.43 × 10−4 (±7 × 10−5), implying less lattice distortion. Interestingly, we observed different PbI2 orientation distributions between the target film and the reference. The target film has a secondary peak at an azimuth angle of 30°, while the PbI2 in the reference film is oriented in the out-of-plane direction (Fig. S20, ESI†). However, a further study is required to investigate the origin of this orientation difference and its effect on the film properties.
Fig. 2(B) illustrates steady-state photoluminescence (PL) spectra of the target and reference films. The target film shows an identical PL peak position (790 nm) to that of the reference film, which provides further evidence that both reference and target films have very similar chemical composition, despite being formed using different precursor routes. The PL emission intensity of the target film increased by a factor of about 2.5 compared to that of the reference film, which is consistent with the improved crystallinity and implies the suppression of nonradiative recombination. Time-resolved photoluminescence (TRPL) measurements were performed to further investigate the nonradiative carrier recombination of the perovskite thin films. The PL decay curves reveal a significantly prolonged carrier lifetime in the target film (Fig. 2(C)). Carrier lifetimes were calculated by fitting the PL decay curves to the biexponential equation Y = A1exp(−t/τ1) + A2exp(−t/τ2). The τ1 component is related to the trap-assisted nonradiative recombination, and the τ2 component is correlated to radiative recombination in the bulk perovskite.49 The reference film presented a τ1 lifetime of 0.5 μs and τ2 of 3.3 μs (mean lifetime 2.5 μs), while the target film showed significantly longer lifetimes with a τ1 of 5.6 μs and τ2 of 14.6 μs (mean lifetime 12.8 μs), respectively. We further investigated the charge carrier lifetime of the films on the microscale (10 μm × 10 μm) by performing time-resolved confocal PL lifetime mapping measurements (Fig. 2(D) and (E)). Compared with the reference film (blue region), the target film exhibited a much longer lifetime (green region). Furthermore, the target film presented a more uniform PL lifetime distribution, while the reference film showed some dispersed dark areas (as shown in the histogram of Fig. S21), ESI.† The increased PL lifetime and uniformity further accentuate the enhanced optoelectronic performance and effective suppression of non-radiative recombination when the PbAc2 route is used.
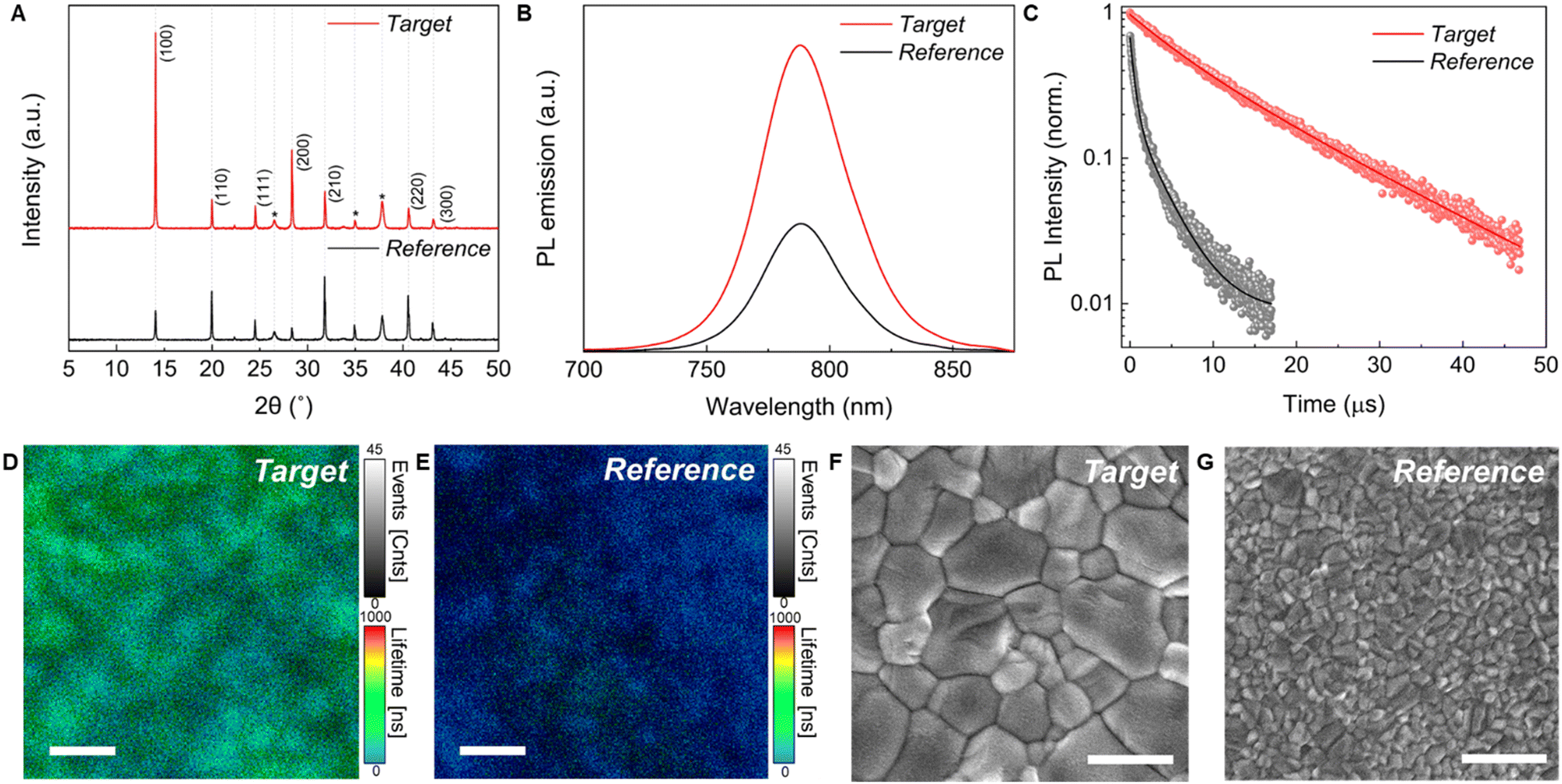 | ||
| Fig. 2 Characterization of the FA0.83Cs0.17Pb(I0.9Br0.1)3 films (both films were prepared by spin coating.) (A) XRD patterns of the reference (black, PbI2 route) and target (red, A-Ac-FACs route) FA0.83Cs0.17Pb(I0.9Br0.1)3 films. Peaks marked with * are assigned to FTO. (B) Steady-state photoluminescence (PL) spectra of the FA0.83Cs0.17Pb(I0.9Br0.1)3 films. (C) Time-resolved photoluminescence (TRPL) of the FA0.83Cs0.17Pb(I0.9Br0.1)3 films. (D and E) Time-resolved confocal PL lifetime maps of the (D) target and (E) reference FA0.83Cs0.17Pb(I0.9Br0.1)3 films. Scale bar, 2 μm. (F and G) Top-view scanning electron microscopy (SEM) images of the (F) target and (G) reference FA0.83Cs0.17Pb(I0.9Br0.1)3 films. Scale bar, 1 μm. | ||
We performed scanning electron microscopy (SEM) to study the morphologies of the perovskite films. Top view images are shown in Fig. 2(F) and (G). The reference and target films both show compact, uniform and pinhole-free surfaces, but with significantly larger apparent grain sizes in the target films. Moreover, XRD refinement results also show a larger crystallite size in the target film (target: 488 ± 137 nm vs. reference: 106 ± 14 nm). Cross-sectional SEM images are shown in Fig. S22 (ESI†). Both films show monolithic grains from the top to the bottom. Compared with the reference film (∼500 nm), the target film has a lesser film thickness (∼400 nm) due to the lower precursor concentration dictated by solubility limitations. Atomic force microscopy (AFM) images were recorded to further investigate the uniformity of both perovskite films (Fig. S23, ESI†). The apparent grain size in AFM images is consistent with the SEM results. The root mean square (RMS) roughness was obtained from large area AFM images (30 μm × 30 μm) to reveal reliable surface roughness information. Compared with the reference film (RMS = 28.4 nm), the roughness of the target film (RMS = 19.3 nm) is reduced by almost 30%. The smooth surface of our FACs-perovskite films made via the PbAc2 route is similar to a previous report on MA-based perovskites prepared from PbAc2.22
Photovoltaic performance of the PSCs and PSMs
We subsequently fabricated n–i–p structured PSCs incorporating FA0.83Cs0.17Pb(I0.9Br0.1)3. We used a chemical bath deposition method (CBD) to fabricate the SnO2 layer.50,51 i-Butylammonium bromide (i-BABr) was spin-coated on top of both perovskite layers to passivate surface defects.52 Spiro-OMeTAD was used as the hole transporting material. Fig. 3(A) and (B) show current density (J)–voltage (V) characterization of the champion reference and target devices under both reverse and forward scan directions. The champion device for the PbAc2 route showed a maximum PCE of 20.3% with an open-circuit voltage (VOC) of 1.15 V, a short-circuit current density (JSC) of 22.3 mA cm−2 and a fill factor of 79.1% in the reverse scan direction, which also has almost the same J–V curve in both scan directions with negligible hysteresis. The champion reference device exhibited a maximum PCE of 19.7% with a VOC of 1.07 V, a JSC of 22.9 mA cm−2 and a fill factor of 80.4% in the reverse scan direction. The statistic results of the device performance parameters are shown in Fig. 3(C). The target devices exhibited higher PCEs overall due to significant improvements in VOC and FF, which is consistent with the PL and TRPL results, while the reference device with a thicker perovskite active layer showed a higher JSC.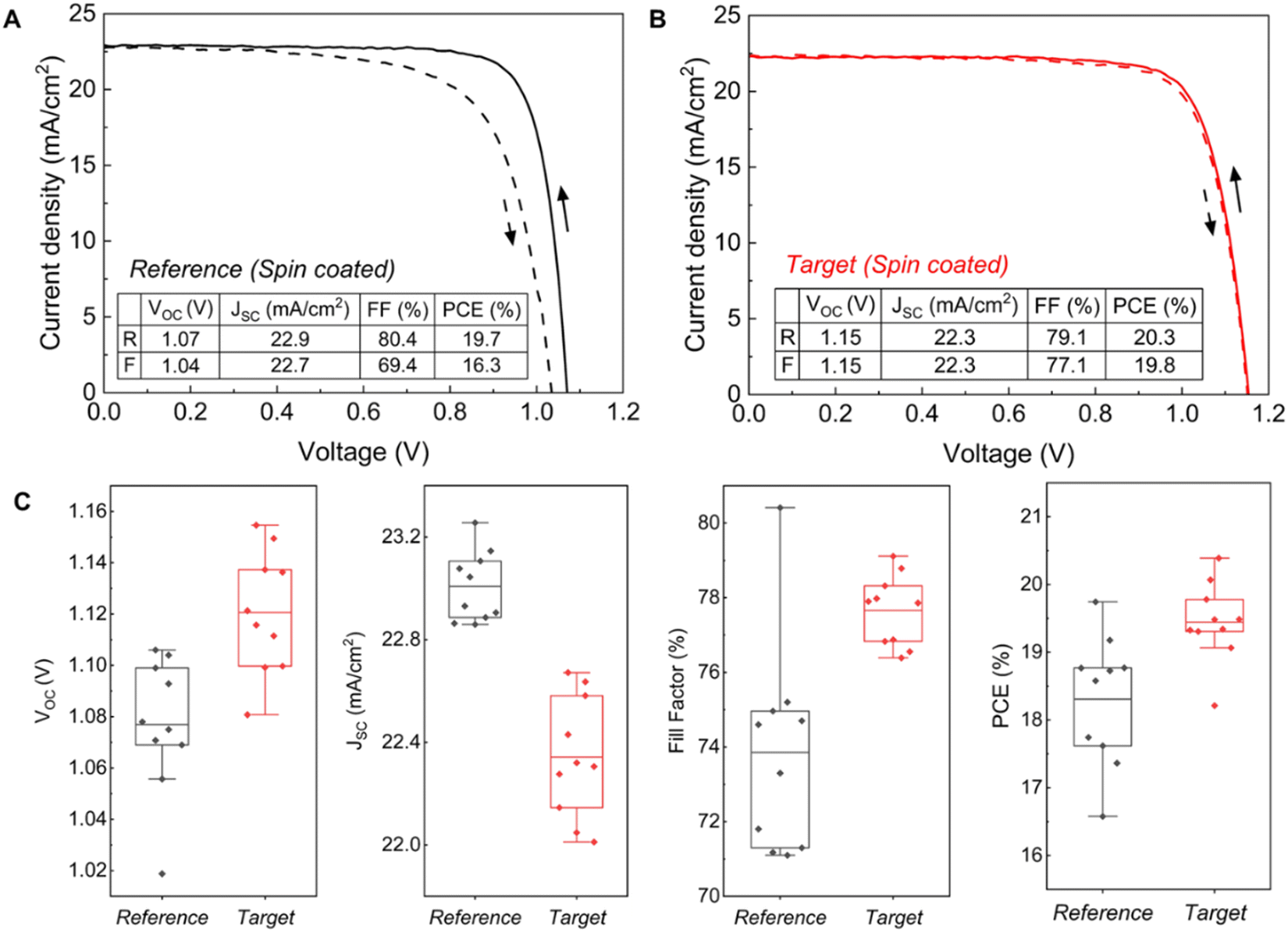 | ||
| Fig. 3 Device performance comparison of FA0.83Cs0.17Pb(I0.9Br0.1)3 perovskite solar cells spin coated using the PbI2 route (reference, black) and PbAc2 route (target, red). (A and B) J–V characteristics of champion (A) reference and (B) target FA0.83Cs0.17Pb(I0.9Br0.1)3 devices (mask area: 0.16 cm2). (C) Statistic results of device parameters (reverse scan direction). | ||
To demonstrate the potential of our lead acetate route for scalable fabrication of PSCs, we prepared perovskite active layers by blade coating in an ambient atmosphere on 10 cm × 10 cm substrates, which were then cut into 16 smaller pieces (2.5 cm × 2.5 cm) for the following deposition of i-BABr and spiro-OMeTAD. Fig. 4(A) represents a PCE of 21.0% of one of our best blade coated devices with a VOC of 1.13 V, JSC of 22.7 mA cm−2 and fill factor of 81.8% in the reverse scan direction, which is the highest reported efficiency for PSCs from non-halide lead sources. We also measured the external quantum efficiency (EQE) of the champion device (Fig. S24, ESI†). The integrated JSC is in good agreement with the JSC measured from J–V curves at 1 sun. The stabilized power output of the devices reaches 20.6% after 60 s (Fig. 4(B)). To evaluate the uniformity of the blade-coated perovskite films, we plotted the PCE distribution of 16 subcells across the full area as shown in Fig. S25 (ESI†). The average PCE of the 16 devices is 20.3% with a standard deviation of 0.46%, which emphasizes the scalability of our method.
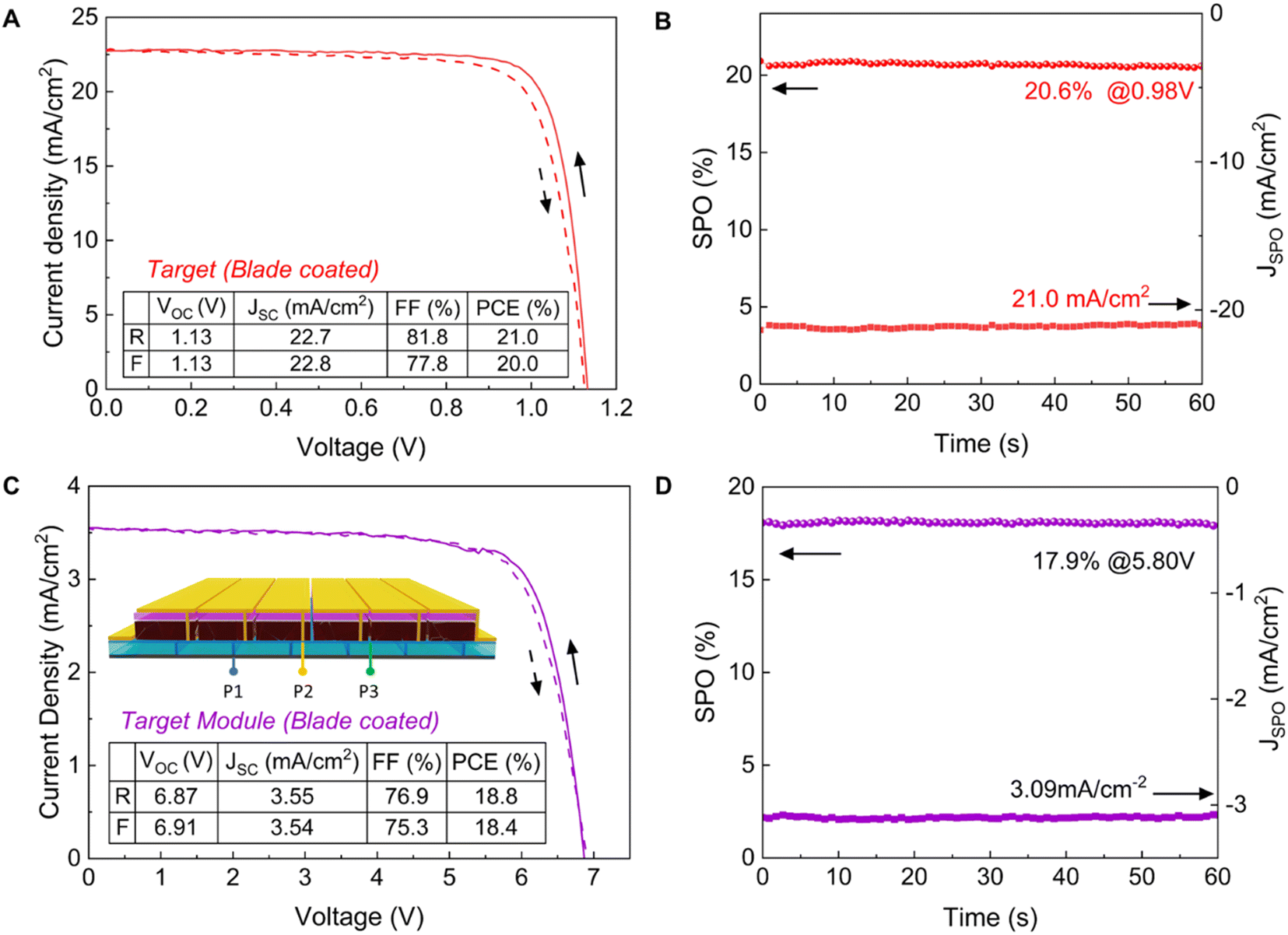 | ||
| Fig. 4 Device characterization of the blade coated FA0.83Cs0.17Pb(I0.9Br0.1)3 perovskite solar cell and solar module. (A) J–V characteristics of champion blade coated target FA0.83Cs0.17Pb(I0.9Br0.1)3 devices (mask area: 0.16 cm2). (B) Stabilized power output (SPO) of champion FA0.83Cs0.17Pb(I0.9Br0.1)3 devices. (C) Champion J–V characteristics of the blade coated FA0.83Cs0.17Pb(I0.9Br0.1)3 mini-module with a mask area of 10 cm2. (D) SPO of a champion mini-module. | ||
To further examine the scalability of our PbAc2-based precursor approach, we fabricated mini solar modules on 5 cm × 5 cm substrates with an aperture area of 10 cm2 following a previously reported method.51 A sketch of the top view of the module and metal mask is shown in Fig. S26 (ESI†). The J–V curves of the champion module under both reverse and forward scans are shown in Fig. 4(C). The inset shows the schematic diagram of a six subcell series-connected module. The module showed a maximum PCE of 18.8% with negligible hysteresis. The SPO of this module over 60 seconds reached 17.9% (Fig. 4(D)).
Stability of the PSCs
To assess the stability of the devices, we first measured the inherent stability of the perovskite active layer by performing a time-dependent in situ XRD measurement. Perovskite thin films were thermally aged at 130 °C in a N2 environment. High-resolution XRD patterns were measured every 5 hours. Films were cooled to room temperature before each measurement. Generally, the appearance of the PbI2 peak is indicative of perovskite decomposition. Quantifying the amount of PbI2 can be used to judge how much decomposition has occurred. Our XRD data show the PbI2 peak (2θ at 12.7°) which increases in intensity over time (Fig. 5(A) and (B)). Differences in PbI2 peak intensity can however also stem from differences in PbI2 orientation. According to the GIWAXS measurement, PbI2 in the target film exhibits populations with 2 orientations, the (001) plane preferentially oriented at χ = 0° and ∼30°, whereas in the reference film, the PbI2 phase only has (001) out-of-plane texture (χ = 0°). The XRD measurements only show a subset of PbI2 population, which is in the out-of-plane direction, rather than the entire PbI2 phase population. We therefore conducted the estimation of PbI2 intensity in all directions (azimuth angle from −90° to 90°) based on the PbI2 pole figure measured with GIWAXS. We noted that the PbI2 orientation distribution does not change after thermal aging (Fig. S27, ESI†), and hence used the PbI2 pole figure from fresh films (Fig. S20, ESI†) to estimate the PbI2 intensity. Fig. 5(C) shows the PbI2 intensity evolution after correction, suggesting faster perovskite decomposition in the reference film.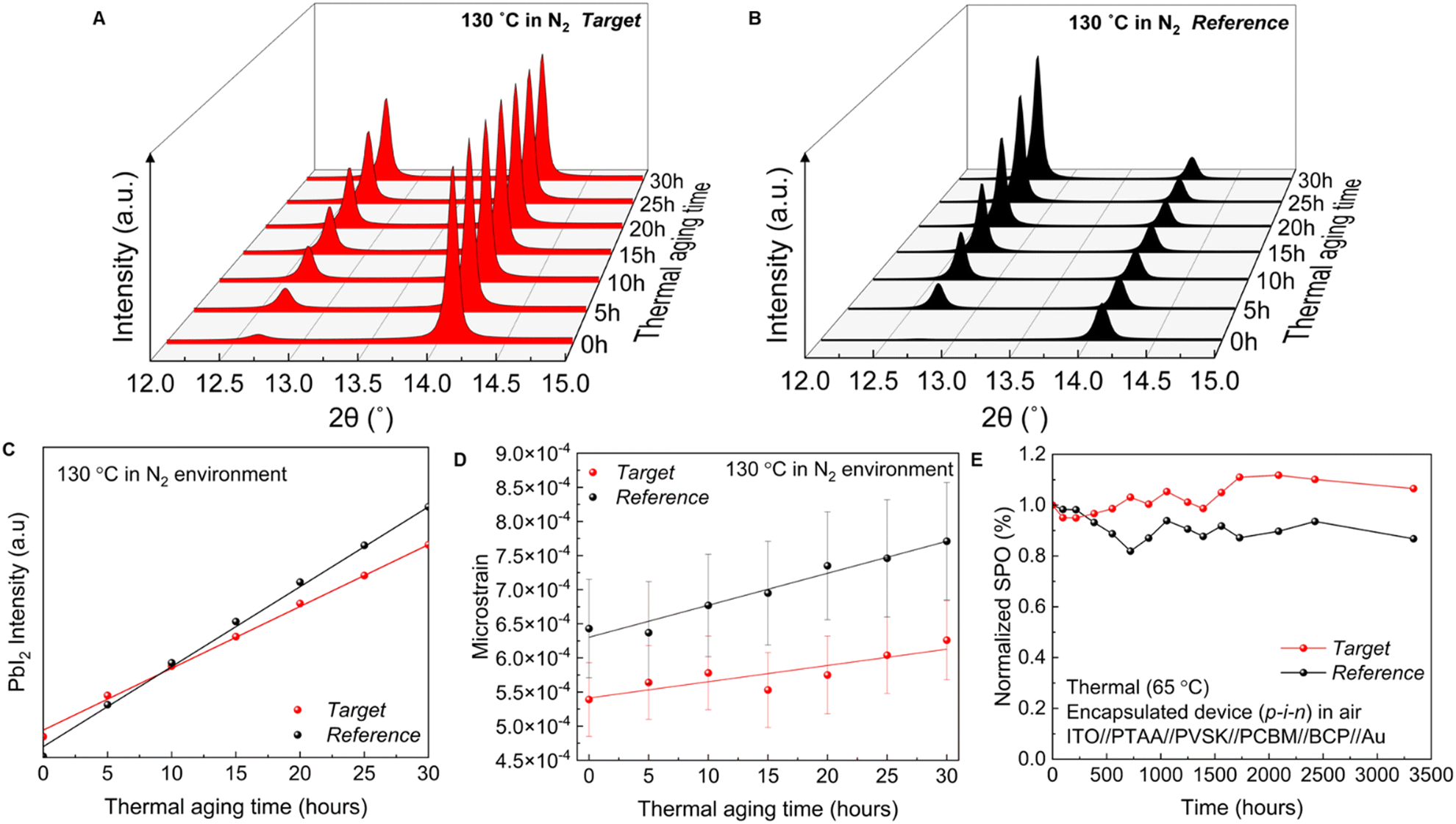 | ||
| Fig. 5 Stability comparison between FA0.83Cs0.17Pb(I0.9Br0.1)3 spin coated using the PbI2 route (reference, black) and PbAc2 (target, red). (A and B) In situ XRD measurement of the target (A) and reference (B) perovskite films. Films were thermally aged at 130 °C in a N2 environment. (C) and (D) PbI2 peak intensity (C) and microstrain evolution (D) of perovskite films. (E) Thermal stability of inverted PSCs (p–i–n) (encapsulated device aged at 65 °C in an ambient atmosphere under dark conditions). | ||
We further calculated the microstrain evolution to evaluate stability. As shown in Fig. 5(D), our target film has a smaller initial microstrain which increases at a slow rate, further indicating its improved stability. As mentioned above, FACs-perovskites are more stable than MA-based perovskites when PbI2 is used as the lead source.2,12 To determine if the same trend is observed with the PbAc2 route we compared the thermal stability of MAPb(I0.9Br0.1)3 and FA0.83Cs0.17Pb(I0.9Br0.1)3 at 130 °C in a dark nitrogen environment. As seen in Fig. S28 (ESI†), the edges of MAPb(I0.9Br0.1)3 films begin to turn yellow after 5 hours of aging, indicating the degradation of perovskite and formation of PbI2. The films became completely yellow after 30 hours, while FA0.83Cs0.17Pb(I0.9Br0.1)3 films prepared via the A-Ac-FACs-route remained black without any observable colour change.
Finally, we studied the stability of complete PSC devices. A thermal stability test was first performed by exposing the encapsulated devices to a temperature of 65 °C in a dark ambient air atmosphere (Fig. 5(E)). To avoid the thermal stability being compromised by the instability of spiro-OMeTAD at elevated temperatures, an inverted (p–i–n) device structure was employed. The target cell retained its initial PCE for more than 3300 h, whereas the PCE of the reference device decreased by around 15% over this time. We further studied the operational stability of both devices under continuous LED light illumination (100 mW cm−2). Non-encapsulated devices were aged in an environment of N2 at room temperature without an active cooling system. We replaced spiro-OMeTAD with PTAA in the n–i–p structure device for the test. Under maximum power point (MPP) testing (Fig. S29, ESI†), the target device shows a T80 of 327 h, while the reference device only has a T80 of 152 h.
Conclusion
The chemical side-reactions that occur in PbAc2-based precursor solutions, and the chemical by-products formed that give rise to non-perovskite phases, were identified in this work. Introducing NH4+ as the volatile cation was shown to offer a simple and versatile approach to avoid these detrimental side reactions, yielding pure FACs perovskite thin films via the PbAc2 route. Importantly, high-quality perovskite films are produced from this precursor solution using industrially scalable blade coating techniques to give high efficiency perovskite single cells (21.0%, mask area 0.16 cm2) and modules (18.8%, mask area 10 cm2). Moreover, the PbAc2-based PSC devices produced in this work display improved device performance and stability compared with the devices prepared via the conventional PbI2 route.Author contributions
Conceptualization, J. Z. and U. B.; methodology, J. Z., S. O. F., D. P. M., W. M., J. L. and U. B.; formal analysis, J. Z., S. O. F., D. P. M., Q. L., P. L., J. M., W. L. T., C. W., A. S. R. C., B. T. and A. D. S.; investigation, J. Z., S. O. F., D. P. M., Q. L., J. M,. W. L. T, B. T., and H. Y.; writing – original draft, J. Z.; writing – review and editing, J. Z., S. O. F., D. P. M., W. M., W. L. T., J. M., A. S. R. C., A. D. S., J. L. and U. B.; visualization, J. Z., Q. L., W. M., J. L. and U. B.; supervision, W. M., J. L. and U. B., and funding acquisition C. R. M., J. L., Y.-B. C. and U. B.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was financially supported by the Australian Research Council through the Centre of Excellence in Exciton Science (CE170100026) and additional grants (DP160104575 and LE170100235). The authors acknowledge financial support from the Australian Government through the Australian Renewable Energy Agency (ARENA) and the Australian Centre for Advanced Photovoltaics (ACAP). The authors acknowledge the Marie Skłodowska-Curie grant agreement SAMA no 101029896. The authors acknowledge the National Natural Science Foundation of China (22075221, 52002302, and 91963209), the Hubei Provincial Natural Science Foundation (2020CFB172), and the Foshan Xianhu Laboratory of the Advanced Energy Science and Technology Guangdong Laboratory (XHD2020-001). The authors also acknowledge Dr Doojin Vak (CSIRO) for his help in fabricating the blade-coated perovskite thin films. The authors acknowledge Siqi Deng and Yvonne Hora for the assistance in part of the visual representations. Synchrotron X-ray scattering experiments were performed at the SAXS/WAXS beamline at the Australian Synchrotron, part of ANSTO. This work was performed in part at the Melbourne Centre for Nanofabrication (MCN) in the Victorian Node of the Australian National Fabrication Facility (ANFF). The authors acknowledge the use of facilities within the Monash Centre for Electron Microscopy (MCEM), Monash X-Ray Platform (MXP).References
- Q. Dong, Y. Fang, Y. Shao, P. Mulligan, J. Qiu, L. Cao and J. Huang, Science, 2015, 347, 967–970 CrossRef CAS PubMed.
- D. P. McMeekin, G. Sadoughi, W. Rehman, G. E. Eperon, M. Saliba, M. T. Hörantner, A. Haghighirad, N. Sakai, L. Korte, B. Rech, M. B. Johnston, L. M. Herz and H. J. Snaith, Science, 2016, 351, 151–155 CrossRef CAS PubMed.
- W. Nie, H. Tsai, R. Asadpour, J.-C. Blancon, A. J. Neukirch, G. Gupta, J. J. Crochet, M. Chhowalla, S. Tretiak, M. A. Alam, H.-L. Wang and A. D. Mohite, Science, 2015, 347, 522–525 CrossRef CAS PubMed.
- D. Shi, V. Adinolfi, R. Comin, M. Yuan, E. Alarousu, A. Buin, Y. Chen, S. Hoogland, A. Rothenberger, K. Katsiev, Y. Losovyj, X. Zhang, P. A. Dowben, O. F. Mohammed, E. H. Sargent and O. M. Bakr, Science, 2015, 347, 519–522 CrossRef CAS PubMed.
- S. D. Stranks, G. E. Eperon, G. Grancini, C. Menelaou, M. J. P. Alcocer, T. Leijtens, L. M. Herz, A. Petrozza and H. J. Snaith, Science, 2013, 342, 341–344 CrossRef CAS PubMed.
- National Renewable Energy Laboratory (NREL), Best research-cell efficiency chart (2022); http://www.nrel.gov/pv/cell-efficiency.html.
- N. J. Jeon, J. H. Noh, Y. C. Kim, W. S. Yang, S. Ryu and S. I. Seok, Nat. Mater., 2014, 13, 897–903 CrossRef CAS PubMed.
- M. Xiao, F. Huang, W. Huang, Y. Dkhissi, Y. Zhu, J. Etheridge, A. Gray-Weale, U. Bach, Y.-B. Cheng and L. Spiccia, Angew. Chem., Int. Ed., 2014, 53, 9898–9903 CrossRef CAS PubMed.
- W. S. Yang, B.-W. Park, E. H. Jung, N. J. Jeon, Y. C. Kim, D. U. Lee, S. S. Shin, J. Seo, E. K. Kim, J. H. Noh and S. I. Seok, Science, 2017, 356, 1376–1379 CrossRef CAS PubMed.
- N. Pellet, P. Gao, G. Gregori, T.-Y. Yang, M. K. Nazeeruddin, J. Maier and M. Grätzel, Angew. Chem., Int. Ed., 2014, 53, 3151–3157 CrossRef CAS PubMed.
- N. J. Jeon, J. H. Noh, W. S. Yang, Y. C. Kim, S. Ryu, J. Seo and S. I. Seok, Nature, 2015, 517, 476–480 CrossRef CAS PubMed.
- S.-H. Turren-Cruz, A. Hagfeldt and M. Saliba, Science, 2018, 362, 449–453 CrossRef CAS PubMed.
- J. J. Yoo, S. Wieghold, M. C. Sponseller, M. R. Chua, S. N. Bertram, N. T. P. Hartono, J. S. Tresback, E. C. Hansen, J.-P. Correa-Baena, V. Bulović, T. Buonassisi, S. S. Shin and M. G. Bawendi, Energy Environ. Sci., 2019, 12, 2192–2199 RSC.
- Q. Jiang, Y. Zhao, X. Zhang, X. Yang, Y. Chen, Z. Chu, Q. Ye, X. Li, Z. Yin and J. You, Nat. Photonics, 2019, 13, 460–466 CrossRef CAS.
- G. Yang, Z. Ren, K. Liu, M. Qin, W. Deng, H. Zhang, H. Wang, J. Liang, F. Ye, Q. Liang, H. Yin, Y. Chen, Y. Zhuang, S. Li, B. Gao, J. Wang, T. Shi, X. Wang, X. Lu, H. Wu, J. Hou, D. Lei, S. K. So, Y. Yang, G. Fang and G. Li, Nat. Photonics, 2021, 15, 681–689 CrossRef CAS.
- W. A. Dunlap-Shohl, Y. Zhou, N. P. Padture and D. B. Mitzi, Chem. Rev., 2019, 119, 3193–3295 CrossRef CAS PubMed.
- M. A. Green, A. Ho-Baillie and H. J. Snaith, Nat. Photonics, 2014, 8, 506–514 CrossRef CAS.
- Y. Zhang, S. Seo, S. Y. Lim, Y. Kim, S.-G. Kim, D.-K. Lee, S.-H. Lee, H. Shin, H. Cheong and N.-G. Park, ACS Energy Lett., 2020, 5, 360–366 CrossRef CAS.
- W. Mao, J. Zheng, Y. Zhang, A. S. R. Chesman, Q. Ou, J. Hicks, F. Li, Z. Wang, B. Graystone, T. D. M. Bell, M. U. Rothmann, N. W. Duffy, L. Spiccia, Y. B. Cheng, Q. Bao and U. Bach, Angew. Chem., Int. Ed., 2017, 56, 12486–12491 CrossRef CAS PubMed.
- Y. Guo, K. Shoyama, W. Sato, Y. Matsuo, K. Inoue, K. Harano, C. Liu, H. Tanaka and E. Nakamura, J. Am. Chem. Soc., 2015, 137, 15907–15914 CrossRef CAS PubMed.
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338, 643–647 CrossRef CAS PubMed.
- W. Zhang, M. Saliba, D. T. Moore, S. K. Pathak, M. T. Hörantner, T. Stergiopoulos, S. D. Stranks, G. E. Eperon, J. A. Alexander-Webber, A. Abate, A. Sadhanala, S. Yao, Y. Chen, R. H. Friend, L. A. Estroff, U. Wiesner and H. J. Snaith, Nat. Commun., 2015, 6, 6142 CrossRef CAS PubMed.
- N. Ahn, D.-Y. Son, I.-H. Jang, S. M. Kang, M. Choi and N.-G. Park, J. Am. Chem. Soc., 2015, 137, 8696–8699 CrossRef CAS PubMed.
- K. Hwang, Y.-S. Jung, Y.-J. Heo, F. H. Scholes, S. E. Watkins, J. Subbiah, D. J. Jones, D.-Y. Kim and D. Vak, Adv. Mater., 2015, 27, 1241–1247 CrossRef CAS PubMed.
- Y. Deng, C. H. Van Brackle, X. Dai, J. Zhao, B. Chen and J. Huang, Sci. Adv., 2019, 5, eaax7537 CrossRef CAS PubMed.
- J. Li, J. Dagar, O. Shargaieva, M. A. Flatken, H. Köbler, M. Fenske, C. Schultz, B. Stegemann, J. Just, D. M. Többens, A. Abate, R. Munir and E. Unger, Adv. Energy Mater., 2021, 11, 2003460 CrossRef CAS.
- X. Dai, S. Chen, H. Jiao, L. Zhao, K. Wang, Z. Ni, Z. Yu, B. Chen, Y. Gao and J. Huang, Nat. Energy, 2022, 7, 923–931 CrossRef CAS.
- K. Xiao, Y.-H. Lin, M. Zhang, R. D. J. Oliver, X. Wang, Z. Liu, X. Luo, J. Li, D. Lai, H. Luo, R. Lin, J. Xu, Y. Hou, H. J. Snaith and H. Tan, Science, 2022, 376, 762–767 CrossRef CAS PubMed.
- G. Yang, Z. Ni, Z. J. Yu, B. W. Larson, Z. Yu, B. Chen, A. Alasfour, X. Xiao, J. M. Luther, Z. C. Holman and J. Huang, Nat. Photonics, 2022, 16, 588–594 CrossRef CAS.
- S. Chen, X. Dai, S. Xu, H. Jiao, L. Zhao and J. Huang, Science, 2021, 373, 902–907 CrossRef CAS PubMed.
- A. E. Williams, P. J. Holliman, M. J. Carnie, M. L. Davies, D. A. Worsley and T. M. Watson, J. Mater. Chem. A, 2014, 2, 19338–19346 RSC.
- A. Mutlu, T. Yeşil, D. Kiymaz and C. Zafer, ACS Appl. Energy Mater., 2021, 4, 47–60 CrossRef CAS.
- G. Tong, D.-Y. Son, L. K. Ono, H.-B. Kang, S. He, L. Qiu, H. Zhang, Y. Liu, J. Hieulle and Y. Qi, Nano Energy, 2021, 87, 106152 CrossRef CAS.
- Y. Zhou, A. Najar, J. Zhang, J. Feng, Y. Cao, Z. Li, X. Zhu, D. Yang and S. F. Liu, ACS Appl. Mater. Interfaces, 2022, 14, 28729–28737 CrossRef CAS PubMed.
- M. Wang, C. Fei, M. A. Uddin and J. Huang, Sci. Adv., 2022, 8, eabo5977 CrossRef CAS PubMed.
- Z. Wang, J. Jin, Y. Zheng, X. Zhang, Z. Zhu, Y. Zhou, X. Cui, J. Li, M. Shang, X. Zhao, S. Liu and Q. Tai, Adv. Energy Mater., 2021, 11, 2102169 CrossRef CAS.
- H. Xie, J. Xu, C. Gao, J. Zhang, C. Gao and X. Liu, Sol. Energy, 2021, 222, 212–218 CrossRef CAS.
- M. Du, X. Zhu, L. Wang, H. Wang, J. Feng, X. Jiang, Y. Cao, Y. Sun, L. Duan, Y. Jiao, K. Wang, X. Ren, Z. Yan, S. Pang and S. Liu, Adv. Mater., 2020, 32, 2004979 CrossRef CAS PubMed.
- Y. Deng, S. Xu, S. Chen, X. Xiao, J. Zhao and J. Huang, Nat. Energy, 2021, 6, 633–641 CrossRef CAS.
- D. Luo, L. Zhao, J. Wu, Q. Hu, Y. Zhang, Z. Xu, Y. Liu, T. Liu, K. Chen, W. Yang, W. Zhang, R. Zhu and Q. Gong, Adv. Mater., 2017, 29, 1604758 CrossRef PubMed.
- J. A. Aguiar, S. Wozny, T. G. Holesinger, T. Aoki, M. K. Patel, M. Yang, J. J. Berry, M. Al-Jassim, W. Zhou and K. Zhu, Energy Environ. Sci., 2016, 9, 2372–2382 RSC.
- X. Wang, Y. Fan, L. Wang, C. Chen, Z. Li, R. Liu, H. Meng, Z. Shao, X. Du, H. Zhang, G. Cui and S. Pang, Chem, 2020, 6, 1369–1378 CAS.
- E. J. Juarez-Perez, L. K. Ono and Y. Qi, J. Mater. Chem. A, 2019, 7, 16912–16919 RSC.
- W. T. M. Van Gompel, R. Herckens, G. Reekmans, B. Ruttens, J. D’Haen, P. Adriaensens, L. Lutsen and D. Vanderzande, J. Phys. Chem. C, 2018, 122, 4117–4124 CrossRef CAS.
- B. G. Cox, Acids and Bases: Solvent Effects on Acid-Base Strength, Oxford University Press, Oxford, 2013 Search PubMed.
- Y.-H. Lin, N. Sakai, P. Da, J. Wu, H. C. Sansom, A. J. Ramadan, S. Mahesh, J. Liu, R. D. J. Oliver, J. Lim, L. Aspitarte, K. Sharma, P. K. Madhu, A. B. Morales-Vilches, P. K. Nayak, S. Bai, F. Gao, C. R. M. Grovenor, M. B. Johnston, J. G. Labram, J. R. Durrant, J. M. Ball, B. Wenger, B. Stannowski and H. J. Snaith, Science, 2020, 369, 96–102 CrossRef CAS PubMed.
- W. Zhang, S. Pathak, N. Sakai, T. Stergiopoulos, P. K. Nayak, N. K. Noel, A. A. Haghighirad, V. M. Burlakov, D. W. deQuilettes, A. Sadhanala, W. Li, L. Wang, D. S. Ginger, R. H. Friend and H. J. Snaith, Nat. Commun., 2015, 6, 10030 CrossRef CAS PubMed.
- S. Zhang, S. Wu, R. Chen, W. Chen, Y. Huang, H. Zhu, Z. Yang and W. Chen, J. Phys. Chem. Lett., 2019, 10, 2898–2903 CrossRef CAS PubMed.
- L. Krückemeier, B. Krogmeier, Z. Liu, U. Rau and T. Kirchartz, Adv. Energy Mater., 2021, 11, 2003489 CrossRef.
- J. J. Yoo, G. Seo, M. R. Chua, T. G. Park, Y. Lu, F. Rotermund, Y.-K. Kim, C. S. Moon, N. J. Jeon, J.-P. Correa-Baena, V. Bulović, S. S. Shin, M. G. Bawendi and J. Seo, Nature, 2021, 590, 587–593 CrossRef CAS PubMed.
- T. Bu, J. Li, H. Li, C. Tian, J. Su, G. Tong, L. K. Ono, C. Wang, Z. Lin, N. Chai, X.-L. Zhang, J. Chang, J. Lu, J. Zhong, W. Huang, Y. Qi, Y.-B. Cheng and F. Huang, Science, 2021, 372, 1327–1332 CrossRef CAS PubMed.
- S. Gharibzadeh, B. Abdollahi Nejand, M. Jakoby, T. Abzieher, D. Hauschild, S. Moghadamzadeh, J. A. Schwenzer, P. Brenner, R. Schmager, A. A. Haghighirad, L. Weinhardt, U. Lemmer, B. S. Richards, I. A. Howard and U. W. Paetzold, Adv. Energy Mater., 2019, 9, 1803699 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ee01634f |
| This journal is © The Royal Society of Chemistry 2023 |