
Near-unity electrochemical conversion of nitrate to ammonia on crystalline nickel porphyrin-based covalent organic frameworks†
Fang
Lv‡
ab,
Mingzi
Sun‡
 c,
Yongpan
Hu‡
ab,
Jie
Xu
ab,
Wei
Huang
ab,
Na
Han
*ab,
Bolong
Huang
*ce and
Yanguang
Li
*abd
c,
Yongpan
Hu‡
ab,
Jie
Xu
ab,
Wei
Huang
ab,
Na
Han
*ab,
Bolong
Huang
*ce and
Yanguang
Li
*abd
aInstitute of Functional Nano & Soft Materials (FUNSOM), Soochow University, Suzhou, 215123, China. E-mail: hanna@suda.edu.cn
bJiangsu Key Laboratory for Advanced Negative Carbon Technologies, Soochow University, Suzhou, 215123, China. E-mail: yanguang@suda.edu.cn
cDepartment of Applied Biology and Chemical Technology, The Hong Kong Polytechnic University, Hung Hom, Kowloon, Hong Kong SAR, China. E-mail: bhuang@polyu.edu.hk
dMacao Institute of Materials Science and Engineering (MIMSE), MUST-SUDA Joint Research Center for Advanced Functional Materials, Macau University of Science and Technology, Taipa 999078, Macau SAR, China
eResearch Centre for Carbon-Strategic Catalysis, The Hong Kong Polytechnic University, Hung Hom, Kowloon, Hong Kong SAR, China
First published on 23rd November 2022
Abstract
Electrochemical nitrate reduction, which has attracted rapidly increasing attention over recent years, can potentially enable the indirect fixation of atmospheric N2 as well as the efficient removal of nitrate from industrial wastewater. It is, however, limited by the lack of efficient and low-cost electrocatalysts available so far. To address this challenge, we here demonstrate a two-dimensional nickel porphyrin-based covalent organic framework (COF) as a potential candidate for the first time. The product has a highly ordered molecular structure with abundant square-shaped nanopores. In neutral solution, the reduction of nitrate ions at different concentrations from ammonia is realized with a great selectivity of ∼90% under a mild overpotential, a remarkable production rate of up to 2.5 mg h−1 cm−2, a turnover frequency of up to 3.5 s−1, and an intrinsic stability that is best delivered under pulse electrolysis. This cathodic reaction can also be coupled with the oxygen evolution reaction to enable full-cell electrolysis at high efficiency. Theoretical computations indicate that nickel centers can stably adsorb nitrate, and facilitate its subsequent reduction by lowering the energy barrier of the rate-determining step.
Broader contextThe selective electrochemical nitrate reduction reaction (NO3RR) to ammonia may play two essential roles within the artificial nitrogen cycle. When powered using renewable electricity, it can enable the indirect fixation of atmospheric N2 without emissions; it can also remove nitrates from industrial wastewater, and provide a much needed remedy to the global environmental and water security challenges that we face today. Unfortunately, the NO3RR involves multistep electron transfer and can yield a spectrum of reduction products, giving rise to a limited reaction selectivity. Copper represents the most studied catalyst, but generally requires a large overpotential in order to attain satisfactory NH3 selectivity and production rates. Here, we demonstrate transition metal macrocycles as a potential candidate system. As a proof of concept, we incorporate nickel porphyrin into two-dimensional covalent organic frameworks in order to maximize the catalytic activity and efficiency. In a neutral solution, our catalyst exhibits great selectivity that is close to unity, an excellent turnover frequency, and intrinsic stability. This work offers significant insights to extend promising electrocatalysts for nitrate reduction. |
Introduction
Nitrogen is an indispensable ingredient for all living organisms. It exists in many different forms in the atmosphere, water, soils, and biota, which are naturally interconverted via the nitrogen cycle.1,2 The development of the Haber–Bosch process more than a century ago enabled the artificial conversion of atmospheric N2 to NH3, which since then has made ammonia fertilizers widely available.3 Unfortunately, this industrial process is heavily energy intensive and has a massive carbon footprint. Direct NH3 synthesis from electrochemical N2 reduction under mild conditions is highly appealing, but is a formidable challenge and suffers from poor reaction rates (of the order of ∼1 nmol s−1) owing to the chemical inertness of N2.4–7 By comparison, NH3 synthesis from the nitrate/nitrite reduction reaction (NO3RR or NO2RR, respectively) is more kinetically favorable, and has recently attracted rapidly growing attention.8–10 The nitrate or nitrite feed sources can be derived from N2via plasma treatments, thereby enabling the indirect fixation of atmospheric N2;11,12 in addition, the reaction can be powered using renewable electricity from solar and wind sources, and is potentially emission-free. Moreover, the NO3RR or NO2RR offers a viable solution for removing nitrates and nitrites in eutrophic water bodies produced via over-fertilization or other anthropogenic activities and may mitigate the global water security challenge that we face today.13–15 However, the NO3RR or NO2RR to NH3 involves multistep electron transfer that generally compromises the reaction selectivity.16 There have been increasing calls for the development of efficient NO3RR or NO2RR electrocatalysts at low costs.Among different candidates, Cu-based catalysts have received the most attention and have been investigated in a number of recent studies.17–19 They do, however, generally demand a large overpotential in order to attain satisfactory NH3 selectivity and production rates.9,20 On the other hand, transition metal macrocycles with discrete metal centers (in particular, Fe, Co, and Ni) have found applications for a range of important electrochemical processes (e.g., the oxygen reduction reaction and the CO2 reduction reaction),21–24 although they have never been considered for the NO3RR or NO2RR to the best of our knowledge. Their diverse and well-defined molecular structures offer a unique opportunity to be used as a model catalyst and to understand structure–activity correlations. It has also been suggested that the isolated metal centers may suppress undesirable N–N coupling and inhibit the side reaction pathway.25 Unfortunately, transition metal macrocycles are often subject to strong intermolecular π–π stacking, and form microsized crystals with low surface areas, which adversely impair their attainable activities.26 Incorporating transition metal macrocycles within two-dimensional (2D) conjugated covalent organic frameworks (COFs) provides a promising strategy to address the above issue. The porous structure of COFs could significantly enhance the surface accessibility of active sites, and their molecular conjugation may also facilitate rapid intralayer and interlayer electron transfer to the active sites during electrochemical reactions.
To this end, we here report a 2D nickel porphyrin-based COF (NiPr-TPA-COF) for the efficient electrochemical NO3RR to NH3 in a neutral medium. NiPr-TPA-COF features a highly crystalline framework and has abundant mesoporosity. It exhibits great electrocatalytic activity and an NH3 selectivity of ∼90%. A remarkable NH3 production rate of up to 2.5 mg h−1 cm−2 and a turnover frequency (TOF) of 3.5 s−1 are measured. Density functional theory (DFT) calculations suggest that the NiPr moieties can stably adsorb NO3−, and enable efficient electron transfer for its further reduction with a decreased energy barrier.
Results and discussion
NiPr-TPA-COF was synthesized from the Schiff base condensation reaction between 5,10,15,20-tetrakis(4-aminophenyl)porphinato nickel and terephthalaldehyde (TPA) in the presence of 6 M acetic acid as the catalyst (see ESI† for details). The slow and reversible crosslinking of the amine and aldehyde precursors catalyzed by acetic acid triggers a self-correction mechanism that is proven essential for high product crystallinity.27–29 In addition, we also prepared metal-free H2Pr-TPA-COF as a control by replacing the NiPr precursor with H2Pr under otherwise identical conditions.
Fig. 1a schematically depicts the structural topology of NiPr-TPA-COF, which consists of the A–A stacking of 2D molecular layers with a square shaped porosity, which is confirmed via its powder X-ray diffraction (XRD) pattern. The sharp diffraction peaks indicate long-range structural ordering and high product crystallinity (Fig. 1b). The most intense peak located at 3.45° is assignable to (100) diffraction and corresponds to an average pore width of 2.5 nm, in good agreement with the proposed structure. The molecular structure of NiPr-TPA-COF was further interrogated using a multitude of spectroscopic analyses. Compared with the two starting monomers, both the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration (1697 cm−1) and the N–H stretching vibration (3300–3500 cm−1) disappear in the Fourier-transform infrared (FT-IR) spectrum of NiPr-TPA-COF. A new peak at 1617 cm−1 is instead observed in line with previous results,30,31 attesting to the complete condensation and the formation of the imine linkage (Fig. 1c). Raman analysis corroborates the formation of the imine linkage via its characteristic signal at 1587 cm−1 (Fig. S1, ESI†).32 Moreover, the imine functionalities are also reflected from the 13C solid-state nuclear magnetic resonance (NMR) spectrum of NiPr-TPA-COF, which shows a resonance peak at δ = 158.6 ppm along with other signals from the porphyrin (Fig. S2, ESI†).33 The X-ray photoelectron spectroscopy (XPS) of NiPr-TPA-COF supports the presence of Ni centers in the divalent state (Fig. S3, ESI†).34 Spectroscopic characterization of H2Pr-TPA-COF reveals a similar molecular structure except for the transition metal centers, as summarized in Fig. S4 (ESI†).
O stretching vibration (1697 cm−1) and the N–H stretching vibration (3300–3500 cm−1) disappear in the Fourier-transform infrared (FT-IR) spectrum of NiPr-TPA-COF. A new peak at 1617 cm−1 is instead observed in line with previous results,30,31 attesting to the complete condensation and the formation of the imine linkage (Fig. 1c). Raman analysis corroborates the formation of the imine linkage via its characteristic signal at 1587 cm−1 (Fig. S1, ESI†).32 Moreover, the imine functionalities are also reflected from the 13C solid-state nuclear magnetic resonance (NMR) spectrum of NiPr-TPA-COF, which shows a resonance peak at δ = 158.6 ppm along with other signals from the porphyrin (Fig. S2, ESI†).33 The X-ray photoelectron spectroscopy (XPS) of NiPr-TPA-COF supports the presence of Ni centers in the divalent state (Fig. S3, ESI†).34 Spectroscopic characterization of H2Pr-TPA-COF reveals a similar molecular structure except for the transition metal centers, as summarized in Fig. S4 (ESI†).
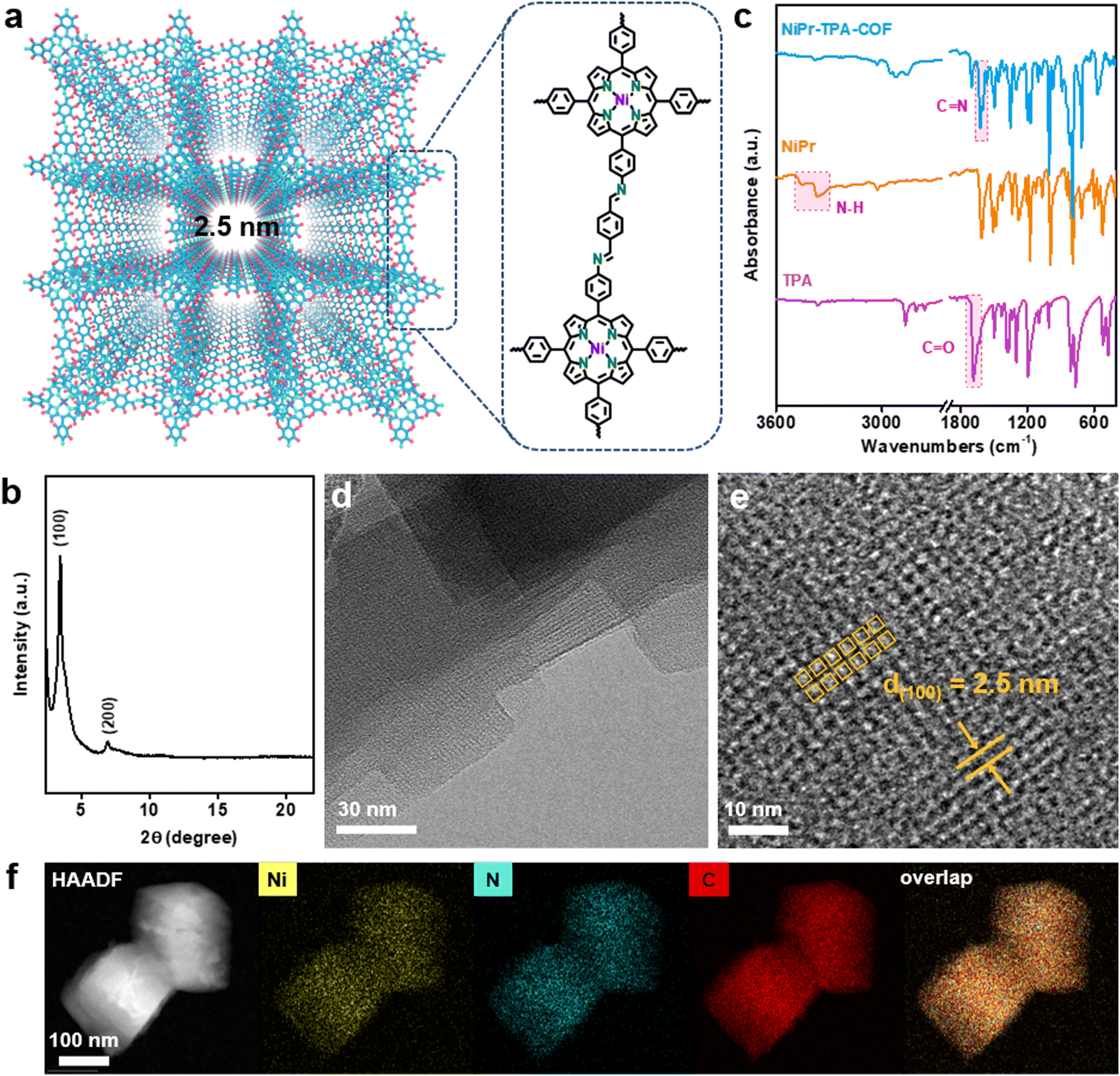 | ||
| Fig. 1 Structural characterization of NiPr-TPA-COF. (a) Schematic topology of NiPr-TPA-COF and its constituent units. (b) XRD pattern of NiPr-TPA-COF. (c) FT-IR spectra of NiPr-TPA-COF, NiPr, and TPA. (d) TEM image, (e) HR-TEM image, and (f) EDS elemental mapping of NiPr-TPA-COF. | ||
NiPr-TPA-COF was subsequently subjected to microscopic imaging. Under low-resolution transmission electron microscopy (TEM), the product was unveiled to consist of square nanoflakes that are further stacked together (Fig. 1d). The individual nanoflakes have a lateral size of ∼100 nm, and exhibit smooth edges and sharp corners. The square shape reflects the inherent four-fold symmetry of the molecular structure. Under high-resolution TEM, evident square fringes are observed with a d-spacing of 2.5 nm. These nanoflakes are determined to be terminated with (001) planes. Periodic mesoporous channels run perpendicularly through nanoflakes and correspond to the light contrast regions in Fig. 1e. Energy dispersive spectroscopy (EDS) elemental mapping in Fig. 1f shows the uniform spatial distribution of C, N, and Ni species and hence the homogeneity of the composite. The porous nature of NiPr-TPA-COF is additionally confirmed via BET analysis, which measures a large surface area of 899 m2 g−1 and a pore volume of 0.49 cm3 g−1 (Fig. S5, ESI†). Altogether, the above results unambiguously support that the highly crystalline 2D COF is formed via imide linkages and with the successful incorporation of NiPr moieties. Its ordered and porous structure facilitates fast mass transport and charge transport, which are essential for efficient electrocatalysis.
We next evaluated the electrochemical performance of NiPr-TPA-COF for the NO3RR using a two-compartment H-cell (see ESI† for details). The catalyst powder was dropcaste onto a 1 × 1 cm carbon fiber paper electrode at a mass loading of 1 mg cm−2. Two control samples, i.e., metal-free H2Pr-TPA-COF and the NiPr monomer were brought into the comparison side by side. We initially attempted to carry out the NO3RR in 1 M KOH, like done in many other studies.17,35,36 NiPr-TPA-COF delivered a large current density and an NH3 selectivity that is superior to most competitors under similar conditions (Fig. S6, ESI†).17,36,37 However, we surprisingly discovered that the current collector itself was electrochemically active in the alkaline medium (which will be the subject of a future study) and strongly interfered with our measurements (Fig. S7, ESI†). Therefore, all the experiments were carried out in a neutral solution where the activity of the bank current collector was minimized (Fig. S8, ESI†).
Fig. S9 (ESI†) depicts the polarization curves of NiPr-TPA-COF in Ar-saturated 0.5 M K2SO4 with and without 0.1 M KNO3. In the absence of KNO3, only the hydrogen evolution reaction (HER) takes place, which does not exhibit any appreciable current density until less than −1.5 V (versus the saturated calomel electrode or SCE, the same hereinafter). The addition of KNO3 positively displaces the polarization curve. The current density starts to take off at −0.8 V and increases to 46 mA cm−2 at −1.5 V, which is the first indication of the great NO3RR activity of NiPr-TPA-COF. By contrast, both H2Pr-TPA-COF and NiPr have negligible cathodic responses in the presence of KNO3 within the same potential region.
In order to identify and quantify the reduction products, chronoamperometric (i–t) analysis was conducted at different working potentials. The amount of NH3 production was measured via the indophenol blue method using ultraviolet-visible (UV-Vis) spectrophotometry (Fig. S10, ESI†).38 Its corresponding faradaic efficiency was derived for different samples and plotted with respect to the working potential, as shown in Fig. 2a. It is worth noting that both NiPr-TPA-COF and NiPr have a remarkable NH3 selectivity between −1.3 V and −1.5 V. In particular, the former exhibits a great value of >80% over the potential region examined, and delivers a maximum selectivity of ∼90% at −1.38 V, which is superior to most single atom catalysts (SACs)25,39 and some Cu-based electrocatalysts previously reported under similar conditions.40 The NH3 faradaic efficiency of NiPr starts to decline at less than −1.4 V owing to competition from the HER. These two samples share the same NiPr moieties that are believed to be catalytically active sites. Metal-free H2Pr-TPA-COF has much inferior selectivity of <30%, which clearly manifests the critical role of Ni2+ centers during the NO3RR.
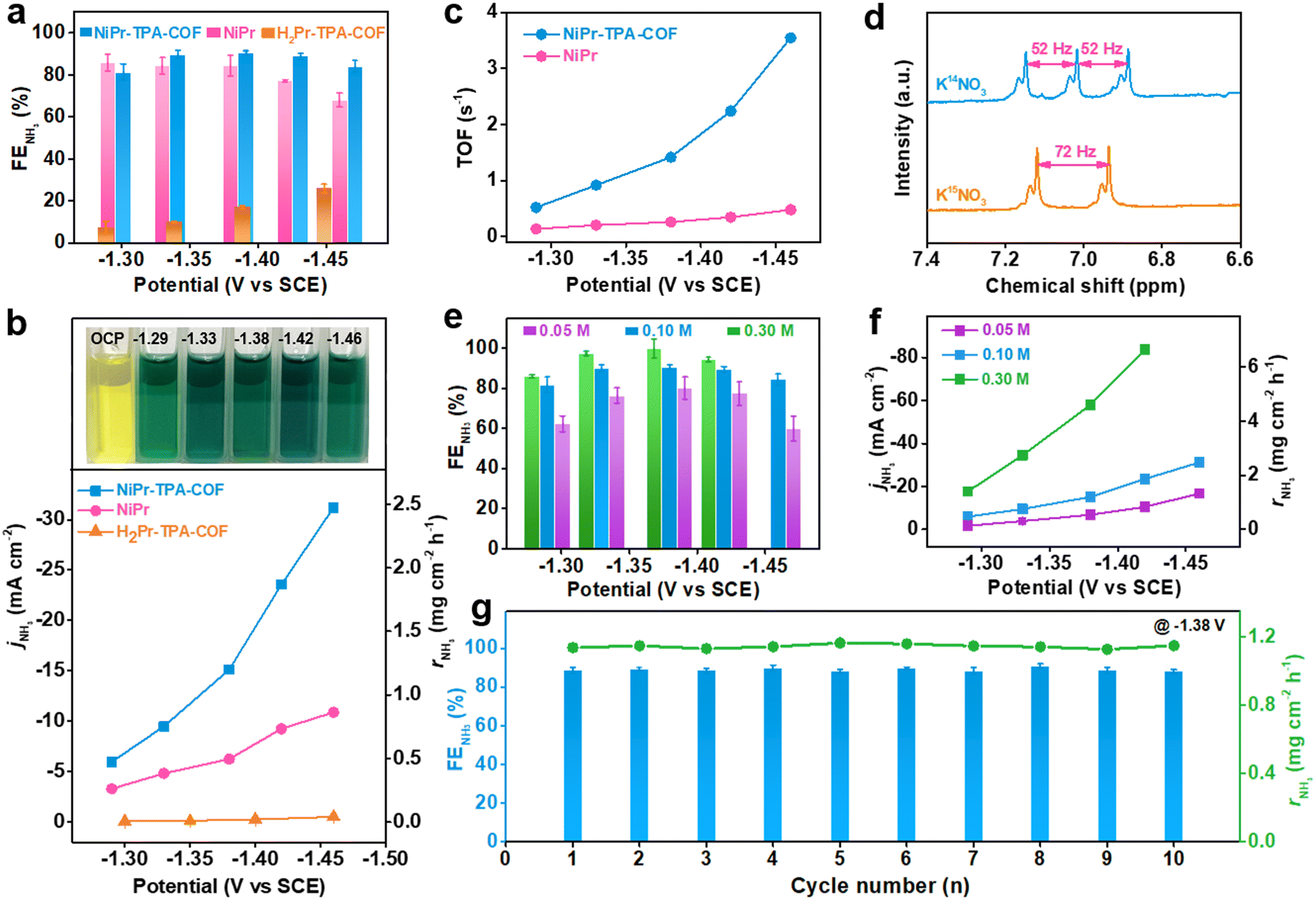 | ||
| Fig. 2 Electrochemical NO3RR performance of NiPr-TPA-COF. (a) NH3 faradaic efficiency and (b) NH3 partial current density and production rate of NiPr-TPA-COF in comparison with those of H2Pr-TPA-COF and NiPr; the top illustration in (b) shows the diluted catholyte after the NO3RR at different potentials and with the addition of indophenol indicator. (c) TOF values of NiPr-TPA-COF and NiPr. (d) 1H NMR spectra of NH3 using K14NO3 or K15NO3 for the NO3RR. (e) NH3 faradaic efficiency and (f) NH3 partial current density and production rate of NiPr-TPA-COF in 0.5 M K2SO4 with the addition of different KNO3 concentrations. (g) Cycling test of NiPr-TPA-COF at −1.38 V showing the average NH3 faradaic efficiency and production rate within each cycle. | ||
The potential-dependent NH3 partial current density and production rate were then calculated. They are observed to rise continuously with the increasing overpotential (Fig. 2b). At −1.46 V, the NH3 partial current density of NiPr-TPA-COF reaches 31.2 mA cm−2, and its NH3 production rate reaches 2.5 mg cm−2 h−1. These values are ∼3 times larger than those of NiPr and >60 times larger than those of H2Pr-TPA-COF. They are also orders of magnitude larger than the reported rates for electrochemical N2 reduction to NH3. Moreover, we estimated the TOF by normalizing the NH3 partial current density over the amount of surface accessible Ni sites (Fig. S11, ESI†). It was derived as 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 754 h−1 (or 3.5 s−1) on NiPr-TPA-COF and 1717 h−1 (or 0.5 s−1) on NiPr (Fig. 2c), reflecting the significantly higher site-specific activity when NiPr moieties are incorporated within the molecular frameworks. Among the very few studies that reported TOF values for the NO3RR to NH3, Ni nanoparticles were previously disclosed to have a TOF value of ∼6 h−1 (or 0.0017 s−1) at −0.5 V vs. RHE.41 The orders-of-magnitude higher TOF value observed for our NiPr-TPA-COF highlights its extraordinary activity. To confirm the chemical origin of NH3, 15N-labeled isotope experiments were carried out on NiPr-TPA-COF (Fig. 2d). The 1H NMR spectrum of normal 14NH3 features a triplet with a coupling constant of 52 Hz. When K15NO3 is used instead of K14NO3, the 1H NMR spectrum of its reduction product exhibits a distinct doublet with a coupling constant of 72 Hz that is characteristic of 15NH3.42 These results support that KNO3 rather than other possible nitrogen contaminants is reduced during the NO3RR to yield NH3 at high rates.
754 h−1 (or 3.5 s−1) on NiPr-TPA-COF and 1717 h−1 (or 0.5 s−1) on NiPr (Fig. 2c), reflecting the significantly higher site-specific activity when NiPr moieties are incorporated within the molecular frameworks. Among the very few studies that reported TOF values for the NO3RR to NH3, Ni nanoparticles were previously disclosed to have a TOF value of ∼6 h−1 (or 0.0017 s−1) at −0.5 V vs. RHE.41 The orders-of-magnitude higher TOF value observed for our NiPr-TPA-COF highlights its extraordinary activity. To confirm the chemical origin of NH3, 15N-labeled isotope experiments were carried out on NiPr-TPA-COF (Fig. 2d). The 1H NMR spectrum of normal 14NH3 features a triplet with a coupling constant of 52 Hz. When K15NO3 is used instead of K14NO3, the 1H NMR spectrum of its reduction product exhibits a distinct doublet with a coupling constant of 72 Hz that is characteristic of 15NH3.42 These results support that KNO3 rather than other possible nitrogen contaminants is reduced during the NO3RR to yield NH3 at high rates.
Our catalyst can operate under a range of KNO3 concentrations. Running the reaction in 0.05 M KNO3 slightly compromises the NH3 selectivity, and reduces the NH3 production rate of NiPr-TPA-COF as expected. But even so, it still exhibits an NH3 selectivity of ∼80% within −1.3 and −1.4 V and a production rate of ∼1.3 mg cm−2 h−1 at −1.46 V (Fig. 2e and f). This capability of working with low-concentration KNO3 indicates the great potential of our catalyst for converting wastewater nitrate (typically 0.01–0.1 M) into valuable NH3. On the other hand, increasing the KNO3 concentration to 0.3 M in the electrolyte boosts both the selectivity (close to unity) and activity (6.6 mg cm−2 h−1 at −1.42 V) of NiPr-TPA-COF. There is no sign of catalyst poisoning at high substrate concentrations, which is a concern when using Cu-based materials.13,43,44 Our NiPr-TPA-COF can also enable an efficient NO2RR to NH3 in 0.5 M K2SO4 with 0.1 M KNO2 (Fig. S13, ESI†). Its high NH3 selectivity (∼100% at −1.33 V) and great activity (5.3 mg cm−2 h−1 or 67 mA cm−2 at −1.44 V) also render it one of the best NO2RR electrocatalysts in neutral solution. Moreover, the effect of the catalyst loading was explored and is summarized in Fig. S12 (ESI†). Replacing the Ni centers with Co centers in CoPr-TPA-COF results in a lower NH3 selectivity and production rate (Fig. S14 and S15, ESI†). In addition to a high activity and selectivity, operation stability is an important metric for the evaluation of catalysts. We find that when NiPr-TPA-COF is biased at a relatively negative working potential (e.g., −1.38 V), its cathodic current density is subjected to a rapid increase in the first 100–200 s and then gradually declines afterward (Fig. S16, ESI†). Such a dynamic evolution of the current density reflects an activation-to-deactivation process that appears common to NO3RR electrolysis.43 The accumulation of side reduction products near the catalyst is believed to block its active sites for a selective NO3RR, which might explain the gradual but considerable deactivation.45 Nevertheless, this deactivation causes no damage to the catalyst since its original activity can be fully recovered when the electrolyte is refreshed. We attempted to eliminate the deactivation process using pulse electrolysis, which has been demonstrated as an efficient tool for surface poison removal or catalyst regeneration.46–51 The running program was set to consist of alternating reduction steps at −1.38 V for 1000 s and oxidation steps at +0.4 V for 1 s. During the cathodic step, the NO3RR takes place at the working electrode; during the anodic step, the side reduction products accumulated on the catalyst surface are quickly oxidized. In this way, our catalyst can be periodically regenerated without sacrificing current density or NH3 selectivity. Its current density goes back to the initial level after the anodic step. As a result, long-term electrolysis can be carried out without interruption for ∼20000 s (Fig. S17, ESI†). During our measurements, the cathodic current density at the reduction steps stays at ∼20 mA cm−2, and the average NH3 faradaic efficiency is ∼73% (Fig. 2g). XRD, FT-IR and TEM characterization of the catalyst after long-term electrolysis revealed no discernible structural change on different scales, further attesting to the great stability of the catalyst (Fig. S18, ESI†).
In order to gain further insight into the great performance of NiPr-TPA-COF, DFT calculations were carried out to reveal the distinct electronic structures in comparison with metal-free H2Pr-TPA-COF (Fig. S19, ESI†). The projected partial density of states (PDOSs) analysis shows that its Ni-3d orbitals are located close to the Fermi level (EF), providing evidence for the high catalytic activity (Fig. 3a). PDOS analysis also exhibits a peak at Ev −1.30 eV (where Ev denotes 0 eV) that overlaps considerably with both N-2p and C-2p orbitals, which may enable fast site-to-site electron transfer from the surrounding C and N sites to the Ni center. When comparing the N-2p orbitals in NiPr-TPA-COF and H2Pr-TPA-COF, it comes to our attention that the Ni center activates nearby N sites (N–CN3), as reflected by their evidently increased electron density near EF (Fig. 3b). The N sites far away from the metal center and with a coordination number (CN) of two (N–CN2) are not affected. Similarly, the Ni center also influences nearby C sites in the porphyrin, and increases their electron density near EF (Fig. 3c). Even at the benzene linker, carbon atoms adjacent to the imine group show upshifted C-2p orbitals relative to those connecting to H. These results reveal a gradual downshifting trend of the C-2p orbitals in NiPr-TPA-COF that forms an efficient electron path for electrolysis. Moreover, the PDOS data for NO3 adsorption are analyzed in order to disclose the interaction between the catalyst and the reactant (Fig. 3d). The 2p orbitals of free NO3− ions show a sharp peak at Ev −1.70 eV, which is close to the Ni-3d peaks. This can enable efficient electron transfer from NiPr-TPA-COF to NO3− with a small energy barrier. Once NO3− ions are adsorbed, we observe that the strong interaction noticeably downshifts the 2p orbitals of NO3*, supporting the electron transfer from the Ni site. Accordingly, the Ni-3d orbitals are also modulated. The good orbital overlap between them ensures the stable adsorption of NO3* for further reduction. This also supports that the Ni center is the catalytic site for the NO3RR.
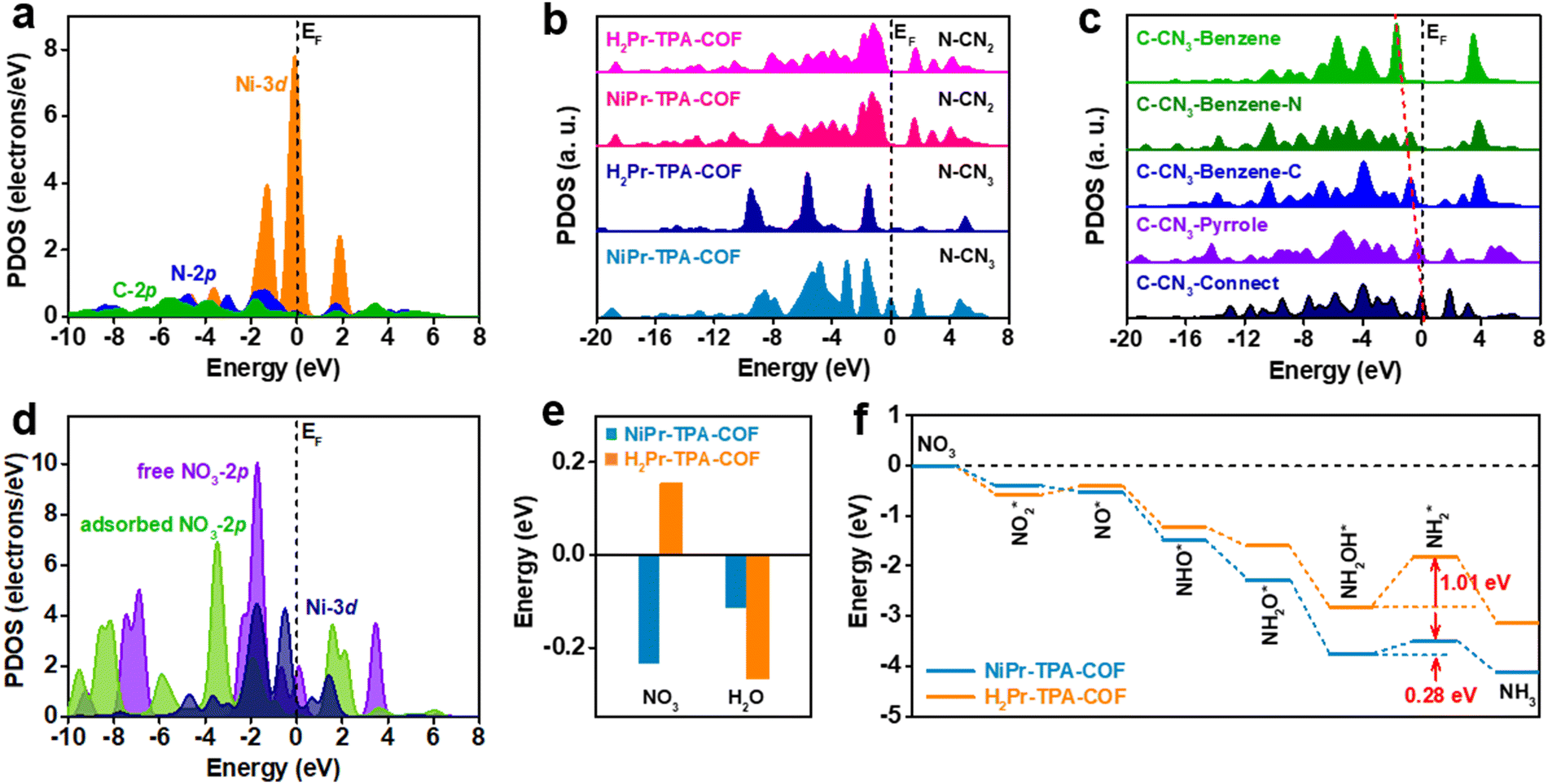 | ||
| Fig. 3 DFT simulations of the catalyst electronic structures and NO3RR pathway. (a) PDOSs for NiPr-TPA-COF. (b) Site-dependent PDOSs of N-2p orbitals in NiPr-TPA-COF and H2Pr-TPA-COF. (c) Site-dependent PDOSs for C-2p orbitals in NiPr-TPA-COF. (d) PDOSs for free NO3 and NO3 adsorbed on NiPr-TPA-COF. (e) Adsorption energies of NO3 and H2O on NiPr-TPA-COF and H2Pr-TPA-COF. (f) Energetic reaction pathways of the NO3RR to NH3 on NiPr-TPA-COF and H2Pr-TPA-COF. | ||
Furthermore, we calculated the adsorption energies of NO3 and H2O on the two catalysts (Fig. 3e). It is worth noting that the adsorption of NO3 on NiPr-TPA-COF (−0.23 eV) is spontaneous owing to its strong binding to the Ni center as explained above, while the adsorption of NO3 on metal-free H2Pr-TPA-COF (0.15 eV) is endothermic, which may hinder its subsequent reduction. This confirms the enhanced binding strength of N-containing intermediates on Ni sites, which are significant for guaranteeing the stable fixation of intermediates for ammonia formation. The adsorption of H2O is thermodynamically favorable on both NiPr-TPA-COF (−0.11 eV) and H2Pr-TPA-COF (−0.27 eV), but its stronger binding on the latter may enhance the competing HER process and lower the NO3RR efficiency. The mechanism of nitrate reduction is complicated, and involves 9 electron transfers in the process with valence state changes.52,53Fig. 3f depicts the simulated full reduction pathway from nitrate to ammonia.54 Nitrate is first adsorbed on the catalyst surface, and dissociates to NO2* and then NO*. Subsequently, NO* undergoes three proton-coupled electron transfers and is converted to NH2OH* before further reduction to NH3 and final desorption from the catalyst surface. We find that the overall reaction trend is downhill on NiPr-TPA-COF with a total energy of −4.1 eV for NO3RR.
Compared with H2Pr-TPA-COF, it has a stronger affinity towards all the intermediates except for NO2*. The reduction step from NH2OH* to NH2* is identified to be the rate-determining step for both catalysts, and its energy barrier is lowered from 1.01 eV on H2Pr-TPA-COF to 0.28 eV on NiPr-TPA-COF. In addition to its favorable electronic structure, we believe that the highly ordered and porous structure of NiPr-TPA-COF may facilitate mass transport, i.e., the diffusion in of reactants (NO3− and H2O) and the diffusion out of products (NH3 and OH−). Its conjugated molecular framework may also form conductive pathways for efficient electron transfer to the active centers. All these factors combined lead to the great NO3RR activity and selectivity observed experimentally on NiPr-TPA-COF.
Having established NiPr-TPA-COF as a promising NO3RR electrocatalyst, we then explored its potential in the coupled electrolysis of the NO3RR and the oxygen evolution reaction (OER). A two-electrode configuration was assembled by pairing the NiPr-TPA-COF cathode with a piece of IrO2-coated Ti mesh (IrO2@Ti) as the anode (Fig. 4a). Fig. 4b presents the polarization curve without iR compensation. It starts to take off at 2.4 V and delivers 34 mA cm−2 at 3.5 V. A lithium-ion battery was then used to power the two-electrode cell. Under the voltage of ∼3.4 V (Fig. 4c), the full reaction can be driven at a current density of 23 mA cm−2 (Fig. 4d). The average NH3 faradaic efficiency and energy efficiency were measured to be 91% and 14.5%, respectively.
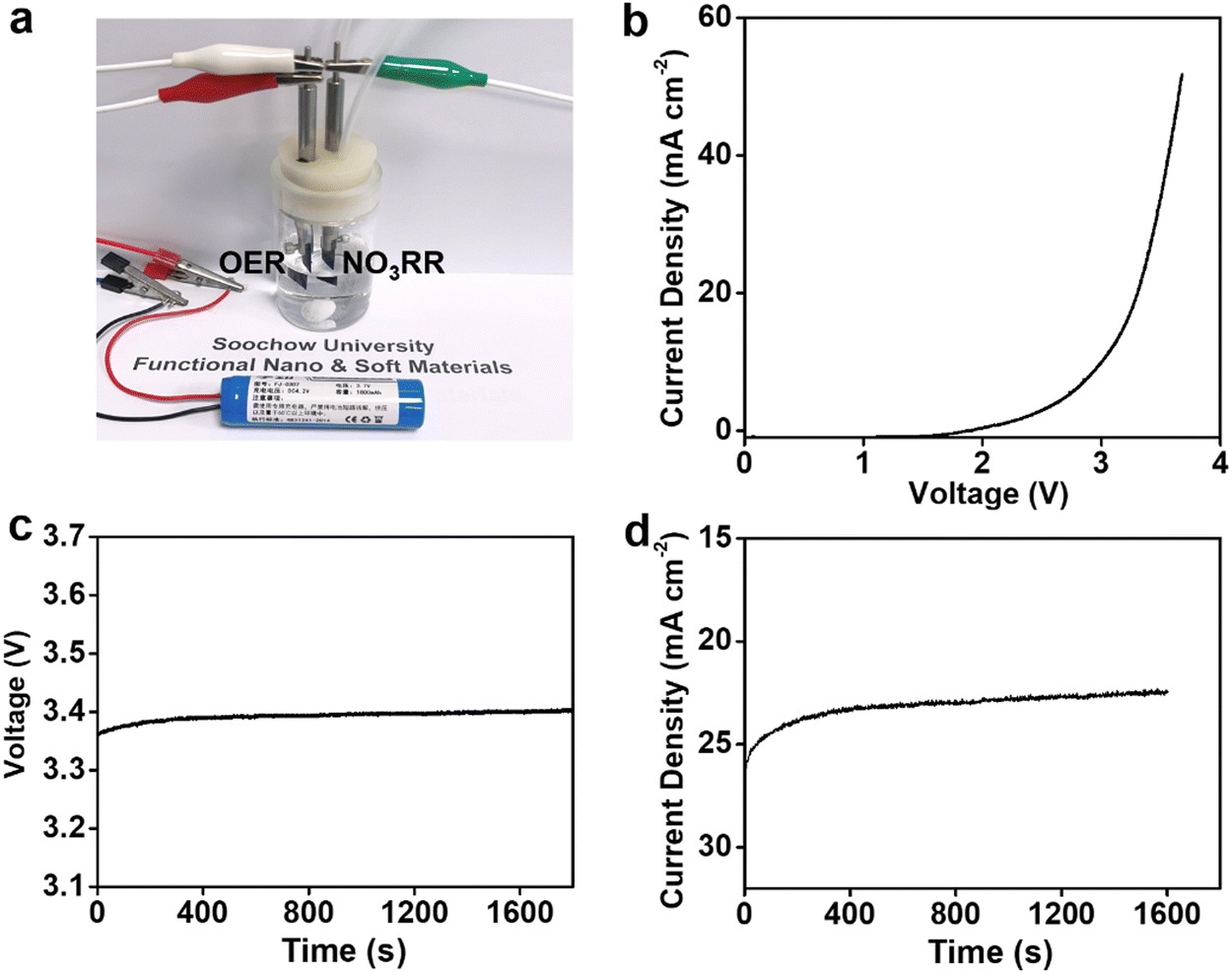 | ||
| Fig. 4 Full-cell NO3RR-OER electrolysis. (a) Photograph showing the setup for battery-powered NO3RR–OER electrolysis. (b) Full-cell polarization curve. (c) Voltage change and (d) current evolution for full-cell electrolysis. | ||
Achieving a high current density for electrochemical catalysis has been a significant challenge for the practical application of electrocatalysts. Although there have been great efforts for the NO3RR, most electrocatalysts still cannot break through a current density of 200 mA cm−2, which strongly hinders their further practical applications for NH3. For the NO3RR, previous research has proposed that the adsorption-energy scaling relations are the key factor that causes the tradeoff between the partial current density and the faradaic efficiency.55 In the more negative potential, the competitive hydrogen evolution reaction (HER) process causes shifting of the faradaic efficiency, which limits the current density of the NO3RR and the production rate of NH3.56 For transition metal alloys, the surface roughness of the catalyst materials also limits the current density due to a lower mass transfer.57 These studies indicate that further optimization studies are still needed in the future to achieve an even higher current density.
The experiment here demonstrates the practical viability of the battery-driven NO3RR using our catalyst, even though both the current density and the energy efficiency are not fully optimized. Future optimization can be approached by carrying out the coupled reaction in an alkaline solution since both the NO3RR and OER are intrinsically more active in alkaline solution than in neutral solution. We may also replace IrOx/C with more active NiFeOx catalysts for the OER in the alkaline solution. To further improve the energy efficiency, the sluggish OER half-reaction can be substituted with the electrochemical oxidation of alcohols such as benzyl alcohol and glycerol. These alternative reactions are thermodynamically more favorable and kinetically faster and can yield oxidation products with higher economic values and in more condensed forms. Therefore, in the future, this work will inspire more related research to achieve practical applications of the NO3RR.
Conclusions
In summary, here we reported the first demonstration of COF-based NO3RR electrocatalysis to NH3. NiPr-TPA-COF was prepared via the reversible Schiff base condensation reaction between amine-terminated NiPr and TPA. The product featured great structural crystallinity and abundant mesoporosity, as characterized using a multitude of spectroscopic and microscopic techniques. When assessed as the electrocatalyst for the NO3RR in neutral solution, NiPr-TPA-COF exhibited a near-unity NH3 selectivity, a great NH3 production rate of up to 2.5 mg h−1 cm−2, and a TOF of 3.5 s−1 at −1.46 V. These values were superior to single atom catalysts and Cu-based materials under similar conditions. Moreover, NiPr-TPA-COF showed great stability that could be periodically recovered and was further improved under pulse electrolysis. DFT simulations revealed that NiPr-TPA-COF had unique electronic structures that enhanced its interaction with NO3, and facilitated electron transfer during electrocatalysis. The presence of the Ni center lowered the activation energy barrier of the rate-determining step considerably. Finally, we demonstrated the coupled electrolysis of the NO3RR and the OER by combining NiPr-TPA-COF as the cathode catalyst and IrO2@Ti as the anode catalyst. When powered using a lithium-ion battery, the two-electrode cell stably delivered a current density of 23 mA cm−2 and a great NH3 selectivity of 91%. Our study here showcases the great potential of transition metal macrocycles for selective the NO3RR to NH3. The catalyst reported here consists of Earth-abundant elements (C, H, and N) and a small fraction of Ni, and is potentially more cost-effective than other candidate catalysts.Author contributions
Y. Li proposed the concept and designed all experimental studies. Y. Hu, W. Huang, and F. Lv carried out materials synthesis, characterization, and electrochemical NO3RR measurements. J. Xu performed the TEM analysis. B. Huang and M. Sun designed and performed DFT calculations. All authors contributes to the preparation of the manuscript. F. Lv, M. Sun and Y. Hu contributed equally in this work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge support from the National Natural Science Foundation of China (U2002213, 52161160331 and 21902114), the Science and Technology Development Fund Macau SAR (0077/2021/A2), the Collaborative Innovation Center of Suzhou Nano Science and Technology, the 111 Project, Joint International Research Laboratory of Carbon-Based Functional Materials and Devices, the National Natural Science Foundation of China/Research Grants Council (RGC) of Hong Kong Joint Research Scheme Project (N_PolyU502/21), the funding for Projects of Strategic Importance of The Hong Kong Polytechnic University (Project Code: 1-ZE2V), Research Institute for Smart Energy (RISE) and Research Institute for Intelligent Wearable Systems (RI-IWEAR) of the Hong Kong Polytechnic University.References
- N. Gruber and J. N. Galloway, Nature, 2008, 451, 293–296 CrossRef CAS PubMed
.
- J. N. Galloway, A. R. Townsend, J. W. Erisman, M. Bekunda, Z. Cai, J. R. Freney, L. A. Martinelli, S. P. Seitzinger and M. A. Sutton, Science, 2008, 320, 889–892 CrossRef CAS
- H. Liu, Chin. J. Catal., 2014, 35, 1619–1640 CrossRef CAS
- B. H. R. Suryanto, H. L. Du, D. Wang, J. Chen, A. N. Simonov and D. R. MacFarlane, Nat. Catal., 2019, 2, 290–296 CrossRef CAS
- J. Choi, B. H. Suryanto, D. Wang, H. L. Du, R. Y. Hodgetts, F. M. Ferrero Vallana, D. R. MacFarlane and A. N. Simonov, Nat. Commun., 2020, 11, 5546 CrossRef CAS PubMed
- G. Soloveichik, Nat. Catal., 2019, 2, 377–380 CrossRef CAS
- F. Lv, S. Zhao, R. Guo, J. He, X. Peng, H. Bao, J. Fu, L. Han, G. Qi, J. Luo, X. Tang and X. Liu, Nano Energy, 2019, 61, 420–427 CrossRef
- P. H. van Langevelde, I. Katsounaros and M. T. M. Koper, Joule, 2021, 5, 290–294 CrossRef
- Y. Wang, C. Wang, M. Li, Y. Yu and B. Zhang, Chem. Soc. Rev., 2021, 50, 6720–6733 RSC
- Y. Zeng, C. Priest, G. Wang and G. Wu, Small Methods, 2020, 4, 2000672 CrossRef CAS
- Y. Ren, C. Yu, L. Wang, X. Tan, Z. Wang, Q. Wei, Y. Zhang and J. Qiu, J. Am. Chem. Soc., 2022, 144, 10193–10200 CrossRef CAS PubMed
- J. Sun, D. Alam, R. Daiyan, H. Masood, T. Zhang, R. Zhou, P. J. Cullen, E. C. Lovell, A. R. Jalili and R. Amal, Energy Environ. Sci., 2021, 14, 865–872 RSC
- S. Garcia-Segura, M. Lanzarini-Lopes, K. Hristovski and P. Westerhoff, Appl. Catal., B, 2018, 236, 546–568 CrossRef CAS
- A. Bhatnagar and M. Sillanpää, Chem. Eng. J., 2011, 168, 493–504 CrossRef CAS
- F. Y. Chen, Z. Y. Wu, S. Gupta, D. J. Rivera, S. V. Lambeets, S. Pecaut, J. Y. T. Kim, P. Zhu, Y. Z. Finfrock, D. M. Meira, G. King, G. Gao, W. Xu, D. A. Cullen, H. Zhou, Y. Han, D. E. Perea, C. L. Muhich and H. Wang, Nat. Nanotechnol., 2022, 17, 759–767 CrossRef CAS PubMed
- V. Rosca, M. Duca, M. T. de Groot and M. T. M. Koper, Chem. Rev., 2009, 109, 2209–2244 CrossRef CAS PubMed
- Y. Wang, A. Xu, Z. Wang, L. Huang, J. Li, F. Li, J. Wicks, M. Luo, D. H. Nam, C. S. Tan, Y. Ding, J. Wu, Y. Lum, C. T. Dinh, D. Sinton, G. Zheng and E. H. Sargent, J. Am. Chem. Soc., 2020, 142, 5702–5708 CrossRef CAS
- Y. Wang, W. Zhou, R. Jia, Y. Yu and B. Zhang, Angew. Chem., Int. Ed., 2020, 59, 5350–5354 CrossRef CAS
- G. F. Chen, Y. Yuan, H. Jiang, S. Y. Ren, L. X. Ding, L. Ma, T. Wu, J. Lu and H. Wang, Nat. Energy, 2020, 5, 605–613 CrossRef CAS
- W. Jung and Y. J. Hwang, Mater. Chem. Front., 2021, 5, 6803–6823 RSC
- S. Yang, Y. Yu, X. Gao, Z. Zhang and F. Wang, Chem. Soc. Rev., 2021, 50, 12985–13011 RSC
- S. Dey, B. Mondal, S. Chatterjee, A. Rana, S. Amanullah and A. Dey, Nat. Rev. Chem., 2017, 1, 0098 CrossRef CAS
- F. Lv, N. Han, Y. Qiu, X. Liu, J. Luo and Y. Li, Coord. Chem. Rev., 2020, 422, 213435 CrossRef CAS
- N. Han, Y. Wang, L. Ma, J. Wen, J. Li, H. Zheng, K. Nie, X. Wang, F. Zhao, Y. Li, J. Fan, J. Zhong, T. Wu, D. J. Miller, J. Lu, S. T. Lee and Y. Li, Chem, 2017, 3, 652–664 CAS
- Z.-Y. Wu, M. Karamad, X. Yong, Q. Huang, D. A. Cullen, P. Zhu, C. Xia, Q. Xiao, M. Shakouri, F. Y. Chen, J. Y. T. Kim, Y. Xia, K. Heck, Y. Hu, M. S. Wong, Q. Li, I. Gates, S. Siahrostami and H. Wang, Nat. Commun., 2021, 12, 2870 CrossRef CAS
- L. Sun, V. Reddu, A. C. Fisher and X. Wang, Energy Environ. Sci., 2020, 13, 374–403 RSC
- J. L. Segura, M. J. Mancheño and F. Zamora, Chem. Soc. Rev., 2016, 45, 5635–5671 RSC
- L. Cusin, H. Peng, A. Ciesielski and P. Samorì, Angew. Chem., Int. Ed., 2021, 133, 14356–14370 CrossRef
- W. Huang, W. Luo and Y. Li, Mater. Today, 2020, 40, 160–172 CrossRef CAS
- H. L. Nguyen, C. Gropp and O. M. Yaghi, J. Am. Chem. Soc., 2020, 142, 2771–2776 CrossRef CAS PubMed
- Y. Li, L. Guo, Y. Lv, Z. Zhao, Y. Ma, W. Chen, G. Xing, D. Jiang and L. Chen, Angew. Chem., Int. Ed., 2021, 133, 5423–5429 CrossRef
- W. Dai, F. Shao, J. Szczerbiński, R. McCaffrey, R. Zenobi, Y. Jin, A. D. Schlüter and W. Zhang, Angew. Chem., Int. Ed., 2016, 128, 221–225 CrossRef
- S. Lin, C. S. Diercks, Y. B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D. Kim, P. Yang and O. M. Yaghi, Science, 2015, 349, 1208–1213 CrossRef CAS
- L. Sun, M. Lu, Z. Yang, Z. Yu, X. Su, Y. Q. Lan and L. Chen, Angew. Chem., Int. Ed., 2022, 134, e202204326 Search PubMed
- X. Deng, Y. Yang, L. Wang, X. Z. Fu and J. L. Luo, Adv. Sci., 2021, 8, 2004523 CrossRef CAS
- J. Li, G. Zhan, J. Yang, F. Quan, C. Mao, Y. Liu, B. Wang, F. Lei, L. Li, A. W. M. Chan, L. Xu, Y. Shi, Y. Du, W. Hao, P. K. Wong, D. Wang, J. Shi-Xue, L. Zhang and J. C. Yu, J. Am. Chem. Soc., 2020, 142, 7036–7046 CrossRef CAS
- J. Yang, H. Qi, A. Li, X. Liu, X. Yang, S. Zhang, Q. Zhao, Q. Jiang, Y. Su, L. Zhang, J. Li, Z. Tian, W. Liu, A. Wang and T. Zhang, J. Am. Chem. Soc., 2022, 144, 12062–12071 CrossRef CAS
- P. L. Searle, Analyst, 1984, 109, 549–568 RSC
- W. D. Zhang, H. Dong, L. Zhou, H. Xu, H. R. Wang, X. Yan, Y. Jiang, J. Zhang and Z.-G. L. Gu, Appl. Catal., B, 2022, 317, 121750 CrossRef CAS
- J. Wang, T. Feng, J. Chen, V. Ramalingam, Z. Li, D. M. Kabtamu, J. H. He and X. Fang, Nano Energy, 2021, 86, 106088 CrossRef CAS
- P. Gao, Z. H. Xue, S. N. Zhang, D. Xu, G. Y. Zhai, Q. Y. Li, J. S. Chen and X. H. Li, Angew. Chem., Int. Ed., 2021, 133, 20879–20884 CrossRef
- A. C. Nielander, J. M. McEnaney, J. A. Schwalbe, J. G. Baker, S. J. Blair, L. Wang, J. G. Pelton, S. Z. Andersen, K. Enemark-Rasmussen, V. Čolić, S. Yang, S. F. Bent, M. Cargnello, J. Kibsgaard, P. C. K. Vesborg, I. Chorkendorff and T. F. Jaramillo, ACS Catal., 2019, 9, 5797–5802 CrossRef CAS
- K. S. Rajmohan and R. Chetty, ECS Trans., 2014, 59, 397–407 CrossRef CAS
- H. Xu, Y. Ma, J. Chen, W. X. Zhang and J. Yang, Chem. Soc. Rev., 2022, 51, 2710–2758 RSC
- D. Reyter, D. Bélanger and L. Roué, Electrochim. Acta, 2008, 53, 5977–5984 CrossRef CAS
- C. W. Lee, N. H. Cho, K. T. Nam, Y. J. Hwang and B. K. Min, Nat. Commun., 2019, 10, 3919 CrossRef PubMed
- R. Casebolt, K. Levine, J. Suntivich and T. Hanrath, Joule, 2021, 5, 1987–2026 CrossRef CAS
- H. S. Jeon, J. Timoshenko, C. Rettenmaier, A. Herzog, A. Yoon, S. W. Chee, S. Oener, U. Hejral, F. T. Haase and B. Roldan Cuenya, J. Am. Chem. Soc., 2021, 143, 7578–7587 CrossRef CAS
- L. Gao, L. Ding and L. Fan, Electrochim. Acta, 2013, 106, 159–164 CrossRef CAS
- T. Liu, J. Wang, X. Yang and M. Gong, J. Energy Chem., 2021, 59, 69–82 CrossRef CAS
- C. Kim, L. C. Weng and A. T. Bell, ACS Catal., 2020, 10, 12403–12413 CrossRef CAS
- Y. Wang, C. Wang, M. Li, Y. Yu and B. Zhang, Chem. Soc. Rev., 2021, 50, 6720–6733 RSC
- M. Duca and M. T. M. Koper, Energy Environ. Sci., 2012, 5, 9726–9742 RSC
- P. Li, Z. Jin, Z. Fang and G. Yu, Energy Environ. Sci., 2021, 14, 3522–3531 RSC
- Q. Gao, H. S. Pillai, Y. Huang, S. Liu, Q. Mu, X. Han, Z. Yan, H. Zhou, Q. He, H. Xin and H. Zhu, Nat. Commun., 2022, 13, 2338 CrossRef CAS PubMed
- X. Deng, Y. Yang, L. Wang, X.-Z. Fu and J.-L. Luo, Adv. Sci., 2021, 8, 2004523 CrossRef CAS
- O. Q. Carvalho, R. Marks, H. K. K. Nguyen, M. E. Vitale-Sullivan, S. C. Martinez, L. Árnadóttir and K. A. Stoerzinger, J. Am. Chem. Soc., 2022, 144, 14809–14818 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ee02647c |
| ‡ These three authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |