
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBeneficial vs. inhibiting passivation by the native lithium solid electrolyte interphase revealed by electrochemical Li+ exchange†
Gustavo M.
Hobold
 ,
Kyeong-Ho
Kim
and
Betar M.
Gallant
*
,
Kyeong-Ho
Kim
and
Betar M.
Gallant
*
Department of Mechanical Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA. E-mail: bgallant@mit.edu
First published on 6th April 2023
Abstract
Despite being a leading candidate to meet stringent energy targets of Li-ion batteries, the lithium (Li) metal anode has yet to achieve Coulombic efficiency (CE) requirements for long cycle life (>99.9%). These limitations derive from the native solid electrolyte interphase (SEI) which, among multiple functions, stabilizes and protects deposited Li. The SEI also plays a critical role in regulating Li+ exchange between the electrolyte and the electrode, but quantification of this effect has been non-straightforward, and a general relationship between Li+ exchange and CE has not been clearly elucidated to date. Using electrochemical impedance spectroscopy and voltammetry, we report self-consistent Li+ exchange values of native SEIs over a range of relevant electrolytes with CE spanning 78.0% to >99%. CE and its retention at high rates are found to be positively correlated with the rate of SEI Li+ exchange. Additionally, SEI Li+ exchange rates increased during cycling in high-CE electrolytes, in some cases by an order of magnitude to exceed 10 mA cm−2, whereas for low-CE electrolytes they remained low (<1 mA cm−2), revealing a chemistry-dependent picture of SEI evolution with often-complex dynamics. The evolution in Li+ exchange unique to high-CE electrolytes also provides insights into the role and effectiveness of the formation cycle on Cu current collectors upon the first plating step. Altogether, these findings indicate that Li+ exchange governs several key processes related to Li deposition and cycling efficiency. Consequently, its quantification can help to guide future high-CE electrolyte design, particularly targeting high rates (>1 mA cm−2).
Broader contextLithium (Li) metal batteries can in theory upgrade the capacity of today's Li-ion battery anodes by 10-fold, but their commercialization is hindered by low Coulombic efficiency (CE, <99.9%), particularly when high rate charge/discharge is required. Efforts in addressing these challenges have focused on engineering new battery electrolytes capable of generating a native solid electrolyte interphase (SEI) at the Li surface that is sufficiently protective to suppress unwanted side reactions. However, the protection provided by the SEI can also bottleneck and passivate Li against Li+ exchange with the electrolyte, a phenomenon only understood qualitatively and that has still-unclear effects on CE. Here, we make use of electrochemical techniques to precisely quantify Li+ exchange in SEIs spanning a broad range of electrolyte formulations and reveal the relationship between this SEI-derived property and CE. The results unambiguously demonstrate that fast Li+ exchange is a desirable and necessary property of an SEI to minimize its breakage and promote improved cycling, especially when the battery is cycled at high charge/discharge rates. Broadly, our findings indicate that current electrolyte intervention strategies targeting electrolyte design or artificial interface engineering are most effective if they enable fast Li+ exchange. |
Introduction
The lithium (Li) metal anode is the most compelling alternative to today's graphite anodes for meeting stringent gravimetric energy targets (>500 Wh kg−1) in Li-ion batteries, as it allows a theoretical 10-fold upgrade in capacity (3860 mAh g−1vs. 372 mAh g−1) at comparable electrode potential. Unlike graphite, Li reversibility in liquid electrolytes, as quantified by Coulombic efficiency (CE) – the ratio between the stripping and plating capacity onto a non-Li substrate such as Cu – still falls below the >99.9% required for >1000 cycles, although electrolytes exceeding 99% CE for at least a portion of cycle life are increasingly common.1,2 The Li anode also displays rate capability well below the requirements for fast charge (>2 C at >99.9% CE).3 These gaps have motivated extensive efforts towards increasing CE through electrolyte and interface design.4By definition, cycling inefficiencies associated with the Li anode reflect a loss of cyclable Li0 inventory from the cell. These inefficiencies can manifest in two ways: electrolyte reduction with concerted consumption of Li0 to form the solid electrolyte interphase (SEI), and as disconnected metallic Li0 that becomes electronically stranded during cycling.5–7 Typically, capacity loss due to SEI formation, rather than to metallic Li0, predominates at higher CE where deposition is more uniform and less porous.5,8 Once formed, an imperfect SEI may invite further non-uniform Li/plating stripping2 and SEI breakage, which necessitates its sustained reformation over subsequent cycles. Thus, the Li-electrolyte interface that forms spontaneously on the electrode as the cell cycles, henceforth referred to as “native SEI”, not only inherits its composition from the electrolyte, but can also develop morphological features that depend on the cell's cycling history.
Critically, the SEI also regulates transport of Li+ ions between electrolyte and electrode. As such, the presence of an SEI can have profound effects on transport and kinetics at the interface. For instance, cyclic voltammetry on microelectrodes at ultrafast scan rates (e.g., > 10 V s−1, Fig. 1, left), which are fast enough to continuously disrupt the native SEI, have found that the intrinsic kinetics of Li plating/stripping, as parametrized by a formal exchange current j0,9 can be as high as ∼40 mA cm−2 in carbonate-based electrolytes.10 However, chemical Li+ exchange rates, as determined by nuclear magnetic resonance (NMR) measurements on a pristine SEI soaked in similar carbonate electrolytes, have been reported to be substantially lower at ∼0.5–3 mA cm−2.11 These lower values correspond to charge-transfer resistances up to ∼100-fold10 higher compared to an SEI-less interface imposed by ultrafast scan rates, indicating that the SEI can substantially bottleneck Li+ exchange.
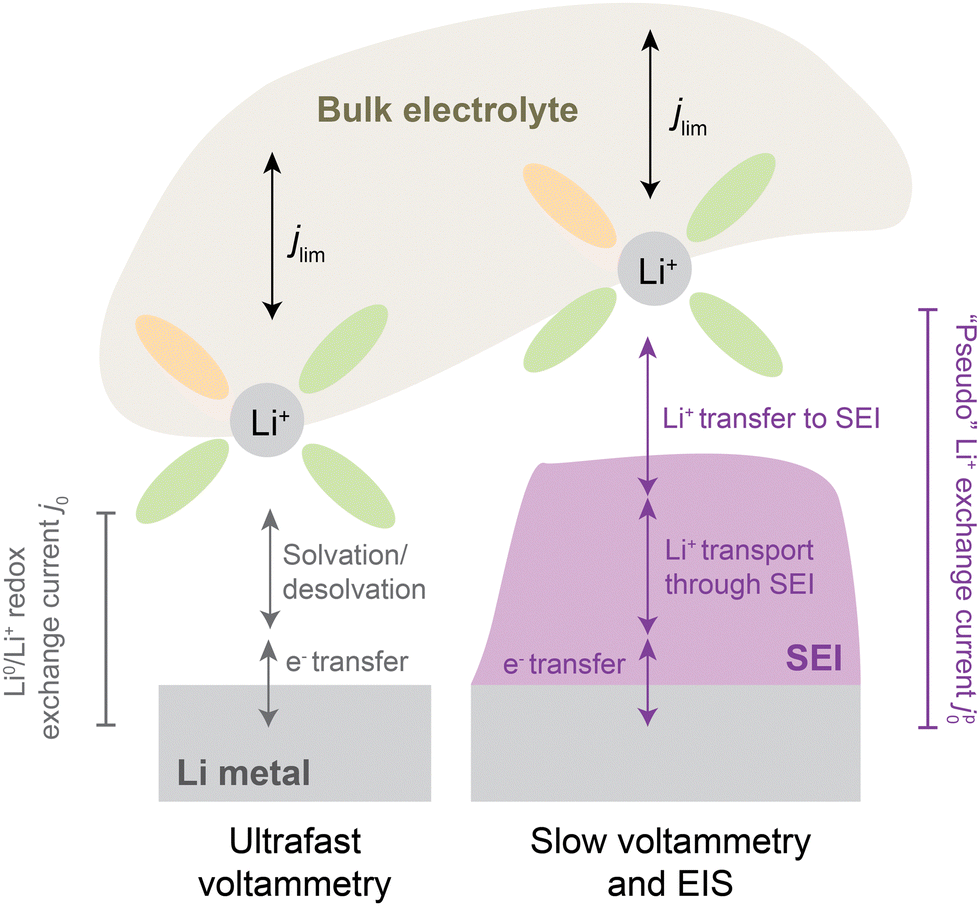 | ||
| Fig. 1 Illustration of processes involved in Li0/Li+ redox from bulk electrolyte to the Li metal electrode, without (left) and with (right) an SEI present. At left, j0 refers to a classically defined9 exchange current density [mA cm−2] that reflects the intrinsic kinetics of electron transfer coupled to solvation and ion transfer of Li+ to form Li0. It is probed experimentally by ultrafast voltammetry, typically using microelectrodes. With an SEI present, electron transfer, solvation and Li+ transfer become impeded by an SEI, which itself has electrolyte-dependent properties. Experiments under such conditions report on an apparent or “pseudo”-exchange current density regulated by an SEI, herein denoted jp0. The above schematic assumes negligible electronic conductivity throughout the SEI and makes no assumptions about its microstructure or charge-transport mechanisms, other than being dominated by Li+ as charge carrier. | ||
Prior attempts to quantify j0 electrochemically under realistic battery operating conditions, i.e., with an SEI present, have focused largely on carbonate-based electrolytes (Table S1, ESI†). In those studies, numerical values of j0 were inconsistent. For example, reported values of j0 varied by over one order of magnitude for 1 M LiClO4 in propylene carbonate (PC) across different studies,12,13 which may relate in part to how the Li electrode was prepared and/or aged, as well as which experimental technique was used. Despite some scatter in the data, however, meaningful differences across electrolytes can begin to be discerned. j0 was reported to range between 0.1–0.3 mA cm−2 (0.1 M)14,15 or 0.95–1.8 mA cm−2 (1 M)14,15 for LiClO4 in PC, indicating that j0 can be sensitive to salt concentration. Similarly, j0 was found to have a lower range in 1 M LiPF6 PC (0.3–2.2 mA cm−2)16,17 than 1 M LiBF4 PC (0.5–7.5 mA cm−2),16,17 suggesting that the anion also influences j0. Exchange current values have also been reported to a much more limited extent in ethers. In those solvents, j0 again exhibited sensitivity to the salt anion, ranging between 1.4–3.6 mA cm−2 for 1 M LiPF6, LiClO4, LiBF4 and LiAsF6 in 2-methyl-tetrahydrofuran (2-Me-THF) in increasing order.17 Similarly, the relative volume fraction of ether blends can affect j0, reported to range between 2.5–7 mA cm−2 for 1 M LiAsF6 in blends of diethyl ether (DEE) and tetrahydrofuran (THF).18 Despite these important fundamental efforts, the values reported by different studies are still significantly different from each other, even when reported for the same class of electrolytes, as highlighted in Fig. S1a–c, ESI.† Such variation makes it challenging to ascertain which Li+ exchange values are applicable to practical Li battery conditions.
Whether any clear and universal relationship between j0 and CE exists has remained even less certain. This is largely because comprehensive data for j0, as well as concurrently measured CE, are missing across a wide range of electrolytes. When existing, reports on the effect of j0 on CE are often based on limited datasets composed almost exclusively of isolated electrolyte systems. Moreover, prior reports have presented conflicting interpretations of whether higher j0 is beneficial19–22 (hence desired) or detrimental17,23–25 (hence to be avoided) for promoting Li reversibility, altogether leading to contrasting conclusions on the role of Li+ exchange (e.g., Fig. S1d, ESI†). For example, experiments in tetraethylene glycol dimethyl ether (TEGDME)-based electrolytes led to a conclusion that higher j0 leads to lower CE.24 In contrast, other studies have reported that incorporation of additives such as FEC19 or SiCl421 into a carbonate electrolyte increases both j0 and CE. Similarly, NMR-based observations have reported that the beneficial role of additives such as FEC or LiNO3 may derive from faster chemical exchange of Li+ in the SEI.11,26 As noted, one challenge in generalizing these observations arises, in part, from variable measurement and analysis protocols, e.g., NMR,11,26 microelectrode17,23,25vs. coin cells,19–22 and/or slow (<100 mV s−1)17,19,21,22vs. fast (>100 mV s−1)23,25 scan rates (Table S1 and Fig. S1, ESI†) used by different groups.
In this context, there is a significant need for quantification of Li+ exchange under practical battery conditions and elucidation of its dependence on electrolyte chemistry, testing protocol, and rate. In this study, Li+ exchange at the Li anode is systematically quantified using cyclic voltammetry (CV) at slow scan rates (1 mV s−1) and electrochemical impedance spectroscopy (EIS), both of which intentionally allow an SEI to develop natively (Fig. 1, right). In order to span a diverse set of SEI chemistries of interest, we employ a selection of historically-relevant and more modern, high-performance electrolytes. We interpret the measured exchange rates in the framework of a “pseudo”-exchange current, jp0, that represents the total rate of Li+ exchange on the electrode and acknowledges the significance of changing surface areas of Li deposits during cycling. Indeed, the pseudo-exchange current jp0 reveals a transient evolution of Li+ exchange that helps explain observed historical discrepancies in exchange current reported by different studies. Importantly, jp0 is found to vary considerably across electrolytes, with an unambiguous positive correlation with CE consistent across different measurement protocols. In addition, tracking of jp0 evolution upon cycling reveals unexpected dynamic behaviors that distinguish low-CE from high-CE electrolytes and allow for new insights into the evolution of electrochemically-active surface area and SEI quality upon cycling. We anticipate that these findings can provide a framing for future development of high-rate and high-CE electrolytes by considering how to maximize the SEI rates of Li+ exchange.
Measuring Li+ exchange through impedance spectroscopy
Representative electrolytes were selected for analysis based on their historical success in improving the Li CE, and as a result span a range of reported CE values.4 The electrolytes (Fig. 2a) included 1.5 M LiAsF6 in 2-Me-THF (CE = 78.0%); conventional and fluorinated carbonates of 1 M LiClO4 in PC (81.9%), 1 M LiPF6 in EC/DEC (93.8%), 1 M LiFSI in FEC (95.3%) or 1 M LiPF6 in EC/DMC (96.9%); and higher-concentration fluorinated carbonates of 2 M and 7 M LiFSI in FEC (97.5% and 98.2%, respectively). Finally, two additive-containing electrolytes were examined: 1 M LiTFSI in DOL/DME and 2 M LiFSI/1 M LiTFSI in DOL/DME, each containing 3 wt% LiNO3 (99.0% and 99.3%, respectively). Altogether, these choices of electrolyte avoid biases that can emerge from isolated systems composed only of specific solvents (e.g., ethers) or salts (e.g., LiFSI), and instead incorporate a large variety of compositions, with 8 different solvents and 6 different salts. All CE values were measured using a pre-formation/reservoir cycling protocol following Adams et al.27 as described further in the Experimental methods (Fig. S2, ESI†). These values generally agree well with prior literature reports (Table S2 and Fig. S3, ESI†), with small variations likely explained by the difference in CE protocols between the original studies and the methodology used herein.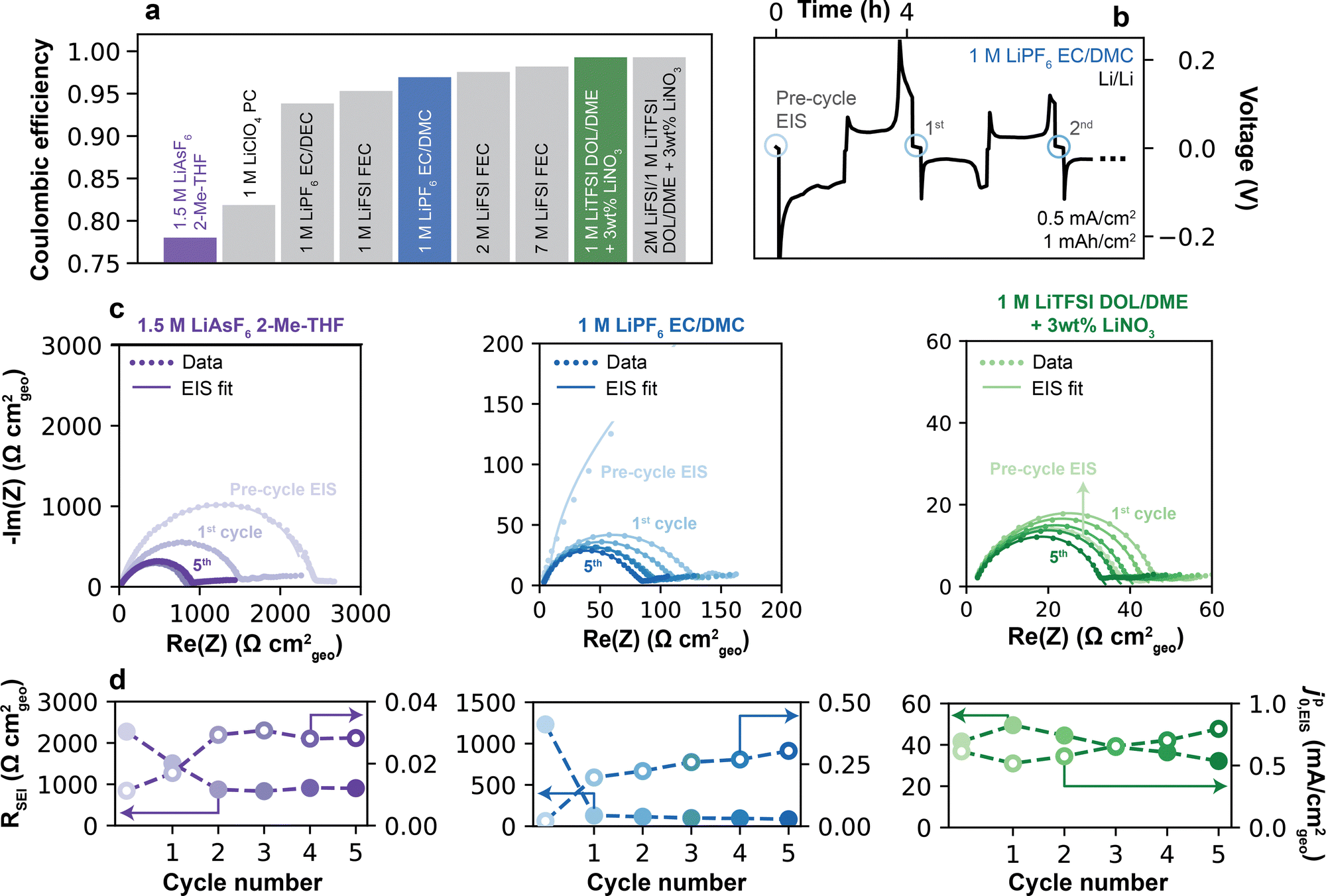 | ||
| Fig. 2 Measuring Li+ exchange by electrochemical impedance spectroscopy. (a) CE of electrolytes considered in this study, measured at 0.5 mA cm−2 using a pre-formation/reservoir cycling protocol (Fig. S2, ESI†). (b) Representative galvanostatic plating/stripping cycles in a Li/Li cell in 1 M LiPF6 EC/DMC at 0.5 mA cm−2 and 1 mAh cm−2 plating capacity. Blue circles indicate the timesteps at which EIS was collected following a 5 min. rest at OCV. (c) Nyquist plots of Li/Li cells collected pre- and post-cycling in three representative electrolytes: 1.5 M LiAsF6 2-Me-THF, 1 M LiPF6 EC/DMC, and 1 M LiTFSI DOL/DME + 3 wt% LiNO3. Markers indicate raw data, while solid lines represent fits to the high-frequency data points (>20 Hz) using an equivalent RC-Warburg circuit. (d) SEI resistance and pseudo-exchange current density, jp0,EIS, were obtained from fitting EIS spectra in (c). | ||
Before examining the proposition that SEI can be a relevant factor that limits, and thus determines, electrochemical measures of Li+ exchange, we first examined possible alternative relationships between CE and two key bulk electrolyte properties. First, Li+ diffusivity in the bulk electrolyte (D), which governs the mass transport limiting current (jlim, Fig. 1) upon Li plating,2,9,28 was measured using diffusion-ordered spectroscopy (DOSY) NMR. Values of D ranged from 8.90 × 10−8 cm2 s−1 for 7 M LiFSI FEC to 3.42 × 10−6 cm2 s−1 for 1 M LiTFSI DOL/DME + 3 wt% LiNO3 (Fig. S4, ESI†). However, no clear relationship with CE was observed. Additionally, we also examined the 7Li chemical shift in liquid NMR, which has been shown to reflect the degree of contact-ion pairing when compared between electrolytes of alike solvent (e.g., varying anion chemistry and/or concentration in the same solvent).29 Under such circumstances, 7Li chemical shift can correlate with CE for beneficial anions30 because contact-ion pairing affects the SEI composition and properties. Regardless, when compared over electrolytes of dissimilar composition, 7Li chemical shift did not reveal any correlation with CE (Fig. S4, ESI†) because the chemical environment of the Li+ ion is more diverse across electrolytes, making chemical shift unrepresentative of SEI composition. These observations illustrate how intrinsic properties of the bulk electrolyte are not universally predictive of CE. Rather, SEI properties may be hypothesized to serve as more general and reliable descriptors.
Considering the presence of the SEI, Li+ exchange currents were first measured using EIS analysis in symmetric Li/Li cells. Fig. 2b shows a typical protocol. Following cell assembly, an initial impedance spectrum, denoted by an open blue circle, was conducted in the pristine state (“Pre-cycle EIS”) after a 5 h rest at OCV, sufficient to establish an intact SEI. This interphase includes artifacts from the preparation and handling of the electrodes.31 Next, a complete plating/stripping cycle was conducted (0.5 mA cm−2, 1 mA h cm−2). Following a 5 min. rest at OCV, which is sufficient to re-form a native SEI32 but short enough to avoid substantial aging effects (Fig. S5, ESI†)31,33 and CE losses (Fig. S6, ESI†), a second impedance spectrum was collected. This process was repeated subsequently for additional plating/stripping + EIS cycles. Fig. 2c shows EIS data over five such cycles for three representative electrolytes (1.5 M LiAsF6 in 2-Me-THF, 1 M LiPF6 in EC/DMC, and 1 M LiTFSI in DOL/DME + 3% LiNO3) corresponding to the colored bars in Fig. 2a, with the comprehensive data set for all electrolytes shown in the ESI† (Fig. S7). In general, the EIS responses took the form of a single, high-frequency semi-circle with a lower-frequency tail. The semi-circle was fitted by a simple equivalent circuit34 comprising a parallel RC element associated with charge-transfer and a capacitance-Warburg impedance element associated with Li+ diffusion across the interphase (see ESI† Note S1 for details).
An exchange current value was extracted from the EIS fitting by jp0,EIS= kT/(eRSEI), where k is the Boltzmann constant, T is temperature (297 K), e is the elementary charge and RSEI is the total SEI resistance. The discussion will first address the high frequency semi-circle from the 20 kHz to 20 Hz region of the EIS spectra (Experimental methods and ESI† Note S1), and will later examine qualitative features observed in the low frequency region (<20 Hz). The parameters RSEI [Ω cm2geo] and jp0,EIS [mA cm−2geo, hereafter also denoted simply as mA cm−2] are normalized to the geometric surface area of the Li electrode, given that the true surface area of plated Li is unknown and may change over the electrode's use. To emphasize this point, we employ the superscript “p” to denote a “pseudo”-exchange current. As we will show, jp0,EIS still has significant physical meaning and provides insights into the properties and behavior of the SEI.
For the majority of studied electrolytes, the pre-cycle EIS spectrum was largest in magnitude. This is attributable to the presence of a foreign surface film formed during cell preparation, which can occur even in a clean glovebox environment.35,36 Consequently, the measured impedance from an as-assembled cell is unlikely to fully represent a dynamically-formed SEI relevant to sustained use of the Li anode. Cycling was therefore next performed in order to expose fresh Li, which inevitably led to an increase in the surface area of the electrode. Subsequent plating/stripping + EIS iterations consistently yielded smaller semi-circles, attributed to breakage of the original surface film and formation of the native SEI. Notably, after 5 cycles the RSEI magnitudes varied significantly across electrolytes (Fig. 2d), being highest for 1.5 M LiAsF6 in 2-Me-THF (825.8 Ω cm2 on cycle 5), followed by 1 M LiPF6 in EC/DMC (82.6 Ω cm2) and 1 M LiTFSI DOL/DME + 3% LiNO3 (31.3 Ω cm2). Correspondingly, the jp0,EIS values across these three electrolytes showed an increasing trend of 0.03, 0.30 and 0.79 mA cm−2, respectively.
Fig. 3a shows the high-frequency jp0,EIS measured over 50 cycles for each electrolyte. Over this longer cycling regime, two distinct behaviors in jp0,EIS evolution were observed, which we denote by “Type 1” and “Type 2”. In Type 1 electrolytes, jp0,EIS remained reasonably stable over 50 cycles, as with 1.5 M LiAsF6 2-Me-THF, 1 M LiClO4 PC, 1 M LiPF6 EC/DEC, 1 M LiFSI FEC, and 1 M LiPF6 EC/DMC (data for an additional Type 1 electrolyte, 1 M LiFSI PC, is shown in Fig. S8, ESI†). Owing to the largely unchanging jp0,EIS, a single representative value in each electrolyte can be identified. In Type 2 electrolytes, however, a substantial increase in jp0,EIS was observed with cycling, as with 2 M LiFSI FEC, 7 M LiFSI FEC, 1 M LiTFSI DOL/DME + 3 wt% LiNO3, and 2 M LiFSI 1 M LiTFSI DOL/DME + 3 wt% LiNO3. In some cases, jp0,EIS increased by up to two orders of magnitude, e.g., 2 M LiFSI FEC increased from 0.25 to >25 mA cm−2; however, in no case did jp0,EIS decrease. These variations in jp0,EIS are summarized in Fig. 3b, where the average per-cycle change in jp0,EIS highlights the increase seen by Type 2 electrolytes, but not by their Type 1 counterparts. Because of these changes, it is challenging to identify a singular representative value of jp0,EIS in those systems. Interestingly, Type 1 electrolytes corresponded to those in the lower CE range (Fig. S8b and S9, ESI†), which have porous and high aspect ratio Li deposition morphology (Fig. 3c), namely 1.5 M LiAsF6 2-Me-THF (CE = 78.9%) to 1 M LiPF6 EC/DMC (CE = 96.9%). Owing to these Li morphologies, CE in this range can also be influenced by formation of inactive Li0.5 Past this threshold, CE is dominated by the SEI,5 and all electrolytes displaying Type 2 (increasing jp0,EIS) behavior exhibited compact Li deposition morphology (Fig. 3c). We note that jp0,EIS for 1 M LiPF6 EC/DMC converges to 0.45 ± 0.04 mA cm−2, which is in excellent agreement with the steady-state values obtained for chemical Li+ exchange measured recently via solid-state NMR (0.47 mA cm−2) on the native SEI in the same electrolyte.11 The converged values obtained in other carbonate electrolytes (for instance, 0.30 mA cm−2 for 1 M LiClO4 PC) were also within the same order of magnitude (typically <1 mA cm−2) as values noted in earlier works by EIS (Table S1, ESI†).
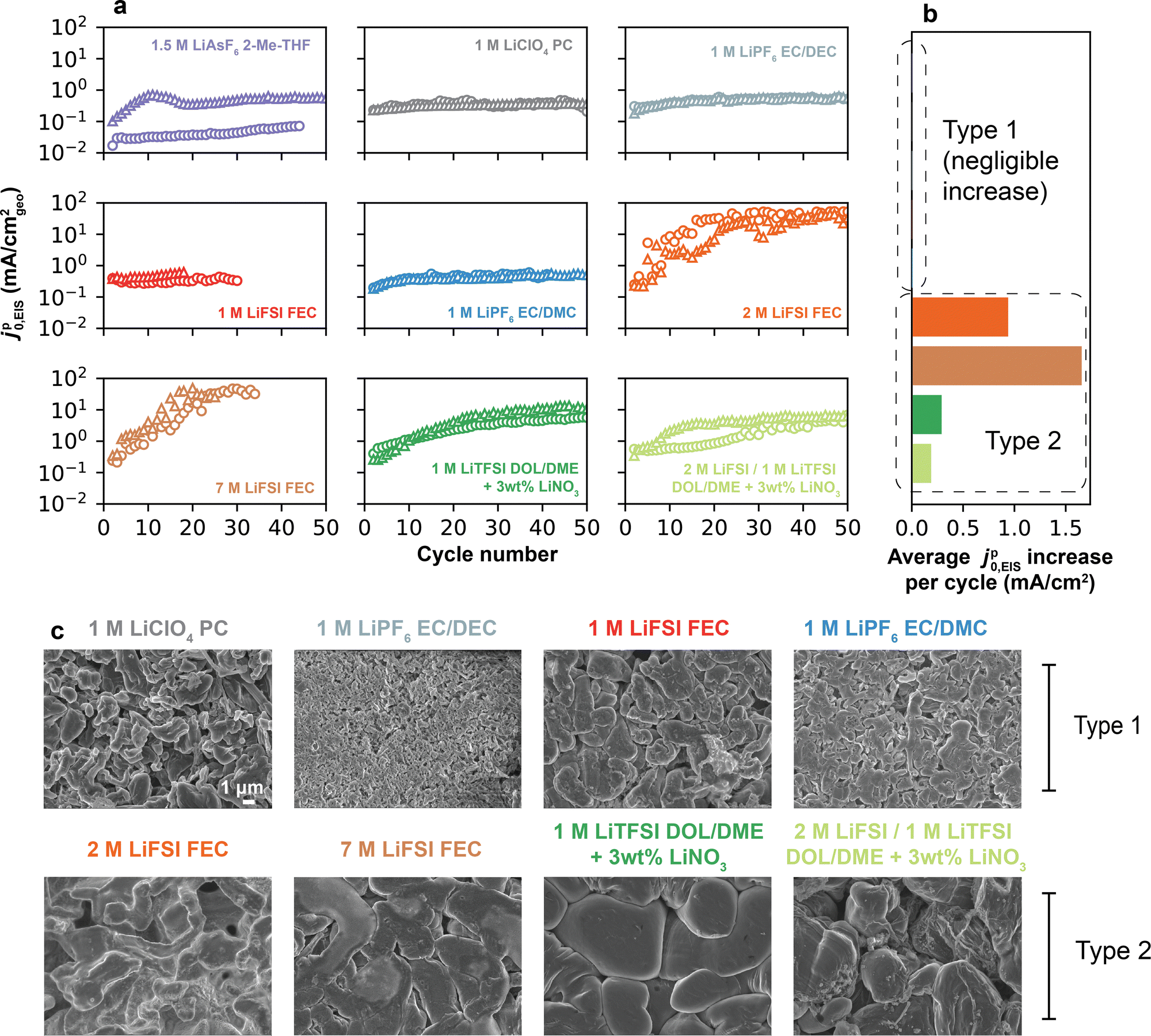 | ||
| Fig. 3 Cycle-dependent Li+ exchange on Li anodes. (a) jp0,EIS fitted from EIS spectra as a function of galvanostatic cycle number (1 mAh cm−2 plating/stripping at 0.5 mA cm−2) in different electrolytes. Data were collected up to 50 cycles, or until apparent cell short-circuit (e.g., 1 M LiFSI and 7 M LiFSI in FEC). Triangular and circular markers denote replicates. Poor reproducibility for 1.5 M LiAsF6 2-Me-THF was found to be due to its low CE and chemical instability between 2-Me-THF and LiAsF6. (b) Average per-cycle variation in jp0,EIS calculated from data in (a), distinguishing Type 1 from Type 2 electrolytes. (c) SEM images of the plated Li morphology acquired after 5 cycles in each electrolyte. | ||
Changes in the above-measured jp0,EIS over cycling and their variation across electrolytes can have two origins: (1) an increase in the intrinsic rate of Li+ exchange of the SEI, such as from differences in chemical composition and its evolution over cycling; (2) an increase in the electrochemically-active area for Li+/Li0 redox due to surface roughening. Both may occur in practice. While (1) is challenging to probe, and will be analyzed later through voltammetry, (2) is undoubtedly a significant factor given the nonuniform nature of Li deposition. As observed by Lee et al., low-CE electrolytes display a several-fold higher electrolyte-contact surface area compared to high-CE electrolytes after just a single cycle,37 leading to higher porosity as well as formation of inactive Li0 and SEI Li+.5 Curiously, the absence of any decreasing jp0,EIS values in Fig. 3a suggests that the accumulation of these inactive materials does not suppress total Li+ exchange rates, even in low-CE electrolytes. Additionally, a qualitative comparison between scanning electron microscopy (SEM) images of plated Li electrodes after 5 cycles between Type 1 and Type 2 electrolytes confirms that the Type 1 electrolytes display higher surface area and more irregular deposits (Fig. 3c).38 Thus, the higher degree of porosity and surface area in the lower-CE Type 1 electrolytes do not translate into an increase in the rate of Li+ exchange upon cycling. This observation stands in contrast to higher-CE Type 2 electrolytes which, despite developing less SEI over a comparable number of cycles, display significant increases in total Li+ exchange rates. In other words, unlike in lower-CE (Type 1) electrolytes, the additional surface created in higher-CE (Type 2) electrolytes is active, as it allows Li+ exchange between the electrolyte and the underlying Li metal even if it forms to a comparatively lesser extent.
We further explored whether the high-frequency Li+ exchange continued to increase unabatedly in high-CE electrolytes. Interestingly, jp0,EIS for the DOL/DME-based electrolytes (exhibiting Type 2 behavior) further increased for approximately ten additional cycles, but did eventually stabilize around 60 cycles, e.g., 8.3 mA cm−2 for 2 M LiFSI 1 M LiTFSI DOL/DME + 3 wt% LiNO3 and 5.7 mA cm−2 for 1 M LiTFSI DOL/DME + 3 wt% LiNO3 (Fig. 4a). This stable value was, however, much higher compared to low-CE electrolytes (e.g., 0.39 mA cm−2 for 1 M LiClO4 PC, Fig. 4a). Although the underlying reasons for this behavior are not fully clear, a possible explanation is as follows (Fig. 4b). In low-CE electrolytes, the residual SEI has low Li+ exchange values, thus provides little advantage for preferential nucleation of freshly-deposited Li in subsequent cycles. Because the SEI is not effectively re-utilized, subsequent Li plating requires re-establishment of the entirety of the SEI on each cycle, leading to lower CE. This regrowth process forms Li deposits and SEI to a similar extent cycle-to-cycle for a fixed plating/stripping capacity. In contrast, in high-CE electrolytes, SEI residuals from previous cycles may remain active for Li plating, and can be built-upon; hence, each cycle is history-dependent. Indeed, Fig. 4c shows that the Li morphology of Type 2 electrolytes remains compact and electronically-percolated even after extended cycling, whereas Type 1 electrolytes continue to develop highly porous Li microstructures. These observations are consistent with cryo-EM evidence that demonstrate substantial necking and loss of active deposits upon stripping in low-CE electrolytes, whereas high-CE electrolytes show reusable SEI “scaffolds” or “exoskeletons” that can be refilled on cycling.39,40 The formation of these SEI structures has been demonstrated to be assisted by beneficial electrolyte components such as LiNO3,40 which lead to fast chemical exchange of Li+.26 We hypothesize that the eventual stabilization of Li+ exchange in the high-CE electrolytes reflects a point at which the growth of new SEI scaffolds has completed, possibly due to their percolation throughout the electrode. Overall, these findings indicate that “passivation”, a commonly used term to describe the function of an SEI,6,33,41,42 has substantially different meaning depending on the system: while all SEIs at least minimally passivate Li against chemical reactivity with the electrolyte, some SEIs are inhibiting with respect to Li+ exchange specifically, whereas others permit it.
 | ||
| Fig. 4 Li+ exchange over extended galvanostatic cycling. (a) jp0,EIS as a function of cycle number of select low- and high-CE electrolytes, demonstrating eventual stabilization of Li+ exchange for the latter. (b) Illustration of a possible mechanism consistent with the observed dynamically-increasing Li+ exchange values in high-CE electrolytes vs. stability of these values in low-CE electrolytes. Bold outlines on deposits denote new SEI formation during the indicated cycle. (c) SEM images of the plated Li morphology acquired after 5 and 25 cycles in select Type 1 and Type 2 electrolytes. (d-e) Evolution of the EIS spectra in (d) Type 1 (1 M LiClO4 PC) and (e) Type 2 (2 M LiFSI/1 M LiTFSI DOL/DME + 3 wt% LiNO3) electrolytes, highlighting the emergence of a low frequency tail in the latter. (f) Li+ exchange rates calculated for the Type 1 and Type 2 electrolyte, considering the effect of the high-CE electrolyte's low frequency tail. | ||
Distinctions were also observed in the qualitative features of the impedance responses between Type 1 and Type 2 electrolytes over cycling, particularly at low frequencies. In Type 1 electrolytes, the EIS spectra remained largely unchanged during cycling, consistent with their stable Li+ exchange rates, and persisted as a single semi-circle (e.g., 1 M LiClO4 PC, Fig. 4d), making the high-frequency fitting descriptive over extended cycling conditions. Type 2 electrolytes, on the other hand, displayed a much more dynamic impedance response as shown in Fig. 4e. In initial cycling stages, their spectra showed the expected high frequency semi-circle with a small low frequency tail. In later cycles, this inner semi-circle reduced significantly in magnitude, underlying the increase of Li+ exchange in Type 2 electrolytes discussed so far. Additionally, the original low-frequency tail developed into a larger, distinct, second semi-circle-like feature. When accounting for the tail growth, jp0,EIS was lower than if this feature was not considered (Fig. 4f and Fig. S10, ESI†), indicating that low frequency features may hinder Li+ exchange through an additional mechanism as they develop, partly countering the decreasing compact SEI impedance at high frequency. These low frequency features are often associated with a secondary effect related to tortuous Li+ diffusion in the electrolyte through porous electrodes,34,43,44 which is not directly derived from SEI chemistry as is the higher-frequency semi-circle. As seen in Fig. 3c, the morphology of plated Li in Type 2 electrolytes is porous yet compact enough that Li+ transport through its pores may be constricted, thus possibly underlying formation of the low frequency tail. Through more accurate EIS modeling and impedance imaging45 in future studies, these features may reveal more details of the morphological evolution of the SEI.
Determining Li+ exchange by cyclic voltammetry
Cyclic voltammetry (CV) measurements were performed on Cu/Li cells in order to measure SEI Li+ exchange under conditions closer to the initial SEI formation not accessible by the previously-discussed EIS protocol. CV measurements typically cycle a smaller amount of Li per scan (<1 mA h cm−2, Fig. S11, ESI†) compared to the galvanostatic cycles performed for EIS. More importantly, CVs also allow full stripping of the underlying active and electronically-percolated Li at the end of each scan, leaving only the SEI behind. Altogether, these features unique to CVs decrease the effects originating from the galvanostatic cycling relating to the increase in active surface area in Li/Li cells, and thus offer a distinct way of measuring SEI Li+ exchange. In this context, Li+ exchange can be determined from CV during a plating/stripping step by evaluating the current–voltage slope around the Li0/Li+ equilibrium potential.9 For the voltammetry analysis presented herein, coin cells were used to best capture conditions in a typical Li battery with limited electrolyte volume (100 μL) and also to provide direct correspondence with the conditions used for EIS. In the adopted protocol (Fig. 5a), 11 continuous CV cycles were conducted in Cu/Li coin cells at 1 mV s−1 between −0.2 V and 1 V, with full stripping of Cu occurring on the end of each sweep. A single CV scan provides one data point for pseudo-Li+ exchange current, which we here term jp0,CV (ESI† Note S2). To further compare values on a freshly deposited Li reservoir, 1 mAh cm−2 of Li was next galvanostatically plated at 0.5 mA cm−2, followed by another set of 11 CV cycles from −0.2 V to 0.2 V. Finally, a full galvanostatic stripping step was conducted to 1 V to yield back a formed Cu current collector, followed by a final set of 11 CVs. An extended analysis of jp0,CV over a larger number of CV scans and galvanostatic cycles will be discussed later.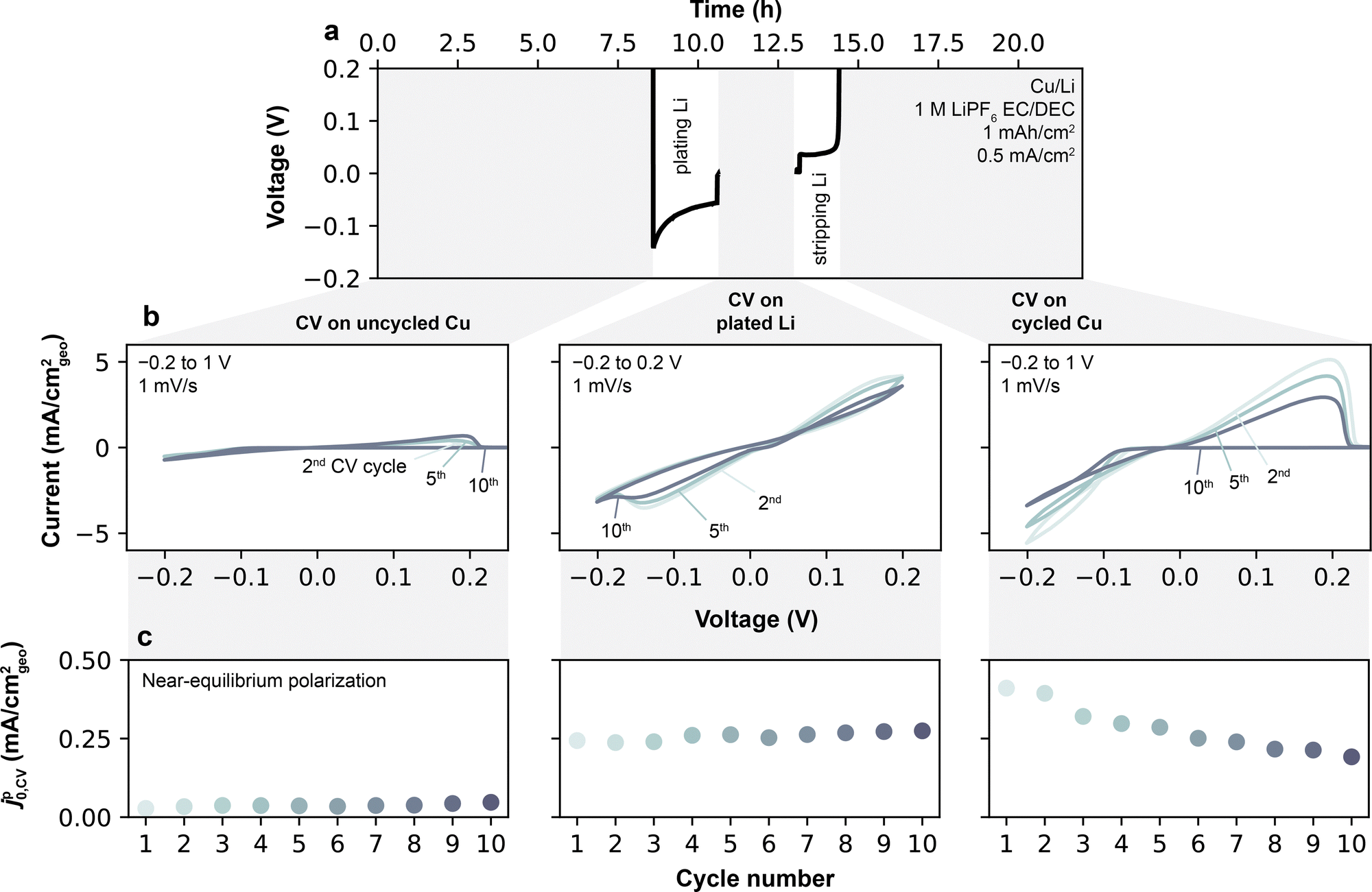 | ||
| Fig. 5 Substrate-dependent cyclic voltammetry and extracted jp0,CV of Li plating and stripping. (a) Procedure for obtaining jp0,CV as a function of electrode condition (on Cu or plated Li), shown for an exemplar electrolyte, 1 M LiPF6 EC/DEC. The shaded gray regions denote periods of continuous CV scans. (b) Representative CV sweeps, from which jp0,CV was obtained on each reverse scan. Following such CV scans on initially-pristine Cu, a Li reservoir of 1 mA h cm−2 was plated galvanostatically as shown in (a), and CV cycling continued (second gray region and middle column). After 11 such CV cycles, Li was fully stripped, and an additional set of 11 CV cycles was conducted on the remaining formed Cu substrate. (c) jp0,CV obtained from fitting CV data to the linear current–potential relationship near equilibrium as a function of CV cycle number. | ||
The current–voltage curves obtained on pristine Cu (Fig. 5b) in an exemplar 1 M LiPF6 EC/DEC electrolyte show a typical behavior of metal deposition onto an inert substrate. On the forward scan, in which Li is first plated on Cu, an overpotential of ∼100 mV is required to initiate Li plating on all cycles. On the reverse scan, Li continued to be plated on Cu until voltage exceeded the equilibrium potential (E0, the voltage at which j = 0), after which Li was stripped from Cu. Under these conditions, the Cu/Li cell behaves effectively as a symmetric cell, owing to the Li previously plated during the forward scan, and hence displays a symmetric potential-current relationship. At low plating/stripping overpotential windows of the reverse scan (typically <20 mV, see Fig. S12, ESI†), the magnitudes of the anodic and cathodic currents are small (<0.2 mA cm−2), and the current exhibits a linear response to overpotential. The slope of this response is proportional to jp0,CV by j = jp0,CV(F/RT)Ew,corrected,9 where Ew,corrected is the measured potential corrected for ohmic losses and E0 (Experimental methods, ESI† Note S2). Fitting jp0,CV at the linear limit avoids reliance on more complex kinetic models such as Tafel extrapolation, Butler–Volmer or Marcus–Hush formalisms that may be unphysical for interphase-dominated processes,9 and avoids features that may appear at high overpotentials due to asymmetries in the quantities of excess Li between each electrode.
Li+ exchange for the pristine, Li-plated and fully-stripped Cu current collectors determined by this method are shown in Fig. 5c. In 1 M LiPF6 EC/DEC, jp0,CV is low for the pristine Cu electrode (0.02 mA cm−2), but increases substantially and stabilizes to an average of 0.26 mA cm−2 after Li is plated on the electrode. After Li is stripped away, jp0,CV remains more similar to that of the Li-plated electrode than the pristine Cu electrode, i.e. is initially 0.40 mA cm−2 in the 1st CV cycle before stabilizing around 0.19 mA cm−2 in the 11th cycle. Altogether, these results confirm that Li plating behaviors are strongly dependent on surface conditioning and its history. Thus, in the following, we first examine the cross-electrolyte results on plated Li (middle column of Fig. 5b) for direct comparison with the EIS data, and later return to compare the differences between uncycled and cycled Cu.
Comparable voltammetry analysis on plated Li was performed in all other electrolytes. Fig. 6a shows Tafel plots for the three electrolytes highlighted in Fig. 2, where a steeper j–Ew,corrected relationship near equilibrium indicates higher jp0,CV. For all electrolytes, the j–Ew,corrected relationship shows typical Tafel behavior at high overpotentials, i.e., linear log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) j–Ew,corrected behavior as seen in Fig. 6a. This behavior could be fitted well by Butler–Volmer9 and Marcus–Hush–Chidsey46 models at potentials below 100 mV (Fig. S13, ESI†), even though such models do not consider an SEI. However, it is unclear at present whether this agreement carries any physical meaning.
j–Ew,corrected behavior as seen in Fig. 6a. This behavior could be fitted well by Butler–Volmer9 and Marcus–Hush–Chidsey46 models at potentials below 100 mV (Fig. S13, ESI†), even though such models do not consider an SEI. However, it is unclear at present whether this agreement carries any physical meaning.
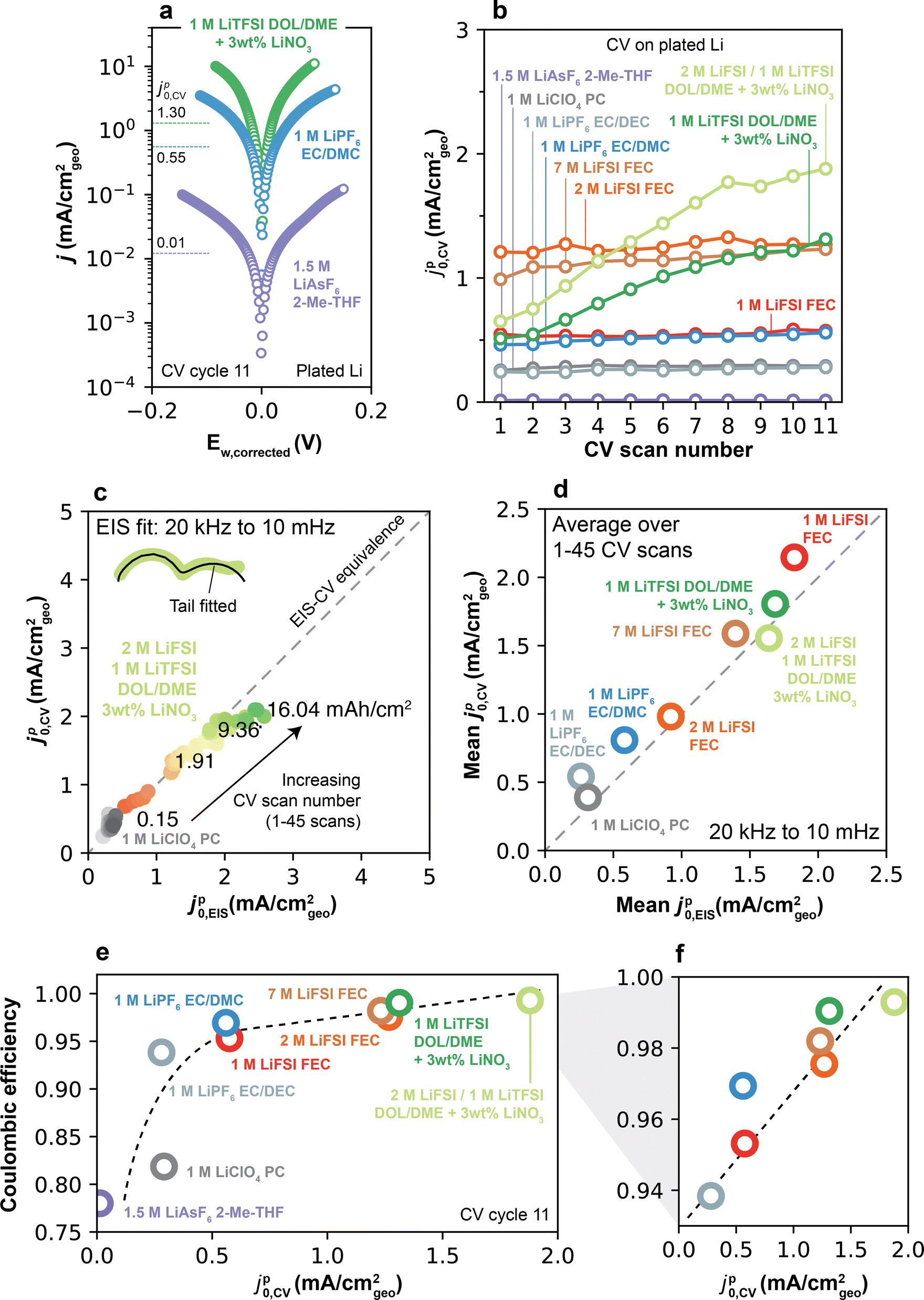 | ||
| Fig. 6 Electrolyte-dependent Li+ exchange captured by voltammetry on plated Li. (a) Tafel plot of select electrolytes measured by CV at 1 mV s−1 on 1 mAh cm−2 of plated Li. Annotated values indicate the jp0,CV value measured from the corresponding Tafel plot. (b) jp0,CV as a function of CV scan number on plated Li for all electrolytes. (c) Equivalence between Li+ exchange measured by CV and EIS, plotted as jp0,CVvs. jp0,EIS measured in the same Li/Li cell at the same cycling history and cycled capacities. jp0,EIS is calculated using the full frequency spectrum (from 20 kHz to 10 mHz). (d) Mean jp0,CV and jp0,EIS shown for all electrolytes and averaged over CV scans 1–45, measured as in (c). See ESI† Note S3 for all details on the experiment relating to (c) and (d). (e) CE–jp0,CV relationship of the electrolytes considered in this study, where jp0,CV was obtained from the 11th scan number in (b), and CE was measured by the PNNL protocol (Fig. 2a). (f) Magnification of (e) in a higher CE range. Dashed lines in (e–f) are included for visual aid and are not fit to the data. | ||
The evolution of jp0,CV as a function of CV scan number across all electrolytes is shown in Fig. 6b. First-scan values of jp0,CV ranged from very low (0.01 mA cm−2 for 1.5 M LiAsF6 2-Me-THF) to 1.21 mA cm−2 in 2 M LiFSI FEC. In the low-CE electrolytes, jp0,CV remained largely constant as a function of CV scan number. In comparison, higher-CE electrolytes displayed a small but persistent increase in jp0,CV after each CV scan (Fig. S14, ESI†), consistent with EIS findings (Fig. 3). Among Type 2 electrolytes, DOL/DME-based systems displayed the highest increase in jp0,CV, contrasting with jp0,EIS, in which 2 M and 7 M LiFSI FEC electrolytes showed the highest increase in jp0,EIS. These differences are attributable to the different cycling capacities accessed by EIS and CV experiments, which can lead to distinct active surface areas across these experiments.
To more systematically evaluate this point, additional experiments were designed to measure jp0,CV and jp0,EIS in the same Li/Li cell, ensuring measurements were taken under equivalent cycled capacities (see ESI† Note S3 for detailed methods). Under these conditions, exchange values from both methods display excellent quantitative correspondence (Fig. 6c and d for per-cycle and mean values, respectively). This agreement emphasizes that these self-consistent values of Li+ exchange hold physical meaning, as they emerge to govern Li plating/stripping regardless of measurement technique. These experiments also revealed that the low-frequency EIS features typical of Type 2 electrolytes (Fig. 4) can have a substantial impact on charge transfer, such that disregarding their effect in the calculation of jp0,EIS leads to mismatches with jp0,CV after only 3–7 mA h cm−2 of cycled capacity (∼15 CV scans and ∼3 galvanostatic cycles, Fig. S15, ESI†).
Notably, returning to the values for Li+ exchange on plated Li in Fig. 6a and b (i.e., CVs after a single galvanostatic plating step), a direct and positive correlation was observed between jp0,CV and CE at the end of all scans (Fig. 6e and f). This relationship shows clearly that fast Li+ exchange is a key property that distinguishes high- vs. low-CE electrolytes.
Emergence of the jp0–CE relationship from SEI formation
In order to examine the origin of the jp0–CE relationship, we now return to compare jp0 on the Cu substrate before and after significant amounts of Li have been cycled (Fig. 5c). As mentioned previously, a distinct attribute of measurements on Cu is that they allow full stripping of the underlying Li at the end of each scan, therefore leaving behind only the native SEI, which in turn eliminates effects from accumulation of active Li over several cycles or CV scans. Fig. 7a shows jp0,CV over the first 11 CV scans on pristine Cu for all electrolytes. Given that unformed Cu surfaces typically exhibit low current densities, the total amount of charge plated/stripped was small (e.g., ∼0.03–0.3 mAh cm−2 per cycle, Fig. S11, ESI†). In general, jp0,CV values exhibited some scatter depending on the electrolyte but were typically low (<1 mA cm−2). After the 1 mAh cm−2 galvanostatic cycle (Fig. 7b), an evident upgrade in Li+ exchange was observed in some, but not all, electrolytes. This is further depicted in Fig. 7c and d, which summarize jp0,CV on cycle 11 on the pre- and post-conditioned Cu, but now plotted as a function of CE. First, we observe that no correlation was found on Cu prior to the 1 mAh cm−2 plating step (Fig. 7c), which is contrasted with post-plated Cu, where a strong monotonic trend was observed linking jp0,CV and CE. This finding shows that, similar to the trends on Li, higher-CE electrolytes are also more effective at modifying the Cu-electrolyte interface. Importantly, this effect persists even after full galvanostatic stripping of the underlying Li. In contrast, low-CE electrolytes display minimal difference in Li+ exchange between pre-plated and stripped Cu, indicating that SEI formation on Cu is ineffective in these electrolytes even after a 1 mAh cm−2 cycle. The quantitative CE–jp0,CV monotonic relationship specific to post-stripped Cu is fully summarized in Fig. 7e. We also investigated the evolution of jp0,CV over 100 CV scans in-between galvanostatic cycles, mimicking the methodology used for EIS in Fig. 3 and 4. Expectedly, even when subjected to repeated 1 mAh cm−2 plating/stripping cycles, Type 1 electrolytes showed the expected stable behavior and could not reach Li+ exchange rates comparable to those achieved after just a single cycle in Type 2 electrolytes (Fig. S16, ESI†).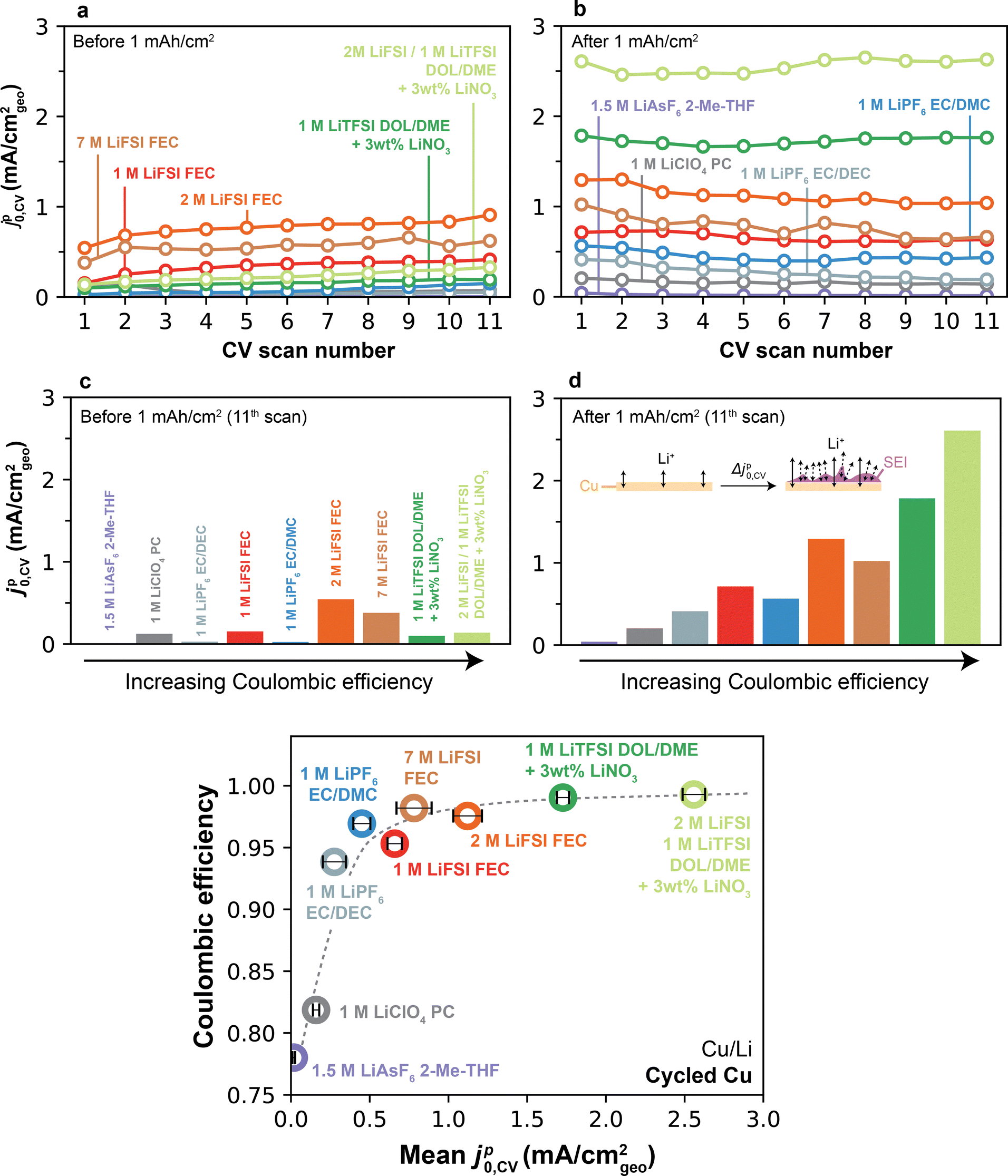 | ||
| Fig. 7 Effect of the Cu formation cycle on Li+ exchange and CE. (a) jp0,CV as a function of CV scan number on pristine Cu. (b) jp0,CV as a function of CV scan number on cycled Cu. (c) jp0,CV measured on the 11th CV scan on Cu prior to a 1 mAh cm−2 galvanostatic formation cycle. (d) jp0,CV measured on the 11th CV scan following the 1 mAh cm−2 galvanostatic formation cycle. Inset represents an illustration of a possible mechanism for the observed upgrade in Li+ exchange after Cu formation. The protocol in Fig. 5 was used to collect all data. Color legend in (d) as in (a–c). (e) Li CE vs. jp0,CV measured on cycled Cu after the 1 mAh cm−2 formation cycle, averaged over the subsequent 11 CV cycles. Error bars denote standard deviation over all 11 CV cycles on cycled Cu. | ||
The previous correlations relate jp0,CV measured on a per-cycle basis to a single-valued CE measured separately in each electrolyte. However, CE itself can change dynamically during cycling. To most rigorously examine the connection between dynamic Li+ exchange and dynamic CE, we exploited the fact that CV measurements enable the calculation of CE and Li+ exchange concomitantly on each scan. This jp0,CV–CE relationship is shown in Fig. 8a for 1 M LiPF6 EC/DEC (Type 1 electrolyte) and Fig. 8b for 2 M LiFSI 1 M LiTFSI DOL/DME + 3 wt% LiNO3 (Type 2 electrolyte); these data were obtained from the same CV scans underlying Fig. 7a and b. Focusing first on the Type 1 electrolyte (Fig. 8a), a monotonic increase in per-cycle CE was observed with increasing jp0,CV from cycles 1–10. Following a 1 mA h cm−2 galvanostatic cycle, jp0,CV exhibited a step-change increase to higher values, with accompanying improvements in CE. A similar behavior was observed in the Type 2 electrolyte (Fig. 8b), though expectedly with much higher CE and jp0,CV. These results agree with our previous findings on the relationship between these two parameters, but it also highlights a subtle additional insight: CE is also closely coupled to Li+ exchange on a per-cycle basis. In this context, the lower first-cycle CE typically found in anode-free batteries27,47 can be physically explained by an inability of the uncycled Cu to accommodate fast Li+ exchange. This jp0,CV–CE coupling is further explored in ESI† Note S4, which shows experimental evidence of a strong relationship between jp0,CV and CE over 100 continuous CV scans.
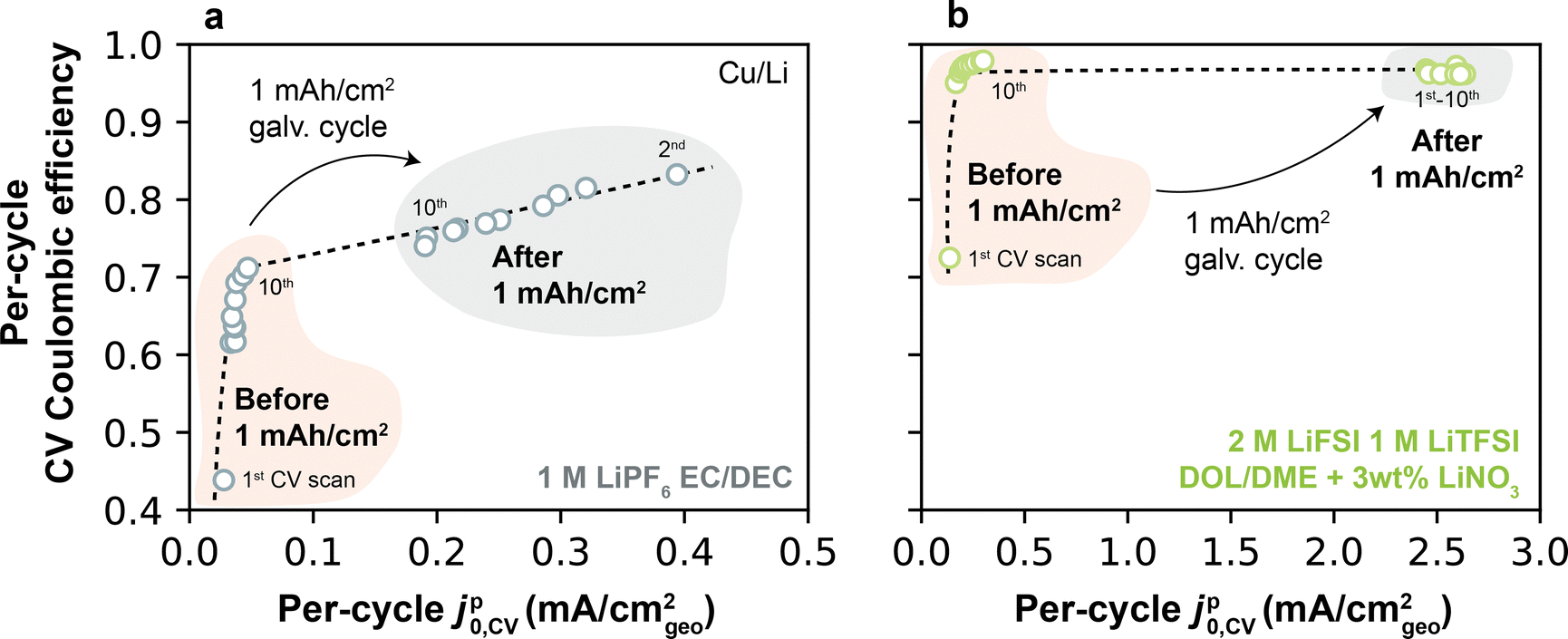 | ||
| Fig. 8 Per-cycle CE and jp0,CV obtained by CV on Cu, before and after a full 1 mA h cm−2 galvanostatic cycle in (a) 1 M LiPF6 EC/DEC; and (b) 2 M LiFSI 1 M LiTFSI DOL/DME + 3 wt% LiNO3. | ||
Implications of jp0 on Li rate capability
Finally, we examined the implications of Li+ exchange on the rate capability (here defined as CE vs. cycling current density) attainable in different electrolytes. As extensively discussed, Li+ exchange quantifies the facility by which an SEI exchanges Li+ from the electrolyte to the electrode. Thus, in addition to its relationship to CE, we hypothesized that the rate of SEI Li+ exchange defines the maximum cycling current density j at which Li can be cycled without substantial decline in CE. For this analysis, we focused on the Li+ exchange obtained by EIS (jp0,EIS) which was measured between galvanostatic cycles, a protocol that is most relevant to how CE is typically measured. The Type 1 and Type 2 electrolytes tested herein exhibit two distinct regimes: Type 1 electrolytes show jp0,EIS < 1 mA cm−2, a value that is in the range of typical cycling currents used in Li anode studies (0.1–2 mA cm−2). Type 2 electrolytes, however, can sustain jp0,EIS exceeding 10 mA cm−2, a value well beyond typical cycling currents.In this context, Fig. 9 shows CE as a function of current density (see Fig. S2, ESI,† for protocol) for select electrolytes spanning Type 1 and Type 2 behaviors. The CE in Type 1 carbonate electrolytes (1 M LiPF6 EC/DEC and 1 M LiClO4 PC) is less stable with increasing j, showing general decline at high j when compared to the respective CE values at j < 0.5 mA cm−2. This behavior is most evident in 1 M LiClO4 PC, in which CE decreases from 87.1% to 76.9% between 0.2 mA cm−2 and 2 mA cm−2. On the other hand, the Type 2 electrolytes (2 M LiFSI FEC and DOL/DME-based electrolytes) have CE values invariant with cycling current j. These observations can be interpreted in light of the applied current with respect to the SEI Li+ exchange capability. In 1 M LiClO4 PC and 1 M LiPF6 EC/DEC, jp0,EIS never exceeds 0.25 mA cm−2 and 0.55 mA cm−2, respectively; these electrolytes displayed substantial loss of CE when the applied current density exceeds these values. On the other hand, the rate-independence observed by the Type 2 electrolytes may be attributed to the extreme Li+ exchange rates that SEI in those systems can tolerate, which can easily exceed 5 mA cm−2 (Fig. 3 and 4). Interestingly, Type 2 electrolytes exhibit variable Li+ exchange under stress conditions (such as continued cycling), a factor that may enhance their ability to cycle well at high rate compared to Type 1 electrolytes. Because growth of SEI surface area increases total Li+ exchange, it is possible that non-uniform Li deposition is not necessarily detrimental and may even be beneficial for high cycling currents in these electrolytes, as long as such deposits remain electronically-percolated. We further note that, when jp0 is very high, other rate-limiting factors may become relevant before the SEI becomes the major bottleneck. For instance, jp0 can reach values >5 mA cm−2 in Type 2 electrolytes; under these conditions, mass transport at the bulk electrolyte may become limiting prior to SEI Li+ exchange.2 Further studies on this interplay, and transition between limiting regimes,4 are expected to be fruitful.
 | ||
| Fig. 9 CE as a function of rate for select Type 1 (low and stable jp0) and Type 2 (high and increasing jp0) electrolytes. | ||
Conclusion
Total Li+ exchange through natively-formed Li SEIs was quantified in a selection of low- and high-CE electrolytes using two electrochemical techniques, EIS and CV. These methods offer unique but complementary approaches for quantification of Li+ exchange rates, and proved self-consistent in each given electrolyte, providing confidence in the physicality of the obtained values. Low-CE electrolytes typically displayed stable and modest rates of Li+ exchange (<1 mA cm−2), i.e., on the order of typical battery cycling currents. On the other hand, high-CE electrolytes exhibited higher total Li+ exchange rates that further increased over cycling. A tight positive correlation was found between CE and Li+ exchange even when accounting for these dynamic effects, thus demonstrating unambiguously that electrolytes that enable fast Li+ exchange correlate with high CE. Our results also revealed that Li+ exchange is more sluggish on uncycled vs. cycled Cu, with correspondingly lower CE on pristine Cu, providing insights into underlying processes occurring during the first plating step. Looking forward, these findings may support new frameworks for electrolyte design to maximize SEI phases that promote facile Li+ exchange, once such phases can be better identified with chemical specificity. These strategies may include, for example, designing electrolytes that promote formation of SEI phases reported to possess higher Li+ ionic conductivity and diffusivity – for example Li2O or Li2S – compared to more ionically impeding phases.48,49 There are also great opportunities to combine quantitative chemical analysis of the SEI, such as by titration-based methods,5,50,51 with exchange current measurements to develop stronger correlations between SEI composition and function. In addition to SEI composition, the leading LiNO3-based electrolytes studied herein have also been shown to form SEI scaffolds/exoskeletons that can be optimized through the initial SEI formation cycle40,52 which, as demonstrated in this study (Fig. 4), leads to an enhancement of Li+ exchange. As such, maximization of jp0 can also guide the advancement of SEI formation protocols and surface engineering of Cu, which can have profound effects on both Li+ exchange and reversibility.Experimental methods
Materials preparation
Dimethyl carbonate (DMC, Acros Organics), 1,3-dioxolane (DOL), 2-methyl-tetrahydrofuran (2-Me-THF, MilliporeSigma), 1,2-dimethoxyethane (DME, Tokyo Chemical Industry), and fluoroethylene carbonate (FEC, Tokyo Chemical Industry) solvents were dried under molecular sieves (4 Å, MilliporeSigma) for at least 36 h before use. Lithium bis(fluorosulfonyl)imide (LiFSI, Arkema), lithium bis(trifluoromethanesulfonyl)imide (LiTFSI, Tokyo Chemical Industry) and lithium nitrate (LiNO, Tokyo Chemical Industry) salts were dried at 110 °C in a vacuum glass oven (Buchi) before use. Lithium hexafluoroarsenate(V) (LiAsF6, Strem) was used as-received. Battery-grade 1 M LiPF6 ethylene carbonate (EC)/diethyl carbonate (DEC) and 1 M LiPF6 EC/DMC (1:1 vol%, MilliporeSigma) were used as-received. All other electrolytes were prepared in an Ar-filled glovebox (MBraun, <0.1 ppm O2, <0.1 ppm H2O). Li metal foil (0.75 mm, Alfa Aesar) was rolled and cut into ∅15 mm diameter disks immediately before use. Cu foil (MTI) was punched to ∅15 mm diameter, acid-washed with 1 M H2SO4 for 1 h, cleaned with deionized (DI) water (MilliQ, >18.2 MΩ/cm), rinsed with acetone and immediately dried at room temperature for at least 8 h under antechamber vacuum. Coin cell components (CR2032, MTI) were cleaned with ethanol followed by DI water and dried before use. Glass fiber separators (Whatman QM-A) were punched to ∅16 mm diameter and dried in a vacuum glass oven at 130 °C before use. Microporous polymeric separators (Celgard 2325 and 3501) were punched to ∅20 mm diameter and dried under antechamber vacuum for at least 8 h before use.
Cell assembly
CR2032 coin cells were assembled inside an Ar-filled glovebox (Mbraun, <0.1 ppm H2O, <0.1 ppm O2). Li strips were rolled into thin films, punched to ∅15 mm diameter, then pressed into stainless steel current collectors and used as counter/reference electrode for all measurements. For measurements in Li/Li cells, another identical piece of Li film pressed into a stainless steel current collector was used as the working electrode. For measurements in Cu/Li cells, Cu was used as the working electrode with a stainless steel current collector. For general electrochemical measurements, one Whatman glass fiber, soaked in 100 μL of the desired electrolyte, was used per cell. For SEM characterization, two separators (either Celgard 2325 or 3501) were used per cell, along with a total of 50 μL of the desired electrolyte. The assembled CR2032 was closed with a wave spring, crimped and taken outside the glovebox for testing.Coulombic efficiency
Coulombic efficiency (CE) measurements were conducted as described by Adams et al.27 in Cu/Li cells. All measurements include a single formation cycle, plating 4 mA h cm−2 Li on Cu and then stripping until all reversible Li is depleted to 1 V at 0.5 mA cm−2. A Li reservoir of 4 mA h cm−2 is then plated onto Cu, and 1 mA h cm−2 from the reservoir is cycled 10 times, after which the reservoir is completely stripped to 1 V. Between each half-cycle, the cell rests at open-circuit voltage (OCV) for 1 min. CE is determined as the ratio of the total amount of Li stripped to the total amount plated, excluding the formation cycle. For all measurements, the formation cycle was performed at 0.5 mA cm−2 but current density for reservoir cycling was performed at the indicated current density. Fig. S2, ESI,† demonstrates the CE measurement protocol. All CE measurements were conducted on a battery cycler (BST, MTI).Electrochemical impedance spectroscopy
Electrochemical impedance spectroscopy (EIS) measurements were conducted in Li/Li cells that were taken outside of the glovebox immediately after cell assembly. EIS spectra were collected on Biologic MPG or VSP-3 potentiostats with a sine-wave excitation amplitude of 5 mV around OCV, with a typical frequency range of 10 mHz to 20 kHz, with at least 10 points collected per decade. Before the initial EIS measurement, the cell rested at OCV for 5 h.After the spectra were collected, the single semi-circle (typically corresponding to frequencies >20 Hz, unless noted otherwise) was fit to an equivalent resistor-capacitance-Warburg circuit, where the RC impedance represents the Li0/Li+ redox charge-transfer resistance and the Warburg impedance represents diffusional transport across the interphase.34 The circuit was fit to data using a custom Python script that employs a least-square method. The total resistance to interface exchange (RSEI) was taken as the sum of the Warburg coefficient (W1) and the charge-transfer resistance Rct to capture all SEI-relevant processes including charge transfer (kinetics) and transport, which are physically coupled in a thin but finite SEI. The equivalent EIS exchange current (jp0,EIS) is then inversely proportional to the total SEI resistance (RSEI)9 such that
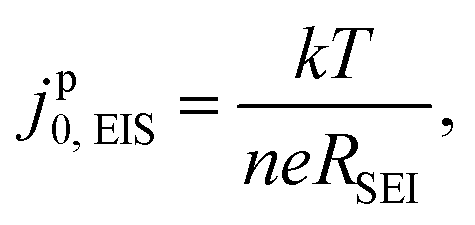
Cyclic voltammetry
Cyclic voltammetry (CV) data were acquired in two-electrode coin cells using Biologic MPG or VSP-3 potentiostats, with a typical scan rate of 1 mV s−1 unless otherwise mentioned. For experiments where Cu was the working electrode (i.e., pristine or fully-stripped Cu), the measurement started at OCV (typically ∼3 V for pristine Cu and ∼0 V for fully-stripped Cu) and voltage was scanned from OCV to −0.2 V, and then from −0.2 V to 1 V, cycling within the voltage window for the desired number of cycles. For experiments where Li was the working electrode (i.e., plated Li), the measurement starts at OCV (∼0 V) and voltage was scanned from OCV to −0.2 V, and then between −0.2 V to 0.2 V, for the desired number of cycles. All CV cycles were conducted continuously. In order to obtain jp0,CV, the CV data was processed such that, first, the cell voltage was iR-compensated and corrected such that the potential at zero current was set to zero (i.e., Ew,corrected (j = 0) = 0 V). Then, data in the low-overpotential regime, where the current response is linear with overpotential, was used to obtain a representative exchange current jp0,CV such that j = jp0,CV (F/RT) Ew,corrected, thus avoiding the use of more complex kinetic models or Tafel interpolation at high overpotentials that may yield unphysical results for interphase-dominated processes.9 More information on the CV analysis is presented in ESI† Note S2.Author contributions
G. M. H. and B. M. G. conceptualized the study and analyzed the data; G. M. H. performed experiments, acquired and processed the data; K. H. K. acquired SEM images of cycled electrodes. B. M. G. supervised data acquisition and processing; G. H. M. and B. M. G. wrote, edited and reviewed all drafts of the manuscript.Conflicts of interest
None to report.Acknowledgements
The authors acknowledge a collaborative grant from the Sagol Weizmann-MIT Bridge Program. This work made use of the MRSEC Shared Experimental Facilities at MIT, supported by the National Science Foundation under award number DMR-1419807. This work also made use of the Department of Chemistry Instrumentation Facility (DCIF) at MIT.References
- H. Wang, Z. Yu, X. Kong, S. C. Kim, D. T. Boyle, J. Qin, Z. Bao and Y. Cui, Joule, 2022, 6, 588–616 CrossRef CAS
.
- J. Zheng, M. S. Kim, Z. Tu, S. Choudhury, T. Tang and L. A. Archer, Chem. Soc. Rev., 2020, 49, 2701–2750 RSC
- P. Albertus, S. Babinec, S. Litzelman and A. Newman, Nat. Energy, 2017, 3, 16–21 CrossRef
- G. M. Hobold, J. Lopez, R. Guo, N. Minafra, A. Banerjee, Y. Shirley Meng, Y. Shao-Horn and B. M. Gallant, Nat. Energy, 2021, 6, 951–960 CrossRef CAS
- C. Fang, J. Li, M. Zhang, Y. Zhang, F. Yang, J. Z. Lee, M. H. Lee, J. Alvarado, M. A. Schroeder, Y. Yang, B. Lu, N. Williams, M. Ceja, L. Yang, M. Cai, J. Gu, K. Xu, X. Wang and Y. S. Meng, Nature, 2019, 572, 511–515 CrossRef CAS PubMed
- A. B. Gunnarsdottir, C. V. Amanchukwu, S. Menkin and C. P. Grey, J. Am. Chem. Soc., 2020, 142, 20814–20827 CrossRef CAS PubMed
- Y.-C. Hsieh, M. Leißing, S. Nowak, B.-J. Hwang, M. Winter and G. Brunklaus, Cell. Rep. Phys. Sci., 2020, 1, 100139 CrossRef CAS
- C. Fang, B. Lu, G. Pawar, M. Zhang, D. Cheng, S. Chen, M. Ceja, J.-M. Doux, H. Musrock, M. Cai, B. Liaw and Y. S. Meng, Nat. Energy, 2021, 6, 987–994 CrossRef CAS
-
A. J. Bard and L. R. Faulkner, Electrochemical methods: fundamentals and applications, John Wiley & Sons, Hoboken, 2007 Search PubMed
- D. T. Boyle, X. Kong, A. Pei, P. E. Rudnicki, F. Shi, W. Huang, Z. Bao, J. Qin and Y. Cui, ACS Energy Lett., 2020, 5, 701–709 CrossRef CAS
- A. B. Gunnarsdóttir, S. Vema, S. Menkin, L. E. Marbella and C. P. Grey, J. Mater. Chem. A, 2020, 8, 14975–14992 RSC
- J. N. Butler, D. R. Cogley and J. C. Synnott, J. Phys. Chem., 2002, 73, 4026–4027 CrossRef
- M. W. Verbrugge and B. J. Koch, J. Electroanal. Chem., 1994, 367, 123–129 CrossRef CAS
- S. G. Meibuhr, J. Electrochem. Soc., 1970, 117, 56 CrossRef CAS
- R. F. Scarr, J. Electrochem. Soc., 1970, 117, 295–299 CrossRef CAS
- S. G. Meibuhr, J. Electrochem. Soc., 1971, 118, 1320–1322 CrossRef CAS
- R. Tao, X. Bi, S. Li, Y. Yao, F. Wu, Q. Wang, C. Zhang and J. Lu, ACS. Appl. Mater. Interfaces, 2017, 9, 7003–7008 CrossRef CAS PubMed
- K. S. Aojula, J. D. Genders, A. D. Holding and D. Pletcher, Electrochim. Acta, 1989, 34, 1535–1539 CrossRef CAS
- C. C. Su, M. He, J. Shi, R. Amine, J. Zhang and K. Amine, Angew. Chem., 2020, 59, 18229–18233 CrossRef CAS PubMed
- X. Fan, L. Chen, O. Borodin, X. Ji, J. Chen, S. Hou, T. Deng, J. Zheng, C. Yang, S. C. Liou, K. Amine, K. Xu and C. Wang, Nat. Nanotechnol., 2018, 13, 715–722 CrossRef CAS PubMed
- Q. Zhao, Z. Tu, S. Wei, K. Zhang, S. Choudhury, X. Liu and L. A. Archer, Angew. Chem., 2018, 57, 992–996 CrossRef CAS PubMed
- X.-Q. Zhang, X. Chen, L.-P. Hou, B.-Q. Li, X.-B. Cheng, J.-Q. Huang and Q. Zhang, ACS Energy Lett., 2019, 4, 411–416 CrossRef CAS
- F. Shi, A. Pei, D. T. Boyle, J. Xie, X. Yu, X. Zhang and Y. Cui, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 8529–8534 CrossRef CAS PubMed
- Y. Liu, X. Xu, M. Sadd, O. O. Kapitanova, V. A. Krivchenko, J. Ban, J. Wang, X. Jiao, Z. Song, J. Song, S. Xiong and A. Matic, Adv. Sci., 2021, 8, 2003301 CrossRef CAS PubMed
- F. Shi, A. Pei, A. Vailionis, J. Xie, B. Liu, J. Zhao, Y. Gong and Y. Cui, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 12138–12143 CrossRef CAS PubMed
- R. May, K. J. Fritzsching, D. Livitz, S. R. Denny and L. E. Marbella, ACS Energy Lett., 2021, 6, 1162–1169 CrossRef CAS
- B. D. Adams, J. Zheng, X. Ren, W. Xu and J. G. Zhang, Adv. Energy Mater., 2017, 8, 1702097 CrossRef
- P. Bai, J. Li, F. R. Brushett and M. Z. Bazant, Energy Environ. Sci., 2016, 9, 3221–3229 RSC
- Y. Chen, Z. Yu, P. Rudnicki, H. Gong, Z. Huang, S. C. Kim, J. C. Lai, X. Kong, J. Qin, Y. Cui and Z. Bao, J. Am. Chem. Soc., 2021, 143, 18703–18713 CrossRef CAS PubMed
- Z. Yu, H. Wang, X. Kong, W. Huang, Y. Tsao, D. G. Mackanic, K. Wang, X. Wang, W. Huang, S. Choudhury, Y. Zheng, C. V. Amanchukwu, S. T. Hung, Y. Ma, E. G. Lomeli, J. Qin, Y. Cui and Z. Bao, Nat. Energy, 2020, 5, 526–533 CrossRef CAS
- D. Aurbach and A. Zaban, J. Electroanal. Chem., 1993, 348, 155–179 CrossRef CAS
- M. Odziemkowski and D. E. Irish, J. Electrochem. Soc., 2019, 140, 1546–1555 CrossRef
- D. T. Boyle, W. Huang, H. Wang, Y. Li, H. Chen, Z. Yu, W. Zhang, Z. Bao and Y. Cui, Nat. Energy, 2021, 6, 487–494 CrossRef CAS
- A. Zaban, E. Zinigrad and D. Aurbach, J. Phys. Chem., 1996, 100, 3089–3101 CrossRef CAS
- S. P. Kühn, F. Pfeiffer, M. Bela, U. Rodehorst, D. Weintz, M. Stan, M. Baghernejad, M. Winter and I. Cekic-Laskovic, J. Power Sources, 2022, 549, 232118 CrossRef
- D. Aurbach and A. Zaban, J. Electroanal. Chem., 1994, 365, 41–45 CrossRef CAS
- J. Z. Lee, T. A. Wynn, M. A. Schroeder, J. Alvarado, X. Wang, K. Xu and Y. S. Meng, ACS Energy Lett., 2019, 4, 489–493 CrossRef CAS
- J. Qian, W. A. Henderson, W. Xu, P. Bhattacharya, M. Engelhard, O. Borodin and J. G. Zhang, Nat. Commun., 2015, 6, 6362 CrossRef CAS PubMed
- Y. Li, W. Huang, Y. Li, A. Pei, D. T. Boyle and Y. Cui, Joule, 2018, 2, 2167–2177 CrossRef CAS
- B. Han, X. Li, Q. Wang, Y. Zou, G. Xu, Y. Cheng, Z. Zhang, Y. Zhao, Y. Deng, J. Li and M. Gu, Adv. Mater., 2022, 34, 2108252 CrossRef CAS PubMed
- E. Peled and S. Menkin, J. Electrochem. Soc., 2017, 164, A1703–A1719 CrossRef CAS
- S. K. Heiskanen, J. Kim and B. L. Lucht, Joule, 2019, 3, 2322–2333 CrossRef CAS
- D. D. Macdonald, Electrochim. Acta, 2006, 51, 1376–1388 CrossRef CAS
- C. Criado, P. Galán-Montenegro, P. Velásquez and J. R. Ramos-Barrado, J. Electroanal. Chem., 2000, 488, 59–63 CrossRef CAS
- J. Song and M. Z. Bazant, Phys. Rev. Lett., 2018, 120, 116001 CrossRef CAS PubMed
- Y. Zeng, R. Smith, P. Bai and M. Z. Bazant, 2014, arXiv:1407.5370.
- J. Xiao, Q. Li, Y. Bi, M. Cai, B. Dunn, T. Glossmann, J. Liu, T. Osaka, R. Sugiura, B. Wu, J. Yang, J.-G. Zhang and M. S. Whittingham, Nat. Energy, 2020, 5, 561–568 CrossRef CAS
- R. Guo and B. M. Gallant, Chem. Mater., 2020, 32, 5525–5533 CrossRef CAS
- S. Lorger, K. Narita, R. Usiskin and J. Maier, Chem. Commun., 2021, 57, 6503–6506 RSC
- E. J. McShane, A. M. Colclasure, D. E. Brown, Z. M. Konz, K. Smith and B. D. McCloskey, ACS Energy Lett., 2020, 5, 2045–2051 CrossRef CAS
- G. M. Hobold and B. M. Gallant, ACS Energy Lett., 2022, 7, 3458–3466 CrossRef CAS
- B. Han, X. Li, S. Bai, Y. Zou, B. Lu, M. Zhang, X. Ma, Z. Chang, Y. S. Meng and M. Gu, Matter, 2021, 4, 3741–3752 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ee04203g |
| This journal is © The Royal Society of Chemistry 2023 |