
Isomerization strategy on a non-fullerene guest acceptor for stable organic solar cells with over 19% efficiency†
Zhenyu
Chen
ab,
Jintao
Zhu
 c,
Daobin
Yang
*ab,
Wei
Song
*ab,
Jingyu
Shi
ab,
Jinfeng
Ge
ab,
Yuntong
Guo
a,
Xinyu
Tong
ab,
Fei
Chen
c and
Ziyi
Ge
*ab
c,
Daobin
Yang
*ab,
Wei
Song
*ab,
Jingyu
Shi
ab,
Jinfeng
Ge
ab,
Yuntong
Guo
a,
Xinyu
Tong
ab,
Fei
Chen
c and
Ziyi
Ge
*ab
aZhejiang Engineering Research Center for Energy Optoelectronic Materials and Devices, Ningbo Institute of Materials Technology and Engineering, Chinese Academy of Sciences, Ningbo 315201, China. E-mail: yangdaobin@nimte.ac.cn; songwei@nimte.ac.cn; geziyi@nimte.ac.cn
bCenter of Materials Science and Optoelectronics Engineering, University of Chinese Academy of Sciences, Beijing 100049, China
cDepartment of Chemical and Environmental Engineering, University of Nottingham Ningbo China, Ningbo 315100, China
First published on 13th June 2023
Abstract
The strategy of isomerization plays a simple and effective role in optimizing the molecular configurations and improving the performance of binary organic solar cells (OSCs). However, the effect of isomerization in guest materials on ternary OSCs has rarely been reported, and their structure–property relationships are not yet clearly understood. Herein, two large π-conjugated isomers, QX-α and QX-γ, with different orientations of their fused thiophene-rings were designed and synthesized to investigate the influence of isomers in non-fullerene guest acceptors on the photovoltaic properties in a D18:N3 host system. Compared to QX-γ, QX-α demonstrated a stronger dipole moment, a more ordered stacking, and a higher surface energy due to the presence of S⋯N non-covalent interactions. As a result, the OSCs device based on D18:N3:QX-α achieved the higher efficiency of 19.33%, while the device based on D18:N3:QX-γ exhibited an efficiency of only 18.30%. Remarkably, the flexible OSC based on D18:N3:QX-α produced an outstanding PCE of 18.01%, which is a record PCE for flexible OSCs. In addition, the ternary device showed a significant increase in efficiency retention from 49% to 87% after 476 h of storage in a N2-filled glove box at 85 °C compared to the binary device. Moreover, the extrapolated T80 lifetime of the D18:N3:QX-α-based ternary device was as high as over 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h in a glove box at room temperature. The results indicate that rational tuning of the atomic orientation can be an effective way to construct non-fullerene guest acceptors for achieving highly efficient and stable OSCs.
000 h in a glove box at room temperature. The results indicate that rational tuning of the atomic orientation can be an effective way to construct non-fullerene guest acceptors for achieving highly efficient and stable OSCs.
Broader contextSolution-processed organic solar cells (OSCs) have attracted great attention in the field of green energy technology due to their promising advantages of flexibility, portability, and transparency. The careful design and synthesis of novel photovoltaic materials are the key issues for the development of higher power conversion efficiencies (PCEs), and in this regard the ternary strategy has become one of the most important methods to further improve the PCE of OSCs. In this work, two Y6-derivatives with isomeric central electron-deficient cores, QX-α and QX-γ, with different orientations of their fused thiophene-rings were designed and synthesized to investigate the influence of the isomers in non-fullerene guest acceptors on their photovoltaic properties in the host system of D18:N3. The PCE of the D18:N3:QX-α-based rigid device was as high as 19.33%, while the D18:N3:QX-γ-based rigid device only exhibited a PCE of 18.30%, which is comparable to the standard binary device. The enhanced PCE could be attributed to the more balanced hole/electron mobilities, more efficient charge collection, less charge carrier recombination, and lower energy losses of the D18:N3:QX-α-based ternary device. Remarkably, the flexible OSC based on D18:N3:QX-α produced an outstanding PCE of 18.01%, which is a record PCE for flexible OSCs. In addition, the ternary device maintained over 97% of its initial PCE for over 2200 h with an extrapolated T80 of over 17000 h after storage in a N2-filled glove box.
|
1. Introduction
Solution-processed organic solar cells (OSCs) have attracted great attention in the field of green energy technology due to their promising advantages of flexibility, portability, and transparency.1,2 In recent years, with the development of new materials and the optimization of device fabrication technology, the power conversion efficiencies (PCEs) of OSCs have been rapidly improved.3,4 Currently, the highest PCEs of single-junction OSCs have exceeded 19%,5–8 and almost all the high efficiencies have been achieved in ternary devices. Therefore, the ternary strategy has become one of the most important methods to improve the performance of OSCs.9,10Isomers are considered as two or more different substances with the same molecular formula but different chemical and physical properties.11 Recently, the strategy of isomerization has been considered as an effective way to improve the efficiency of OSCs by rational tuning of the molecular structures.12 To date, a large number of studies on isomer effects have been conducted focused on the host materials of binary OSCs,13–16 but only a few works have been reported on ternary OSCs.17 Lim reported that two diketopyrrolopyrrole (DPP)-based isomers, o-DPPPhCN and p-DPPPhCN, could be introduced as guest acceptors into the host system of P3HT:PC61BM.18 The para-cyano-substituted p-DPPPhCN showed a positive effect on the PCE of the ternary OSCs, improving it from 2.07% to 2.48%. Recently, Lu and co-workers constructed two fused-ring acceptor isomers, m-BAIDIC and p-BAIDIC, with nonlinear and linear molecular conformations, respectively.19 When the linear p-BAIDIC was incorporated into the PM6:BO-4Cl binary system, an improvement of the PCE from 16.9% to 17.6% was observed, which was attributed to the simultaneous enhancements of the open-circuit voltage (VOC), short-circuit current density (JSC), and fill factor (FF). However, these PCEs still lag far behind the current efficiency. Therefore, there is a great room for the further development of novel isomers to clearly understand the relationship between the structure and property, contributing to the achievement of high-performance OSCs.
In 2019, Zou et al. developed a powerful non-fullerene acceptor Y6 and realized an over 15% efficiency in OSCs.20 Since then, numerous Y6-derivatives have been reported, rapidly pushing up the efficiency of OSCs.21,22 Currently, the isomerization of Y6-derivatives has become an effective way to improve the performance of binary OSCs.23–25 In particular, almost all the Y6-derivatives have been developed by employing isomeric end-capping groups or side chains,26–30 but the isomerization of the central electron-deficient core in Y6-derivatives has never been investigated or reported. In addition, the low glass transition temperatures (Tg) and high diffusion properties of the Y6-derivatives lead to unstable morphologies in blend films.31,32 Therefore, there is an urgent need to increase the Tg of the Y6-derivatives.
According to our previous work, the thienyl-substituted quinoxaline unit was chosen as the central electron-deficient core, which could provide a higher VOC and better film morphology.33 Moreover, the rotatable thiophene-groups were fused to enhance the rigidity of the molecules and extend the π-conjugation skeleton.34 Herein, two Y6-derivatives with isomeric central electron-deficient cores, QX-α and QX-γ, with different orientations of their fused thiophene-rings were designed and synthesized to investigate the influence of the isomers in non-fullerene guest acceptors on their photovoltaic properties. As we know, S⋯N intramolecular non-covalent interactions have become a popular means to modulate the optoelectronic performances of organic semiconductors.35,36 Bo and Huang et al. reported that S⋯N non-covalent interactions could endow a more planar molecular conformation and ordered stacking, which resulted in increased PCEs of the tested OSCs.37,38 Compared to previous studies, more eigenproperties of the materials, such as dipole moment and surface energy, were investigated in this work. The relationship between the chemical structure–material's eigenproperty–photovoltaic performance was carefully investigated and revealed. As a result, the PCEs of the D18:N3:QX-α-based rigid and flexible devices were as high as 19.33% and 18.01%, respectively, while the D18:N3:QX-γ-based rigid and flexible devices only exhibited PCEs of 18.30% and 16.99%, which were comparable to the standard binary device. The enhanced PCE was attributed to the more balanced hole/electron mobilities, more efficient charge collection, less charge carrier recombination, and lower energy losses of the D18:N3:QX-α-based ternary device. In addition, the efficiency retention of the D18:N3:QX-α-based ternary device after 476 h of storage in a N2-filled glove box at 85 °C was significantly increased, from 49% to 87%, compared to the D18:N3-based binary device. Remarkably, the ternary device maintained over 97% of the initial PCE for over 2200 h with an extrapolated T80 over 17000 h after storage in a N2-filled glove box.
2. Results and discussion
The molecular structures of the Y6-derivative N3 and the two new isomeric non-fullerene acceptors QX-α and QX-γ are shown in Fig. 1a, and the detailed synthetic routes of the two isomers are provided in Schemes S1 and S2 in the ESI.† The chemical structures of the intermediates and two objective compounds were carefully confirmed by NMR and high-resolution time-of-flight-mass (TOF-MS) spectroscopy, and are given in Fig. S1–S8 (ESI†). Both QX-α and QX-γ had good solubility in common organic solvents, such as chloroform and chlorobenzene. The UV–vis absorption spectra of D18, N3, QX-α, and QX-γ in neat films are shown in Fig. 1b. QX-α and QX-γ exhibited similar absorption profiles with the maximum absorption peaks at 805 and 803 nm, respectively, which were blue-shifted by around 27 nm from that of N3. Consequently, the optical bandgaps of QX-α and QX-γ were slightly wider than that of N3 (1.37 vs. 1.33 eV). The energy levels of the non-fullerene acceptors were evaluated by cyclic voltammetry (CV) measurements, and the corresponding data and CV curves are shown in Fig. 1c and Fig. S9 (ESI†), respectively. The highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) energy levels of QX-α were −5.64 and −3.88 eV, while the HOMO and LUMO values of QX-γ were −5.58 and −3.90 eV, respectively. The energy levels were in agreement with the density functional theory (DFT) calculations (Fig. S10, ESI†), indicating that these experimental results are reliable. Furthermore, both QX-α and QX-γ displayed significant differences in energy levels with respect to the donor D18 and the acceptor N3. The cascaded energy level alignments were favorable for efficient charge transfer at the D–A interface to reduce energy losses, thereby improving the VOC.39,40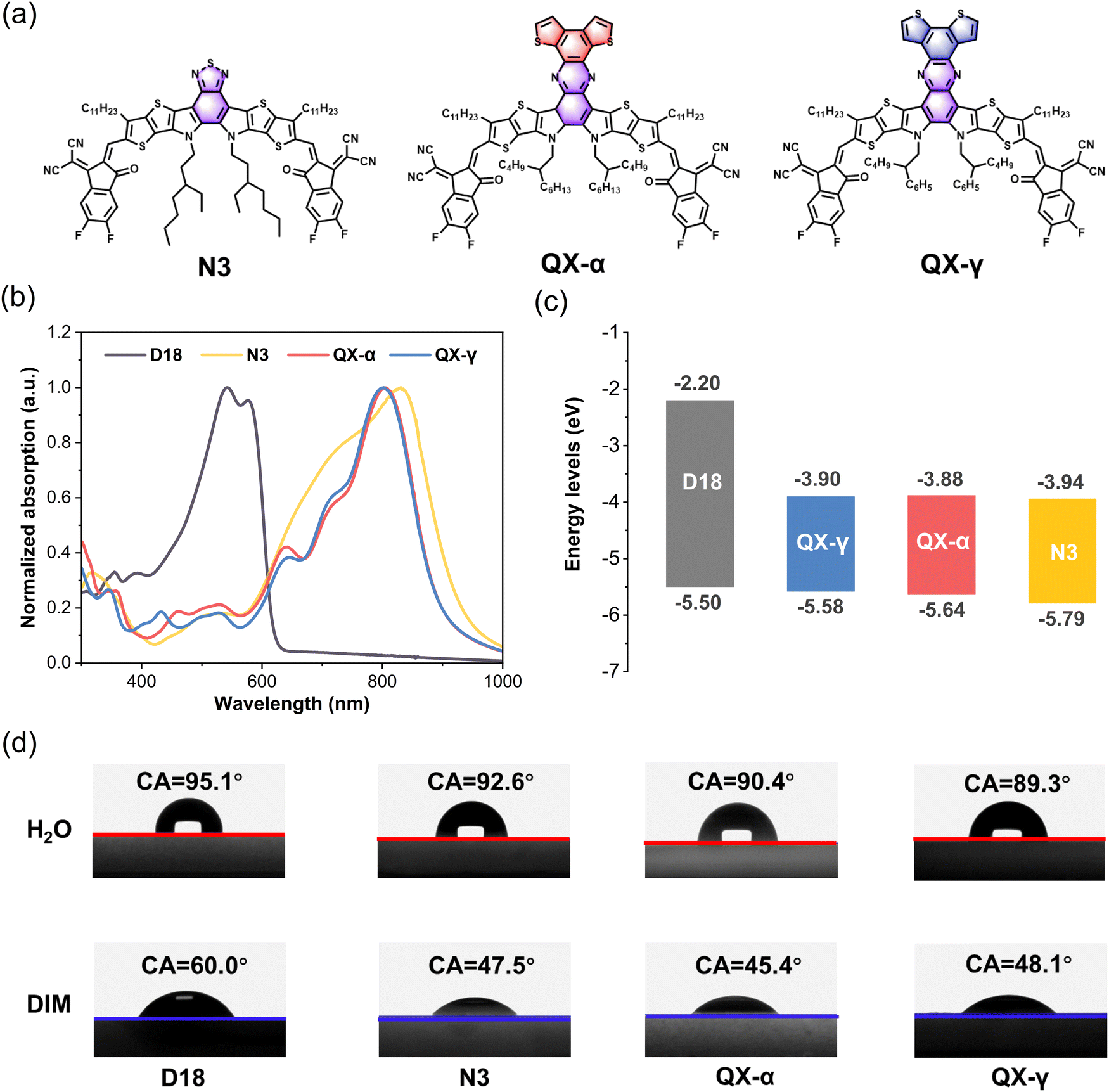 | ||
| Fig. 1 (a) Molecular structures of the non-fullerene acceptors N3, QX-α, and QX-γ. (b) Normalized UV-vis absorption spectra of D18, N3, QX-α, and QX-γ in neat films. (c) Energy level diagrams of D18,47 N3, QX-α, and QX-γ. (d) Contact angles of D18, N3, QX-α, and QX-γ thin films by applying deionized water (H2O) and diiodomethane (DIM) liquid drops. | ||
The thermal stabilities of the non-fullerene acceptors were measured by thermogravimetric analysis (TGA). As shown in Fig. S11 (ESI†), QX-α and QX-γ displayed similar decomposition temperatures (5% weight loss) at around 348 °C, which were higher than that of N3 (333 °C), indicating that the large π-conjugated quinoxaline cores endowed the molecules with higher thermal stability. In addition, differential scanning calorimetry (DSC) was carried out to evaluate the crystallization process and intermolecular interactions, as displayed in Fig. S12 (ESI†). Both QX-α and QX-γ had similar melting endothermic peaks at 336 °C and 339 °C, respectively, which were obviously higher than that of N3 (320 °C). However, the melting enthalpy of QX-α was significantly larger than that of QX-γ (50.15 vs. 35.39 J g−1), indicating the stronger intermolecular interaction in QX-α.
The contact angles (CAs) of water and diiodomethane liquid drops were determined to explore the compatibility of D18, N3, QX-α, and QX-γ, as shown in Fig. 1d and Table S1 (ESI†), and the corresponding surface energies (γ) were calculated by the Owens–Wendt–Rabel–Kaelble method.41,42 The γ values of D18, N3, QX-α, and QX-γ were 28.60, 35.71, 36.81, and 35.35 mN m−1, respectively. QX-α had a higher γ value than QX-γ due to its larger polarity, as evidenced by thin-layer chromatography (TLC) analysis (Fig. S13, ESI†). Moreover, the degree of molecular miscibility could be estimated by the Flory–Huggins interaction parameter χ, 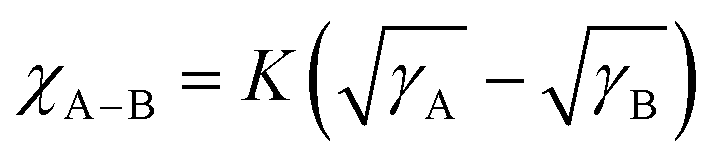 ,2 where γA and γB are the γ values of compounds A and B.43 The χ parameters were calculated to be 0.3944 K, 0.5170 K, 0.3564 K, 0.0083 K, and 0.0010 K for D18:N3, D18:QX-α, D18:QX-γ, N3:QX-α, and N3:QX-γ, respectively, indicating that the two isomers possessed excellent miscibility and compatibility with the host acceptor N3. In addition, to point out where the introduced guest acceptors QX-α and QX-γ were located in the ternary system (D18:N3:A1), the wetting coefficient (ω) of the third component A1 in the mixture of D18:N3 was calculated according to Young's equation,44
,2 where γA and γB are the γ values of compounds A and B.43 The χ parameters were calculated to be 0.3944 K, 0.5170 K, 0.3564 K, 0.0083 K, and 0.0010 K for D18:N3, D18:QX-α, D18:QX-γ, N3:QX-α, and N3:QX-γ, respectively, indicating that the two isomers possessed excellent miscibility and compatibility with the host acceptor N3. In addition, to point out where the introduced guest acceptors QX-α and QX-γ were located in the ternary system (D18:N3:A1), the wetting coefficient (ω) of the third component A1 in the mixture of D18:N3 was calculated according to Young's equation,44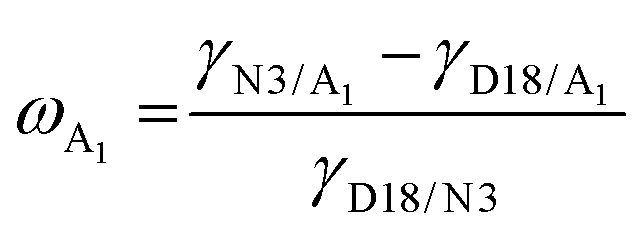 . The ωQX-α and ωQX-γ were calculated to be −1.03 and −0.42 (Table S1, ESI†), respectively, suggesting that QX-α lay in the domain of N3 (if ωA1 < −1), while QX-γ was located at the interfaces between D18 and N3 (if −1 < ωA1 < 1). Consequently, the results indicate that QX-α created an N3:QX-α acceptor alloy within the D18:N3:QX-α-based blend film, which is beneficial for improving the PCE.45,46
. The ωQX-α and ωQX-γ were calculated to be −1.03 and −0.42 (Table S1, ESI†), respectively, suggesting that QX-α lay in the domain of N3 (if ωA1 < −1), while QX-γ was located at the interfaces between D18 and N3 (if −1 < ωA1 < 1). Consequently, the results indicate that QX-α created an N3:QX-α acceptor alloy within the D18:N3:QX-α-based blend film, which is beneficial for improving the PCE.45,46
To gain further insights into the effects of the isomers on the electronic properties, DFT calculations were performed on QX-α and QX-γ. As shown in Fig. 2a and b, an S⋯N non-covalent interaction could be observed in the optimized molecular configuration of QX-α (3.13 Å), but was absent in QX-γ. Moreover, both QX-α and QX-γ had similar directions of the dipole moment, but the dipole moment of QX-α was two times higher than that of QX-γ (3.64 vs. 1.18 D). This suggests that QX-α had significantly stronger dipole–dipole interactions, which could endow QX-α with a more ordered stacking and higher hole mobility.48 This presumption could be confirmed by grazing-incidence wide-angle X-ray scattering (GI-WAXS). As shown in Fig. S14 and Table S2 (ESI†), both QX-α and QX-γ neat films exhibited predominant face-on orientations with a π–π stacking peak at qz = 1.74 Å−1 (d = 3.62 Å) and a lamellar stacking peak at qXY = 0.35 Å−1 (d = 17.9 Å) along the out-of-plane (OOP) and in-plane (IP) directions, respectively. However, the crystal coherence length (CCL) of the π–π stacking peak (010) in QX-α was higher than that of QX-γ (24.2 vs. 22.8 Å). This result shows that QX-α had more ordered stacking than QX-γ.
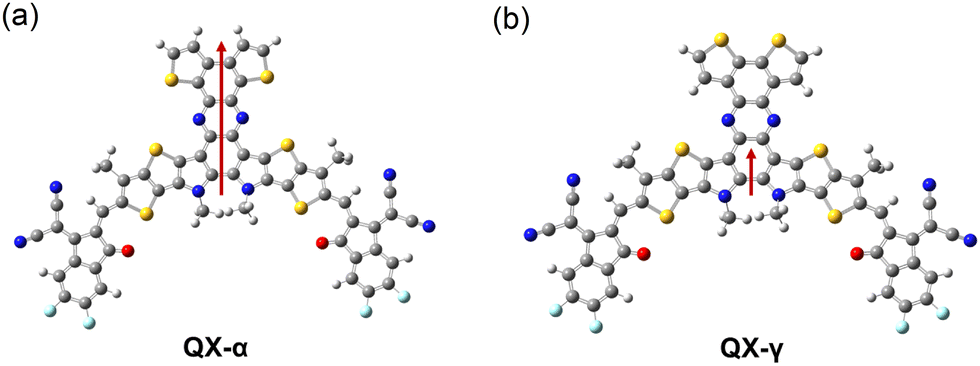 | ||
| Fig. 2 Dipole moments of QX-α (a) and QX-γ (b). | ||
To explore the photovoltaic properties of QX-α and QX-γ, a series of OSCs with a conventional device architecture of ITO/PEDOT:PSS/D18:acceptor/PDINN/Ag were fabricated, and the chemical structures of the used materials are given in Fig. S15 (ESI†). First, the binary devices with D18 as the donor and the isomer as the acceptor were investigated, and the J–V curves are shown in Fig. S16 (ESI†), and the corresponding data are listed in Table 1. The D18:QX-α-based device exhibited a moderate PCE of 14.30% with a VOC of up to 0.944 V, a JSC of 21.67 mA cm−2, and an FF of 0.699, while a higher PCE of 15.72% was observed in the D18:QX-γ-based device due to its improved JSC and FF. Although the electron mobility (μe) of the D18:QX-γ-based device was lower than that of the D18:QX-α system (4.09 × 10−4vs. 4.42 × 10−4 cm2 V−1 s−1, Fig. S17 (ESI†) and Table 1), a more balanced electron/hole mobility (μe/μh) could be found (1.25 vs. 1.37) in the D18:QX-γ-based device, which was available for the enhancement of JSC and FF.
| BHJ | V oc (V) | J SC/JEQESC (mA cm−2) | FF | PCEa (%) | μ h/10−4 (cm2 V−1 s−1) | μ e/10−4 (cm2 V−1 s−1) | μ e/μh |
|---|---|---|---|---|---|---|---|
| a Average values of 15 individual cells are given in parentheses. | |||||||
| D18:QX-α (1:1.3) |
0.944 | 21.67/21.04 | 69.9 | 14.30 (14.14 ± 0.10) | 3.24 | 4.42 | 1.37 |
| D18:QX-γ (1:1.3) |
0.935 | 23.16/22.01 | 72.6 | 15.72 (15.10 ± 0.65) | 3.26 | 4.09 | 1.25 |
| D18:N3 (1:1.3) |
0.832 | 27.98/26.73 | 78.0 | 18.16 (17.84 ± 0.17) | 3.35 | 4.22 | 1.26 |
| D18:N3:QX-α (1:1.1:0.2) |
0.862 | 27.86/26.80 | 80.5 | 19.33 (19.01 ± 0.13) | 4.54 | 4.58 | 1.01 |
| D18:N3:QX-γ (1:1.1:0.2) |
0.853 | 27.39/26.40 | 78.3 | 18.30 (17.81 ± 0.21) | 4.14 | 5.07 | 1.22 |
However, the trend of PCEs in the ternary devices was reversed when QX-α and QX-γ were introduced into the host system of D18:N3, respectively, as shown in Fig. 3a, Fig. S18 (ESI†), Table 1, and Table S3 (ESI†). Impressively, the ternary device based on D18:N3:QX-α achieved a champion PCE of 19.33% with a VOC of 0.862 V, an excellent JSC of 27.86 mA cm−2, and a high FF of 0.805, which is one of the highest PCEs for single-junction OSCs reported to date. In contrast, when incorporating QX-γ, the ternary device based on D18:N3:QX-γ only gave a slightly higher PCE of 18.30% with a VOC of 0.853 V, JSC of 27.39 mA cm−2, and FF of 0.783, as compared to the binary device (PCE = 18.16%). Compared to the binary device, both the ternary devices exhibited significant improvements in VOC from 0.832 V to 0.853/0.862 V, owing to their cascaded energy level alignments. Notably, the D18:N3:QX-α-based device showed increases in JSC and FF with respect to the D18:N3:QX-γ-based device. As show in Table 1 and Fig. S17 (ESI†), the D18:N3:QX-α-based device possessed the highest hole mobility of 4.54 × 10−4 cm2 V−1 s−1 and the most balanced electron/hole mobilities with a ratio of 1.01, which could facilitate charge transport, leading to the higher FF. As shown in Fig. 3b and Table 1, the calculated JSC values from the external quantum efficiency (EQE) spectra were in good agreement with the JSC obtained from the J–V curves. The EQE spectra showed that all the devices exhibited similar and strong photoresponses in the spectral range of 350–900 nm. However, the D18:N3:QX-γ-based device had slightly lower EQE values in the 500 to 850 nm range than the other devices, thereby giving a lower JSC.
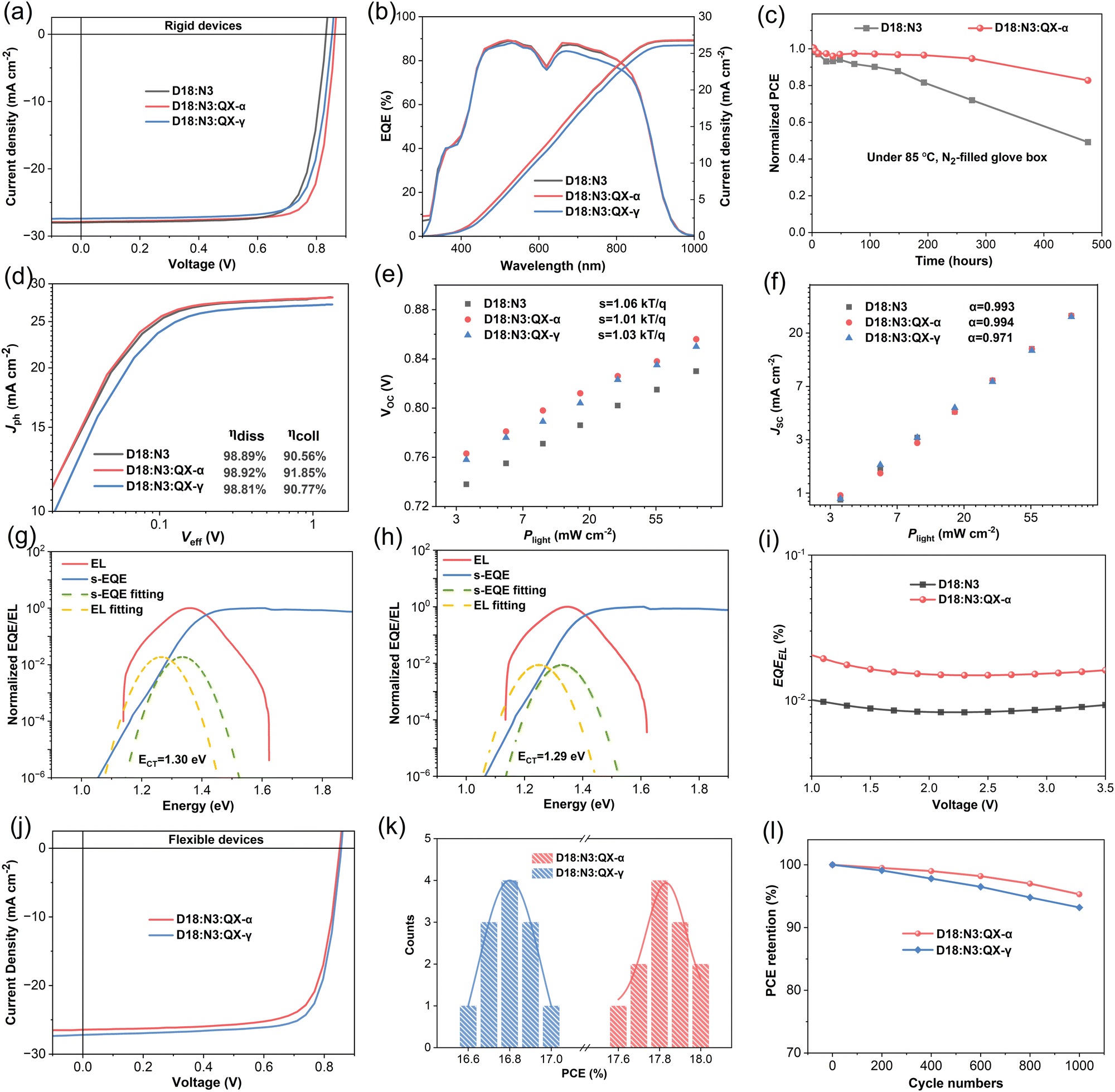 | ||
| Fig. 3 (a) J–V curves of the optimized OSCs fabricated by D18:N3, D18:N3:QX-α, and D18:N3:QX-γ. (b) EQE curves of the corresponding devices. (c) Normalized PCEs of the unencapsulated OSCs based on D18:N3, and D18:N3:QX-α under 85 °C in a N2-filled glove box. (d) Photocurrent density (Jph) versus effective bias (Veff) characteristics of the corresponding devices. VOC (e) and JSC (f) depending on the variation of the light intensity. FTPS-EQE and EL spectra of the optimized OSCs fabricated by D18:N3 (g) and D18:N3:QX-α (h). (i) EQEEL curves of the corresponding devices. (j) J–V curves of the flexible OSCs based on D18:N3:QX-α and D18:N3:QX-γ. (k) Histogram of the PCEs for 12 flexible OSCs. (l) PCE retentions of the flexible OSCs measured after bending with different cycle numbers at a curvature radius of 2 mm. | ||
To understand the exciton-dissociation (ηdiss) and charge-collection (ηcoll) properties of the OSCs, plots of the photocurrent density (Jph) versus effective voltage (Veff) were measured, as depicted in Fig. 3d. All the devices exhibited very similar ηdiss values of around 98.85%, indicating that there was nearly no difference in the exciton-dissociation efficiency of these devices. In contract, the D18:N3:QX-α-based device showed a slightly higher ηcoll of 91.85% compared to the 90.56% and 90.77% for the D18:N3 and D18:N3:QX-α-based devices, respectively. To investigate the charge-recombination mechanism of the OSCs, the dependences of VOC and JSCversus the light intensity (Plight) were carefully characterized. As shown in Fig. 3e, the S values were 1.06, 1.01, and 1.03kT/q (where k, T, and q are the Boltzmann constant, the Kelvin temperature, and the elementary charge, respectively) for the D18:N3, D18:N3:QX-α, and D18:N3:QX-γ-based devices, demonstrating that the introduction of QX-α into the host system could mitigate the trap-assisted recombination state. As shown in Fig. 3f, the α values of the D18:N3 and D18:N3:QX-α-based devices (0.993 and 0.994, respectively) were slightly higher than that of the D18:N3:QX-γ-based devices (0.971). Consequently, the D18:N3:QX-α-based device had more efficient charge collection, and weaker trap-assisted and bimolecular recombination, which is beneficial for improving the Jsc and FF. The morphology of these blend films should be the main reason for the results, which will be discussed later.
To understand the excited state dynamics of the three blend films, femtosecond transient absorption (fs-TAS) spectroscopy was performed, and the corresponding spectra are shown in Fig. S19 (ESI†). After excitation (at a wavelength of 500 nm), the ground-state bleach (GSB) signal of the donor D18 at 545 and 590 nm could be observed in the D18:N3, D18:N3:QX-α, and D18:N3:QX-γ-based blend films, which was consistent with the absorption feature of D18. As shown in Fig. S20 (ESI†), the decay time of the D18:N3:QX-α-blend film (0.64 ps) was shorter than that of the D18:N3 and D18:N3:QX-γ-blend films with values of 0.67 and 0.68 ps, respectively, demonstrating that fastest electron transfer occurred from the donor to acceptors in the D18:N3:QX-α-blend film.
To understand the origin of the impressive performance of the devices, the microstructures of these blend films were carefully investigated. The surface morphologies of the blend films were measured by tapping-mode atomic force microscopy (AFM). As shown in Fig. 4a–c, all the blend films based on D18:N3, D18:N3:QX-α, and D18:N3:QX-γ exhibited uniform and smooth surfaces with root mean square (RMS) roughness values of 0.88, 0.86, and 0.83 nm, respectively. In the phase images (Fig. S21, ESI†), fiber-like domains with proper sizes could be clearly observed in these blend films, which facilitated the achievement of a high JSC and FF.49,50 To determine the domain size, photoinduced force microscopy (PiFM) was carried out. As shown in Fig. 4d–i, N3 in the D18:N3:QX-γ blend film had an average domain size of 24.6 nm, which was slightly higher than that of the D18:N3 (22.5 nm) and D18:N3:QX-α blend films (22.6 nm). These result demonstrated that there was greater aggregation of N3 in the D18:N3:QX-γ blend film, leading to the lower JSC. As shown in Fig. 4j–l, Fig. S22 (ESI†), and Table S4 (ESI†), the crystallinity and orientation of the blend films were investigated by GI-WAXS. All the blend films displayed very strong face-on orientations with a π–π stacking peak at qz = 1.73 Å−1 (d = 3.63 Å) and a lamellar stacking peak at qXY = 0.29 Å−1 (d = 21.7 Å) along the OOP and IP directions, respectively, which is beneficial for charge transport in OSCs.51,52 The CCLs of the π–π stacking peaks for the three blend films were around 28.5 Å, while the D18:N3:QX-γ blend film possessed a slightly higher CCL value (82.68 Å) of the lamellar stacking peak than those of the D18:N3 (77.54 Å)and D18:N3:QX-γ blend films (73.92 Å). Consequently, the fiber-like morphologies, proper domain sizes, and strong face-on orientations of the blend films resulted in a high JSC and FF of the OSCs.
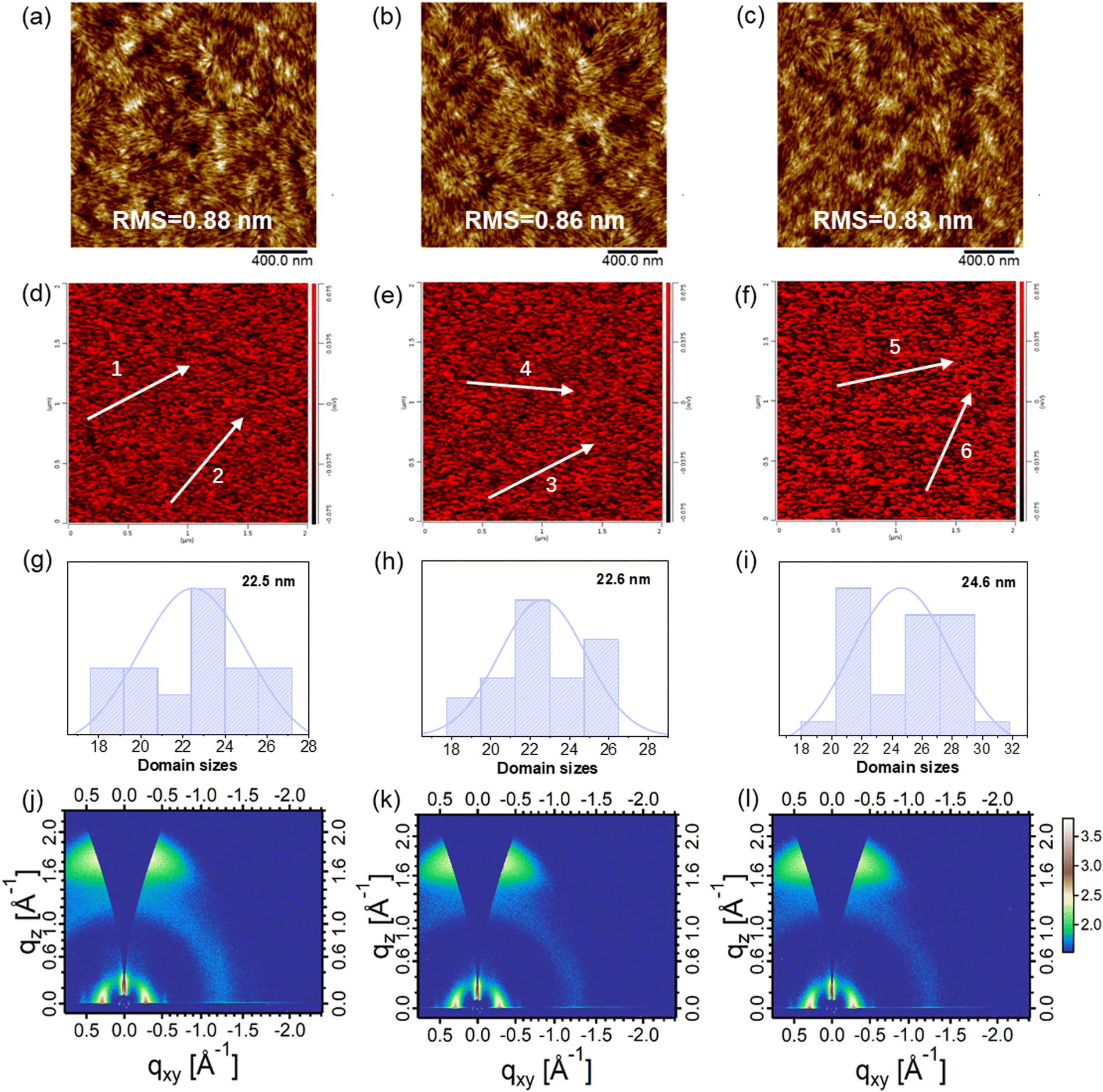 | ||
| Fig. 4 Height images obtained by tapping-mode AFM for the blend films based on D18:N3 (a), D18:N3:QX-α (b), and D18:N3:QX-γ (c). PiFM images at a wavenumber of 1276 cm−1 (representing N3) and the line profiles along the white arrows used to obtain the domain sizes of N3 in the blended films based on D18:N3 (d and g), D18:N3:QX-α (e and h), and D18:N3:QX-γ (f and i). 2D GI-WAXS patterns of the blend films based on D18:N3 (j), D18:N3:QX-α (k), and D18:N3:QX-γ (l). | ||
To explain the significantly enhanced VOC of the ternary OSCs, the detailed energy losses (Eloss) were measured. The FTPS-EQE, EL spectra, and EQEEL curves of the devices are given in Fig. 3g–i, the corresponding data are summarized in Table S5 (ESI†). Based on the Shockley–Queisser (SQ) limit model, the Eloss in solar cells can be divided into the three parts as shown in the following equations:53,54
| Eloss = Eg – qVoc |
| = (Eg −qVSQoc) + (qVSQoc − qVradoc) + (qVradoc − qVoc) |
| = ΔE1 + ΔE2 + ΔE3. |
Compared to the binary device, the ΔE2 value of the D18:N3:QX-α-based ternary device was decreased by 0.017 eV. Moreover, the value of the D18:N3:QX-α-based device was twice as high as that of the D18:N3-based device (Fig. 3i), representing ΔE3 value of 0.221 and 0.239 eV, due to the non-radiative recombination, respectively, thereby an 0.018 eV lowered ΔE3 could be observed in the ternary device. Consequently, the Eloss of the ternary device was obviously lower than that of the binary device (0.587 vs. 0.553 eV), resulting in its increased VOC. These results indicate that the introduction of QX-α into the binary system of D18:N3 could reduce not only the non-radiative recombination energy loss, but also the radiative recombination energy loss below the bandgap. The cascaded energy level alignments should be the main reason for reducing the energy losses.40
Flexible devices have attracted much attention as a promising power source for wearable electronics.55–57 Here, a conventional device architecture of PET/Ag grids/D-PH1000/PEDOT:PSS/active layer/PDINN/Ag was fabricated.58 As shown in Fig. 3j–k, the flexible device based on D18:N3:QX-γ exhibited a high PCE of 16.99% with a VOC of 0.851 V, a JSC of 26.44 mA cm−2, and an FF of 0.755. However, when using D18:N3:QX-α as the active layer, the flexible device achieved an outstanding PCE of 18.01% with a VOC of 0.856 V, a JSC of 27.21 mA cm−2, and an FF of 0.773, which was the highest value reported so far for flexible OSCs (Tables S6 and S7, ESI†). The enhanced PCE could be attributed to the improved JSC and FF, which were consistent with the previous rigid OSCs. Moreover, after 1000 continuous bending cycles with a radius of curvature (r) of 2 mm, the PCEs of the two flexible OSCs maintained 95.3% and 93.2% of their initial values for D18:N3:QX-α and D18:N3:QX-γ, respectively, as shown in Fig. 3l. This demonstrates that the two flexible ternary OSCs have high flexural strength.
In addition to the improvement of the PCE, the stability of OSCs is critical for commercial applications.59,60 As shown in Fig. 3c, the D18:N3:QX-α-based ternary device maintained 87% of its initial efficiency after 476 h of storage at 85 °C in a N2-filled glove box, indicating its excellent thermal stability. However, without the introduction of QX-α, the binary device retained only 49% of its starting PCE due to a significant reduction in VOC and FF. Under continuous illumination from an LED with 100 mW cm−2, the D18:N3:QX-α-based ternary device could retain 78% of its initial PCE after 200 h of illumination, which was obviously higher than that of the binary device (60%), as shown in Fig. S23 (ESI†). More importantly, in a long-term stability test, as depicted in Fig. S24 (ESI†), the ternary device retained over 97% of the initial PCE after being stored in a N2-filled glove box at room temperature for over 2200 h with an extrapolated T80 lifetime of over 17000 h, indicating its extremely high stability. These results demonstrate that QX-α could endow the ternary device with a high stability, which should be attributed to its strong intermolecular interactions and the acceptor alloy formed with N3.
3. Conclusion
Two large π-conjugated isomers, QX-α and QX-γ, with different orientations of their fused thiophene-rings were designed and synthesized to investigate the influence of the isomers in non-fullerene guest acceptors on the photovoltaic properties of the D18:N3 host system. QX-α and QX-γ possessed similar absorption spectra; however, QX-α exhibited a larger dipole moment, more ordered π–π stacking, and a higher surface energy as compared to QX-γ. In addition, when QX-α was introduced into the binary system of D18:N3, the ternary device showed higher and more balanced hole/electron mobilities, more efficient charge collection, and much lower energy losses than that of the binary device, resulting in obviously improved VOC and FF values. Compared with the D18:N3:QX-γ-based ternary device, the D18:N3:QX-α-based ternary device exhibited higher hole mobility, more balanced hole/electron mobilities, more efficient charge collection, and lower charge recombination, yielding significantly increased JSC and FF values. As a result, the PCE of the D18:N3:QX-α-based OSCs was as high as 19.33%, while the D18:N3:QX-γ-based device only exhibited a PCE of 18.30%, which was comparable to the standard binary device with a PCE of 18.16%. In addition, a record PCE of 18.01% could be achieved in flexible OSCs based on D18:N3:QX-α. Moreover, the ternary device showed a significant increase in efficiency retention from 49% to 87% after being stored at 85 °C for 476 h when QX-α was incorporated. These results demonstrate that the isomerization strategy can be an effective way to construct non-fullerene guest acceptors to achieve highly efficient and stable ternary OSCs.Author contributions
Z. C. conceived the idea and synthesized the two molecular acceptors. J. Z. and F. C. performed the detailed energy losses (Eloss) measurement. Z. C., J. S., Y. G. and X. T. fabricated the rigid devices and finished the characterization. W. S. fabricated flexible devices and finished basic characterizations of the flexible devices. D. Y and Z. C. drafted the manuscript. D. Y., J. G. and Z. G. revised the paper. Z. G supervised this project.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. U21A20331), the National Science Fund for Distinguished Young Scholars (No. 21925506), and Zhejiang Provincial Natural Science Foundation of China (No. LQ22E030013).References
- S. Park, T. Kim, S. Yoon, C. W. Koh, H. Y. Woo and H. J. Son, Adv. Mater., 2020, 30, 2002217 CrossRef.
- K. Jiang, J. Zhang, C. Zhong, F. R. Lin, F. Qi, Q. Li, Z. Peng, W. Kaminsky, S.-H. Jang, J. Yu, X. Deng, H. Hu, D. Shen, F. Gao, H. Ade, M. Xiao, C. Zhang and A. K.-Y. Jen, Nat. Energy, 2022, 7, 1076–1086 CrossRef.
- Y. Liu, B. Liu, C.-Q. Ma, F. Huang, G. Feng, H. Chen, J. Hou, L. Yan, Q. Wei, Q. Luo, Q. Bao, W. Ma, W. Liu, W. Li, X. Wan, X. Hu, Y. Han, Y. Li, Y. Zhou, Y. Zou, Y. Chen, Y. Li, Y. Chen, Z. Tang, Z. Hu, Z.-G. Zhang and Z. Bo, Sci. China: Chem., 2022, 65, 224–268 CrossRef CAS.
- Y. Liu, B. Liu, C.-Q. Ma, F. Huang, G. Feng, H. Chen, J. Hou, L. Yan, Q. Wei, Q. Luo, Q. Bao, W. Ma, W. Liu, W. Li, X. Wan, X. Hu, Y. Han, Y. Li, Y. Zhou, Y. Zou, Y. Chen, Y. Liu, L. Meng, Y. Li, Y. Chen, Z. Tang, Z. Hu, Z.-G. Zhang and Z. Bo, Sci. China: Chem., 2022, 65, 1457–1497 CrossRef CAS.
- L. Zhu, M. Zhang, J. Xu, C. Li, J. Yan, G. Zhou, W. Zhong, T. Hao, J. Song, X. Xue, Z. Zhou, R. Zeng, H. Zhu, C.-C. Chen, R. C. I. MacKenzie, Y. Zou, J. Nelson, Y. Zhang, Y. Sun and F. Liu, Nat. Mater., 2022, 21, 656–663 CrossRef CAS.
- H. Chen, S. Y. Jeong, J. Tian, Y. Zhang, D. R. Naphade, M. Alsufyani, W. Zhang, S. Griggs, H. Hu, S. Barlow, H. Y. Woo, S. R. Marder, T. D. Anthopoulos, I. McCulloch and Y. Lin, Energy Environ. Sci., 2023, 16, 1062–1070 RSC.
- M. Deng, X. Xu, Y. Duan, L. Yu, R. Li and Q. Peng, Adv. Mater., 2023, 35, 2210760 CrossRef CAS PubMed.
- J. Gao, N. Yu, Z. Chen, Y. Wei, C. Li, T. Liu, X. Gu, J. Zhang, Z. Wei, Z. Tang, X. Hao, F. Zhang, X. Zhang and H. Huang, Adv. Sci., 2022, 9, 2203606 CrossRef CAS.
- N. Gasparini, A. Salleo, I. McCulloch and D. Baran, Nat. Rev. Mater., 2019, 4, 229–242 CrossRef.
- Z. Chen, W. Song, K. Yu, J. Ge, J. Zhang, L. Xie, R. Peng and Z. Ge, Joule, 2021, 5, 2395–2407 CrossRef CAS.
- T. Umeyama and H. Imahori, Acc. Chem. Res., 2019, 52, 2046–2055 CrossRef CAS.
- Z. Luo, T. Liu, H. Yan, Y. Zou and C. Yang, Adv. Funct. Mater., 2020, 30, 2004477 CrossRef CAS.
- X. Shen, X. Lai, H. Lai, T. Zhao, Y. Zhu, M. Pu, H. Wang, P. Tan and F. He, Macromolecules, 2022, 55, 6384–6393 CrossRef CAS.
- Z. Jia, Q. Ma, L. Meng, J. Zhang, S. Qin, Z. Chen, X. Li, J. Zhang, J. Li, Z. Zhang, Z. Wei, Y. Yang and Y. Li, Adv. Funct. Mater., 2022, 32, 2204720 CrossRef CAS.
- H. Wang, H. Chen, W. Xie, H. Lai, T. Zhao, Y. Zhu, L. Chen, C. Ke, N. Zheng and F. He, Adv. Funct. Mater., 2021, 31, 2100877 CrossRef CAS.
- Z. Luo, T. Liu, J. Oh, R. Ma, J. Miao, F. Ni, G. Zhang, R. Sun, C. Zhang, Z. Chen, Y. Zou, J. Min, C. Yang, H. Yan and C. Yang, Adv. Funct. Mater., 2022, 32, 2203200 CrossRef CAS.
- D. Deng, H. Zhang, Q. Wu, M. A. Adil, C. Yang, J. Zhang and Z. Wei, J. Mater. Chem. C, 2020, 8, 7519–7526 RSC.
- E. Lim, Appl. Sci., 2021, 11, 755 CrossRef CAS.
- G. Cai, Z. Chen, M. Li, Y. Li, P. Xue, Q. Cao, W. Chi, H. Liu, X. Xia, Q. An, Z. Tang, H. Zhu, X. Zhan and X. Lu, Adv. Sci., 2022, 9, 2103428 CrossRef CAS.
- J. Yuan, Y. Zhang, L. Zhou, G. Zhang, H.-L. Yip, T.-K. Lau, X. Lu, C. Zhu, H. Peng, P. A. Johnson, M. Leclerc, Y. Cao, J. Ulanski, Y. Li and Y. Zou, Joule, 2019, 3, 1140–1151 CrossRef CAS.
- Q. He, P. Ufimkin, F. Aniés, X. Hu, P. Kafourou, M. Rimmele, C. L. Rapley and B. Ding, SusMat, 2022, 2, 591–606 CrossRef CAS.
- J. Ge, L. Xie, R. Peng and Z. Ge, Adv. Mater., 2023, 35, 2206566 CrossRef CAS.
- D. Mo, H. Chen, J. Zhou, L. Han, Y. Zhu, P. Chao, N. Zheng, Z. Xie and F. He, J. Mater. Chem. A, 2020, 8, 23955–23964 RSC.
- X. Zhao, Q. An, H. Zhang, C. Yang, A. Mahmood, M. Jiang, M. H. Jee, B. Fu, S. Tian, H. Y. Woo, Y. Wang and J.-L. Wang, Angew. Chem., Int. Ed., 2023, 62, e202216340 CAS.
- H. Yu, M. Pan, R. Sun, I. Agunawela, J. Zhang, Y. Li, Z. Qi, H. Han, X. Zou, W. Zhou, S. Chen, J. Y. L. Lai, S. Luo, Z. Luo, D. Zhao, X. Lu, H. Ade, F. Huang, J. Min and H. Yan, Angew. Chem., Int. Ed., 2021, 60, 10137–10146 CrossRef CAS PubMed.
- H. Lai, X. Lai, Z.-Y. Chen, Y. Zhu, H. Wang, H. Chen, P. Tan, Y. Zhu, Y. Zhang and F. He, CCS Chem., 2023, 5, 1118–1129 CrossRef CAS.
- M. Luo, Y. Chen, J. Liang, J. Zhou, D. Yuan, Z. Zhang, X. Liu, L. Zhang, Z. Xie and J. Chen, ACS Appl. Mater. Interfaces, 2022, 14, 35985–35996 CrossRef CAS.
- X. Kong, C. Zhu, J. Zhang, L. Meng, S. Qin, J. Zhang, J. Li, Z. Wei and Y. Li, Energy Environ. Sci., 2022, 15, 2011–2020 RSC.
- S. Jeong, J. Park, Y. Ji, Y. Cho, B. Lee, M. Jeong, S. Jung, S. Yang, Y. Zhang, S.-J. Yoon and C. Yang, J. Mater. Chem. A, 2023, 11, 4703–4716 RSC.
- Z. Luo, R. Ma, Z. Chen, Y. Xiao, G. Zhang, T. Liu, R. Sun, Q. Zhan, Y. Zou, C. Zhong, Y. Chen, H. Sun, G. Chai, K. Chen, X. Guo, J. Min, X. Lu, C. Yang and H. Yan, Adv. Energy Mater., 2020, 10, 2002649 CrossRef CAS.
- Y. Liang, D. Zhang, Z. Wu, T. Jia, L. Lüer, H. Tang, L. Hong, J. Zhang, K. Zhang, C. J. Brabec, N. Li and F. Huang, Nat. Energy, 2022, 7, 1180–1190 CrossRef CAS.
- C. Sun, J.-W. Lee, C. Lee, D. Lee, S. Cho, S.-K. Kwon, B. J. Kim and Y.-H. Kim, Joule, 2023, 7, 416–430 CrossRef CAS.
- Z. Chen, J. Ge, Y. Guo, M. Zhao, J. Shi, Y. Qiu, E. Zhou and Z. Ge, ACS Energy Lett., 2022, 7, 3432–3438 CrossRef CAS.
- S. Dey, Small, 2019, 15, 1900134 CrossRef.
- M. Liu, X. Han, H. Chen, Q. Peng and H. Huang, Nat. Commun., 2023, 14, 2500 CrossRef CAS.
- P. Yan, D. Yang, H. Wang, S. Yang and Z. Ge, Energy Environ. Sci., 2022, 15, 3630–3669 RSC.
- S. Pang, X. Zhou, S. Zhang, H. Tang, S. Dhakal, X. Gu, C. Duan, F. Huang and Y. Cao, ACS Appl. Mater. Interfaces, 2020, 12, 16531–16540 CrossRef CAS.
- X. Wang, H. Lu, Y. Liu, A. Zhang, N. Yu, H. Wang, S. Li, Y. Zhou, X. Xu, Z. Tang and Z. Bo, Adv. Energy Mater., 2021, 11, 2102591 CrossRef CAS.
- L. Xie, A. Lan, Q. Gu, S. Yang, W. Song, J. Ge, R. Zhou, Z. Chen, J. Zhang, X. Zhang, D. Yang, B. Tang, T. Wu and Z. Ge, ACS Energy Lett., 2023, 8, 361–371 CrossRef CAS.
- F. Meng, Y. Qin, Y. Zheng, Z. Zhao, Y. Sun, Y. Yang, K. Gao and D. Zhao, Angew. Chem., Int. Ed., 2023, 62, e202217173 CrossRef CAS.
- D. K. Owens and R. C. Wendt, J. Appl. Polym. Sci., 1969, 13, 1741–1747 CrossRef CAS.
- D. H. Kaelble, J. Adhes., 1970, 2, 66–81 CrossRef CAS.
- L. Ye, H. Hu, M. Ghasemi, T. Wang, B. A. Collins, J.-H. Kim, K. Jiang, J. H. Carpenter, H. Li, Z. Li, T. McAfee, J. Zhao, X. Chen, J. L. Y. Lai, T. Ma, J.-L. Bredas, H. Yan and H. Ade, Nat. Mater., 2018, 17, 253–260 CrossRef CAS PubMed.
- M. Sumita, K. Sakata, S. Asai, K. Miyasaka and H. Nakagawa, Polym. Bull., 1991, 25, 265–271 CrossRef CAS.
- F. Liu, L. Zhou, W. Liu, Z. Zhou, Q. Yue, W. Zheng, R. Sun, W. Liu, S. Xu, H. Fan, L. Feng, Y. Yi, W. Zhang and X. Zhu, Adv. Mater., 2021, 33, 2100830 CrossRef CAS.
- A. D. de Zerio and C. Müller, Adv. Energy Mater., 2018, 8, 1702741 CrossRef.
- Q. Liu, Y. Jiang, K. Jin, J. Qin, J. Xu, W. Li, J. Xiong, J. Liu, Z. Xiao, K. Sun, S. Yang, X. Zhang and L. Ding, Sci. Bull., 2020, 65, 272–275 CrossRef CAS.
- D. Yang, Y. Jiao, L. Yang, Y. Chen, S. Mizoi, Y. Huang, X. Pu, Z. Lu, H. Sasabe and J. Kido, J. Mater. Chem. A, 2015, 3, 17704–17712 RSC.
- L. Zhu, M. Zhang, J. Xu, C. Li, J. Yan, G. Zhou, W. Zhong, T. Hao, J. Song, X. Xue, Z. Zhou, R. Zeng, H. Zhu, C.-C. Chen, R. C. I. MacKenzie, Y. Zou, J. Nelson, Y. Zhang, Y. Sun and F. Liu, Nat. Mater., 2022, 21, 656–663 CrossRef CAS PubMed.
- T. Liu, L. Huo, S. Chandrabose, K. Chen, G. Han, F. Qi, X. Meng, D. Xie, W. Ma, Y. Yi, J. M. Hodgkiss, F. Liu, J. Wang, C. Yang and Y. Sun, Adv. Mater., 2018, 30, 1707353 CrossRef PubMed.
- Y. Ma, M. Zhang, S. Wan, P. Yin, P. Wang, D. Cai, F. Liu and Q. Zheng, Joule, 2021, 5, 197–209 CrossRef CAS.
- T. Zhang, Y. Xu, H. Yao, J. Zhang, P. Bi, Z. Chen, J. Wang, Y. Cui, L. Ma, K. Xian, Z. Li, X. Hao, Z. Wei and J. Hou, Energy Environ. Sci., 2023, 16, 1581–1589 RSC.
- J. Yao, T. Kirchartz, M. S. Vezie, M. A. Faist, W. Gong, Z. He, H. Wu, J. Troughton, T. Watson, D. Bryant and J. Nelson, Phys. Rev. Appl., 2015, 4, 014020 CrossRef.
- D. Yang, Y. Wang, T. Sano, F. Gao, H. Sasabe and J. Kido, J. Mater. Chem. A, 2018, 6, 13918–13924 RSC.
- K. Fukuda, K. Yu and T. Someya, Adv. Energy Mater., 2020, 10, 2000765 CrossRef CAS.
- Y. Sun, T. Liu, Y. Kan, K. Gao, B. Tang and Y. Li, Small Sci., 2021, 1, 2100001 CrossRef.
- G. Zeng, W. Chen, X. Chen, Y. Hu, Y. Chen, B. Zhang, H. Chen, W. Sun, Y. Shen, Y. Li, F. Yan and Y. Li, J. Am. Chem. Soc., 2022, 144, 8658–8668 CrossRef CAS PubMed.
- W. Song, K. Yu, J. Ge, L. Xie, R. Zhou, R. Peng, X. Zhang, M. Yang, Z. Wei and Z. Ge, Matter, 2022, 5, 1877–1889 CrossRef CAS.
- Q. Burlingame, M. Ball and Y.-L. Loo, Nat. Energy, 2020, 5, 947–949 CrossRef.
- X. Xu, D. Li, J. Yuan, Y. Zhou and Y. Zou, EnergyChem, 2021, 3, 100046 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ee01164j |
| This journal is © The Royal Society of Chemistry 2023 |