
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDeciphering electrochemical interactions in metal–polymer catalysts for CO2 reduction†
Xingyu
Wang
 a,
Sanjubala
Sahoo
bc,
Jose
Gascon
d,
Mikhail
Bragin
e,
Fangyuan
Liu
b,
Julia
Olchowski
a,
Samuel
Rothfarb
a,
Yuankai
Huang
a,
Wenjun
Xiang
a,
Pu-Xian
Gao
bc,
S. Pamir
Alpay
bc and
Baikun
Li
*a
a,
Sanjubala
Sahoo
bc,
Jose
Gascon
d,
Mikhail
Bragin
e,
Fangyuan
Liu
b,
Julia
Olchowski
a,
Samuel
Rothfarb
a,
Yuankai
Huang
a,
Wenjun
Xiang
a,
Pu-Xian
Gao
bc,
S. Pamir
Alpay
bc and
Baikun
Li
*a
aDepartment of Civil and Environmental Engineering, University of Connecticut, Storrs, Connecticut 06269-3037, USA. E-mail: baikun.li@uconn.edu
bInstitute of Materials Science, University of Connecticut, Storrs, Connecticut 06269-3136, USA
cDepartment of Materials Science and Engineering, University of Connecticut, Storrs, Connecticut 06269-3136, USA
dDepartment of Chemistry, University of Connecticut, Storrs, Connecticut 06269-3037, USA
eDepartment of Electrical and Computer Engineering, University of Connecticut, Storrs, Connecticut 06269-3037, USA
First published on 8th August 2023
Abstract
Polymers play a critical role in catalyst design to stabilize metal nanoparticles on the cathode for electrochemical carbon dioxide reduction reaction (CO2RR). However, electrochemical interactions between the metal and polymer complex remain unclear due to the lack of quantitative analysis of catalytic process variations tailored by such structure modifications on the cathode surface. In this study, we investigate the effects of polymer physical binding on cathode surface polarity, intermediate adsorption, and the barriers of CO2RR. We examine the resultant selectivity, taking into account mass transport and charge transfer. Especially, we select polytetrafluoroethylene (PTFE) as the model polymer to minimize ion flux interference, since the structure of PTFE, with the absence of ionic groups for ion transport, exhibits unmatched physiochemical performance. By utilizing PTFE, we ensure the integrity of our observations, enabling a precise analysis of the effects of polymer physical binding on the performance and selectivity of CO2RR. In addition, a comprehensive multiscale simulation-experiment tandem analysis is conducted for the PTFE–Cu complex to identify the mass and charge transfer processes. Our analysis offers a mechanistic foundation for different CO2RR pathways through both dynamic processes and molecular mechanisms. Our study reveals an unusual shift of surface reaction mechanism induced by direct mass transport alternation and indirect charge transfer from the redistribution of H+/CO2 adsorption on the cathode surface. Specifically, our modeling results demonstrate a significant enhancement in the binding energy of CO2 (from −0.31 eV to −0.38 eV) and critical intermediates involved in CH4 generation (from −1.56 eV to −1.63 eV) upon the addition of PTFE. Our experimental findings validate these results by revealing a 29.9% reduction in surface charge when 10% PTFE is introduced, in comparison to pristine Cu. This binding energy increment and surface charge reduction reinforces the CO2 reduction process, modifies the CO2RR pathway, and ultimately enhances the average CH4 production by 10%. It is worth noting that despite a 32.26% increase in ohmic resistance, the benefits of PTFE addition persist and lower the energy barrier from 1.14 eV to 0.68 eV during CO protonation. Our findings unveil a novel approach for polymer binding in metal design, leading to simpler and more effective materials compared to the intricate polymer encapsulation for CO2RR.
Broader contextElectrochemical CO2 reduction is a key strategy of the carbon removal portfolio considering its profitability towards recyclable fuel energy and carbon products. With the goal of acquiring a highly efficient CO2 reduction reaction (CO2RR) without utilizing noble metals and complicated methods, we employ an integration of multiscale simulations and judiciously designed experiments, and rationalize the necessity of polymer binding catalysts for potential scale-up application. Polymer-binding catalysts distinctly contribute to electrochemical CO2 reduction and promote reaction efficiency compared with conventional metal catalysts. In this study, polytetrafluoroethylene (PTFE) was selected as the target because of its wide application as a polymer binder backbone in almost all the cost-efficient modern catalyst designs. As non-conductive materials, polymers could generate a membrane-like effect, alter distributions of molecules and charges, redefine quantitative profiles of mass and charge transfer, and consequently revolutionize our view of their role in the electrochemical processes. Our novel quantitative multiscale analysis of CO2RR reveals substantial electrocatalytic enhancement through physical metal–polymer binding, altering molecular surface adsorption and polarity on the cathode, while maintaining robust catalytic performance across diverse potentials. With a multiscale-experiment analysis tool specifically designed to decipher the complexity of the metal–polymer junction, our study innovatively discovered coherent interactions between polymer–metal catalytic structures and surface reaction processes, which ushers in new pathways of simple metal–polymer cathode design and analysis for a wide range of affordable and efficient CO2 reduction devices. |
Introduction
Electrochemical carbon dioxide reduction reaction (CO2RR) is a prospective pathway to drive down the net cost of carbon removal while achieving carbon neutrality with potential commercialization opportunity.1 Recent studies focus on the description and implementation of CO2RR using highly reactive material systems at the catalytic reaction interface.2,3 The long reaction process chains of the CO2RR are characterized by multiple stages of electron transfer under both equilibrium and non-equilibrium states,4–7 where organic polymers have been introduced into cathode materials as they can stabilize electrocatalysis and even influence the product selectivity.8,9 Metal–polymer cathode designs exhibit high performance in terms of catalytic activity and faradaic efficiency.2,3,10,11 However, understanding of polymer-induced cathodic activity and CO2RR surface reaction processes is still unclear. Given the essential role of mass/charge transfer in the overall efficiency of CO2RR process, the lack of mass and charge transfer profiles at the complex metal–polymer junctional interface poses a major obstacle for new material design.Upon the incorporation in the modern CO2RR cathode design, polymers are often required to be electrochemically stable during the reduction of CO2. Although a special group of ionic polymers (ionomers) has been recently introduced as the catalyst binder to tune the kinetics of electrochemical CO2RR through specific noncovalent interactions with CO2 molecules,8,12 the ionic transport could dominate the surface process due to the ion exchange and conduction effects. Hence, the influences towards the adsorption and electron transfer of CO2 mainly come from the interference of the ionic groups attached onto the polymers. Whereas in electrochemical CO2RR systems, polymer groups provide the major functionality for cathodes to enable the exceptional gas permeability and stability under high voltages, keep the adsorption of dissolved ionic groups onto the cathode surface, and reduce the overpotential caused by surface polarization.13 Current knowledge about quantification of surface charge and polymer participation in electrochemical processes is still limited, hindering comprehension and rationalization of polymer group functionality in the CO2RR process. We target in this study polymers without ionic modifications to avoid the interference from ion flux caused by ionomer materials and fundamentally elucidate the CO2RR mass/charge transfer process.
Development of new metal–polymer cathode materials can be roughly divided into two categories according to the catalyst-polymer interactions, polymer encapsulation and polymer binding.9,14 Although encapsulation could potentially bolster the polymer engagement in the metal-dominated catalytic process and strengthen catalytic activity compared to polymer binding, it requires complicated synthesis steps and delicate condition controls to ensure the modified catalytic structures with good conductivity and proper energy gap.15,16 At the current stage, polymer binding is a better option as it separates the fabrication of organics and metals, reduces the complexity of material design, and assures the flexibility of the target oriented CO2RR system configurations. Polytetrafluoroethylene (PTFE) has been commonly recognized as an essential and intrinsic polymer binder for cathode fabrication due to its high hydrophobicity and physical stability under various cathodic potentials, making it a preferred choice in the design of porous or gas diffusion cathodes.17,18 PTFE has been utilized to prevent cathodic flooding and reduce the thickness of the diffusion layer wetted by electrolyte.2,19 Furthermore, PTFE serves as a crucial backbone for various ionomers used in electrochemistry and membrane science (e.g., Nafion20), and its exceptional mechanical and chemical performance makes it the most effective material in CO2 reduction reaction applications. Given the variable electrolyte environments in an electrolysis system, no material has been proven as a good alternative of PTFE. However, the catalytic effects of PTFE and other polymers (e.g., polyvinyl alcohol and polyvinylidene fluoride) have received limited attention,21–23 which primarily attributes to the limited understanding regarding the relationships between the intrinsic electrochemical properties (e.g., polarity) of polymers and the kinetics of the CO2 reduction reaction beyond the premise of polymer stability.24 Consequently, the accuracy of the cathode performance comparison is compromised without considering the impact of PTFE. The evaluation of PTFE, a vital polymer binder in cathode fabrication, necessitates a thorough consideration of its impact on both catalytic efficiency and cathode performance. Due to the exceptional properties inherent to PTFE, comprehending and assessing its role is of critical significance in order to augment overall system performance further assumes.
Polymers can crystallize by partially re-aligning the molecular chain structure, and thus they could drastically change the cathode surface morphology and alter the surface electrochemical processes (e.g., mass/charge transfer loss, binding orders) on the cathode–electrolyte interface.25,26 As a result, the polymer binders in the catalyst may not be electrochemically stable as were assumed due to the formation of capacitor-like double layer structure containing both faradaic and non-faradaic processes.27 Moreover, the magnitude of the interfacial dipole moment from the molecules attached on the metal surface can affect the metal function deterministic to the energy barriers of electron transfer in heterogeneous catalytic reactions (e.g., CO2RR).28,29 Therefore, as the direct measurement and characterization of interfacial dipole, cathode surface polarity becomes the center stage of our study to elucidate the essential mechanisms of the polymers (e.g., PTFE) that alter the selectivity and efficiency of CO2RR reaction through the surface processes and catalytical barriers.
In this study, we elucidate the modifications in mass and charge transfer introduced by PTFE through two interwoven steps. First, from a modeling perspective, we redesign the multiscale paradigm to evaluate dynamic CO2RR surface electrochemical reaction process via characterizing mass and charge transfer barriers of metal–polymer influence from both atomistic and continuum perspectives (Fig. 1b). Density Functional Theory (DFT) enables us to illustrate the PTFE effect through adsorption and reaction pathway calculations, including binding and barrier energies,30–32 while electrochemistry identifies the corresponding reaction rates and CO2RR polarization curves to determine the ideal reaction performance associated with the DFT calculations.7,33 Furthermore, transport modeling provides insights into ion distribution profiles and the supply of the two pivotal reactants-CO2 and H+, towards the cathode.34,35 Secondly, from an experimental perspective, we extensively explore the catalyst's performance in the presence of PTFE, which includes Cu yielding multiple products and Sn and Ag yielding single product (HCOOH and CO, respectively).1 These well-crafted experiments validate the modeling findings obtained from theoretical multiscale simulations. Through the seamless integration of these two pivotal stages, our study demonstrates the catalytic effects of polymers in conjunction with metals, supported by comparisons of catalysts with and without PTFE, both in theoretical modeling and experiments.
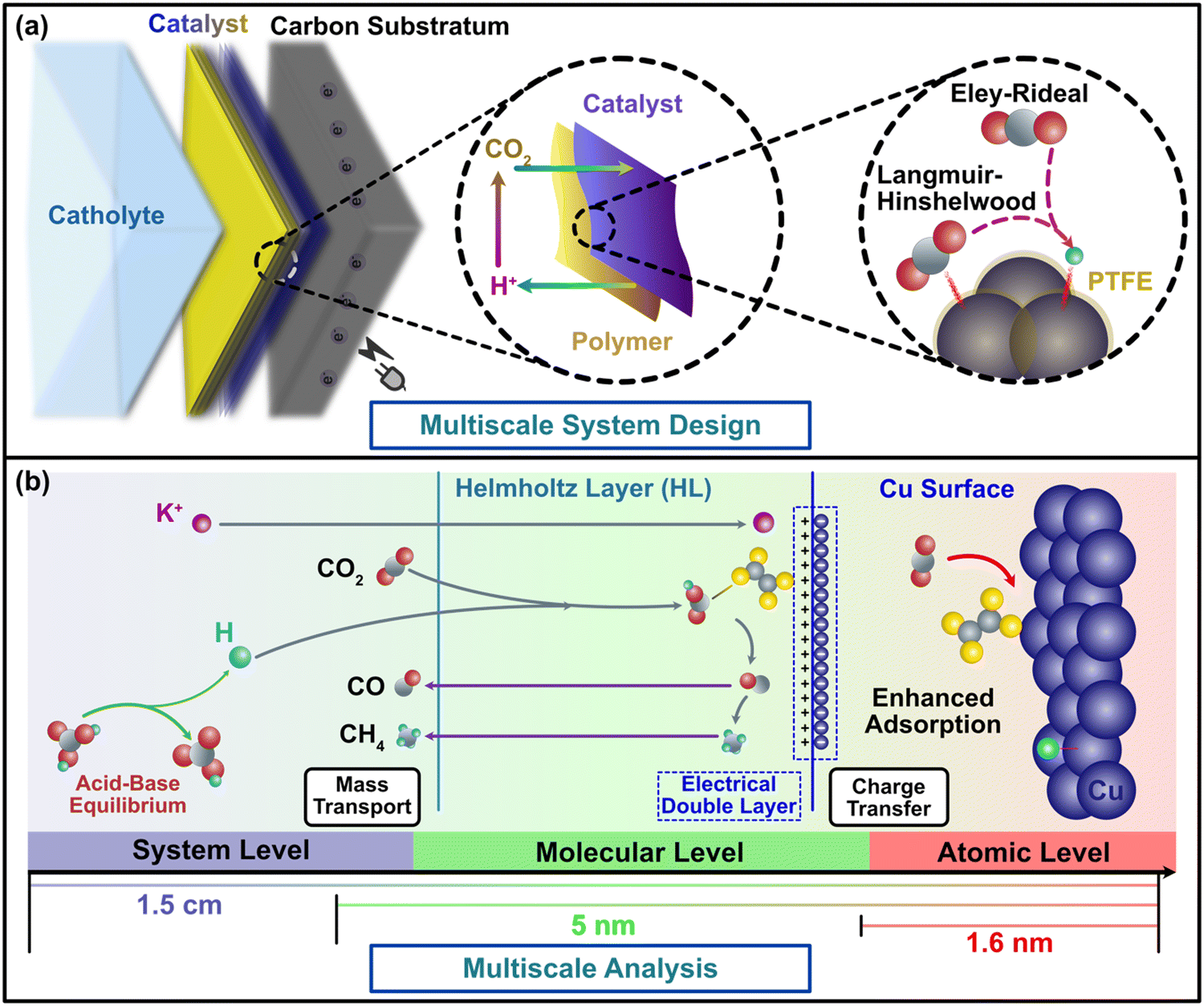 | ||
| Fig. 1 Multiscale demonstration and theoretical analysis of a CO2RR cathodic system with metal–polymer catalyst. (a) Multiscale diagram of CO2RR cathode and ambient environment on the cathode–electrolyte interface. The design of the metal–polymer complex catalyst system can bolster the robustness of the cathode and influence the mass and charge transfer process of CO2, H+, and electrons on cathode surface. (b) The three levels of the multiscale theoretical diagram associated with each scale of the material system in (a). The system level describes the transport and distribution of molecules in the bulk solution as well as the region close to the cathode surface (from 5 nm to 1.5 cm). The molecular level is critical because all the ambient environmental factors (e.g., concentration, pH, cations, temperature) would influence CO2 reduction through this layer. In this model level, the mass transport and charge transfer barriers and the surface reaction processes were defined. These processes in the molecular level function as the bridge between the distribution described by the macroscopic model and reactivity described by the quantum-mechanical model. This model level functions between 1.6 nm and 5 nm. The atomistic level uses quantum mechanics to calculate the free energies and barrier energies on the cathode surface. Free energies were used in the microkinetic model to determine the reaction kinetics, while the rate limiting steps were determined via the barrier energies obtained from the various reaction pathways. The binding energies of reactants, intermediates, and products in CO2RR were calculated to interpret the stability of adsorbed species. This model level describes the reaction in the atomistic scope within 1.6 nm from the cathode surface. | ||
Using our novel scheme integrating the structure–process–property multiscale model and a judiciously designed metal/PTFE experiment, our study reveals that the crystallization of polymer binders on a metal catalyst can change the cathode surface polarity and alter the molecular adsorption of reactants and intermediates. These modifications consequently lead to a shift in the surface reaction mechanisms. We identify possible pathways of CO2 reduction through the analysis of mass transport and charge transfer. By revealing a critical surface polarity-induced interaction overlooked in previous studies,10,17,18 our findings provide an unprecedented quantitative analysis illustrating the drastic influence of polymer binders (e.g., PTFE) on surface reaction process of cathodes, unfold the formerly unrecognized role of surface reaction kinetics related to polymer binders on catalyst performance, and consequently formulate new avenues for future design of CO2RR cathode materials.
Results and discussion
Polymer-induced CO2 discrepancy during mass transfer of surface reaction processes
Relative to metal-only cathode materials, metal–polymer cathode materials exhibit inconsistent surface polarity due to the notably lower dielectric constants of the polymer coating on the metal surface. Although surface polarity is a crucial cathode property, it has not been fully articulated in previous CO2RR studies as a result of the difficulty in associating it with intrinsic catalytic structure of the cathode material.9 This gap can be addressed using the electrochemical analysis in our multiscale paradigm (Fig. 1), which bridges the discontinuity of conventional multiscale studies and provides a feasible path to determine the complex metal–polymer interactions. Conventional understanding infers that cathode polarity influences CO2RR efficacy in two aspects.28,29 Firstly, due to the redistribution of the active sites on the catalyst, CO2 is immensely influenced by polarization and more likely to adhere to the electrode surface. Secondly, subsided double layer capacitance with low polarity mitigates the potential drop in the Helmholtz layer located around 5 nm away from the catalyst surface (Fig. S2a, ESI†), and raises the charge transfer rate. Concurrently, surface polarity is tuned by modifying the Cu cathodes using PTFE in the experimental design of this study, where a clear trend of surface coverage alternation has been found from Cu dominance (blue in Fig. 2a) to PTFE–Cu mixture (yellow in Fig. 2a). The results of contact angle and surface charge measurements confirm the direct correlation between surface polarity and the PTFE amount mixed with Cu nanoparticles as the higher amount of PTFE are associated with larger contact angle (contact angle grows from 26° to ∼90°, Fig. 2c), while the mixing of PTFE drastically changes the cathode surface charge (Fig. 2d). Furthermore, the PTFE-covered surface exhibits a relatively smoother pattern and has larger particle cluster size compared to the pristine Cu cathode, which manifests the bridging effects of PTFE between Cu nanoparticles and the lower availability of active sites for catalytic processes of CO2RR.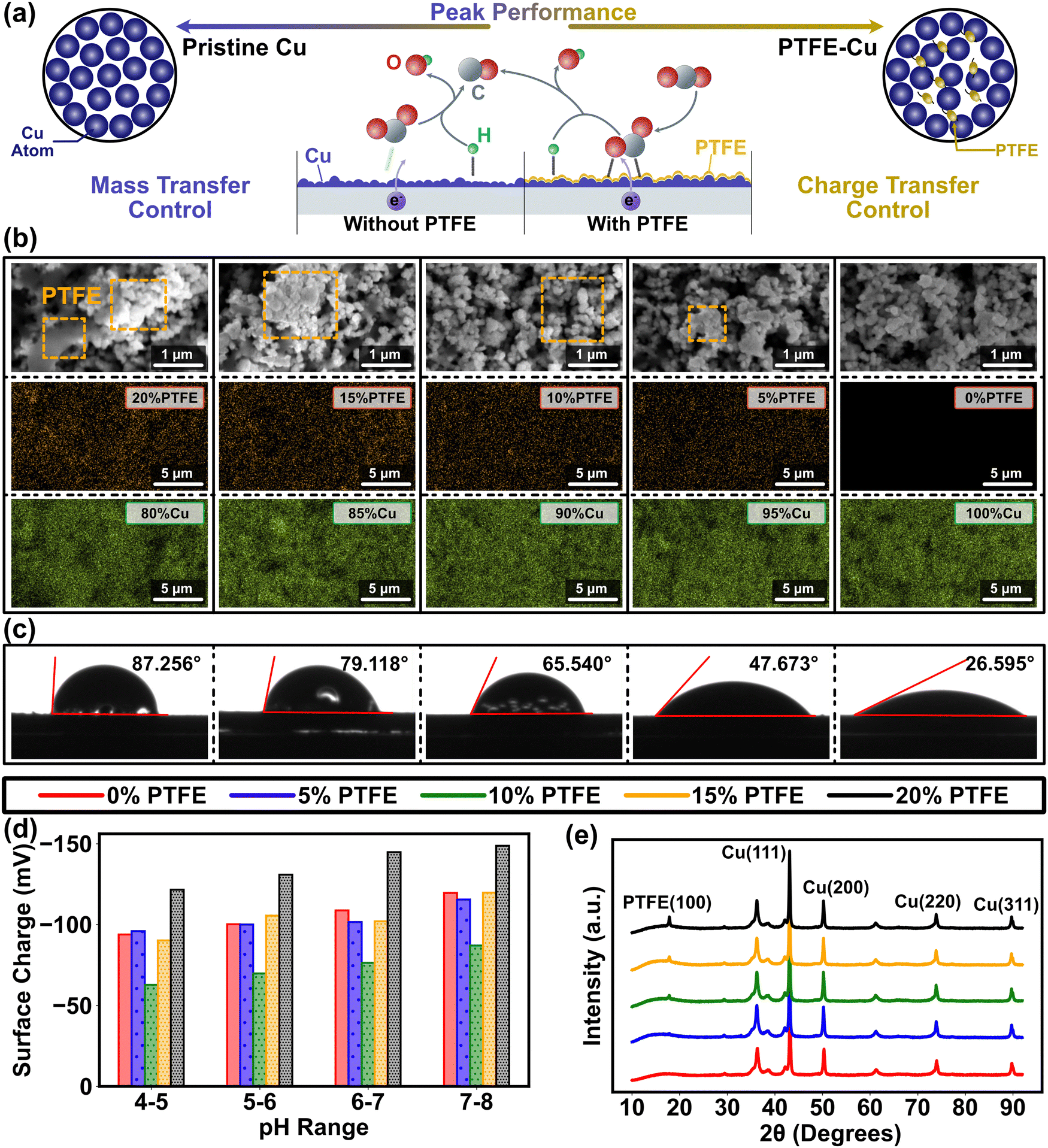 | ||
| Fig. 2 Illustration of the impact of Cu and PTFE on CO2RR cathode morphology. (a) Diagram of Cu and PTFE impact on CO2RR process. The alternation of surface polarity tilts the equilibrium of the molecular distribution in the Helmholtz layer away from polarization, thereby boosting the CO2 concentration on the cathode surface. (b) SEM images (top) and EDAX elemental analysis (bottom) of pristine Cu cathode (green for Cu element) and PTFE–Cu cathode (orange for F element) with the PTFE ratios from 5% to 20%, indicating the even distribution of metal and polymer on the cathode. The PTFE is marked by orange dash square in the SEM images. The corresponding SEM images of EDAX mappings have been included in Fig. S27 (ESI†). (c) Water contact angles of pristine Cu cathode and PTFE–Cu cathode with the PTFE ratios from 5% to 20%, which show the influence of different ratio of PTFE on the Cu catalyst polarity. (d) Variation of the cathode surface charge with the ratios of Cu to PTFE and the pH of bulk solution. (e) The XRD results of the pristine Cu and PTFE–Cu cathodes with the PTFE ratio from 5% to 20%. PTFE (100) peak rises with the increase in the PTFE content. | ||
An explicit understanding of PTFE for CO2 reduction necessitates an investigation into the composition and coverage of PTFE on the Cu catalyst surface. To address this requirement, we employed energy-dispersive X-ray spectroscopy (EDAX) and analyzed the composition and elemental distribution of the cathode (Fig. 2b). Such analysis is crucial for determining the functionality and effectiveness of PTFE in facilitating the CO2 reduction process. The results demonstrated a consistent and uniform distribution of both PTFE and Cu throughout the entire range of PTFE ratios. Furthermore, when combined with the scanning electron microscopy (SEM) results, we observed that PTFE does not cover the entire surface of Cu (Fig. 2b), which allows for active spaces suitable for CO2 reduction. In addition, the experimental results of PTFE ratio obtained from our study aligns closely with the theoretical values we had set (Table S1, ESI†). Characterization through X-ray diffraction (XRD) and SEM points towards crystallization in the PTFE–Cu catalysts as opposed to the pristine Cu, suggesting an influence on both non-faradaic mass transport and faradaic electron transfer during interfacial reaction processes (Fig. 2b and e). Due to their distinct structure and physiochemical compositions, Cu and PTFE are usually weakly bounded without involving chemical interaction, as suggested in prior studies and applications.36–39 The physical binding and the presence of weak interactions between Cu and PTFE observed in this study are consistent with our initial expectations. This outcome aligns logically with our objective, confirming the effectiveness of PTFE in facilitating the desired binding interactions on the cathode.
In the context of physically interaction between PTFE and catalyst metal, hydrophobicity plays a critical role in the polymer binder of PTFE during the process of molecular mass transfer on the PTFE/Cu surfaces and interfaces. This hydrophobic nature of PTFE creates more efficient CO2 throughput pathways towards metal catalysts compared to ion transport, particularly with respect to cations for the cathode, leading to improved catalytic activity and overall performance in the system. For heterogeneous catalytical process, mass transfer of gaseous and aqueous reagents onto solid cathode surface is the major limitation of electrochemical CO2RR at the triple-phase (gas–liquid–solid) catalytic interface, because of the barriers generated by gaseous reactant dissolution and molecular transport in electrolyte.34 In addition, the proportion and attachment of CO2 and cations (e.g., H+, K+ in this study) on the cathode surface can be substantially influenced by the potentials applied (Theory S.1.1 and Fig. S1, ESI†). Because the reagents at the large scale (in the bulk solution) are well mixed with CO2 continuously pumped into in the experimental system, we assume that all the molecules and ions are distributed in a relatively constant manner in the simulation. Our simulation results indicate that the accumulation of H+ and K+ in the Helmholtz layer under the negative applied potential renders a dense layer with opposite charge and contributes to an electric potential drop near the electrode surface according to Gouy–Chapman theory (Fig. S2b and c, ESI†). In addition, a concurrent descend of CO2 concentration is found in the Helmholtz layer, which can be interpreted as a decline of CO2 adsorption free energy under high ionic strength (Eq. 6) and lead to a near-zero status at the junction of the cathode surface and the Helmholtz layer (<−0.6 V, Fig. 1b and Fig. S2d, ESI†). A previous study of Ag catalysts displayed a similar trend and attributed this phenomena to the steric effect,40 in which most of the space on the cathode surface was occupied when the cations possess more affinity under the negative potential.
Steric effects on the cathode surface during non-faradaic mass transport reveal a dominance of positively charged polar cations over uncharged non-polar CO2 molecules (Fig. S2, ESI†). Given the coexistence of polar (K+, H+, and H2O) and non-polar (CO2) molecules, the dynamic mass transfer of CO2RR is controlled by the balance between electrostatic forces and van der Waals forces on the cathode.41–45 Lower surface polarity results in the smaller cathode surface occupied by the cations. In fact, the simulation results indicate that the surface concentrations of K+ and H+ are suppressed at low cathodic polarity as the PTFE amount rises (Fig. S3a and b, ESI†). Simultaneously, the corresponding Nernstian loss merely dwindles by 0.0056 V (Fig. S4, ESI†), indicating that PTFE scarcely affect the equilibrium reactant concentration on the cathode.46 These results further underscore that the conventional understanding (i.e., PTFE alters CO2 surface coverage) falls short in explaining the reagent distributions governed by steric effects. A more profound relationship exists between mass transport and charge transfer, which is intricately linked to the surface polarity and adsorption induced by PTFE.
Charge transfer partiality instigated from metal–polymer cathode surface polarity
The bonding and interaction of CO2 with catalysts plays a fundamental role in determining CO2 conversion and kinetics on conventional metal catalysts, which directly influence the charge transfer process. In the context of our study, it is important to consider how the presence of PTFE may impact the charge transfer in the CO2RR process. By examining the bonding and interactions of reactants and intermediates, we can understand the broader implications of PTFE on the charge transfer dynamics in CO2RR. This consideration becomes vital in elucidating the overall mechanism and efficiency of the CO2 conversion process facilitated by the PTFE-modified cathodes.To explore charge distribution and transfer in the context of CO2RR, we employed the first-principle computations based on DFT. These computations allowed us to describe molecular interactions and reactions involved in the CO2RR process.47–51 The application of such methods to physically bind metal–polymer cathodes has been limited due to the relatively weak interactions and the complexity of molecular profiles at organic–inorganic junctions (Fig. 1a). Moreover, some previous studies had adopted continuum models with DFT to elucidate the basic kinetics of the (quasi-)equilibrium reaction for metal–polymer junctions, which are still in their infancy for predicting CO2RR (Fig. S1, ESI†).34,52–56 We addressed this issue by incorporating PTFE effects in a bottom-up strategy starting from the atomistic level.
In our DFT study to explore metal–polymer interactions, Cu emerges as a standout candidate among all CO2 reduction catalysts. Its unique ability to generate multiple products through distinct pathways provides a valuable opportunity to investigate the influence of polymers on charge transfer phenomena.1 We specifically selected the Cu (111) surfaces due to their exceptional stability and widespread occurrence, ensuring reliable and relevant experimental conditions (Theory S1.3, ESI†).57 Considering a relatively weak non-faradaic mass transport of CO2, the charge transfer towards CO2 should be closely related to the two dominant cations (K+ and H+) in the Helmholtz layer via different surface mechanisms. Unlike K+ that does not react in CO2RR systems, H+ acts as a major electron acceptor competing with CO2 molecules in the faradaic process involving electron transfer.58 Although binding energies of CO2 and H+ are approximately similar during the adsorption process (Fig. S5 and S6, ESI†), a strong binding of *COOH compared to *H (* represents binding to the catalyst) indicates that the initial step of CO2RR involving the first proton–electron pair transfer and COOH formation is thermodynamically more favorable than the hydrogen evolution reaction (HER) with a lower value of *H2 energy barrier (EB, −0.05 eV) compared to that of the *COOH EB (−1.88 eV) and reaction pathway (Fig. S5–S7, ESI†).59 The charge transfer coefficient (α) clearly manifests this trend, in which the onset of electron transfer toward CO2 fosters α on the cathode (Fig. 3a) and shifts the energy of protonated *H⋯CO2 at the transition state by 1.89 eV compared to that of the pre-protonation state (Fig. 3b and Fig. S7, ESI†). Such a tendency leads to the dissociation of the *OC–OH bond, which has a relatively low barrier energy to generate *CO (0.89 eV, Fig. 3b and Fig. S8, ESI†). Our findings of pristine Cu are in strong concurrence with other pioneering studies conducted earlier in this field.30–32,60,61 The projected density of states (DOS) shows that CO2 protonation leads to an upward shift of the C-p and O-p DOS to ∼4 eV with empty orbitals (Fig. 3e–g). Similar results are observed at the transition state (*H⋯CO2, Fig. 3d) with slight delocalization of the C-p and O-p peaks (Fig. 3h). However, the scenario of DOS becomes different during the second electron transfer of CO2 reduction that involves the dissociation of *COOH to *CO⋯*OH. The high adsorption energy stabilizes COOH on the Cu(111) surface and leads to strong hybridization between the Cu-d and O-p states near the Fermi level (−4 eV to −2 eV, Fig. 3i), which further facilitates the appearance of additional peaks at higher energy ranges (2–4 eV, Fig. 3j) and mitigates the barrier energy.
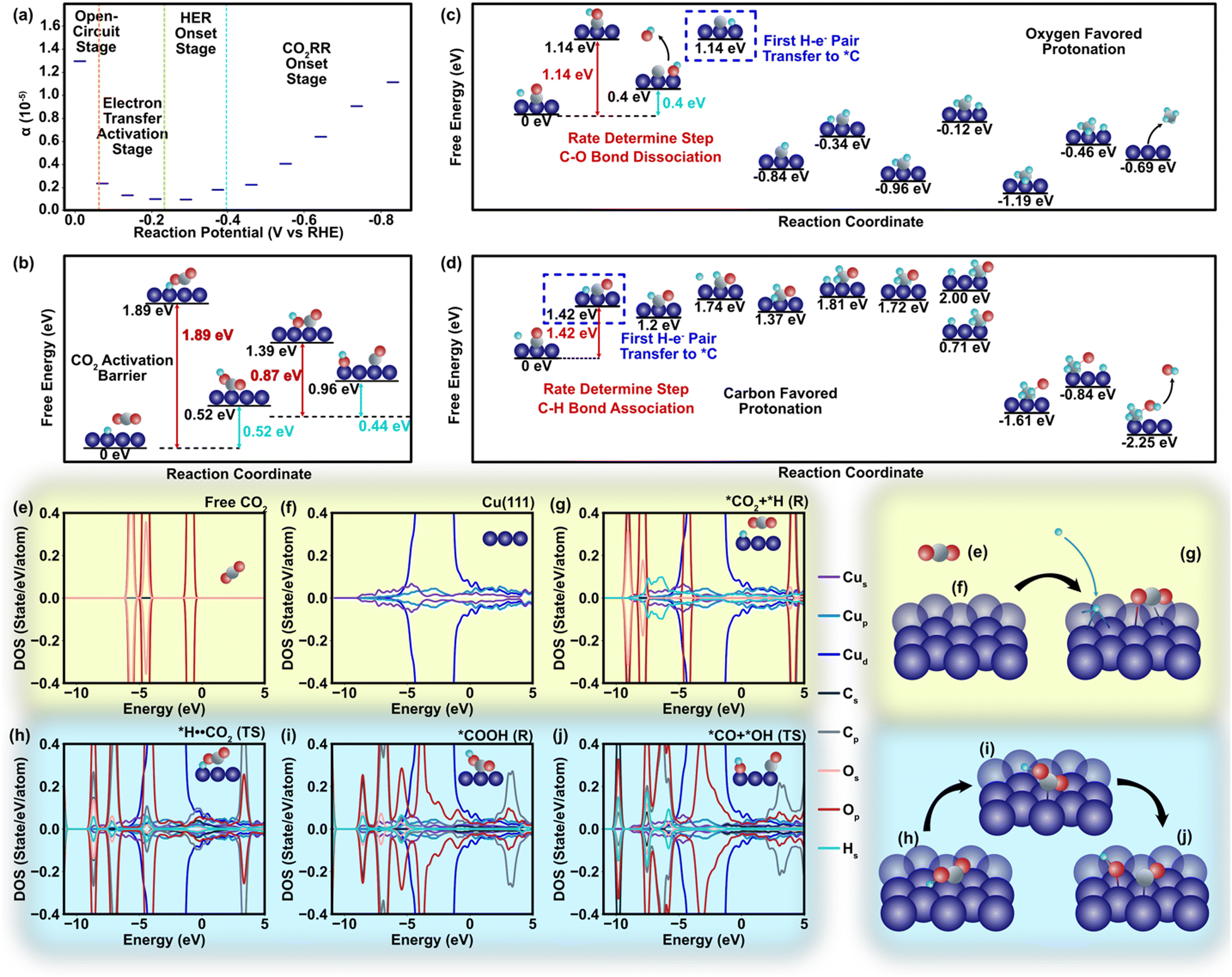 | ||
| Fig. 3 Atomistic energies and density of state regarding to the charge transfer process. (a) Equilibrium charge transfer coefficient alternation in different stages of the HER/CO2RR reaction. (b) Free energy and barrier energy profile of the reaction from CO2 to CO (Table S2, ESI†). The energy barrier of *CO2 protonation is 1.88 eV. As an initial step, the CO2 is adsorbed on to the catalyst surface. Several symmetrically inequivalent sites of Cu(111) surface are considered for the CO2 adsorption along with various CO2 orientations to determine the stable adsorption configuration. The atop site, bridge site, and the hollow sites are considered among the surface sites, whereas the parallel and perpendicular orientations are considered among the molecular orientation. The deformation of CO2 happens during activation and favors protonation of oxygen atoms due to their stronger electronegativity. The CO2 is first protonated to form the adsorbed COOH which thereafter dissociates and forms the adsorbed CO and the OH group. (c) The free energy and barrier energy profile of the protonation of CO. This figure describes the free and barrier energies of CH4 generation associated with oxygen favored protonation. In addition to the outset of CO protonation, further protonation of the reaction intermediates towards methane formation lowers energy barriers with values of 0.75 eV, 0.5 eV, 0.83 eV, and 0.73 eV, respectively, for the reactions in Mechanism I (Table S3, ESI†). (d) The free energy and barrier energy profile of the protonation of CO. This figure describes the Mechanism II for CH4 generation associated with carbon-favored protonation, leading to the energy barrier values of 1.42 eV, 0.44 eV, 0.28 eV, and 0.77 eV (Table S4, ESI†). (e) The site-projected density of states (DOS) for the free CO2. (f) the DOS for the pristine Cu(111) surface. For the free CO2 (e), the projected DOS is primarily located within an energy range of −6 eV to −2 eV and remains occupied. (g) The DOS for the absorbed CO2 and H+ as reactants on Cu surface. (h) The DOS for the transition state of *H⋯CO2 on Cu surface. (i) The DOS for *COOH after the CO2 protonation. (j) The DOS for the *CO and *OH. The Fermi level is set to zero eV and is leveled as the vertical black line. | ||
The incorporation of PTFE into Cu enhances the cathode's surface properties, such as surface polarity and CO2 adsorption characteristics. These alterations considerably affect the association and dissociation processes of atomistic bonds, thereby influencing the charge transfer dynamics occurring on the cathode surface during CO2RR (Table S5, ESI†). We observed that despite the relatively weak physical binding between PTFE and Cu, the addition of PTFE has a substantial impact on the binding energies of CO2 and intermediates. In a more detailed manner, the effect of PTFE is demonstrated at the atomistic level, where a monomer (TFE) of PTFE is introduced in close proximity to the co-adsorbed reactant (CO2) and several reaction intermediates, including COOH, HCOO, CH2O, CHO, and CH4 (Fig. 4). The CO2 binding is enhanced due to the presence of TFE (from −0.31 eV to −0.38 eV), indicating a strong promotion effect on CO2 reduction. This agrees with previous findings that PTFE bolsters the CO2 binding.62,63 Further, the presence of TFE results in a less charge donation of CO2 to its surrounding compared to that of without TFE (qC = +1.98e and qO = +1.12e). This is due to the high electronegativity of fluorine atoms located close to CO2. Among the reaction intermediates, COOH, HCOO, and CHO display enhanced binding in the presence of TFE (Fig. 4). It should be noted that presence of TFE weakens the CH4 binding with Cu, indicating that CH4 could be readily desorbed after its formation.
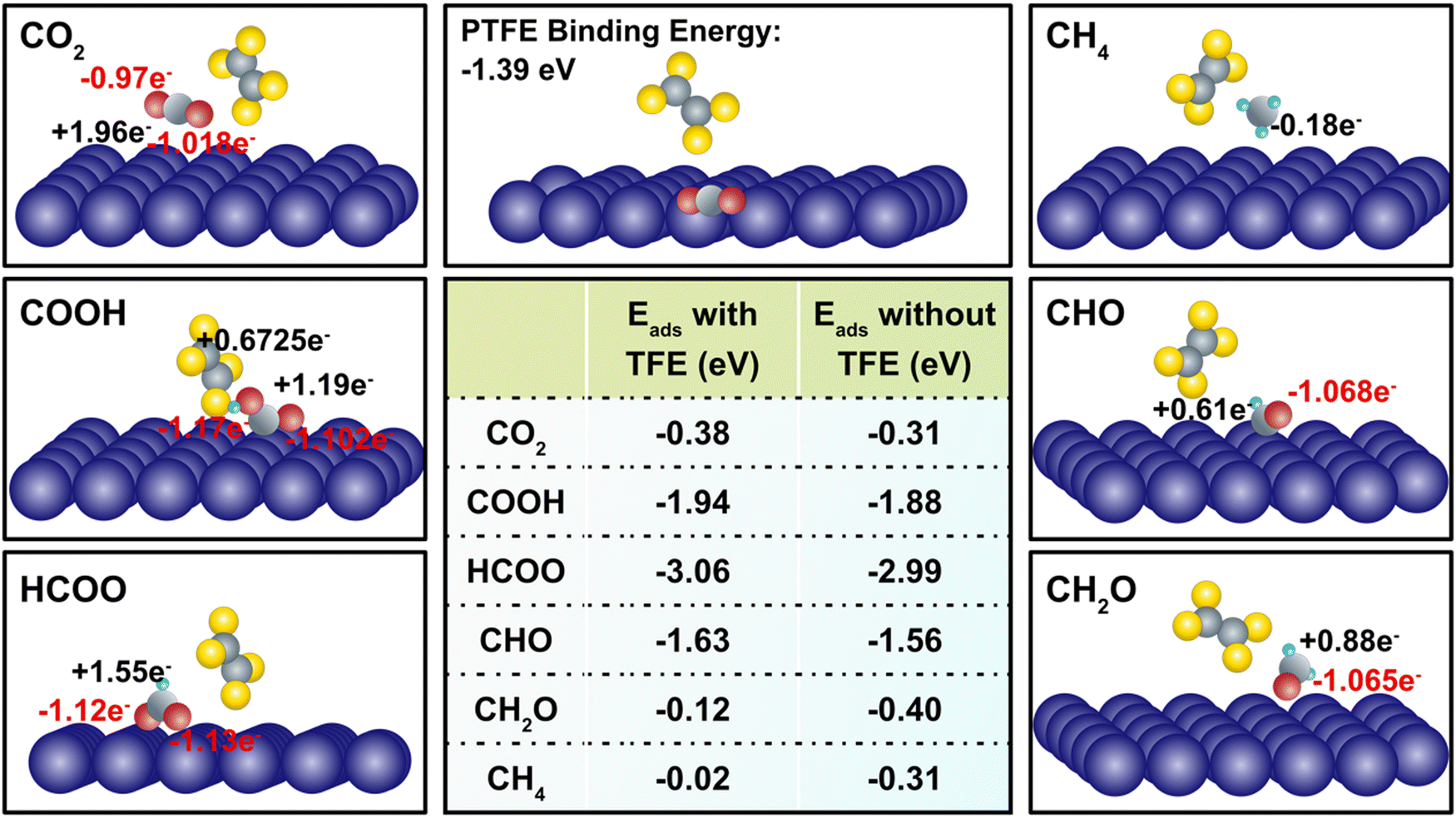 | ||
| Fig. 4 Ab initial calculation of binding energy (in the table), and charge distribution (black and red numbers) of CO2 and important intermediates, including COOH, HCOO, CHO, CH2O, and CH4. For the charge distribution, the black positive numbers in each diagram represent the charge on carbon atom, while the red negative numbers represent the charge on oxygen atom. | ||
The introduction of PTFE into Cu resulted in two notable changes in adsorption when comparing the effects of PTFE on binding energies. First, the addition of H+ to the carbon atom of CO2 and CO enhances the chemisorption of intermediates such as HCOO (from −2.99 eV to −3.06 eV) and CHO (from −1.56 eV to −1.63 eV) (Fig. 4), thereby promoting the reaction pathway towards the generation of CH4. Second, as more H+ is added, PTFE radically reduces the binding energy. For example, as CHO is transformed into CH2O, the binding energy drops from −0.4 eV to −0.12 eV. Remarkably, the binding energy of CH4 with PTFE (−0.02 eV) is even lower than that of H2 (−0.22 eV), indicating a massive promotion of CH4 production. Overall, these observations emphasize the profound influence of PTFE on the binding energies of reactants and intermediates, underscoring its role in modulating the charge transfer and favorably affecting the CO2 reduction process.
In addition to the aforementioned observations regarding the effects of PTFE on the binding energies of CO2 and intermediates on Cu, the CO2 protonation process and the relative abundance of H+ and CO2 in the Helmholtz layer are central in the CO2 reduction mechanism. The protonation of carbon and oxygen atoms during CO2RR is associated with different energy barriers and reaction pathways. A previous study had revealed that the Eley–Rideal (ER) mechanism is more favorable than the Langmuir–Hinshelwood (LH) mechanism when H+ was added to oxygen atoms.64 Specifically, the results of CO2RR current density test imply that the charge transfer coefficient rises in the CO2 activation stage (Fig. 3a), which signifies the existence of a fast charge-transfer-induced water dissociation to provide H+ and support H–O–C–O bond association. Therefore, the key step is the proton–electron-induced oxygen dissociation from CO2 molecules.65 Given the oxygen-favored protonation process, the ER mechanism is more likely to be dominant during CO2 activation and protonation, which is consistent with the results of cation domination from the mass transport simulation (Fig. S2, ESI†). According to the ER mechanism, CO2 molecules can directly form H–O–C–O bond without pre-bonding onto the cathode in advance, and thus the steric effect limitation (K+ and H+ dominate the cathode surface) can be eliminated. Moreover, although conventional polymer groups are not expected to participate in the electron transfer processes, they can still affect the destination of electrons transfer through adsorption of reactants and intermediates by changing the surface polarity, where ER-induced mechanism (Mechanism I) and LH-induced mechanism (Mechanism II) for CH4 generation is also galvanized.56,66 Adsorption sensitivity of the cathode surface towards CO, a vital CO2RR intermediate, plays a critical role in the protonation process (Fig. S9, ESI†). From the DFT-calculated barrier energies we can perceive that Mechanism I favors oxygen protonation (Fig. S10, ESI†). *CO is first deoxygenated with an energy barrier of 1.14 eV as the rate determining step (Fig. 3c). In contrast, the carbon atom on *CO overcomes an energy barrier of 0.68 eV and is protonated prior to the oxygen atom in Mechanism II, inferring a more favorable path compared to the ER at this step (Fig. 3d and Fig. S11, ESI†). The EB of CO protonation for Mechanism II indicates the second electron transfer is the rate-determining step (Fig. 3d). The surface stability contributed by protonation raises the energy barrier of C–O dissociation (EB: 2.06 eV) under this mechanism as opposed to *CO generation (EB: 0.89 eV). Although the mechanism of direct charge transfer for CO2RR at the metal–polymer physical interface remains unclear, our study unveils that surface polarity of cathode can influence the charge transfer to CO2 indirectly through mass transport and protonation process.
Overall, in the context of various pathways related to the CO2 protonation process, the metal and the polymer may exhibit distinct preferences towards ER-induced mechanism (Mechanism I) and LH-induced mechanism (Mechanism II) due to their varying polarities and affinities towards reactants and intermediates. In fact, from the analysis of energy barriers in our multiscale paradigm, a pathway of aldehyde (CHO) in Mechanism II is more favorable than methylidyne (CH) in Mechanism I for CH4 formation given that Mechanism II has a lower EB (0.67 eV) than Mechanism I (1.14 eV) (Fig. 3c and d). Subsequent reactions also stipulate the higher preferability for Mechanism II, which has lower energy profiles than Mechanism I (Fig. S12–S14, ESI†). Relative to the CO2 activation stage, the C atom protonation in Mechanism I and CO protonation in Mechanism II have higher energy barriers (EB) (Fig. 5c and d), requiring excessive H+ in HL to promote the forward reaction rate (Theory S2, ESI†).
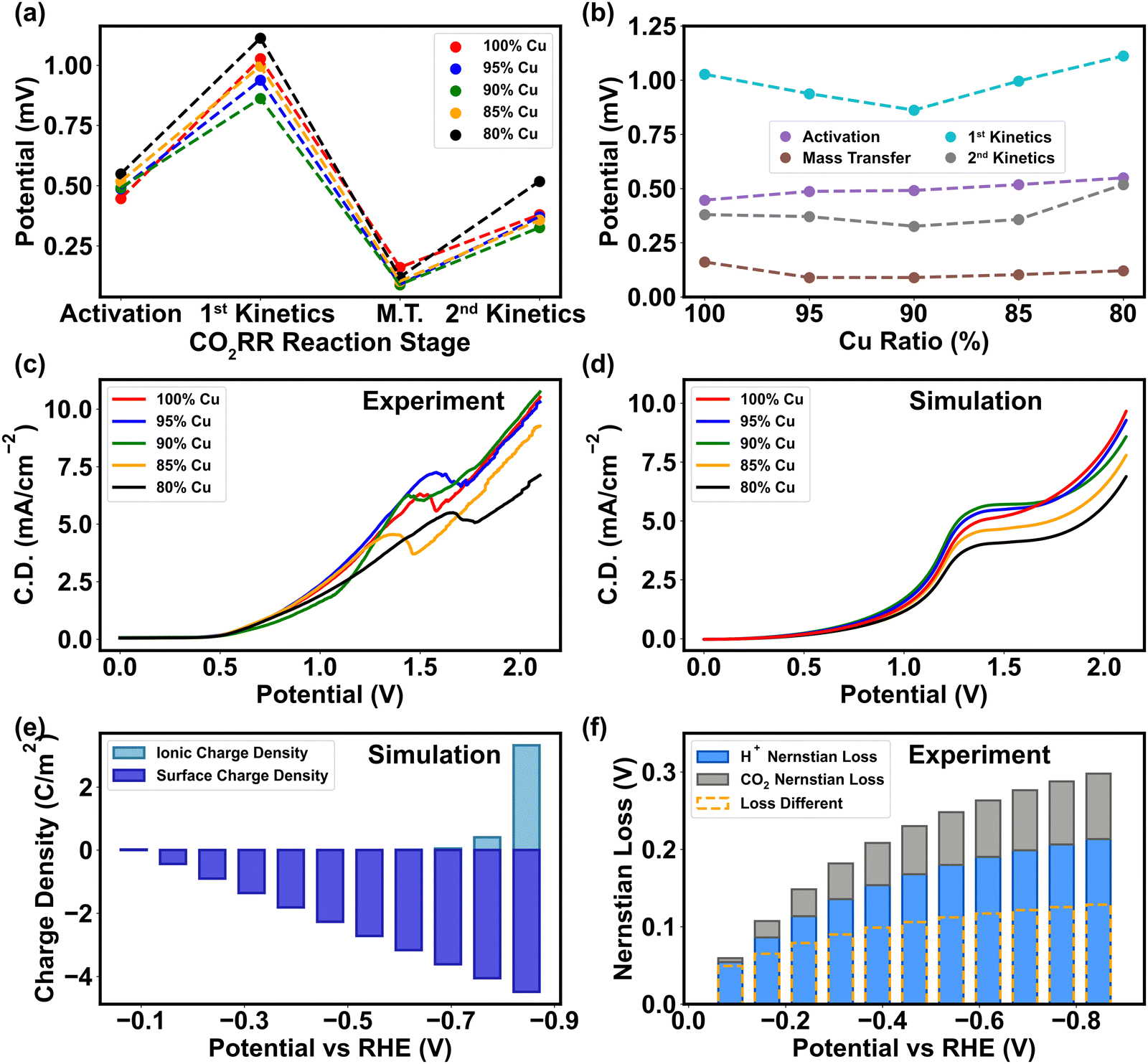 | ||
| Fig. 5 Quantitative analysis of polarization curve, charge transfer barrier, and mass transfer barrier under the impact of CO2/H+, potential, and PTFE. (a) Potential being consumed in four stages of CO2RR, including activation (before CO2RR onset), 1st kinetics (Mainly CO generation), mass transport (M.T.) limitation, and 2nd kinetics (mainly CH4 generation). (b) Comparison of potential alternation in different stages under different ratios of Cu from 100% to 80% (mixed with PTFE). (c) Experimental total polarization curve of CO2RR from 0 V to 2.1 V on the cathode surface with response current density (C.D.). The sweeping step is 0.005 V. (d) Simulation of total polarization curve from multiscale analysis. (e) Simulated results of surface charge density and ionic charge density. The accumulation of ionic charge density manifests the dynamic cation-induced concentration polarization, hence further lessening the electric potential in Helmholtz layer. (f) Variation of Nernstian losses contributed by HER and CO2RR according to the current density obtained from CO2RR experiments under potentials between 0 V and −0.8 V vs. RHE. | ||
Dual effect of mass and charge transfer in a CO2RR system with metal–polymer cathode
The preceding section elucidates the pivotal roles of binding energy and charge transfer in determining the pathway of CO2RR, with the relative abundance of H+ and CO2 being the crucial factors influencing CO2 protonation process. Consequently, both mass transport and charge transfer become essential in shaping CO2 reduction selectivity, as they control the concentration of H+ and CO2 in the vicinity of the cathode. The mass discrepancy of CO2 near the cathode surface in the mass transport simulation (Fig. S2–S4, ESI†) implies that the essence of mass transfer is the re-balance of CO2 in the Helmholtz layer. Compared to the pristine Cu, the PTFE–Cu surface attracts higher concentration of CO2 due to larger surface polarity, thereby minimizing the CO2 mass transport loss (Fig. 5a and b). The experimental polarization curve under different ratios manifested that 5–15% of PTFE in Cu overcomes the mass transport barriers encountered in the pristine Cu, indicating that PTFE immensely improves the efficiency of CO2 transport as the cathodic potential ascends (Fig. 5c). The PTFE ratio of 5% displays a similar trend compared to the pristine Cu, meaning that small surface polarity change does not affect the CO2 mass transport efficiency (Fig. 5c). Furthermore, the experimental results of polarization curve show that extra polarity (>15% PTFE) causes an even higher activation loss (Fig. 5a and b) and ohmic loss (Fig. S15, ESI†), manifesting the shift of the major reaction barrier from mass transport to charge transfer, which is consistent with the simulation of the charge transfer coefficient (Fig. 3a) and surface charge characterization (Fig. 2c). Due to the cathode morphology alternation, 10–15% PTFE renders a uniform surface by filling the gap of Cu nanoparticles and linking them together to promote the mass transfer efficiency (Fig. 2b). However, higher PTFE coverage (>15%) invades the regular and continuous Cu cluster structure, compromising the electron transfer across the catalytic interface (Fig. 2b and 4c).In parallel, the charge discrepancy at the cathode–electrolyte interface is also discovered. The experimental polarization curve illustrates that the onset potential of CO2RR ascends with higher amount of PTFE (Fig. 5c), meaning that the activation limitation during charge transfer rises with higher surface polarity (Fig. 5b), which is consistent with the simulation results (Fig. 5d). In addition, in the heterogeneous double layer zone (Fig. 1b), the cathodic surface charge density (ρs) varies linearly with the potential applied, while the ionic charge density (ρi) in the Helmholtz layer only procures a minor alternation under low cathodic potential (>−0.7 V vs. RHE) (Fig. 5e). This phenomenon is in align with a high concentration polarization near the cathode surface before the onset of CO2RR, where the cation-induced ionic charge density rapidly surges and thereby reduces the cathodic potential in the higher-polarity Helmholtz layer (Fig. S2a, ESI†). The results of the onset potential and surface charge tests demonstrate that the charge transfer in CO2RR is more sensitive to the alternation of cathodic potential rather than intrinsic surface polarity (Fig. 2c and 4b).
Based on charge and mass analyses, we confirm that the polymer exerts its influence on metal catalytic functions through two main mechanisms: the binding energy modification of reactants and intermediates, and the surface polarity adjustment. These mechanisms, in turn, modulate the mass transport during CO2RR. The direct impacts on charge transfer are evident, particularly during the activation stage of CO2, as this phase is intertwined with the adsorption of these molecules. Moreover, our experimental results indicate that with the same flow rate and partial pressure of CO2 into the catholyte solution, CO2 concentration remains the same in the bulk solution, while pH drops mildly throughout the entire testing period, indicating that H+ prompts more Nernstian loss than CO2RR (Fig. 5f). Hence, the change in chemical composition on the cathode surface is mainly caused by H+ rather than CO2. In the low potential range (<1 V vs. Ag/AgCl) where HER dominates, the relatively high HER efficiency (∼80%, Fig. 6a) is driven by the ρs–ρi discrepancy.67 In the high potential range (>1.5V vs. Ag/AgCl) where the CO2 reduction dominates, a higher rate of electron transfer towards CO2 undermines the charge-transfer induced water dissociation, and thus the polarization on the cathode surface is attenuated (Fig. 5e).
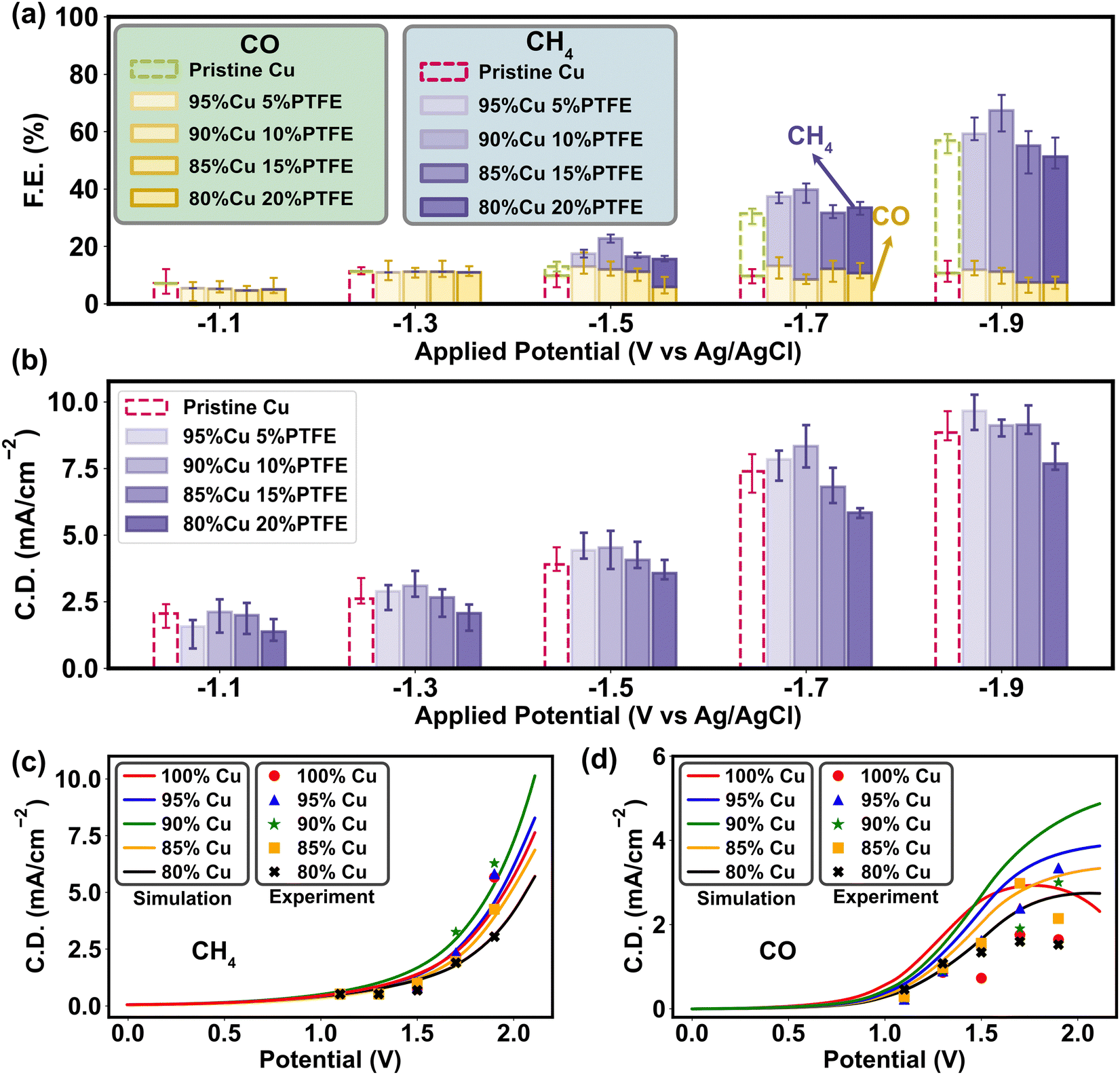 | ||
| Fig. 6 Product analysis of CO2RR with metal–polymer catalysts. (a) Comparison of the faradaic efficiency (F.E.) under different potentials and PTFE amounts during CO2RR. (b) Comparison of the current density (C.D.) under different potentials and PTFE amounts during CO2RR. (c) Simulated polarization curve and experimental validation of response current density of CH4 under different potentials and PTFE amounts. (d) Simulated polarization curve and experimental validation of response current density of CO under different potentials and PTFE amounts. | ||
As discussed above, rebalancing CO2 and H+ involves a dynamic interplay between mass transfer-induced steric effects (molecular distribution) and charge transfer-associated binding energy (molecular adsorption). With the addition of PTFE, these interactions can be fine-tuned, which influence the relative abundance of CO2 and H+ species. Given the combined effects of mass and charge transfer, CO2 still overthrows H+ as the major electron acceptor with higher faradaic efficiency (Fig. S16, ESI†) despite the decline of concentration under more negative potentials (<−0.6 V),67 implying that the electrons transferred to CO2 in the inner Helmholtz layer may not contact directly with the catalyst. The multiscale simulation reveals a possible pathway through the ER mechanism (Mechanism I) as H+ cumulates on the cathode surface (Fig. S2c, ESI†), and the experimental results demonstrate that the cumulation of K+ exerts a significant impact on CO2 reduction (Fig. S2b and S17, ESI†). Previous studies postulated this circumstance as the result of complex many-body correlations and the mutual interaction of chemical species.40 We find that the presence of metal–polymer catalyst surface redistributes the charges on the CO2 molecules (Fig. S18, ESI†) and promotes the activation of CO2. Under the structure of catalyst-cation-CO2 triple connection, the cations act as a bridge for the electron transfer, avoiding the intrinsic high electron conductive barrier of PTFE polymer (Fig. S19 and S20, ESI†). Consequently, the conduction of cations in the outermost electron layer exerts considerate impact on the electron transfer efficiency, and reactive alkali metal atoms (e.g., Na+, K+) are more competent to promote CO2 activation. Our result further proves that a shorter vertical distance (1.16 Å) between K+ (+0.84e−) and CO2 promotes a greater charge transfer from cation to CO2 than does a longer vertical distance (1.89 Å) for Na+ (+0.80e−) to CO2 (Fig. S19 and S20, ESI†). CO2RR tests were conducted in both 0.1 M KHCO3 and 0.1 M NaHCO3 solutions as an evaluation in this study. The presence of K+ in the reaction outperforms Na+ in both current density and faradaic efficiency (Fig. S17, ESI†), indicating the importance of cations as the bridge for the charge transfer in CO2RR process.
Experimental demonstration of CO2RR pathway shift induced by polymers
To ascertain the mass–charge transfer modulation introduced by PTFE and its consequent impact on CO2RR selectivity, we conducted a series of experimental tests. The results of water contact angle and surface charge (Fig. 2b and c) reveal that the surface polarity rises with higher amount of PTFE, while the overall polarization curves under different PTFE ratios exhibit a similar limitation of mass transport and charge transfer compared to the simulation results (Fig. 5c and d). As we dive into the faradaic efficiency results of CO and CH4 generation in CO2RR tests, the effects of PTFE on CO2RR activation and protonation are further confirmed through the multiscale analysis (Fig. 5a). We specifically focus on the CO/CH4 products due to their straightforward composition and short reaction chain. These products are easily testable through both experimental validation and multiscale simulations, enabling us to gain a thorough understanding of how surface polarity influences the selectivity and efficiency of CO2RR. Given the primarily mediating role of PTFE that involves the processes like CO2 adsorption and H+ redistribution through surface polarity modification, the influence of PTFE on the final performance may not manifest as readily as with direct catalytic materials like Cu. To clarify these trends, we investigated the faradaic efficiency (FE) statistically for both CH4 and CO. The results exhibited completely opposite patterns, in which CH4 generation reaches its peak at 10% PTFE, while CO production is suppressed the most compared to the pristine Cu catalyst (Table S6, ESI†). At the activation potential (∼−1.0 V vs. Ag/AgCl), the PTFE–Cu cathode possesses a lower faradaic efficiency but a higher current density, implying that incorporating PTFE may stimulate the CO2 reduction (Fig. 6b), especially the CH4 generation at the ascending potentials, since PTFE aggregates CO2 molecules in the Helmholtz layer and alleviates the mass transfer limitation by increasing the CO2 binding energy on the catalyst and improving the relative abundance of CO2/H+ (Fig. 4 and 5b). The electrochemical impedance spectroscopy (EIS) tests also prove that the PTFE–Cu surfaces within 5–15% PTFE loadings have the lowest diffusion resistance in 0.1 M KHCO3 solution, while the charge transfer resistance is similar to the pristine Cu surface (Fig. S21 and S22, ESI†). Further increment of PTFE ratio (>15%) escalates both charge transfer and diffusion resistance (Fig. S21 and S22, ESI†), which is attributed to the curtailment of active sites and is consistent with the FE results (Fig. 6a and 5b). The simulated polarization curves and the experimental results exhibit a similar trend of CH4 current density (Fig. 6c). In contrast, the experimental CO polarization curve exhibits a relatively lower current density compared to the simulated results (Fig. 6d), meaning that in the reaction occurring in the experimental system, the EB of CO2 protonation is higher than the H–O bond association (1.89 eV). Because the mass transport limitation in the overall CO2RR polarization curve was found at around 1.5 V vs. Ag/AgCl in our experimental system (Fig. 5c), we infer that the mass transport of CO2 becomes the major limitation of the whole reaction system.To assess the impact of PTFE on metal catalyst performance, we further expanded our experimental investigation to include two catalysts (Sn and Ag) yielding single product. Sn is known to mainly produce HCOOH,68 while Ag exclusively produces CO.69 The results obtained from the Sn catalyst exhibited a similar trend to that of Cu (Fig. S23a, ESI†). Under the addition of 10% PTFE, the Sn cathode had the best performance, indicating that the inclusion of PTFE enhanced the protonation of CO2 during the reaction and improved catalytic activity. However, with an increase in the PTFE ratio to 15% and 20% in the Sn catalyst, the FE dropped below 80%, which is caused by the higher cathodic resistance and blockage of active sites due to the higher PTFE content. The higher resistance likely hampers the charge transfer and diminishes the overall efficiency of the CO2 reduction process. A similar trend was observed in the Ag catalyst test, where the incorporation of 5% and 10% PTFE outperformed the pristine Ag cathode, leading to a higher CO yield (Fig. S23b, ESI†). However, increasing the amount of PTFE beyond this point resulted in higher resistance, limiting further enhancements in reaction efficiency. Through the examination of Sn and Ag catalysts, we provide additional evidence to reinforce our findings concerning the impact of PTFE and explicate the influence of PTFE on the protonation of CO2 and the subsequent catalytic performance.
The consistent trend observed in the experiments of both the Cu catalyst yielding multiple products and the Sn and Ag catalysts yielding single product underscores the enhanced CO2 reduction selectivity and efficiency with the addition of PTFE. These findings further emphasize the pivotal role played by surface adsorption and polarity in the design of CO2RR catalysts. As the polarization curve results for CO and CH4 display different correspondences with the multiscale simulation, it is critical to control mass transport towards the catalyst surface so as to tune the CO2RR selectivity.70–72 In the Helmholtz layer, excessive accumulation of H+ is suppressed on the PTFE–Cu cathode surface, since a lower polarity minimizes cathodic double layer capacitance amplifies surface affinity towards CO2 (Fig. 5e). A previous study had discussed the implicit involvement of H+ and the consequent pH sensitivity on the pristine Cu,64 whereas our study illuminates that with the lower cathode surface polarity induced by PTFE polymer, the H+ can also be tuned by changing the surface mass transport properties to enhance CO2 reduction (Fig. S24, ESI†).73,74 Therefore, according to our study, the charge transfer dynamics are enormously affected by the changes in molecular adsorption of reactants and intermediates. The metal–polymer catalyst influences the reaction selectivity through modifying the relative abundance of H+ and CO2 on the surface. This effect is particularly important, as it occurs in conjunction with variations in the mass transport barrier due to surface polarity.
In the context of Cu–PTFE cathode, our study has unveiled another aspect related to surface adsorption and polarity. Specifically, we observed an intriguing disparity between the charge transfer coefficient and current density during CO protonation in our experiment (Fig. 5a and 6b). This discrepancy leads to a decline in the cathodic exchange current density of CO2RR and signifies a disruption of the equilibrium state. This finding can be induced in two ways. First, the conversion of molecules alters the species coverage. The high binding energy of CO (−1.03 eV) intensifies the accumulation effect and reduces the active sites prior to CO protonation (Fig. S5 and S6, ESI†). Second, H+ is redistributed over the course of the CO2RR and fluctuates the rate constant at the high free energy barrier of protonation.75
Conclusion
Our multiscale simulation-experiment approach offers an in-depth generic understanding of the catalytic impact of PTFE on metal catalysts, extending beyond the conventional comprehension of hydrophobicity alone on metal catalyst surfaces. This rigorous investigation is accomplished through five key steps:27,40,56,761. We characterized the distribution and interaction of Cu–PTFE, elucidating the physical bonding and weak interaction, as well as the even spatial distribution of Cu and PTFE on the cathode. This step provided valuable insights into the physical and chemical properties of the Cu–PTFE interface.
2. Being cognizant of the Cu–PTFE characterization, we further explored the functional attributes of PTFE within the cathode system, considering factors such as hydrophobicity, CO2 adsorption, surface polarity, and various other pertinent aspects. We found that polymer binding with metal catalyst changes the polarity of cathode surface, reshaping the distribution of H+ and CO2 adjacent to the cathode surface affects charge transfer towards CO2.
3. Having gained an in-depth understanding of PTFE functionality, we proceeded to examine the alternation in charge transfer processes influenced by PTFE, in which although the polymer does not participate in the electron transfer directly, it can influence the reaction through the adjustment of reactant adsorption and intermediate adsorption.
4. Along with charge transfer, we further investigated the mass transfer resulting from the addition of PTFE. Our study reveals that the CO2/K+/H+ distributions on the cathode surface in the Helmholtz layer can be altered through PTFE-induced mass transport. With the existence of PTFE, the increment of CO2 permeability escalates the forward reaction rate, while the decrement of cations reduces the cathodic polarization, enabling lower overpotential for the activation of CO2 molecules.
5. As the final step, we expanded conventional evaluation experiment protocols to thoroughly validate our newly uncovered discoveries. Our primary conclusion from the Cu–PTFE experiment is designed to illustrate the PTFE-induced selectivity alteration, which aligns well with the results obtained from our modeling analysis. Subsequently, the Sn/Ag–PTFE experiments were conducted to eliminate any interference caused by the broad product spectrum observed in Cu. These experiments validated the changes of PTFE-induced performance and assessed the accuracy and universality of our findings across a wide range of catalysts.
Furthermore, our DFT and DOS results reveal that different pathways of protonation from pro-oxygen to pro-carbon in the CO2RR can be controlled by the surface polarity, which requires further study with advanced in situ techniques to prove. These vital findings transcend the conventional cognitions about polymer functions in the CO2RR cathode design and strongly indicate that physically bound polymers can influence the efficiency and selectivity of CO2RR on metal catalysts, and thus providing a scientific support for more flexible material combination strategies.
In summary, we report a unique and comprehensive multiscale simulation-experiment tandem study and demonstrate that the structure-induced crystallization of metal–polymer cathode surface polarity alters the CO2RR interfacial processes and CO2/cation distributions, which can simultaneously affect the mass and electron transfer by changing the adsorption process. Our findings substantiate that the polymers in metal catalysts do not simply tune the heterogeneous catalytic reaction by changing the surface area or surface morphology, but rather optimize polymer-induced cathode surface properties (e.g., polarity) and shift the catalyst performance by changing surface mass transport and reaction process. We believe that this revolutionary discovery can refocus further CO2RR studies and unify various single-parameter explorations through multiscale structure–process analysis, presenting a new vision for the complex correlation between heterogeneous catalytic interface and CO2 reduction efficiency. Our results offer practical guidance for tuning catalyst materials to modulate the reaction pathways of CO2 reduction through the polymer material in the state-of-the-art CO2RR cathode designs.
Experimental procedures
CO2RR cathode material preparation and thermal treatment
The CO2RR cathode was consisting of Cu, Sn, and Ag nanoparticles (NPs), as well as polytetrafluoroethylene (PTFE) to ensure large relative surface area for the reaction efficiency. Each of the catalyst nanoparticles (Cu, Sn, and Ag NPs) was measured to be 25 mg in quantity (Sigma-Aldrich, 40–60 nm) were measured out and dispersed into Hexane and sonicated for 40 minutes. Then the mixture was washed with ethanol and redisperse into Hexane. For Cu NPs, after complete drying under 85 °C, the particles were heated under 185 °C for 6 hours to remove all the oxides and impurities.77 Two types of cathodes were fabricated and compared, pristine Cu cathode and PTFE–Cu cathode. To fabricate the cathode, 10 mg of Cu nano particles was dispersed into 1 mL isopropanol following with 1h of sonication. The mixture was then sprayed onto carbon electrodes with dimension of 0.5 cm × 0.5 cm and 3 cm × 3 cm prepared in advance. The fabrication of the PTFE–Cu cathode, 0.5 mg, 1 mg, 1.5 mg, and 2 mg PTFE (Sigma-Aldrich) was mixed individually with 9.5 mg, 9 mg, 8.5 mg, and 8 mg of Cu NPs while dispersing into the 1 mL of isopropanol. The fabrication of Sn and Ag cathode also follows the same procedures above on the same size of carbon electrode. The loading of each cathode was controlled to be 1 mg cm−2. After the electrodes became completely dry, the electrodes were pressed with thermoheater (The MAXX Clam Heat Press by Stahls) under 150 °C for 2 minutes to stabilize the structure. Each type of cathode was made in triplicate for the duplicate test to reduce the system errors. The cathode and anode electrodes were separated by 0.182 mm Nafion proton exchange membrane (Fuel-cell Store).Experimental setups and operations for CO2RR
The CO2RR system in this study was set as the continuous flow mode to investigate the comprehensive process from activation to reaction equilibrium in the 0.1 M KHCO3 solution at a pH of 6.8 and saturated with CO2. The size of both cathodic and anodic chambers was set to be 3 cm × 3 cm × 2.5 cm (Fig. S25, ESI†). Before starting to apply voltage, the cathodic chamber was first pumped with CO2 gas for 30 minutes at a flow rate of 40 sccm till the solution becomes CO2 saturated. Then the CO2 flow was set to be 10 sccm during the reduction experiments to reduce the impact of hydraulic disturbance in the solution. The anodic chamber (3 cm × 3 cm × 2.5 cm) was left open air to keep it being under air pressure for oxygen evolution. The anode of the reaction system was a 1.5 cm × 1.5 cm Pt foil (Sigma-Aldrich). The cathodic chamber was sealed from the outer environment for the collection of the gases generated. The CO2RR system was operated continuously for 5 hours, during which the gas to be tested was collected into a sample bag (volume: 0.6 L) every 1 h for the Gas chromatography (GC) measurement. The liquid sample in the cathodic chamber (∼9 mL) was extracted and saved at the end of every CO2RR experiment for the test of nuclear magnetic resonance (NMR) to determine the generation of organic carbon products (e.g., methanol) in the solution. Each experiment with the same cathode material and running time was operated three times to ensure consistency.Physical and electrochemical characterization of the cathode
The crystal structure of all types of electrodes were characterized by X-ray diffraction (XRD) (D2 PHASER, Bruker Corp., scan range from 10° to 92° (2θ), Cu Kα source, λ = 0.1542 nm). The sharp diffraction peaks of Cu nanoparticles78 (JCPDS: 85-1326) and PTFE particles79 are detectable. The surface morphology and coverage of Cu/PTFE were characterized by scanning electron microscopy (SEM). The samples were mounted onto SEM stubs using double sided carbon tape and were then sputter coated with gold/palladium (80:20, E5100, Polaron) and examined using a scanning electron microscope (Teneo LVSEM, FEI) at 10 kV. X-ray Energy Dispersive Spectroscopy (EDS) was performed at 15 kV using Oxford Aztec Energy Microanalysis System with X-Max 80 Silicon Drift Detector to determine elemental distribution on the surface. The electrochemical characterization of cathodes was conducted along with Pt as the counter electrode and a saturated Ag/AgCl electrode as the reference electrode. To characterize the surface capacity of the cathodes prepared, cyclic voltammetry (CV) was acquired within the range of −1.8 V to 0 V vs. Ag/AgCl at the scan rate from 10 to 100 mV s−1 in the 0.1 M KHCO3 solution after purging CO2 for 30 minutes with a flow rate of 40 sccm. The electrochemical impedance spectroscopy (EIS) was performed in the same solution. The frequency of EIS spectra was from 0.1 Hz to 106 Hz with amplitude of 0.01 V. The polarization curve was measured using linear sweep voltammetry (LSV). The range of LSV was set to be 0 V to 2.1 V, and the scan rate was 0.005 V s−1. The response current was collected every 0.001 s with 40 sccm CO2 flow. The water contact angle was also measured using Ossila Contact Angle Goniometer to determine the surface polarity of the cathode with different ratio of PTFE. Moreover, the surface charge of all the cathodes tested in our experiment was measured by Anton Paar SurPASS. The testing electrolyte was prepared with 0.1 M HCl standard solution, in which the measurements were conveyed from pH 4 to pH 7.5 by titration using 0.1 M NaOH. The flow pressure of testing solution is 500 mbar. Each sample was tested 4 times at every pH value to get the average and minimize the system error.Measurement of gas and liquid products from the CO2RR system
The gas products collected in the sample bag were measured by injecting the gas sample into gas chromatography (GC, Agilent 6890). The detection was carried out by thermal conductivity detection (TCD). Each gas sample was 250 μL and the results of each test were recorded when the gas was stably generated with constant flow reading on the flowmeter. The gas samples were taken every 1 hour for five times throughout each CO2RR experiment. The liquid samples were collected after the CO2RR system was operated for 5 hours. In each experiment, 9 mL catholyte liquid was extracted from the cathodic chamber using a syringe. To ensure the consistency of the liquid sample collection in each test, we only collected the catholyte solution once the reaction system fully stopped running, which was inferred when there's no gas bubble generating from the cathode surface. The liquid samples were stored in the fridge under the temperature of 4 °C before analysis. The liquid products were analyzed through H-NMR (Agilent NMR Spectrometer 300 mHz) to detect both liquid products from CO2RR and to make sure there is no leaching of Cu from the cathode. The samples for NMR tests were prepared by mixing 0.56 mL catholyte sample (90% V/V) with 0.06 mL deuterium oxide (D2O, 10% V/V, Sigma-Aldrich). The liquid sample was first run for 64 cycles in the NMR machine to roughly determine the existence of products, and then run for 576 cycles to precisely determine the quantitative amount of each product in the liquid sample.Implementation of the electrochemistry-centered structure–process multiscale analysis
The multiscale analysis was implemented to incorporate models at three different levels: the system level to simulate continuum molecular transport, the molecular level to simulate electrochemical properties, and the atomistic level to calculate adsorption and free energies. To annex each level while preserving the scientific meaning and accuracy, we innovatively absorbed the ideology of doubly connected edge list (DCEL) data structure into our multiscale methods, which preserves the output results from one model level and use them as input parameters for other levels (Fig. S26, ESI†). The system and molecular levels were performed within COMSOL Multiphysics in which the data collection and unit conversions were achieved by python script. Calculation of electrochemical losses and barriers were implemented using python library of numpy and scipy. The atomistic level simulation was delivered through Vienna ab initio Simulation Package (VASP) that determines the functional properties of materials accurately.80–82 All the details of multiscale model theory, implementation, and calculations can be found in ESI.†Author contributions
B. L. supervised this project. X. W. conducted materials synthesis and electrochemical characterizations. X. W. and J. O. conducted electrochemical CO2RR experiments. X. W. conducted continuum model (system level) and electrochemical model (molecular level) calculations. S. S. carried out quantum/atomistic DFT calculation (atomistic level). F. L. carried out cathode catalyst structural characterizations. S. R. performed the experiments of Sn-PTFE and Ag-PTFE cathodes. X. W. performed gas chromatography and nuclear magnetic resonance analysis. X. W. and Y. H. carried out electrochemical impedance spectroscopy of cathodes and CO2RR systems. W. X. performed cathode surface charge measurement. J. G. provided help in theoretical analysis of modeling results. M. B. provided help in quantitative methods of modeling and calculations. P. G. provided help in material development and characterization. P. A. provided help in result analysis. X. W. and B. L. wrote the manuscript. All authors discussed the results and assisted during manuscript preparation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Science Foundation (NSF) Smart and Connected Communities (SCC) Project (Award No.: ECCS-2018492) and Research Experience for Undergraduates (REU) Project (Award No.: 2051084). J. G. acknowledges support from the NSF awards 2203567 and 1757634. F. L. and P. G. acknowledge the support by the University of Connecticut CARIC program, and the NSF (Award No. IIP 1919231). The electron microscopy studies are performed using the facilities in the UConn/Thermo Fisher Scientific Center for Advanced Microscopy and Materials Analysis (CAMMA). The authors thank B. Javidi for the assistant with manuscript preparation.References
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- Z. Xing, L. Hu, D. S. Ripatti, X. Hu and X. Feng, Nat. Commun., 2021, 12, 136 CrossRef CAS PubMed.
- P. An, L. Wei, H. Li, B. Yang, K. Liu, J. Fu, H. Li, H. Liu, J. Hu, Y.-R. Lu, H. Pan, T.-S. Chan, N. Zhang and M. Liu, J. Mater. Chem. A, 2020, 8, 15936–15941 RSC.
- A. Rendón-Calle, S. Builes and F. Calle-Vallejo, Curr. Opin. Electrochem., 2018, 9, 158–165 CrossRef.
- S. Garg, M. Li, A. Z. Weber, L. Ge, L. Li, V. Rudolph, G. Wang and T. E. Rufford, J. Mater. Chem. A, 2020, 8, 1511–1544 RSC.
- K. Manthiram, B. J. Beberwyck and A. P. Alivisatos, J. Am. Chem. Soc., 2014, 136, 13319–13325 CrossRef CAS PubMed.
- A. H. Motagamwala and J. A. Dumesic, Chem. Rev., 2021, 121, 1049–1076 CrossRef CAS PubMed.
- L. Zhang, Z. Wei, S. Thanneeru, M. Meng, M. Kruzyk, G. Ung, B. Liu and J. He, Angew. Chem., 2019, 131, 15981–15987 CrossRef.
- T. L. Soucy, W. S. Dean, J. Zhou, K. E. Rivera Cruz and C. C. L. McCrory, Acc. Chem. Res., 2022, 55, 252–261 CrossRef CAS PubMed.
- S. Reichman, T. Duvdevani, A. Aharon, M. Philosoph, D. Golodnitsky and E. Peled, J. Power Sources, 2006, 153, 228–233 CrossRef CAS.
- J. H. Lee, S. Kattel, Z. Xie, B. M. Tackett, J. Wang, C.-J. Liu and J. G. Chen, Adv. Funct. Mater., 2018, 28, 1804762 CrossRef.
- D. M. Koshy, S. A. Akhade, A. Shugar, K. Abiose, J. Shi, S. Liang, J. S. Oakdale, S. E. Weitzner, J. B. Varley, E. B. Duoss, S. E. Baker, C. Hahn, Z. Bao and T. F. Jaramillo, J. Am. Chem. Soc., 2021, 143, 14712–14725 CrossRef CAS PubMed.
- S. Adhikari, M. K. Pagels, J. Y. Jeon and C. Bae, Polymer, 2020, 211, 123080 CrossRef CAS.
- J. Lee, Y. Ahn and D. Kim, Macromol. Res., 2019, 27, 175–181 CrossRef CAS.
- E. Villemin, Y. C. Ong, C. M. Thomas and G. Gasser, Nat. Rev. Chem., 2019, 3, 261–282 CrossRef CAS.
- Y. Liu and C. C. L. McCrory, Nat. Commun., 2019, 10, 1683 CrossRef PubMed.
- S. Liu, K. Wippermann and W. Lehnert, Int. J. Hydrogen Energy, 2021, 46, 14687–14698 CrossRef CAS.
- L. Li, X. Li, Y. Sun and Y. Xie, Chem. Soc. Rev., 2022, 51, 1234–1252 RSC.
- T. Burdyny and W. A. Smith, Energy Environ. Sci., 2019, 12, 1442–1453 RSC.
- K. A. Mauritz and R. B. Moore, Chem. Rev., 2004, 104, 4535–4586 CrossRef CAS PubMed.
- G. S. Avcioglu, B. Ficicilar and I. Eroglu, Int. J. Hydrogen Energy, 2016, 41, 10010–10020 CrossRef CAS.
- S. Cheng, H. Liu and B. E. Logan, Environ. Sci. Technol., 2006, 40, 364–369 CrossRef CAS PubMed.
- Y. He, Y. L. Wu, X. X. Zhu and J. N. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 11527–11536 CrossRef CAS PubMed.
- I. M. Simeon, K. Herkendell, D. Pant and R. Freitag, Chem. Eng. J. Adv., 2022, 10, 100246 CrossRef CAS.
- W. Zhang, Y. Wu, Q. Bao, F. Gao and J. Fang, Adv. Energy Mater., 2014, 4, 1400359 CrossRef.
- Z. Liang, Q. Zhang, L. Jiang and G. Cao, Energy Environ. Sci., 2015, 8, 3442–3476 RSC.
- S. Ringe, E. L. Clark, J. Resasco, A. Walton, B. Seger, A. T. Bell and K. Chan, Energy Environ. Sci., 2019, 12, 3001–3014 RSC.
- H. J. Lee, A. C. Jamison and T. R. Lee, Acc. Chem. Res., 2015, 48, 3007–3015 CrossRef CAS PubMed.
- N. Giovambattista, P. G. Debenedetti and P. J. Rossky, J. Phys. Chem. B, 2007, 111, 9581–9587 CrossRef CAS PubMed.
- W. Luo, X. Nie, M. J. Janik and A. Asthagiri, ACS Catal., 2016, 6, 219–229 CrossRef CAS.
- K. Jiang, R. B. Sandberg, A. J. Akey, X. Liu, D. C. Bell, J. K. Nørskov, K. Chan and H. Wang, Nat. Catal., 2018, 1, 111–119 CrossRef CAS.
- Q. Zhao, J. M. P. Martirez and E. A. Carter, J. Am. Chem. Soc., 2021, 143, 6152–6164 CrossRef CAS PubMed.
- S. C. Shen, E. Khare, N. A. Lee, M. K. Saad, D. L. Kaplan and M. J. Buehler, Chem. Rev., 2023, 123, 2242–2275, DOI:10.1021/acs.chemrev.2c00479.
- J. C. Bui, E. W. Lees, L. M. Pant, I. V. Zenyuk, A. T. Bell and A. Z. Weber, Chem. Rev., 2022, 122, 11022–11084, DOI:10.1021/acs.chemrev.1c00901.
- F. P. G. de Arquer, C.-T. Dinh, A. Ozden, J. Wicks, C. McCallum, A. R. Kirmani, D.-H. Nam, C. Gabardo, A. Seifitokaldani, X. Wang, Y. C. Li, F. Li, J. Edwards, L. J. Richter, S. J. Thorpe, D. Sinton and E. H. Sargent, Science, 2020, 367, 661–666, DOI:10.1126/science.aay4217.
- B. Kim, F. Hillman, M. Ariyoshi, S. Fujikawa and P. J. A. Kenis, J. Power Sources, 2016, 312, 192–198 CrossRef CAS.
- Q. Wang, H. Dong, H. Yu and H. Yu, J. Power Sources, 2015, 279, 1–5 CrossRef CAS.
- P. An, L. Wei, H. Li, B. Yang, K. Liu, J. Fu, H. Li, H. Liu, J. Hu, Y.-R. Lu, H. Pan, T.-S. Chan, N. Zhang and M. Liu, J. Mater. Chem. A, 2020, 8, 15936–15941 RSC.
- Z. Xing, X. Hu and X. Feng, ACS Energy Lett., 2021, 6, 1694–1702 CrossRef CAS.
- D. Bohra, J. H. Chaudhry, T. Burdyny, E. A. Pidko and W. A. Smith, Energy Environ. Sci., 2019, 12, 3380–3389 RSC.
- Q. Sun and Z. Liu, Front. Chem. China, 2011, 6, 164 CrossRef.
- D. Kopač, B. Likozar and M. Huš, Appl. Surf. Sci., 2019, 497, 143783 CrossRef.
- A. Alhajaj, N. M. Dowell and N. Shah, Energy Proc., 2013, 37, 2552–2561 CrossRef CAS.
- A. Pavlišič, M. Huš, A. Prašnikar and B. Likozar, J. Cleaner Prod., 2020, 275, 122958 CrossRef.
- M. Huš, D. Kopač, N. S. Štefančič, D. L. Jurković, V. D. B. C. Dasireddy and B. Likozar, Catal. Sci. Technol., 2017, 7, 5900–5913 RSC.
- J. Lyu, V. Kudiiarov and A. Lider, Batteries, 2020, 6, 9 CrossRef CAS.
- A. Wuttig, Y. Yoon, J. Ryu and Y. Surendranath, J. Am. Chem. Soc., 2017, 139, 17109–17113 CrossRef CAS PubMed.
- A. Goyal, G. Marcandalli, V. A. Mints and M. T. M. Koper, J. Am. Chem. Soc., 2020, 142, 4154–4161 CrossRef CAS PubMed.
- T. K. Todorova, M. W. Schreiber and M. Fontecave, ACS Catal., 2020, 10, 1754–1768 CrossRef CAS.
- F. Studt, I. Sharafutdinov, F. Abild-Pedersen, C. F. Elkjær, J. S. Hummelshøj, S. Dahl, I. Chorkendorff and J. K. Nørskov, Nat. Chem, 2014, 6, 320–324 CrossRef CAS PubMed.
- T. Cheng, H. Xiao and W. A. Goddard, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1795–1800 CrossRef CAS PubMed.
- L.-C. Weng, A. T. Bell and A. Z. Weber, Energy Environ. Sci., 2020, 13, 3592–3606 RSC.
- F. Bidault, D. J. L. Brett, P. H. Middleton and N. P. Brandon, J. Power Sources, 2009, 187, 39–48 CrossRef CAS.
- L.-C. Weng, A. T. Bell and A. Z. Weber, Energy Environ. Sci., 2019, 12, 1950–1968 RSC.
- Z. Yang, D. Li, L. Xing, H. Xiang, J. Xuan, S. Cheng, E. H. Yu and A. Yang, ACS Sustainable Chem. Eng., 2021, 9, 351–361 CrossRef CAS.
- S.-J. Shin, H. Choi, S. Ringe, D. H. Won, H.-S. Oh, D. H. Kim, T. Lee, D.-H. Nam, H. Kim and C. H. Choi, Nat. Commun., 2022, 13, 5482 CrossRef CAS PubMed.
- L. Vitos, A. V. Ruban, H. L. Skriver and J. Kollár, Surf. Sci., 1998, 411, 186–202 CrossRef CAS.
- T. Möller, T. N. Thanh, X. Wang, W. Ju, Z. Jovanov and P. Strasser, Energy Environ. Sci., 2021, 14, 5995–6006 RSC.
- M. Karamad, V. Tripkovic and J. Rossmeisl, ACS Catal., 2014, 4, 2268–2273 CrossRef CAS.
- Q. Chang, J. H. Lee, Y. Liu, Z. Xie, S. Hwang, N. S. Marinkovic, A.-H. A. Park, S. Kattel and J. G. Chen, JACS Au, 2022, 2, 214–222 CrossRef CAS PubMed.
- X. Nie, M. R. Esopi, M. J. Janik and A. Asthagiri, Angew. Chem., 2013, 125, 2519–2522 CrossRef.
- A. Constantinou, S. Barrass and A. Gavriilidis, Ind. Eng. Chem. Res., 2014, 53, 9236–9242 CrossRef CAS.
- M. Li, Z. Zhu, M. Zhou, X. Jie, L. Wang, G. Kang and Y. Cao, J. Membr. Sci., 2021, 627, 119232 CrossRef CAS.
- T. Cheng, H. Xiao and W. A. Goddard, J. Am. Chem. Soc., 2016, 138, 13802–13805 CrossRef CAS PubMed.
- D. Strmcnik, M. Uchimura, C. Wang, R. Subbaraman, N. Danilovic, D. van der Vliet, A. P. Paulikas, V. R. Stamenkovic and N. M. Markovic, Nat. Chem., 2013, 5, 300–306 CrossRef CAS PubMed.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, Energy Environ. Sci., 2010, 3, 1311–1315 RSC.
- Y. Hori, A. Murata and R. Takahashi, J. Chem. Soc., Faraday Trans. 1, 1989, 85, 2309–2326 RSC.
- T. Tsujiguchi, Y. Kawabe, S. Jeong, T. Ohto, S. Kukunuri, H. Kuramochi, Y. Takahashi, T. Nishiuchi, H. Masuda, M. Wakisaka, K. Hu, G. Elumalai, J. Fujita and Y. Ito, ACS Catal., 2021, 11, 3310–3318 CrossRef CAS.
- C. Kim, T. Eom, M. S. Jee, H. Jung, H. Kim, B. K. Min and Y. J. Hwang, ACS Catal., 2017, 7, 779–785 CrossRef CAS.
- L. Lin, T. Liu, J. Xiao, H. Li, P. Wei, D. Gao, B. Nan, R. Si, G. Wang and X. Bao, Angew. Chem., Int. Ed., 2020, 59, 22408–22413 CrossRef CAS PubMed.
- W. Lin, K. M. Stocker and G. C. Schatz, J. Am. Chem. Soc., 2017, 139, 4663–4666 CrossRef CAS PubMed.
- H. Song, Y. C. Tan, B. Kim, S. Ringe and J. Oh, ACS Appl. Mater. Interfaces, 2021, 13, 55272–55280 CrossRef CAS PubMed.
- G. E. Parsons, G. Buckton and S. M. Chatham, Int. J. Pharm., 1992, 83, 163–170 CrossRef CAS.
- P. Salvi, P. Nelli, M. Villa, Y. Kiros, G. Zangari, G. Bruni, A. Marini and C. Milanese, Int. J. Hydrogen Energy, 2011, 36, 7816–7821 CrossRef CAS.
- J. O. Bockris and Z. Nagy, J. Chem. Educ., 1973, 50, 839 CrossRef CAS.
- M. R. Singh, J. D. Goodpaster, A. Z. Weber, M. Head-Gordon and A. T. Bell, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E8812–E8821 CrossRef CAS PubMed.
- Y. Mun, S. Lee, A. Cho, S. Kim, J. W. Han and J. Lee, Appl. Catal., B, 2019, 246, 82–88 CrossRef CAS.
- Y. H. Zhao, Z. Horita, T. G. Langdon and Y. T. Zhu, Mater. Sci. Eng., A, 2008, 474, 342–347 CrossRef.
- A. Ramalho and J. C. Miranda, Wear, 2005, 259, 828–834 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- S. Sahoo, D. R. Dekel, R. Maric and S. P. Alpay, ACS Catal., 2021, 11, 2561–2571 CrossRef CAS.
- S. Bamonte, S. Shubhashish, H. Khanna, S. Shuster, S. J. B. Rubio, S. L. Suib, S. P. Alpay and S. Sahoo, ACS Appl. Mater. Interfaces, 2022, 14, 27799–27813 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ee01647a |
| This journal is © The Royal Society of Chemistry 2023 |