
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePowering the hydrogen future: current status and challenges of anion exchange membrane fuel cells
Jonghyun
Hyun
a and
Hee-Tak
Kim
 *ab
*ab
aDepartment of Chemical and Biomolecular Engineering, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 34141, Republic of Korea. E-mail: heetak.kim@kaist.ac.kr
bAdvanced Battery Center, KAIST Institute for the NanoCentury, Korea Advanced Institute of Science and Technology (KAIST), 335 Gwahangno, Yuseong-gu, Daejeon 34141, Republic of Korea
First published on 2nd November 2023
Abstract
Hydrogen energy and related technologies are essential for combating climate change and meeting the growing energy demands. Despite being considered a key technology, proton exchange membrane fuel cells (PEMFCs) are yet to overcome their dependence on noble-metal catalysts, hindering progress toward a hydrogen economy. Recently, anion exchange membrane fuel cells (AEMFCs) have emerged as promising alternatives to PEMFCs owing to the use of inexpensive metals for both the catalysts and bipolar plates in alkaline environments. Although the AEMFC technology has achieved tremendous progress in materials development, several chronic problems need to be addressed. This review presents a comprehensive overview of the critical issues in the development of AEMFCs and the previous efforts made to resolve them. In addition, a comparative analysis between PEMFCs and AEMFCs is performed to diagnose the current status of AEMFCs from a practical perspective and to propose potential avenues for future development.
 Jonghyun Hyun | Jonghyun Hyun joined the Korea Advanced Institute of Science and Technology (KAIST) in 2014 to pursue both his bachelor's and master's degrees in chemical and biomolecular engineering. Currently, he is engaged in his PhD research, under the guidance of Professor Hee-Tak Kim. His primary research expertise lies in AEM fuel cells and AEM water electrolysis, focusing on enhancing performance and durability through a comprehensive understanding of interfacial phenomena between catalysts and ionomers. Additionally, he is dedicated to optimizing the design of catalyst layers and MEAs to achieve further advancements in these technologies. |
 Hee-Tak Kim | Professor Hee-Tak Kim is a full professor at the Department of Chemical Engineering and serves as the head of the advanced battery center and the Frontier Research Lab at KAIST. His primary research expertise lies in the design of electrochemical energy devices, which encompass fuel cells, water electrolysis and secondary batteries. Through the integration of electrochemical engineering, materials engineering, and interfacial engineering, his research group delves into innovative concepts and efficient designs aimed at addressing crucial challenges within this domain. |
Broader contextHydrogen, as a clean energy vector, has become increasingly crucial in combating climate change. In the hydrogen value chain, fuel cells play a vital role in harnessing hydrogen and converting chemical energy into electricity, which can be used in various industries, including transportation, industrial, and power generation. Although proton exchange membrane fuel cells (PEMFCs) have been the most studied type of fuel cell, anion exchange membrane fuel cells (AEMFCs) have recently gained attention due to their use of inexpensive materials. The recent remarkable developments in anion exchange membranes (AEMs) and ionomers (AEIs) have significantly improved the performance and durability of AEMFCs, shedding light on the path forward. However, the current status falls short of practical applicability, demanding a significant leap beyond the current level. In this article, we present a comprehensive review of AEMFCs, comparing them with PEMFCs using objective indicators. We summarize the problems associated with AEMFCs from a material and system perspective and suggest improvements accordingly. This review will provide insights for researchers and contribute to the progress of AEMFCs. |
1. Introduction
Hydrogen is the most desired ecofriendly energy vector for preventing climate change and meeting the rising global energy demand. Fuel cells are electrochemical devices that can convert the chemical energy of hydrogen to electricity and are regarded as key enablers of hydrogen technologies. Among the many types of fuel cells, proton exchange membrane fuel cells (PEMFCs) are the most widely used, as exemplified by stationary fuel cells (FCgen® and FCwave® from Ballard™) and fuel cell electric vehicles (Nexo from Hyundai and Mirai from Toyota), because of their superior power performance and acceptable durability. PEMFC technology is grounded on fast oxygen reduction and hydrogen oxidation reactions catalyzed by noble-metal catalysts, such as platinum (Pt) coupled with a proton exchange membrane (PEM). However, in terms of commercialization, PEMFC technology still suffers from high system costs.1,2 There are many reasons for this limitation, but the Achilles’ heel for such a price increase is the acidic environment in PEMFCs due to the use of proton exchange membranes and ionomers. In general, an acidic environment is corrosive to materials, thereby compelling the use of noble-metal catalysts and perfluorinated ionomers that are stable under acidic conditions as well as acid-resistant bipolar plates.Anion exchange membrane fuel cells (AEMFCs) have recently emerged as cost-effective alternatives to PEMFCs. AEMFCs are a type of fuel cell that use anion exchange membranes (AEMs) as electrolytes to facilitate the migration of hydroxide ions (OH−). This technology originates from the alkaline fuel cell, which was first developed in the 1960s in response to NASA's Gemini, Apollo, and Space Shuttle projects.3 The alkaline fuel cell technology that utilizes highly concentrated alkaline solution as an electrolyte has evolved into AEMFC technology by replacing the liquid electrolyte with an AEM. However, it was not until the early 2000s that the technology started gaining significant attention as a potential alternative to PEMFCs. Since the early 2000s, there have been significant research and development efforts into AEMFCs, with numerous academic and industrial groups working to improve the efficiency and durability of this technology. There have been several demonstration projects and research consortia focused on AEMFCs in recent years. AEMFCs offer several advantages over other types of fuel cells, including their ability to use non-precious metal (NPM) catalysts, hydrocarbon membranes, and cheap metal bipolar plates, which significantly reduce the cost of the fuel cells.
AEMFCs constitute a relatively new technology that is currently under development. As with any new field, several critical challenges have been faced and overcome. One of the most significant challenges to date has been the achievement of anion exchange membrane stability under highly alkaline conditions.4,5 Researchers in this field have been working hard to overcome these challenges, and significant progress has been made in recent years. Based on these efforts, state-of-the-art AEMFCs can now deliver remarkable power performances that surpass those of ordinary PEMFCs with durability of over 2000 h.6 Several manufacturers (AFC Energy and HydroLite) are now working on commercializing AEMFCs for use in stationary power generation and other applications. However, practical applications of AEMFCs still remain challenging owing to various instabilities and degradation modes that are not yet clearly identified and understood, in addition to a myriad of requirements for optimizing fuel cell systems for specific applications. Similar to other types of fuel cells, the development of electrochemically active and durable catalysts is indispensable for commercialization. Furthermore, there is a high demand for precise water and thermal management to maintain optimal operating conditions.7,8
The present review provides an overview of AEMFC technology, covering its key features such as structures, materials, and cost-effectiveness, as well as a comparative analysis with PEMFCs to assess the current status, strengths, and weaknesses. We then delve into the critical issues concerning AEMFCs, including their power performance and durability, and provide detailed and organized accounts of their causes, such as membrane degradation, catalyst activity and stability, and water management challenges. We also highlight the practical challenges presented by these issues and discuss their implications for future developments in AEMFC technologies. Furthermore, we identify unexplored areas within this technology sector and suggest some research directions and solutions for practical development of AEMFCs. It is expected that a timely review could stimulate more researchers to develop the technology and provide the necessary impetus for future progress.
2. Key features of AEMFCs and drivers of AEMFC development
The structure of AEMFCs, as shown in Fig. 1a, is similar to that of PEMFCs. Both types of fuel cells have a membrane electrode assembly (MEA) as their key component. The MEA consists of a membrane and a pair of catalyst layers (CLs) and diffusion layers within a single cell. The single-cell structure is repeated in a stack, like in the PEMFCs, to deliver high power and a high working voltage. However, the materials used for the AEMFC MEA are different because the operating environment of the AEMFC is alkaline, contrary to the acidic environment of the PEMFC, which offers the possibility of reducing the fuel cell cost. NPM-based oxygen reduction reaction (ORR) catalysts are used in AEMFCs, which is in contrast to the dominantly used Pt-based catalysts in PEMFCs. Protons and hydroxide ions migrate in opposite directions in PEMFCs and AEMFCs, respectively, resulting in different electro-osmosis behaviors. In AEMFCs, water is consumed at the cathode, unlike water generation at the cathode in PEMFCs, leading to different water distributions in the MEAs and thereby requiring different water management strategies.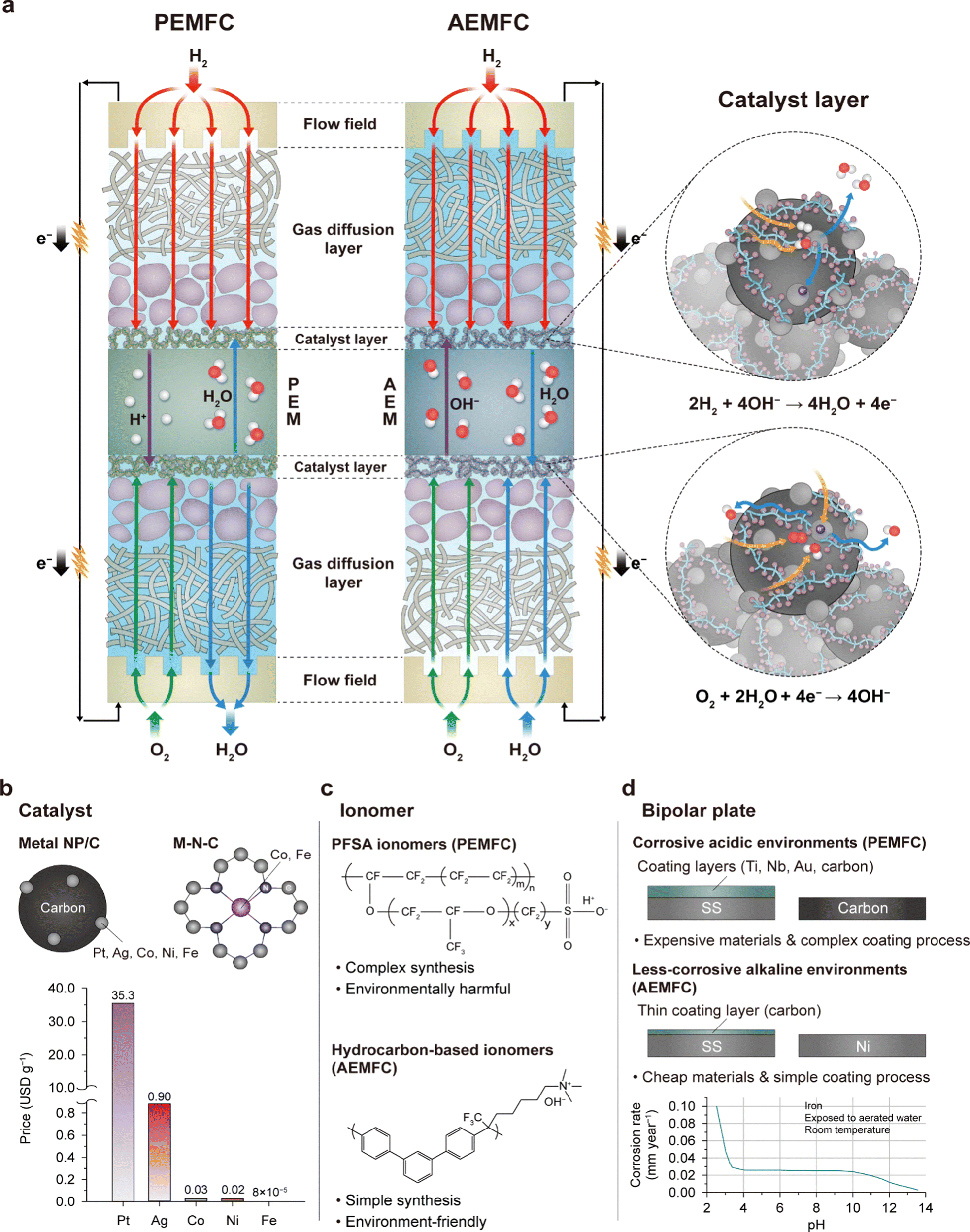 | ||
| Fig. 1 Potential cost-reduction with AEMFCs. (a) Schematic illustrations of the structures and electrochemical reactions of PEMFCs and AEMFCs. Three reasons for cost savings via switching from PEMFCs to AEMFCs: (b) catalyst, (c) ionomer, and (d) bipolar plate. The metal prices in (b) were calculated by referring to the London Metal Exchange.9 The corrosion rate of iron with pH is reprinted from ref. 10. | ||
Various factors contribute to the cost-effectiveness of AEMFCs. One such factor is the use of less-corrosive NPMs, which are expected to have high catalytic activities owing to the alkaline environment. Carbon-supported metal nanoparticles and N-coordinated metal catalysts are being studied parallelly as catalysts (Fig. 1b). Regarding the former, carbon modification technologies such as the fluorination of the carbon support surface11 attract interest because of their critical role in minimizing water flooding on the anode side of AEMFCs. While Pt is mainly being used at the single-cell level, Ag,12,13 Co,14 Ni,15 and Fe metals16 or alloys17,18 have been studied as catalysts for AEMFCs. Indeed, the prices of NPMs are 2–5 orders of magnitude lower than that of Pt, as demonstrated in the price comparison plot in Fig. 1b. This has the potential to significantly reduce the cost of fuel cells.
The second aspect of cost savings is the utilization of hydrocarbon-based ionomers (Fig. 1c). Under acidic conditions, hydrocarbon-based PEMs are easily degraded primarily through hydrolytic reactions;19,20 they degrade much faster in Fenton's reagent than perfluorosulfonic acid (PFSA) membranes.21,22 Moreover, their insufficient mechanical stabilities under repeated exposure to humid and dry conditions make them unsuitable for dynamic PEMFC operations.23 Therefore, the industry has standardized the use of PFSA membranes to ensure durability of PEMFCs. However, in alkaline environments, the degradation of hydrocarbon ionomers can be mitigated.4 Indeed, state-of-the-art hydrocarbon AEMs and ionomers (AEIs) have demonstrated long-term operations in AEMFCs.6,24,25 Compared to the complex and harmful synthesis process of the PFSA ionomer, the synthesis of hydrocarbon ionomers is generally simpler and milder, which makes them more cost-effective.5,26 Although the fluorinated backbone of the PFSA ionomer provides high chemical stability that is sufficient for operations exceeding 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h, it takes a long time to biodegrade, requiring expensive postprocessing after use. For reference, fluorinated polyacrylates, another type of fluorinated polymer, have an estimated biodegradation half-life of approximately 1200–1700 years.27,28 In contrast, the biodegradability of hydrocarbon ionomers is relatively faster, which makes them a more ecofriendly option.
000 h, it takes a long time to biodegrade, requiring expensive postprocessing after use. For reference, fluorinated polyacrylates, another type of fluorinated polymer, have an estimated biodegradation half-life of approximately 1200–1700 years.27,28 In contrast, the biodegradability of hydrocarbon ionomers is relatively faster, which makes them a more ecofriendly option.
One additional driver of cost reduction is the use of low-cost metal bipolar plates, including those made of materials like stainless steel or nickel. This is in contrast to the use of thick acid-resistant surface-coated metals or highly graphitic carbon bipolar plates in PEMFCs owing to the corrosive nature of the Nafion ionomer. As seen in Fig. 1d, the corrosion rate of iron reduces as the pH increases, indicating the possibility of using inexpensive stainless steel as the bipolar plate in AEMFCs. The commercial carbon-coated stainless steel 316L has been shown to maintain a low internal contact resistance (<20 mΩ cm2) in the AEMFC environment,29 but an additional coating process is necessary in PEMFCs as their internal contact resistances exceed 200 mΩ cm2 due to surface corrosion.30,31
3. Performance and durability of AEMFCs compared with PEMFCs
The foundation of fuel cell commercialization is based on achieving sufficient performance and durability. Previous papers document the performance, durability, and milestones for AEMFCs.63–65 Although significant progress has been made in improving their performance and durability, there are still considerable gaps for meeting practical demands. For objective assessments of the current status of AEMFCs, their performance and durability are compared with those of state-of-the-art PEMFCs in Fig. 2.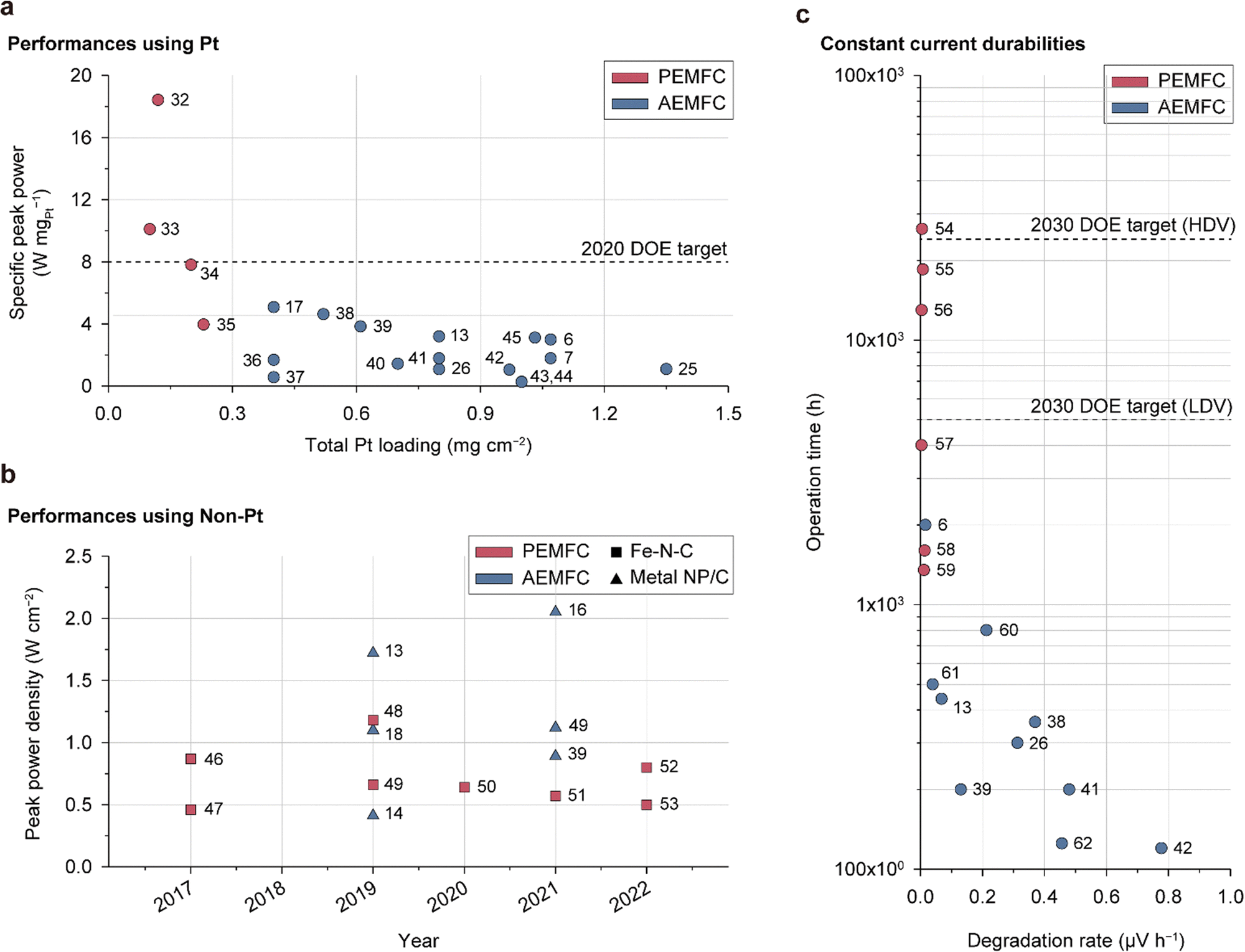 | ||
| Fig. 2 Performance and durability comparisons of state-of-the-art PEMFCs and AEMFCs. (a) Specific peak power density versus total Pt loading for fuel cells containing Pt-based cathodes: peak power density is standardized by the total Pt loading in both electrodes (Table 1).6,7,13,17,25,26,32–45 (b) Peak power densities of fuel cells containing non-Pt catalyst-based cathodes over the past 6 years (Table 2).13,14,16,18,39,46–53 (c) Fuel cell operation time versus degradation rate: the fuel cells are operated under the constant current mode (Table 3).6,13,26,38,39,41,42,54–62 | ||
Fig. 2a compares the specific peak power densities reported for Pt-based AEMFC and PEMFC single cells, which are obtained by dividing the peak power density by the amount of Pt catalyst used (Table 1). One notable feature here is that AEMFCs use significantly larger amounts of Pt (>0.3 mgPt cm−2) compared to PEMFCs (<0.3 mgPt cm−2). Furthermore, the specific peak power of AEMFCs is less than that of PEMFCs. The peak power density of AEMFCs is comparable to that of PEMFCs, which is attributed to the use of a larger amount of Pt catalyst. Considering the good ORR kinetics of Pt in the alkaline environment,66,67 the relatively low specific peak power of the AEMFC suggests low Pt utilization. This motivates further research on the CL structure, water management, CO2 poisoning, and catalyst/AEI interface with the aim of augmenting Pt utilization.
Fig. 2b shows the peak power densities of NPM catalysts (metal nanoparticles supported on carbon (metal NP/C) and Fe–N–C) at the PEMFC and AEMFC cathodes reported over the past 6 years (Table 2). Despite being in the early stages of research, a few AEMFCs based on NPM catalysts have exhibited a notable power performance of over 1.5 W cm−2, which is far higher than that of PEMFCs.13,16 Remarkably, a Fe–N–C catalyst-based AEMFC was reported with a power density of over 2.0 W cm−2 and durability of more than 150 h,16 demonstrating the potential for cost-effective AEMFC development and stimulating further research. However, the use of pure O2, excessive supply of reaction gases, and high catalyst loading are still challenges for NPM-based AEMFCs. Due to the low activity site density of NPM catalysts, large amounts of the catalysts are required, leading to thick CLs and making optimal design difficult for both PEMFCs and AEMFCs.
Durability is a crucial factor for practical applications of fuel cells. However, until recently, the alkaline stabilities of AEMs and AEIs were too low to enable long-term operation. The discovery of the ether-cleavage degradation pathway and subsequent development of ether-free ionomers have achieved significant strides in improving the alkaline stabilities of AEMs and AEIs. However, the durability of AEMFCs is still lacking, as evidenced by the comparison of the lifetimes of AEMFCs and PEMFCs under constant current operation, as published recently (Fig. 2c and Table 3). While PEMFCs have various durability evaluation protocols tailored to their specific purposes, AEMFCs do not have a standardized protocol and are typically evaluated through constant current operation.
Several PEMFCs exhibit durabilities of 10000 h or more, exceeding the 2030 DOE target for high-performance vehicles (HDVs),68 while the longest lifespan of any reported AEMFC to the best of our knowledge has been 2000 h.6 Despite the good stabilities of metal catalysts under alkaline conditions and recent developments in alkali-stable AEMs and AEIs, present AEMFCs do not meet the target durability level, highlighting the need for intensive research directed at enhancing durability. The reasons for the rapid degradation of AEMFCs are described and discussed in Section 5.
4. Challenges and solutions for improving the power performance of AEMFCs
Power performance is an essential characteristic of a fuel cell as it is closely linked to its energy conversion efficiency and stack/system size. It is typically evaluated using a polarization curve that provides information on various polarization components. The dependency on operating conditions is the basis of an optimal fuel cell system design, considering heat management and the balance of the plant. Regarding the power performances of AEMFCs, most studies have focused on MEA development, while very little attention has been devoted to stack- or system-level designs. In the pursuit of enhancing the power performance of the MEA, key materials such as AEMs, AEIs, and catalysts have been studied extensively. However, the CLs and interfaces are less explored despite their strong impact on the power performance. Moreover, only a few studies have reported quantitative analyses of the polarization curve; therefore, the factors affecting the power performance have not been understood fully.The power performance of AEMFCs is largely dictated by the properties of the catalyst, AEM, AEI, and gas-diffusion layer. Fast ORR/hydrogen oxidation reaction (HOR) kinetics of catalysts and high OH− conductivity of the AEM/AEI materials can ensure higher power performance under the given operating conditions. Moreover, the composition and structure of the CL are of great importance for enhancing the power performance since the electrochemical reactions in AEMFCs occur at triple-phase boundaries (TPBs) at which the gas feed, catalyst, and ionomer coexist. Furthermore, several chronic problems, such as strong ionomer adsorption on the catalyst surface, severe water imbalance between the two electrodes, and formation of carbonate species by CO2, are crucial to the power performance of AEMFCs.
In this section, we review the challenges related to enhancing the power performance and the approaches to address them. We first focus on the issues concerning materials and then cover some issues pertaining to power performance, such as water flooding and CO2 poisoning. To explain the drawbacks from a practical perspective, the current status of ionomer and catalyst developments for AEMFCs is compared with those of PEMFCs. The efforts regarding MEA design for power performance enhancement are also summarized.
4.1. Material development
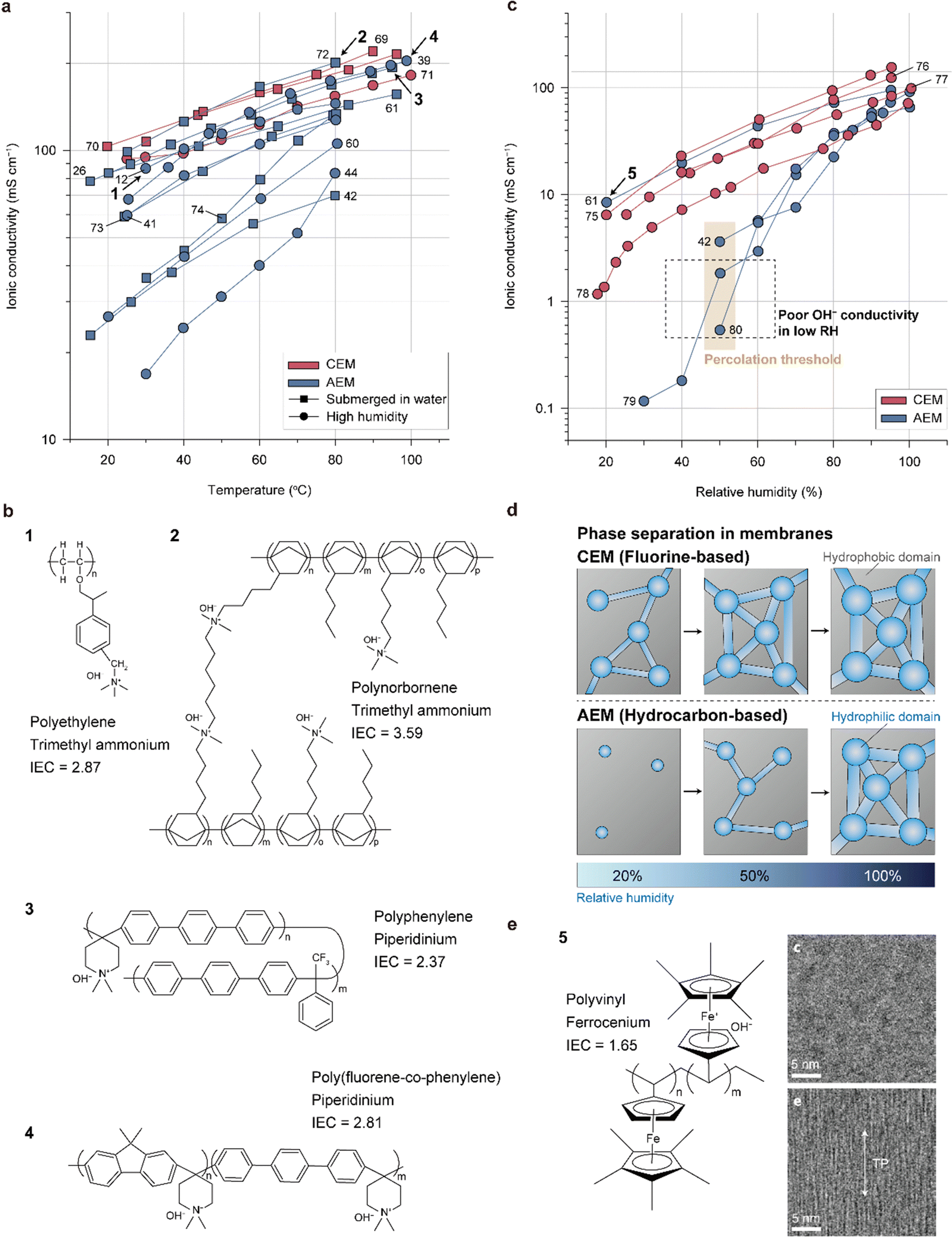 | ||
| Fig. 3 Ionic conductivities and structures of the representative membranes. (a) Ionic conductivities of state-of-the-art CEMs and AEMs as functions of temperature measured under highly humid environments or in liquid water (Table 4).12,26,39,41,42,44,60,61,69–74 (b) Chemical structures of representative AEMs with similar ionic conductivities as CEMs. (c) Ionic conductivities of state-of-the-art CEM and AEM as functions of the relative humidity (RH, Table 5).42,61,75–80 (d) Schematic showing the evolution of the phase-separated morphology with increasing RH for the CEM and AEM. (e) Chemical structure of polyvinyl ferrocenium (left) and cross-sectional transmission electron microscopy (TEM) images of the polyvinyl ferrocenium membranes with unaligned (upper right) and aligned (lower right) structures in the through-plane direction. Cross-sectional TEM images are adapted with permission.61 The membranes were stained with tungstate to clearly observe the phase-separation morphology. In the images, the dark and bright regions correspond to the hydrophilic and hydrophobic domains, respectively. | ||
Fig. 3b and Table 5 provide information on the structures, types of backbones, types of cationic groups, and ion exchange capacities (IECs) of the four highly conductive AEMs (polyethylene with hexyl TMA side groups, polynorbornene with hexyl TMA side groups, polyphenylene with piperidinium side groups, and poly(fluorene-co-phenylene) with piperidinium side groups). The ionic conductivity of each AEM is indicated in Fig. 3a. The polyethylene-based polymer (1), developed by Varcoe's group, stands as one of the pioneering materials for highly conductive AEMs.12 The AEMFC utilizing this polyethylene-based polymer exhibited an impressive power density of 2.55 W cm−2 with a Pt/C cathode and a notable 1.72 W cm−2 with an Ag/C cathode. Moreover, when driven at 0.6 A cm−2 for 440 h, the AEMFC demonstrated a mere 7% voltage loss, showcasing its exceptional durability. Ongoing research on polyethylene-based AEMs92–94 continues to inspire numerous subsequent developments in the field. The polynorbornene polymer (2) features a remarkably high IEC (3.59), which accounts for its high conductivity. Normally, an increase in the IEC leads to excessive water swelling, resulting in reduced mechanical stability. To address this issue, the polynorbornene AEM is partially cross-linked by linking the side groups, thereby controlling its swelling behavior. This AEM was first reported by Huang et al.95 and has been the subject of numerous studies by various researchers.24,72,96 The AEMFC using the polynorbornene-based AEM exhibits the highest power density (3.5 W cm−2) achieved to date97 and exceptionally high durability (2000 h).6 The superior power performance is attributed not only to its high ionic conductivity but also to its fast water transport properties; the water generated at the anode readily moves to the cathode through the AEM, achieving a well-balanced water distribution inside the MEA. This prevents drying of the cathode even at remarkably high current densities (∼10 A cm−2). The polyphenylene with piperidinium side groups (3), which was first reported by Yan's group, also has a high IEC (2.37) and low water swelling ratio (<20%).26 The low water swelling ratio is attributed to its rigid and hydrophobic polyphenylene backbone structure and high molecular weight. A single cell employing the polyphenylene AEM maintains a hydrogen gas cross-over of less than 10 sccm even after 5000 wet/dry cycles, demonstrating its high mechanical strength. Lee's group synthesized poly(fluorene-co-phenylene) with piperidinium side groups (4)39 and fabricated its reinforced membrane with a porous polyethylene support.38 The reinforced membrane exhibited a significant reduction in water uptake and swelling ratio as well as improved mechanical strength, leading to an impressive durability of over 300 h and a low voltage degradation rate (0.37 mV h−1). Furthermore, the copolymer was found to be a promising candidate for use as an AEI binder because the polyfluorene segment mitigates strong absorption of the phenylene segment on the Pt surface, which causes a drastic decrease in HOR activity due to its nonrotatable properties. The AEMFC employing the copolymer as both AEM and AEI delivers a high power density of 2.4 W cm−2.
Similar to conventional CEMs, the ionic conductivities of AEMs are impacted by the relative humidity (RH) since the level of dissociation of the cation–anion groups and mobility of OH− increase with higher water content in the AEMs. Although the AEMs mentioned earlier demonstrate exceptional conductivities when fully hydrated, they have lower ion conductivities than CEMs at low RHs, as depicted in Fig. 3c. The significant decrease in ionic conductivity at low RHs for the AEMs compared to the perfluorinated CEMs can be attributed to the morphologies of the water channels inside the AEMs (Fig. 3d). With increasing RH, the hydrophilic domain of the CEM or AEM expands through hydration and becomes more connected to the nearby domains, eventually forming well-connected channels for proton transport at a certain RH known as the “percolation threshold.” As observed for hydrocarbon-based CEMs,98,99 ionomers with rigid backbones form narrower water channels of AEMs compared to perfluorinated CEMs, resulting in percolation thresholds at higher RH and a less-connected water channel structure (Fig. 3d). Although increasing the IEC can improve this issue, this approach is practically limited by the resulting decrease in mechanical properties.
Alignment of the water channel in the thickness direction can significantly improve the ionic conductivity at low RH.100 This was recently achieved with an AEM having a polyvinyl backbone and ferrocenium cationic group, whose structure is shown in Fig. 3e (indicated by 5).61 The AEM showed similar or even higher ionic conductivity than the CEM. The ferrocenium moiety aligns with the direction of the external magnetic field owing to its paramagnetic property, presenting a possibility for aligning the water channels in the thickness direction, which is the most preferred morphology for shortening the OH− transport pathways. The phase-separation morphologies of the AEMs prepared with and without an external magnetic field are compared in Fig. 3e; the dark regions corresponding to the hydrophilic ferrocenium domains are aligned and connected in the through-plane direction. This result motivates future research for achieving high ionic conductivity under low RH conditions by aligning the water channels and achieving percolation at a much lower RH.
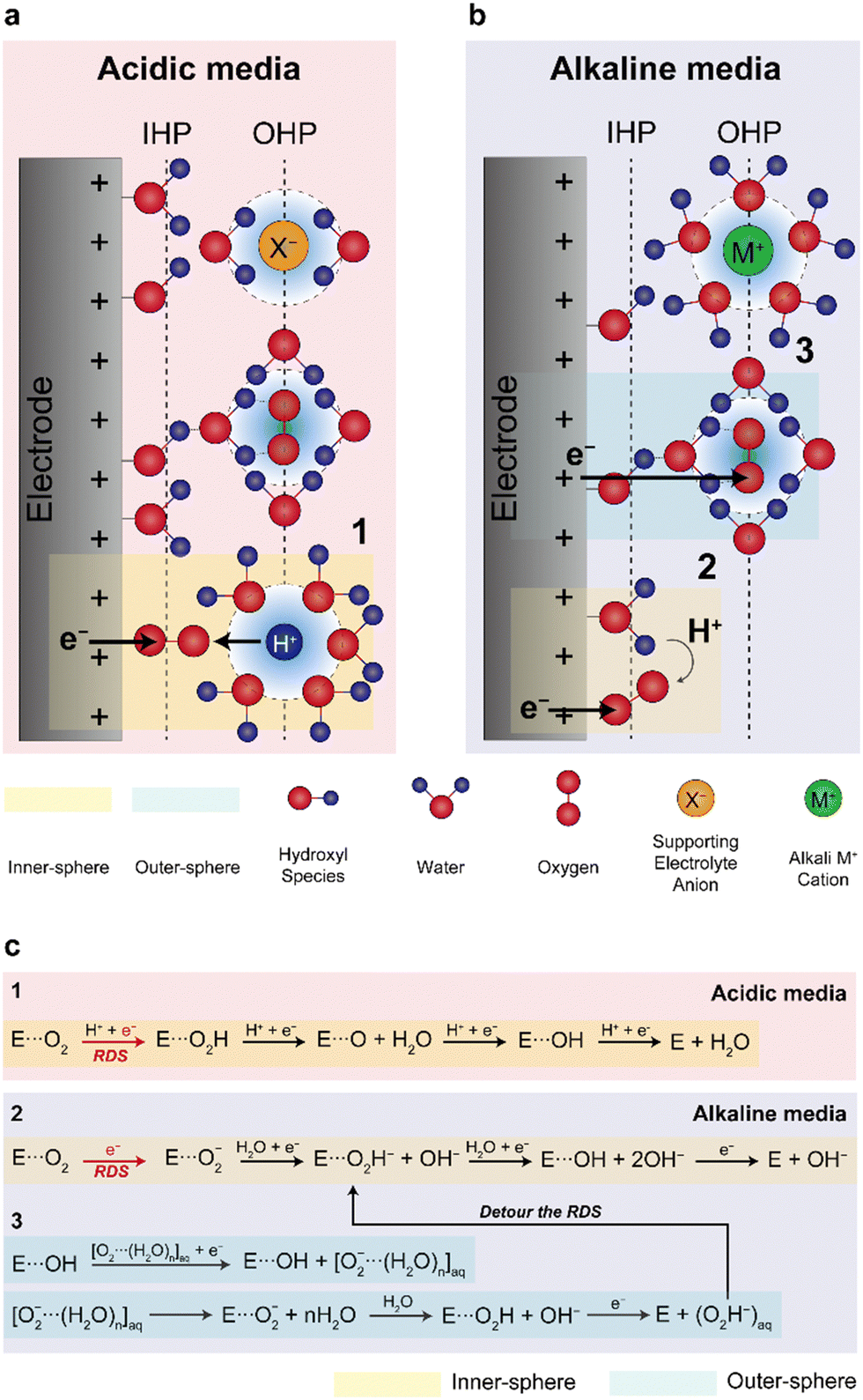 | ||
| Fig. 4 Proposed inner- and outer-sphere electron transfers for the ORR in alkaline media. Schematic illustrations of the double-layer structures in (a) acidic and (b) alkaline media during the ORR and (c) reaction mechanisms in the acidic and alkaline media. (a) and (b) are adapted with permission.67 | ||
Despite Pt-based catalysts exhibiting higher ORR activities under alkaline conditions compared to acidic environments, NPM catalysts have garnered more attention as ORR catalysts for AEMFCs owing to their potentially lower cost. Previous studies have reported that NPM catalysts may have higher ORR activities compared to Pt-based catalysts, presenting the possibility of replacing Pt catalysts with NPM catalysts in AEMFCs. Fig. 5 presents a comparison of the ORR Tafel slope and half-wave potentials for some recently reported NPMs and conventional Pt/C catalysts; the figure clearly shows that typical NPM catalysts have higher half-wave potentials and lower Tafel slopes for ORRs in half-cell tests, demonstrating the potential for cost-effective AEMFCs (Table 6).
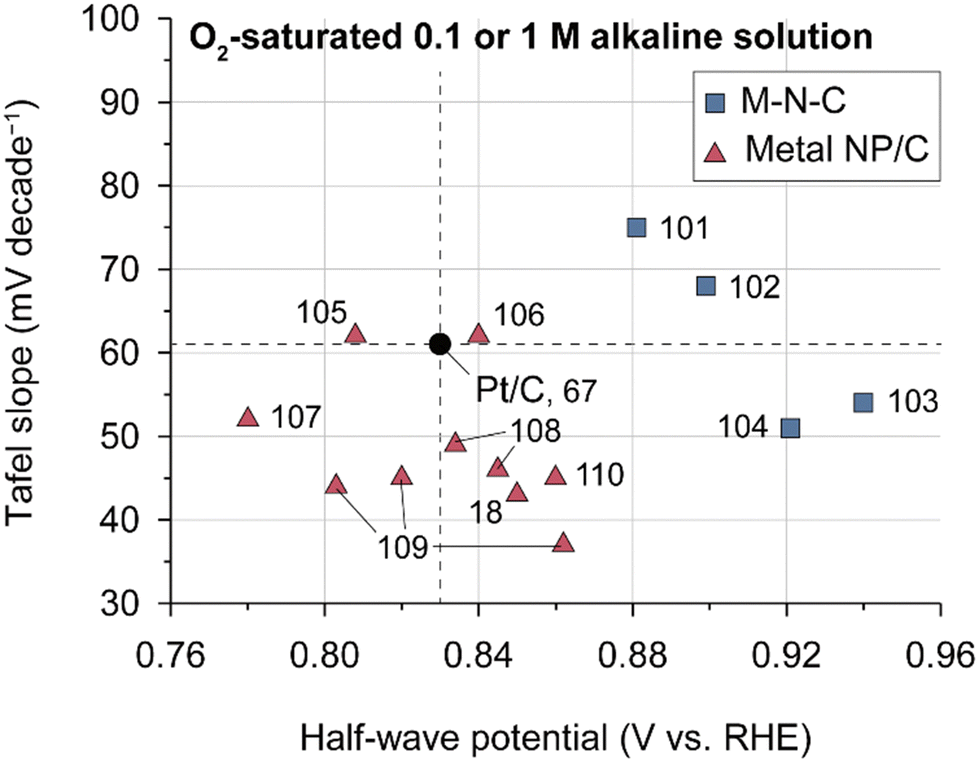 | ||
| Fig. 5 ORR performances of state-of-the-art NPM catalysts. Tafel slopes and half-wave potentials of various NPM ORR catalysts. The performances were evaluated for 0.1 or 1 M aqueous alkali solution in the RDE system (Table 6).18,67,101–110 | ||
Among NPM catalysts, manganese oxide-based catalysts have emerged as a promising alkaline ORR catalyst and have been a subject of intensive investigation. MnO2 exhibits diverse crystal structures, including α, β, γ (1D-tunneled), δ (2D layered), and λ (3D spinel), and the catalytic activity varies depending on the crystal structure (δ- ≈ α- > γ- > λ- > β-MnO2).111,112 In the case of δ-MnO2, cation intercalation takes place, and ORR activity changes with the type of intercalated cation.112–114 It was found that Ni doping is effective in increasing the catalytic activity of manganese oxide catalysts.115–117 A recent research unveiled the correlation between MnO2 degradation and HO2− generation, offering valuable insights for advancing the durability of the MnO2 catalyst.118
However, the high ORR activities of the NPM catalysts measured in half cells do not necessarily translate to high single-cell power performances. In a half-cell, the CL readily forms a two-phase interface of electrolyte and catalyst; therefore, the ORR activity is not highly sensitive to the CL structure, whereas in a single cell, the TPB involving the electrolyte, catalyst, and gas, which sensitively changes with the CL structure determines the power performance.
In the half-cell test with a rotating disk electrode (RDE), the current density of the ORR is less than 10 mA cm−2 owing to the low O2 solubility of the liquid electrolyte. In contrast, single cells can operate at high current densities on the order of a few amperes per square centimeter because of the significant supply of O2 gas.119 Indeed, Riasse et al. demonstrated that the RDE analysis is not capable of assessing fuel cell performances beyond the high potential regime, in contrast to gas diffusion electrode (GDE) and differential cell (DC) methods.120
High-current-density operation is achieved only when the catalyst has enough surface area or turnover frequency (TOF). NPM catalysts suffer from low active-site densities and low TOFs, so a larger amount of catalyst is required to enable high-current-density operation. The high catalyst loading for NPM catalysts leads to additional problems; the thickness of the CL becomes large, increasing the mass transport limitation due to the longer O2 transport pathway. In addition, the thick CL increases the electron and ion pathways, causing an increase in the Ohmic resistance. NPM-based CL structures have rarely been studied, and this lack of understanding makes it difficult to design high-performance NPM CLs. A recent review121 and related research.122 emphasize the importance of NPM-based CL design.
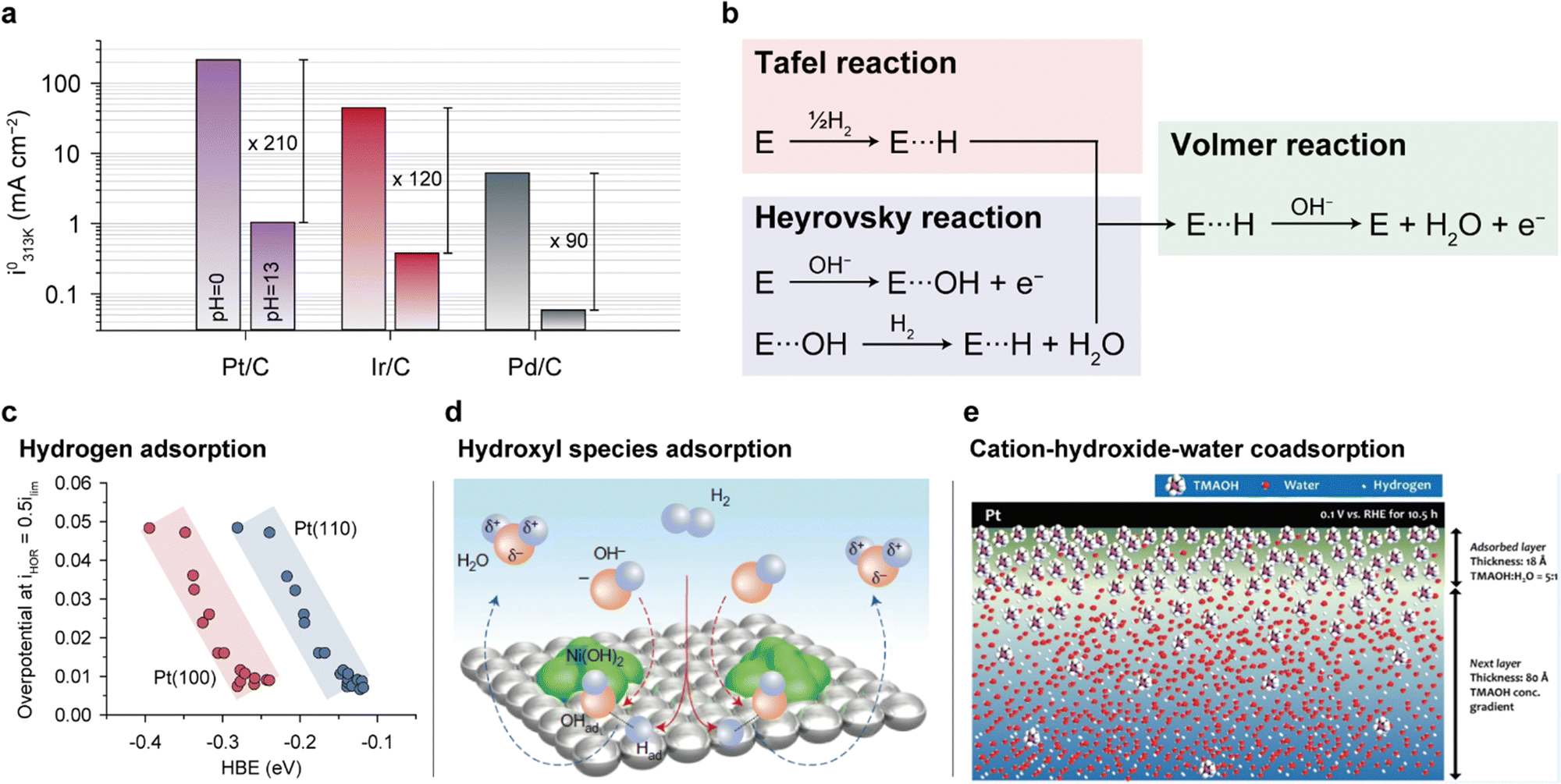 | ||
| Fig. 6 HOR mechanisms and performance determinants for anode catalysts. (a) HOR/HER exchange current densities of carbon-supported Pt, Ir, and Pd at 313 K at 0 and 13 pH. (b) Two proposed HOR pathways. Three parameters governing the HOR performance: (c) hydrogen adsorption, (d) hydroxyl species adsorption, and (e) cation-hydroxide-water co-adsorption. The HOR/HER exchange current densities in (a) are reprinted with permission.123 The half-wave overpotential versus HBE plot in (c) is reprinted with permission.124 Schematic representation of the HOR process on Ni(OH)2/Pt(111) in (d) and the cation–hydroxide–water co-adsorbed layer in (e) are adapted with permission.125,126 | ||
Although it is still unclear which of the two reaction pathways dominate the HOR, previous studies have provided significant results supporting both mechanisms. Yan's group systematically compared the HOR activities of Pt at various pH values, demonstrating that the HOR half-wave potentials (Fig. 6c) and HOR overpotentials monotonically increase with greater HBE, supporting the Tafel–Volmer mechanism.124 Gasteiger's group demonstrated that oxophilicity is not a critical descriptor of the HOR as Pt exhibits greater HOR activity compared to Ir and Pd at pH 13, despite having the highest oxophilicity for Ir.123 They found that the difference in HBEs between pH 0 and 13 (ΔEbinding = 12.5–13.5 kJ mol−1) follows the Arrhenius equation 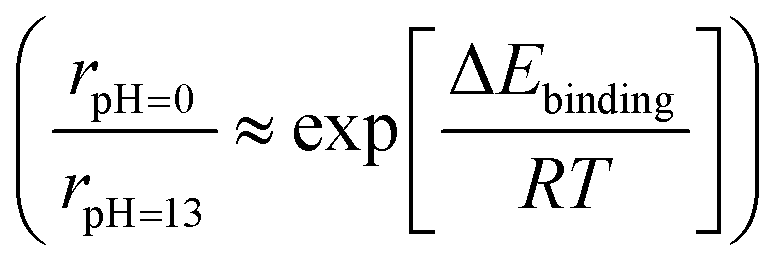 and that this difference successfully explains the 120–180 times lower exchange current densities at pH 13 compared to those at pH 0 (Fig. 6a), supporting the Tafel–Volmer mechanism.
and that this difference successfully explains the 120–180 times lower exchange current densities at pH 13 compared to those at pH 0 (Fig. 6a), supporting the Tafel–Volmer mechanism.
In contrast, Markovic's group observed an increase in HOR activity with increasing oxophilicity for metal catalysts (Ir > Ru > Pt).125 Moreover, they demonstrated increased HOR kinetics by introducing Ni(OH)2 nanoparticles on the Pt surface, which promotes the dissociation of water into hydrogen and hydroxyl intermediates (Fig. 6d). On the basis of their findings, they proposed that the adsorption energy of OHads could be a key descriptor of the HOR, supporting the Heyrovsky–Volmer mechanism.
Kim's group presented a different view on the HOR catalytic activity by considering the interactions between the ionomer and catalyst.126 They suggested that the cationic groups of AEIs strongly adsorbed on the Pt catalyst surface hinder diffusion of H2, thereby lowering the HOR activities (Fig. 6e). The HOR activity of the Pt catalyst decreases with an increase in the absorbance of the methyl peak in the infrared reflection absorption spectroscopy data, indicating the adsorption of TMA (typical cationic group of AEIs) on the Pt surface. Furthermore, neutron reflectance analysis demonstrates that when a voltage of 0.1 V vs. RHE is applied to the Pt catalyst in 0.1 M TMAOD/D2O electrolyte (hydrogen is converted to deuterium in TMAOH/H2O), an 18 Å TMA-OH-water coadsorption layer forms on the surface after 10.5 h. The high cation–hydroxide concentration in the coadsorbed layer restricts H2 access to the catalyst surface, thereby restraining HOR activity (Fig. 6e).
HOR catalysts have received less attention in the context of AEMFCs compared to ORR catalysts owing to their higher activities. It is worth noting that Pt/C catalysts exhibit fast HOR kinetics in alkaline environments and that other precious-metal catalysts such as Ir, Pd,127 Ru, and their alloys are not superior to the Pt-based catalysts, as demonstrated in the comparison of HOR activities among Pt/C, Ni/C, and other precious-metal catalysts (Fig. 7 and Table 7). Among NPMs, nickel-based catalysts are under active investigation for their high activity. A partially oxidized Ni surface proves to be an ideal structure for the HOR since H strongly adsorbs on Ni metal and weakly adsorbs on Ni oxide.128,129 Various strategies including functionalization,130 core–shell structures,136,139 alloys (Ni0.95Cu0.05),140 and carbon supports (carbon nanotubes (CNTs) and N, S-doped carbon),137,138 have been employed to modify nickel-based catalysts for their use as a HOR catalyst. However, their HOR activity remains approximately two orders of magnitude lower than that of Pt/C catalysts.
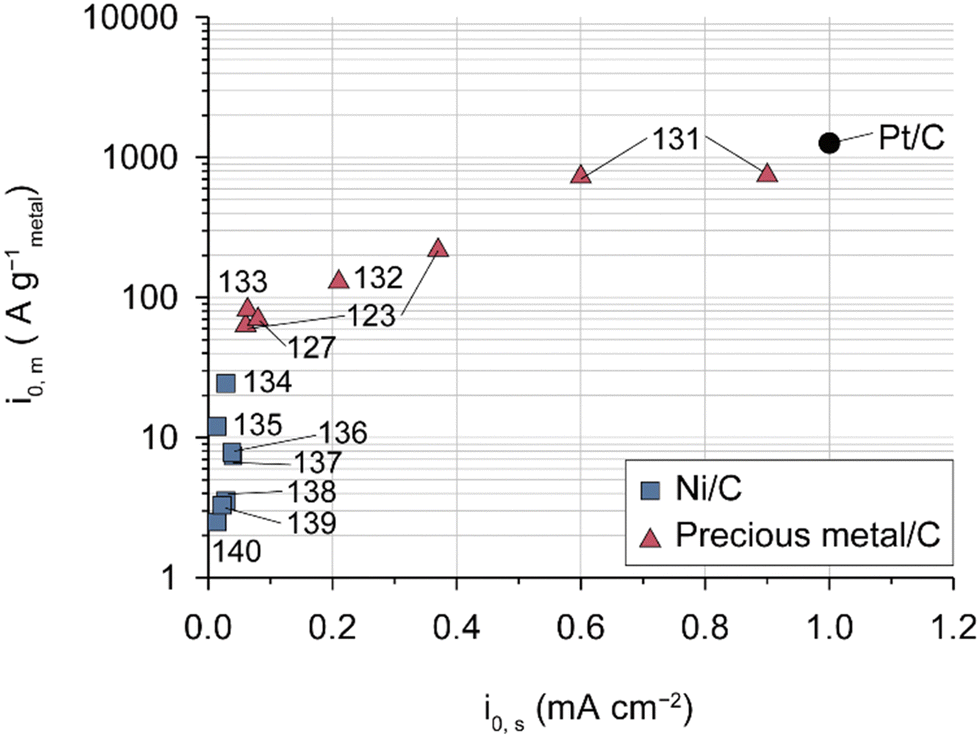 | ||
| Fig. 7 HOR activities measured using the half cell with state-of-the-art catalysts. Mass- and area-specific exchange current densities of various HOR catalysts evaluated for 0.1 or 1.0 M aqueous alkali solution in a RDE system (Table 7).123,127,131–140 | ||
Practical anode CLs in AEMFCs typically have higher precious-group metal (PGM) catalyst loadings of over 0.3 mg cm−2 compared to PEMFCs (<0.1 mg cm−2).6,25,45 This difference can be attributed not only to the relatively low HOR activities of PGM catalysts under alkaline conditions but also to the adsorption of AEIs on the catalysts, which is exacerbated by water flooding in the anode CLs (as described in the next section). Although PtRu/C is conventionally used instead of Pt/C because PtRu reduces the adsorption energy with AEIs more effectively than Pt,40,141 this does not completely solve the problem of limiting H2 diffusion caused by AEI adsorption. Therefore, while emphasizing the importance of research on non-precious metal-based HOR catalysts, it is essential to consider the problem of AEI adsorption on the precious-metal catalyst surface and the inhibition of H2 supply due to water flooding, especially for low-loading precious-metal catalysts on the anode.
4.2. Water management
Despite the use of highly active ORR catalysts and highly conductive AEMs and AEIs, the power performances of AEMFCs are often limited by water imbalance in the MEA. This is attributed to the simultaneous water generation at the anode and water consumption at the cathode (Fig. 8a). When water cross-over from the anode to cathode is not as fast as the generation or depletion of water, the anode can become flooded and the cathode can become dried out. The difference in water content between the two electrodes increases with current or operation time, exacerbating the water flooding and drying problems. Water imbalance on both electrodes during actual AEMFC operation has been observed using neutron beams (Fig. 8b) or synchrotron X-rays (Fig. 8c). While this issue also occurs in PEMFCs, it is not as significant as that in AEMFCs owing to their different stoichiometries; in PEMFCs, two water molecules are generated per four electrons (O2 + 4H+ + 4e− → 2H2O), whereas in AEMFCs, four water molecules are generated per four electrons (2H2 + 4OH− → 4H2O + 4e−). Therefore, at a fixed current density, the water generation rate at the AEMFC anode is two times faster than that at the PEMFC cathode. Additionally, water consumption at the cathode exacerbates the water imbalance.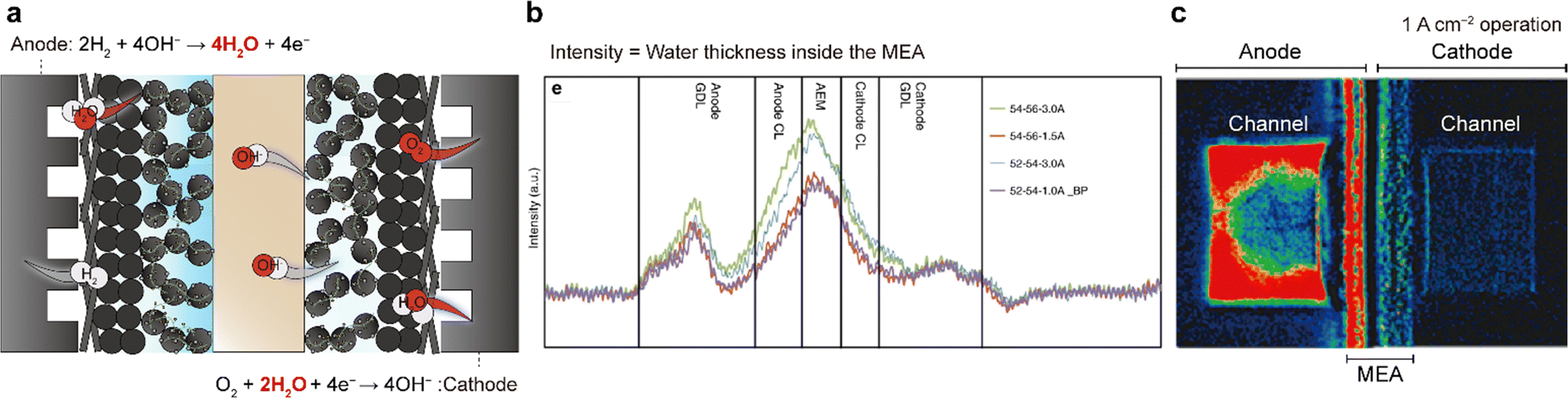 | ||
| Fig. 8 Water imbalance issue in AEMFCs. (a) Schematic of water flooding at the anode and drying out at the cathode. (b) Water distribution plots inside the MEA extracted from high-resolution neutron radiographic images. (c) In situ synchrotron X-ray colored images during single-cell operation using the constant current mode at 1 A cm−2 under fully wetted conditions. The water density decreases from red to blue. Panels (b) and (c) are reprinted with permission.8,142 | ||
Water imbalance in the MEA causes serious problems; flooded water fills the interstitial pores in the anode CL, blocking the supply of H2 molecules to the catalyst surface. Besides, the swelling of the AEI phase in the anode CL leads to pore clogging, which worsens the H2 transport problem. While the water depletion at the cathode CL can facilitate diffusion of O2 molecules, drying of the cathode leads to increased Ohmic resistance due to the ionic conductivity loss of the AEI binder. The mass transport resistance of the cathode CL can be augmented because of the limited supply of H2O.
Various attempts have been made to achieve balanced water distribution within the MEA. Peng et al. showed that the cell performance can be dramatically improved by systematically controlling the temperature of the cell and humidifier.8 On the left side of Fig. 9a, the micro-X-ray computed tomography images of an anode CL at open-circuit voltage (OCV, upper) and at a current density of 0.2 A cm−2 (lower) are compared. This technique enables real-time observation of the internal structural changes of the CL during cell operation. When a current of 0.2 A cm−2 was applied, the pores inside the CL, which initially appeared as dark regions, became bright. These results demonstrate the swelling of AEI binders during cell operation and clogging of the pores between the Pt/C particles. To inhibit excess ionomer swelling, the RH of the anode feed was lowered, and a 3-fold increase in the entire range of current densities of the AEMFC was achieved (right side of Fig. 9a).
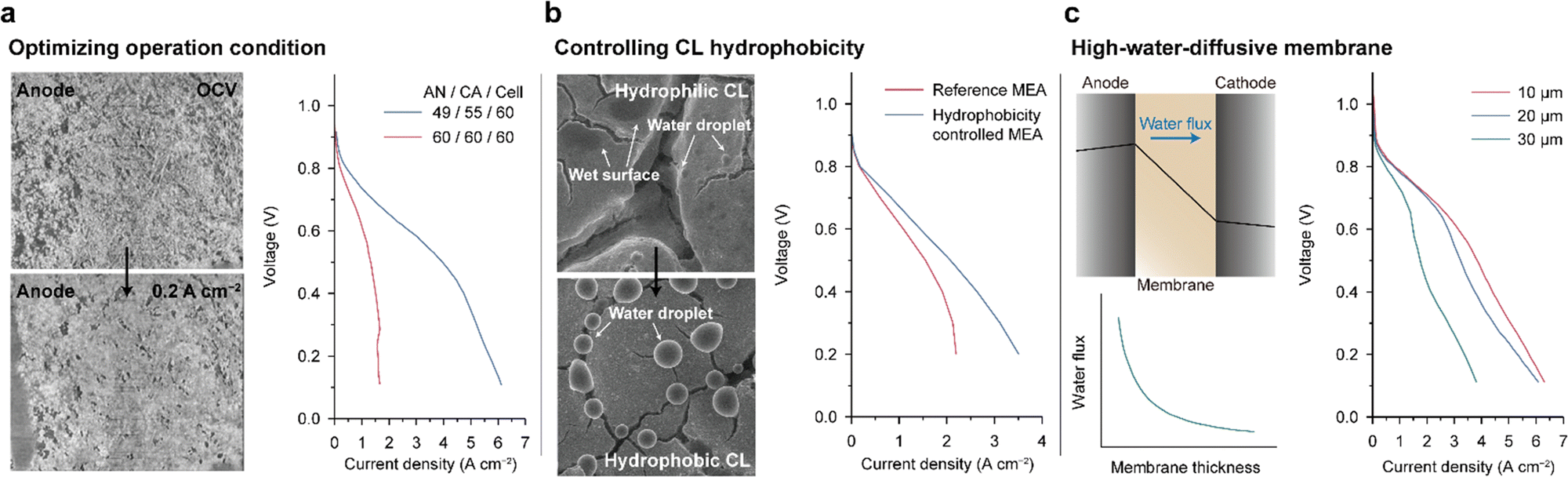 | ||
| Fig. 9 Strategies for mitigating water imbalance. (a) Operando micro-X-ray computed tomography scans of the interface between the gas diffusion layer and CL of PtRu/C anodes at open-circuit voltage (OCV) (left upper) and 0.2 A cm−2 (left lower) under 100% RH. The i–V curves of the fully humidified and optimal humid conditions are displayed on the right. (b) SEM images of the CL surfaces containing a hydrophilic (left upper) and a hydrophobic (left lower) carbon additive under fully humidified conditions. The i–V curves of the control and optimal MEAs paired with the hydrophobic anode CL and hydrophilic cathode CL (right). (c) Predicted water distribution and direction of water flux inside the MEA during cell operation (left upper), water flux versus membrane thickness plot (left lower), and three i–V curves for MEAs with different membrane thicknesses (right). Panels (a) and (b) are adapted with permission.8,143 The water flux plot and i–V curves in (c) are reprinted from ref. 144 and 97, respectively. | ||
Another approach to mitigate water imbalance is controlling the hydrophobicity of the CL. Hyun et al. introduced oxygen- and fluorine-functionalized carbon additives in the CL to produce hydrophilic and hydrophobic CLs, respectively.143 They observed water condensation on the CL surfaces using environmental scanning electron microscopy (SEM). As shown in Fig. 9b, the hydrophilic CL surface was wet or partly covered with water droplets with very small contact angles (upper), which is in contrast to water droplets with large contact angles formed on the hydrophobic CL surface (lower). The hydrophobicity-controlled MEA paired with the hydrophilic cathode CL and hydrophobic anode CL showed higher current densities compared to the control MEA and alleviated the limiting current behaviors in the high-current-density regime (Fig. 9b). These results suggest that the hydrophilic CL retains water and that the hydrophobic CL drains water, thus mitigating the water imbalance.
The water imbalance can also be reduced by introducing a membrane with high water flux. During cell operation, the accumulated water in the anode CL diffuses to the cathode because of a concentration gradient, which is denoted as “water back-diffusion” (upper left in Fig. 9c). The water back-diffusion is facilitated using a membrane with higher diffusivity. Accordingly, a polynorbornene-based AEM with high water diffusivity resulted in superior power performance. Mandal et al. decreased the thickness of the polynorbornene-based AEM using light cross-linking with the hexyl spacer diamine (1 of Fig. 3b).97 As the membrane thickness decreased, the water flux across the membrane increased exponentially (lower left in Fig. 9c).144 As shown on the right side of Fig. 9c, the power performance was enhanced with decreasing membrane thickness, and by using a 10 μm-thick AEM, a current density exceeding 6 A cm−2 was achieved.
Recently, a self-regulating anode CL was suggested by Zhang et al. for better water management at the anode.145 PtRu/C catalysts have a tendency to aggregate more than Pt/C, resulting in the formation of a larger pore structure when used as a CL. The researchers combined PtRu/C and Pt/C CLs into one anode CL with distinct layers, creating smaller pores from the gas diffusion layer to the AEM. The CLs with this gradient nanopore structure exhibit efficient water transport through the capillary effect. They were characterized by rapid moisture drainage even under high humidity conditions and demonstrated high power performance at 100% RH.
The significance of regulating water distribution within the MEA is apparent from its substantial influence on the cell performance. However, in practical applications like light-duty vehicles, where the distribution of water in the MEA changes rapidly due to the dynamic operating conditions, prompt responses are essential. Thus, a more dynamic evaluation method that models real driving conditions beyond the i–V polarization curve and incorporates material development with MEA design is necessary.
Additionally, the water distribution within the CL significantly impacts the durability of AEMFC as reported previously.146 The performance loss of AEMFCs can be classified into two categories: recoverable loss and permanent loss. The former arises from temporary limitation in H2 (anode) and OH− (cathode) supply caused by water flooding and water drying, respectively. When the water shortage in cathode exacerbates, the OH− concentration in cathode increases, leading to the degradation of the cation group in the AEI (further details are provided in Section 5.2.2). Notably, Hyun et al. demonstrated that mitigating water imbalance through carbon modifications can help achieve stable operation for 1000 h, which is in contrast to the short lifetime (200 h) of the control MEA.143 Various carbon modification methods including fluorination11 can be used in this approach.
4.3. CO2 poisoning
CO2 poisoning is the most critical problem of an AEMFC from the practical aspect. CO2 can chemically react with OH−, forming carbonate species (note the equations in Fig. 10a). The carbonate formation reaction occurs spontaneously and is quite fast; merely 400 ppm of CO2 from ambient air can significantly lower the OH− concentration in the AEM and AEI. Accumulation of the carbonate species greatly reduces the AEMFC performance or destabilizes the OCV. CO2 poisoning essentially complicates the manufacturing process of the AEMFC. Before cell assembly, AEM- and AEI-containing CLs should be submerged in an alkaline solution to replace the carbonate anions with OH−. However, despite these efforts, the AEM and CLs are inevitably exposed to CO2 again during single-cell assembly.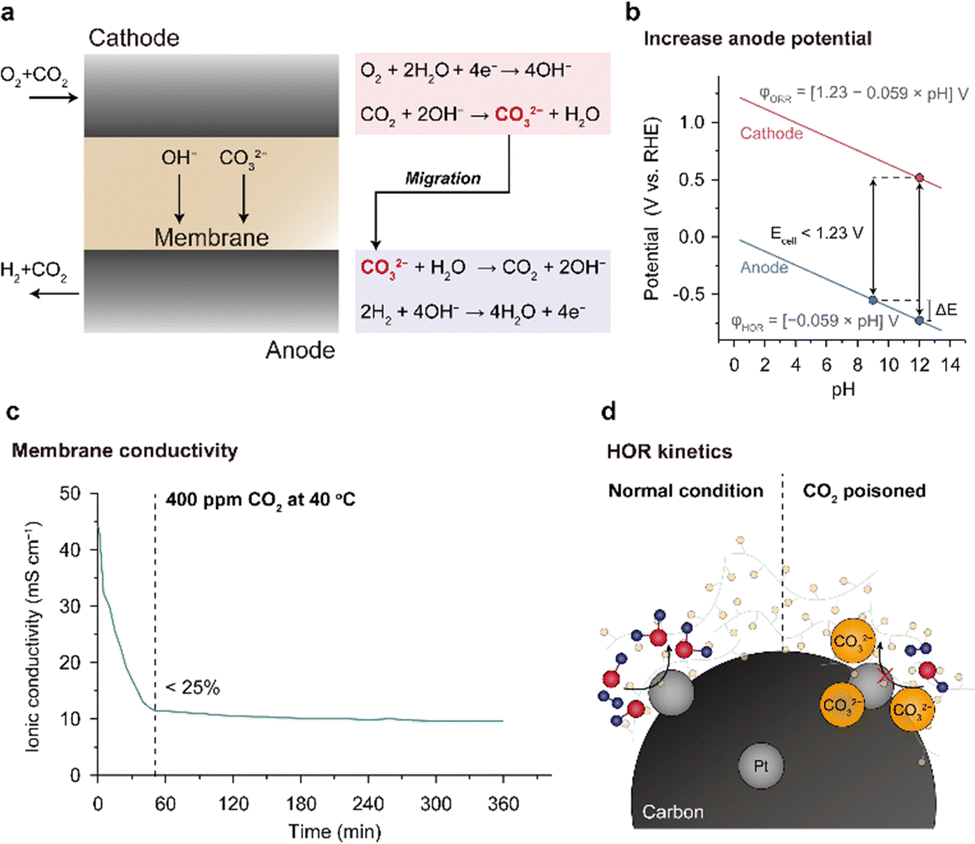 | ||
| Fig. 10 CO2 poisoning in AEMFCs. (a) Carbonate species formation and removal processes at the cathode and anode. Three reasons for AEMFC performance degradation due to CO2 poisoning: (b) reduced cell voltage by increase of anode potential, (c) decreased ionic conductivity of the membrane by carbonate species formation, and (d) kinetic loss in the HOR due to blocking of the catalyst surface by the carbonate species. The plot for ionic conductivity versus time is reprinted with permission.147 | ||
CO2 poisoning also makes cell operation with ambient air challenging. As shown in Fig. 10a, the CO2 injected into the cathode reacts with the OH− of the AEI in the CL and is converted to CO32−. The carbonate species migrates from the cathode to anode driven by the electric field and accumulates at the anode CL. Some carbonate species are also reconverted to CO2 and emitted. If the AEMFC is operated under a constant voltage with constant reactant gas flow, the carbonate concentrations of the cathode, membrane, and anode CL can converge to steady-state values. However, CO2 poisoning is hard to control in dynamic operations, often leading to a local increase in the carbonate concentration in the MEA. This means that sophisticated power control is required for AEMFCs used in transportation applications.
The carbonate species in the MEA decreases the performance of the AEMFC in three ways: (1) increasing the anode potential, (2) decreasing the ionic conductivity of the AEM, and (3) decreasing the HOR kinetics of the anode catalyst. In the absence of CO2 in the AEMFC, it can be assumed that both the anode and cathode have the same pH and that the theoretical voltage is 1.23 V (Fig. 10b). However, when CO2 poisoning starts and the carbonate species accumulate at the anode, the pH of the anode becomes lower than that of the cathode. Since the potential of the anode decreases linearly with pH, the theoretical voltage of the AEMFC decreases (<1.23 V). For this reason, when the AEMFC is assembled under ambient air, its OCV is often low and unstable.
The ionic conductivity of the AEM is decreased by carbonate formation because of the reduced OH− concentration in the AEM. OH− molecules can migrate rapidly in the AEM under an electric field via the Grotthuss mechanism, whereas the migration of CO32− molecules is slower than that of OH− because they does not migrate via the Grotthuss mechanism but through the vehicle mechanism. As shown in Fig. 10c, the ionic conductivity of the AEM decreases exponentially to 25% of the initial value within 1 h when exposed to ambient air. Under the cell operating environment, the ionic conductivity of the AEM is not expected to be significantly reduced because the carbonate species are removed from the anode. However, in the idle mode, when the MEA is exposed to the atmosphere, carbonate species will be readily generated inside the AEM and AEI, causing rapid performance degradation upon restart.
The last negative effect of CO2 poisoning on AEMFC performance is reduction of HOR catalyst activity (Fig. 10d). The high concentration of carbonate species near the HOR catalyst surface may block the access of OH− to the surface of the HOR catalyst and increase the charge transfer resistance.148,149 Although there are no studies reporting the direct measurement of charge transfer resistance, Mustain's group was able to derive it by setting up an environment that can neglect the resistive component from the cathode ORR side and by subtracting the Nernst voltage loss due to the pH increase of the anode and Ohmic overvoltage from the total voltage drop on the anode side.148
Various approaches have been attempted to suppress carbonate formation. Mustain's group controlled various operation parameters of the AEMFC (current density, temperature, and anode/cathode feed flow rate) as well as conducted electrochemical analyses and real-time probing of CO2 emissions from the cathode and anode. With increasing current density, the Ohmic resistance of the AEM reduced, indicating reduced CO2 in the MEA (1 of Fig. 11a). This was supported by the increased CO2 concentration in the anode exhaust and decreased CO2 concentration in the cathode exhaust. Increasing the cell temperature also mitigates CO2 poisoning. With increasing temperature, the solubility of CO2 in water decreases, the conversion rate of carbonate to CO2 increases, and the transportation of CO2 and OH− become easier. Owing to these combined effects, the cell overpotential caused by CO2 decreases with temperature (2 of Fig. 11a). The flow rate of the feed gas also impacts carbonation in a cell. The CO2 overpotential decreases with the flow rate of the anode feed (3 of Fig. 11a), whereas it increases with the flow rate of the cathode feed (4 of Fig. 11a). Intuitively, a higher flow rate of the anode gas can release CO2 more efficiently from the cell, and a higher flow rate of the cathode gas will lead to increased amount of CO2 entering the cell, resulting in more significant carbonation. In addition, the hydration level of the cell affects CO2 poisoning.149 Increasing the amount of water inside the cathode reduces the basicity of the electrolyte in the CL, which leads to a decrease in CO2 solubility. Under optimized conditions, the AEMFC shows a stable i–V polarization curve without any catastrophic voltage losses.
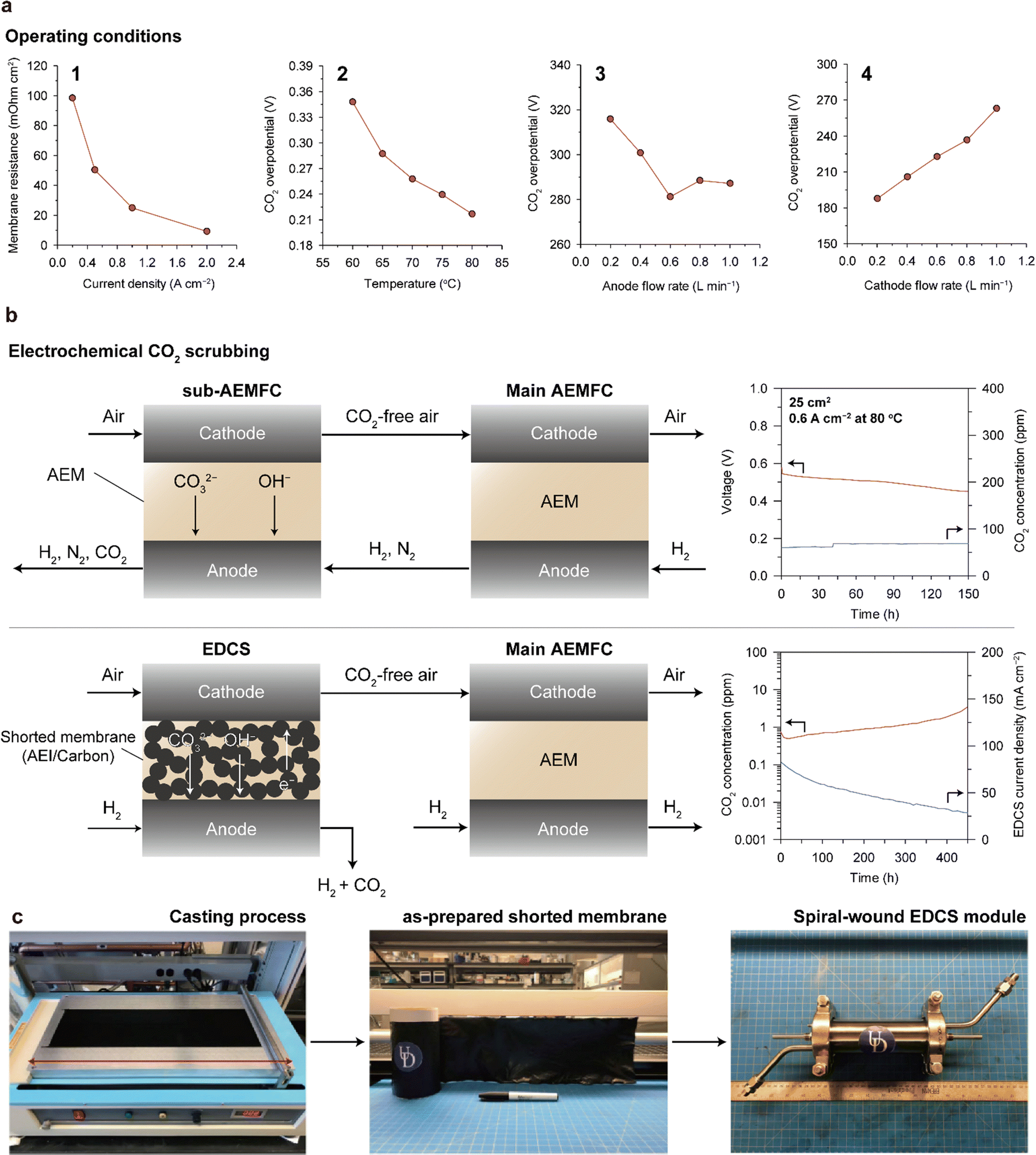 | ||
| Fig. 11 Strategies for mitigating CO2 poisoning. (a) Change in membrane resistance or CO2 overpotential with adjustment of the operating conditions, including current density, temperature, anode feed flow rate, and cathode feed flow rate (reprinted with permission from ref. 148 and 149). (b) Structure and durability of the sub-AEMFC (top)150 and EDCS (bottom)151 as an electrochemical CO2 scrubbing system. (c) Overall fabrication process of the spiral-wound EDCS module. Bottom of panels (b) and (c) are adapted with permission from ref. 151. | ||
The fine-tuning of the operating environment of the AEMFC could significantly mitigate carbonation inside the MEA; however, it is not compatible with dynamic operations of practical fuel cells. Instead, CO2 scrubbing systems are more effective and straightforward for solving the carbonation problem. The currently available CO2 capture systems are mostly based on reversible CO2 adsorption and desorption on absorbent materials with a regeneration process.152,153 However, this type of chemical CO2 scrubbing is hard to combine with AEMFC systems because of the insufficient CO2 adsorption capacity and time-consuming regeneration step. Therefore, in the AEMFC technology sector, an electrochemically driven CO2 scrubbing system that can continuously remove CO2 has attracted interest.
One approach is using the sub-AEMFC as a CO2 filter. The overall configuration is shown at the top of Fig. 11b; the cathode outlet of the sub-AEMFC is connected to the cathode inlet of the main AEMFC, and the anode outlet of the main AEMFC is connected to the anode inlet of the sub-AEMFC. The CO2-free effluent from the sub-AEMFC is supplied to the main AEMFC, and the residual H2 from the main AEMFC is injected into the sub-AEMFC. The reactions in the sub-AEMFC are identical to those in the main AEMFC, as described in Fig. 10a. The CO2 from the atmosphere combines with OH− to form carbonate anions at the cathode of the sub-AEMFC. The carbonate anions are transported to the anode, converted to CO2, and released into the atmosphere with the anode effluent. Zheng et al. implemented a pair of sub- and main-AEMFC systems and demonstrated that a sub-AEMFC with a size of 5 cm2 can reduce the CO2 concentration from 400 to 240 ppm.150 The 25 cm2 sub-AEMFC can operate for over 150 h with less than 100 ppm CO2 emission (upper right of Fig. 11b). Furthermore, they showed that the main AEMFC exhibits the same i–V polarization curves when tested with CO2-free air and the exhaust from a 50 cm2 sub-AEMFC that operates under ambient air.
Another approach is adopting an electrochemically driven CO2 separator (EDCS) developed by Yan's group.151 The EDCS is based on carbonate chemistry similar to the sub-AEMFC, but it employs a shorted membrane to induce internal current. The shorted membrane comprising carbon and AEIs enables transport of anions (OH− and carbonate species) and electrons concurrently. The internal shorting accelerates the ORR and HOR in the EDCS, enhancing the efficiency of CO2 removal from the cathode air feed. The filtering ability of the EDCS is remarkable; it can lower the CO2 concentration in the air feed at 1000 sccm from 400 to <1 ppm using only 5 sccm of H2 gas. Moreover, the EDCS showed 99% CO2 removal capacity for 450 h (lower right of Fig. 11b). The strong advantage of the EDCS is that it does not require any external circuitry and the degree of CO2 removal from the air feed can be adjusted by varying the flow rate of the H2 injected into the EDCS. Furthermore, both the cast-made shorted membrane and spiral-wound configurations of the EDCS (Fig. 11c) are compatible with conventional manufacturing processes, thereby increasing the practicality of the EDCS. However, EDCS inevitably consumes a certain amount of hydrogen, which decreases fuel efficiency; system level energy efficiency should be studied in more detail.
As described in this section, the injection of CO2 causes catastrophic performance loss to the AEMFC. Therefore, this problem must be resolved before using the AEMFC as a power source.65,154 However, CO2 removal technology is a long-standing challenge that has not been completely solved in other electrochemical devices, such as zinc–air and Li–air batteries, and the complex carbonate chemistry increases the difficulty of research. Nevertheless, the sub-AEMFC and EDCS are promising candidates for CO2 removal systems, and their further research is encouraged. In addition, membrane CO2 filtration can be a viable option to consider.155 Alongside the infiltration of CO2 from outside into AEMFCs, CO2 can be generated inside the MEA via electrochemical oxidation of carbon materials. This issue cannot be fully addressed even when a CO2 scrubbing system is employed. Therefore, it should be also considered for achieving long-term operation of AEMFCs.
4.4. MEA design
The HOR and ORR occur at electrochemically active surface areas (ECSAs) of the catalyst surfaces where the transportation of electrons, ions, and molecules (reactants, and products) is available, conventionally denoted as TPB. Since a thin ionomer layer in contact with the catalysts enables the passage of ions and molecules, the two-phase interface of the ionomer/catalyst can form a TPB with pores and serve as an ECSA. To improve the performance of the AEMFC with a limited amount of catalyst, the ECSA should be augmented. Therefore, a CL design that directs uniform ionomer distribution and a porous CL structure is required. In the case of nonuniform ionomer distribution, locally concentrated ionomers on the catalyst surface block the access of reaction gases and also result in electrically isolated catalysts. In addition, the ionomer-deficient portion of the CL limits the migration of OH− ions. Since the reactant and product gases pass through the pores of the CL, it should have high porosity for facile mass transport. In addition to porosity, the size of the pores is also important. Small pores can cause water flooding, which restricts gas access, and pores that are too large reduce the connectivity of the CL. Designing an optimal CL poses a formidable challenge as it necessitates satisfying multiple conditions concurrently. Detailed discussions on this topic can be found in recent comprehensive reviews.156,157Only a few studies have been conducted on the CL structure of the AEMFC despite its importance. Carlson et al. changed the CL structure by varying the ionomer–Pt/C ratio to 0.2, 0.6, and 1.0 and analyzing the cell performances comparatively.158 The SEM images of the CL surfaces varied with the ionomer content, as shown on the left side of Fig. 12a. As the ionomer content increased, the pores disappeared, and a thick ionomer film was observed at a ratio of 1.0. The highest cell performance was achieved at an intermediate ratio of 0.6 because an adequate amount of ionomer ensures facile ion and mass transport. The CL with a lower ionomer content (0.2) has the lowest performance due to limited OH− transport in the CL. At a higher ratio of 1.0, the thick ionomer film hinders the reactant molecules from reaching the catalyst surface, limiting the cell performance, as indicated by the large overpotential in the high-current regime. The ionomer content is a critical design parameter that determines CL performance, and it should be carefully adjusted considering the interplay between ionomer and catalyst materials. The optimal ionomer content should vary depending on the size and surface properties of the catalysts because the catalyst surface area–ionomer volume ratio and ionomer–catalyst interaction influence the ionomer distribution and pore structure of the CL. Moreover, the influence of ionomer content on mass transport may change depending on the hydrophilicity of catalyst particles. When the catalyst is prone to water flooding, ionomer content needs to be reduced to provide sufficient interstitial pore volume. Understanding of the ionomer–catalyst interaction and the consequent CL structure is necessary to advance AEMFC CL technology.
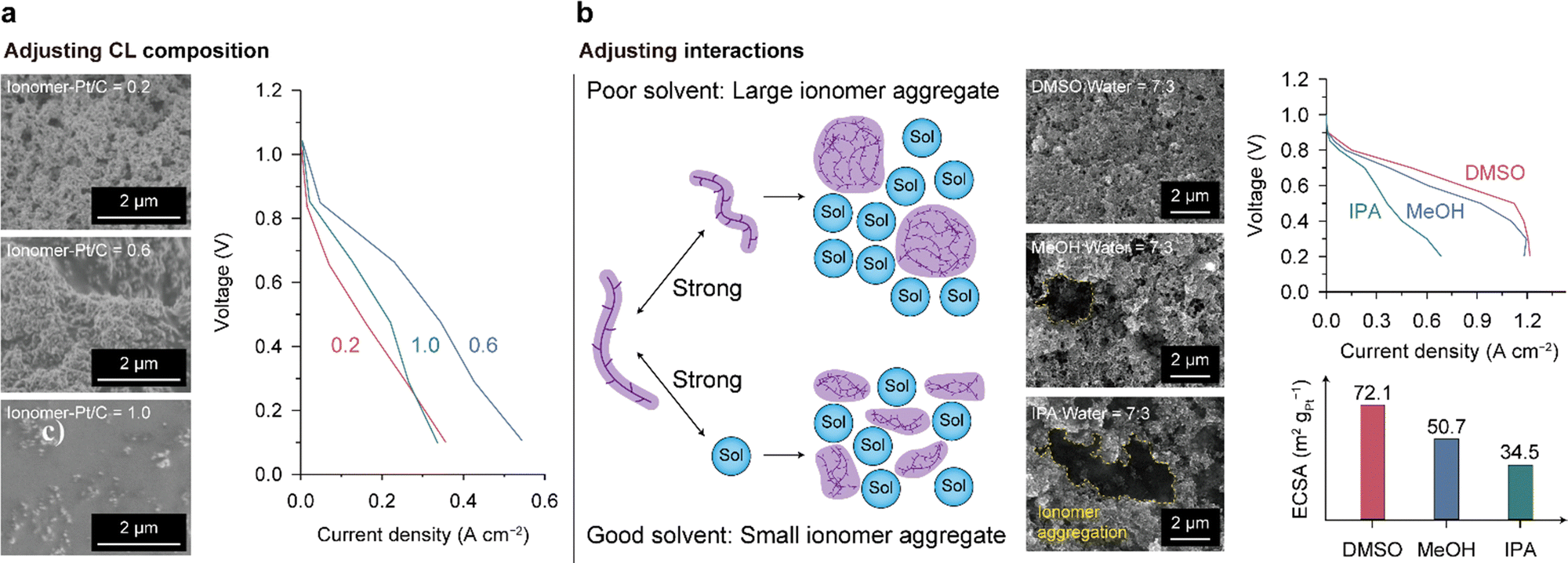 | ||
| Fig. 12 Structural control of the AEMFC CL. (a) Adjustment of the ionomer content: surface SEM images of the CLs with different ionomer contents (ionomer-Pt/C ratio of 0.2 (top), 0.6 (middle), and 1.0 (bottom)) and comparison of the i–V polarization curves of the CLs. (b) Adjustment of the interactions between the ionomer and solvent: schematic of the size of ionomer aggregates in dispersion according to ionomer–solvent interaction, surface SEM images of the CLs fabricated with different solvents (DMSO (top), MeOH (middle), and IPA (bottom)), i–V polarization curves, and ECSA values for the CLs. Data in (a) and (b) are reprinted with permission from ref. 36 and 156, respectively. | ||
Hyun et al. investigated the effects of the solvent of ionomer dispersion on controlling the CL structure and demonstrated the correlation between the resulting CL structure and cell performance.36 The size of the ionomer aggregate in dispersion can be controlled by adjusting the ionomer–solvent interactions (Fig. 12b). If the ionomer–solvent interactions are weaker than the ionomer–ionomer interactions, the ionomer chains tend to aggregate. In contrast, stronger ionomer–solvent interactions disperse the ionomer aggregates to smaller dimensions. They found that the aggregate size of the QPC-TMA (poly(9-(6-(trimethylammoniumbromide)-hexyl)-9H-carbazole-co-1,1,1-trifluoroisopropane)) AEI in dispersion could be decreased in the order of isopropanol (IPA) > methanol (MeOH) > dimethylsulfoxide (DMSO). As shown in Fig. 12b, the DMSO-based CL has a uniform and porous structure, whereas the MeOH- and IPA-based CLs show clumpy ionomer aggregates on the surface and pore clogging, indicating nonuniform ionomer distribution in the CLs. The power performance was enhanced by increasing the uniformity of ionomer distribution. This study also established a strong correlation between the ECSA and power performance.
5. Challenges and solutions for improving AEMFC durability
To achieve long-lasting AEMFCs, it is crucial that the materials used in the MEA exhibit chemical, mechanical, and electrochemical stabilities under operating conditions. Furthermore, the various layers and interfaces in AEMFCs must possess mechanical robustness. To date, research has primarily focused on the durability of materials, with limited studies conducted on the mechanical durability of CLs or diffusion layers, and on the degradation or failure modes of stacks or systems. This section provides a summary of the degradation modes of the materials, describes the resulting decline in MEA performance, and discusses the underlying mechanisms and strategies to address these issues.5.1. Catalysts
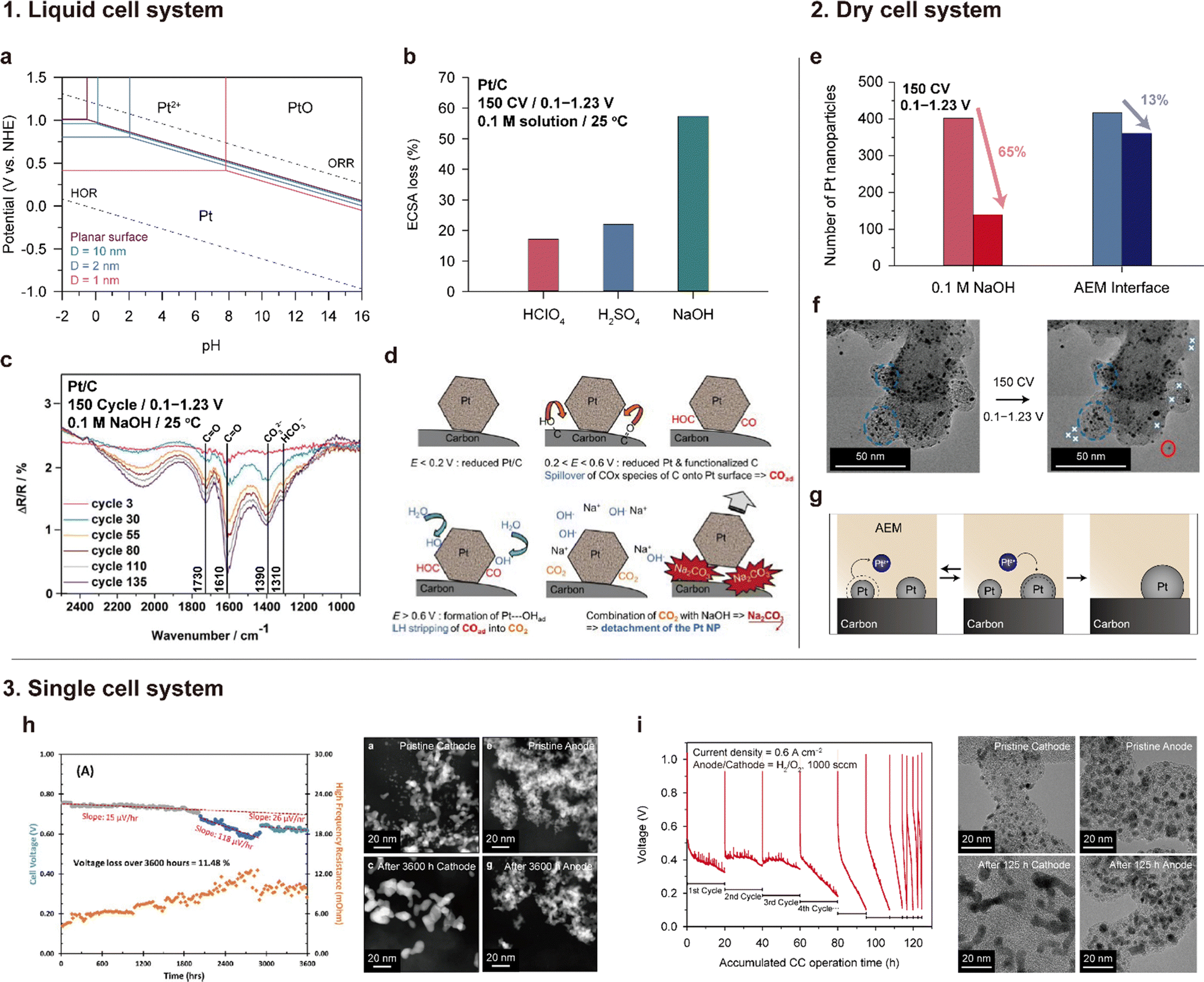 | ||
| Fig. 13 Degradation of the Pt/C catalyst under alkaline conditions. 1. Severe Pt detachment in an alkaline solution: (a) Pourbaix diagram of Pt for different sizes. (b) ECSA loss after 150 CV in various 0.1 M aqueous solutions (CV is performed in the RDE setup at room temperature, and the voltage range is 0.1–0.23 V vs. RHE). (c) Fourier-transform infrared (FTIR) spectrum of the Pt/C catalyst after 150 voltage cycles in the range of 0.1–1.23 V vs. RHE in a 0.1 M NaOH at room temperature. (d) Pt detachment mechanism in an alkaline solution. Panel (a) is adapted with permission from ref. 159. Panels (b), (c), and (d) are reprinted with permission from ref. 160 and 161. 2. Ostwald ripening of Pt at the interface with an AEM. (e) Number of Pt NPs after 150 voltage cycles in 0.1 M NaOH and an AEM/CL interface. (f) TEM images before and after the degradation test. (g) Ostwald ripening and redeposition of Pt at the AEM interface. Panels (e) and (f) are adapted with permission from ref. 162. 3. Pt degradation in a single cell. (h) and (i) Voltage profiles during 0.6 A cm−2 of constant current operation (left) and TEM images of the cathode (Pt/C) and anode (PtRu/C) catalysts before and after durability tests (right). Panels (h) and (i) are reprinted with permission from ref. 6 and 163. | ||
In their subsequent work, Chatenet's group discovered the formation of carbonate species on the Pt/C catalyst during voltage cycling.161,164Fig. 13c shows the Fourier-transform infrared (FTIR) spectra of the Pt/C surface, which change with voltage cycling in a 0.1 M NaOH solution. The bands at 1310 and 1390 cm−1 are assigned to carbonate ions (CO32−) and bicarbonate ions (HCO3−), respectively, and the bands at 1610 and 1730 cm−1 are linked to the oxygen-containing surface groups on carbon. The intensities of these four FTIR bands increase over the CV cycles (Fig. 13c), which suggests the formation of oxygen groups on the carbon support and carbonate species on the Pt/C surface. Based on this finding, they suggested that Pt detachment from the carbon support is the major degradation mechanism of Pt/C catalysts in alkaline solutions, as shown in Fig. 13d. The Pt/C stably exists under <0.2 V potential. When the potential gradually increases, carbon corrosion/functionalization starts occurring in the 0.2–0.6 V potential range, and CO and/or COH molecules are adsorbed on the Pt surface through back spillover. Over 0.6 V, the OH molecules are adsorbed subsequently because of the positive charge and/or oxidization of the Pt. Then, Pt-adsorbed CO and OH react together and form CO2 (Pt⋯COads + Pt⋯OHads + OH− → CO2 + H2O + 2Pt + e−). Another scenario for CO2 formation is that the functional group on the carbon support directly reacts with the Pt-adsorbed OH (Cn⋯COads + Pt⋯OHads + OH− → CO2 + H2O + Pt + Cn + e−). As a final step, the CO2 molecules chemically react with OH− to form carbonate precipitates, such as Na2CO3 on the Pt/C surface, releasing Pt from the carbon support.
Lafforgue et al. investigated the stability of the Pt/C catalyst surrounded by the AEM phase using transmission electron microscopy (TEM) analysis.162 In this work, the Pt/C and AEM-containing ink were deposited on a Lacey-carbon TEM gold grid, which served as both a working electrode and platform for TEM observations. Note that accelerated stress testing (AST) was conducted in a “dry cell”, where the electrolyte consists of only a solid AEM. In contrast to the significant loss of ECSA (65%) observed for Pt/C exposed to 0.1 M NaOH solution, Pt/C surrounded by the AEM exhibited only a 13% ECSA loss after 150 cycles of CV (as illustrated in Fig. 13e). As depicted in Fig. 13f, Pt detachment did not occur significantly; instead, minor Ostwald ripening occurred in Pt/C under the AEM environment. In a liquid environment, detached or dissolved Pt can be washed away from its location, but in a solid AEM environment, it can stay around and redeposit onto Pt/C (as depicted in Fig. 13g). Additionally, even if carbonate ions are formed, precipitates do not form because they pair with the cationic groups of the AEM.
Only a few research groups have reported the stability of the Pt/C cathode and PtRu/C anode catalysts under actual operation in AEMFCs. One such study by Hassan et al. revealed that after 2000 h of constant current operation at 0.6 A cm−2, Pt nanoparticles in the cathode were severely agglomerated (Fig. 13h).6 Hyun et al. also found that Pt nanoparticles in the cathode had aggregated to form worm-like shapes after operating for 125 h at a current density of 0.6 A cm−2 (Fig. 13i).163 In contrast to Pt/C catalysts, PtRu/C catalysts used in the anode CL did not exhibit any aggregation in both studies.
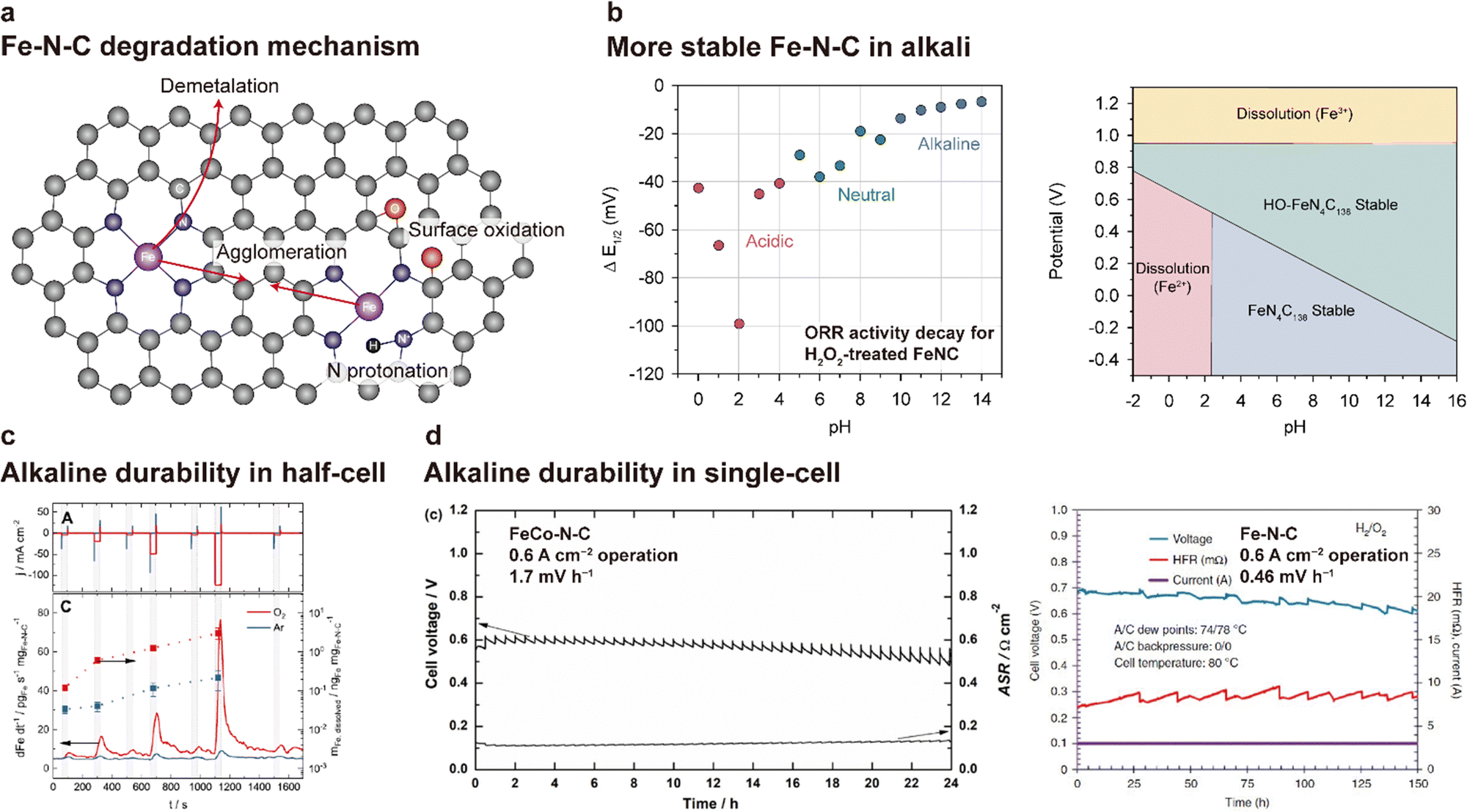 | ||
| Fig. 14 Degradation of Fe–N–C catalysts in alkaline media. (a) Schematic of the degradation mechanism of the Fe–N–C catalyst. (b) Difference in E1/2 values of the Fe–N–C catalyst before and after H2O2 treatment (left). H2O2 treatment was conducted in a pH-adjusted solution containing 1 wt% H2O2 for 2 h at 50 °C. Pourbaix diagram of FeN4C138, HO-FeN4C138, and their dissolution states (Fe2+ and Fe3+) at 298.15 K (right). Plots are reprinted with permission from ref. 165 and 166. (c) Comparison of Fe–N–C demetalation between Ar- and O2-saturated alkaline (0.1 M NaOH) environments. The current density profile (top) and corresponding Fe dissolution rate and amount of dissolved Fe (bottom). The plot in (c) is adapted with permission from ref. 167. (d) Alkaline durability test results for the AEMFCs using FeCo–N–C (left) and Fe–N–C (right) are reprinted with permission from ref. 168 and 169, respectively. | ||
Owing to the recent interest in the use of NPM catalysts for AEMFCs, the durability of Fe–N–C-based catalysts in alkaline environments is being investigated. Bae and Chung et al. demonstrated that the Fe–N–C catalyst is more stable in an alkaline environment than in an acidic environment.165 The stability was assessed by monitoring changes in the half-wave potential for ORRs after H2O2 treatment. As shown on the left side of Fig. 14b, the change in the half-wave potential was smaller at higher pH. Under acidic conditions, H2O2 that is formed via the two-electron pathway of ORR reacts with Fe2+ ions, releasing OH˙ radicals. The OH˙ radical results in surface oxidation of the carbon of Fe–N–C, reducing the electrical conductivity, inducing demetalation due to structural collapse, and decreasing the TOF due to a change in the electrical structure. Under acidic conditions, the surface oxidation is significant because the OH˙ radical is stably maintained. In contrast, under alkaline conditions, the formation of the ferryl ion species (FeO2+), which is less reactive with carbon, is more preferred over the OH˙ radical, resulting in higher stability of carbon compared to that under acidic conditions. In addition, the dissolved Fe2+ and Fe3+ are readily precipitated in their hydroxide forms (Fe(OH)2 and Fe(OH)3, respectively), preventing OH˙ radical formation. Holby et al. investigated the stability of a Fe–N–C catalyst through density functional theory (DFT) calculations.166 As shown on the right side of Fig. 14b, the FeN4C138 catalyst is stable in the pH range of 3–16, whereas Fe2+ dissolution could occur below pH 2 in an operating voltage range of OCV–0.4 V.
In contrast to the reports mentioned above, some studies have reported the degradation of Fe–N–C catalysts under alkaline environments. Recently, Ku et al. reported that a Fe–N–C catalyst can be dissolved due to the occurrence of ORR.167 Comparison of Fe dissolution from the Fe–N–C catalyst between Ar- and O2-saturated 0.1 M NaOH solution (Fig. 14c) shows that the dissolution rate is much faster in the presence of O2. Another important feature is that Fe dissolution begins to occur as soon as the ORR current flows and that it is proportional to the current density. On the basis of these observations, they proposed that Fe dissolution is induced by the instability of the coordinated Fe during the Fe3+/Fe2+ redox reaction during the course of the ORR. The instability of Fe–N–C was also observed under actual AEMFC operation. As shown in Fig. 14d (left), an AEMFC using the FeCo–N–C catalyst for the cathode CL showed a degradation rate of 1.7 mV h−1 at 0.6 A cm−2.168 Similarly, an AEMFC utilizing a silica-templated porous Fe–N–C catalyst exhibited a degradation rate of 0.46 mV h−1 during 150 h of 0.6 A cm−2 operation (Fig. 16d (right)).16 Taken together, the electrochemical stabilities of Fe–N–C catalysts under alkaline conditions are still the subject of debate and the degradation pathways of Fe–N–C catalysts are not fully clear, highlighting the need for additional research.
| Fuel cell type | Pt loading (anode/cathode, mgPt cm−2) | Peak power density (W cm−2) | Operating conditionsa | Ref. |
|---|---|---|---|---|
| a Cell temperature, humidity conditions (anode/cathode), reaction gas, and back-pressure are presented in order. | ||||
| PEMFC | 0.05/0.07 | 2.21 | 80 °C, 100% RH, H2/O2, 150 kPaabs | 32 |
| 0.01/0.09 | 1.01 | 80 °C, 100% RH, H2/O2, 150 kPaabs | 33 | |
| 0.1/0.1 | 1.56 | 80 °C, 100% RH, H2/O2, 150 kPaabs | 34 | |
| 0.1/0.13 | 0.909 | 80 °C, 100% RH, H2/O2, 150 kPaabs | 35 | |
| AEMFC | 0/0.4 | 2.03 | 95 °C, 88/97 °C, H2/O2, 250 kPa | 17 |
| 0.26/0.26 | 2.4 | 80 °C, 75%/100% RH, H2/O2, 1.3 bar | 38 | |
| 0.28/0.33 | 2.34 | 80 °C, 75%/100% RH, H2/O2, 1.3 bar | 39 | |
| 0.4/0.4 | 2.55 | 80 °C, 92% RH, H2/O2 | 13 | |
| 0.448/0.672 | 3.21 | 80 °C, 70/74 °C, H2/O2 | 45 | |
| 0.467/0.6 | 3.2 | 80 °C, 72/74 °C, H2/O2 | 6 | |
| 0.473/0.6 | 1.9 | 60 °C, 45/46 °C, H2/O2 | 7 | |
| 0.4/0.4 | 1.42 | 60 °C, 56/58 °C, H2/O2 | 41 | |
| 0.2/0.2 | 0.67 | 80 °C, 100% RH, H2/O2 | 36 | |
| 0.1/0.6 | 1.0 | 80 °C, 100% RH, H2/O2, 285 kPa | 40 | |
| 0.4/0.4 | 0.86 | 95 °C, 95/97 °C, H2/O2, 0.1 MPag | 26 | |
| 0.5/0.6 | 1.26 | 80 °C, 100%/50% RH, H2/O2, 147.5 kPa | 25 | |
| 0.4/0.0026 | 0.224 | 40 °C, 80% RH, H2/O2 | 37 | |
| 0.47/0.5 | 1.01 | 60 °C, 51/56 °C, H2/O2, 0.1 MPa at the anode | 42 | |
| 0.5/0.5 | 0.261 | 60 °C, 100% RH, H2/O2 | 43 | |
| 0.5/0.5 | 0.25 | 60 °C, 100% RH, H2/O2 | 44 | |
| Fuel cell type | Catalyst type | Peak power density (W cm−2) | Operating conditionsa | Ref. |
|---|---|---|---|---|
| a Cell temperature, humidity conditions (anode/cathode), reaction gas, and back-pressure are presented in order. | ||||
| PEMFC | FeNC | 0.87 | 80 °C, 100% RH, H2/O2, 1/2 bar | 46 |
| FeNC | 0.46 | 80 °C, H2/O2, 1 bar | 47 | |
| FeNC | 1.18 | 80 °C, 100% RH, H2/O2, 2.5 bar | 48 | |
| FeNC | 0.66 | 80 °C, 100% RH, H2/O2, 1 bar | 49 | |
| CoNC | 0.64 | 80 °C, 100% RH, H2/O2, 1 bar | 50 | |
| FeNC | 0.57 | 80 °C, 100% RH, H2/O2, 1 bar | 51 | |
| FeNC | 0.8 | 80 °C, 100% RH, H2/O2, 1 bar | 52 | |
| AEMFC | Ag/C | 1.72 | 80 °C, 92% RH, H2/O2 | 13 |
| Mn-Co/C | 1.1 | 60 °C, 100% RH, H2/O2, 0.1 MPa | 18 | |
| Co/C | 0.412 | 60 °C, 100% RH, H2/O2 | 14 | |
| FeNC | 2.05 | 80 °C, 70/75 °C, H2/O2, 200 kPa | 16 | |
| FeNC | 1.12 | 60 °C, H2/O2, 1 bar | 53 | |
| Co/C | 0.891 | 80 °C, H2/O2, 0.5 bar | 39 | |
| Fuel-cell type | Operation time (h) | Degradation rate (μV−1) | Operating conditionsa | Ref. |
|---|---|---|---|---|
| a Current density, cell temperature, humidity conditions (anode/cathode), and reaction gas are presented in order. | ||||
| PEMFC | 26300 |
0.005 | 0.8 A cm−2, 70 °C, 100% RH, H2/air | 54 |
| 18500 |
0.0075 | 0.29 A cm−2, 71–72 °C, 100% RH, H2/air | 55 | |
| 12860 |
0.0035 | 0.26 A cm−2, 60–65 °C, 0%/60–75% RH, H2/air | 56 | |
| 4000 | 0.0031 | 0.4 A cm−2, 60 °C, 100% RH, H2 + CO2/air | 57 | |
| 1600 | 0.0133 | 0.25 A cm−2, 200 °C, H2 + CO2 + CO + N2/air | 58 | |
| 1350 | 0.011 | 0.4 A cm−2, 80 °C, 100% RH, H2/air | 59 | |
| AEMFC | 2000 | 0.0154 | 0.6 A cm−2, 75 °C, 72/74 °C, H2/O2 | 6 |
| 800 | 0.2125 | 0.2 A cm−2, 80 °C, 100% RH, H2/O2 | 60 | |
| 500 | 0.0393 | 0.5 A cm−2, 120 °C, 40% RH, H2/O2 | 61 | |
| 440 | 0.068 | 0.6 A cm−2, 70 °C, 92% RH, H2/air (CO2 free) | 13 | |
| 360 | 0.37 | 0.6 A cm−2, 70 °C, 68.5/72 °C, H2/O2 | 38 | |
| 300 | 0.3128 | 0.5 A cm−2, 95 °C, 94/95 °C, H2/air (CO2 free) | 26 | |
| 200 | 0.13 | 0.2 A cm−2, 70 °C, 90%/100% RH, H2/O2 | 39 | |
| 200 | 0.48 | 0.3 A cm−2, 70 °C, 68/70 °C, H2/O2 | 41 | |
| 125 | 0.456 | 0.2 A cm−2, 80 °C, 100% RH, H2/air (CO2 free) | 62 | |
| 120 | 0.7776 | 0.6 A cm−2, 60 °C, H2/O2 | 42 | |
| Membrane type | Chemical structure | IEC (meq g−1) | Conductivity measurement environment | Ref. |
|---|---|---|---|---|
| CEM | PFSA | — | Water | 69 |
| Ferrocyanide-coordinated polymer/phosphotungstic acid | 1.48 | 70 | ||
| PFSA/sulfonated CNT | 0.95 | High humidity | 71 | |
| AEM | Poly(norbornene) with TMA | 3.59 | Water | 72 |
| Poly(aryl piperidinium) | 2.37 | 26 | ||
| Poly(vinylferrocene) | 1.65 | 61 | ||
| Poly(arylene piperidinium) | 2.34 | 73 | ||
| Fluoropoly(olefin) with TMA | 1.12 | 42 | ||
| Partial diallyl bisphenol A poly(arylene ether) with TMA | 2.16 | 74 | ||
| Poly(arylimidazoliums) | 2.86 | 95% RH | 44 | |
| Low-density polyethylene with TMA | 2.87 | 12 | ||
| Poly(arylene ether sulfone) with imidazolium | 2.2 | 100% RH | 60 | |
| Polyphenylene/polysulfone with TMA | 2.3 | 41 | ||
| Poly(fluorenyl aryl piperidinium) | 2.81 | 39 | ||
| Membrane type | Chemical structure | IEC (meq g−1) | Conductivity measurement temperature | Ref. |
|---|---|---|---|---|
| CEM | Polyphenylene with sulfonate | 2.4 | 80 °C | 75 |
| PFSA/reduced graphene oxide | 1.5 | 76 | ||
| PFSA/reduced graphene oxide | 0.84 | 77 | ||
| PFSA | 0.93 | 78 | ||
| AEM | Cu2+-cross-linked chitosan | 1.6 | 25 °C | 79 |
| Polyethylene glycol with imidazolium | 1.96 | 80 °C | 80 | |
| Fluoropoly(olefin) with TMA | 1.12 | 42 | ||
| Poly(vinylferrocene) | 1.65 | 61 | ||
| Catalyst type | Half-wave potential (V vs. RHE) | Tafel slope (mV decade−1) | Measurement condition | Ref. |
|---|---|---|---|---|
| Co–N–C | 0.881 | 75 | O2-saturated 1 M KOH | 101 |
| Fe–N–C | 0.899 | 68 | 102 | |
| Fe–N–C | 0.94 | 54 | 103 | |
| FeCl1N4/CNS | 0.921 | 51 | 104 | |
| Co3O4, Co NP/N-doped graphene | 0.808 | 62 | 105 | |
| CoO NP/N-doped carbon | 0.84 | 62 | 106 | |
| FexN NP/N-doped graphene | 0.78 | 52 | 107 | |
| CoMn2O4/C | 0.834 | 49 | 108 | |
| MnCo2O4/C | 0.845 | 46 | ||
| Ni3N/C | 0.803 | 44 | 109 | |
| Fe3N/C | 0.82 | 45 | ||
| Co3N/C | 0.862 | 37 | ||
| MnCo/C | 0.85 | 43 | 18 | |
| MnCo2O3/C | 0.86 | 45 | 110 | |
| Pt/C | 0.83 | 61 | O2-saturated 0.1 M NaOH | 67 |
| Catalyst type | Specific exchange current density (mA cm−2) | Mass exchange current density (A gmetal−1) | Activity measured environment | Ref. |
|---|---|---|---|---|
| Ir3PdRu6/C | 0.9 | 740 | H2-saturated 0.1 M KOH | 131 |
| Ir9Ru/C | 0.6 | 728 | ||
| Ir/C | 0.37 | 218 | 0.1 M NaOH, pH2 ≈ 100 kPaabs | 123 |
| Pd/C | 0.06 | 63 | ||
| Pd/C | 0.08 | 70.5 | H2-saturated 0.1 M KOH | 127 |
| Ir/C | 0.21 | 128.6 | 132 | |
| Ru/C | 0.063 | 82 | H2-saturated 0.1 M NaOH | 133 |
| Ni/C | 0.028 | 24.41 | H2-saturated 0.1 M KOH | 134 |
| Ni3N/C | 0.014 | 12 | 135 | |
| Ni (core)@N-doped carbon (shell)/C | 0.038 | 7.84 | 136 | |
| Ni/S-doped C | 0.04 | 7.4 | 137 | |
| Ni/N-doped CNT | 0.028 | 3.54 | 138 | |
| Ni (core) @boron nitride (shell)/C | 0.022 | 3.3 | H2-saturated 0.1 M NaOH | 139 |
| Ni0.95Cu0.05/C | 0.014 | 2.5 | 140 |
5.2. AEMs and AEIs
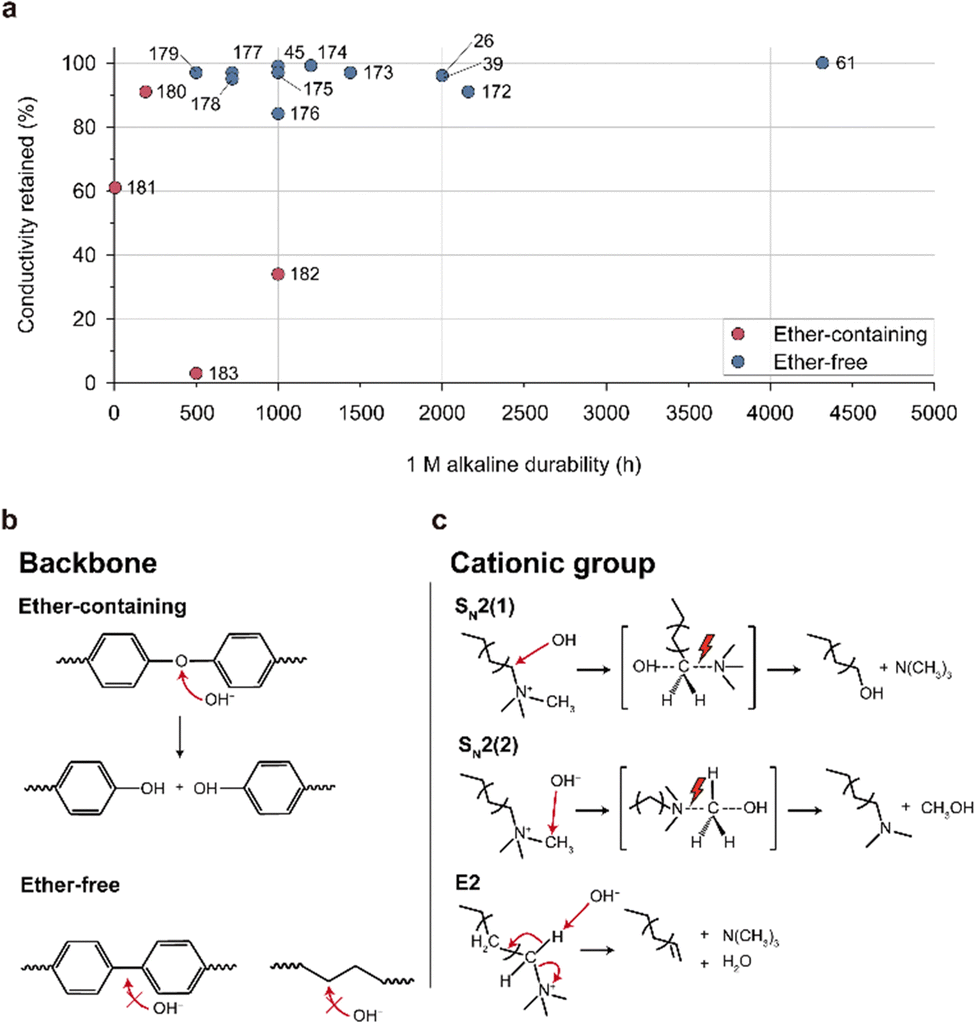 | ||
| Fig. 15 Alkaline stability and degradation mechanism of the AEM. (a) Alkaline stability of ether-containing and ether-free AEMs in a 1 M alkaline solution (Table 8).26,39,45,61,172–183 Alkaline degradation pathways for the (b) backbone structures (ether-containing and ether-free) and (c) cationic group (hexylTMA) in AEMs. | ||
| AEM type | Chemical structure | Alkaline stability test condition | Test time (h) | Conductivity retention (%) | Ref. |
|---|---|---|---|---|---|
| Ether-free | Poly(vinylferrocene) | 1 M NaOH, 95 °C | 4320 | 100 | 61 |
| Poly(aryl-co-aryl piperidinium) | 1 M KOH, 80 °C | 2160 | 91 | 172 | |
| Poly(aryl piperidinium) | 1 M NaOH 100 °C | 2000 | 96 | 26 | |
| Poly(fluorenyl aryl piperidinium) | 1 M NaOH 80 °C | 2000 | 96 | 39 | |
| Poly(terphenylene) with TMA | 1 M KOH 95 °C | 1440 | 98 | 173 | |
| Poly(norbornene) with TMA | 1 M KOH 80 °C | 1200 | 99 | 174 | |
| Poly(norbornene) with TMA | 1 M NaOH 80 °C | 1000 | 99 | 45 | |
| Poly(fluoroalkylene-co-phenylene) with TMA | 1 M KOH 80 °C | 1000 | 97 | 175 | |
| Poly(olefin) with TMA | 1 M KOH 80 °C | 1000 | 84 | 176 | |
| Poly(fluorene) with TMA | 1 M KOH 80 °C | 720 | 97 | 177 | |
| Poly(ethylene) with imidazolium | 1 M KOH 80 °C | 720 | 95 | 178 | |
| Poly(fluoroalkylene-co-phenylene) with TMA | 1 M KOH 80 °C | 500 | 97 | 179 | |
| Ether-containing | Poly(phenylene oxide) with piperidium | 1 M KOH 90 °C | 192 | 91 | 180 |
| Poly(sulfone) with TMA | 1 M KOH 50 °C | 6 | 61 | 181 | |
| Poly(arylene ether sulfone ketone-co-phenylene) with TMA | 1 M KOH 60 °C | 1000 | 34 | 182 | |
| Partially fluorinated poly(arylene ether-co-phenylene) with TMA | 1 M KOH 80 °C | 500 | 3 | 183 | |
In recent years, new-generation AEMs, which have an ether-free backbone such as polyphenylene and polyolefins, have been proposed and their enhanced alkaline stabilities have been reported. The delocalized electron densities of these ether-free AEMs enable them to endure nucleophilic OH− attacks (Fig. 15b). Indeed, the ether-free AEMs demonstrate exceptional durability in 1 M alkaline solution (>1000 h operation while retaining >90% of their initial conductivities) (Fig. 15a). However, the use of an ether-free backbone does not completely prevent the decrease in ionic conductivity because of the degradation of the cationic groups that impart ionic conductivity. The alkaline degradation pathway for the representative cationic group, hexylTMA, is presented in Fig. 15c. The hexylTMA group is commonly degraded by nucleophilic substitution (SN2) and Hofmann elimination (E2). In the SN2 reaction, the OH− molecule attacks the carbon atom linked to the nitrogen atom in the cationic group, forming the hydroxyl group. At this moment, the OH− molecule can attack both the backbone side (SN2(1)) and methyl side (SN2(2)) carbons connected to the nitrogen atom; thus, the two pathways are possible. For the E2 reaction, the OH− molecule attacks the hydrogen atom of the carbon from the hexyl group and forms H2O; in this case, the C–H and C–N bonds break and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond forms simultaneously.
C double bond forms simultaneously.
To enable facile conduction of OH−, the cationic groups in AEMs must be both positively charged and highly polarized. This polarization leads to the localization of electrons near the cation groups. As a result, the nucleophilic OH− ion can attack the electron-deficient site, which can ultimately lead to decomposition of the cationic group. To address this issue, we suggest that the best cationic structures are those that are highly polarized with low pKb values and that can also sterically hinder OH− access to the electron-deficient sites. Additionally, incorporating a structure capable of minimizing the localization of electrons would also be preferable.
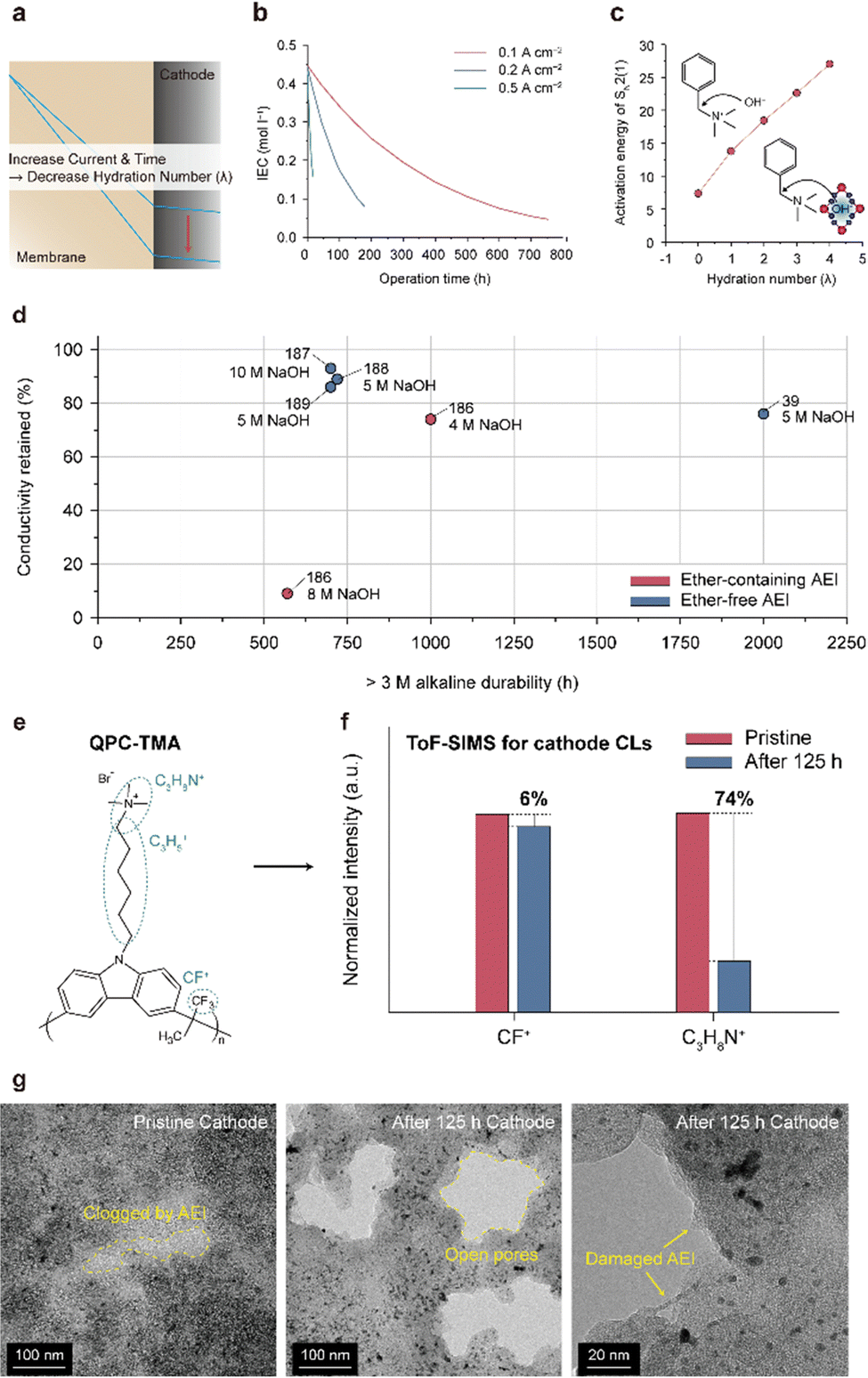 | ||
| Fig. 16 Decomposition of the AEI at the cathode CL owing to the highly alkaline environment. (a) Changes in water concentrations at the cathode and membrane with operation time or current. (b) Predicted IEC of the AEI at the cathode during constant current operation at various current densities. (c) Correlation between the activation energy of SN2(1) reaction and hydration number of OH−. Panels (b) and (c) are reprinted with permission from ref. 184 and 185, respectively. (d) Retention of ionic conductivity under storage in a highly concentrated alkaline solution for various AEIs (Table 9).39,186–189 (e)–(g) Degradation of QPC-TMA ionomer in the cathode CL during constant current single-cell operation. (e) Chemical structure of the QPC-TMA. (f) ToF-SIMS intensities of CF+ and C3H8N+ and (g) TEM images of the cathode CL before and after 125 h of operation at 0.6 A cm−2. Panels (e)–(g) are reprinted with permission from ref. 163. | ||
| Chemical structure | Alkaline stability test condition | Storage time (h) | Conductivity retention (%) | Ref. |
|---|---|---|---|---|
| Poly(fluorenyl aryl piperidinium) | 5 M NaOH 80 °C | 2000 | 76 | 39 |
| Partially fluorinated poly(arylene ether-co-phenylene) with TMA | 4 M KOH 80 °C | 1000 | 74 | 186 |
| Partially fluorinated poly(arylene ether-co-phenylene) with TMA | 8 M KOH 80 °C | 570 | 9 | |
| Poly(olefin) with TMA | 10 M NaOH 80 °C | 700 | 93 | 187 |
| Poly(aryl piperidinium) | 5 M NaOH 80 °C | 720 | 89 | 188 |
| Quaternary ammonium-containing poly(olefins) with chain extenders | 5 M NaOH 80 °C | 700 | 87 | 189 |
The deterioration of the cathode AEI during actual AEMFC operation was experimentally verified for a CL based on the QPC-TMA ionomer, whose structure is given in Fig. 16e. Hyun et al. investigated the relative amounts of the cationic group (C3H8N+) and a main-chain moiety (CF+) of the AEI in the cathode CL before and after 125 h of AEMFC operation at 0.6 A cm−2 using ToF-SIMS analysis.163 As shown in Fig. 16f, the CF+ intensity decreased by only 6%, whereas the C3H8N+ intensity reduced by 74% after the operation, indicating significant degradation of the side-chain cationic group compared to the main chain. The TEM images of the cathode CL (Fig. 16g) demonstrate that the QPC-TMA phase, which clogs the interstitial pores between the Pt/C particles, was damaged after 125 h of cell operation, forming pores. In the high-magnification image, damaged QPC-TMA residues were identified at the perimeters of the pores.
In addition to chemical degradation under an alkaline environment, electrochemical reactions are another cause of AEI degradation at the cathode. Maurya et al. reported the electrochemical oxidations of phenyl-group-containing AEIs.190 As shown in Fig. 17a, the cell voltage gradually decreased at a current density of 0.6 A cm−2 and 80 °C, followed by immediate recovery after alkaline solution replenishment by injection of 1 M KOH solution into the cell. The arrows in Fig. 17a indicate injection of the alkaline solution. The gain achieved through the replenishment process is regarded as a recoverable performance loss. However, despite the replenishment step, the initial cell voltage decreased gradually, as indicated by the dashed line, which is considered a permanent performance loss. The other interesting finding of this work is the decreased lifetime of the AEMFC with decreasing operating current (Fig. 17b), which implies that the cell durability is lower at higher cell voltages, which contrasts with the previous perception that the degradation of the cathode AEI becomes more severe with increasing operating current due to cathode dehydration.184 The 1H NMR analysis for the cathode AEI (BPN ionomer) before and after 75 h of operation at 0.9 V (Fig. 17c) shows that the phenylene moiety is oxidized and converted to phenol after operation. Based on these findings, they suggested that the permanent performance loss originates from phenyl oxidation (Fig. 17d). The phenyl groups of the AEI backbone adsorb onto the Pt surface and are electrochemically oxidized to phenol owing to the high potential at the ORR catalyst. The hydroxyl group of the phenol drags the TMA+/OH− pair and reduces the pH at the ORR catalyst/AEI interface, resulting in decreased ORR kinetics in the AEMFC (Fig. 17e).
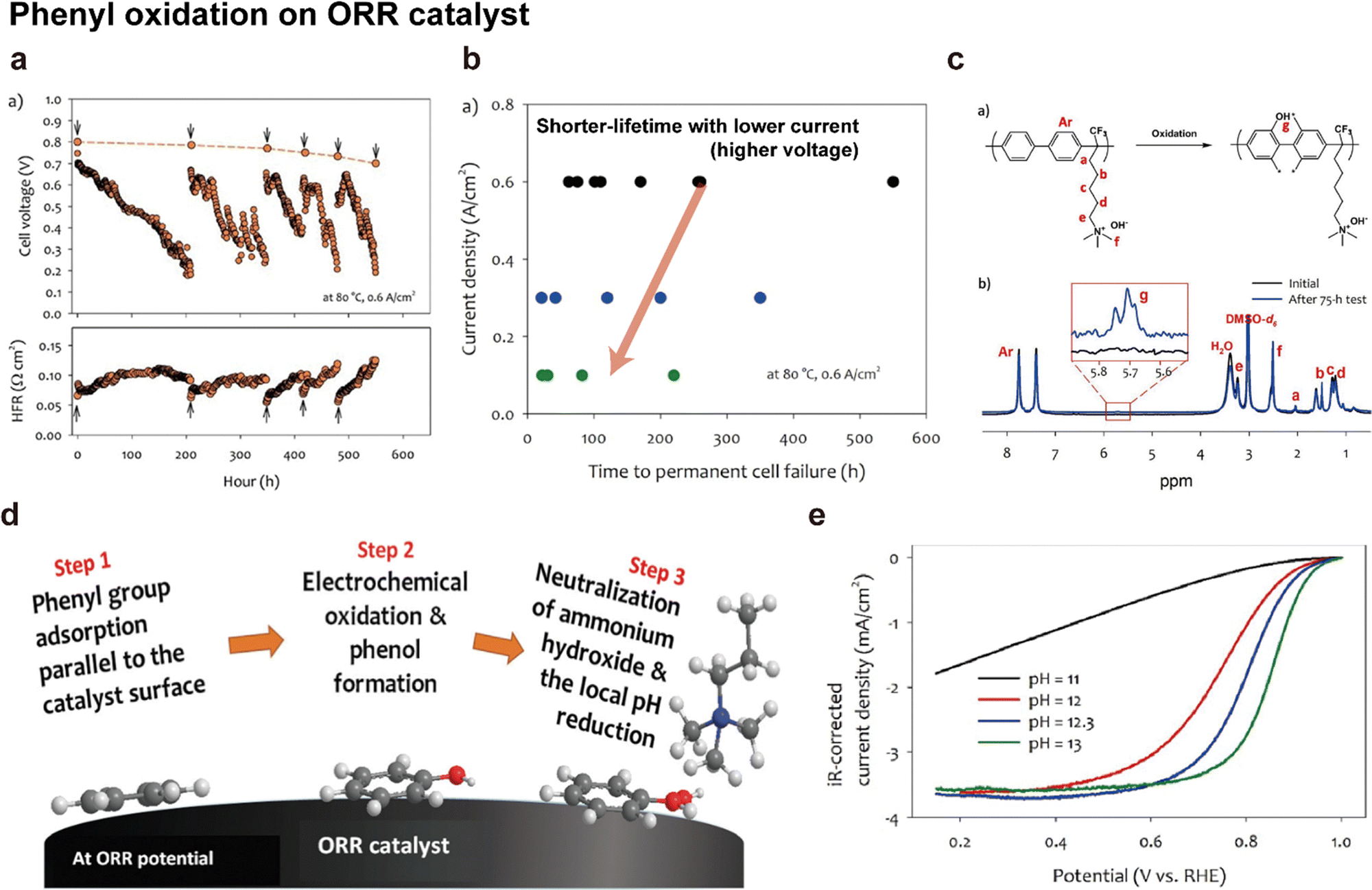 | ||
| Fig. 17 Permanent performance degradation of AEMFCs caused by oxidation of the phenylene backbone of the AEI on ORR catalyst. (a) Cell voltage and high frequency resistance changes under operation at 0.6 A cm−2. (b) Correlation between cell operating current density and time to permanent cell failure for an AEMFC MEA. The MEAs were constructed using m-TPN AEM, quaternized poly(fluorene) (FLN) AEI, and Pt/C for the cathodes and PtRu/C for the anode. (c) Structural changes in the quaternized poly(biphenylene) (BPN) ionomer due to phenyl oxidation on the cathode catalyst surface (top) and 1H NMR spectra of the BPN ionomer before and after 75 h of holding test at 0.9 V. (d) Schematic illustration of the phenyl oxidation process on ORR catalyst. (e) ORR voltammograms of the Pt polycrystalline RDE in pH-controlled NaOH solutions. The rotating speed and scan rate were 1600 rpm and 20 mV s−1, respectively. Panels (a)–(e) are adapted with permission from ref. 190 and 146, respectively. | ||
5.3. Mechanical durability of the AEMFC
In addition to chemical durability, mechanical durability is an essential factor for achieving long-lasting AEMFCs. In the PEMFC field, the mechanical durability of the membrane is a critical issue, and it is enhanced by either designing an ionomer structure or incorporating a reinforcing element, such as a porous support and filler. To assess the mechanical durability of CEMs, the essential work of fracture and tensile tests were suggested and demonstrated.191 Considering the repeated volume expansion and shrinkage of the AEM during dynamic operation, mechanical failure of the AEM can include the formation of pinholes or cracks in the AEM and delamination between the AEM and CL (Fig. 18a). For PEMFC MEAs, CLs strongly bind with the membrane via thermal lamination as an external reinforcement; however, the thermal lamination method does not always work optimally with AEMs. The mechanical disintegration of the CLs can be an important degradation mode in AEMFCs; however, this issue has not been investigated yet.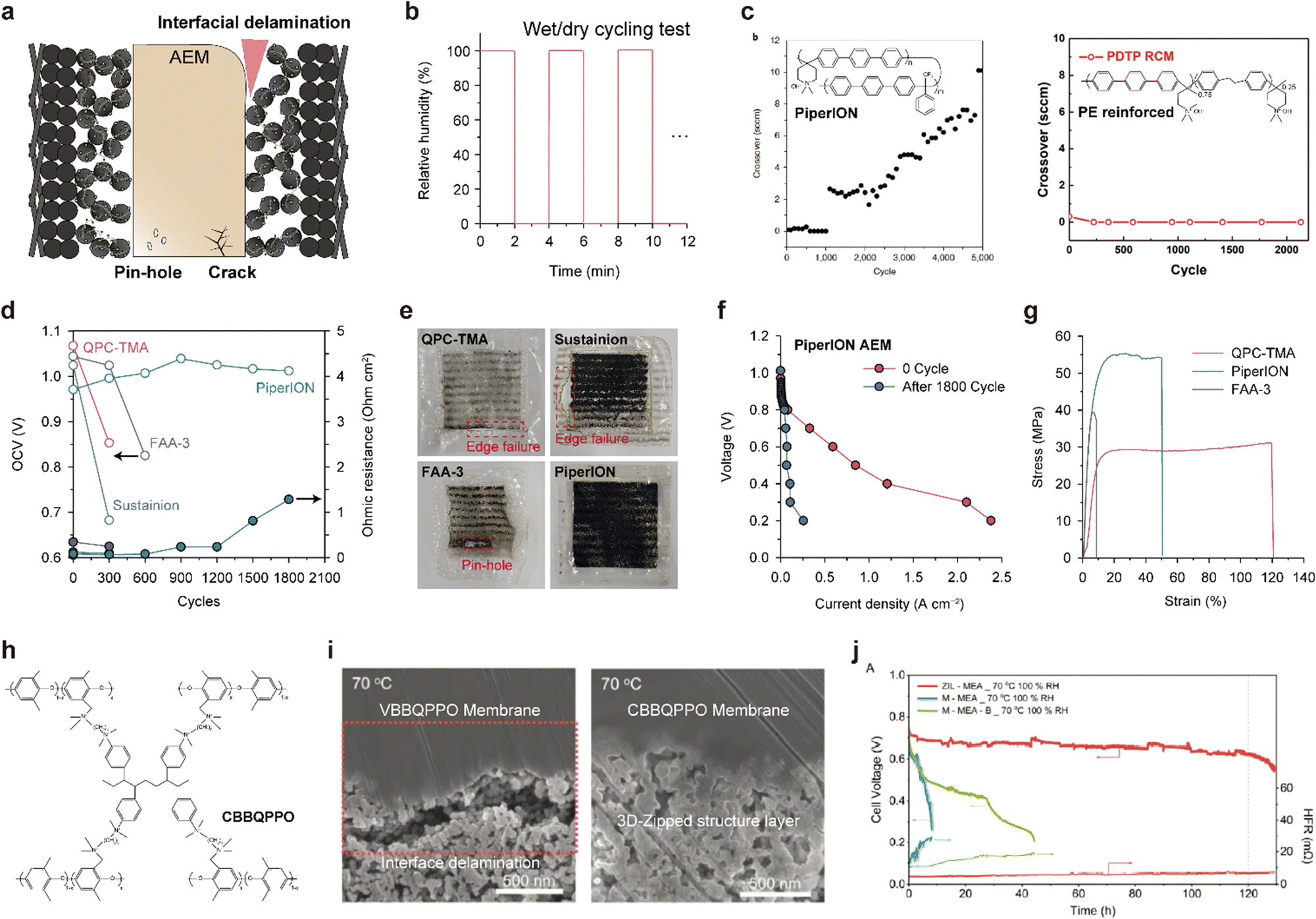 | ||
| Fig. 18 Mechanical instability of the AEMFC MEA. (a) Mechanical deterioration modes for the AEMFC MEA. (b) Humidity changes under wet/dry cycling test. (c) Hydrogen cross-over changes with wet/dry cycling for the MEA using PipeION AEM (left) and a reinforced membrane (PDTP-RCM) with a porous polyethylene film (right). Data in (c) are adapted with permission from ref. 26 and 38. (d) Changes in OCV and Ohmic resistance with the wet/dry cycle number for the QPC-TMA, PiperION, FAA-3, and Sustainion AEMs. (e) Optical images of the membranes after wet/dry cycling tests. (f) i–V polarization curves of the MEA with the PiperION AEM before and after 1800 wet/dry cycles. (g) Stress–strain curves of QPC-TMA, PiperION, and FAA-3 AEMs. (h) Chemical structure of a 3D-zipped ionomer (CBBQPPO). (i) SEM images of the membrane/CL interface for the MEAs using the control (without 3D-zipped process, VBBQPPO) and 3D-zipped ionomers. (j) Voltage changes upon operation for the MEAs using the control (M-MEA and M-MEA-B) and 3D-zipped ionomers (ZIL-MEA). Data in (h)–(j) are reprinted with permission from ref. 192. | ||
The most commonly used durability test protocol for evaluating the mechanical stability of a fuel cell is wet/dry cycling that consists of repeating stages of fully humid and dry conditions every 2 min (Fig. 18b). Although originally developed for PEMFCs, the test can also be applied to AEMFCs. Fig. 18c shows the change in hydrogen cross-over with the wet/dry cycle number for an MEA with PiperION AEM26 and one with a composite membrane consisting of a porous polyethylene membrane and the PDTP-RCM ionomer.38 The reinforced membrane exhibited superior durability, and hydrogen cross-over barely occurred over 2000 wet/dry cycles.
Fig. 18d shows the changes in OCV and Ohmic resistance for MEAs using various AEMs (QPC-TMA, Sustainion, FAA-3, and PiperION) as functions of the wet/dry cycle number (our data that has not been published yet). Except for PiperION, all AEMs exhibited a drop in OCV below 0.9 V within 600 cycles. The significant decrease in the OCV implies that H2 and O2 gases pass through the AEMs owing to their mechanical damage. A post-mortem analysis (Fig. 18e) revealed membrane tearing at the edge of the CL for the QPC-TMA and Sustainion AEMs and pinholes for FAA-3. In contrast, the PiperION AEM did not show any notable membrane failure. Although the PiperION-based MEA maintained the initial OCV value, the Ohmic resistance abruptly rose after 1200 cycles, and the i–V power performance was significantly reduced after 1800 cycles, as shown in Fig. 18f. Such degradation can be caused by delamination of the AEM/CL interface or disintegration of the CL. The superior wet/dry cycling stability of PiperION can be attributed to its high ultimate stress compared with other AEMs (Fig. 18g).
Xu's group also reported interfacial delamination between the AEM and CL and used a thermally cross-linked AEM (CBBQPPO) (Fig. 18h) to achieve a robust interface.192 For the reference membrane where a 3D-zipped structure is not applied, the interface was delaminated after AEMFC operation at 70 °C (Fig. 18i left). In contrast, when the thermally cross-linked AEM and AEI binder were used, the AEM/CL interface was stably maintained after AEMFC testing (Fig. 18i right). The strong AEM/CL interface resulting from the cross-linking enables over 120 h of operation at 0.6 A cm−2 while the other MEAs without 3D-zipped interfaces show rapid voltage losses.
6. Outlook
Significant progress has been made in the development of AEMs and AEIs, thereby greatly improving the performances of AEMFCs. This has increased expectations for AEMFCs as viable alternatives to PEMFCs. However, from a commercialization perspective, AEMFCs still require substantial leaps forward in increasing their cost-competitiveness, durability, and reliability. Current AEMFC studies mainly deal with materials and MEAs; however, successful system development requires delicate water and thermal management, which is missing.Although achieving higher power performance is desirable, it is essential to note that AEMFCs require significant amounts of Pt compared to PEMFCs. This negates the inherent incentive for cost-competitiveness. To address this issue, it is necessary to design a CL that maximize the ECSA or to develop catalysts with higher electrochemical activities. Mitigating the water imbalance is also necessary to augment the ECSA. Additionally, iterative MEA designs with lower amounts of Pt catalyst are required to identify factors that limit their performance. In the PEMFC technology sector, optimal CL structures have been designed by tuning the catalyst/ionomer compositions and their distribution. The remarkably high specific power density of PEMFCs (>8 W cm−2 mgPt−1) resulting from long-term accumulated efforts in optimizing the CL structure can motivate further research on the CL designs of AEMFCs.
An intuitive way to increase the cost competitiveness of AEMFCs is to use non-Pt catalysts as the ORR catalysts as they have higher ORR activities in bases than acids. However, the design of a CL exploiting non-Pt catalysts presents a lot of challenges. Owing to the low volumetric density of electrochemically active sites, the CL thickness can be quite large at the catalyst loading level required for high power performance, resulting in large electrical resistances and/or mass transport resistances. The large and inhomogeneous sizes of the current non-Pt catalysts hinder the expansion of ionomer/catalyst contact. Therefore, more full-cell-level studies on cathode CLs based on non-Pt catalysts are needed.
Regarding the use of non-precious-metal catalysts for the anode, current HOR catalysts do not deliver high enough activities to replace Pt/C catalysts. Given the susceptibility of AEMFC anodes to water flooding, a physical structure that facilitates efficient drainage of water and gas diffusion must be considered in addition to the HOR activity. Carbon supports with hydrophobic surface modifications143 or mesoporous structures (CNovel™ from Toyo Tanso)193,194 can be good options. Additionally, the problem of strong adsorption of the AEI binder on the surface of the HOR catalyst should be addressed. The structural design of the AEI based on the calculation of the binding energy to the catalyst surface and tuning the AEI distribution in the CL can be effective approaches.
Durability is the biggest hurdle for AEMFCs from a practical standpoint, but it has not been fully emphasized in favor of presenting high power performances in a competitive manner. In sharp contrast to the durability of PEMFCs (over 10000 h in a stack), AEMFCs have delivered a maximum of 2000 h of operation at the single-cell level. In our opinion, the most critical problem is the insufficient understanding of the key factors causing degradation during cell operation. To the best of our knowledge, the durability of AEMFCs has only been evaluated at constant current. However, considering the operating environments of real fuel cells, the current values are bound to vary depending on the power requirements. At this time, the water distribution of the MEA changes dynamically, and the degradation behavior also changes. Therefore, it is necessary to evaluate the durability of AEMFCs in an environment where the current or RH value changes, and it is also necessary to develop a standard protocol for this purpose.
Another overlooked aspect of durability is the mechanical deterioration of materials and components, which is more important in automotive applications. Crack and pinhole formation in membranes during repeated swelling/drying of the AEM and AEI at startup/shutdown can lead to sudden death. The interfacial delamination of the AEM/CL and disintegration of the CL can result in a drop in cell performance. However, in contrast to the chemical durability of AEMFCs, the mechanical durability has not been the subject of intensive investigations, probably owing to the use of the constant current mode in durability tests, which may not lead to significant mechanical degradation. A test protocol that reflects dynamic operation of AEMFCs would hence be necessary to motivate research on mechanical durability.
CO2 poisoning presents a potential risk for fuel cell system development. Even though electrochemical CO2 scrubbing systems appear promising, it is questionable whether the scrubbing system is durable enough because it shares a lot of degradation mechanisms with AEMFCs. The consumption of H2 fuel in the scrubbing system results in a loss of fuel efficiency in the total AEMFC system. A techno-economic analysis would be needed to assess the cost-effectiveness of an AEMFC paired with a CO2 remover. A new strategy to resolve the CO2 poisoning problem in an efficient and cost-effective manner will have a great impact on AEMFC technology.
Acquiring fabrication technologies for the membranes, catalysts, CLs, and MEAs are important milestones for advancing AEMFC technology. For example, the state-of-the-art CLs for AEMFCs conventionally employ the spray-coating method, because it enables the use of small amounts of catalysts and ionomers. For PEMFCs, spray coating was used in the early stages of MEA research; however, current PEMFC CLs are manufactured mainly by slurry-casting methods, which are compatible with the roll-to-roll process. Very recently, our group presented a slurry cast CL for AEMFCs, initiating the research on CL manufacturing.195 It is essential that the fabrication technology keeps pace with materials development to ensure the successful advancement of AEMFC technology.
Considering the fuel cells for transport applications significantly require low humidity and high-temperature operation to minimize the need for radiators and balance of plant, research focusing on such harsh operating conditions is highly meaningful. However, few studies reported low humidity and high-temperature operation of AEMFCs.61,196 More research would be needed to explore the possibility of simplifying the system by enabling low-humidity or high-temperature operations.
In summary, AEMFCs have significant potential to power the hydrogen future owing to their high cost competitiveness. Current strides in material developments have encouraged the community to move forward. However, to make AEMFCs practical, the next step should be achieving sufficient durability, incorporating non-precious-metal catalysts, and improving reliability by addressing problems such as CO2 poisoning and water imbalance.
Author contributions
Jonghyun Hyun: conceptualization, investigation, visualization, and writing – original draft; Hee-Tak Kim: supervision, funding acquisition, project administration, and writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Agency for Defense Development by the Korean Government (UD2200061D), the Technology Innovation Program (20011712) funded by the Ministry of Trade, Industry and Energy (MOTIE, Korea), and the Korea Institute of Energy Technology Evaluation and Planning (KETEP) and the Ministry of Trade, Industry and Energy (MOTIE, Korea) of the Republic of Korea (No. 20223030040220).References
- K. Kodama, T. Nagai, A. Kuwaki, R. Jinnouchi and Y. Morimoto, Nat. Nanotechnol., 2021, 16, 140–147 CrossRef CAS PubMed.
- D. A. Cullen, K. C. Neyerlin, R. K. Ahluwalia, R. Mukundan, K. L. More, R. L. Borup, A. Z. Weber, D. J. Myers and A. Kusoglu, Nat. Energy, 2021, 6, 462–474 CrossRef CAS.
- F. Bidault and P. H. Middleton, 4.07 - Alkaline fuel cells: Theory and application, Comprehensive Renewable Energy, Elsevier, 2012, pp. 179–202 Search PubMed.
- S. Noh, J. Y. Jeon, S. Adhikari, Y. S. Kim and C. Bae, Accounts Chem. Res., 2019, 52, 2745–2755 CrossRef CAS PubMed.
- W. H. Lee, Y. S. Kim and C. Bae, ACS Macro Lett., 2015, 4, 814–818 CrossRef CAS PubMed.
- N. Ul Hassan, M. Mandal, G. R. Huang, H. A. Firouzjaie, P. A. Kohl and W. E. Mustain, Adv. Energy Mater., 2020, 10, 2001986 CrossRef CAS.
- T. J. Omasta, A. M. Park, J. M. LaManna, Y. F. Zhang, X. Peng, L. Q. Wang, D. L. Jacobson, J. R. Varcoe, D. S. Hussey, B. S. Pivovar and W. E. Mustain, Energy Environ. Sci., 2018, 11, 551–558 RSC.
- X. Peng, D. Kulkarni, Y. Huang, T. J. Omasta, B. Ng, Y. W. Zheng, L. Q. Wang, J. M. LaManna, D. S. Hussey, J. R. Varcoe, I. V. Zenyuk and W. E. Mustain, Nat. Commun., 2020, 11, 3561 CrossRef CAS PubMed.
- https://www.londonstockexchange.com/(accessed 13 April, 2023).
- DOE Fundamentals Handbook, Chemistry, Volume 1 of 2.
- M. Chatenet, S. Berthon-Fabry, Y. Ahmad, K. Guerin, M. Colin, H. Farhat, L. Frezet, G. Zhang and M. Dubois, Adv. Energy Mater., 2023, 13, 2204304 CrossRef CAS.
- L. Q. Wang, J. J. Brink, Y. Liu, A. M. Herring, J. Ponce-Gonzalez, D. K. Whelligan and J. R. Varcoe, Energy Environ. Sci., 2017, 10, 2154–2167 RSC.
- L. Q. Wang, X. Peng, W. E. Mustain and J. R. Varcoe, Energy Environ. Sci., 2019, 12, 1575–1579 RSC.
- M. Sharma, J. H. Jang, D. Y. Shin, J. A. Kwon, D. H. Lim, D. Choi, H. Sung, J. Jang, S. Y. Lee, K. Y. Lee, H. Y. Park, N. Jung and S. J. Yoo, Energy Environ. Sci., 2019, 12, 2200–2211 RSC.
- R. Ren, C. Ge, Q. Li, G. Wang, L. Xiao, J. Lu and L. Zhuang, J. Power Sources, 2023, 556, 232439 CrossRef CAS.
- H. Adabi, A. Shakouri, N. Ul Hassan, J. R. Varcoe, B. Zulevi, A. Serov, J. R. Regalbuto and W. E. Mustain, Nat. Energy, 2021, 6, 834–843 CrossRef CAS.
- Y. R. Xue, L. Shi, X. R. Liu, J. J. Fang, X. D. Wang, B. P. Setzler, W. Zhu, Y. S. Yan and Z. B. Zhuang, Nat. Commun., 2020, 11, 5651 CrossRef CAS PubMed.
- Y. Wang, Y. Yang, S. F. Jia, X. M. Wang, K. J. Lyu, Y. Q. Peng, H. Zheng, X. Wei, H. Ren, L. Xiao, J. B. Wang, D. A. Muller, H. D. Abruna, B. O. E. Hwang, J. T. Lu and L. Zhuang, Nat. Commun., 2019, 10, 1506 CrossRef PubMed.
- G. Meyer, C. Perrot, G. Gebel, L. Gonon, S. Morlat and J. L. Gardette, Polymer, 2006, 47, 5003–5011 CrossRef CAS.
- C. Genies, R. Mercier, B. Sillion, R. Petiaud, N. Cornet, G. Gebel and M. Pineri, Polymer, 2001, 42, 5097–5105 CrossRef CAS.
- K. Miyatake, H. Zhou, T. Matsuo, H. Uchida and M. Watanabe, Macromolecules, 2004, 37, 4961–4966 CrossRef CAS.
- Y. Chikashige, Y. Chikyu, K. Miyatake and M. Watanabe, Macromolecules, 2005, 38, 7121–7126 CrossRef CAS.
- S. J. Hong, H. Y. Jung, S. J. Yoon, K.-H. Oh, S.-G. Oh, Y. T. Hong, D. M. Yu and S. So, J. Power Sources, 2022, 551, 232221 CrossRef CAS.
- D. P. Leonard, M. Lehmann, J. M. Klein, I. Matanovic, C. Fujimoto, T. Saito and Y. S. Kim, Adv. Energy Mater., 2023, 13, 2203488 CrossRef CAS.
- D. P. Leonard, S. Maurya, E. J. Park, L. D. Manriquez, S. Noh, X. F. Wang, C. Bae, E. D. Baca, C. Fujimoto and Y. S. Kim, J. Mater. Chem. A, 2020, 8, 14135–14144 RSC.
- J. H. Wang, Y. Zhao, B. P. Setzler, S. Rojas-Carbonell, C. Ben Yehuda, A. Amel, M. Page, L. Wang, K. Hu, L. Shi, S. Gottesfeld, B. J. Xu and Y. S. Yan, Nat. Energy, 2019, 4, 392–398 CrossRef CAS.
- Y. Huang, Y. H. Pan, W. W. Wang, L. Jiang and Y. Dan, Mater. Design, 2019, 162, 285–292 CrossRef CAS.
- M. H. Russell, W. R. Berti, B. Szostek and R. C. Buck, Environ. Sci. Technol., 2008, 42, 800–807 CrossRef CAS PubMed.
- S. Proch, M. Stenstrom, L. Eriksson, J. Andersson, G. Sjoblom, A. Jansson and J. Westlinder, Int. J. Hydrogen Energy, 2020, 45, 1313–1324 CrossRef CAS.
- K. H. Cho, W. G. Lee, S. B. Lee and H. Jang, J. Power Sources, 2008, 178, 671–676 CrossRef CAS.
- K. Feng, Y. Shen, J. M. Mai, D. A. Liu and X. Cai, J. Power Sources, 2008, 182, 145–152 CrossRef CAS.
- Q. Zhang, S. Dong, P. Shao, Y. Zhu, Z. Mu, D. Sheng, T. Zhang, X. Jiang, R. Shao, Z. Ren, J. Xie, X. Feng and B. Wang, Science, 2022, 378, 181–186 CrossRef CAS PubMed.
- Z. P. Zhao, Z. Y. Liu, A. Zhang, X. X. Yan, W. Xue, B. S. Peng, H. L. Xin, X. Q. Pan, X. F. Duan and Y. Huang, Nat. Nanotechnol., 2022, 17, 968–975 CrossRef CAS PubMed.
- Z. Y. Chen, C. Hao, B. W. Yan, Q. Y. Chen, H. Y. Feng, X. Q. Mao, J. M. Cen, Z. Q. Tian, P. Tsiakaras and P. K. Shen, Adv. Energy Mater., 2022, 12, 2201600 CrossRef CAS.
- A. Avid, J. L. Ochoa, Y. Huang, Y. C. Liu, P. Atanassov and I. V. Zenyuk, Nat. Commun., 2022, 13, 6349 CrossRef CAS PubMed.
- J. Hyun, J. Y. Jeon, G. Doo, J. Jung, S. Choi, D. H. Lee, D. W. Lee, J. Kwen, W. Jo, C. Bae and H. T. Kim, Chem. Eng. J., 2022, 427, 131737 CrossRef CAS.
- R. J. Gao, J. Wang, Z. F. Huang, R. R. Zhang, W. Wang, L. Pan, J. F. Zhang, W. K. Zhu, X. W. Zhang, C. X. Shi, J. Lim and J. J. Zou, Nat. Energy, 2021, 6, 614–623 CrossRef CAS.
- C. Hu, J. H. Park, H. M. Kim, H. H. Wang, J. Y. Bae, M. L. Liu, N. Y. Kang, K. S. Yoon, C. D. Park, N. Chen and Y. M. Lee, J. Mater. Chem. A, 2022, 10, 6587–6595 RSC.
- N. J. Chen, H. H. Wang, S. P. Kim, H. M. Kim, W. H. Lee, C. Hu, J. Y. Bae, E. S. Sim, Y. C. Chung, J. H. Jang, S. J. Yoo, Y. B. Zhuang and Y. M. Lee, Nat. Commun., 2021, 12, 2367 CrossRef CAS PubMed.
- S. Maurya, S. Noh, I. Matanovic, E. J. Park, C. N. Villarrubia, U. Martinez, J. Han, C. Bae and Y. S. Kim, Energy Environ. Sci., 2018, 11, 3283–3291 RSC.
- M. S. Cha, J. E. Park, S. Kim, S. H. Shin, S. H. Yang, S. J. Lee, T. H. Kim, D. M. Yu, S. So, K. M. Oh, Y. E. Sung, Y. H. Cho and J. Y. Lee, J. Mater. Chem. A, 2022, 10, 9693–9706 RSC.
- L. Zhu, X. Peng, S. L. Shang, M. T. Kwasny, T. J. Zimudzi, X. D. Yu, N. Saikia, J. Pan, Z. K. Liu, G. N. Tew, W. E. Mustain, M. Yandrasits and M. A. Hickner, Adv. Funct. Mater., 2019, 29, 1902059 CrossRef.
- H. Cao, J. Pan, H. R. Zhu, Z. Sun, B. W. Wang, J. L. Zhao and F. Yan, Adv. Sci., 2021, 8, 2101744 CrossRef CAS PubMed.
- J. T. Fan, S. Willdorf-Cohen, E. M. Schibli, Z. Paula, W. Li, T. J. G. Skalski, A. T. Sergeenko, A. Hohenadel, B. J. Frisken, E. Magliocca, W. E. Mustain, C. E. Diesendruck, D. R. Dekel and S. Holdcroft, Nat. Commun., 2019, 10, 2306 CrossRef PubMed.
- M. Mandal, G. Huang, N. Ul Hassan, W. E. Mustain and P. A. Kohl, J. Mater. Chem. A, 2020, 8, 18386 RSC.
- H. T. Chung, D. A. Cullen, D. Higgins, B. T. Sneed, E. F. Holby, K. L. More and P. Zelenay, Science, 2017, 357, 479–483 CrossRef CAS PubMed.
- W. Wang, W. H. Chen, P. Y. Miao, J. Luo, Z. D. Wei and S. L. Chen, ACS Catal., 2017, 7, 6144–6149 CrossRef CAS.
- X. Wan, X. F. Liu, Y. C. Li, R. H. Yu, L. R. Zheng, W. S. Yan, H. Wang, M. Xu and J. L. Shui, Nat. Catal., 2019, 2, 259–268 CrossRef CAS.
- H. Zhang, H. T. Chung, D. A. Cullen, S. Wagner, U. I. Kramm, K. L. More, P. Zelenay and G. Wu, Energy Environ. Sci., 2019, 12, 2548–2558 RSC.
- X. H. Xie, C. He, B. Y. Li, Y. H. He, D. A. Cullen, E. C. Wegener, A. J. Kropf, U. Martinez, Y. W. Cheng, M. H. Engelhard, M. E. Bowden, M. Song, T. Lemmon, X. S. Li, Z. M. Nie, J. Liu, D. J. Myers, P. Zelenay, G. F. Wang, G. Wu, V. Ramani and Y. Y. Shao, Nat. Catal., 2020, 3, 1044–1054 CrossRef CAS.
- L. Jiao, J. K. Li, L. L. Richard, Q. Sun, T. Stracensky, E. R. Liu, M. T. Sougrati, Z. P. Zhao, F. Yang, S. C. Zhong, H. Xu, S. Mukerjee, Y. Huang, D. A. Cullen, J. H. Park, M. Ferrandon, D. J. Myers, F. Jaouen and Q. Jia, Nat. Mater., 2021, 20, 1385–1391 CrossRef CAS PubMed.
- H. Xie, X. H. Xie, G. X. Hu, V. Prabhakaran, S. Saha, L. Gonzalez-Lopez, A. H. Phakatkar, M. Hong, M. L. Wu, R. Shahbazian-Yassar, V. Ramani, M. I. Al-Sheikhly, D. E. Jiang, Y. Y. Shao and L. B. Hu, Nat. Energy, 2022, 7, 281–289 CrossRef CAS.
- J. Lilloja, E. Kibena-Poldsepp, A. Sarapuu, J. C. Douglin, M. Kaarik, J. Kozlova, P. Paiste, A. Kikas, J. Aruvali, J. Leis, V. Sammelselg, D. R. Dekel and K. Tammeveski, ACS Catal., 2021, 11, 1920–1931 CrossRef CAS PubMed.
- S. J. C. Cleghorn, D. K. Mayfield, D. A. Moore, J. C. Moore, G. Rusch, T. W. Sherman, N. T. Sisofo and U. Beuscher, J. Power Sources, 2006, 158, 446–454 CrossRef CAS.
- R. Sharma, P. Morgen, S. Chiriaev, P. B. Lund, M. J. Larsen, B. Sieborg, L. Grahl-Madsen and S. M. Andersen, ACS Appl. Mater. Inter., 2022, 14, 49658–49671 CrossRef CAS PubMed.
- L. Dubau, L. Castanheira, M. Chatenet, F. Maillard, J. Dillet, G. Maranzana, S. Abbou, O. Lottin, G. De Moor, A. El Kaddouri, C. Bas, L. Flandin, E. Rossinot and N. Caque, Int. J. Hydrogen Energy, 2014, 39, 21902–21914 CrossRef CAS.
- X. Cheng, L. Chen, C. Peng, Z. W. Chen, Y. Zhang and Q. B. Fan, J. Electrochem. Soc., 2004, 151, A48–A52 CrossRef CAS.
- A. Kannan, A. Kabza and J. Scholta, J. Power Sources, 2015, 277, 312–316 CrossRef CAS.
- M. W. Fowler, R. F. Mann, J. C. Amphlett, B. A. Peppley and P. R. Roberge, J. Power Sources, 2002, 106, 274–283 CrossRef CAS.
- K. H. Lee, D. H. Cho, Y. M. Kim, S. J. Moon, J. G. Seong, D. W. Shin, J. Y. Sohn, J. F. Kim and Y. M. Lee, Energy Environ. Sci., 2017, 10, 275–285 RSC.
- X. Liu, N. Xie, J. D. Xue, M. Y. Li, C. Y. Zheng, J. F. Zhang, Y. Z. Qin, Y. Yin, D. R. Dekel and M. D. Guiver, Nat. Energy, 2022, 7, 329–339 CrossRef CAS.
- H. Peng, Q. Li, M. Hu, L. Xiao, J. Lu and L. Zhuang, J. Power Sources, 2018, 390, 165–167 CrossRef CAS.
- D. R. Dekel, J. Power Sources, 2018, 375, 158–169 CrossRef CAS.
- A. Serov, I. V. Zenyuk, C. G. Arges and M. Chatenet, J. Power Sources, 2018, 375, 149–157 CrossRef CAS.
- S. T. Thompson, D. Peterson, D. Ho and D. Papageorgopoulos, J. Electrochem. Soc., 2020, 167, 084514 CrossRef CAS.
- N. Ramaswamy and S. Mukerjee, Adv. Phys. Chem., 2012, 2012, 491604 Search PubMed.
- N. Ramaswamy and S. Mukerjee, J. Phys. Chem. C, 2011, 115, 18015–18026 CrossRef CAS.
- J. Marcinkoski, DOE Advanced Truck Technologies, 2019, 1–31 Search PubMed.
- C. Y. Ahn, J. Ahn, S. Y. Kang, O. H. Kim, D. W. Lee, J. H. Lee, J. G. Shim, C. H. Lee, Y. H. Cho and Y. E. Sung, Sci. Adv., 2020, 6, eaaw0870 CrossRef CAS PubMed.
- X. Liu, Y. Li, J. D. Xue, W. K. Zhu, J. F. Zhang, Y. Yin, Y. Z. Qin, K. Jiao, Q. Du, B. W. Cheng, X. P. Zhuang, J. X. Li and M. D. Guiver, Nat. Commun., 2019, 10, 842 CrossRef PubMed.
- C. S. Yin, J. J. Li, Y. W. Zhou, H. N. Zhan, P. F. Fang and C. Q. He, ACS Appl. Mater. Inter., 2018, 10, 14026–14035 CrossRef CAS PubMed.
- M. Mandal, G. Huang, N. Ul Hassan, W. E. Mustain and P. A. Kohl, J. Mater. Chem. A, 2020, 8, 17568–17578 RSC.
- J. S. Olsson, T. H. Pham and P. Jannasch, Adv. Funct. Mater., 2018, 28, 1702758 CrossRef.
- X. L. Gao, L. X. Sun, H. Y. Wu, Z. Y. Zhu, N. Xiao, J. H. Chen, Q. Yang, Q. G. Zhang, A. M. Zhu and Q. L. Liu, J. Mater. Chem. A, 2020, 8, 13065–13076 RSC.
- J. Miyake, R. Taki, T. Mochizuki, R. Shimizu, R. Akiyama, M. Uchida and K. Miyatake, Sci. Adv., 2017, 3, eaao0476 CrossRef PubMed.
- W. Jia, B. B. Tang and P. Y. Wu, ACS Appl. Mater. Inter., 2017, 9, 22620–22627 CrossRef CAS PubMed.
- Y. Kim, K. Ketpang, S. Jaritphun, J. S. Park and S. Shanmugam, J. Mater. Chem. A, 2015, 3, 8148–8155 RSC.
- Y. Sone, P. Ekdunge and D. Simonsson, J. Electrochem. Soc., 1996, 143, 1254–1259 CrossRef CAS.
- M. L. Wu, X. Zhang, Y. Zhao, C. P. Yang, S. S. Jing, Q. S. Wu, A. Brozena, J. T. Miller, N. J. Libretto, T. P. Wu, S. Bhattacharyya, M. N. Garaga, Y. G. Zhang, Y. Qi, S. G. Greenbaum, R. M. Briber, Y. S. Yan and L. B. Hu, Nat. Nanotechnol., 2022, 17, 629–636 CrossRef CAS PubMed.
- W. Yu, J. Zhang, X. Liang, X. L. Ge, C. P. Wei, Z. J. Ge, K. Y. Zhang, G. Li, W. J. Song, M. A. Shehzad, L. Wu and T. W. Xu, J. Membr. Sci., 2021, 634, 119404 CrossRef CAS.
- P. Atkins, P. W. Atkins and J. de Paula, Atkins’ physical chemistry, Oxford University Press, 2014 Search PubMed.
- W. H. Lee, E. J. Park, J. Han, D. W. Shin, Y. S. Kim and C. Bae, ACS Macro Lett., 2017, 6, 566–570 CrossRef CAS PubMed.
- H. S. Dang and P. Jannasch, J. Mater. Chem. A, 2017, 5, 21965–21978 RSC.
- J. Pan, C. Chen, Y. Li, L. Wang, L. S. Tan, G. W. Li, X. Tang, L. Xiao, J. T. Lu and L. Zhuang, Energy Environ. Sci., 2014, 7, 354–360 RSC.
- J. Pan, J. J. Han, L. Zhu and M. A. Hickner, Chem. Mater., 2017, 29, 5321–5330 CrossRef CAS.
- H. S. Dang and P. Jannasch, Macromolecules, 2015, 48, 5742–5751 CrossRef CAS.
- L. Zhu, J. Pan, Y. Wang, J. J. Han, L. Zhuang and M. A. Hickner, Macromolecules, 2016, 49, 815–824 CrossRef CAS.
- K. H. Lee, J. Y. Chu, A. R. Kim, H. G. Kim and D. J. Yoo, J. Membr. Sci., 2021, 634, 119435 CrossRef CAS.
- H. Chen, R. Tao, K. T. Bang, M. H. Shao and Y. Kim, Adv. Energy Mater., 2022, 12, 2200934 CrossRef CAS.
- N. Chen and Y. M. Lee, Prog. Polym. Sci., 2021, 113, 101345 CrossRef CAS.
- N. J. Chen and Y. M. Lee, Trends Chem., 2022, 4, 236–249 CrossRef CAS.
- A. Chakraborty, I. Salam, M. Choolaei, J. Lee, C. Crean, D. K. Whelligan, R. Bance-Soualhi and J. R. Varcoe, Mater. Adv., 2023, 4, 2099–2105 RSC.
- S. Willdorf-Cohen, A. Zhegur-Khais, J. Ponce-Gonzalez, S. Bsoul-Haj, J. R. Varcoe, C. E. Diesendruck and D. R. Dekel, ACS Appl. Energy Mater., 2023, 6, 1085–1092 CrossRef CAS PubMed.
- S. Haj-Bsoul, J. R. Varcoe and D. R. Dekel, J. Electroanal. Chem., 2022, 908, 116112 CrossRef CAS.
- G. Huang, M. Mandal, X. Peng, A. C. Yang-Neyerlin, B. S. Pivovar, W. E. Mustain and P. A. Kohl, J. Electrochem. Soc., 2019, 166, F637–F644 CrossRef CAS.
- M. Lehmann, D. Leonard, J. C. Zheng, L. L. He, X. M. Tang, X. C. Chen, K. H. Lim, S. Maurya, Y. S. Kim and T. Saito, ACS Appl. Energy Mater., 2023, 6, 1822–1833 CrossRef CAS.
- M. Mandal, G. Huang, N. Ul Hassan, X. Peng, T. L. Gu, A. H. Brooks-Starks, B. Bahar, W. E. Mustain and P. A. Kohl, J. Electrochem. Soc., 2019, 167, 054501 CrossRef.
- K. A. Mauritz and R. B. Moore, Chem. Rev., 2004, 104, 4535–4585 CrossRef CAS PubMed.
- D. W. Shin, M. D. Guiver and Y. M. Lee, Chem. Rev., 2017, 117, 4759–4805 CrossRef CAS PubMed.
- J. Hyun, G. Doo, S. Yuk, D. H. Lee, D. W. Lee, S. Choi, J. Kwen, H. Kang, R. Tenne, S. G. Lee and H. T. Kim, ACS Appl. Energy Mater., 2020, 3, 4619–4628 CrossRef CAS.
- P. Q. Yin, T. Yao, Y. Wu, L. R. Zheng, Y. Lin, W. Liu, H. X. Ju, J. F. Zhu, X. Hong, Z. X. Deng, G. Zhou, S. Q. Wei and Y. D. Li, Angew. Chem., Int. Ed., 2016, 55, 10800–10805 CrossRef CAS PubMed.
- W. J. Jiang, L. Gu, L. Li, Y. Zhang, X. Zhang, L. J. Zhang, J. Q. Wang, J. S. Hu, Z. D. Wei and L. J. Wan, J. Am. Chem. Soc., 2016, 138, 3570–3578 CrossRef CAS PubMed.
- D. N. Wang, Y. L. Wu, Z. G. Li, H. Pan, Y. Q. Wang, M. S. Yang and G. X. Zhang, Chem. Eng. J., 2021, 424, 130401 CrossRef CAS.
- Y. H. Han, Y. G. Wang, R. R. Xu, W. X. Chen, L. R. Zheng, A. J. Han, Y. Q. Zhu, J. Zhang, H. B. Zhang, J. Luo, C. Chen, Q. Peng, D. S. Wang and Y. D. Li, Energy Environ. Sci., 2018, 11, 2348–2352 RSC.
- J. Guo, S. Gadipelli, Y. C. Yang, Z. N. Li, Y. Lu, D. J. L. Brett and Z. X. Guo, J. Mater. Chem. A, 2019, 7, 3544–3551 RSC.
- X. Peng, T. J. Omasta, E. Magliocca, L. Q. Wang, J. R. Varcoe and W. E. Mustain, Angew. Chem., Int. Ed., 2019, 58, 1046–1051 CrossRef CAS PubMed.
- H. Yin, C. Z. Zhang, F. Liu and Y. L. Hou, Adv. Funct. Mater., 2014, 24, 2930–2937 CrossRef CAS.
- Y. Yang, Y. Xiong, M. E. Holtz, X. Feng, R. Zeng, G. Chen, F. J. DiSalvo, D. A. Muller and H. D. Abruña, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 24425–24432 CrossRef CAS PubMed.
- R. Zeng, Y. Yang, X. Feng, H. Li, L. M. Gibbs, F. J. DiSalvo and H. D. Abruña, Sci. Adv., 2022, 8, eabj1584 CrossRef CAS PubMed.
- Y. Yang, R. Zeng, Y. Xiong, F. J. DiSalvo and H. D. Abruna, Chem. Mater., 2019, 31, 9331–9337 CrossRef CAS.
- F. Cheng, Y. Su, J. Liang, Z. Tao and J. Chen, Chem. Mater., 2010, 22, 898–905 CrossRef CAS.
- S. Kosasang, N. Ma, P. Wuamprakhon, N. Phattharasupakun, T. Maihom, J. Limtrakul and M. Sawangphruk, Chem. Commun., 2018, 54, 8575–8578 RSC.
- X. Zhong, M. Oubla, X. Wang, Y. Huang, H. Zeng, S. Wang, K. Liu, J. Zhou, L. He, H. Zhong, N. Alonso-Vante, C.-W. Wang, W.-B. Wu, H.-J. Lin, C.-T. Chen, Z. Hu, Y. Huang and J. Ma, Nat. Commun., 2021, 12, 3136 CrossRef CAS PubMed.
- F. Moureaux, P. Stevens and M. Chatenet, Electrocatalysis, 2013, 4, 123–133 CrossRef CAS.
- J. Vondrák, B. Klápště, J. Velická, M. Sedlaříková, J. Reiter, I. Roche, E. Chainet, J. F. Fauvarque and M. Chatenet, J. New Mater. Electrochem. Syst., 2005, 8, 209–212 Search PubMed.
- A. C. Garcia, J. J. Linares, M. Chatenet and E. A. Ticianelli, Electrocatalysis, 2014, 5, 41–49 CrossRef CAS.
- I. Roche, E. ChaÎnet, M. Chatenet and J. Vondrák, J. Phys. Chem. C, 2007, 111, 1434–1443 CrossRef CAS.
- F. D. Speck, P. G. Santori, F. Jaouen and S. Cherevko, J. Phys. Chem. C, 2019, 123, 25267–25277 CrossRef CAS.
- T. Lazaridis, B. M. Stuhmeier, H. A. Gasteiger and H. A. El-Sayed, Nat. Catal., 2022, 5, 363–373 CrossRef CAS.
- R. Riasse, C. Lafforgue, F. Vandenberghe, F. Micoud, A. Morin, M. Arenz, J. Durst and M. Chatenet, J. Power Sources, 2023, 556, 232491 CrossRef CAS.
- M. M. Hossen, M. S. Hasan, M. R. I. Sardar, J. bin Haider, Mottakin, K. Tammeveski and P. Atanassov, Appl. Catal., B, 2023, 325, 121733 CrossRef CAS.
- N. Bevilacqua, T. Asset, M. A. Schmid, H. Markotter, I. Manke, P. Atanassov and R. Zeis, J. Power Source Adv., 2021, 7, 100042 CrossRef CAS.
- J. Durst, A. Siebel, C. Simon, F. Hasche, J. Herranz and H. A. Gasteiger, Energy Environ. Sci., 2014, 7, 2255–2260 RSC.
- W. C. Sheng, Z. B. Zhuang, M. R. Gao, J. Zheng, J. G. G. Chen and Y. S. Yan, Nat. Commun., 2015, 6, 5848 CrossRef CAS PubMed.
- D. Strmcnik, M. Uchimura, C. Wang, R. Subbaraman, N. Danilovic, D. van der Vliet, A. P. Paulikas, V. R. Stamenkovic and N. M. Markovic, Nat. Chem., 2013, 5, 300–306 CrossRef CAS PubMed.
- J. H. Dumont, A. J. Spears, R. P. Hjelm, M. Hawley, S. Maurya, D. G. Li, G. C. Yuan, E. B. Watkins and Y. S. Kim, ACS Appl. Mater. Inter., 2020, 12, 1825–1831 CrossRef CAS PubMed.
- R. Sgarbi, H. Doan, V. Martin and M. Chatenet, Electrocatalysis, 2023, 14, 267–278 CrossRef CAS.
- A. G. Oshchepkov, A. Bonnefont, V. N. Parmon and E. R. Savinova, Electrochim. Acta, 2018, 269, 111–118 CrossRef CAS.
- A. G. Oshchepkov, G. Braesch, A. Bonnefont, E. R. Savinova and M. Chatenet, ACS Catal., 2020, 10, 7043–7068 CrossRef CAS.
- H. Doan, I. Kendrick, R. Blanchard, Q. Jia, E. Knecht, A. Freeman, T. Jankins, M. K. Bates and S. Mukerjee, J. Electrochem. Soc., 2021, 168, 084501 CrossRef CAS.
- H. Wang and H. D. Abruña, J. Am. Chem. Soc., 2017, 139, 6807–6810 CrossRef CAS PubMed.
- J. Zheng, Z. B. Zhuang, B. J. Xu and Y. S. Yan, ACS Catal., 2015, 5, 4449–4455 CrossRef CAS.
- J. Ohyama, T. Sato, Y. Yamamoto, S. Arai and A. Satsuma, J. Am. Chem. Soc., 2013, 135, 8016–8021 CrossRef CAS PubMed.
- W. Y. Ni, T. Wang, P. A. Schouwink, Y. C. Chuang, H. M. Chen and X. L. Hu, Angew. Chem., Int. Ed., 2020, 59, 10797–10801 CrossRef CAS PubMed.
- W. Y. Ni, A. Krammer, C. S. Hsu, H. M. Chen, A. Schuler and X. L. Hu, Angew. Chem., Int. Ed., 2019, 58, 7445–7449 CrossRef CAS PubMed.
- J. Wang, X. Dong, J. Liu, W. Z. Li, L. T. Roling, J. P. Xiao and L. H. Jiang, ACS Catal., 2021, 11, 7422–7428 CrossRef CAS.
- F. L. Yang, X. Bao, Y. M. Zhao, X. W. Wang, G. Z. Cheng and W. Luo, J. Mater. Chem. A, 2019, 7, 10936–10941 RSC.
- Z. B. Zhuang, S. A. Giles, J. Zheng, G. R. Jenness, S. Caratzoulas, D. G. Vlachos and Y. S. Yan, Nat. Commun., 2016, 7, 10141 CrossRef CAS PubMed.
- L. J. Gao, Y. Wang, H. B. Li, Q. H. Li, N. Ta, L. Zhuang, Q. Fu and X. H. Bao, Chem. Sci., 2017, 8, 5728–5734 RSC.
- O. V. Cherstiouk, P. A. Simonov, A. G. Oshchepkov, V. I. Zaikovskii, T. Y. Kardash, A. Bonnefont, V. N. Parmon and E. R. Savinova, J. Electroanal. Chem., 2016, 783, 146–151 CrossRef CAS.
- I. Matanovic and Y. S. Kim, Curr. Opin. Electrochem., 2023, 38, 101218 CrossRef CAS.
- D. Choi, J. H. Jang, D. W. Lee, Y. S. Kang, H. Jin, K. Y. Lee and S. J. Yoo, J. Power Sources, 2021, 494, 229738 CrossRef CAS.
- J. Hyun, W. Jo, S. H. Yang, S. H. Shin, G. Doo, S. Choi, D. H. Lee, D. W. Lee, E. Oh, J. Y. Lee and H. T. Kim, J. Power Sources, 2022, 543, 231835 CrossRef CAS.
- G. Camera-Roda, A. Cardillo, V. Loddo, L. Palmisano and F. Parrino, Membranes, 2014, 4, 96–112 CrossRef PubMed.
- J. Zhang, Y. Liu, W. Zhu, Y. Pei, Y. Yin, Y. Qin, X. Li and M. D. Guiver, Cell Rep. Phys. Sci., 2021, 2, 100377 CrossRef CAS.
- W. E. Mustain, M. Chatenet, M. Page and Y. S. Kim, Energy Environ. Sci., 2020, 13, 2805–2838 RSC.
- N. Ziv, A. N. Mondal, T. Weissbach, S. Holdcroft and D. R. Dekel, J. Membr. Sci., 2019, 586, 140–150 CrossRef CAS.
- Y. W. Zheng, T. J. Omasta, X. Peng, L. Q. Wang, J. R. Varcoe, B. S. Pivovar and W. E. Mustain, Energy Environ. Sci., 2019, 12, 2806–2819 RSC.
- Y. W. Zheng, G. Huang, L. Q. Wang, J. R. Varcoe, P. A. Kohl and W. E. Mustain, J. Power Sources, 2020, 467, 228350 CrossRef CAS.
- Y. W. Zheng, G. R. Huang, M. Mandal, J. R. Varcoe, P. A. Kohl and W. E. Mustain, J. Electrochem. Soc., 2021, 168, 024504 CrossRef CAS.
- L. Shi, Y. Zhao, S. Matz, S. Gottesfeld, B. P. Setzler and Y. S. Yan, Nat. Energy, 2022, 7, 238–247 CrossRef CAS.
- G. Pellegrini, R. Strube and G. Manfrida, Energy, 2010, 35, 851–857 CrossRef CAS.
- S. E. Renfrew, D. E. Starr and P. Strasser, ACS Catal., 2020, 10, 13058–13074 CrossRef CAS.
- T. Wang, L. Shi, J. Wang, Y. Zhao, B. P. Setzler, S. Rojas-Carbonell and Y. Yan, J. Electrochem. Soc., 2019, 166, F3305–F3310 CrossRef CAS.
- Z. Liang, F. Yang, Y. Li, J. Tang, D. R. Dekel and X. He, Sep. Purif. Technol., 2022, 289, 120713 CrossRef CAS.
- J. Zhang, W. Zhu, T. Huang, C. Zheng, Y. Pei, G. Shen, Z. Nie, D. Xiao, Y. Yin and M. D. Guiver, Adv. Sci., 2021, 8, 2100284 CrossRef CAS PubMed.
- Y. Yang, C. R. Peltier, R. Zeng, R. Schimmenti, Q. Li, X. Huang, Z. Yan, G. Potsi, R. Selhorst, X. Lu, W. Xu, M. Tader, A. V. Soudackov, H. Zhang, M. Krumov, E. Murray, P. Xu, J. Hitt, L. Xu, H.-Y. Ko, B. G. Ernst, C. Bundschu, A. Luo, D. Markovich, M. Hu, C. He, H. Wang, J. Fang, R. A. DiStasio, L. F. Kourkoutis, A. Singer, K. J. T. Noonan, L. Xiao, L. Zhuang, B. S. Pivovar, P. Zelenay, E. Herrero, J. M. Feliu, J. Suntivich, E. P. Giannelis, S. Hammes-Schiffer, T. Arias, M. Mavrikakis, T. E. Mallouk, J. D. Brock, D. A. Muller, F. J. DiSalvo, G. W. Coates and H. D. Abruna, Chem. Rev., 2022, 122, 6117–6321 CrossRef CAS PubMed.
- A. Carlson, P. Shapturenka, B. Eriksson, G. Lindbergh, C. Lagergren and R. W. Lindstrom, Electrochim. Acta, 2018, 277, 151–160 CrossRef CAS.
- L. Tang, X. Li, R. C. Cammarata, C. Friesen and K. Sieradzki, J. Am. Chem. Soc., 2010, 132, 11722–11726 CrossRef CAS PubMed.
- A. Zadick, L. Dubau, N. Sergent, G. Berthome and M. Chatenet, ACS Catal., 2015, 5, 4819–4824 CrossRef CAS.
- C. Lafforgue, A. Zadick, L. Dubau, F. Maillard and M. Chatenet, Fuel Cells, 2018, 18, 229–238 CrossRef CAS.
- C. Lafforgue, M. Chatenet, L. Dubau and D. R. Dekel, ACS Catal., 2018, 8, 1278–1286 CrossRef CAS.
- J. Hyun, S. H. Yang, G. Doo, S. Choi, D. H. Lee, D. W. Lee, J. Kwen, W. Jo, S. H. Shin, J. Y. Lee and H. T. Kim, J. Mater. Chem. A, 2021, 9, 18546–18556 RSC.
- C. Lafforgue, F. Maillard, V. Martin, L. Dubau and M. Chatenet, ACS Catal., 2019, 9, 5613–5622 CrossRef CAS.
- G. Bae, M. W. Chung, S. G. Ji, F. Jaouen and C. H. Choi, ACS Catal., 2020, 10, 8485–8495 CrossRef CAS.
- E. F. Holby, G. F. Wang and P. Zelenay, ACS Catal., 2020, 10, 14527–14539 CrossRef CAS.
- Y. P. Ku, K. Ehelebe, A. Hutzler, M. Bierling, T. Bohm, A. Zitolo, M. Vorokhta, N. Bibent, F. D. Speck, D. Seeberger, I. Khalakhan, K. J. J. Mayrhofer, S. Thiele, F. Jaouen and S. Cherevko, J. Am. Chem. Soc., 2022, 144, 9753–9763 CrossRef CAS PubMed.
- K. Kisand, A. Sarapuu, J. C. Douglin, A. Kikas, A. Treshchalov, M. Kaeaerik, H. M. Piirsoo, P. Paiste, J. Aruvaeli, J. Leis, V. Kisand, A. Tamm, D. R. Dekel and K. Tammeveski, ACS Catal., 2022, 12, 14050–14061 CrossRef CAS.
- C. H. Choi, H. K. Lim, M. W. Chung, G. Chon, N. R. Sahraie, A. Altin, M. T. Sougrati, L. Stievano, H. S. Oh, E. S. Park, F. Luo, P. Strasser, G. Drazic, K. J. J. Mayrhofer, H. Kim and F. Jaouen, Energy Environ. Sci., 2018, 11, 3176–3182 RSC.
- K. Kumar, L. Dubau, M. Mermoux, J. K. Li, A. Zitolo, J. Nelayah, F. Jaouen and F. Maillard, Angew. Chem., Int. Ed., 2020, 59, 3235–3243 CrossRef CAS PubMed.
- P. G. Santori, F. D. Speck, J. Li, A. Zitolo, Q. Y. Jia, S. Mukerjee, S. Cherevko and F. Jaouen, J. Electrochem. Soc., 2019, 166, F3311 CrossRef CAS.
- N. Chen, C. Hu, H. H. Wang, J. H. Park, H. M. Kim and Y. M. Lee, J. Membr. Sci., 2021, 638, 119685 CrossRef CAS.
- E. J. Park, C. B. Capuano, K. E. Ayers and C. Bae, J. Power Sources, 2018, 375, 367–372 CrossRef CAS.
- M. Mandal, G. Huang and P. A. Kohl, J. Membr. Sci., 2019, 570, 394–402 CrossRef.
- H. Ono, T. Kimura, A. Takano, K. Asazawa, J. Miyake, J. Inukai and K. Miyatake, J. Mater. Chem. A, 2017, 5, 24804–24812 RSC.
- L. Zhu, X. D. Yu, X. Peng, T. J. Zimudzi, N. Saikia, M. T. Kwasny, S. F. Song, D. I. Kushner, Z. S. Fu, G. N. Tew, W. E. Mustain, M. A. Yandrasits and M. A. Hickner, Macromolecules, 2019, 52, 4030–4041 CrossRef CAS.
- W. H. Lee, A. D. Mohanty and C. Bae, ACS Macro Lett., 2015, 4, 453–457 CrossRef CAS PubMed.
- W. You, E. Padgett, S. N. MacMillan, D. A. Muller and G. W. Coates, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 9729–9734 CrossRef CAS PubMed.
- A. M. A. Mahmoud, A. M. M. Elsaghier, K. Otsuji and K. Miyatake, Macromolecules, 2017, 50, 4256–4266 CrossRef CAS.
- H. S. Dang and P. Jannasch, J. Mater. Chem. A, 2016, 4, 11924–11938 RSC.
- A. D. Mohanty, Y. B. Lee, L. Zhu, M. A. Hickner and C. Bae, Macromolecules, 2014, 47, 1973–1980 CrossRef CAS.
- R. Akiyama, N. Yokota, E. Nishino, K. Asazawa and K. Miyatake, Macromolecules, 2016, 49, 4480–4489 CrossRef CAS.
- N. Yokota, M. Shimada, H. Ono, R. Akiyama, E. Nishino, K. Asazawa, J. Miyake, M. Watanabe and K. Miyatake, Macromolecules, 2014, 47, 8238–8246 CrossRef CAS.
- D. R. Dekel, I. G. Rasin and S. Brandon, J. Power Sources, 2019, 420, 118–123 CrossRef CAS.
- D. R. Dekel, M. Arnar, S. Willdorf, M. Kosa, S. Dhara and C. E. Diesendruck, Chem. Mater., 2017, 29, 4425–4431 CrossRef CAS.
- A. M. A. Mahmoud and K. Miyatake, J. Mater. Chem. A, 2018, 6, 14400–14409 RSC.
- M. Zhang, C. R. Shan, L. Liu, J. Y. Liao, Q. Chen, M. Zhu, Y. G. Wang, L. A. An and N. W. Li, ACS Appl. Mater. Inter., 2016, 8, 23321–23330 CrossRef CAS PubMed.
- X. M. Du, H. Y. Zhang, Y. J. Yuan and Z. Wang, J. Power Sources, 2021, 487, 229429 CrossRef CAS.
- M. Zhang, J. L. Liu, Y. G. Wang, L. A. An, M. D. Guiver and N. W. Li, J. Mater. Chem. A, 2015, 3, 12284–12296 RSC.
- S. Maurya, A. S. Lee, D. G. Li, E. J. Park, D. P. Leonard, S. Noh, C. Bae and Y. S. Kim, J. Power Sources, 2019, 436, 226866 CrossRef CAS.
- E. Moukheiber, C. Bas and L. Flandin, Int. J. Hydrogen Energy, 2014, 39, 2717–2723 CrossRef CAS.
- X. Liang, X. L. Ge, Y. B. He, M. Xu, M. A. Shehzad, F. M. Sheng, R. Bance-Soualhi, J. J. Zhang, W. S. Yu, Z. J. Ge, C. P. Wei, W. J. Song, J. L. Peng, J. R. Varcoe, L. Wu and T. W. Xu, Adv. Sci., 2021, 8, 2102637 CrossRef CAS PubMed.
- Y. J. Lee, H. E. Kim, E. Lee, J. Lee, S. Shin, H. Yun, E. J. Kim, H. Jung, H. C. Ham, B. J. Kim and H. Lee, Adv. Energy Mater., 2021, 11, 2102970 CrossRef CAS.
- Y. J. Lee, H. E. Kim, H. Oh, H. Yun, J. Lee, S. Shin, H. Lee and B. J. Kim, ACS Nano, 2022, 16, 2988–2996 CrossRef CAS PubMed.
- J. Hyun, D. W. Lee, E. Oh, H. Bae, J. Park, G. Doo and H.-T. Kim, J. Power Sources, 2023, 573, 233161 CrossRef CAS.
- J. C. Douglin, J. R. Varcoe and D. R. Dekel, J. Power Sources Adv., 2020, 5, 100023 CrossRef.
| This journal is © The Royal Society of Chemistry 2023 |