
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Analysis of micro- and nanoscale heterogeneities within environmentally relevant thin films containing biological components, oxyanions and minerals using AFM-PTIR spectroscopy†
Deborah
Kim
 and
Vicki H.
Grassian
*
and
Vicki H.
Grassian
*
Department of Chemistry and Biochemistry, University of California San Diego, La Jolla, CA 92093, USA. E-mail: vhgrassian@ucsd.edu
First published on 1st February 2023
Abstract
Minerals in groundwater interact with various chemical and biological species including organic matter, proteins, and prevalent oxyanions, resulting in surface coatings and thin films of these different components. Surface interactions and the surface adsorption of these components on both oxide and oxyhydroxide iron surfaces have been widely investigated using a variety of spectroscopic methods. Despite these numerous studies, there still remains uncertainty with respect to interactions between these individual components, as well as heterogeneities and phase segregations within these thin films. In this study, we investigate mixtures containing Fe-containing minerals, proteins, and oxyanions to better understand surface interactions and phase segregation using Atomic Force Microscopy PhotoThermal Infrared (AFM-PTIR) spectroscopy. The results of this study show that AFM-PTIR spectroscopy can identify both nano- and microscale heterogeneities present within these thin films that are difficult to discern with other more conventional techniques such as ATR-FTIR spectroscopy due to phase segregation and mineral surface interactions. Overall, AFM-PTIR spectroscopy provides insights into multi-component environmental films that are difficult to uncover using other methodologies. This method has the potential to differentiate between bound and unbound toxic species as well as biological components, including environmental DNA, which can be used to assess the fate and transport of these species in the environment.
Environmental significanceHeterogeneities of complex environmental interfaces and environmental thin films are important to understand, yet difficult to probe. Here we investigate heterogeneities within environmentally relevant thin films containing biological components, oxyanions, and minerals to determine the distribution of different interactions through the examination of small scale structural and chemical heterogeneities. Utilizing Atomic Force Microscopy-Infrared (AFM-PTIR) spectroscopy, micro- and nanoscale heterogeneities are delineated through localized point spectra and larger area chemical mapping. These results provide new information on spatial scales that have not been previously discerned and suggest important insights can be gained concerning phase segregation, mineral interactions, bioavailability, and the transport of different biological and chemical species in the environment. |
Introduction
Groundwater contains a myriad of different biological and chemical species – both natural and anthropogenic – including organic matter, proteins, oxyanions, and chemical contaminants. Over the past few decades, the interactions of each of these components have been examined in laboratory studies with commonly found minerals in the environment.1–4 These studies provide great insight into molecular level interactions at geochemical surfaces.5,6 Several factors that impact these mineral interactions including the nature of the mineral, specific mineral surface planes, adsorbed water and solution pH and ionic strength. All of these alter the dynamics of the surface adsorption process for mineral surfaces.7 However, few studies have probed multi-component systems, as competitive interactions between different species can drive the surface chemistry of mineral surfaces.8–13In the environment, biological components represent an important fraction of these different species and range from free amino acids to proteins, to environmental DNA (eDNA) and to entire organisms such as bacteria.10 Bovine Serum Albumin (BSA) has been chosen in several studies as a representative model protein due to its stability in the environment as well as its natural state at circumneutral pH.14 BSA has a molar mass of 66 kDa and an isoelectric point of 4.5 to 5.0, which is within the range of many other proteins extracted from groundwater.15,16
Besides biological components, the interaction of oxyanions, including phosphate, sulfate and carbonate, and mineral surfaces have been widely investigated.17–20 The protonation states of these anions change as a function of solution pH and can impact the coordination to the surface, whether outer versus inner sphere and mono- or bi-dentate. Sulfate speciation on oxide surfaces has, in particular, been widely studied by vibrational spectroscopy due to its affinity to adsorb onto mineral surfaces.21 Changes in the vibrational modes upon adsorption provide insights into the decrease in symmetry, change in protonation state and nature of the coordination mode. Past studies have reported sulfate speciation to be highly sensitive to factors such as pH, dehydration, and ionic strength, which has resulted in many proposed mechanisms of sulfate coordination onto iron-containing surfaces, such as goethite.22–28 Sulfate can also block sites for surface adsorption of other species present, which can affect the availability of trace metals in the environment.15 Additionally, interactions between biological species like BSA, and phosphate, another commonly studied oxyanion, have been investigated, but BSA interactions with sulfate have yet been explored.29 These different interactions can result in the formation of different complexes that have vibrationally distinct spectral features.30
In this study, we investigate thin films of BSA protein, sodium sulfate, and goethite to determine the degree of heterogeneity in these films. Dried, thin films on the order of several hundred nanometers thick have been prepared by forming suspensions of different environmentally-relevant components at circumneutral pH. While more typical vibrational techniques like ATR-FTIR spectroscopy can provide spectral characterization of the average surface interactions occurring within these films, further insights into the micro- and nanoscale heterogeneities are explored with AFM-PTIR spectroscopy. In particular, phase segregation and different mineral interactions are probed through localized point spectra and larger area chemical mapping. These results provide information on spatial scales that have not been previously explored and suggest insights can be gained concerning phase segregation, mineral interactions, bioavailability, and transport of different biological and chemical species in the environment. Given that this method has the potential to differentiate between bound and unbound species, it can potentially be used to assess not only the fate but also the transport of such species in the environment.
Materials and methods
Sample preparation and source of materials
In these studies, samples for both AFM-PTIR spectroscopy and complementary ATR-FTIR spectroscopy were prepared for analysis for both techniques. Stock solutions were made for each component at a concentration of 1 mg mL−1 for BSA (Sigma Aldrich) and sodium sulfate (Fisher Scientific) and 0.3 mg mL−1 for goethite (Alfa Aesar) in Milli-Q water. Single component thin films were prepared by drop-casting 1 mL aliquots of each solution onto an AMTIR (Amorphous Material Transmitting Infrared Radiation) crystal for ATR-FTIR analysis and 5 μL onto a cleaned silica wafer for AFM-PTIR analysis.For AFM-PTIR and ATR-FTIR analysis, solutions were allowed to dry for 5 hours and 3 hours, respectively; the differences in the drying times were dependent on the differences in the volume of solution as well as the surface area coverage of each substrate. For the multi-component thin films, mixtures containing a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume ratios, or 1:1:1 volume ratios for two- and three-component systems, respectively, were prepared with the components of interest from the initial stock solutions. For goethite containing samples, the suspension was probe-sonicated for 15 seconds, inverted, and then repeated once more to facilitate the mixing of goethite particles with the solution. Post sonication, the solution turned into an opaque, yellow color, indicating proper mixing of the nanoparticles. Once the mixtures were prepared, the same procedure for drop-casting and drying was applied for thin film formation, as adapted in previous studies, ensuring comparable film deposition between both substrates. All of the data collected and presented here have been selected as the most representative from a triplicate set of measurements that have been taken from at least three different samples and regions. In addition, there are no high safety hazards that were encountered during the course of this sample preparation.
:1 volume ratios, or 1:1:1 volume ratios for two- and three-component systems, respectively, were prepared with the components of interest from the initial stock solutions. For goethite containing samples, the suspension was probe-sonicated for 15 seconds, inverted, and then repeated once more to facilitate the mixing of goethite particles with the solution. Post sonication, the solution turned into an opaque, yellow color, indicating proper mixing of the nanoparticles. Once the mixtures were prepared, the same procedure for drop-casting and drying was applied for thin film formation, as adapted in previous studies, ensuring comparable film deposition between both substrates. All of the data collected and presented here have been selected as the most representative from a triplicate set of measurements that have been taken from at least three different samples and regions. In addition, there are no high safety hazards that were encountered during the course of this sample preparation.
Atomic force microscopy-infrared (AFM-PTIR) spectroscopy
AFM-PTIR spectroscopy is a microspectroscopic technique that utilizes a sharp AFM probe tip which is typically on the order of tens of nanometers in diameter, that is usually at the end of a cantilever. For infrared measurements, this technique is based on the principle of photothermal induced resonance, where the IR absorption signal is transduced from the cantilever oscillation upon photothermal expansion of the sample in a nondestructive and nonperturbative manner.31,32 Using a nanoIR2 microscopy system (Bruker, Anasys – Santa Barbara, CA) equipped with a mid-IR tunable quantum cascade laser (QCL), samples were analyzed after drying onto a silica wafer. Images, PTIR spectra, and spectral maps were collected in primarily contact mode using gold-coated silicon nitride probes, with tip radii of ∼30 nm, a resonant frequency of 75 kHz and a spring constant of 1 to 7 N m−1. PTIR spectra were collected at a resolution of 5 cm−1. For point spectra, the data is plotted as absorbance, which is proportional to the change in the photodiode as a result of the cantilever deflection, as a function of wavenumber. Absorbances in these PTIR spectra is related to the deflection of the cantilever upon sample absorption of IR light. For imaging and chemical mapping, the scan rate was set to 0.2 Hz. AFM images and IR maps were prepared using the Gwyddion software and the spectra presented were co-averaged from 5 spectra and were filtered using a 10-point, 3rd order polynomial, Savitzky–Golay algorithm.Attenuated total reflectance fourier transform infrared (ATR-FTIR) spectroscopy
ATR-FTIR spectroscopy is based upon the principle of total internal reflection between an optically dense and an optically rare medium.8 For these experiments, an AMTIR crystal, an internal reflecting element, that is housed in a horizontal, Teflon-coated flow cell (Pike Technologies) configured for multiple reflections is used. Specifically, an AMTIR crystal was chosen for its high effective path length, resulting in high spectral sensitivity. It should be noted that while the flow cell can hold 500 μL when capped, when open, it can hold up to a solution volume of 1 mL. Dried, thin films were prepared as described previously and after drying was complete, infrared spectra were averaged over 100 scans at a resolution of 4 cm−1 over a spectral range from 725 to 4000 cm−1.Results and discussion
Comparison of AFM-PTIR spectra of single component BSA, sulfate, and goethite thin films
AFM-PTIR spectra of thin films of BSA, sulfate and goethite are shown in Fig. 1. There is overall agreement with these single component nanoscale point spectra and ATR-FTIR spectra collected ATR-FTIR spectra which can be found in (ESI) Fig. S1.† | ||
| Fig. 1 (a) AFM-PTIR spectra of dried, thin films of BSA, sodium sulfate, and goethite on a silica wafer substrate, respectively. (b) AFM images are shown in both height (left) and deflection (right) modes. The PTIR spectra on and off of each of these thin films are shown. (c) The black line marked on each AFM height image indicates where the height profile was taken. | ||
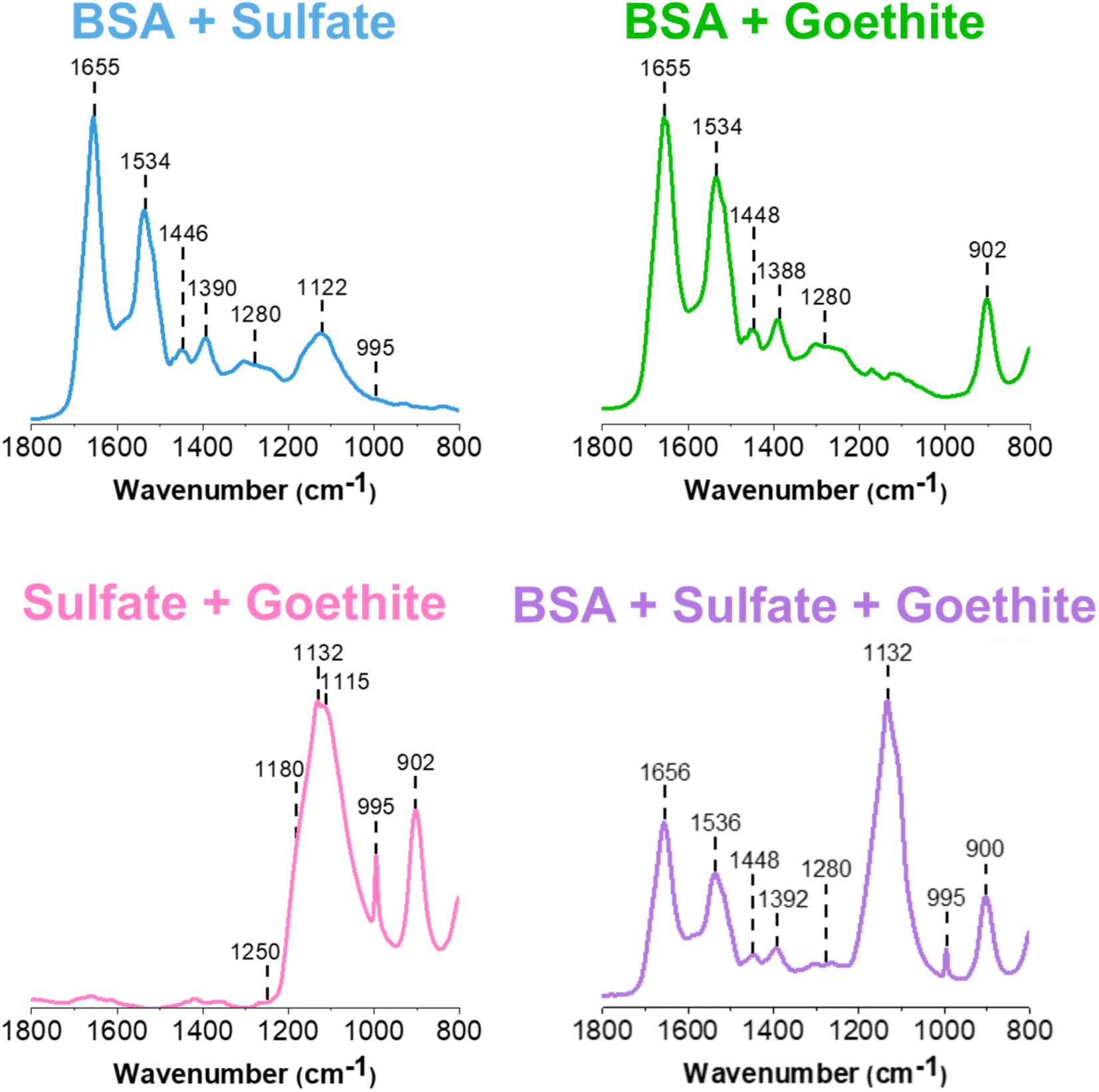
The amide I and II vibrational modes, representative of proteins, are observed at ca. 1655 cm−1 and 1540 cm−1, respectively, in the AFM-PTIR spectra.33,34 It is seen in the sulfate spectrum that there is a strong absorption band at 1132 cm−1, which corresponds to the υ3 asymmetric sulfate stretching vibration. Sulfate speciation and symmetry are influenced by environmental factors such as phase and hydration state. In its aqueous form, sulfate oxyanions are tetrahedral in geometry, belonging to the Td symmetry. Within these oxyanions, there is a nondegenerate, symmetric vibration, υ1, and a triply degenerate, asymmetric vibration, υ3, in the spectral region from ca. 975 cm−1 to 1100 cm−1. Here, the υ1 band is IR active at 994 cm−1, whereas the υ3 band is IR active at 1132 cm−1.23,27,35,36
The peak positions shown in Fig. 1 are attributed to a dehydrated film. In particular, Fig. 1a shows a broad, asymmetric peak at 1132 cm−1 with a shoulder at 1168 cm−1 and a sharp peak at 994 cm−1. These changes are related to the distortion of the tetrahedral geometry as well as the shift to 994 cm−1, for the nondegenerate υ1 band, as sodium sulfate becomes more crystalline.23,35,36 For the goethite thin film, the ATR-FTIR spectrum shows two sharp peaks present at 805 cm−1 and 900 cm−1.37 These bands are attributed to the in/out-of-plane bending modes of the bulk surface hydroxyls within the iron oxyhydroxide. The AFM-PTIR spectrum shows peaks at the same wavenumbers, but the full peak at 805 cm−1 cannot be seen due to limited range of the laser being used, starting at 800 cm−1. The vibrational peak assignments of these spectra are summarized in Table 1.
| Wavenumber (cm−1) | Vibrational peak assignment | |
|---|---|---|
| BSA | 1654 to 1656 | Amide I |
| 1534 to 1540 | Amide II | |
| Sulfate | 1132 to 1140 | Asymmetric sulfate stretch (υ3) |
| 994 to 995 | Symmetric sulfate stretch (υ1) | |
| Goethite | 805 | In-plane O–H bend |
| 900 | Out-of-plane O–H bend |
Further chemical analysis using AFM-PTIR spectroscopy of these single component thin films are shown in Fig. 1B. It should be noted that AFM-PTIR spectra are not identical to the ATR-FTIR spectra shown in Fig. S1.† For example, in the BSA spectra, differences in the 1655/1540 cm−1 peak ratio, i.e. the ratio in intensity of the Amide I/Amide II band can be observed. Since AFM-PTIR point spectra probe the “local” environment, it can give slightly different spectral measurements compared to the bulk ATR-FTIR spectra. The two techniques also operate under different principles, one being a photothermal technique and the other, an optical absorption technique. This can result in peaks that differ some in intensity and frequency. However, it is important to recognize that they are in good agreement with each other.
In addition to point spectra, AFM-PTIR spectroscopy offers physical insight into these films as shown in Fig. 1c and d. As can be seen by the height images in Fig. 1c, these films range in thickness from several hundreds to thousands of nanometers. Images are shown in both height and deflection mode, and two-point spectra were taken on each 5 × 5 μm image-one spectrum on the film and the other on the substrate. In addition, the morphologies of each of the thin films are seen to be distinct from each other. While the BSA film is smooth and has a monotonic increase in height, the sodium sulfate film is over a smaller region and is thicker on the order of 1.4 μm at the highest point. The BSA film has a root mean square roughness of 240 nm, and can also be qualitatively examined by the smoothness of the line scan across the region. It should also be made clear that the BSA height profile begins at 25 nm in the film. The height profile increases steadily in thickness and the line is straight, contrasted to that of the sulfate film where there are significantly tall features with a more jagged profile. This suggests that aggregates in the sulfate image are a result of sodium sulfate crystallization as the film dried out from an initial, aqueous solution.38 For the goethite thin film, individual rods can be resolved in the goethite thin film, consistent with the known morphology of goethite particles.37,39 A closer look at individual goethite nanoparticles can be found in Fig. S2.† One consistent behavior between all three films is the gradual increase in film thickness from the edge of the film towards the center due to the drop-cast and drying methods was used to prepare these films.
Fig. 2 shows an analysis of a BSA thin film as a function of thickness across the region of interest. Although the BSA thin film appears smooth and homogenous, a larger image size was specifically chosen to measure point spectra with increasing film thickness across the film. Fig. 2b shows a compilation of 15 point spectra taken across the 30 × 30 μm film, as a function of wavenumber, absorbance, and thickness. The limit of detection as well as detection saturation can be determined to be ∼50 nm and ∼400 nm, respectively. This was determined by comparing the signal-to-noise ratios (SNR) of groups of spectra. For example, the limit of detection for this particular film was determined by a significant decrease in the SNR, whereas the detection saturation was determined by a significant increase in SNR.40,41 Furthermore, these estimates can be quantified by plots that show the relationship between increasing signal and BSA film thickness. Fig. 3c shows how these plots are nonlinear. Instead of the signal increasing linearly with increasing thickness, the signal increases monotonically with film thickness initially and then saturates.42 The dashed lines in these plots represent linear fits for film thicknesses below 400 nm, prior to the plateau observed for film thicknesses above ca. 400 nm. The differences in the slopes of each line directly correlates with the relative intensity of each of the vibrational modes analyzed.
 | ||
| Fig. 2 Analysis of a thin film of 1 mg mL−1 BSA. The 2D height and corresponding deflection images are shown in (a). The AFM image is taken at the edge of the film in order to show the clear contrast between on and off the film. (b) Is a compilation of 15 AFM-PTIR spectra plotted as a function of absorbance, wavenumber, and film thickness. The relationship between the absorbance intensity of the different vibrational bands found in BSA is shown as a function of thickness in (c). The darker, dotted lines represent a linear fit to the portion of the spectra below 400 nm as the signal saturates at ca. 400 nm. The signal-to-noise ratio falls below 3 at a thickness of 50 nm. | ||
 | ||
| Fig. 3 Normalized ATR-FTIR spectra of two and three-component thin films of BSA, sodium sulfate, and goethite. | ||
ATR-FTIR spectroscopy of multi-component thin films
For multi-component thin films, mixtures were prepared in solution and then dried to form thin films containing different components (see Experimental methods for more detail on sample preparation). The ATR-FTIR spectra of these different films are shown in Fig. 3. Spectral signatures of each of the components are clearly identified, as confirmed by the peak assignments presented in Table 1.First, it is important to establish any similarities and differences between the single component spectra and these two and three component spectra. The BSA and sulfate spectrum (blue) presented in Fig. 3 contains the characteristic features of its two components. BSA, as indicated by the 1655 cm−1 and 1534 cm−1 bands, and sulfate, as indicated by the 1122 cm−1 band. Compared to the reference single component BSA and sodium sulfate spectra presented in Fig. S1,† the major difference is in the shift and broadening of the sulfate band, which shifts from 1132 cm−1 to 1122 cm−1 and the relatively weak presence of the 995 cm−1 band. Given the IR spectrum shown in Fig. 3, it is difficult to support the presence of the same, sharp 995 cm−1 band. However, its inconsistent presence in sulfate-containing systems suggest that it is most likely related to changes in the symmetry of the anion, protonation state and intermolecular interactions. While the interaction of BSA and sodium sulfate has not been studied extensively, spectrally, the speciation of sodium sulfate, such as its crystallization, appears to be affected by the presence of the protein. Furthermore, the broadening suggests there may be some interaction between the negatively charged sulfate anion with positively charged lysine and arginine residues on the outer surface of the protein. Hydrogen bonding and other non-ionic interactions most likely comprise these interactions.43–45
Comparing the BSA and goethite spectrum (light green) in Fig. 3 to that of BSA and goethite alone (Fig. S1†), there are no differences in the relative peak intensities and ratio of Amide I/Amide II in the BSA region. The presence of goethite is indicated by the 900 cm−1 peak and the ca. 800 cm−1 peak shifts below the region of the laser used for these measurements. Overall, this assessment aligns with what has been proposed by Schmidt et al., where it was suggested that a deconvolution of the Amide I and Amide II peaks has indicated BSA adsorption onto goethite as a result of conformational changes, that cannot be distinguished within the broad band.15 Furthermore, past studies have also shown BSA adsorption onto iron-containing mineral oxide surfaces from conformational changes, mainly via electrostatic interactions.46–48
For the sodium sulfate and goethite spectrum (pink), there are some notable changes in the sulfate region compared to the BSA/sodium sulfate spectrum discussed above. For example, there is clear evidence for a disruption in the sodium sulfate symmetry, as seen by the sharp feature at 995 cm−1. Another change is the relative enhancement of the intensity of the sulfate peak at 1132 cm−1, along with broadening of the peak compared to the reference sulfate spectrum presented in Fig. S1.† This broadening serves as evidence of coordinated sulfate to goethite nanoparticles, as the band is more symmetric than asymmetric. Previous studies have associated sulfate adsorption onto goethite with monodentate or bidentate bridging mechanisms, which is accompanied by the splitting of the major v3 symmetric stretch. While these bands are not clearly resolved, there is an emergence of shoulders in the spectra at 1115 cm−1 and 1180 cm−1 along with a major 1132 cm−1 band which suggests a combination of sulfate crystallization as well as inner-sphere bridging occurring at the surface.23,36
Observing an inner-sphere binding mechanism at a circumneutral pH may appear to be inconsistent with past studies which have concluded that sodium sulfate adsorption in aqueous conditions follows an inner-sphere binding mechanism in acidic environments, and an outer-sphere binding mechanism as pH increases. However, the reported findings here are from dried, thin films of sulfate and goethite at a circumneutral pH, which is a non-acidic environment. For dried films such as those that are presented in this manuscript, the effect of dehydration on potential interactions must be considered.23 Paul et al. has reported that hydrated mineral surfaces can become more acidic as a function of drying, indicating that the dehydration can result in a potential acidification of the goethite thin film. It was proposed that this is a result of the dissociation of water adsorption on polarizable metal cation sites, which, in this case, would be Fe.24,49 Therefore, this subsequent proton accumulation is likely responsible for creating a slightly more acidic environment, allowing for such binding to occur. The differences between coordinated and non-coordinated sulfate can be clearly identified from differences in broadness and presence of shoulders between the ATR-FTIR spectra of sodium sulfate and sodium sulfate/goethite in Fig. S1† and 3 respectively. However, within the sodium sulfate/goethite thin film, there could also exist free, “uncoordinated sulfate” (used as a general term here to distinguish from surface coordinated and crystalline sulfate) within different microenvironments present in the film. This serves as evidence as to why utilizing microspectroscopic tools such as AFM-PTIR spectroscopy is crucial to further exploring these heterogeneities.
The three component ATR-FTIR spectrum (purple) shown in Fig. 3, clearly shows each of the three components as evident by distinct absorption bands: BSA at 1656 cm−1, sulfate at 1132 cm−1, and goethite at 900 cm−1. The most prominent difference between this multicomponent spectrum compared to both of its single and two component systems is again the clear enhancement of the sulfate peak indicating interactions with goethite as noted above as well as crystallization of sodium sulfate, as discussed for the two-component spectrum of sulfate and goethite. The shape and intensity of the sulfate region centered at 1132 cm−1 does not appear to resemble exactly the two component systems but instead appears to be a close combination of the different spectra, suggesting that specific interactions may be occurring in these films. The ATR-FTIR technique can provide important chemical information on the macroscopic adsorbate–mineral interactions, including the type of coordination occurring on the surface of goethite. However, this technique is limited in detecting the heterogeneities present across the system, which are features that can only be exploited via micro-spectroscopic probes such as AFM-PTIR spectroscopy, increasing the level of chemical complexity that can be achieved.
AFM-PTIR spectroscopy of a BSA and sodium sulfate thin film
In Fig. 4, data collected for a thin film of BSA and sodium sulfate with AFM-PTIR spectroscopy are shown. The height and deflection image in Fig. 4a show an interesting tee-shaped aggregate that has arranged itself onto a smoother and thinner underlying film. This aggregate is the main contributor to the tallest feature of this image, with a thickness of 525 nm. This appears to be crystalline with jagged edges, of varying sizes and shapes, that resemble those previously observed from the sodium sulfate thin film. In this 5 × 5 μm image, a total of 4 point spectra were taken – 3 on the film itself and one on the substrate. | ||
| Fig. 4 AFM-PTIR images, point spectra and spectral maps of a BSA and sodium sulfate thin film. (a) Shows the height and deflection image of the 5 × 5 μm region and (b) represents the height profile across the region. (c) Shows point spectra taken across the surface, where the colors of the dots on the deflection image correspond to line colors of the corresponding PTIR spectra. In particular, Film 1, Film 2 and Film 3 seem to suggest different films. Spectral heat maps are seen in (d), at representative wavenumbers of sulfate at 1155 cm−1 and BSA at 1650 cm−1. | ||
In Fig. 4, each PTIR spectrum shows various combinations of BSA and sodium sulfate spectral features. However, the point spectra shown in Fig. 4c show much more variation in not only the intensity of the peaks but also the shape and ratio between these two components. For spectral mapping, 1155 cm−1 was selected because it was the common band present in all of the sulfate-attributed spectra, that also included the 1130 cm−1, which has been shown to be representative of both crystalline and non-coordinated sulfate. Maps at representative wavenumbers confirm that the T-shaped aggregate is indeed sulfate while the smoother areas of the film are BSA.
Each of the point spectra taken on the film provides unique information. The dark blue point spectrum indicates a region that is only BSA, which is consistent as the red “hot” spot on the 1650 cm−1 map as well as the position on the smooth film as visualized by the deflection image. However, the remaining two spectra (light blue and teal) are taken on different parts of the sulfate aggregate. While the light blue spectrum shows a singular, sharp peak at 1155 cm−1, the teal spectrum shows a very broad double peak. The differences in relative peak intensity, peak shift, and peak shape in the sulfate band suggest changes in sulfate speciation and interaction in the presence of and with BSA, respectively. AFM-PTIR spectroscopy allows for infrared mapping by tuning and fixing the laser at a particular wavenumber of interest as it scans across a particular region. While the regions color-coded in red indicate high relative absorbance, the regions color-coded in blue indicate low to no absorbance at the particular wavenumber of interest. Using this information, a heat map is generated.
From this information, we can deduce the presence of an interaction between these two components, as discussed previously from similar observations made from sulfate peak broadening and lack of variability in the reference sulfate spectra, as seen in ATR-FTIR spectra presented in Fig. 1. The physical, crystalline features on this film suggest the presence of sodium sulfate crystallinity. However, the absence of a strong, sharp 995 cm−1 peak, which has been used to identify sodium sulfate crystallinity, is clear. While this result appears to be contradictory to previous statements, the absence of this peak can be attributed to three potential reasons: (1) there are weaker interactions present, that do not alter the morphological features of the sodium sulfate crystals or that preserves the phase; (2) AFM-PTIR spectroscopy is unable to detect detailed physical differences between crystalline sulfate and uncoordinated sulfate and; (3) sulfate crystallinity is much more complex than is currently known, and should not absolutely be defined only from a chemical perspective, such as the presence of the 995 cm−1 peak, but rather as shifts in the peak location and shape, as a whole. Moreover, it is important to highlight this heterogeneity that is detected via AFM-PTIR spectra, differing vastly from what could be discerned from the ATR-FTIR spectrum in Fig. 3. There is also spectral evidence of BSA in each of those points, which suggests two possible modes of microphase segregation: (1) sodium sulfate crystals have arranged themselves on top of the BSA film and (2) there are smaller aggregates of BSA embedded in the larger, sodium sulfate crystals.
AFM-PTIR spectroscopy of a BSA and goethite thin film
The ability to take localized, point spectra on nanometer length scales allows the investigation of heterogeneity of mineral-containing thin films. BSA and goethite films are shown in Fig. 5. The images and line scan in Fig. 5a and b show the presence of goethite as well as an increase in the height image from 200 nm to 600 nm, respectively. This increase in thickness can be attributed directly to the presence and aggregation of rod-like goethite nanoparticles, which can be resolved in the corresponding deflection image and offers more contrast of the region of interest. It can be seen that the goethite nanoparticles cluster in different regions including on the edge of the thin film, giving insight into some of the nanoscale phase segregation occurring in these thin films. | ||
| Fig. 5 AFM-PTIR images, point spectra and spectral maps of a BSA and goethite thin film. (a) Shows the height and deflection image of the 5 × 5 μm region as well as the height profile in (b). (c) Represents the point spectra taken both on and off the film. The colors of the dots present on the deflection image correspond to the location in which the point spectra were taken. Additional spectra presented are reference PTIR spectra. AFM-PTIR maps are shown in (d), at representative wavenumbers of goethite at 900 cm−1 and BSA at and 1640 cm−1. Examples of protein-rich and mineral-rich regions are also shown in (d). | ||
Fig. 5c shows point spectra both on and off the film; the presence of the bands from 1390 cm−1 to 1655 cm−1 and at 900 cm−1 indicates BSA and goethite, respectively. Using the peak positions identified in each spectrum, infrared maps of the same 5 × 5 μm region were generated, as shown in Fig. 5d. Here, we are able to confirm that the taller features on the film are attributed to goethite aggregation, as indicated by the red “hot” spots from the 900 cm−1 map. The 1650 cm−1 map represents BSA, and we see how BSA appears to concentrate specifically in regions where goethite aggregation is present, but not homogenously across the entire surface. In comparison with the BSA FTIR spectrum, the light green AFM-PTIR spectrum shows no qualitative difference in the peak shape, peak shift, or Amide I/Amide II ratio but allows for us to conclude that this adsorption process is heterogenous, with specific areas present that are richer in BSA.
AFM-PTIR spectroscopy of a sodium sulfate and goethite thin film
It is evident from ATR-FTIR spectroscopy that sulfate coordination and crystallization can occur in multicomponent microenvironments. In order to further probe the heterogenous nature of this interaction, AFM-PTIR spectroscopy was employed as shown in Fig. 6.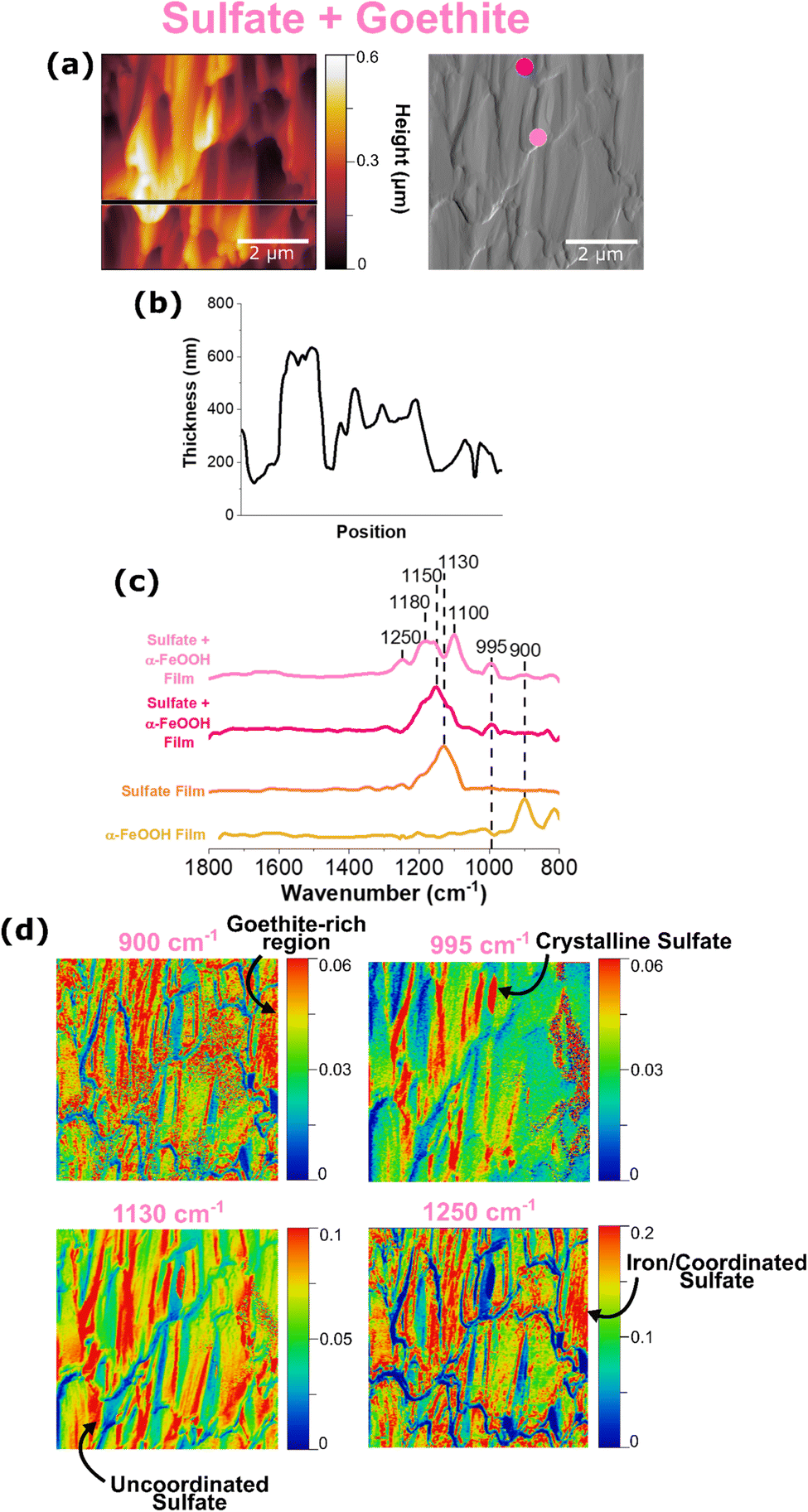 | ||
| Fig. 6 AFM-PTIR images, point spectra and spectral maps of a sodium sulfate and goethite thin film. (a) Shows the height and deflection image of the 5 × 5 μm region and the height profile in (b). Point spectra taken across the film are seen (c), where the colors of the dots on the deflection image correspond to spectra. PTIR spectral maps are shown in (d), at representative wavenumbers of goethite at 900 cm−1, crystalline sulfate at 995 cm−1, uncoordinated sulfate at 1130 cm−1 and iron-coordinated sulfate at 1250 cm−1. | ||
The height and deflection image in Fig. 6a show random aggregations, that appear to resemble a combination of sodium sulfate crystals due to its jagged edges, as well as goethite nanoparticles due to the rod-like shape. It should be noted that the height profile does not start at 0 nm because this image was collected in the center of a sulfate-goethite thin film, rather than the edge, where the bare substrate is simultaneously exposed. For this thin film, point spectra were taken at 2 different regions, as denoted by the light and dark pink dots on the deflection image. Similar to what has been observed (vide supra), each point spectrum represents sulfate in different environments. While the light pink spectrum represents uncoordinated sulfate, the dark pink spectrum provides evidence for adsorbed sulfate with inner-sphere coordination to goethite nanoparticles. This evidence is crucial in understanding the heterogenous existence of both forms of sulfate within a thin film, confirming the presence of both micro- and nano-environments in these films.
It is important to note how the distribution of surface-coordinated sulfate at 1250 cm−1 and uncoordinated sulfate at 1130 cm−1 differ. The prevalence of the 1130 cm−1 free sulfate map across the entire region indicates the presence of free sulfate evenly distributed across the surface. The distribution of coordinated sulfate as indicated by the 1250 cm−1 spectral map aligns with the distribution of goethite nanoparticles, confirming a physical interaction between the two species. In addition, a chemical interaction between the two is evidenced by discussions reported by previous studies, of strong electrostatic interactions between sulfate and iron oxide surfaces.17,22,24 Therefore, it is important to examine the sodium sulfate and goethite ATR-FTIR spectra in Fig. 3 and compare them to the AFM-PTIR spectra shown above.
The spectrum shown in light pink has a singular peak that is well in agreement with what is known for non-coordinated sulfate. It should be noted that there is a slight upward shift in the wavenumber, compared to the AFM-PTIR spectrum for a reference sodium sulfate thin film as shown in Fig. 1a. This shift can suggest outer-sphere mechanisms or other, non-coordinating interactions that are causing slights shifts to the molecular symmetry of the free sulfate molecules. The ATR-FTIR spectrum shown in Fig. 3 is an average of many spectra taken across the bulk, thin film surface, which serves as an explanation for any differences between that and the localized PTIR spectra seen in Fig. 6. The differences in each of the localized PTIR spectra taken across this region is shown to highlight the heterogeneity of the interactions of sulfate within the sample.
Previous studies have utilized the relationship between molecular vibrational modes and molecular symmetry to specify the potential inner-sphere binding mechanisms that can occur in these systems. Upon coordination to Fe, the number of bands the υ3 asymmetric band splits into can help determine whether or not monodentate bridging or bidentate bridging can occur.23 Looking closely at the spectrum shown in dark pink, there are well-resolved shoulders 1100 cm−1, 1180 cm−1, and 1250 cm−1, which is representative of the υ3 band splitting into three indicating bidentate binuclear bridging, where the two Fe ions are each coordinated to an oxygen atom in the sulfate molecule.23,25 In addition, the strong IR activity of the nondegenerate υ1 band at 995 cm−1 lines also suggest the presence of crystalline sulfate, which can be deduced as the large, jagged features seen in the AFM image.36 However, the various peaks shown in dark pink is slightly more complex, as it can correspond to a few different binding mechanisms. For example, there exists a peak at 1180 cm−1, which has also been suggested by Peak et al. to be the result of a hydrogen bond between a sulfate and a monodentate sulfate.23 In addition, the PTIR spectrum also shows a 1250 cm−1 peak, which has been hypothesized as a change in sulfate speciation, from mono or bidentate bridging sulfate to mono or bidentate bridging bisulfate.22,24,27,50 While AFM-PTIR spectroscopy has proven to be a unique and valuable technique in identifying the presence of multiple sulfate-goethite interactions, it is important to note that further investigation is needed to explicitly identify every type of sulfate-goethite interaction. Although the overlapping peaks are difficult to delineate from one another, changes in point spectra at differing locations on the film confirm the presence of heterogenous sulfate behavior. A summary of these sulfate interactions is shown in Table 2.
| Wavenumber (cm−1) | Type of sulfate | Figure reference | |
|---|---|---|---|
| AFM-PTIR | ATR-FTIR | ||
| 995, 1130 | 994, 1132 | Crystalline sulfate | Fig. 1, S1,† 3, 6 and 7 |
| 1130–1155 | 1122, 1130 | Free, uncoordinated sulfate; outer-sphere sulfate | Fig. 1, S1,† 3, 4, 6 and 7 |
| 1100–1180, 1250 | 1115–1180, 1250 | Inner-sphere sulfate | Fig. 1, S1,† 3, 6 and 7 |
AFM-PTIR spectroscopy of a BSA, sodium sulfate, and goethite thin film
Data collected for a multi-component thin film in which all three components are present are shown in Fig. 7. As can be seen by the height profiles presented in Fig. 7b, this multi-component film contains features that are smooth with randomly dispersed rougher, aggregates. | ||
| Fig. 7 AFM-PTIR images, point spectra and spectral maps of a BSA, sodium sulfate and goethite thin film. (a) Shows the height and deflection image of the 5 × 5 μm region and the height profile in (b). Point spectra taken across the film are seen in (c), where the colors of the dots on the deflection image correspond to the line color for each of the spectra. AFM-PTIR maps are shown in (d), at representative wavenumbers of goethite at 900 cm−1, crystalline sulfate at 995 cm−1, non-coordinated sulfate at 1130 cm−1, coordinated sulfate at 1260 cm−1, and BSA at 1655 cm−1. | ||
Similar to the two-component systems shown previously, heterogeneity in the AFM-PTIR spectra can be seen by the differences in the shape and frequencies observed in the sulfate region, as seen by the light and dark purple spectra. The purple ATR-FTIR spectrum of the multi-component system presented in Fig. 3 shows a very broad and intense sulfate spectrum, compared to what is observed in Fig. 7b. The variation between the intensity ratios of these bands across this region not only indicates heterogeneity, but also emphasizes the importance of spatially, well-resolved techniques such as AFM-PTIR spectroscopy in providing data to detect these differences occurring on the micro- and nanoscale level.
In order to further probe the sulfate region, three different spectral regions were selected to map this 5 × 5 μm region. Consistent with the previous maps, crystalline sulfate arising from sulfate symmetry distortion was represented at 995 cm−1, non-coordinated sulfate at 1130 cm−1, and coordinated sulfate at 1260 cm−1. On the microscale level, all three maps appear to look very similar; however, it is important to focus on the nanoscale features of these maps. There are significantly more red-colored spots on the top region of this image for the 995 cm−1 spectral map, indicating localization of crystalline sulfate. This differs from the 1130 cm−1 map, representative of uncoordinated sulfate, which is more present in the bottom half of the region. We can also see the similarities between the 900 cm−1 and 1260 cm−1 map, which is consistent with what is expected since the latter map represents coordinated sulfate onto iron nanoparticles, as indicated by the former. From the prominent 1260 cm−1 peak in the AFM-PTIR spectra as well as these maps matching up to goethite-rich areas, it can be concluded that sodium sulfate interacts with goethite via bidentate binuclear bridging.22 Furthermore, the red, hot spots in the BSA map at 1655 cm−1 are consistent with regions rich in BSA that form smooth domains within these films.
Conclusion
In this study, environmentally relevant thin films containing biological components, oxyanions, and minerals were analyzed via a novel approach utilizing micro-spectroscopic probes. While vibrational techniques such as ATR-FTIR spectroscopy provide hints of these different interactions, it is important to recognize the added value of micro- and nanoscale techniques. This study shows AFM-PTIR spectroscopy to be a useful measurement tool in the distribution of interactions occurring along mineral surfaces in the environment, through examinations of micro-structural heterogeneities. This study has depicted that by using AFM-PTIR spectroscopy complemented with ATR-FTIR spectroscopy, the complex nature of the chemistry occurring in these systems can be unraveled one at a time, to better understand the various environmental processes these species undergo in the environment. However, limitations in delineating overlapping spectral peaks, has indicated the need for further experimental development and utilization of other analytical tools to accurately determine the exact mechanisms and phase transitions occurring in these thin film. With more information on the chemistry occurring at the micro- and nanoscale level, a greater understanding of environmental processes can be achieved.Conflicts of interest
The authors claim no known conflict of interest to declare.Acknowledgements
The research reported here was funded in whole or in part by the Army Research Office/Army Research Laboratory via grant #W911NF-19-1-0078 to the University of California, San Diego. Any errors and opinions are not those of the Army Research Office or Department of Defense and are attributable solely to the author(s).References
- E. K. Coward, T. Ohno and D. L. Sparks, Environ. Sci. Technol., 2019, 53, 642–650 CrossRef CAS PubMed.
- A. Bhattacharyya, M. P. Schmidt, E. Stavitski and C. E. Martínez, Org. Geochem., 2018, 115, 124–137 CrossRef CAS.
- Y. S. Jun, B. Lee and G. A. Waychunas, Environ. Sci. Technol., 2010, 44, 8182–8189 CrossRef CAS.
- D. Kim and V. H. Grassian, ACS Earth Space Chem., 2022, 6, 81–89 CrossRef CAS.
- G. E. Brown, A. L. Foster and J. D. Ostergren, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 3388–3395 CrossRef CAS.
- G. E. Brown, V. E. Henrich, W. H. Casey, D. L. Clark, C. Eggleston, A. Felmy, D. W. Goodman, M. Grȁtzel, G. Maciel, M. I. McCarthy, K. H. Nealson, D. A. Sverjensky, M. F. Toney and J. M. Zachara, Chem. Rev., 1999, 99, 77–17 CrossRef CAS PubMed.
- J. D. Kubicki, L. M. Schroeter, M. J. Itoh, B. N. Nguyen and S. E. Apitz, Geochim. Cosmochim. Acta, 1999, 63, 2709–2725 CrossRef CAS.
- I. A. Mudunkotuwa, A. Al Minshid and V. H. Grassian, Analyst, 2014, 139, 870–881 RSC.
- I. A. Mudunkotuwa and V. H. Grassian, Environ. Sci.: Nano, 2015, 2, 429–439 RSC.
- I. Sit, H. Wu and H. Grassian Vicki, Annu. Rev. Anal. Chem., 2021, 14, 489–514 CrossRef CAS PubMed.
- H. Wu, L. Huang, A. Rose and V. H. Grassian, Environ. Sci.: Nano, 2020, 7, 3783–3793 RSC.
- H. Wu, N. I. Gonzalez-Pech and V. H. Grassian, Environ. Sci.: Nano, 2019, 6, 489–504 RSC.
- X. Lin, G. Wei, X. Liang, J. Liu, L. Ma and J. Zhu, Minerals, 2021, 11, 1–17 Search PubMed.
- B. E. Givens, Z. Xu, J. Fiegel and V. H. Grassian, J. Colloid Interface Sci., 2017, 493, 334–341 CrossRef CAS PubMed.
- M. P. Schmidt and C. E. Martínez, Environ. Sci. Technol., 2018, 52, 4079–4089 CrossRef CAS PubMed.
- W. X. Schulze, Biogeosciences, 2005, 2, 75–86 CrossRef CAS.
- J. D. Kubicki, K. D. Kwon, K. W. Paul and D. L. Sparks, Eur. J. Soil Sci., 2007, 58, 932–944 CrossRef CAS.
- K. D. Kwon and J. D. Kubicki, Langmuir, 2004, 20, 9249–9254 CrossRef CAS PubMed.
- J. D. Kubicki, K. W. Paul, L. Kabalan, Q. Zhu, M. K. Mrozik, M. Aryanpour, A. M. Pierre-Louis and D. R. Strongin, Langmuir, 2012, 28, 14573–14587 CrossRef CAS.
- E. J. Elzinga and D. L. Sparks, J. Colloid Interface Sci., 2007, 308, 53–70 CrossRef CAS.
- C. M. Eggleston, S. Hug, W. Stumm, B. Sulzberger and M. D. S. Afonso, Geochim. Cosmochim. Acta, 1998, 62, 585–593 CrossRef CAS.
- S. J. Hug, J. Colloid Interface Sci., 1997, 188, 415–422 CrossRef CAS.
- D. Peak, R. G. Ford and D. L. Sparks, J. Colloid Interface Sci., 1999, 218, 289–299 CrossRef CAS PubMed.
- K. W. Paul, M. J. Borda, J. D. Kubicki and D. L. Sparks, Langmuir, 2005, 21, 11071–11078 CrossRef CAS PubMed.
- H. Wijnja and C. P. Schulthess, J. Colloid Interface Sci., 2000, 229, 286–297 CrossRef CAS PubMed.
- X. Wang, Z. Wang, D. Peak, Y. Tang, X. Feng and M. Zhu, ACS Earth Space Chem., 2018, 2, 387–398 CrossRef CAS.
- R. L. Parfitt and R. S. C. Smart, Soil Sci. Soc. Am. J., 1978, 42, 48–50 CrossRef CAS.
- C. E. Martinez, A. W. Kleinschmidt and M. A. Tabatabai, Biol. Fertil. Soils, 1998, 26, 157–163 CrossRef CAS.
- Z. Xu and V. H. Grassian, J. Phys. Chem. C, 2017, 121, 21763–21771 CrossRef CAS.
- J. Han, M. Kim and H. M. Ro, Environ. Chem. Lett., 2020, 18, 631–662 CrossRef CAS.
- A. Dazzi and C. B. Prater, Chem. Rev., 2017, 117, 5146–5173 CrossRef CAS PubMed.
- A. Dazzi, C. B. Prater, Q. Hu, D. Bruce, J. F. Rabolt and C. Marcott, Appl. Spectrosc., 2012, 66, 1365–1384 CrossRef CAS PubMed.
- J. Grdadolnik and Y. Maréchal, Biopolym. - Biospectroscopy Sect., 2001, 62, 54–67 CrossRef CAS.
- M. P. Schmidt and C. E. Martínez, Environ. Sci. Technol., 2018, 52, 4079–4089 CrossRef CAS.
- R. L. Parfitt, Am. Mineral., 1965, 50, 132–147 Search PubMed.
- X. N. Feng, H. N. Chen, Y. M. Luan, S. H. Tan, S. F. Pang and Y. H. Zhang, Chem. Phys., 2014, 430, 78–83 CrossRef CAS.
- D. M. Cwiertny, G. J. Hunter, J. M. Pettibone, M. M. Scherer and V. H. Grassian, J. Phys. Chem. C, 2009, 113, 2175–2186 CrossRef CAS.
- C. Rodriguez-Navarro, E. Doehne and E. Sebastian, Cem. Concr. Res., 2000, 30, 1527–1534 CrossRef CAS.
- G. Rubasinghege, P. K. Kyei, M. M. Scherer and V. H. Grassian, J. Colloid Interface Sci., 2012, 385, 15–23 CrossRef CAS.
- S. Kenkel, A. Mittal, S. Mittal and R. Bhargava, Anal. Chem., 2018, 90, 8845–8855 CrossRef CAS PubMed.
- G. Ramer, V. A. Aksyuk and A. Centrone, Anal. Chem., 2017, 89, 13524–13531 CrossRef CAS PubMed.
- S. Morsch, S. Lyon, S. Edmondson and S. Gibbon, Anal. Chem., 2020, 92, 8117–8124 CrossRef CAS PubMed.
- L. D. Thompson, M. W. Pantoliano and B. A. Springer, Biochemistry, 1994, 33, 3831–3840 CrossRef CAS PubMed.
- M. Yu, T. Zhang, W. Zhang, Q. Sun, H. Li and J. P. Li, Front. Mol. Biosci., 2021, 7, 1–13 Search PubMed.
- I. B. Ustunol, E. K. Coward, E. Quirk and V. H. Grassian, Environ. Sci.: Nano, 2021, 8, 2811–2823 RSC.
- S. Servagent-Noinville, M. Revault, H. Quiquampoix and M. H. Baron, J. Colloid Interface Sci., 2000, 221, 273–283 CrossRef CAS PubMed.
- M. S. C. Barreto, E. J. Elzinga and L. R. F. Alleoni, Sci. Rep., 2020, 10, 1–13 CrossRef.
- F. Liu, X. Li, A. Sheng, J. Shang, Z. Wang and J. Liu, Environ. Sci. Technol., 2019, 53, 10157–10165 CrossRef CAS.
- M. M. Mortland and K. V. Raman, Clays Clay Miner., 1968, 16, 393–398 CrossRef.
- R. L. Parfitt, J. Chem. Soc., Faraday Trans., 1976, 73, 796–802 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3em00005b |
| This journal is © The Royal Society of Chemistry 2023 |