DOI:
10.1039/D2EY00107A
(Review Article)
EES Catal., 2023,
1, 103-116
Evolution of singlet oxygen in peroxymonosulfate activation: a review
Received
18th December 2022
, Accepted 23rd January 2023
First published on 24th January 2023
Abstract
Given that sulfate-radical-based advanced oxidation processes (SR-AOPs) have attracted great attention in recent years, PMS activation has been reckoned to be an alternative method to the Fenton process in environmental remediation. Hence, in this review, we deliberately retrospect and study the recent progress using singlet oxygen (1O2) in SR-AOPs since 2016. First, the fundamental principles and means of characterization of 1O2 are carefully presented. Several categories of activators, such as metal-free catalysts (e.g., carbon tubes and graphene), metal-based (i.e., the elements Co and Mo), and other metallic-based catalysts (i.e., the elements Fe, Mn, and noble metals), are then specifically proposed. Under these circumstances, the mechanisms of 1O2 formation via direct electron transfer, self-decomposition, superoxide radical mediation, oxygen vacancy, perxoymonosulfate radical recombination, etc., have been deliberately summarized in sections. Notably, the research gaps and perspectives for 1O2 in environmental remediation have been critically put forward. Hopefully, this review can offer detailed and theoretical guidance for researchers participating in the study of 1O2 in SR-AOPs.
Broader context
As peroxymonosulfate (PMS) activation has received great attention in recent years, researchers have revealed that not only sulfate radicals (SO4˙−) and hydroxyl radicals (˙OH) are involved the removal of organic contaminants (OCs); singlet oxygen (1O2) also plays a leading role under some circumstances. Due to the complexity of 1O2 formation during sulfate-radical-based advanced oxidation processes (AOPs), and the fact that topic has been less-discussed, it is imperative to discuss the formation mechanism of 1O2 intensively. In this review, we summarize several formation mechanisms based on various nanomaterials and point out the research gaps and perspectives. It is expected that this review will provide some guidance for the future development of 1O2 in AOPs.
|
Introduction
Facing the thriving and prosperous human society are emerging, recalcitrant, and persistent organic contaminants (OCs), which create potential and subtle risks to water and food security, as well as human health. Typically, these OCs are widespread in wastewater; thus, feasible and efficient wastewater treatment technology is of paramount importance to be developed. Advanced oxidation processes (AOPs) are known to be promising choices to remove these adverse pollutants via the production of efficient reactive oxidizing species (ROSs), including hydroxyl radicals (˙OH) and sulfate radicals (SO4˙−).1,2 Among the AOPs, the sulfate-radical-based AOPs (SR-AOPs) have attracted great attention. They are primarily driven by peroxymonosulfate (PMS, KHSO5) and persulfate (PS, K2S2O8) activation.3,4 Specific illustrations of PMS and PS activation to generate SO4˙− and ˙OH radicals are provided in eqn (1) and (2).| | | Mn+ + HSO5− → Mn+1 + SO4˙− + OH− | (1) |
| | | Mn+ + S2O8− → Mn+1 + SO4˙− + SO42− | (2) |
Moreover, in recent years, SR-AOPs have become more convincing and better recognized by researchers than the conventional Fenton process due to the greater stability of the oxidants in solid powder form, facile transportation of common chemicals used in the process, and a more broadly adaptable pH region (pH 2.0–8.0). In contrast, the hydroxyl-radical-dominated Fenton process is usually effective under only acidic conditions. However, SR-AOPs would be hampered by the aquatic environment, such as inorganic anions (e.g., HCO3−, Cl−, and H2PO4−) and natural organic matter (NOM) in bodies of water.5–7 Nevertheless, more research has indicated that singlet oxygen (1O2) is emerging as a predominant ROS in SR-AOPs. The formation of 1O2 occurs mainly through a non-radical pathway, whereas the formation of SO4˙− and ˙OH radicals occurs via a radical pathway.8–12 In comparison to SO4˙− and ˙OH radicals, singlet oxygen possesses advantages: (1) higher selectivity toward electron-rich organic complexes owing to its electrophilic attributes,13,14 so as to preferentially degrade pharmaceuticals and endocrine-disrupting compounds (EDCs) in the presence of inorganic ions and organic compounds13,14 and disinfect toxic pathogens (e.g., E. coli and the MS-2 bacteriophage);15,16 (2) higher resistance to frequently-used free radical scavengers, such as methanol and tert-butanol. Principally, the generation of 1O2 initially involves a photosensitization effect using photo-induced energy transfer to molecular oxygen (O2); additionally, superoxide radicals (O2˙−) often act as intermediates of the singlet oxygen. Alternatively, activating PMS is considered to be a feasible and green chemical process for 1O2 production.
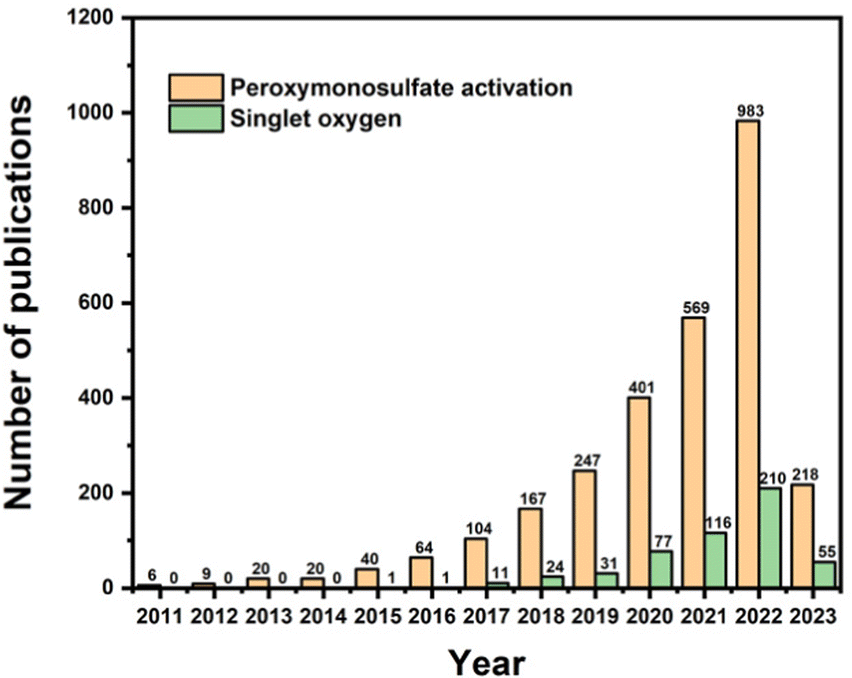
Many correlative studies regarding the production of 1O2 in PMS activation have been reported. It has been summarized that SO4˙− and ˙OH radicals can be obtained by the activation of PMS using ultraviolet (UV) light, heat, and transition metals,17,18etc., whereas singlet oxygen is primarily generated by metal-free catalysts (e.g., carbonaceous materials).19–21 Considering the above information, the interest in PMS activation has increased dramatically, as can be evidently deduced from the number of publications since 2011 depicted in Fig. 1 (from the Scopus database). Specifically, the number of publications concerning PMS activation has grown continuously, and the number of publications concerning singlet oxygen in PMS activation has doubled since 2016. However, it can be observed that before 2016, most research focused on the generation of SO4˙− and ˙OH radicals, while comprehensive analysis of 1O2 formation during PMS activation has rarely been reported. Additionally, current studies rarely summarize the recent progress in 1O2 formation in PMS activation systems. Thus, after a brief introduction of 1O2, this review critically discusses the performance of various types of catalysts and proposes a deep insight into the possible mechanism. Finally, the research gaps and future perspectives are indicated in the last section. This review will provide more theoretical support for future research progress in PMS activation for wastewater treatment.
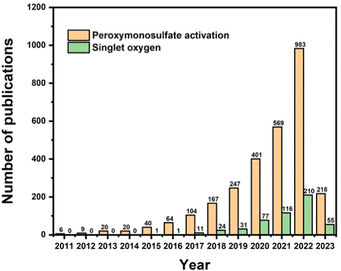 |
| | Fig. 1 Number of papers published on peroxymonosulfate activation and singlet oxygen in PMS activation in the Scopus database. | |
Singlet oxygen in PMS activation
Characters of singlet oxygen
Compared to the SO4˙− (E0 = 2.5–3.1 V) and ˙OH (E0 = 2.8 V) radicals, singlet oxygen, as a mild ROS, possesses a lower redox potential (E0 = 2.2 V).22 Consequently, many scientists have found that singlet oxygen could be resistant to background water matrices, such as NOM and inorganic ions, but its mineralization rates for total organic carbon (TOC) and chemical oxygen demand (COD) are not as good as those of SO4˙− and ˙OH radicals. For the generation of 1O2, several approaches have been summarized: (1) photosensitized excitation of oxygen;23,24 (2) PDS/PMS activation;8,25 (3) periodate;13 and (4) reaction of H2O2 with NaClO.26 Due to its specific properties, singlet oxygen can be applied in tumor therapy,27 organic contaminant degradation,25,28 and pathogenic bacteria inactivation.29
In light of its potential for environmental remediation, the use of 1O2 as a crucial ROS during AOPs and its controversial formation mechanism have been extensively studied. Generally, singlet oxygen can be qualitatively identified using chemical scavenger tests and electron paramagnetic resonance (EPR). Sodium azide (NaN3),30,31 furfuryl alcohol (FFA),13,32 and tryptophan33,34 are commonly adopted as scavengers in chemical probing tests; additionally, spin-trapping agents, such as TEMP, can be used to capture 1O2, forming TEMPO with an intense 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 signal. Moreover, several investigations have showcased the fact that 1O2, as an excited ROS, prefers to approach electron-rich compounds with unsaturated C
:1:1 signal. Moreover, several investigations have showcased the fact that 1O2, as an excited ROS, prefers to approach electron-rich compounds with unsaturated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds and amine and sulfide groups, endowing it with the capability to degrade pharmaceutical and personal care products (PPCPs).5,20,34,35
C bonds and amine and sulfide groups, endowing it with the capability to degrade pharmaceutical and personal care products (PPCPs).5,20,34,35
Metal-free catalysts
Traditionally, the transition metals Fe, Co, and Mn and their oxides have been considered effective activators for the PMS activation process. However, in recent years, metal-free materials have gathered an intensive impetus due to their metal-free nature, sufficient surface area, good biocompatibility, superior stability toward acidic and alkaline conditions, and adjustable electronic and physicochemical characteristics.36,37 More importantly, metal-free catalysts can overcome the drawback of metallic secondary contamination in AOPs. Since Duan et al. discovered that a series of carbonaceous materials, including graphene,38 carbon nanotubes,39 and nanodiamond,40 can act as PMS and/or peroxydisulfate (PDS) activators, this research area has been extensively investigated, and has inspired other researchers involved in this state-of-the-art technology to treat wastewater. In this section, we will talk about the evolution of 1O2 over carbonaceous nanomaterials, for instance, carbon tubes (CNTs), graphene, metal–nitrogen–carbon (M–N–C), single-atom nanomaterials (SACs),41etc. Assuming that in a non-radical reaction system, metal-free catalysts usually serve as an intermediary agent for electron transfer between PMS and organic contaminants, the catalysts contain: (1) delocalized π-electrons;42 (2) defects and vacancies;43 (3) heteroatoms bonded to carbon;44 and (4) pyran-like oxygen functional groups and CO bond.45
Carbon tubes
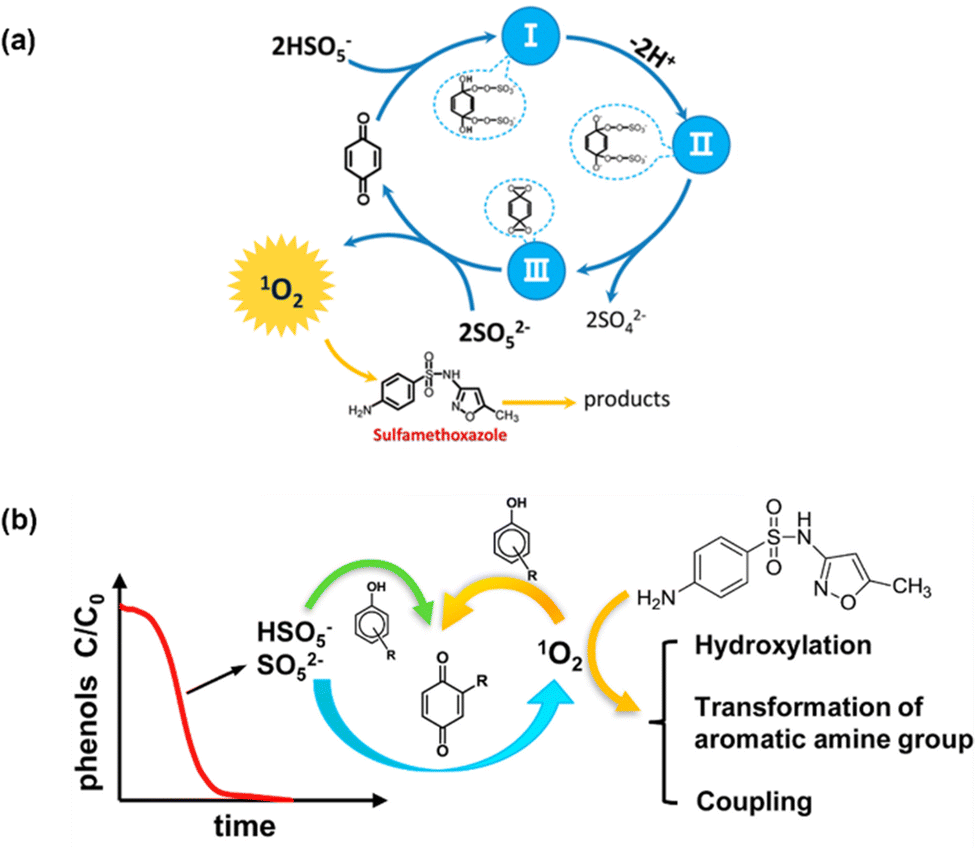
Unlike metallic catalysts, which induce PMS activation relying on SO4˙− and ˙OH radicals, carbonaceous nanomaterials follow three pathways: (1) the radical pathway producing SO4˙− and ˙OH; (2) the non-radical pathway involving complicated complexes, 1O2, and/or direct PMS oxidation; and (3) a combination of radical and non-radical pathways. For example, researchers have also found that organics could mediate PMS activation to produce 1O2; for instance, Zhou et al.46 proposed that the established PMS/benzoquinone (BQ) system demonstrated that BQ could efficiently activate PMS for the degradation of sulfamethoxazole (SMX). In this case, 1O2 played a leading role, and neither ˙OH nor SO4˙− were involved. A possible catalytic mechanism was proposed involving the formation of an intermediate between PMS and BQ, and subsequently the decomposition of this intermediate into 1O2 (Fig. 1a). This mechanism is also referred to in previous works.43,47,48
Zhou et al.49 further employed ten types of phenols as activators in PMS activation systems, including methyl phenol, phenol, methoxy phenols, and dihydroxybenzenes. Consequently, it was found that phenols can effectively activate PMS to generate 1O2 under an alkaline environment (pH 8.5 and 10), with quinone intermediates playing an important role (Fig. 2b). Afterward, Qi et al.50 found that sodium hydroxide itself could act as an activator in PMS activation, and 1O2 stemmed from superoxide radicals (O2˙−), in accordance with eqn (3) and (4). Therefore, in this part, we will systematically discuss the mechanism of the evolution of 1O2 based on carbon-tube-related nanomaterials.
| | | 2O2˙− + 2H+ → 1O2 + H2O2 | (3) |
| | | O2˙− + ˙OH → 1O2 + OH− | (4) |
In particular, the PMS activation efficiency of CNTs is correlated with oxygen functionalities (
i.e., ketonic groups), exposed edge sites, vacancies and heterogeneous atoms doped into the carbon matrix.
51 For example, Yun
et al.52 degraded furfuryl alcohol (FFA) using a CNT/PMS system and proposed singlet oxygenation and mediated electron transfer as plausible nonradical mechanisms. In addition, CNT/PDS systems are complicated; they yielded both SO
4˙
− and
1O
2 as the main ROS catalyzed by quinone groups (
i.e., CNT–C
O).
53 However, some researchers have pointed out that ketonic groups that are initially on a surface or externally introduced onto carbonaceous materials can induce the formation of
1O
2 from PDS without interference of the pH value.
53,54 The presence of C
O bonds in carbon-based catalysts is also reported to enhance the efficiency of PMS activation
via a non-radical pathway.
55,56 For instance, in 2018, Gao
et al.57 fabricated oxygen-doped graphitic carbon nitride (O-CN) and asserted that electron-deficient C atoms were responsible for generating
1O
2. Afterward, Gao
et al. continued to dispel the mist surrounding the generation pathway of
1O
2 involving PMS oxidation over the electron-deficient carbon atoms neighboring graphitic N atoms in nitrogen-doped carbon nanosheets (NCN-900).
58 Distinct from the above-reported mechanism, in their recent work, Sun
et al.59 reported
1O
2 originating from self-decomposition.
Table 1 lists some representative works for the evolution of
1O
2 over carbon tubes, including pure CNTs, modified CNTs, and metallic-atom- and/or metal-oxide-doped CNTs. Correspondingly, Guan
et al.60 utilized CNTs for the degradation of bromophenols (BrPs)
via the nonradical activation of PMS, in which multiple ROS (
i.e., SO
4˙
−, ˙OH, and
1O
2) and PMS-CNT* complexes were the active sites in the PMS/CNT system, unlike in the case of PDS, which involved only nonradical ROS (
i.e., reactive PDS-CNT complexes). Interestingly, Yun
et al.52 drew the same conclusion that the CNTs/PMS system relies on a major radical route (SO
4˙
− and ˙OH)
via electron (from organics) mediation and a minor non-radical route (
i.e.,
1O
2). Heterogenous atom doping is another methodology to manipulate the radical generation pathway. In 2015, Duan
et al.61 for the first time discovered that both radical (
i.e., SO
4˙
− and ˙OH) and non-radical pathways (
i.e., graphitic N) contribute to phenol degradation on N-doped CNTs with PMS activation, which has been intensively studied.
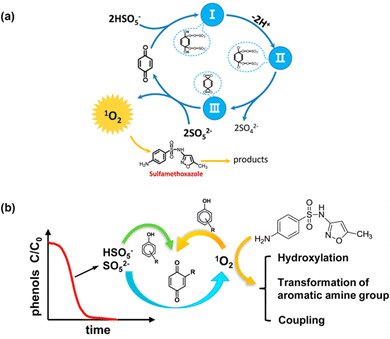 |
| | Fig. 2 (a) The proposed catalytic mechanism in the BQ/PMS system; (b) possible mechanism for several types of phenolic compounds as activators in PMS activation. Copied with permission from ref. 46. Copyright 2018, Elsevier. | |
Table 1 Performance and mechanism for PMS oxidation of pollutants in typical heterogenous systems
| Catalyst |
Target/pollutant |
Degradation rate |
Radicals |
Involved sites |
Ref. |
| Polyimide-modified carbon nanotubes (PI/CNTs) |
Acid Orange 7 (AO7) |
98.9% within 15 min |
1O2 |
N and CO groups |
62
|
|
xFe-N-C |
Phenol |
97% within 10 min |
1O2 |
FeNx sites |
63
|
| Co/NCNT |
Rhodamine B (RhB) |
98% within 7 min |
SO4˙− and 1O2 |
Co species, pyridinic nitrogen, sp2-hybrid carbon, ketonic groups |
64
|
| N/S co-doped ordered mesoporous carbon (NS-CMK-3) |
Acetaminophen (ACT) |
Nearly 100% within 30 min |
1O2 and catalyst-PMS* |
Graphite-N and thiophene-S |
65
|
| Carbon nanotube–magnesium oxide composite (CNTs/MgO) |
Rhodamine B (RhB) |
100% of RhB was degraded in 20 min |
1O2 |
Mg–C and C–O bond, surface-active groups, and oxygen vacancies |
66
|
| Carbon nanotubes (CNTs) |
Bromophenols (BrPs) |
Nearly 100% within 30 min |
SO4˙− and ˙OH, 1O2 |
PMS-CNT* complexes |
62
|
| FeN4-doped carbon nanotubes |
Acid orange 7 (AO7) |
100% within 5 min |
1O2 |
Spin states in FeN4 |
67
|
| Fe3C@BN-C |
Doxycycline hydrochloride (DOX-H) |
91.9% within 120 min |
1O2 |
Fe3C nanoparticles, oxygen functional groups, pyridinic N, pyrrolic N, graphite N, and B, N dopants |
68
|
| Nitrogen-doped carbon nanosheets (NCN-900) |
Bisphenol A |
Complete degradation of bisphenol A within 2 min |
1O2 |
Electron-deficient carbon atoms neighboring graphitic N |
21
|
| Carbon nanotube (CNT) |
Furfuryl alcohol (FFA) |
100% within 60 min |
1O2 and electron |
Electron transfer from organics to PMS |
52
|
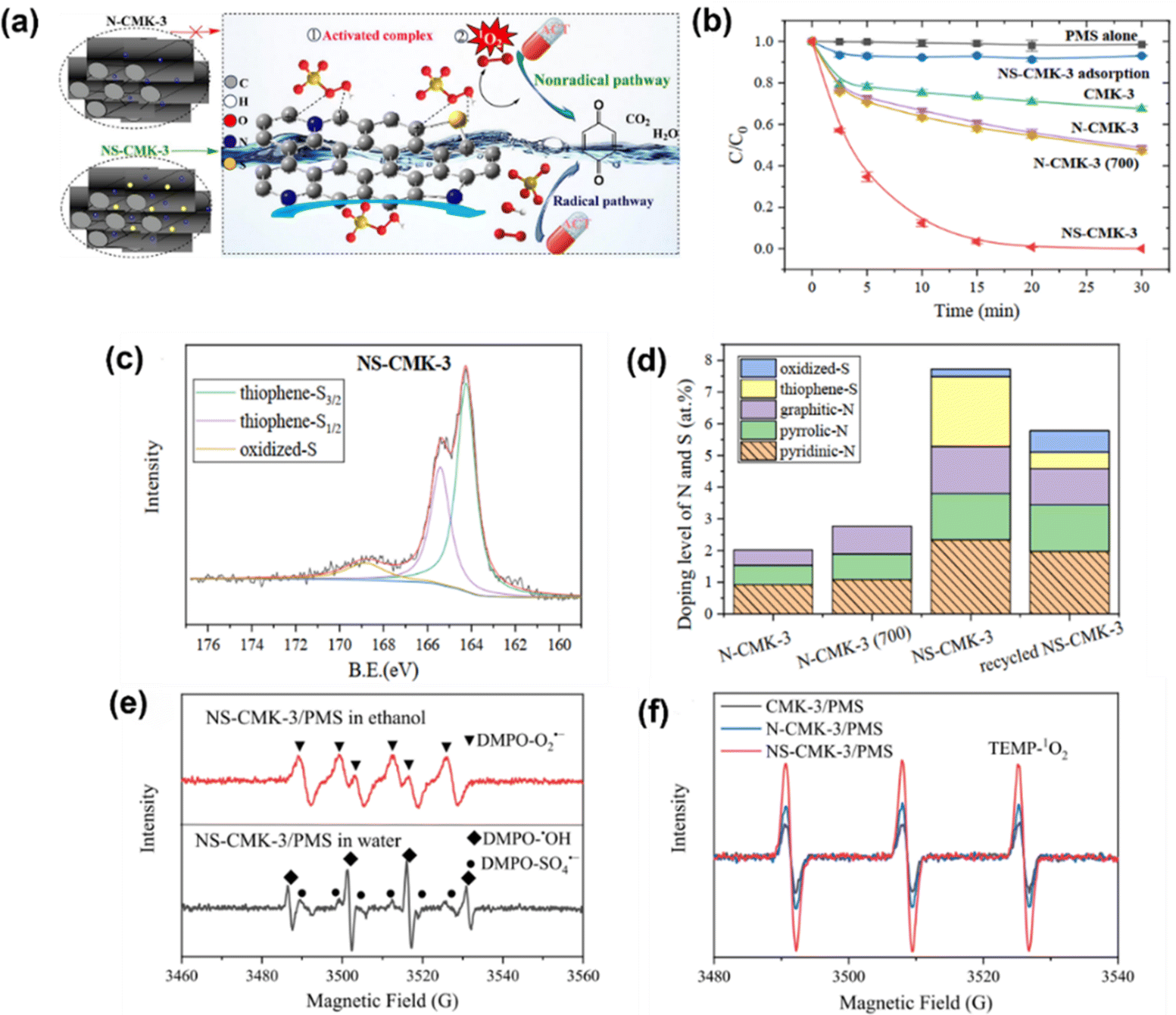
Further, Sun et al.65 prepared N/S co-doped ordered mesoporous carbon (NS-CMK-3) as an activator in PMS activation, which followed a nonradical mechanism. Fig. 3a illustrates the possible mechanism, which is attributed to the synergy of N/S co-doping (i.e., graphite-N and thiophene-S, Fig. 3c and d) contributing to the highly efficient catalytic performance of NS-CMK-3 compared to that of pristine and single N-doped CMK-3 (Fig. 3b). The scavenger tests and EPR spectra (Fig. 3e and f) indicated that NS-CMK-3 triggers two nonradical-dominated oxidations (i.e., singlet oxygen and surface-confined activated PMS). Moreover, Li et al.69 used boron (B)-doped carbon tubes for PMS activation, revealing the intrinsic theory toward the catalytic oxidation of pollutants. The involved ROS, such as SO4˙−, ˙OH, O2˙− and 1O2, were generated after activation by the active sites; additionally, a B-MWNT-PMS* complex was confirmed as an active site via a non-radical pathway and electron transfer mechanism. Although heteroatom doping is well recognized as an efficient protocol because of its capability to modulate the electronic properties of the sp2-hybridized carbon matrix and provide abundant active sites for catalysis,70 the catalytic efficiency of metal-free catalysts is usually impeded by the non-radical process. Regarding this, one promising strategy is to embed transition metal–nitrogen coordination centers into a porous carbon structure via structural engineering.71 In particular, the nitrogen-coordinated iron (FeNx) moiety has aroused particular attraction.72 Thus, for PMS activation, Li et al.73 proposed the use of FeN4-doped carbon nanotubes, which were derived from MIL-101, to degrade acid orange 7 (AO7) in the presence of PMS. The 1O2 stemmed from the self-decomposition of the PMS molecules. In addition, the Fe–N moiety is the active site for the formation of oxygen intermediates. The N atoms in the FeN4 moiety draw electrons from neighboring C atoms, facilitating the production of 1O2. Additionally, Du et al.74 prepared atomically dispersed Fe-Nx site doped N-CNTs derived from ZIF-8; the as-prepared catalyst was used to activate PDS for chloramphenicol (CAP) degradation. Again, a singlet oxygen-dominated process was found in this work, revealing the role of the single-atom site in singlet oxygen evolution (i.e., atomically dispersed Fe-Nx and graphitic N) and offering a new approach for selective removal of trace organic pollutants in complex water matrices.
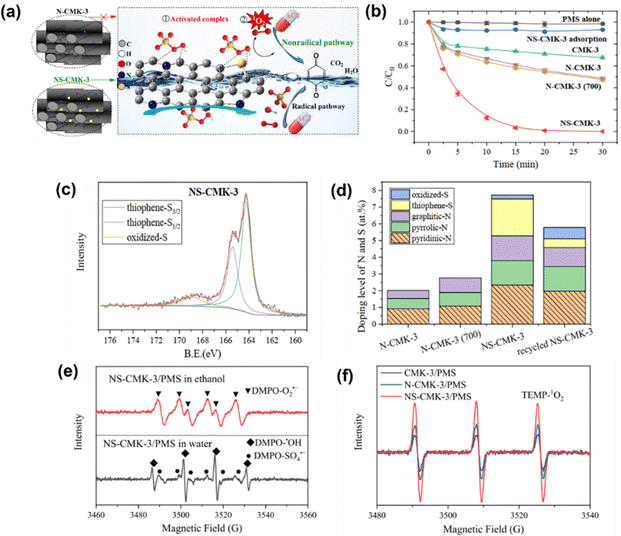 |
| | Fig. 3 (a) Brief illustration of the PMS activation mechanism for N/S co-doped ordered mesoporous carbon (NS-CMK-3). (b) Catalytic performance of acetaminophen (ACT) degradation in various oxidation systems. (c) XPS of S2p for NS-CMK-3, and (d) N and S doping levels of multiple materials. (e) EPR spectra of O2˙−, ˙OH, and SO4˙− and (f) 1O2 in the NS-CMK-3/PMS system. Copied with permission from ref. 65. Copyright 2021, Elsevier. | |
Graphene
Graphene has emerged as a single-atom-thick sheet composed of sp2-hybridized carbon,75 and has received great attention in both the scientific and engineering fields since the first fabrication of this “dream material” through the mechanical exfoliation method in 2004. Due to its high thermal conductivity, good electrical conductivity, abundant specific surface area (SSA), environmentally friendly compatibility and low production cost, graphene has been widely applied in the optical, electronic, biological, and catalytic fields.76–78 In particular, since the discovery of the metal-free activation of persulfates (PMS and PDS) by graphene,38 the mists surrounding carbon catalysis in AOPs have gradually been dispelled. Generally, the PMS and/or PDS activation efficiency are strongly dependent on the inherent complexity of graphene or its derivatives, such as the graphitic degree, surface oxygen groups, and metallic or heterogenous doped atoms.
Hence, in this section, we briefly introduce the graphene-based nanomaterials in SR-AOPs, and systematically unravel their intrinsic nature in catalytic oxidation. Table 2 summarizes some representative materials, including N-doped graphene, single cobalt atoms embedded in N-doped graphene, N,S-doped graphene, etc. Accordingly, it was reported that reduced graphene oxide (rGO) can stimulate PMS to evolve SO4˙− for the degradation of phenolics and dyes; the zigzag edges and ketone (CO) groups at the graphene boundaries are the active sites.38 However, for PDS activation using graphene-like nanosheets (GNS), 1O2 is mainly produced from O2˙−, and the transfer of surface-confined electrons controls the major nonradical oxidation pathway.79 Further, Wang et al.80 found that in the nitrogen-doped graphene (NRGO)/PMS system, NRGO can act not only as a PMS activator but also as an electron transfer mediator. In addition, S and N co-doping exhibits an enhanced synergistic effect for catalysis compared with single doping.81 This reveals that doping rGO at an optimized sulfur loading can effectively break the inertness of carbon systems, activate the sp2-hybridized carbon lattice and facilitate the electron transfer from covalent graphene sheets for PMS activation. A similar result was obtained by Sun et al.,82 in which N doping and additional S doping played pivotal roles in enhancing catalytic performance. Additionally, the nitrogen and boron-co-doped graphene developed by Chen et al.83 shows a synergistic effect between pyridinic N and BC3 (B–C–C–C–pyridinic N), and subsequently has more positively charged carbon atoms as active sites for PMS activation.
Table 2 Performance and mechanism for PMS oxidation of pollutants in typical heterogenous systems
| Catalyst |
Target/pollutant |
Degradation rate |
Radicals |
Involved sites |
Ref. |
| Graphitic N-rich graphene (GNG) |
RhB |
100% RhB removal was achieved within 40 min at 45 °C |
1O2 |
Graphitic N |
84
|
| N-doped graphene |
Phenol |
Nearly 100% within 20 min |
1O2 |
Surface functionalities |
85
|
| Cobalt single atoms embedded in nitrogen-doped graphene (SACo@NG) |
Benzyl alcohol (BzOH) |
Over 90% within 180 min |
1O2, SO4˙− and ˙OH |
Co2+ and Co3+ atoms, as well as the abundant surface N and nucleophilic C = O groups |
86
|
| FeCo@NC |
BPA(bisphenol A) |
Complete degradation within 4 min |
1O2 |
CoN4 site with a single Co atom |
87
|
| Nitrogen-doped graphene (NRGO) |
Sulfamethoxazole (SMX) |
91.7% at 240 min |
1O2 |
Pyrrolic N sites |
80
|
| Nitrogen-doped graphene (N-IrGO) |
Benzophenone-1 (BP-1) |
100% within 60 min |
1O2 |
Graphitic-like nitrogen |
88
|
| Nitrogen–sulfur co-doped industrial graphene (i-rGO-NS) |
Methyl paraben (MP) |
Complete degradation within 5 min |
1O2 |
Graphitic N |
82
|
| Cu-rGO LDH nanohybrid |
Bisphenol A (BPA) |
100% within 40 min |
1O2 |
Electron transfer from PMS to Cu centers |
89
|
| Nitrogen and sulfur co-doped graphene (N, S-G) |
Phenol |
Nearly 100% within 6 min |
1O2 |
Metastable PMS@carbon complex |
90
|
As for single atomic nanomaterials, their high atom utilization efficiency has aroused tremendous interest in the PMS activation process. A Co-based SAC on nitrogen-doped graphene (SACo@NG) material synthesized by Li et al.91 was successfully utilized to selectively oxidize BzOH via PMS activation, and showed superior catalytic performance compared to its counterparts such as NG and CoNP@NG. In the reaction process, the single atomic Co clusters function as active sites via both radical and non-radical pathways, with the latter being preferred. This non-radical pathway is achieved by the adsorption of a BzOH/PMS complex on the surface of SACo@NG followed by electron transfer throughout the carbon matrix.
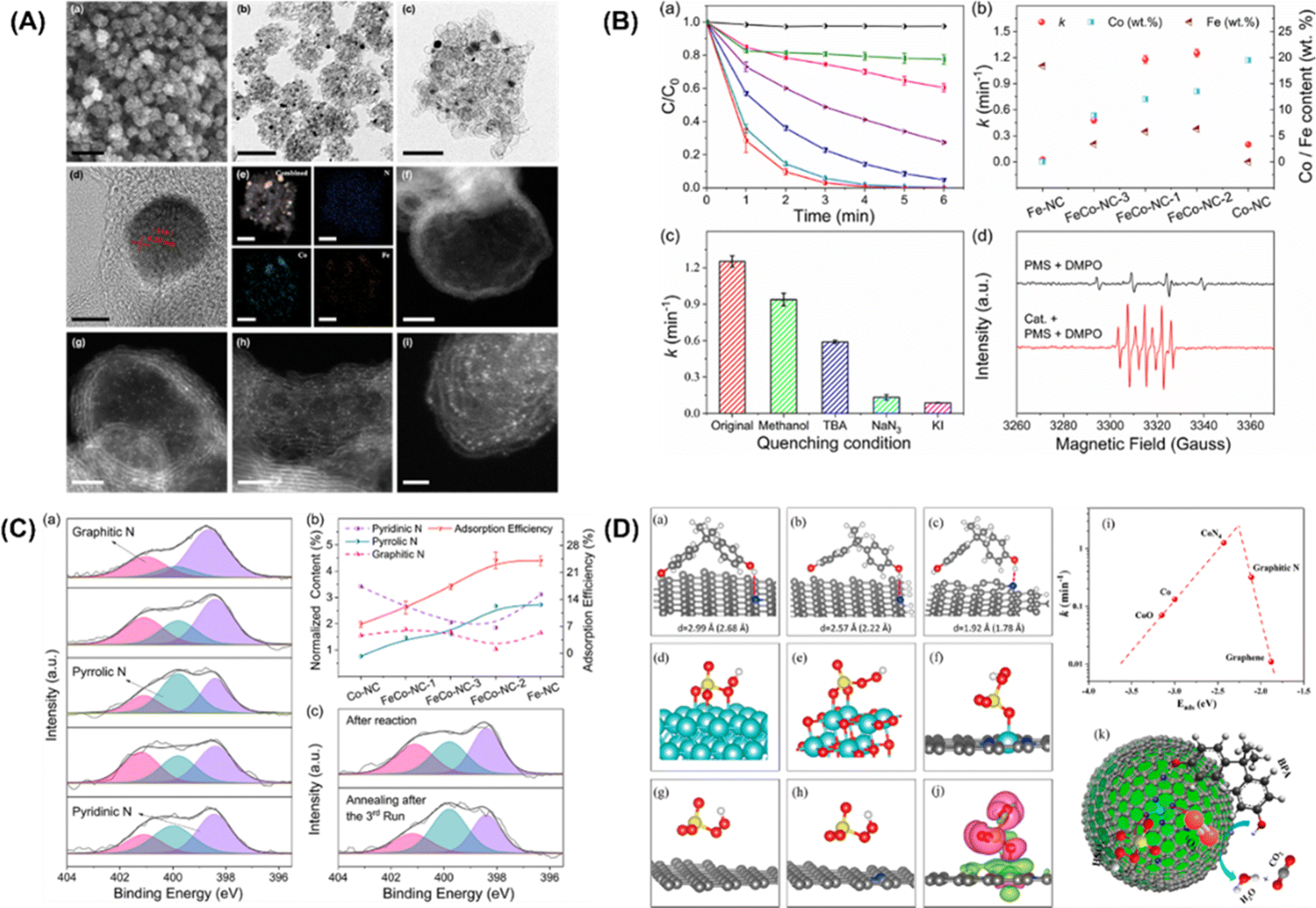
Furthermore, Li et al.92 put forward single cobalt atoms anchored on porous N-doped graphene with dual function in PMS activation for the rapid degradation of bisphenol A (BPA). A nitrogen-doped graphene-coated FeCo bimetallic nanocage (FeCo@NC) was formed using nanospheres of a Prussian blue analog (FeCo PBA) in nitrogen. FESEM, TEM, and HR-TEM images of FeCo-NC-2 (Fig. 4A, inset a–d) showed a large number of hollow graphene spheres, with a small amount of FeCo nanocrystals remaining after acid treatment. The energy dispersive X-ray spectrum (EDX) mapping images of FeCo-NC-2 (Fig. 4A, inset e) illustrate the distribution of N, Fe and Co species in the nanocrystals and the porous graphene shells. The dispersion of individual Co/Fe atoms anchored to porous nitrogen-doped graphene can be clearly observed by HAADF-STEM with aberration correction. As shown in Fig. 4A, inset f–i, the bright spots corresponding to heavy atoms (single Co/Fe atoms) are well dispersed throughout the graphene sphere. In this case, bisphenol A was selected as the target pollutant, and the PMS catalytic performance of the single-Co-atom catalyst was studied. Surprisingly, 100% BPA removal was achieved in 4 minutes using FeCo-NC-2. In addition, the first-order reaction rate constants of FeCo-NC-1 (1.18 min−1), FeCo-NC-2 (1.25 min−1), and FeCo-NC-3 (0.47 min−1) were found to match the relative content of cobalt catalysts (Fig. 4B, inset b). In contrast, the reaction rate constant did not correlate with the content of iron. However, the removal efficiency of BPA by Co-NC with the highest Co content (19.5 wt%) was lower than 73%, indicating that other factors besides the single cobalt atoms may be dominant factors in the Fenton-like catalytic reaction. The scavenger test and EPR spectrum for the determination of free radicals are shown in Fig. 4B (insert C and D), respectively. The results show that the reactive oxygen species (˙OH, SO4˙−) exhibit little impact on the degradation efficiency of BPA, while NaN3 played a crucial role in the degradation of 1O2. Furthermore, KI, as a strong quenching agent of surface-bound free radicals, can quench the reaction, indicating that the reaction relying on 1O2 occurs throughout the whole surface catalytic process. EPR spectra (Fig. 4B, insert D) showed that ˙OH can be produced by hydrolysis of the PMS molecules.
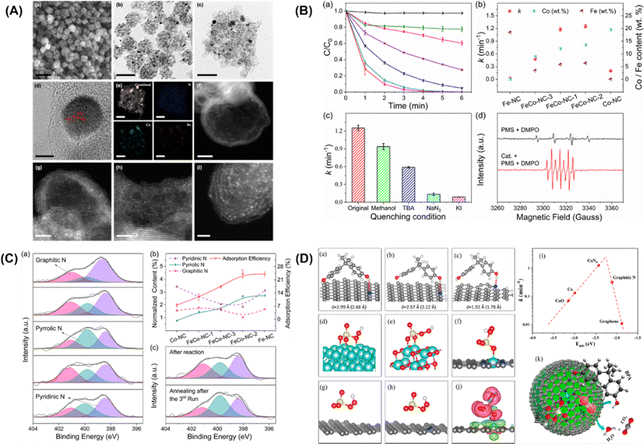 |
| | Fig. 4 (A) FESEM image (a), TEM images (b–c), HRTEM image (d), EDX mappings (e), and HAADF-STEM images of FeCo-NC-2 (f–i). (B) Catalytic performance in BPA degradation in different reaction systems by the leaching solution (a). (b) Reaction rate constants with the contents of Co and Fe using different catalysts. (c) Reaction rate comparison in different quenching conditions. (d) EPR spectra with/without FeCo-NC-2. (C) Determination of the adsorption sites. (a) XPS spectra of N 1s for Fe-NC, FeCo-NC-1, FeCo-NC-2, FeCo-NC-3, and Co-NC. (b) Relationships between BPA adsorption ability and normalized N species content. (c) XPS spectra of N 1s for FeCo-NC-2 after reaction (top) and regeneration by annealing in N2 (bottom). (D) Proposed possible mechanism of the PMS activation reaction. Adsorption configurations of BPA on (a) pyrrolic, (b) pyridinic, and (c) graphitic N-doped graphene. Optimized configurations of PMS adsorbed on (d) Co (0001), (e) CoO (100), (f) CoN4-graphene, (g) graphitic N-doped graphene, and (h) graphene, respectively. (i) Plot of the reaction rate against the adsorption energy of PMS on Co (0001), CoO (100), CoN4-graphene, graphitic N-doped graphene, and graphene. (j) Charge density difference in CoN4-graphene (ρ total−ρ substrate−ρ PMS). (k) Fenton-like reaction mechanism on single-Co-atom catalyst. Copied with permission from ref. 92. Copyright 2018, American Society Chemistry. | |
In addition, an apparent 1O2 signal appeared after FeCo-NC-2 was added to the system, confirming its dominant role in this case. XPS N 1S high-resolution spectra of the prepared catalysts were collected to quantify the type and content of the nitrogen dopant, as shown in Fig. 4C (inset a). Three peaks with binding energies of 398.4 eV, 399.8 eV and 401.1 eV were deconvoluted and attributed to pyridine, pyridine, and graphite N, respectively. The concentration of pyrrole N was observed to be correlated with the BPA adsorption efficiency (Fig. 4C, inset b), suggesting that pyrrole N may act as the adsorption site of BPA. To further confirm this hypothesis, the XPS N1s high-resolution spectra of the catalyst was investigated (Fig. 4C-inset (c)). It indicated that the adsorption of BPA and its corresponding intermediates on the pyrrole N position is the main cause of the gradual inactivation of the mono-Co catalyst. Finally, DFT calculations verified that the electron donor of BPA is the pyrrole N and the electron acceptor is the –OH group. Fig. 4D (inset a–c) shows the optimized configuration of BPA for the adsorption of pyrrole, pyridine and graphene N, where BPA has the optimal adsorption of pyrrole N, the shortest N–H distance (1.78 Å) and the highest adsorption energy (−0.31 eV). In addition, DFT calculations were performed to provide theoretical insight into the activation of PMS at the CoN4 sites containing single atoms of Co. Fig. 4D (inset d–h) reveals the complete relaxation atomic configurations of PMS on Co (0001), CoO (100), CoN4, graphite N, and graphene. Using the PMS adsorption energy descriptor, the relationship with the reaction rate is shown as a volcano plot in Fig. 4D (inset i). Thus, the low-reactivity cobalt metal and cobalt oxide of PMS lead to poisoning of the active site due to their strong binding to PMS. On the other hand, the binding of PMS to graphite N and graphene is too weak to activate the PMS molecules effectively. In addition, from the charge density analysis (Fig. 4D, inset j), it can be seen that there is significant electron transfer between PMS and CoN4, reflecting the chemisorption of PMS at the CoN4 site.
Transition metals
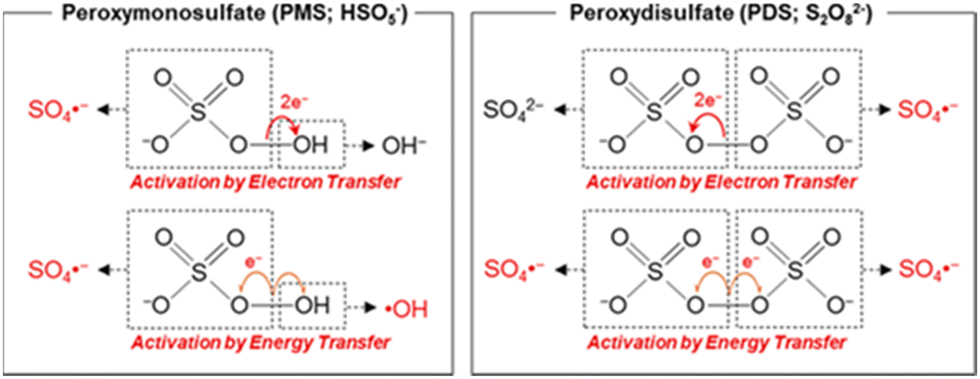
Transition metals with lower valence states, like that of the Fenton regent (i.e., Fe2+ and H2O2), can activate PMS/PDS molecules well according to eqn (1) and (5). Typical PMS and PDS activation by electron transfer and energy transfer are depicted in Fig. 5. In this section, we will discuss the utilization of transition metal ions in SR-AOPs.
| Mn+ + HSO5− → Mn+1 + SO4˙− + OH− (1) |
| | | Mn+ + S2O8− → Mn+1 + SO4˙− + SO42− | (5) |
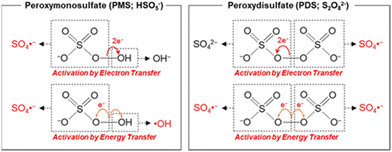 |
| | Fig. 5 Activation of PMS and PDS via electron- and energy-transferring processes. Copied with permission from ref. 91. Copyright 2020, American Chemical Society. | |
Co-based activator
Dionysiou et al.93 reported that Co2+ outperforms most reported transition metal ions, such as Fe2+, Cu2+, Ce3+, Mn2+, and Ni2+, for PMS activation; the intrinsic reason has been ascribed to its highest standard reduction potential among various metallic redox couples, as shown in Table 3. The basic mechanism for the activation of PMS by Co2+ ions is summarized in eqn (6)–(8). In this part, we will list some relevant examples, as comparative studies on the formation of singlet oxygen in elemental Co-based catalysts have progressed greatly.| | | CO2+ + HSO5− → CO3+ + SO4˙− + OH− | (6) |
| | | CO3+ + HSO5− → CO2+ + SO5˙− + H+ | (7) |
| | | SO4˙− + H2O → ˙OH + SO42− + H+ | (8) |
Table 3 Reduction potentials of transition metallic couples
| Metallic redox couple |
E
0 (V) |
| Co3+/Co2+ |
1.92 |
| Fe3+/Fe2+ |
0.77 |
| Ce4+/Ce3+ |
1.72 |
| Mn3+/Mn2+ |
1.54 |
| Cu2+/Cu+ |
0.15 |
| V4+/V3+ |
0.34 |
For instance, Chen et al.94 employed magnetic Co–N-doped carbon hybrid catalysts (Co-NC-x) to treat recalcitrant organic pollutant effluent by using a facile cation exchange and self-reduction method to activate PMS. EPR spectroscopy and scavenger testing demonstrated that SO4˙− and 1O2 were responsible for the degradation of RhB in the Co-NC-850/PMS system; additionally, 1O2 contributed ca. 86.2% to RhB removal. Remarkably, the synergistic effect of Co0 nanoparticles (NPs) and NC on Co-NC-850 acted as active sites to trigger PMS activation, and the direct oxidation of O2˙− by ˙OH played a crucial role for forming 1O2. Zhang et al.35 proposed a CoOOH/PMS system to degrade 2,4-dichlorophenol. Again, 1O2 played a leading role; however, the singlet oxygen was derived from the reaction between PMS molecules and water molecules with the formation of superoxide as an intermediate. Interestingly, Zeng et al.95 utilized oxygen-vacancy-enriched cobalt aluminum hydroxide@hydroxysulfide (CoAl-LDH@CoSx) hollow flowers for the degradation of sulfamethoxazole (SMX) in PMS activation; 1O2 was verified to be dominant ROS responsible for SMX degradation via quenching tests. Mechanism investigation revealed that the oxygen vacancies, redox cycles of Co(II)/Co(III) and S2− as well as sulfate species were responsible for singlet oxygen formation. Similar results were obtained by Dong et al.,95 who employed natural illite microsheets via PMS activation. Accordingly, abundant oxygen vacancies were created by the indistinct lattice boundaries. DFT calculations verified that the oxygen vacancies significantly reduced adsorption energy and accelerated electron transfer, further promoting PMS activation. In addition, the oxygen-vacancy-rich Co3O4/illite exhibited superior catalytic efficiency in a real water matrix, which was ascribed to SO4˙−, ˙OH and 1O2 (generated from oxygen vacancies, eqn (9)–(12)).
| | | Ov + HSO5− → O2˙− + HSO4− | (9) |
| | | O2˙− + H2O → ˙OOH + OH− | (10) |
| | | O2˙− + ˙OOH → 1O2 + HOO− | (11) |
| | | ˙OOH + ˙OOH → 1O2 + H2O2 | (12) |
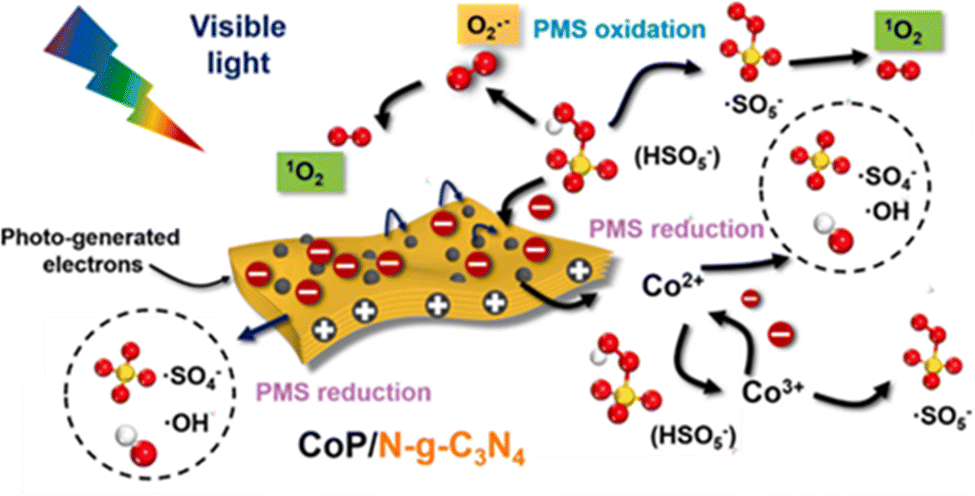
Dong et al.33 reported CoP/N-g-C3N4 nanosheets in which the doping of graphitic N atoms could modulate the electronic properties of the g-C3N4 nanosheets. As previously mentioned, in the N-g-C3N4 nanosheets, the electron-deficient carbon atoms neighboring graphitic N took charge of PMS oxidation in generating 1O2. However, in the visible-light-driven CoP/N-g-C3N4/PMS system, the PMS molecules were preferentially adsorbed onto the electron-deficient carbons near the newly formed NN bonds for PMS reduction (SO4˙− and ˙OH radicals), whereas the CoP nanoparticles were responsible for the PMS oxidation (i.e., 1O2 production) and PMS reduction at the same time. For the ROS, 1O2 plays a leading role, and is derived from SO5˙− and O2˙−. The specifics are shown in Fig. 6.
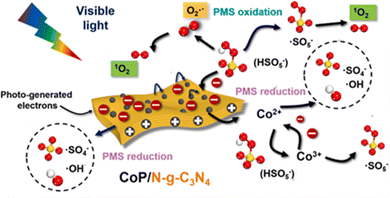 |
| | Fig. 6 The proposed mechanism in the CoP/N-g-C3N4 nanosheets catalyzed visible-light-driven PMS activation. Copied with permission from ref. 33. Copyright 2021, Elsevier. | |
In terms of single atomic materials, it has been clearly elaborated that the Co-N4 site with a single Co atom serves as the active site with optimal binding energy for PMS activation.91 However, Zhan and his group members21 recently found that CoN2+2 could be recognized as the active site and singlet oxygen was the predominant ROS with almost 100% PMS conversion.
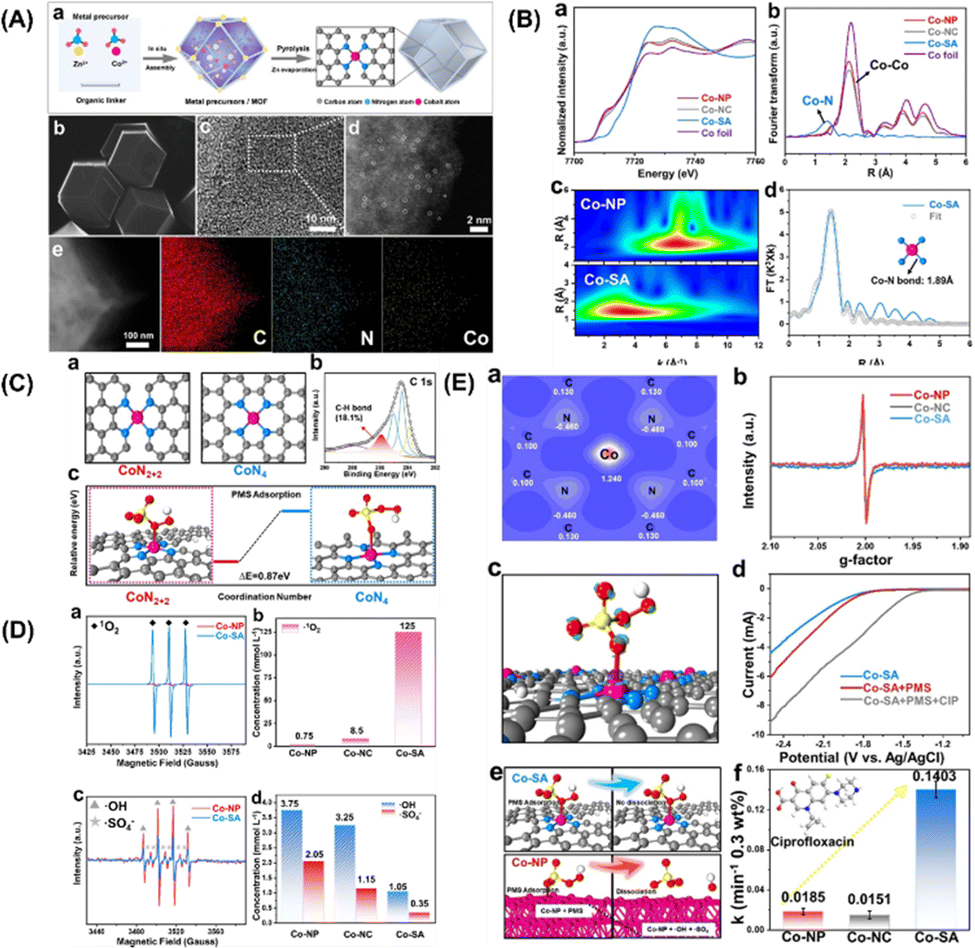
More specifically, this work by Zhan and coworkers focuses on the CoN2+2 monoatomic active sites, which regulate PMS oxidation at the atomic level. Unlike in the case of the rupture of the O–O and/or O–H bonds in PMS, electrons are transferred from PMS to a single Co atom, forming 1O2. Co-SA materials were synthesized via self-assembly and pyrolysis (Fig. 7A, inset a). After the Zn atoms evaporated, the remaining Co atoms were successfully fixed on the N–C support. Co-SA presents a smooth and dodecagonal shape (Fig. 7A, inset b). No obvious aggregation of the finite atoms exists, and individual finite atoms (shown in white circles) are observed fixed throughout the architecture (Fig. 7A, inset c and d). Additionally, mapping of Co-SA revealed the uniform distribution of C, N species (Fig. 7A, inset e). X-ray absorption fine structure (XAFS) analysis suggested the presence of atomic structure of the catalyst. As can be seen from the XANES spectrum (Fig. 7B, inset a), the rising edge of Co-SA is located between that of Co foil and Co-NP, indicating that the average oxidation state of a single Co atom is between Co0 and Co3+. The EXAFS spectrum of Co-SA has a main peak at 1.41 Å, corresponding to the Co–N coordination (Fig. 7B, inset b). Unlike for the Co foil, Co-NP and Co-NC, there is no Co–Co coordination peak at 2.1 Å, indicating the atomic dispersion of Co on the carbon carrier. The wavelet transform (WT) diagram of Co-SA shows that the WT maximum value is 5.1 Å−1, corresponding to Co-NP (Fig. 7B, inset c). In addition, the Fourier transform of the Co-SA K-Edge EXAFS spectrum can be fitted by CoN from the scattering path of the CoN4 structure (Fig. 7B, inset d). Considering the influence of the carbon matrix CoN4 active sites, studies were first computed focusing on the CoN2+2 (CoN4 partially bridged at two adjacent graphite edges) and CoN4 (CoN4 half embedded in a complete graphite layer) sites (Fig. 7C, inset c). The predicted Co–N bond length was 1.88 Å at CoN2+2 and 1.85 Å at CoN4. Based on EXAFS analysis, the CoN bond length at the CoN2+2 site was predicted to be close to 1.89 Å. In addition, a C–H bond was detected, which is consistent with the CoN2+2 configuration (Fig. 7C, inset b).
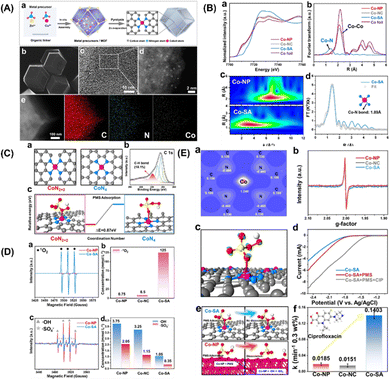 |
| | Fig. 7 (A) (a) Preparation steps of Co-SA. (b)–(e) SEM, TEM, HAADF-STEM and EDS mapping images of Co-SA. (B) (a) Co normalized K-edge XANES spectra and (B) K3-weighted Fourier transform spectra of Co foils and Co-based samples. (c) Wave transformations of Co-NP and Co-SA. (d) EXAFS fitting curve corresponding to Co-SA in R space. (C) Atomic structure of CoN2+2 and CoN4 active sites of Co-SA (a) and XPS spectrum of C of Co-SA (b). The free energy evolution of the active sites for PMS adsorption of CoN2+2 and CoN4 (c). (D) EPR spectrum and quantitative determination of 1O2, ˙OH and SO4˙−. (E) Two-dimensional valence electron density map of the CoN2+2 site in Co-SA (a). EPR spectrum of catalysts (b). The electron density difference diagram (c) of Co-SA and PMS adsorbed at the CoN2+2 site. (d) Linear sweep voltammetry (LSV) curves of Co-SA. (e) Adsorption of PMS on Co-SA and Co-NP surfaces. (f) Degradation efficiency of CIP in different systems. Copied with permission from Ref. 21. Copyright 2021, Wiley. | |
In addition, the adsorption of PMS by the CoN2+2 site is more advantageous in terms of energy than that by CoN4 site (Fig. 7C, inset c). Therefore, it can be inferred from the above results that the configuration of the active Co-SA site is CoN2+2. To identify the ROS generated during PMS activation, EPR was used to directly detect ROS. The intensity of the triplet signal of TEMP-1O2 in the Co-SA system is 37 times higher than that in the Co-NP system (Fig. 7D, inset a). In addition, the 1O2 concentration was quantitatively determined in all three systems (Fig. 7D, inset b). The ratio of 1O2 to produce ROS Co-SA accounted for 98.89%, and SO4˙− and ˙OH radicals were generated in the Co-NP and Co-SA systems (Fig. 7D, inset c), which implies that the major ROS in the Co-NP systems were SO4˙− and ˙OH (Fig. 7D, inset d).
These results show that different reaction pathways and dominant ROS-catalysed activation of PMS on the CoN2+2 active sites. The simulated configuration of the charge properties of the CoN2+2 active sites was determined (Fig. 7E, inset a). The electronic properties of Co-NP and Co-SA were detected using EPR (Fig. 7E, inset b). The EPR signal of Co-NP was strengthened compared to that of Co-SA, revealing a higher concentration of unpaired electrons for Co-NP. Furthermore, the adsorption and reaction of PMS on the prototypical Co-NP and Co-SA surfaces were theoretically modelled, and the results revealed a difference in electron density between PMS and Co-SA (Fig. 7E, inset c). It was found that electrons transfer from PMS to Co single atoms, indicating that the Co single atoms were reduced during the PMS adsorption process. In the LSV analysis, the number of the Co-SA electrodes significantly increased the electron transfer process in the presence of PMS (Fig. 7E, inset d). These results demonstrate that combined Co-NP can be used as an electron donor to reduce the activation of the PMS, and that the body of the molecules tends to transfer electrons to the Co-SA. In the joint Co-NP system, the PMS molecules are adsorbed by the Co-NP system because of the strongly positively charged nanoparticles (Fig. 7E, inset e). The strong interaction between the positively charged Co atom and the negatively charged O atom results in spontaneous dissociation of the adsorbed PMS and the formation of SO4˙− and ˙OH. Thus, the dissociation of PMS on the Co-NP surface is thermally favourable, with no energy barrier. In the Co-SA system, PMS is adsorbed on a single Co atom, and an O atom on the –SO4 side is bonded to the Co atom on the surface, indicating that the Co atom is the active adsorption site regardless of the morphology of Co. The O–O and O–H bonds were not cleaved. The interaction between H and N is too weak for the hydrogen atom to attach stably to the negatively charged N atom. After normalizing the rate constant to the Co concentration, the constants of Co-SA are 10.55 times and 6.27 times those of Co-NP and Co-NC, respectively (Fig. 7E, inset f). In general, Co-SA with CoN2+2 active sites regulates 1O2 production during PMS activation. For comparison, Co-NP and Co-NC were synthesized to study the generation of 1O2 during PMS activation. In the activation using the Co-NP system, the main ROS are SO4˙− and ˙OH radicals. PMS is directly decomposed into SO4˙− and ˙OH on Co nanoparticles, which is mainly attributed to the strong reducibility of the Co adsorption sites. Unlike the traditional catalytic system with free radical generation, the Co-SA activation of PMS involves the oxidation of PMS to 1O2. The spontaneous dissociation of PMS on the Co-SA surface cannot be induced by the charge density of N and the coordination of Co atoms in CoN2+2. However, electron transfer to the Co single atom occurs during the adsorption process of PMS, resulting in 1O2 formation in 98.89% of the generated ROS. The generated singlet oxygen showed effective degradation activity toward several organic pollutants over a wide pH range. This study proposes a strategy to control 1O2 generation through PMS activation.
Mo-based activators
Xing and his group members96 proposed that MoS2 could act as a highly efficient cocatalyst in the Fenton process, mainly driven by the fast circulation of Fe3+/Fe2+ through the exposure of Mo4+ active sites. Relevant research using the element Mo has since progressed greatly.97,98 Xing et al.99 also found that MoS2 could act as a cocatalyst for rapid disinfection of Escherichia coli K-12 (E. coli) and S. aureus. Afterward, with the popularity of PMS activation, Sheng et al.100 further incorporated MoS2 as a cocatalyst in the Fe(II)/PMS system (arising from Fe3+ to Fe2+), accompanied by the yield of a high concentration of SO4˙− and ˙OH radicals, which resulted in effective removal of 2,4,6-trichlorophenol. In addition, Zhou et al.101 employed carbamazepine as a target to unravel the different mechanisms of the MoS2/PMS system and the MoS2/PDS system. Zhang et al.102 observed that the excellent degradation of acetaminophen is due not only to the acceleration of Fe ion circulation by MoS2 but also the formation of a strongly oxidative Mo(VI) peroxo-complex intermediate. Additionally, 1O2 takes charge of the overall process; the 1O2 is derived from the oxidation of MoS2, as presented in eqn (13) and (14).| | | MoS2 + 9HSO5− + 5OH− → MoO42− + 10H+ + 1O2 | (13) |
| | | MoOOH(O2)2− + H2O → MoO42− + 1O2 + H+ | (14) |
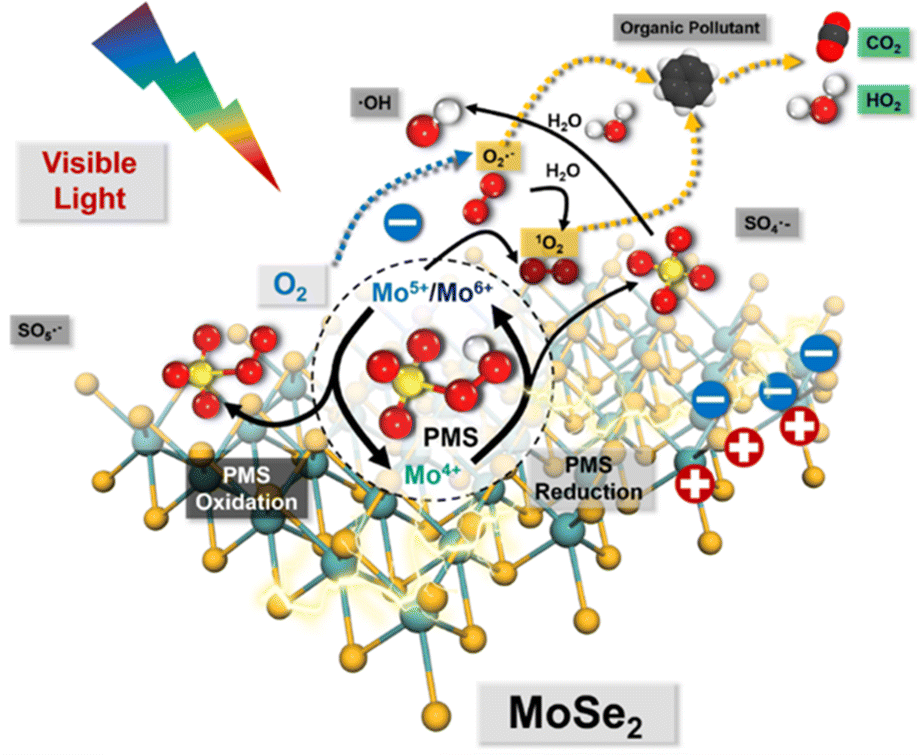
Accordingly, a series of Mo-based catalysts and/or cocatalysts were employed in AOPs (e.g., the Fenton process), such as Mo powder,103,104 MoO2,105 two-dimensional (2D) MoS2,106 and three-dimensional (3D) MoS2 sponge.107 The relevant innovative mechanism for the Fenton process was proposed, which mostly revolves around the following two phenomena: (1) the oxidation process between Fe3+ and Mo4+; (2) the 1O2 stemming from the oxidation–reductive reaction between Mo6+ and O2˙−. However, the mechanism related to 1O2 for SR-AOPs still lacks sufficient exploration. In this context, Dong et al.33 tentatively synthesized bimetallic MoFe/TiO2 nanospheres for solar-light-driven PMS activation. They pointed out that both photo-generated electrons and transition metallic redox couples (i.e., Mo6+/Mo4+, Fe3+/Fe2+ and Mo4+/Fe3+) were responsible for PMS activation. Most importantly, SO4˙−, ˙OH, and SO5˙− participated in the transformation and generation of 1O2. Furthermore, Dong et al.5 used commercial MoSe2 to replace MoS2 to solve the problem of H2S discharge in sewage. In this case, O2˙− is dominant, and 1O2 comes mainly from the transformation of O2˙−. The specific information is shown in Fig. 8 and eqn (15)–(24). If MoSe2 were irradiated with visible light, carriers (i.e., electrons and holes) would be generated (eqn (15)), with photogenerated electrons reacting with oxygen to form O2˙− (eqn (16)). Afterward, Mo4+ reacts with HSO5− to form SO4˙−, Mo5+ and Mo6+ (eqn (17)). After that, the SO4˙− radical leads to the formation of ˙OH radicals, and excess HSO5− tends to produce hydrogen peroxide (eqn (19)), resulting in the reaction of ˙OH free radicals with H2O2 to produce HO2˙− and O2˙− (eqn (20) and (21)). Afterward, excess O2˙− is converted into 1O2. Of note, Mo6+ can oxidize O2˙− to 1O2.106 Accordingly, the 1O2 is mainly derived from the transformation of O2˙− rather than stemming from the previously reported PMS self-decomposition or PMS oxidation. Ultimately, both O2˙− and 1O2 radicals contribute to the degradation of PPCPs.| | | Mo4+ + HSO5− → Mo6+/Mo5+ + SO4˙− + OH− | (17) |
| | | Mo6+/Mo5+ + HSO5− → Mo4+ + SO5˙− + H+ | (18) |
| | | HSO5−(SO5−) + H2O → H2O2 + HSO4−(SO42−) | (19) |
| | | ˙OH + H2O2 → HO2˙− + H2O | (20) |
| | | 2˙O2− + 2H2O → H2O2 + 2OH− + 1O2 | (22) |
| | | Mo6+ + ˙O2− → Mo4+ + 1O2 | (23) |
| | | ˙O2−/1O2 + CBZ → intermediates + CO2 + H2O | (24) |
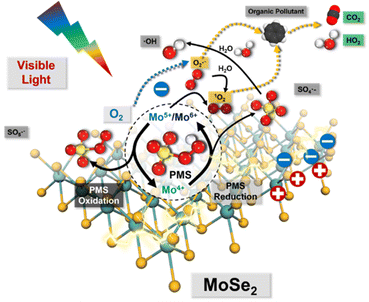 |
| | Fig. 8 Proposed mechanisms for the visible-light-driven MoSe2/PMS system. Copied with permission from ref. 5. Copyright 2021, Elsevier. | |
Other metallic activators
In addition to materials based on the elements Co and Mo, iron-based catalysts are popular in AOPs. Generally, PMS/PDS activation using Fe involves the transformation from Fe2+ to Fe3+ and the generation of ˙OH and SO4˙−. Notably, the concentration of Fe2+ has a crucial impact on PMS/PDS oxidation of organic pollutants. However, the relative progress involving its non-radical pathway is rarely reported. Interestingly, Li et al.108 found that the Fe0/PMS system for the degradation of Rhodamine B (RhB) could be enhanced by the addition of Cu2+, and that the dominant ROSs in the process were 1O2 and O2˙−. Yang et al.109 also confirmed 1O2 and O2˙− to be the dominant species, with ˙OH also coexisting, in the Fe0-montmorillonite/PMS system.
In addition, Mn-based catalysts are considered to be effective PMS/PDS activators with the advantage that Mn is an Earth-abundant element and less toxic than Co.110 In terms of the non-radical pathway, Huang et al.111 revealed that 1O2 was generated in the PMS/MnO2 system under acidic conditions. They also concluded that 1O2 was generated as the primary ROS through the direct oxidation of O2˙− by Mn(IV) and O2˙− recombination, as well as the reaction between O2˙− and metastable manganese intermediates under neutral conditions. Furthermore, metal oxides of perovskites (i.e., ABO3 structure) and spinel (i.e., AB2O4 structure)112 have attracted increasing interest, and the corresponding oxygen vacancies on the surface of the metal or metal oxide account for the generation of 1O2. Due to the facilitated oxygen adsorption on the surface and lattice oxygen vacancies, the rapid generation of O2˙− leads to rapid formation of 1O2, as a greater amount of oxygen vacancies is beneficial to the interfacial electron transfer in PMS activation.113 In addition to the conventional metallic catalysts, Al2O3 and TiO2 loaded with noble metals including Pt, Pd, Au, and Ag can mediate electron transfer from organic pollutants to PMS molecules,114 and subsequently achieve the non-radical oxidation of PMS through 1O2. The degradation efficiency follows the order Pd > Pt ≈ Au ⋙ Ag. Wang et al.115 intensively investigated the catalytic mechanism of PMS. The dispersed Pd particles on the g-C3N4 (Pd/g-C3N4) surface could activate PMS, inducing the formation of 1O2 and O2˙− for BPA degradation. It is worth noting that Pd0 is converted into Pd(II) during the process, and 1O2 is produced viaeqn (25). Table 4 summarizes some other relevant metal-based activators in PMS activation.
Table 4 Performance and mechanism for other metallic-based activators in PMS activation
| Catalyst |
Target/pollutant |
Degradation rate |
Radicals |
Involve sites |
Ref. |
| Fe-Mt-C–H2 |
BPA (25 mg L−1) |
99.3% within 120 min |
1O2 and O2˙− |
Fe0 |
121
|
| Fe–N–C |
BPA (25 mg L−1) |
88% within 120 min |
1O2 |
Metal-N–C catalytic site |
119
|
| MnO2 |
Bisphenol A (BPA) |
94.5% within 10 min |
1
O2 |
Mn4+ and Mn3+ |
111
|
| CuO–CeO2 |
Rhodamine B |
A complete degradation within 60 min |
1
O2 |
Cu2+/Cu+, oxygen vacancies, electron transfer |
122
|
| LaCo0.4Cu0.6O3 |
Phenol |
100% within 12 min |
1O2 |
Cu2+/Cu+, oxygen vacancies |
123
|
| LaNiO3 |
Ofloxacin |
93% within 10 min |
1O2 |
Ni3+/Ni2+, oxygen vacancies |
124
|
| Pd/C3N4 |
BPA |
91% within 60 min |
1O2 |
Pd0 |
114
|
| CuO nanosheet |
AO7 in high salinity wastewater |
95.8% within 30 min |
1O2 |
Cu+/Cu2+ and Fe2+/Fe3+ |
125
|
Factors impacting singlet oxygen in PMS activation
The broad existence of PPCPs, pesticides, and surfactants in the aquatic environment has driven the development of SR-AOPs. The direct and indirect discharge of PPCPs leads to their accumulation in bodies of water and is detrimental to human health and aquatic life. In principle, the treatment efficiency of PMS/PDS activation is influenced by the practical water matrix, including the temperature, pH value, and salinity.4 The general production of SO4˙− and ˙OH radicals can be impeded by several inorganic anions (e.g., Cl−, CO32−/HCO3−, NO3− and SO42−) and by natural organic matter (i.e., fulvic acid and humic acid). In particular, Cl− can efficiently scavenge SO4˙− and ˙OH in SR-AOPs, thereby producing Cl2˙− and Cl˙.116 Generally, the alkalinity of a body of water is tuned by carbonate (CO32−) and bicarbonate (HCO3−) ions, whose concentrations are in the range of 50–200 mg L−1 in real bodies of water.117 It has been found that both CO32− and HCO3− act as radical scavengers in AOPs. Furthermore, the radicals SO4˙− and ˙OH can be suppressed by NO3−, SO42− and phosphate ions, whereas, for 1O2, the inhibitory effect of high salinity is significantly weakened in the non-radical process.117 The insignificant impact of anions on the 1O2-dominated PMS activation process is mainly attributed to the dissymmetry of PMS molecules, which facilitates the self-decomposition of PMS molecules under nucleophilic attack by the anions. Additionally, natural organic matter (NOM) is also a complicated factor in SR-AOPs. Dong et al.5–7 found that the degradation efficiency of PPCPs is not impeded even in the presence of coexisting NOM, which is even self-degraded during the process. Moreover, NOM in aquatic systems acts as a photosensitizer for 1O2 formation rather than a scavenger;118 therefore, the effect of NOM on the 1O2-dominated systems can be ignored.119,120
In addition, the solution pH is another crucial parameter depending on the characteristics of the PMS activators. For instance, in homogeneous activation systems, 1O2 can be directly generated via PMS activation under neutral (6.5 ± 0.3) and alkaline conditions, as the surface hydroxyl groups enhance the chemical binding with PMS molecules. In heterogeneous systems, metal ions can activate PMS utilizing 1O2 over an even broader pH range.93 Overall, the catalyst structure, singlet oxygenation, and electron transfer are crucial factors behind the generation of 1O2.93
Conclusions and perspectives
In summary, we have specifically reviewed the evolution of 1O2 in SR-AOPs. Since 2016, studies of 1O2 in SR-AOPs have attracted dramatic attention. Compared to SO4˙− and ˙OH radicals, 1O2 has advantages, such as its longer lifetime, mild conditions, resistance to inorganic anions (e.g., HCO3−, Cl−, and H2PO4−), NOMs and free-radical scavengers (e.g., methanol and t-butanol), and ability to degrade electron-rich organic complexes. Due to the advantages of 1O2, the 1O2-dominated PMS system is a promising technique in wastewater treatment plants. However, some critical points and perspectives need to be addressed in the future.
• As the degradation efficiency of PMS activation using carbonaceous nanomaterials strongly correlates with the oxygen functionality, the defect degree and the heterogeneous atoms doped in the carbon matrix, the development of advanced carbon-based materials is an effective route to improve SR-AOPs. However, the insufficient oxidation ability of 1O2 is an obstacle to the process, and hence, the emerging M–N–C materials are a reliable choice because of their extremely low metallic leaching. In addition, in the context of the formation of 1O2, the mechanism of PMS-Carbon* is worthy of exploration owing to its complexity.
• Among transition–metal-based materials, single transition metal oxides have more oxygen vacancies and exhibit higher degradation efficiency. Additionally, composites of carbon materials and transition metals provide a new way of thinking in SR-AOPs owing to the synergistic effect between them when the carbonaceous materials act as a mediator.
• 1O2 is insensitive to background inorganic ions (e.g., Cl−, CO32−/HCO3−, NO3−, PO43− and SO42−) and NOM, being more accessible to electron-rich compounds. Hence, 1O2-mediated PMS systems have great potential for the treatment of actual wastewater and the effluents of river water, as well as drinking water, regardless of the background water matrix.
• Most importantly, based on the theoretical foundation, it is more desirable to think more about how to select and design an appropriate material via computational attempts (e.g., machine learning or artificial intelligence) rather than directly conducting experiments in order to save labour and chemical expenses.
Author contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Key R&D Program of China (2022YFE0107900), National Natural Science Foundation of China (21972040), the Program of Introducing Talents of Discipline to Universities (B20031, B16017), Innovation Program of Shanghai Municipal Education Commission (2021-01-07-00-02-E00106), the Science and Technology Commission of Shanghai Municipality (20DZ2250400, 22230780200) and Fundamental Research Funds for the Central Universities.
Notes and references
- C. Dong, M. Xing and J. Zhang, Front. Environ. Chem., 2020, 8, 1 CrossRef PubMed.
- Q. Yan, J. Zhang and M. Xing, Cell. Rep. Phys. Sci., 2020, 1, 100149 CrossRef CAS.
- J. Wang and S. Wang, Chem. Eng. J., 2018, 334, 1502–1517 CrossRef CAS.
- G. Xiao, T. Xu, M. Faheem, Y. Xi, T. Zhou, H. T. Moryani, J. Bao and J. Du, Int. J. Environ. Res. Pub. Health., 2020, 18, 3344 CrossRef PubMed.
- C. Dong, Z. Wang, Z. Ye, J. He, Z. Zheng, X. Gong, J. Zhang and I. M. C. Lo, Appl. Catal., B, 2021, 296, 120223 CrossRef CAS.
- C. Dong, Z. Zheng, Z. Wang, J. He, Z. Ye, X. Gong and I. M. C. Lo, J. Hazard. Mater., 2021, 416, 125891 CrossRef CAS PubMed.
- C. Dong, Z. Zheng, M. A. H. Badsha, J. He and I. M. C. Lo, Environ. Int., 2021, 154, 106572 CrossRef CAS PubMed.
- X. Zheng, X. Niu, D. Zhang, M. Lv, X. Ye, J. Ma, Z. Lin and M. Fu, Chem. Eng. J., 2022, 429, 132323 CrossRef CAS.
- X. Li, L. Wang, Y. Guo, W. Song, Y. Li and L. Yan, Chem. Eng. J., 2022, 450, 138104 CrossRef CAS.
- P. Yang, Y. Long, W. Huang and D. Liu, Appl. Catal., B, 2022, 122245 Search PubMed.
- B. Yu, X. Li, M. He, Y. Li, J. Ding and Y. Zhong, J. Hazard. Mater., 2023, 441, 129940 CrossRef CAS PubMed.
- Y. Qi, X. Zhou and Z. Li, Catalysts, 2022, 12, 1327 CrossRef CAS.
- A. D. Bokare and W. Choi, Environ. Sci. Technol., 2015, 49, 14392–14400 CrossRef CAS PubMed.
- S. Liu, C. Zhao, Z. Wang, H. Ding, H. Deng, G. Yang, J. Li and H. Zheng, Chem. Eng. J., 2020, 386, 124015 CrossRef CAS.
- A. R. Badireddy, E. M. Hotze, S. Chellam, P. Alvarez and M. Wiesner, Environ. Sci. Technol., 2007, 41, 6627–6632 CrossRef CAS PubMed.
- M. Cho, J. Lee, Y. Mackeyev, L. J. Wilson, P. J. Alvarez, J. B. Hughes and J. Kim, Environ. Sci. Technol., 2010, 44, 6685–6691 CrossRef CAS PubMed.
- S. Yang, P. Wang, X. Yang, L. Shan, W. Zhang, X. Shao and R. Niu, J. Hazard. Mater., 2010, 179, 552–558 CrossRef CAS PubMed.
- C. Amor, J. R. Fernandes, M. S. Lucas and J. Peres, Environ. Technol. Innov., 2021, 21, 101183 CrossRef CAS.
- S. Liu, Z. Zhang, F. Huang, Y. Liu, L. Feng, J. Jiang, L. Zhang, F. Qi and C. Liu, Appl. Catal., B, 2021, 286, 119921 CrossRef CAS.
- Y. Gao, Z. Chen, Y. Zhu, T. Li and C. Hu, Environ. Sci. Technol., 2019, 54, 1232–1241 CrossRef PubMed.
- X. Mi, P. Wang, S. Xu, L. Su, H. Zhong, H. Wang, Y. Li and S. Zhan, Angew. Chem., 2021, 133, 4638–4643 CrossRef.
- L. Xie, P. Wang, Y. Li, D. Zhang, D. Shang, W. Zheng, Y. Xia, S. Zhan and W. Hu, Nat. Commun., 2022, 13, 1–11 Search PubMed.
- N. Mehraban and H. Freeman, Mater. Des., 2015, 8, 4421–4456 CrossRef CAS PubMed.
- R. Prieto-Montero, A. Prieto-Castañeda, R. Sola-Llano, A. R. Agarrabeitia, D. García-Fresnadillo, I. López-Arbeloa, A. Villanueva, M. J. Ortiz, S. Moya and V. Martínez-Martínez, Photochem. Photobiol., 2020, 96, 458–477 CrossRef CAS PubMed.
- P. Sun, H. Liu, M. Feng, L. Guo, Z. Zhai, Y. Fang, X. Zhang and V. Sharma, Appl. Catal., B, 2019, 251, 335–345 CrossRef CAS.
- M. Bancirova, Luminescence, 2011, 26, 685–688 CrossRef CAS PubMed.
- M. C. DeRosa and R. Crutchley, Coord. Chem. Rev., 2002, 233, 351–371 CrossRef.
- X. Tian, P. Gao, Y. Nie, C. Yang, Z. Zhou, Y. Li and Y. Wang, Chem. Commun., 2017, 53, 6589–6592 RSC.
- T. Kohn and K. L. J. Es Nelson, Environ. Sci. Technol., 2007, 41, 192–197 CrossRef CAS PubMed.
- A. Sandra Miskoski, P. G. Molina, G. Gunthe, A. L. Zanocco and N. A. Garcia, Photochem. Photobiol., 2005, 81, 325–332 CrossRef.
- J. Yin, Q. Xia and P. Fu, Toxicol. Ind. Health, 2007, 23, 625–631 CrossRef CAS PubMed.
- W. Haag and E. Gassman, Chemosphere, 1984, 13, 631–640 CrossRef CAS.
- C. Dong, Y. Bao, T. Sheng, Q. Yi, Q. Zhu, B. Shen, M. Xing, I. M. Lo and J. Zhang, Appl. Catal., B, 2021, 286, 119930 CrossRef CAS.
- P. Yang, S. Li, L. Xiaofu, A. Xiaojing, D. Liu and W. Huang, Sep. Purif. Technol., 2022, 285, 120288 CrossRef CAS.
- Q. Zhang, D. He, X. Li, W. Feng, C. Lyu and Y. Zhang, J. Hazard. Mater., 2020, 384, 121350 CrossRef CAS PubMed.
- W. Wei, H. Liang, K. Parvez, X. Zhuang, X. Feng and K. Müllen, Angew. Chem., 2014, 126, 1596–1600 CrossRef.
- C. Xing, Y. Xue, B. Huang, H. Yu, L. Hui, Y. Fang, Y. Liu, Y. Zhao, Z. Li and Y. Li, Angew. Chem., 2019, 131, 14035–14041 CrossRef.
- H. Sun, S. Liu, G. Zhou, H. M. Ang, M. O. Tadé and S. Wang, ACS Appl. Mater. Interfaces, 2012, 4, 5466–5471 CrossRef CAS PubMed.
- H. Sun, C. Kwan, A. Suvorova, H. M. Ang, M. O. Tadé and S. Wang, Appl. Catal., B, 2014, 154, 134–141 CrossRef.
- X. Duan, Z. Ao, D. Li, H. Sun, L. Zhou, A. Suvorova, M. Saunders, G. Wang and S. Wang, Carbon, 2016, 103, 404–411 CrossRef CAS.
- B. Lee, E. Gong, M. Kim, S. Park, H. Kim, J. Lee, E. Jung, C. Lee, J. Bok, Y. Jung, Y. Kim, K. Lee, S. Cho, J. Jung, C. Cho, S. Lebègue, K. Nam, H. Kim, S. In and T. Hyeon, Energy Environ. Sci., 2022, 15, 601–609 RSC.
- R. Xiao, Z. Luo, Z. Wei, S. Luo, R. Spinney, W. Yang and D. D. Dionysiou, Curr. Opin. Chem. Eng., 2018, 19, 51–58 CrossRef.
- X. Duan, H. Sun, Z. Ao, L. Zhou, G. Wang and S. Wang, Carbon, 2016, 107, 371–378 CrossRef CAS.
- H. Chen and K. Carroll, Environ. Pollut., 2016, 215, 96–102 CrossRef CAS PubMed.
- Y. Liu, W. Miao, X. Fang, Y. Tang, D. Wu and S. Mao, Chem. Eng. J., 2020, 380, 122584 CrossRef CAS.
- Y. Zhou, J. Jiang, Y. Gao, J. Ma, S.-Y. Pang, J. Li, X.-T. Lu and L.-P. Yuan, Environ. Sci. Technol., 2015, 49, 12941–12950 CrossRef CAS PubMed.
- X. Duan, Z. Ao, L. Zhou, H. Sun, G. Wang and S. Wang, Appl. Catal., B, 2016, 188, 98–105 CrossRef CAS.
- C. Wang, J. Kang, P. Liang, H. Zhang, H. Sun, M. O. Tadé and S. Wang, Environ. Sci.: Nano, 2017, 4, 170–179 RSC.
- Y. Zhou, J. Jiang, Y. Gao, S.-Y. Pang, Y. Yang, J. Ma, J. Gu, J. Li, Z. Wang, L.-H. Wang, L.-P. Yuan and Y. Yang, Water Res., 2017, 125, 209–218 CrossRef CAS PubMed.
- C. Qi, X. Liu, J. Ma, C. Lin, X. Li and H. Zhang, Chemosphere, 2016, 151, 280–288 CrossRef CAS PubMed.
- X. Duan, H. Sun and S. Wang, Acc. Chem. Res., 2018, 51, 678–687 CrossRef CAS PubMed.
- E.-T. Yun, J. H. Lee, J. Kim, H.-D. Park and J. Lee, Environ. Sci. Technol., 2018, 52, 7032–7042 CrossRef CAS PubMed.
- X. Cheng, H. Guo, Y. Zhang, X. Wu and Y. Liu, Water Res., 2017, 113, 80–88 CrossRef CAS PubMed.
- P. Shao, J. Tian, F. Yang, X. Duan, S. Gao, W. Shi, X. Luo, F. Cui, S. Luo and S. Wang, Adv. Funct. Mater., 2018, 28, 1705295 CrossRef.
- Y. Zhang, H. Pan, M. Murugananthan, P. Sun, D. D. Dionysiou, K. Zhang, A. Khan and Y. Zhang, Carbon, 2020, 156, 399–409 CrossRef CAS.
- P. Liang, C. Zhang, X. Duan, H. Sun, S. Liu, M. O. Tade and S. Wang, Environ. Sci.: Nano, 2017, 4, 315–324 RSC.
- Y. Gao, Y. Zhu, L. Lyu, Q. Zeng, X. Xing and C. Hu, Environ. Sci. Technol., 2018, 52, 14371–14380 CrossRef CAS PubMed.
- Y. Gao, Z. Chen, Y. Zhu, T. Li and C. Hu, Environ. Sci. Technol., 2020, 54, 1232–1241 CrossRef CAS PubMed.
- B. Sun, W. Ma, N. Wang, P. Xu, L. Zhang, B. Wang, H. Zhao, K.-Y. A. Lin and Y. Du, Environ. Sci. Technol., 2019, 53, 9771–9780 CrossRef CAS PubMed.
- C. Guan, J. Jiang, C. Luo, S. Pang, Y. Yang, Z. Wang, J. Ma, J. Yu and X. Zhao, Chem. Eng. J., 2018, 337, 40–50 CrossRef CAS.
- X. Duan, H. Sun, Y. Wang, J. Kang and S. Wang, ACS Catal., 2015, 5, 553–559 CrossRef CAS.
- M. Wei, X. Shi, L. Xiao and H. Zhang, J. Hazard. Mater., 2020, 382, 120993 CrossRef CAS PubMed.
- J. Cheng, N. Wei, Y. Wang, Y. Long and G. Fan, Sep. Purif. Technol., 2021, 277, 119441 CrossRef CAS.
- P. Sun, H. Liu, M. Feng, X. Zhang, Y. Fang, Z. Zhai and V. K. Sharma, Sep. Purif. Technol., 2021, 268, 118697 CrossRef CAS.
- Q. Peng, Y. Dai, K. Liu, X. Luo, D. He, X. Tang and G. Huang, J. Mater. Sci.: Mater. Electron., 2020, 55, 11267–11283 CrossRef CAS.
- M. Li, Z. Li, X. Yu, Y. Wu, C. Mo, M. Luo, L. Li, S. Zhou, Q. Liu and N. Wang, Chem. Eng. J., 2022, 431, 133339 CrossRef CAS.
- S. Liang, H.-Y. Niu, H. Guo, C.-G. Niu, C. Liang, J.-S. Li, N. Tang, L.-S. Lin and C.-W. Zheng, Chem. Eng. J., 2021, 405, 126686 CrossRef CAS.
- X. Li, D. Liang, C. Wang and Y. Li, Chemosphere, 2021, 275, 130058 CrossRef CAS PubMed.
- S.-H. Ho, R. Li, C. Zhang, Y. Ge, G. Cao, M. Ma, X. Duan, S. Wang and N. Ren, Water Res., 2019, 159, 77–86 CrossRef CAS PubMed.
- W. Sang, Z. Li, M. Huang, X. Wu, D. Li, L. Mei and J. Cui, Chem. Eng. J., 2020, 383, 123057 CrossRef CAS.
- Y. J. Sa, D.-J. Seo, J. Woo, J. T. Lim, J. Y. Cheon, S. Y. Yang, J. M. Lee, D. Kang, T. J. Shin, H. S. Shin, H. Y. Jeong, C. S. Kim, M. G. Kim, T.-Y. Kim and S. H. Joo, J. Am. Chem. Soc., 2016, 138, 15046–15056 CrossRef CAS PubMed.
- M. Li, Z. Li, X. Yu, Y. Wu, C. Mo, M. Luo, L. Li, S. Zhou, Q. Liu and N. Wang, Chem. Eng. J., 2022, 431, 133339 CrossRef CAS.
- N. Du, Y. Liu, Q. Li, W. Miao, D. Wang and S. Mao, Chem. Eng. J., 2021, 413, 127545 CrossRef CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- M. Allen, V. Tung and R. Kaner, Chem. Rev., 2010, 110, 132–145 CrossRef CAS PubMed.
- J. Wu, W. Pisula and K. Müllen, Chem. Rev., 2007, 107, 718–747 CrossRef CAS PubMed.
- S. Navalon, A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Chem. Rev., 2014, 114, 6179–6212 CrossRef CAS PubMed.
- S. Zhu, C. Jin, X. Duan, S. Wang and S.-H. Ho, Chem. Eng. J., 2020, 393, 124725 CrossRef CAS.
- S. Wang, L. Xu and J. Wang, Chem. Eng. J., 2019, 375, 122041 CrossRef CAS.
- X. Duan, K. O'Donnell, H. Sun, Y. Wang and S. Wang, Small, 2015, 11, 3036–3044 CrossRef CAS PubMed.
- P. Sun, H. Liu, M. Feng, L. Guo, Z. Zhai, Y. Fang, X. Zhang and V. K. Sharma, Appl. Catal., B, 2019, 255, 335–345 CrossRef.
- X. Chen, X. Duan, W.-D. Oh, P.-H. Zhang, C.-T. Guan, Y.-A. Zhu and T.-T. Lim, Appl. Catal., B, 2019, 253, 419–432 CrossRef CAS.
- J. Zhang, P. Chen, W. Gao, W. Wang, F. Tan, X. Wang, X. Qiao and P. K. Wong, Sep. Purif. Technol., 2021, 265, 118474 CrossRef CAS.
- P. Liang, C. Zhang, X. Duan, H. Sun, S. Liu, M. O. Tade and S. Wang, Environ. Sci.: Nano, 2017, 4, 315–324 RSC.
- J. Li, S. Zhao, L. Zhang, S. P. Jiang, S. Z. Yang, S. Wang, H. Sun, B. Johannessen and S. Liu, Small, 2021, 17, 2004579 CrossRef CAS PubMed.
- X. Li, X. Huang, S. Xi, S. Miao, J. Ding, W. Cai, S. Liu, X. Yang, H. Yang and J. Gao, J. Am. Chem. Soc., 2018, 140, 12469–12475 CrossRef CAS PubMed.
- P. Sun, H. Liu, Z. Zhai, X. Zhang, Y. Fang, J. Tan and J. Wu, Chem. Eng. J., 2019, 356, 262–271 CrossRef CAS.
- A. Shahzad, J. Ali, J. Ifthikar, G. G. Aregay, J. Zhu, Z. Chen and Z. Chen, J. Hazard. Mater., 2020, 392, 122316 CrossRef CAS PubMed.
- X. Li, J. Wang, X. Duan, Y. Li, X. Fan, G. Zhang, F. Zhang and W. Peng, ACS Catal., 2021, 11, 4848–4861 CrossRef CAS.
- J. Li, S. Zhao, L. Zhang, S. P. Jiang, S. Z. Yang, S. Wang, H. Sun, B. Johannessen and S. Liu, Small, 2021, 17, 2004579 CrossRef CAS PubMed.
- J. Lee, U. Von Gunten and J.-H. Kim, Environ. Sci. Technol., 2020, 54, 3064–3081 CrossRef CAS PubMed.
- X. Li, X. Huang, S. Xi, S. Miao, J. Ding, W. Cai, S. Liu, X. Yang, H. Yang and J. Gao, J. Am. Chem. Soc., 2018, 140, 12469–12475 CrossRef CAS PubMed.
- G. P. Anipsitakis and D. D. Dionysiou, Environ. Sci. Technol., 2003, 37, 4790–4797 CrossRef CAS PubMed.
- X. Chen, J. Zhou, H. Yang, H. Wang, H. Li, S. Wu and W. Yang, Chemosphere, 2022, 287, 132074 CrossRef CAS PubMed.
- H. Zeng, L. Deng, H. Zhang, C. Zhou and Z. Shi, J. Hazard. Mater., 2020, 400, 123297 CrossRef CAS PubMed.
- M. Xing, W. Xu, C. Dong, Y. Bai, J. Zeng, Y. Zhou, J. Zhang and Y. Yin, Chem, 2018, 4, 1359–1372 CAS.
- C. Dong, W. Fang, Q. Yi and J. Zhang, Chemosphere, 2022, 136205 CrossRef CAS PubMed.
- C. Dong, Y. Bao, M. Xing, M. Anpo and J. Zhang, Appl. Catal., A, 2022, 118963 Search PubMed.
- J. Liu, C. Dong, Y. Deng, J. Ji, S. Bao, C. Chen, B. Shen, J. Zhang and M. Xing, Water Res., 2018, 145, 312–320 CrossRef CAS PubMed.
- B. Sheng, F. Yang, Y. Wang, Z. Wang, Q. Li, Y. Guo, X. Lou and J. Liu, Chem. Eng. J., 2019, 375, 121989 CrossRef CAS.
- H. Zhou, L. Lai, Y. Wan, Y. He, G. Yao and B. Lai, Chem. Eng. J., 2020, 384, 123264 CrossRef CAS.
- Y. Zhang, J. Niu and J. Xu, Chem. Eng. J., 2020, 381, 122718 CrossRef CAS.
- Q. Yi, W. Liu, J. Tan, B. Yang, M. Xing and J. Zhang, Chemosphere, 2020, 244, 125539 CrossRef CAS PubMed.
- J. Ji, R. M. Aleisa, H. Duan, J. Zhang, Y. Yin and M. Xing, Iscience, 2020, 23, 100861 CrossRef CAS PubMed.
- Q. Yi, J. Ji, B. Shen, C. Dong, J. Liu, J. Zhang and M. Xing, Environ. Sci. Technol., 2019, 53, 9725–9733 CrossRef CAS PubMed.
- M. Du, Q. Yi, J. Ji, Q. Zhu, H. Duan, M. Xing and J. Zhang, Chin. Chem. Lett., 2020, 31, 2803–2808 CrossRef CAS.
- L. Zhu, J. Ji, J. Liu, S. Mine, M. Matsuoka, J. Zhang and M. Xing, Angew. Chem., 2020, 132, 14072–14080 CrossRef.
- W. Li, Y. Zhang, P. Zhao, P. Zhou, Y. Liu, X. Cheng, J. Wang, B. Yang and H. Guo, J. Hazard. Mater., 2020, 393, 122399 CrossRef CAS PubMed.
- S. Yang, P. Wu, J. Liu, M. Chen, Z. Ahmed and N. Zhu, Chem. Eng. J., 2018, 350, 484–495 CrossRef CAS.
- S. Taujale, L. R. Baratta, J. Huang and H. Zhang, Environ. Sci. Technol., 2016, 50, 2345–2353 CrossRef CAS PubMed.
- J. Huang, Y. Dai, K. Singewald, C.-C. Liu, S. Saxena and H. Zhang, Chem. Eng. J., 2019, 370, 906–915 CrossRef CAS.
- Z. Chen, S. Bi, G. Zhao, Y. Chen and Y. Hu, Sci. Total Environ., 2020, 711, 134715 CrossRef CAS PubMed.
- C. Li, J. Wu, W. Peng, Z. Fang and J. Liu, Chem. Eng. J., 2019, 356, 904–914 CrossRef CAS.
- S. Lu, G. Wang, S. Chen, H. Yu, F. Ye and X. Quan, J. Hazard. Mater., 2018, 353, 401–409 CrossRef CAS PubMed.
- Y. Wang, D. Cao, M. Liu and X. Zhao, Catal. Commun., 2017, 102, 85–88 CrossRef CAS.
- J. Ma and N. Graham, Water Res., 2000, 34, 3822–3828 CrossRef CAS.
- Z. Li, Y. Sun, W. Huang, C. Xue, Y. Zhu, Q. Wang and D. Liu, J. Environ. Sci., 2020, 88, 46–58 CrossRef CAS PubMed.
- S. Mostafa and F. Rosario-Ortiz, Environ. Sci. Technol., 2013, 47, 8179–8186 CrossRef CAS PubMed.
- Z. Li, D. Liu, Y. Zhao, S. Li, X. Wei, F. Meng, W. Huang and Z. Lei, Chemosphere, 2019, 233, 549–558 CrossRef CAS PubMed.
- S. Zhu, X. Li, J. Kang, X. Duan and S. Wang, Environ. Sci. Technol., 2018, 53, 307–315 CrossRef PubMed.
- Y. Gao, T. Wu, C. Yang, C. Ma, Z. Zhao, Z. Wu, S. Cao, W. Geng, Y. Wang and Y. Yao, Angew. Chem., Int. Ed., 2021, 60, 22513–22521 CrossRef CAS PubMed.
- S. Lu, G. Wang, S. Chen, H. Yu, F. Ye and X. Quan, J. Hazard. Mater., 2018, 353, 401–409 CrossRef CAS PubMed.
- P. Gao, X. Tian, Y. Nie, C. Yang, Z. Zhou and Y. Wang, Chem. Eng. J., 2019, 359, 828–839 CrossRef CAS.
- Z. Li, Y. Sun, W. Huang, C. Xue, Y. Zhu, Q. Wang and D. Liu, J. Environ. Sci., 2020, 88, 46–58 CrossRef CAS PubMed.
- G.-D. Fang, D. D. Dionysiou, Y. Wang, S. R. Al-Abed and D. Zhou, J. Hazard. Mater., 2012, 227, 394–401 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a and
Jinlong
Zhang
a and
Jinlong
Zhang