
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective conversion of polyethylene wastes to methylated aromatics through cascade catalysis†
Jindi
Duan
a,
Hai
Wang
a,
Hangjie
Li
a,
Lujie
Liu
a,
Kai
Fan
b,
Xiangju
Meng
 b,
Zhiguo
Zhang
*a,
Liang
Wang
*a and
Feng-Shou
Xiao
*a
b,
Zhiguo
Zhang
*a,
Liang
Wang
*a and
Feng-Shou
Xiao
*a
aKey Lab of Biomass Chemical Engineering of Ministry of Education, College of Chemical and Biological Engineering, Zhejiang University, Hangzhou 310027, China. E-mail: zhiguo.zhang@zju.edu.cn; liangwang@zju.edu.cn; fsxiao@zju.edu.cn
bKey Lab of Applied Chemistry of Zhejiang Province, Department of Chemistry, Zhejiang University, Hangzhou 310028, China
First published on 7th April 2023
Abstract
Upcycling polyethylene into aromatics has attracted much attention for converting plastic wastes into valuable chemicals, but the general routes strongly depend on harsh conditions, precious metals, and/or wide product distributions. Herein, we report the catalytic conversion of polyethylene to methylated aromatics with high yields over the catalysts of aluminosilicate MFI zeolite nanosheets (s-ZSM-5) and mesoporous MFI zeolite modified with zinc species (Zn/meso-ZSM-5) for cascade reactions of polyethylene depolymerization and olefin aromatization, respectively. Following this route, polyethylene was fully converted into C5+ products yielding 60.1%, of which 76.7% were aromatics at 400 °C, and 93.4% of the collected aromatics were industrially important methylated aromatics, including toluene, xylene, and mesitylene. This strategy can be extended to convert single-use plastics into methylated aromatics, such as polyethylene bottles, shopping bags, food packages, and DKR-310 plastics.
Broader contextThis manuscript focuses on the selective conversion of polyethylene plastic wastes to methylated aromatics through cascade catalysis, which is a great challenge for obtaining valuable platform chemicals from plastic wastes. The previous routes for this conversion rely on the zeolite-catalyzed pyrolysis and platinum-catalyzed direct aromatization. However, the former requires high temperatures and suffers from poor efficiency, and the latter relies on precious metals. This work has significant advances compared with the previous ones, realizing the catalytic conversion of polyethylene to methylated aromatics with high yields. The crucial technique is to employ the catalysts of aluminosilicate MFI zeolite nanosheets (s-ZSM-5) and mesoporous MFI zeolite modified with zinc species (Zn/meso-ZSM-5) for cascade reactions of polyethylene depolymerization and olefin aromatization, respectively. Typically, polyethylene was fully converted into C5+ products yielding 60.1%, of which 76.7% were aromatics at 400 °C, and 93.4% of the collected aromatics were industrially important methylated aromatics. This work introduces a rational design of the catalysts to facilitate the PE-to-aromatics reaction and is expected to be extended to convert more practical single-use plastics into valuable aromatics. |
Introduction
The global production of synthetic and petroleum-based plastics continues to increase due to a huge demand for multiple uses, which reached 368 million tonnes in 2019.1 Such a huge amount corresponds to roughly 7% of carbon resources in crude oil and natural gas produced.2 It is worth noting that over 75% of these are single-use plastics for preserving food and health care systems in the short term, which are then discarded.3–7 Plastic waste is a serious matter of concern due to its disruptive impact on the environment, and only a very small fraction is recycled.8–11 For example, tens of millions of metric tonnes of plastics enter the ocean annually, accumulating in the ecosystem.3 Following these issues, how to deal with the current plastic waste is still unsolved. A general route is burning them to produce heat, but it simultaneously produces CO2 and toxic molecules. More end-of-life management strategies with sustainability are required, but it is challenging.Moving towards a closed-loop life cycle for single-use plastics, mechanical recycling after plastic waste collection and separation has limited success because of the recyclable products with properties that are not as good as the original ones.12,13 In contrast, the chemical conversion of polyolefins into valuable fuels or chemicals is highly desired for realizing an upcycling process. According to this route, gasoline, diesel, olefins, and aromatics have been developed from the selective conversion of polyethylene (PE) and polypropylene (PP).14–19 Particularly, aromatics, such as toluene, xylene, and mesitylene, have attracted considerable attention owing to their importance in the modern chemical industry as platform molecules and fuel additives,20–23 as well as the current gaps between supply and demand. The general process for producing aromatics is naphtha cracking, which normally generates a mixture of mono and polyaromatic hydrocarbons.23 A recent trend is to directly convert PE wastes into aromatics through zeolite-catalyzed pyrolysis, but their selectivity is insufficient with abundant low-value gases (e.g., methane) and polyaromatics because of the high reaction temperature (e.g. 600–700 °C).24–32 Much attention has been focused on reducing the temperature in zeolite-catalyzed PE aromatization, but the efficiency still needs further improvement. For example, Liu et al.33 adopted HZSM-5 for catalyzing the pyrolysis of plastics at 450 °C, obtaining a limited amount of liquid products (∼32.1%) with abundant light gases. In the liquid products, xylene and trimethylbenzene have a fraction of ∼74.3%. Huang et al.34 reported the direct PE aromatization to toluene and xylene over Ga-loaded ZSM-5 at 500 °C, the heavy aromatics appeared as major by-products. At 280 °C, PE was successfully converted to long-chain alkyl aromatics, but this process depends on the use of precious Pt nanoparticle catalysts.35 The sustainable route for producing aromatics from PE wastes with desired features, including high selectivity, free of precious metals, energy-efficient, and mixed-feedstock-agnostic, is highly desired, but it is still unsuccessful yet.
Herein, we have constructed a tandem catalyst system by combining the aluminosilicate MFI zeolite nanosheets (s-ZSM-5, with high activity and selectivity toward PE depolymerization into olefins36) with mesoporous MFI zeolite loaded with ZnOx species (Zn/meso-ZSM-5) for direct conversion of PE to methylated aromatics at a mild temperature (400 °C) relative to the general pyrolysis. By packing the Zn/meso-ZSM-5 catalyst with the s-ZSM-5 catalyst in the reactor, cascade reactions of PE depolymerization and olefin aromatization benefit the selective conversion of PE or PE-rich plastics into methylated aromatics. In the collected C5+ products with a yield of 60.1% from a single run under the full conversion of PE, the aromatic products have a fraction of 76.7%, and 93.4% of them were methylated aromatics, toluene, xylene, and mesitylene. Due to the low temperature, the selectivity to methane was negligible (<0.1%), which is much lower than that reported in the previous pyrolysis process (>15%).37–40
Results
Catalytic PE-to-aromatics conversion
The initial test was performed in the catalytic conversion of PE using the MFI zeolite nanosheets (s-ZSM-5, Fig. S1, ESI†), because of its short b-axis thickness benefiting the rapid molecular diffusion for PE depolymerization.36 In the test with a mixture of the PE feed and s-ZSM-5 zeolite in a fixed bed reactor at 400 °C, the PE was fully converted to light gases (C1–C4) with a selectivity of 45.1% and C5+ products with a selectivity of 54.9% (Fig. 1(a)). In this case, the wax on the catalyst bed was not considered for calculating the product selectivity, which is summarized in Tables S2 and S4 (ESI†). In the C5+ products, the aromatics have a limited fraction of 34.5%, resulting in the one-pass yield of aromatics at 16.6% (Fig. 1(a)). Such performances are similar to those of the previous pyrolysis process with a large amount of undesired gaseous products and a limited amount of aromatics.15–18 In the other hydrocarbon products, the olefins are dominant, therefore, it is expected to convert them through aromatization to improve the yield of the targeted aromatics. A ZnOx-loaded commercial ZSM-5 (Zn/ZSM-5, Si/Al ratio at 12, Zn content at 3.0 wt%, Fig. S2, ESI†), which is well known as a catalyst for the aromatization of methanol, olefins, and alkanes,41–45 was employed. Physically mixing s-ZSM-5 with Zn/ZSM-5 did not increase the yield of C5+ products, but improved the fraction of aromatics to 45.9% in the C5+ mixture (Fig. 1(a)). In this case, the one-pass yield of aromatics was 23.8% with multiple alkyl aromatics (Fig. S3, ESI†). A further attempt was performed by packing the catalysts in a dual-bed manner with Zn/ZSM-5 below s-ZSM-5, resulting in a similar yield of C5+ products at 49.9% to that in the physically mixed catalysts, but the fraction of aromatics was obviously enhanced to 60.4%. These results suggest a significant advantage of spatially separating the two zeolite catalysts (Fig. 1(a)).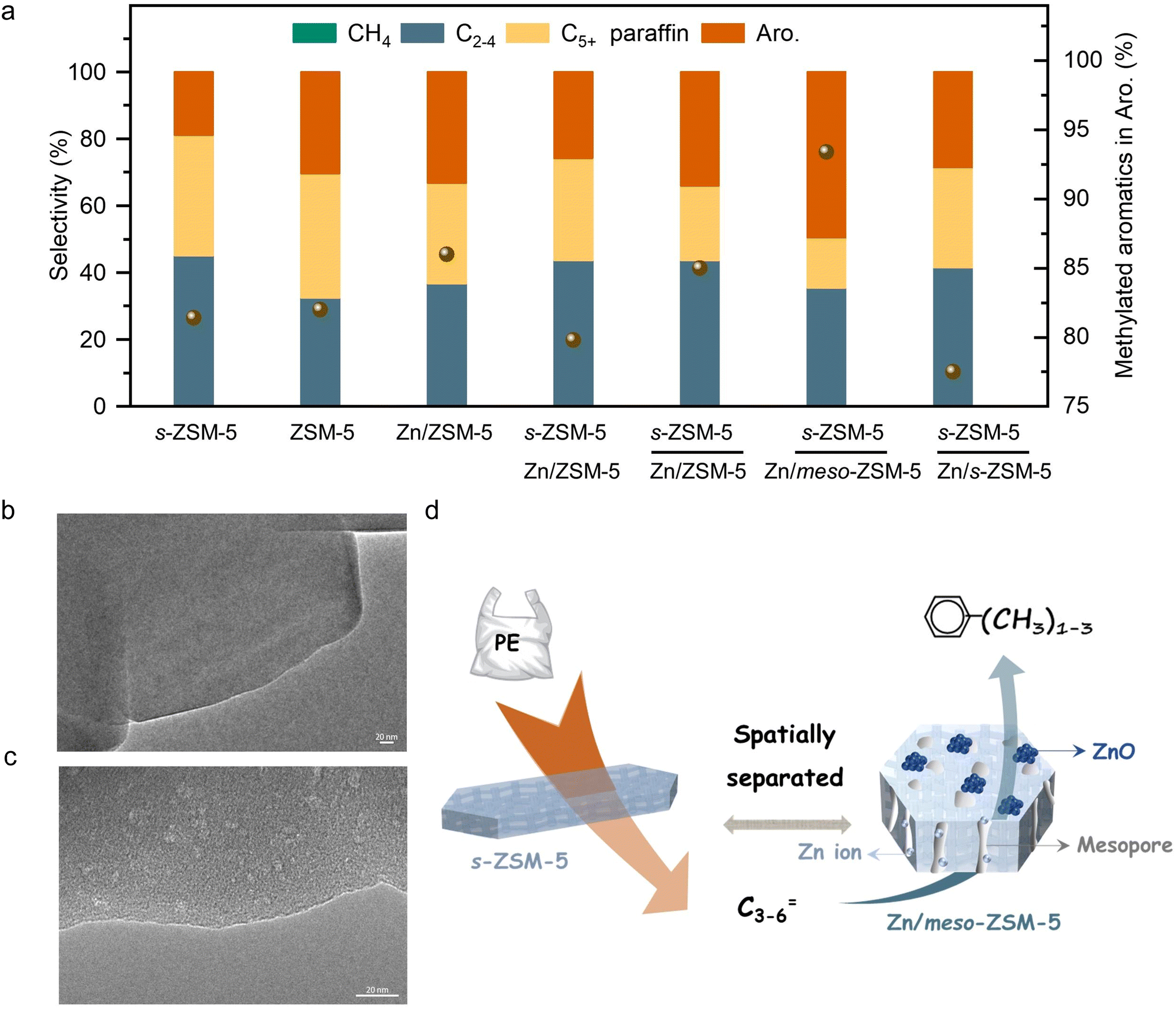 | ||
| Fig. 1 (a) Data characterization of the aromatization of PE over various catalysts. Reaction conditions: the mixture of 500 mg of PE and 100 mg of s-ZSM-5 in the first bed, 400 mg of catalyst for aromatization in the second bed, feed gas of 3.3% H2/29.7% Ar/67% N2, a flow rate of 3 mL min−1, 400 °C, 4 h. * refers to the physically mixed PE/s-ZSM-5 and Zn/ZSM-5. A small amount of C5+ olefins were not separately presented, but included in the paraffin products. TEM images of the (b) ZSM-5 zeolite and (c) meso-ZSM-5 zeolite. (d) Schematic diagram of reaction flow over s-ZSM-5 and Zn/meso-ZSM-5. | ||
Photographs of the spent catalysts in the dual-bed reactor show the s-ZSM-5 bed to be with light color and Zn/ZSM-5 bed with dark color, suggesting distinguished behavior in coke formation over different zeolite catalysts (Fig. S4a, ESI†). The superior coke resistance of s-ZSM-5 zeolite in PE depolymerization has been identified previously,36 therefore, the coke formation on Zn/ZSM-5 should be the control factor for the PE-to-aromatics conversion. We expected to further improve the coking resistance and selectivity to aromatics by optimizing the porosity and acidity.46–52 By an alkaline treatment using NaOH (0.2 M aqueous solution) and pyridine (see ESI† for details), ZSM-5 zeolite with some mesopores (meso-ZSM-5) was obtained. XRD patterns confirmed similar diffractions before and after the alkaline treatment, indicating the well-maintained MFI structure (Fig. S5a, ESI†). N2 sorption of the meso-ZSM-5 showed a profile with a hysteresis loop at 0.4–1.0 assigned to the existence of mesopore, giving the surface area at 370.9 m2 g−1 that exceeds the untreated ZSM-5 zeolite (Fig. S5b and Table S1, ESI†). TEM images provided direct observation of the mesopores (Fig. 1(b), (c) and Fig. S7, ESI†). By combining the Zn/meso-ZSM-5 with s-ZSM-5 in the PE conversion in a dual-bed manner, the yield of C5+ products and the corresponding fraction of aromatics were improved to 60.1% and 76.7%, respectively (Fig. 1(a), (d), and Fig. S8b, ESI†). In this case, the fraction of methylated aromatics including toluene, xylene, and mesitylene was as high as 93.4% in total aromatics, where the other 6.6% was the dominant long-chain aromatics (Fig. 2(a)–(c) and Fig. S10, ESI†). The 13C NMR spectrum of the C5+ product contains signals in the aromatic region (120–150 ppm), most of them corresponding to carbons of unsubstituted rings (Fig. 2(e) and Fig. S11b, ESI†). In addition, the 1H NMR spectrum of the C5+ products showed that most hydrogens were associated with benzene rings of monoaromatics (6.5–7.4 ppm) with an extremely weak signal to polyaromatic rings (7.4–9.0 ppm) (Fig. 2(d) and Fig. S11a, ESI†).
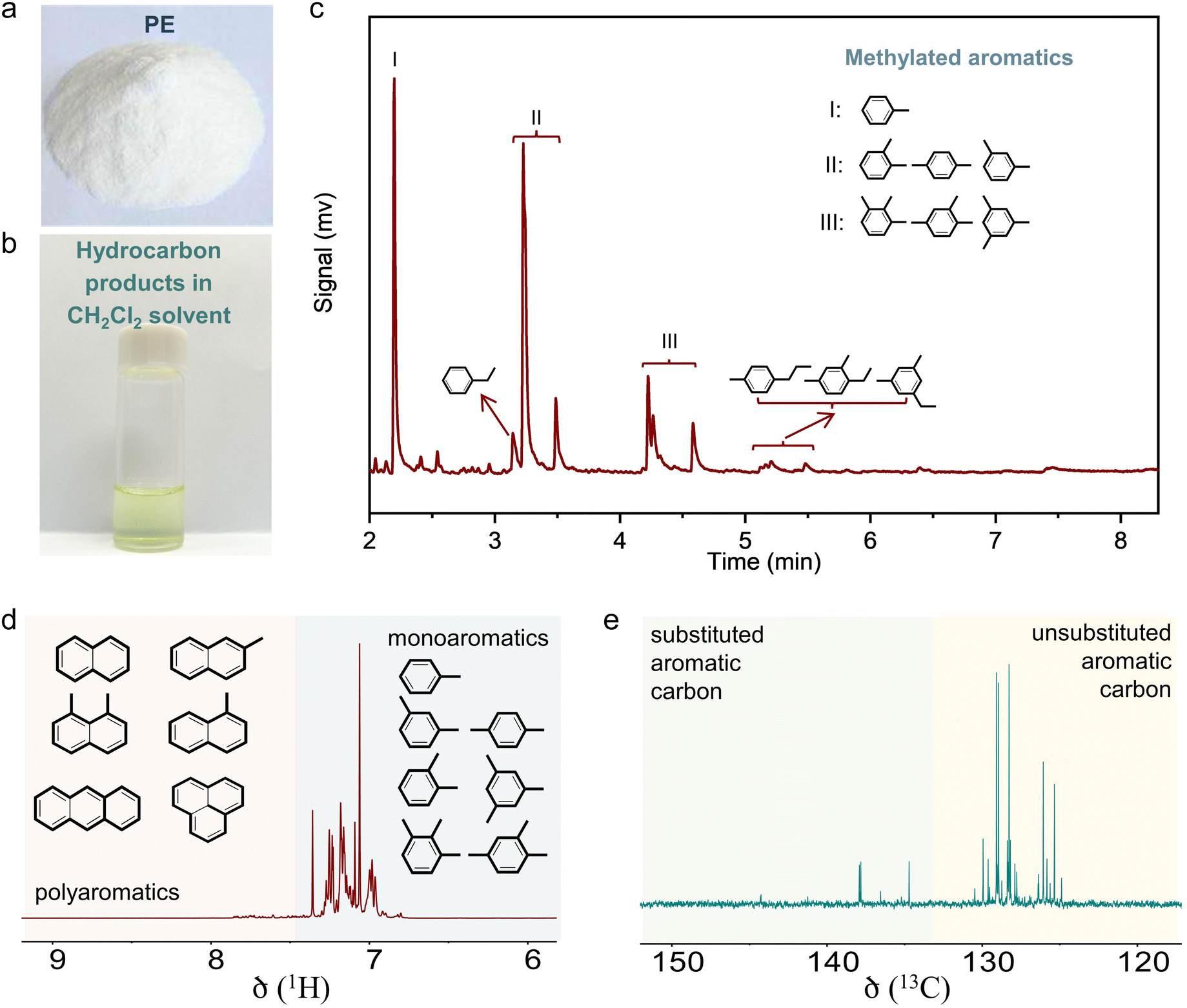 | ||
| Fig. 2 (a) Photographs of the reactant polyethylene. (b) Photographs of the liquid products. (c) GC curves and (d) 1H and (e) 13C NMR spectra analyzing the liquid products. Reaction conditions: the mixture of 500 mg of PE and 100 mg of s-ZSM-5 in the first bed, 400 mg of Zn/meso-ZSM-5 for aromatization in the second bed, feed gas of 3.3% H2/29.7% Ar/67% N2, a flow rate of 3 mL min−1, 400 °C, 4 h. | ||
To clarify the reaction intermediates for aromatization, we show the intermediate products from PE depolymerization over s-ZSM-5 catalysts (Table S2, ESI†), including methane, C2–C4 molecules (olefins dominant, o/p at 10.3), C5+ molecules (C5–C6 with o/p at 1.9, and various aromatics). In this step, some aromatic products were formed (34.5% fraction in the C5+ products), which was unavoidable because of the appropriate temperature (400 °C) and strong acidity of s-ZSM-5 zeolite that would benefit the aromatization of PE. In this case, the aromatics were complex with multiple alkylated hydrocarbons (e.g. C10+).
For the subsequent aromatization step in the cascade catalysis, all the olefins might be converted through aromatization, but the C4–C6 olefins should be dominant because of the obviously decreased selectivity to C4–C6 products compared to that with the s-ZSM-5 catalyzed PE depolymerization test (Table S2, ESI†). This result is in good agreement with the general knowledge that these olefins favor participating in the aromatization more than the lighter olefins (e.g. ethylene and propylene). In the test, C2 and C3 products were dominant alkanes rather than olefins, suggesting that ethylene and propylene also participate in consuming hydrogen from the aromatization step, which promotes the aromatization reaction thermodynamically.46,53,54 Because of the light olefin intermediate, the methylated aromatics were the dominant products, which is in good agreement with the earlier results that the relatively light olefins would benefit the formation of methylated aromatics.55–58
Zeolite structure for aromatization
Fig. S2b (ESI†) shows photographs of spent Zn/meso-ZSM-5 after the PE-to-aromatic conversion, giving the gray color that is much lighter than that of the spent Zn/ZSM-5 without mesopores. Thermogravimetric analysis (TG) was used to roughly quantify the coke amount on the spent zeolites, giving the weight loss assigned to coke burning at 2.1% and 2.7% over Zn/meso-ZSM-5 and Zn/ZSM-5, respectively (Fig. S12a, ESI†). In the temperature-programmed oxidation tests of these catalysts with molecular oxygen (O2-TPO), the spent Zn/meso-ZSM-5 showed CO2 signals (m/z at 44 in the mass spectra detector) at 420 and 510 °C, which is lower than 560 °C in the tests over spent Zn/ZSM-5, indicating the less graphitized coke on Zn/meso-ZSM-5 (Fig. S12b, ESI†). We also performed the Raman characterization of the spent Zn/meso-ZSM-5 (Fig. S12c, ESI†), showing obvious modes at 1380 cm−1 and 1580 cm−1, which are assigned to the D and G signals of coke species. Compared with Zn/meso-ZSM-5, Raman signals of the spent ZSM-5 zeolite were much stronger, confirming the formation of more coke species. These results indicated the importance of mesopores in MFI zeolite for hindering the coke formation, because of the improved mass diffusion, as identified previously.36 We studied the acidity of the meso-ZSM-5 and ZSM-5 zeolites by temperature-programmed desorption of ammonia (NH3-TPD) giving comparable profiles with signals at 240–245 °C and 475–480 °C, suggesting the maintained acidity after introducing mesopores to the ZSM-5 zeolite (Fig. S13a, ESI†). For the ZnOx loaded meso-ZSM-5, the Zn species were uniformly distributed on the zeolite matrix (Fig. S14–S16, ESI†), which eliminated the strong acid sites, as confirmed by the undetectable signal higher than 400 °C in the NH3-TPD profile (Fig. S13b, ESI†). This result suggests the removal of excessive strong acid sites by ZnOx loading, which further improved the coke resistance and increased the selectivity toward aromatics because the aromatization requires mild Brønsted acidity.41–44 Based on these results, the cascade processes of PE depolymerization to olefins and olefin aromatization could be identified in the dual-bed reactor. The MFI zeolite with nanosheet morphology (s-ZSM-5) has a significant advantage in rapid molecular diffusion because of the short b-axis distance, which further motivated the study of Zn/s-ZSM-5 for the aromatization steps (Fig. S17, ESI†). Therefore, we performed the PE conversion over cascade catalysts of s-ZSM-5 and Zn/s-ZSM-5, resulting in the fraction of aromatics in the C5+ products at 48.9%, which is far from the level over s-ZSM-5 and Zn/meso-ZSM-5 catalysts (76.7%), and even lower than that over s-ZSM-5 and Zn/ZSM-5 catalysts (60.4%, Fig. 1(a)).To understand this phenomenon, we directly fed propylene to the Zn/meso-ZSM-5, Zn/ZSM-5, and Zn/s-ZSM-5 catalysts to explore their structure–performance relationship in olefin aromatization. Propylene was employed as a model because its aromatization was more challenging relative to the relatively heavier olefins (e.g., C5+ olefins). Ethylene aromatization was not considered in this test because of the extremely low selectivity in the PE decomposition. The conversion of propylene over Zn/meso-ZSM-5 reached 97.7%, significantly higher than that of Zn/ZSM-5 (89.1%) and Zn/s-ZSM-5 (71.4%) (Fig. 3(a)). Zn/meso-ZSM-5 exhibited higher selectivity to aromatics (47.5%) than that over Zn/ZSM-5 (26.2%) and Zn/s-ZSM-5 (17.8%). We also analyzed the coke amount in TG analysis, giving the weight loss assigned to coke burning at 2.1%, 4.5%, and 1.1% over Zn/meso-ZSM-5, Zn/ZSM-5, and Zn/s-ZSM-5 catalysts, respectively (Fig. 3(b)). The Zn/s-ZSM-5 exhibited the lowest propylene conversion and aromatization selectivity among these three catalysts, because of the insufficient retention time of olefins in the zeolite with nanosheet morphology. For Zn/ZSM-5, the aromatization selectivity was improved compared to that with Zn/s-ZSM-5, but the coking occurred more seriously. As a result, the Zn/meso-ZSM-5 exhibited a balance between coke formation and aromatization.
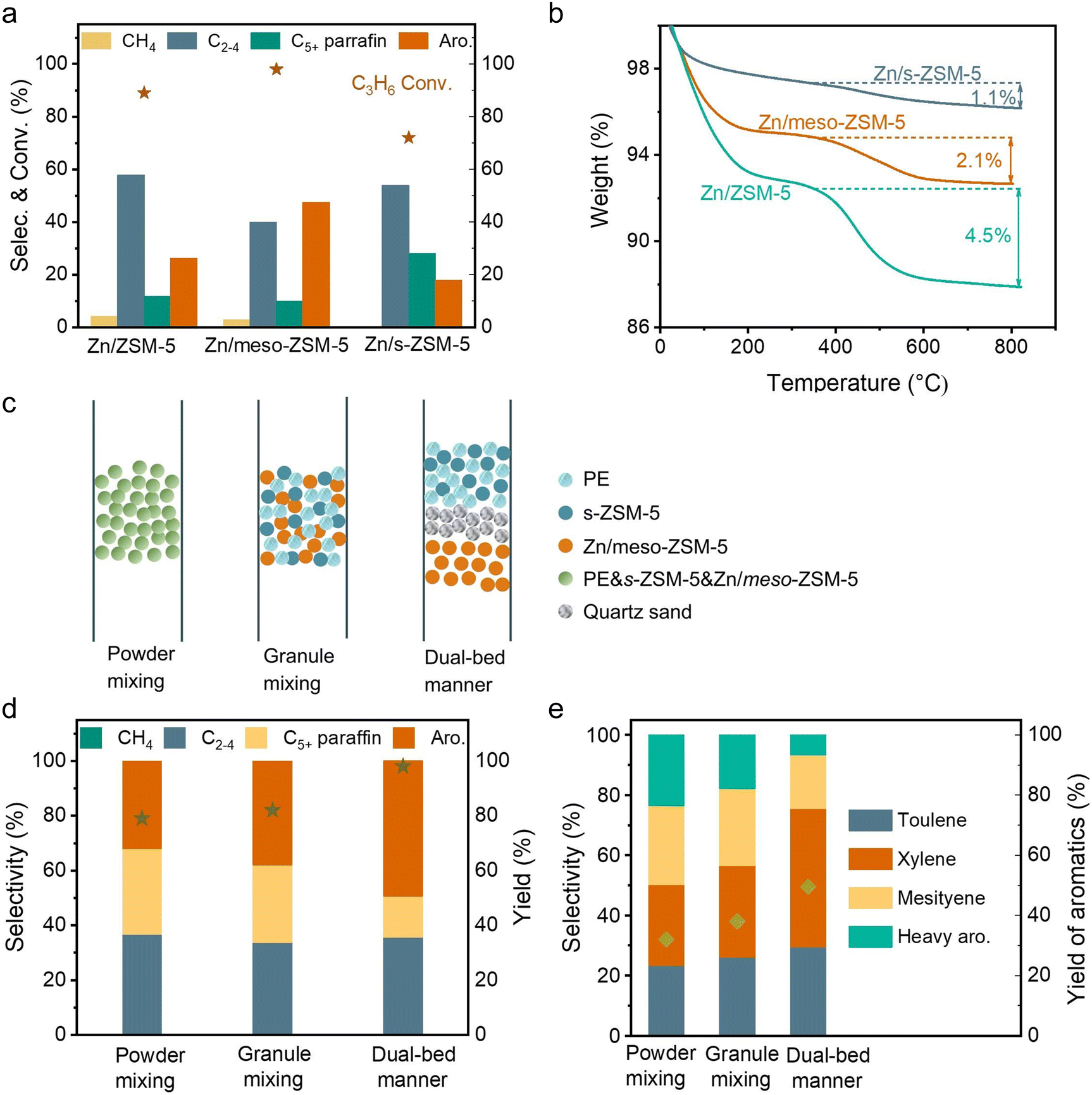 | ||
| Fig. 3 The model of propylene aromatization: (a) conversion of propylene and selectivity of different products. Reaction conditions: 0.4 g of catalyst, feed gas of 10%C3H6/90%N2 at 10 mL min−1, 400 °C, 4 h. (b) TG profiles of different spent catalysts. (c) Modes of the s-ZSM-5 and Zn/meso-ZSM-5 with different proximity. (d) Data characterization of the aromatization of PE in various modes with different proximities between s-ZSM-5 and Zn/meso-ZSM-5 catalysts. (e) Selectivity of different methylated aromatics in aromatic hydrocarbons with different proximities between s-ZSM-5 and Zn/meso-ZSM-5 catalysts. | ||
Catalyst packing manner in the reactor
For cascade catalysis, the catalysts with multiple functions have been extensively explored, and closer is better describing the spatial distribution of different active sites has been regarded general principle in many cases. For example, in the syngas-to-aromatics conversion, the physical mixture of catalysts for syngas conversion and olefin aromatization exhibited better performances than the spatially separated catalysts in dual beds.50 However, we found a different trend in the PE-to-aromatics conversion over s-ZSM-5 and Zn/meso-ZSM-5 catalysts. As shown in Fig. 3(c), the physically mixed s-ZSM-5 and Zn/meso-ZSM-5 in different manners, including the powder and granule mixture (powder mixture, the two zeolite powders were mixed together and then made into granules for the catalysis; granule mixture, the two zeolites were made into granules separately and then mixed in the reactor), showed an obviously low fraction of aromatics in C5+ products (50.5% and 57.2%), compared with the dual-bed catalysts (76.7%, Fig. 3(d) and (e)). These results support the significant advantage of spatially separated s-ZSM-5 and Zn/meso-ZSM-5 catalysts for efficient conversion, while the catalysts with proximity reduced the performances. The reaction proceeds with PE depolymerization to light olefins, olefin oligomerization, cyclization, and dehydrogenation. The PE depolymerization requires strong Brønsted acid sites on s-ZSM-5. The olefin oligomerization and cyclization occur on acid sites of both s-ZSM-5 and Zn/meso-ZSM-5, and the ZnOx benefits the dehydrogenation to produce aromatics (Fig. 4(a)).44,45,50,51 A highly efficient process requires the reactions to occur sequentially over the different active sites, which is a challenge for the physically mixed catalysts. For example, PE would access and depolymerize on Zn/meso-ZSM-5 of the physically mixed catalyst, which competes with the aromatization that resulted in a reduced yield of aromatic products. In addition, Zn/meso-ZSM-5 catalyzed PE depolymerization to form abundant heavy hydrocarbons that favored the production of polycyclic aromatics and long-chain alkyl aromatics rather than methylated aromatics of toluene, xylene, and mesitylene. These catalytic results are summarized in Fig. S4 (ESI†). Following the above steps, we suppose to draw the spatial distribution maps of the multiple active sites. The proximity between zinc sites and the acid sites of meso-ZSM-5 favors promoting the olefin aromatization that relies on ZnOx-optimized acid sites for synergistic cyclization and dehydrogenation. Simultaneously, the spatial separation of strong acidic sites on s-ZSM-5 and ZnOx species on meso-ZSM-5 is necessary to avoid deep hydrogenation to form the undesired saturated alkanes (Fig. S18, ESI†).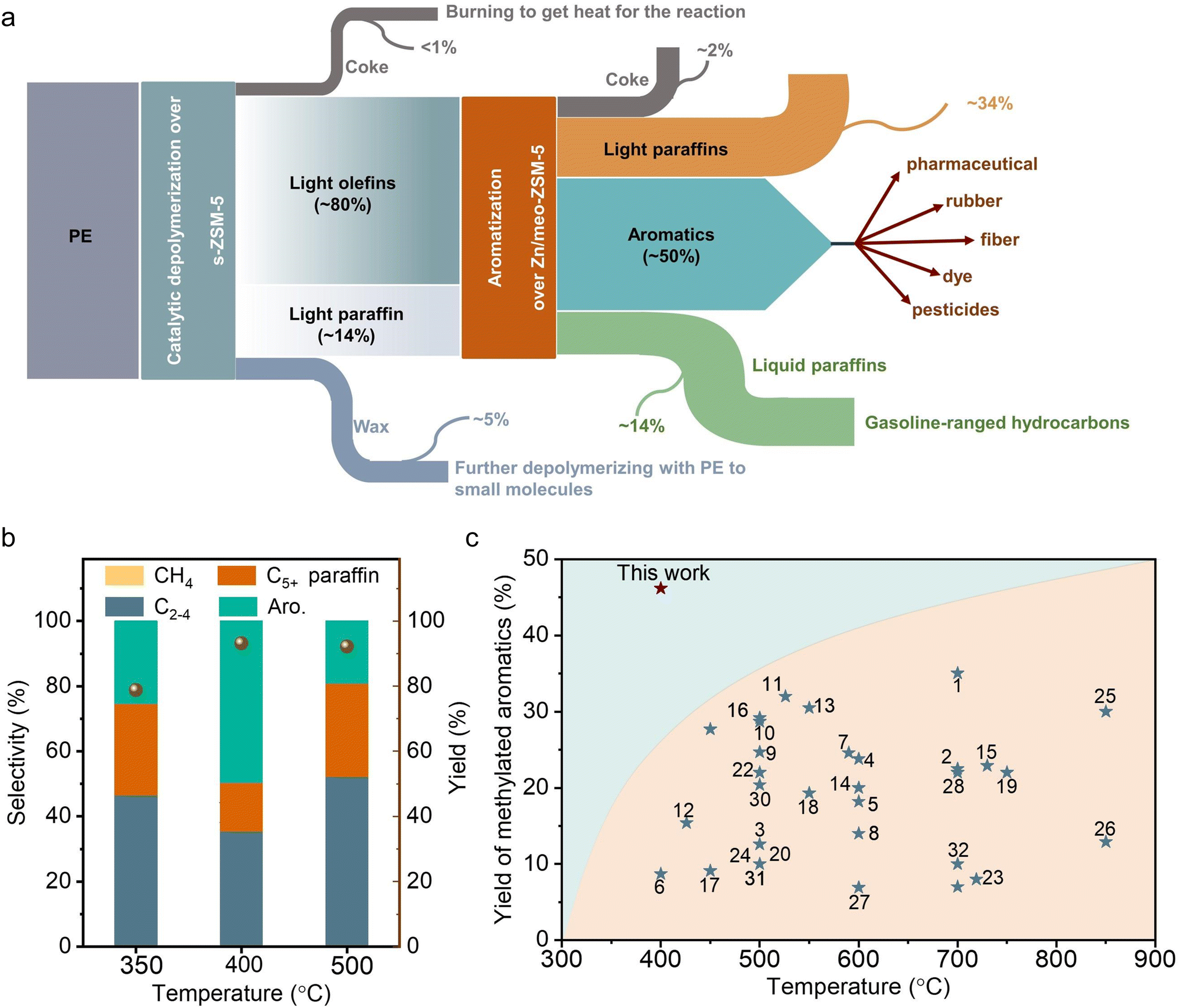 | ||
| Fig. 4 (a) Schematic presentation of the carbon flow in the aromatization of PE over s-ZSM-5 and Zn/meso-ZSM-5 and further utilization of the products. (b) Effect of temperature on PE aromatization over s-ZSM-5 and Zn/meso-ZSM-5. Reaction conditions: the mixture of 500 mg of PE and 100 mg of s-ZSM-5 in the first bed, 400 mg of Zn/meso-ZSM-5 in the second bed, feed gas of 3.3% H2/29.7% Ar/67% N2 at 3 mL min−1, 4 h. (c) The yields of the methylated aromatics at different temperatures for polyethylene aromatization over efficient catalysts in literatures and in this work. | ||
Influence of the reaction conditions
In the previous tests on direct pyrolysis of PE into methylated aromatics, the one-pass yield of methylated aromatics was usually lower than 30.0% at temperatures lower than 600 °C, and could reach ∼35.0% at 700 °C (Fig. 4(c)). In contrast, the one-pass yield of methylated aromatics reached 46.7% at a low temperature of 400 °C over the s-ZSM-5 and Zn/meso-ZSM-5 cascade catalysts, indicating the significant advantage of the cascade reaction system. It is reasonably expected to further improve the yield of methylated aromatics by raising the reaction temperature, but the cascade catalysts showed reduced selectivity to methylated aromatics with the formation of more light alkanes. For example, at 500 °C, the C1–C4 selectivity was 52.3% with selectivity to C5+ products at 47.7%, where the fraction of aromatics was 40.1% in the C5+ products (Fig. 4(b)). Compared with the PE aromatization at 400 °C, the reaction at 500 °C formed more C2–C4 alkane and C5+ hydrocarbon products with less aromatic products. It is a general phenomenon in the aromatization process,59–63 because the higher temperature would cause stronger hydrogen transfer/hydrogenation and cracking that causes a high fraction of C2–C4 alkane to reduce the aromatic yield. In addition, the rapid coke formation at higher temperatures also reduced the catalytic activity for aromatization which caused a lower yield of aromatics. In addition, the aromatic products from the reaction at 500 °C were mixtures of long-chain aromatic and methylated aromatics, which is also different from that at 400 °C with dominant methylated aromatics.We studied thermodynamics for the aromatization of long-chain alkanes (see ESI† for details). The direct conversion of heavy hydrocarbon molecules into methylated aromatics is strongly endothermic, which is unfavorable at mild temperatures (Fig. 5(a)). For example, at 400 °C, the conversion of a long-chain hydrocarbon to xylene [C8nH16n+2 → nC8H10 +(3n + 1) H2, n ≈ 250] results in thermodynamic values of ΔH at 47![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 943 kJ mol−1 and ΔG at 4972 kJ mol−1 (estimated using Benson group contributions for long-chain n-alkanes).35 In contrast, the aromatization of light olefins to methylated aromatics is favorable at 400 °C, as supported by ΔH at 20.8 kJ mol−1 and ΔG at −248.4 kJ mol−1 in the model of butene-to-xylene conversion (2C4H8 → C8H10 + 3H2), and ΔH at −96.4 kJ mol−1 and ΔG at −516.3 kJ mol−1 in the model of propylene-to-xylene conversion (8C3H6 → 3C8H10 + 9H2). In these aromatization processes, the olefin hydrogenation occurred to consume the hydrogen, which thermodynamically contributes to the reaction (e.g., C4H8 + H2 → C4H10, ΔH at −125.8 kJ mol−1, ΔG at −184.7 kJ mol−1) (Fig. S19, ESI†). Considering the thermodynamically favorable feature of hydrogen-participated PE cracking into olefins, the cascade reactions with olefin aromatization make the total reaction occur at 400 °C, which significantly outperforms that for the direct conversion of alkanes into aromatics that usually require higher temperatures. This reaction temperature might enhance the energy conservation compared with direct polyethylene to aromatics (>500 °C), and benefit the formation of a lower proportion of benzene/polyaromatics and a higher proportion of the more valuable methylated products.16,24–27,31,64–67
943 kJ mol−1 and ΔG at 4972 kJ mol−1 (estimated using Benson group contributions for long-chain n-alkanes).35 In contrast, the aromatization of light olefins to methylated aromatics is favorable at 400 °C, as supported by ΔH at 20.8 kJ mol−1 and ΔG at −248.4 kJ mol−1 in the model of butene-to-xylene conversion (2C4H8 → C8H10 + 3H2), and ΔH at −96.4 kJ mol−1 and ΔG at −516.3 kJ mol−1 in the model of propylene-to-xylene conversion (8C3H6 → 3C8H10 + 9H2). In these aromatization processes, the olefin hydrogenation occurred to consume the hydrogen, which thermodynamically contributes to the reaction (e.g., C4H8 + H2 → C4H10, ΔH at −125.8 kJ mol−1, ΔG at −184.7 kJ mol−1) (Fig. S19, ESI†). Considering the thermodynamically favorable feature of hydrogen-participated PE cracking into olefins, the cascade reactions with olefin aromatization make the total reaction occur at 400 °C, which significantly outperforms that for the direct conversion of alkanes into aromatics that usually require higher temperatures. This reaction temperature might enhance the energy conservation compared with direct polyethylene to aromatics (>500 °C), and benefit the formation of a lower proportion of benzene/polyaromatics and a higher proportion of the more valuable methylated products.16,24–27,31,64–67
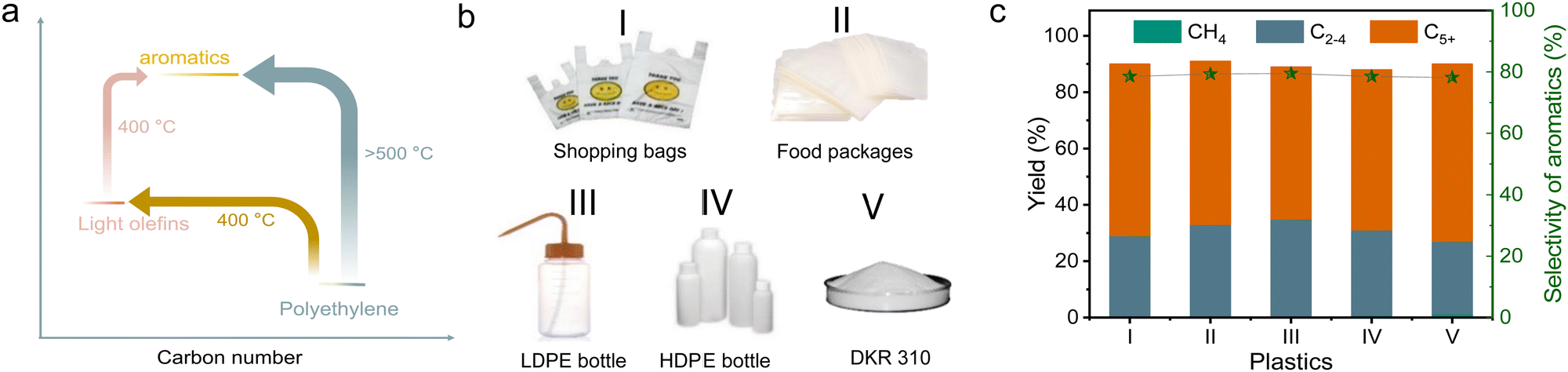 | ||
| Fig. 5 (a) The temperature for the conversion of various molecules with different enthalpy. (b) Photographs of (I) shopping bags, (II) food packages, (III) LDPE bottle, (IV) HDPE bottle, and (V) DKR 310. The component of DKR 310:92.6% PE and 7.4% impurities (the impurities contain 0.4% Fe powder, 3.7% PS, and 3.3% wood). (c) Data characterizing the aromatization of practical plastics and DKR310 in a dual-bed manner over s-ZSM-5 and Zn/meso-ZSM-5 catalysts. Reaction conditions: the mixture of 100 mg of practical plastic and 100 mg of s-ZSM-5 in the first bed, 400 mg of Zn/meso-ZSM-5 for aromatization in the second bed, feed gas of 3.3% H2/29.7% Ar/67% N2 at 3 mL min−1, 400 °C, 4 h. | ||
Aromatization of practically single-use plastic
The cascade route was further used in the upgrading of various practical polyolefin plastics, including supermarket shopping bags, deep-freeze food pouches, low-density polyethylene, and high-density polyethylene, bottles (Fig. 5(b)). The catalyst mixture can effectively convert all these plastic components into various hydrocarbon products with total yields of 89.0–91.5%, and 46.7–51.4% of them were methylated aromatics. We understand that practical plastic wastes usually have impurities such as other kinds of plastics (e.g., polyvinyl chloride and PVC) and inorganic additives. We reasonably introduced PVC to test the performances of cascade catalysts (10% of PVC mixed with 90% of PE), resulting in a high yield of light hydrocarbon products at 96.9%, and 46.8% of them were methylated aromatics (Fig. S20a, ESI†). With the introduction of water to simulate the wet wastes, similar performances were also obtained with the yield of methylated aromatics at 47.5% (Fig. S20b, ESI†).These catalysts also worked efficiently for the conversion of other PE-rich plastics into aromatics, such as DKR 310, which simulates the plastics containing various impurities. In upgrading the DKR 310 plastic with a composition of 92.6% of PE and 7.4% of impurities (0.4% of Fe powder, 3.7% of polystyrene, and 3.3% of wood), the cascade catalysts have the methylated aromatic yield at 49.3% (Fig. 5(c)). Such performance was very similar to that obtained using pure PE, confirming that the impurities in DKR 310 or additives in practical plastics have negligible influence on the catalysis. In the aromatization of polypropylene (PP), accounting for 21% of all global plastics,2 a total yield of collected products was 89.8% (wax not included) with 65.3% selectivity to the C5+ products. Among these C5+ products, 60.5% of them were aromatics, which was much lower than that from PE (Fig. S21a and b, ESI†). To understand the difference in PP conversion, we performed the PP depolymerization test over s-ZSM-5 without Zn/meso-ZSM-5 (Fig. S21c and d, ESI†). As a result, abundant heavier products, including C5–C20 and wax, were obtained, which was different from the equivalent test in PE depolymerization with abundant relative light products (Fig. 1(a)). It is reasonable to imagine that the heavy products from PP depolymerization are not beneficial for the aromatization step in the cascade catalysis.
Recyclability tests on s-ZSM-5 and Zn/meso-ZSM-5 catalysts were performed for the upgrading of PE. After calcination at 500 °C in air, the spent catalyst was fully regenerated with constant performance in the continuous test five times (Fig. S22, ESI†). The XRD and SEM studies on spent s-ZSM-5 and Zn/meso-ZSM-5 catalysts showed that the MFI zeolite structure was well retained during the recycling tests (Fig. S23, ESI†).
Conclusions
In this work, we developed a cascade process for the selective conversion of PE plastics into methylated aromatics, giving the one-pass yield at 60.1% that steadily outperformed the previous pyrolysis techniques. s-ZSM-5 and Zn/meso-ZSM-5 zeolites are crucial for the cascade reactions of PE depolymerization into olefins and then conversion into methylated aromatics. This work provides an ideal route for obtaining methylated aromatics from the various PE-rich plastic wastes, and future work should be focused on engineering the reactor design to realize a continuous reaction, which could further strengthen the vitality for potential utilization.Experimental
Synthesis
Catalytic tests
Catalytic tests were carried out in a fixed-bed reactor involving a quartz tube (internal diameter at 6 mm and 270 mm in length) at constant atmospheric pressure (∼0.1 MPa) with an oven for heating the catalyst bed. PE aromatization was performed by combining the PE depolymerization bed (1st bed) and olefin aromatization bed (2nd bed) in a reaction with a dual-bed manner. In a typical run, 100 mg of s-ZSM-5 catalyst and 500 mg of polyethylene powder were mixed and pressed into particles at 40–60 mesh, which was localized within the upper part of the reaction tube and fixed by quartz wool and quartz sands (40–60 mesh). The 2nd bed contains 400 mg of Zn/meso-ZSM-5 catalyst (40–60 mesh, diluted with 400 mg of quartz sand and fixed by quartz wool). The two beds were separated by quartz sands (40–60 mesh). During catalysis, a feed gas containing 3.3% of H2, 29.7% of Ar, and 67% of N2 was induced with a flow rate of 120 mL h−1. The reaction was performed at 400 (± 5 °C).Author contributions
Jindi Duan: investigation, catalyst preparation, characterization, catalytic tests, thermodynamic calculation, writing – original draft. Hai Wang: the mechanism discussion. Hangjie Li: product analysis and reactor construction. Lujie Liu: the catalyst preparation and characterization. Kai Fan: STEM data curation, formal analysis. Xiangju Meng: helpful discussion. Zhiguo Zhang: initiating and supervising the research, Liang Wang: designing the study, analyzing the data, and writing–reviewing, and editing. Feng-Shou. Xiao: funding acquisition, supervision, writing– review, and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2022YFA1503502) and the National Natural Science Foundation of China (22288101, U21B20101, 22032005, and 22125304). The authors thank Fang Chen for the help in TEM characterization.References
- J. M. Garcia and M. L. Robertson, Science, 2017, 358, 870–872 CrossRef CAS PubMed.
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- M. MacLeod, H. P. H. Arp, M. B. Tekman and A. Jahnke, Science, 2021, 373, 61–65 CrossRef CAS PubMed.
- J. M. Garcia and M. L. Robertson, Science, 2017, 358, 870–872 CrossRef CAS PubMed.
- X. Jie, W. Li, D. Slocombe, Y. Gao, I. Banerjee, S. Gonzalez-Cortes, B. Yao, H. AlMegren, S. Alshihri, J. Dilworth, J. Thomas and T. Xiao, Nat. Catal., 2020, 3, 902–912 CrossRef CAS.
- J. R. Jambeck, R. Geyer, C. Wilcox, T. R. Siegler, M. Perryman, A. Andrady, R. Narayan and K. L. Law, Science, 2015, 347, 768–771 CrossRef CAS PubMed.
- A. J. Martín, C. Mondelli, S. D. Jaydev and J. Perez-Ramirez, Chem, 2021, 7, 1487–1533 Search PubMed.
- A. Chamas, H. Moon, J. Zheng, Y. Qiu, T. Tabassum, J. H. Jang, M. Abu-Omar, S. L. Scott and S. Suh, ACS Sustainable Chem. Eng., 2020, 8, 3494–3511 CrossRef CAS.
- L. M. Persson, M. Breitholtz, I. T. Cousins, C. A. de Wit, M. MacLeod and M. S. McLachlan, Environ. Sci. Technol., 2013, 47, 12619–12622 CrossRef CAS PubMed.
- M. MacLeod, M. Breitholtz, I. T. Cousins, C. A. de Wit, L. M. Persson, C. Rudén and M. S. McLachlan, Environ. Sci. Technol., 2014, 48, 11057–11063 CrossRef CAS PubMed.
- H. P. H. Arp, D. Kühnel, C. Rummel, M. MacLeod, A. Potthoff, S. Reichelt, E. Rojo-Nieto, M. Schmitt-Jansen, J. Sonnenberg, E. Toorman and A. Jahnke, Environ. Sci. Technol., 2021, 55, 7246–7255 CrossRef CAS PubMed.
- A. Mirzaei, J.-H. Kim, H. W. Kim and S. S. Kim, J. Mater. Chem. C, 2018, 6, 4342–4370 RSC.
- I. Vollmer, M. J. F. Jenks, M. C. P. Roelands, R. J. White, T. Harmelen, P. Wild, G. P. van der Laan, F. Meirer, J. T. F. Keurentjes and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2020, 59, 15402–15423 CrossRef CAS PubMed.
- Y. Zhang, D. Duan, H. Lei, E. Villota and R. Ruan, Appl. Energy, 2019, 251, 113337 CrossRef CAS.
- T. Thiounn and R. C. Smith, J. Appl. Polym. Sci., 2020, 58, 1347–1364 CrossRef CAS.
- D. Duan, Z. Feng, X. Dong, X. Chen, Y. Zhang, K. Wan, Y. Wang, Q. Wang, G. Xiao, H. Liu and R. Ruan, Energy, 2021, 232, 121090 CrossRef CAS.
- K. Sun, Q. Huang, Y. Chi and J. Yan, Waste Manage., 2018, 81, 128–137 CrossRef CAS PubMed.
- C. Muhammad, J. A. Onwudili and P. T. Williams, Energy Fuels, 2015, 29, 2601–2609 CrossRef CAS.
- A. M. Elfadly, I. F. Zeid, F. Z. Yehia, A. M. Rabie, M. M. Aboualala and S. E. Park, Int. J. Biol. Macromol., 2016, 91, 278–293 CrossRef CAS PubMed.
- P. Sirous-Rezaei, J. Jae, H. Jeong-Myeong, C. H. Ko, J. M. Kim, J. K. Jeon and Y. K. Park, Green Chem., 2018, 20, 1472–1483 RSC.
- A. Heeres, N. Schenk, I. Muizebelt, R. Blees, B. De Waele, A. J. Zeeuw, N. Meyer, R. Carr, E. Wilbers and H. J. Heeres, ACS Sustainable Chem. Eng., 2018, 6, 3472–3480 CrossRef CAS PubMed.
- A. Zheng, Z. Zhao, S. Chang, Z. Huang, H. Wu, X. Wang, F. He and H. Li, J. Mol. Catal. A: Chem., 2014, 383, 23–30 CrossRef.
- A. Sultana and T. Fujitani, Catal. Commun., 2017, 88, 26–29 CrossRef CAS.
- M.-H. Cho, S.-H. Jung and J.-S. Kim, Energy Fuels, 2010, 24, 1389–1395 CrossRef CAS.
- M. Artetxe, G. Lopez, M. Amutio, G. Elordi, J. Bilbao and M. Olazar, Ind. Eng. Chem. Res., 2013, 52, 10637–10645 CrossRef CAS.
- F. J. Mastral, E. Esperanza, C. Berrueco, M. Juste and J. Ceamanos, J. Anal. Appl. Pyrolysis, 2003, 70, 1–17 CrossRef CAS.
- C. Berrueco, F. J. Mastral, E. Esperanza and J. Ceamanos, Energy Fuels, 2002, 16, 1148–1153 CrossRef CAS.
- M. d R. Hernández, Á. N. García and A. Marcilla, J. Anal. Appl. Pyrolysis, 2007, 78, 272–281 CrossRef.
- P. Gaurh and H. Pramani, Waste Manage., 2018, 77, 114–130 CrossRef CAS PubMed.
- K. Sun, Q. Huang, X. Meng, Y. Chi and J. Yan, Energy Fuels, 2018, 32, 9772–9781 CrossRef CAS.
- S.-H. Jung, M.-H. Cho, B.-S. Kang and J.-S. Kim, Fuel Process. Technol., 2010, 91, 277–284 CrossRef CAS.
- D. Zhang, X. Lin, Q. Zhang, X. Ren, W. Yu and H. Cai, Energy, 2020, 212, 118983 CrossRef CAS.
- X. Zhang, H. Lei, G. Yadavalli, L. Zhu, Y. Wei and Y. Liu, Fuel, 2015, 144, 33–42 CrossRef CAS.
- J. Song, J. Wang, Y. Pan, X. Du, J. Sima, C. Zhu, F. Lou and Q. Huang, J. Environ. Manage., 2022, 322, 116096 CrossRef CAS.
- F. Zhang, M. Zeng, R. D. Yappert, J. Sun, Y.-H. Lee, A. M. LaPointe, B. Peters, M. M. Abu-Omar and S. L. Scott, Science, 2020, 370, 437–441 CrossRef CAS PubMed.
- J. Duan, W. Chen, C. Wang, L. Wang, Z. Liu, X. Yi, W. Fang, H. Wang., H. Wei, S. Xu, Y. Yang, Q. Yang, Z. Bao, Z. Zhang, Q. Ren, H. Zhou, X. Qin, A. Zheng and F.-S. Xiao, J. Am. Chem. Soc., 2022, 144, 14269–14277 CrossRef CAS PubMed.
- P. Gaurh and H. Pramani, Waste Manage., 2018, 71, 86–96 CrossRef CAS PubMed.
- M. S. Renzini, U. Sedran and L. B. Pierella, J. Anal. Appl. Pyrolysis, 2009, 86, 215–220 CrossRef CAS.
- A. Marcilla, M. I. Beltrán and R. Navarro, Appl. Catal., B, 2009, 86, 78–86 CrossRef CAS.
- G. Elordi, M. Olazar, G. Lopez, M. Artetxe and J. Bilbao, Ind. Eng. Chem. Res., 2011, 50, 6061–6070 CrossRef CAS.
- S. S. Arzumanov, A. A. Gabrienko, A. V. Toktarev, Z. N. Lashchinskaya, D. Freude, J. Haase and A. G. Stepanov, J. Phys. Chem. C, 2019, 123, 30473–30485 CrossRef CAS.
- I. I. Ivanova, Y. G. Kolyagin, V. V. Ordomsky, E. V. Asachenko, E. M. Pasynkova and Y. A. Pirogov, J. Mol. Catal. A: Chem., 2009, 305, 47–53 CrossRef CAS.
- Z. N. Lashchinskaya, A. A. Gabrienko, S. S. Arzumanov, A. A. Kolganov, A. V. Toktarev, D. Freude, J. Haase and A. G. Stepanov, ACS Catal., 2020, 10, 14224–14233 CrossRef CAS.
- S. M. T. Almutairi, B. Mezari, P. C. M. M. Magusin, E. A. Pidko and E. J. M. Hensen, ACS Catal., 2012, 2, 71–83 CrossRef CAS.
- E. A. Uslamin, H. Saito, N. Kosinov, E. Pidko, Y. Sekine and E. J. M. Hensen, Catal. Sci. Technol., 2020, 10, 2774 RSC.
- B. Zhao, P. Zhai, P. Wang, J. Li, T. Li, M. Peng, M. Zhao, G. Hu, Y. Yang, Y.-W. Li, Q. Zhang, W. Fan and D. Ma, Chem, 2017, 3, 323–333 CAS.
- T. Xue, H. Liu, Y. Zhang, H. Wu, P. Wu and M. He, Microporous Mesoporous Mater., 2017, 242, 190–199 CrossRef CAS.
- J. Wang, C. Liu, P. Zhu, H. Liu, X. Zhang, Y. Zhang, J. Liu, L. Zhang and W. Zhang, New J. Chem., 2021, 45, 18659 RSC.
- S. Wodarz, N. A. Slaby, M. C. Zimmermann, T. N. Otto, J. Holzinger, J. Skibsted, T. A. Zevaco, S. Pitter and J. Sauer, Ind. Eng. Chem. Res., 2020, 59, 17689–17707 CrossRef CAS.
- Y. Li, M. Wang, S. Liu, F. Wu, Q. Zhang, S. Zhang, K. Cheng and Y. Wang, ACS Catal., 2022, 12, 8793–8801 CrossRef CAS.
- L.-H. Chen, X.-Y. Li, J. C. Rooke, Y.-H. Zhang, X.-Y. Yang, Y. Tang, F.-S. Xiao and B.-L. Su, J. Mater. Chem., 2012, 22, 17381–17403 RSC.
- Z. Jing, Z. Liang, C. Guojing, W. Haiyan, W. Min and M. Jun, Pet. Sci. Technol., 2008, 26, 586–592 CrossRef CAS.
- Y. T. Kim, J. P. Chada, Z. Xu, Y. J. Pagan-Torres, D. C. Rosenfeld, W. L. Winniford, E. Schmidt and G. W. Huber, J. Catal., 2015, 323, 33–44 CrossRef CAS.
- M. Boronat, P. Viruela and A. Corma, J. Phys. Chem. B, 1997, 101, 10069–10074 CrossRef CAS.
- V. R. Choudhary, D. Panjala and S. Banerjee, Appl. Catal., A, 2002, 231, 243–251 CrossRef CAS.
- P. Borges, R. Ramos Pinto, M. A. N. D. A. Lemos, F. Lemos, J. C. Védrine, E. G. Derouane and F. Ramôa Ribeiro, Appl. Catal., A, 2007, 324, 20–29 CrossRef CAS.
- L. Zhang, H. Liu, X. Li, S. Xie, Y. Wang, W. Xin, S. Liu and L. Xu, Fuel Process. Technol., 2010, 91, 449–455 CrossRef CAS.
- A. Ishihara, K. Takai, T. Hashimoto and H. Nasu, ACS Omega, 2020, 5, 11160–11166 CrossRef CAS PubMed.
- G. Zhang, T. Bai, T. Chen, W. Fan and X. Zhang, Ind. Eng. Chem. Res., 2014, 53, 14932–14940 CrossRef CAS.
- H. You, C. Xu, J. Gao, Z. Liu and P. Yan, Catal. Commun., 2006, 7, 554–558 CrossRef CAS.
- B. Dimon, P. Cartraud, P. Magnoux and M. Guisnet, Appl. Catal., A, 1993, 101, 351–369 CrossRef CAS.
- B. Zhang, J. Yao, Y. Wang, W. Gao, Y. Kugue, X. Guo, Y. He, G. Yang and N. TsubakiJ, Chem. Technol. Biotechnol., 2023, 98, 98–105 CrossRef CAS.
- D. S. Fernandes, C. O. Veloso and C. A. Henriques, Catal. Lett., 2020, 150, 738–752 CrossRef CAS.
- K. Sun, N. J. Themelis, A. C. Bourtsalas and Q. Huang, J. Cleaner Prod., 2020, 268, 122038 CrossRef CAS.
- G. Grause, S. Matsumoto, T. Kameda and T. Yoshioka, Ind. Eng. Chem. Res., 2011, 50, 5459–5466 CrossRef CAS.
- S. Pradhan, R. Lloyd, J. K. Bartley, D. Bethell, S. Golunski, R. L. Jenkins and G. J. Hutchings, Chem. Sci., 2012, 3, 2958 RSC.
- M. Stocker, Microporous Mesoporous Mater., 2005, 82, 257–292 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ey00011g |
| This journal is © The Royal Society of Chemistry 2023 |