 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence17O solid state NMR as a valuable tool for deciphering reaction mechanisms in mechanochemistry: the case study on the 17O-enrichment of hydrated Ca-pyrophosphate biominerals†
Ieva
Goldberga
 *a,
Nicholai D.
Jensen
a,
Christèle
Combes
b,
Frédéric
Mentink-Vigier
c,
Xiaoling
Wang
c,
Ivan
Hung
c,
Zhehong
Gan
c,
Julien
Trébosc
d,
Thomas-Xavier
Métro
e,
Christian
Bonhomme
f,
Christel
Gervais
f and
Danielle
Laurencin
*a
*a,
Nicholai D.
Jensen
a,
Christèle
Combes
b,
Frédéric
Mentink-Vigier
c,
Xiaoling
Wang
c,
Ivan
Hung
c,
Zhehong
Gan
c,
Julien
Trébosc
d,
Thomas-Xavier
Métro
e,
Christian
Bonhomme
f,
Christel
Gervais
f and
Danielle
Laurencin
*a
aICGM, Université de Montpellier, CNRS, ENSCM, Montpellier, France. E-mail: ieva.goldberga@umontpellier.fr; danielle.laurencin@umontpellier.fr
bCIRIMAT, Université de Toulouse, CNRS, Toulouse INP – ENSIACET, Toulouse, France
cNational High Magnetic Field Laboratory (NHMFL), Tallahassee, Florida, USA
dUniversité de Lille, CNRS, INRAE, Centrale Lille, Université d'Artois FR2638 – IMEC – Institut Michel Eugène Chevreul, 59000 Lille, France
eIBMM, Université de Montpellier, CNRS, ENSCM, Montpellier, France
fLCMCP, UMR 7574, Sorbonne Université, CNRS, Paris, France
First published on 17th August 2022
Abstract
The possibility of enriching in 17O the water molecules within hydrated biominerals belonging to the Ca-pyrophosphate family was investigated, using liquid assisted grinding (LAG) in the presence of 17O-labelled water. Two phases with different hydration levels, namely triclinic calcium pyrophosphate dihydrate (Ca2P2O7·2H2O, denoted t-CPPD) and monoclinic calcium pyrophosphate tetrahydrate (Ca2P2O7·4H2O, denoted m-CPPT β) were enriched in 17O using a “post-enrichment” strategy, in which the non-labelled precursors were ground under gentle milling conditions in the presence of stoichiometric quantities of 17O-enriched water (introduced here in very small volumes ∼10 μL). Using high-resolution 17O solid-state NMR (ssNMR) analyses at multiple magnetic fields, and dynamic nuclear polarisation (DNP)-enhanced 17O NMR, it was possible to show that the labelled water molecules are mainly located at the core of the crystal structures, but that they can enter the lattice in different ways, namely by dissolution/recrystallisation or by diffusion. Overall, this work sheds light on the importance of high-resolution 17O NMR to help decipher the different roles that water can play as a liquid-assisted grinding agent and as a reagent for 17O-isotopic enrichment.
Introduction
Despite the increasing number of mechanochemical synthetic procedures being developed to prepare molecular and materials systems, the exact details of how reactions occur are still unclear in many cases. Most frequently, ball-milling (BM) reactions are performed in closed jars, where the course of the reaction is followed by stopping the milling process, and analysing the reaction medium using techniques like powder X-ray diffraction (pXRD) and IR/Raman spectroscopies. Yet, such analyses are often insufficient when studying the formation and nature of amorphous or disordered intermediates, as they cannot provide straightforward identification of these species. A contrario, because it is sensitive to the local environment of nuclei, solid-state NMR (ssNMR) is a highly valuable approach for helping determine the nature of amorphous/disordered compounds. As such, it is no surprise that there are an increasing number of examples in which ssNMR has been used to help understand reaction mechanisms in ball-milling.1 Among representative works, one can cite (i) the study by Baxter et al. on the mechanochemical amorphisation of cadmium based ZIFs using 13C, 15N and 113Cd ssNMR;2 and (ii) the studies by MacKenzie and co-workers on the mechanochemical synthesis and also amorphisation of aluminosilicates like mullite, using 27Al ssNMR.3Oxygen-17 stands out as a highly attractive probe for gaining deep insight into the composition of reaction media in ball-milling via ssNMR. Indeed, its chemical shift scale exceeds 1000 ppm (against only ∼200 ppm in the case of 13C). Furthermore, oxygen-17 ssNMR also offers the possibility of reaching complementary information on the local bonding environment of oxygen by measuring the 17O-quadrupolar parameters, as the spin of 17O is 5/2.4 The first evidence of the interest in using high-resolution 17O ssNMR (as well as MAS-DNP – magic angle spinning dynamic nuclear polarisation) to investigate reaction mechanisms in ball-milling was provided by Chen et al., who looked into the formation of chemical bonds between reacting silica and titania particles and provided evidence of the formation of Ti–O–Si bridges after only 3 minutes of milling.5 Subsequently, Leroy et al. demonstrated that 17O ssNMR analyses could provide valuable insight into the structure of Zn-terephthalate phases, isolated in pure form using ball-milling and operando analyses of the reaction medium.6 Lastly, in a recent investigation by Goldberga et al., it was shown that the water molecules within calcium oxalate hydrates could be enriched in 17O, further enabling to investigate the dynamics of the water molecules within these crystalline phases.7 Moreover, the isotopic labelling of the water molecules in 2H and 18O was also performed as part of the same work. This particular isotopic enrichment was used to help rationalise the reaction mechanisms occurring in ball-milling during the isotopic labelling, by showing that the enrichment process predominantly occurred by a dissolution–recrystallisation mechanism.
Beyond enabling a better understanding of ball-milling reactions, the possibility of using mechanochemistry to enrich water molecules in 17O within hydrated materials, like calcium oxalate monohydrate, is very attractive. Many of the minerals found in natural environments contain “water”; however, the exact way in which these molecules can enter, stabilise or affect the properties of different crystal structures is often unclear and requires investigation.8 This is particularly the case for the water molecules within the biomineral phases present in living organisms. For instance, different phases of hydrated calcium pyrophosphates with a general formula of Ca2P2O7·nH2O (with n = 0, 1, 2, 3, and 4) have been reported and studied, some of which are known to be part of pathological calcifications which can form in joints (in diseases like pseudo-gout).9 Even for the most studied dihydrate and tetrahydrate forms, their crystallisation and interconversion still need to be clarified. Thus, high-resolution 17O NMR appears as a desirable tool for looking more specifically at the water molecules in the crystal structures. However, the very high cost of 17O-enriched water (up to more than 2000 € per mL for water enriched at 90% in 17O) makes preparing such compounds prohibitively expensive when using standard precipitation conditions in an aqueous solution. This explains why this analytical technique has never been used so far for studying such phases. Being able to carry out the 17O-isotopic labelling by engaging only microlitre quantities of enriched water, as recently done for calcium oxalate monohydrate (COM) and calcium oxalate dihydrate (COD),7 thus appears highly attractive.
This manuscript aims to investigate how ball-milling can be used to enrich water molecules found in hydrated Ca-pyrophosphates in oxygen-17. The focus was mainly set on the labelling of two crystalline phases (Fig. 1): monoclinic Ca2P2O7·4H2O (m-CPPT, under its β form) and triclinic Ca2P2O7·2H2O (t-CPPD). First, the ball-milling conditions were optimised in view of the enrichment, using various analytical techniques, including pXRD and 31P ssNMR. Then, the isotopic enrichment in 17O was investigated using ssNMR as well as MAS-DNP analyses, and experimental 17O MAS NMR spectra were compared to those calculated from GIPAW-DFT methods. From a more general perspective, how such experiments and characterisations can help inform on reaction mechanisms in ball-milling is discussed.
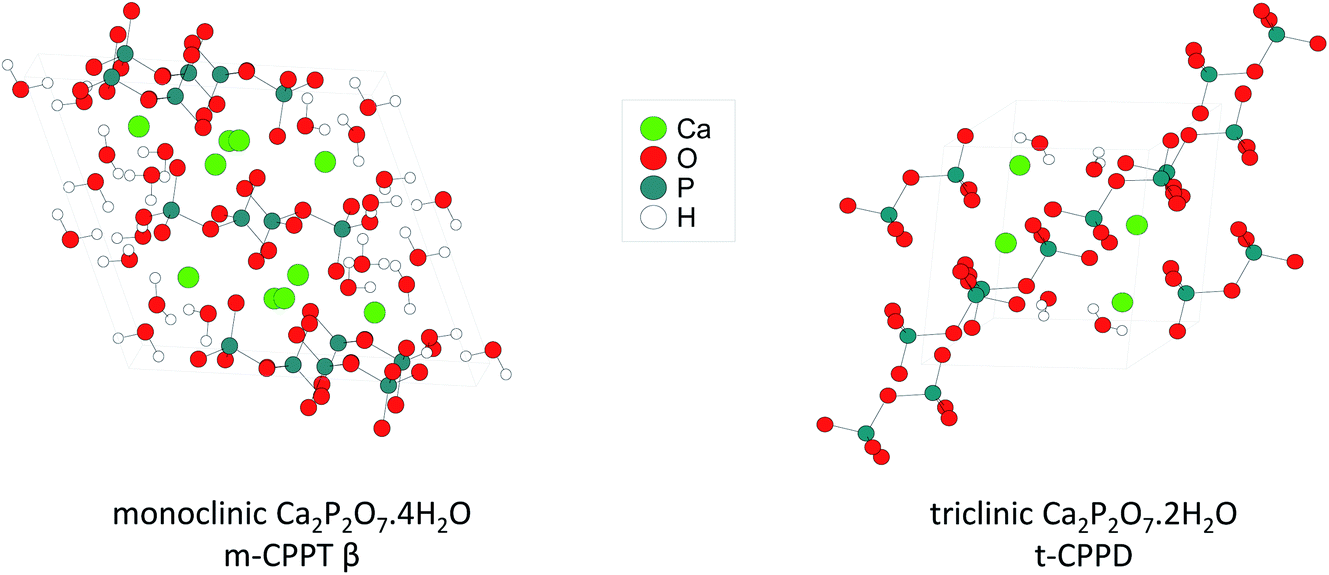 | ||
| Fig. 1 Representation of the crystal structures of the two calcium pyrophosphate hydrates enriched in 17O as part of this work. | ||
Experimental section
Reagents and synthetic equipment
Monoclinic calcium pyrophosphate tetrahydrate (m-CPPT β, Ca2P2O7·4H2O), and triclinic calcium pyrophosphate dihydrate (t-CPPD, Ca2P2O7·2H2O) were synthesised by following or adapting previously published procedures.9 H217O (∼40% or ∼90% 17O enrichment) was purchased from CortecNet and used as received. All milling experiments were carried out in a Retsch Mixer Mill MM400 apparatus operated at room temperature (22 ± 4 °C). Polytetrafluoroethylene (PTFE) balls (10 mm diameter) with a stainless-steel core were purchased from Retsch. Milling jars, beads, and micro-syringes (used to introduce the labelled water) were dried under vacuum before use.Labelling of calcium pyrophosphate tetrahydrate (m-CPPT β)
One PTFE ball with a stainless-steel core was placed in a 10 mL stainless steel grinding jar (with a screw-top lid). Non-labelled m-CPPT β (60 mg, ∼0.2 mmol) was then added. Isotopically enriched water (14 μL, ∼0.8 mmol) was then deposited on the reactor's wall. The jar was closed, sealed with parafilm and subjected to grinding for 5 minutes in the mixer mill operated at 25 Hz. The material was then recovered by gentle scraping of the jar walls and dried under vacuum for 5 minutes to remove excess water. The sample (m = 58 mg) was stored in a parafilmed glass vial placed in a container with molecular sieves at −16 °C until further use. Prior to any characterisation by pXRD, SEM, and 17O NMR spectroscopy, samples were taken out of the freezer and left thawing for 30 minutes.Labelling of calcium pyrophosphate dihydrate (t-CPPD)
One PTFE ball with a stainless-steel core was placed in a 10 mL stainless steel grinding jar (with a screw-top lid), followed by t-CPPD (60 mg, ∼0.2 mmol), and finally, 17O-enriched water (7 μL, ∼0.4 mmol) was deposited on the reactor's wall. The jar was closed, sealed with parafilm and subjected to grinding for 5 minutes in the mixer mill operated at 25 Hz. The material was then recovered by gentle scraping of the jar walls and dried under vacuum (m = 55 mg). The sample was stored under the same conditions as labelled m-CPPT β.Powder X-ray diffraction (pXRD) and scanning electron microscopy (SEM) characterisations
pXRD analyses were performed on an X'Pert MPD diffractometer using CuKα1 radiation (λ = 1.5406 Å) with the operating voltage and current maintained at 40 kV and 25 mA, respectively. X-ray diffractograms were recorded between 5° and 60° in 2θ, with a step size of 0.017° (with count time per step of ∼50 s).SEM analyses were carried out on a Zeiss Evo HD15 scanning electron microscope equipped with an Oxford Instruments X-MaxN SDD 50 mm2 EDXS detector. Before the SEM analyses, samples were deposited on double-sided conducting carbon tape and then metallised with carbon.
31P solid-state NMR experiments
The 31P NMR experiments were performed at 7.0 and 14.1 T on Varian VNMRS spectrometers (ICGM, France), using 3.2 mm HX or HXY probes, operating at 31P Larmor frequencies of 121.44 and 242.82 MHz, respectively. Samples were spun at a frequency of 8 kHz (without temperature regulation). 1H–31P cross-polarisation (CP) MAS (Magic Angle Spinning) NMR experiments were performed at 7.0 T using 1H 90° pulse of 2.5 μs, and a contact time of 3 ms, using a ramped pulse on 1H, and a square pulse on 31P. During acquisition, SPINAL-64 1H decoupling was used.10 Further experimental details are given in Table S1.† A CP-INADEQUATE (Incredible Natural Abundance Double Quantum Transfer Experiment)1131P MAS NMR experiment was also carried out at 14.1 T on the t-CPPD phase after milling. 31P chemical shifts were referenced to hydroxyapatite at 2.8 ppm.17O solid-state NMR experiments
Oxygen-17 solid-state NMR (ssNMR) spectra were recorded at multiple magnetic fields (B0 = 9.4, 14.1, 18.8 and 35.2 T) at different NMR facilities (ICGM in Montpellier, UCCS in Lille and MagLab in Tallahassee). 17O chemical shifts were referenced using tap water at 0.0 ppm (or D2O at −2.7 ppm).17O ssNMR experiments were performed at 9.4 and 14.1 T on Varian VNMRS spectrometers (ICGM, France), using 3.2 mm HX or HXY probes, operating at 17O Larmor frequencies of 54.18 and 81.31 MHz, respectively. Samples were spun at the magic angle, at a frequency of 18 kHz, and with temperature regulated at 0 °C. The 1D 17O MAS (Bloch decay) NMR experiments were performed using a 1.0 μs excitation pulse (which would be a 30° tilt angle on a liquid). The double-frequency sweep (DFS)12 enhancement scheme was used in some of the experiments. The experimental parameters were as follows: DFS pulse of 500 μs (with RF of ∼10 kHz), with a sweep width between 80 and 200 kHz, followed by an excitation pulse of 1.0 μs. SPINAL-64 1H decoupling was applied in some experiments. Other details, such as the RF power used for decoupling, recycle delays, and the number of transients, are reported in Table S1.† An additional DFS-echo 17O MAS NMR experiment was recorded for the m-CPPT β phase at 100 K using the 14.1 T MAS-DNP instrument of the MagLab in Tallahassee (operated without microwave irradiation),13 and spinning at 13 kHz. The echo delay was set to 1 rotor period, the recycle delay used was 1 s, and the number of scans acquired was 4096.
At 18.8 T, 17O MAS NMR spectra were recorded on a Bruker Avance NEO NMR spectrometer (UCCS, France) equipped with a 3.2 mm HX probe operating at 17O and 1H frequencies of 108.46 MHz and 800.12 MHz, respectively. The spinning frequency was controlled at 16 kHz, with the temperature regulated at 0 °C. The 1D 17O MAS (Bloch decay) NMR experiment was performed using a 1.0 μs pulse for excitation (corresponding to a 22.5° tilt angle on a liquid). The recycle delays and number of transients acquired in each experiment can be found in Table S1.†
At 35.2 T, 17O MAS NMR analyses were performed using the SCH magnet14 at the NHMFL (Maglab, USA) on a Bruker Avance NEO NMR spectrometer equipped with a 3.2 mm single-resonance MAS probe operating at a 17O frequency of 203.36 MHz. The spinning frequency was set to 18 kHz with temperature control at +10 °C. The 1D 17O Hahn echo experiment was recorded using one rotor period with a π/2 and π pulse lengths of 5.0 and 10.0 μs, respectively. No 1H decoupling was applied for this experiment. The recycle delay and number of transients acquired can be found in Table S1.†
17O dynamic nuclear polarisation NMR
The MAS-DNP experiments were carried out in Tallahassee on a 14.1 T magnet equipped with the gyrotron operating at 395.17 GHz with 12 W power.13 The temperature for VT/bearing/drive gas was 92/100/105 K and 98 K without microwave irradiation, and the sample was spun at 13 kHz. The labelled m-CPPT β sample was impregnated with a 10 mM solution of AMUPol15 in d8-glycerol/D2O/H2O (6/3/1 %v) (about 15 mg of m-CPPT β impregnated with 9 μL of AMUPol solution), and packed into a 3.2 mm sapphire rotor. It should be noted that impregnation with another radical solution (13 mM TEKPol in 1,1,2,2-tetrachloroethane) did not generate hyperpolarisation on m-CPPT β. Three freeze–thaw cycles were applied to the sample before insertion into the probe. The packed rotor was also prespun using a benchtop spinner at room temperature to ensure stable spinning. The enhancement was εon/off ≈ 25 and the build-up TB ≈ 4 s. For m-CPPT β, the 17O{1H} DNP CPMAS experiments were then performed on a 3.2 mm HXY probe using HCO configuration. The initial 1H CP 90° pulse was 2.5 μs, and a contact time of 1.5 ms with ∼100 kHz RF field was used. SPINAL-64 1H decoupling was applied during acquisition (∼100 kHz). The recycle delay was set to 5 s, and the number of scans acquired was 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 150.
150.
To ensure the signals measured arise from m-CPPT β only, we measured the natural abundance of 17O arising from the DNP matrix alone. 30 μL of the solution was packed into a sapphire rotor which led to an enhancement of 150. Under these conditions, natural abundance 17O signals could be detected, but their intensity was a fraction of the signals measured for the impregnated m-CPPT β.
Lastly, it is worth mentioning that the DFS-echo 17O NMR spectrum of the impregnated m-CPPT β phase was also recorded at 100 K (in the absence of DNP), using a recycle delay of 0.5 s, and acquiring 4096 scans. No significant difference was observed compared to the non-impregnated material.
Computational studies, including 17O GIPAW-DFT calculations of NMR parameters
Geometry optimisations for m-CPPT β and t-CPPD structures were carried out using the crystallographic data reported in the literature, as in our previous experimental/computational study on calcium pyrophosphates, which corresponds to 92 atoms per m-CPPT β structure relaxation, and 34 for t-CPPD.9 First, the missing protons were added to the structures. Atomic positions were then relaxed using the Vienna ab initio simulation package (VASP),16 based on the Kohn–Sham density functional theory (DFT), and using a plane-wave pseudopotential approach with an energy cut-off of 600 eV and 2 × 2 × 2 or 3 × 3 × 3 k-point mesh for m-CPPT β and t-CPPD, respectively. During the geometry optimisation, unit cell parameters were kept fixed to ensure consistency between the experimental and optimised structures. Structural optimisations were performed by relaxing H positions only, as in our previous study on calcium pyrophosphates.9cNMR parameters were calculated for all structures using the QUANTUM-ESPRESSO code,17 keeping the atomic positions equal to the values previously determined using VASP. The Perdew–Burke–Ernzerhof (PBE) generalised gradient approximation was used,18 and the valence electrons were described by norm-conserving pseudopotentials19 in the Kleinman–Bylander form.20 The shielding tensor was computed using the GIPAW approach.21 The wave functions were expanded on a plane wave basis set with a kinetic energy cut-off of 80 Ry. The calculations were done using the same k-space mesh density as for relaxation.
The isotropic chemical shift δiso is defined as δiso ≈ −(σ − σref), where σ is the isotropic shielding and σref is the isotropic shielding for the same nucleus using a hydrate reference system, in a similar way as in our previous work on calcium oxalate monohydrate.7 For these hydrates, the maximum deviation between experimental and DFT-calculated 17O isotropic shifts was found to be 6 ppm.7 The quadrupolar moments (Q) used to calculate the CQ of 17O was −2.558 fm2.22 Calculated values are reported in Table S2 (ESI†).
Results and discussion
Similar liquid-assisted grinding (LAG) conditions as reported previously for the labelling of COM7 were used: 60 mg of calcium pyrophosphate hydrate were milled for just 5 minutes at 25 Hz in the presence of microlitre-quantities of water (∼7 and 14 μL for t-CPPD and m-CPPT β, respectively), using a stainless-steel reactor containing 1 PTFE-coated bead. After the reaction, the products were characterised by various techniques, including pXRD and scanning electron microscopy (SEM). These milling conditions were initially tested using non-labelled water, before switching to 17O-enriched water.As shown in Fig. 2, in the case of m-CPPT β, particles with platelet-morphology were still observed after 5 minutes of milling, and the pXRD pattern essentially resembled that of the starting material, with only some changes in relative intensity of diffraction peaks due to modifications in the particle morphology and packing (thereby affecting the preferential orientation effects apparent in pXRD analyses). Regarding t-CPPD, although pXRD analyses also showed that the compound remained crystalline after the milling, more careful analyses of the baseline of the patterns suggested that small traces of the monoclinic calcium pyrophosphate dihydrate polymorph (Ca2P2O7·2H2O, denoted m-CPPD) may be present, as shown in Fig. S1 (ESI†). Moreover, SEM images revealed a much more pronounced change in particle morphology compared to m-CPPT β.
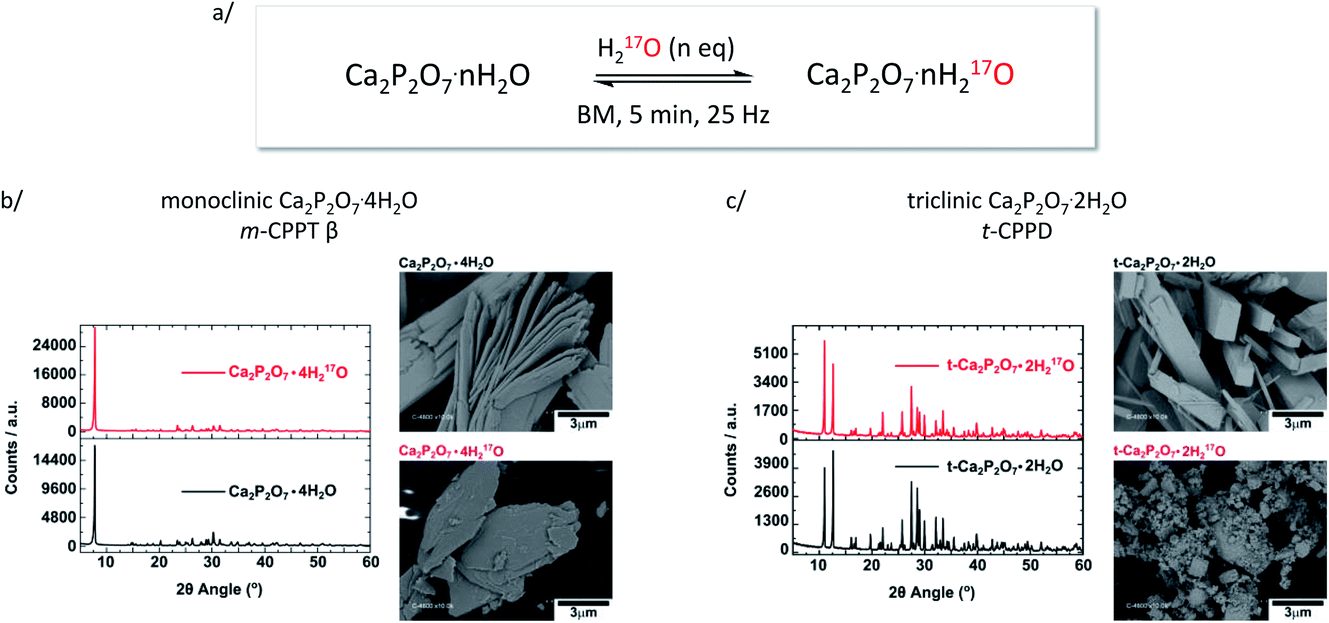 | ||
| Fig. 2 (a) Scheme showing the equilibration conditions used for the isotopic enrichment of water molecules in m-CPPT β and t-CPPD phases using ball-milling (only a fraction of the 17O-enriched water replaces non-labelled water molecules initially present in the lattice); (b) pXRD and SEM analyses of m-CPPT β before and after BM; (c) pXRD and SEM analyses of t-CPPD before and after BM. An expansion of the pXRD patterns of t-CPPD before and after milling is shown in Fig. S1 (ESI†). | ||
As pXRD informs on the crystalline phases present in materials, complementary 31P NMR analyses were carried out under magic angle spinning (MAS). As can be seen in Fig. 3, in the case of m-CPPT β, the spectrum after milling was essentially the same as the one of the starting material. This shows that neither crystalline impurities nor amorphous by-products were formed during the milling. Concerning t-CPPD, in addition to the signals expected for pyrophosphates in a crystalline t-CPPD phase,9c an additional resonance at ∼−9.9 ppm could be observed. A complementary 2D 31P–31P INADEQUATE experiment was performed, showing that this resonance correlates well with another weak-intensity one at ∼−5.9 ppm (see ESI, Fig. S2†). Based on previous 31P MAS NMR analyses of hydrated calcium pyrophosphates,9c both resonances belong to the pyrophosphate unit of the m-CPPD impurity which was detected in pXRD. It is important to highlight that just like for the m-CPPT β phase, no amorphous component could be detected by 31P NMR.
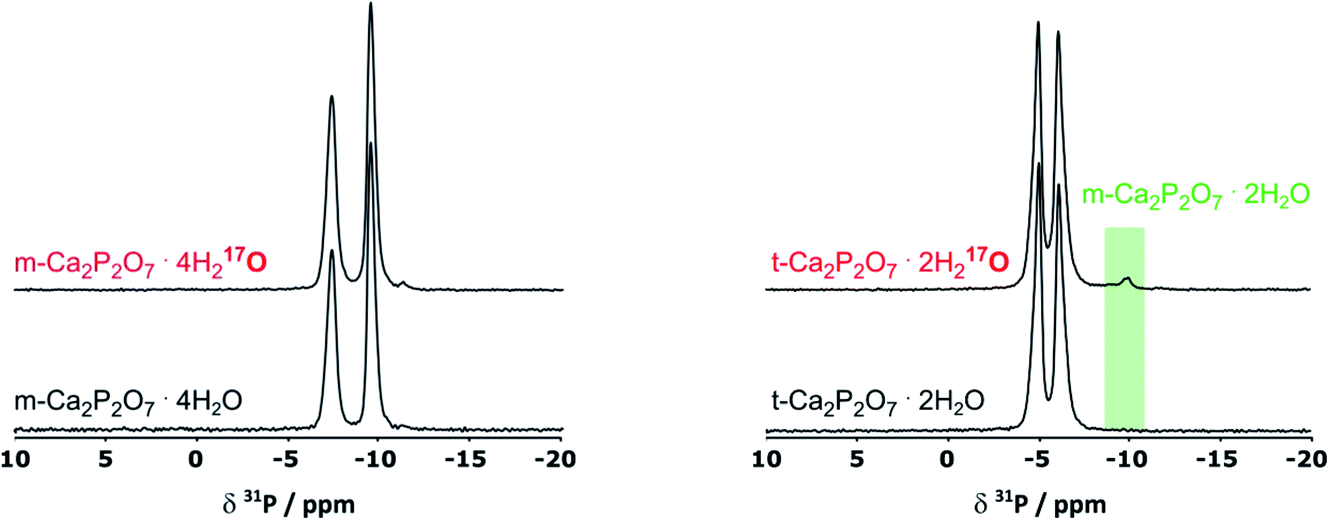 | ||
| Fig. 3 31P CPMAS NMR spectra of m-CPPT β (left) and t-CPPD (right) before and after BM, showing the formation of a new by-product after milling t-CPPD, which was identified as m-CPPD (green-shaded zone). Only the region with isotropic 31P chemical shifts is shown here. | ||
Given that for m-CPPT β and t-CPPD the milling conditions were gentle enough to maintain the crystallinity of the starting materials, with only very limited transformation into other hydrates, 17O NMR analyses were performed. The 17O MAS NMR spectra of the isolated phases are shown in Fig. 4. Due to the quadrupolar nature of oxygen-17, these analyses were performed at different magnetic fields (9.4, 14.1, 18.8 T and 35.2 T). For both compounds, a broad asymmetric 17O NMR signal was observed in just a few hours, which is consistent with what one may expect for water molecules within solid environments.7 This is especially evident when measurements at a lower magnetic field are considered, as the second order quadrupolar lineshape becomes more visible (see Fig. 4, analyses at 9.4 and 14.1 T). Importantly, if the added enriched water had stayed under a “liquid” form in the isolated product (for instance, by forming a weakly-bound layer adsorbed at the surface of the crystallites), a much narrower and symmetric signal, centred around 0 ppm, would have been obtained in 17O NMR. Complementary analyses using DNP were also performed on the m-CPPT β phase, showing that its 17O signature mainly comes from the “core-like” enriched water molecules rather than surface ones (see Fig. 4c). Indeed, the DNP spectra recorded using 1H → 17O CPMAS sequence, in which the 1H are initially excited through the radicals of the impregnating solution (meaning that they are more representative of surface water 17O signature), show a different signature than the direct excitation 17O NMR spectra (recorded with a DFS-echo sequence) obtained at the same field and temperature. Overall, these analyses show that during the liquid-assisted grinding process, the very small amount of labelled water added also plays the role of a reagent, enabling the isotopic enrichment to occur, and leading to an overall exchange with non-labelled water molecules initially present in the hydrated crystal structures. Such information could not have been reached in the absence of isotopic labelling of the water molecules.
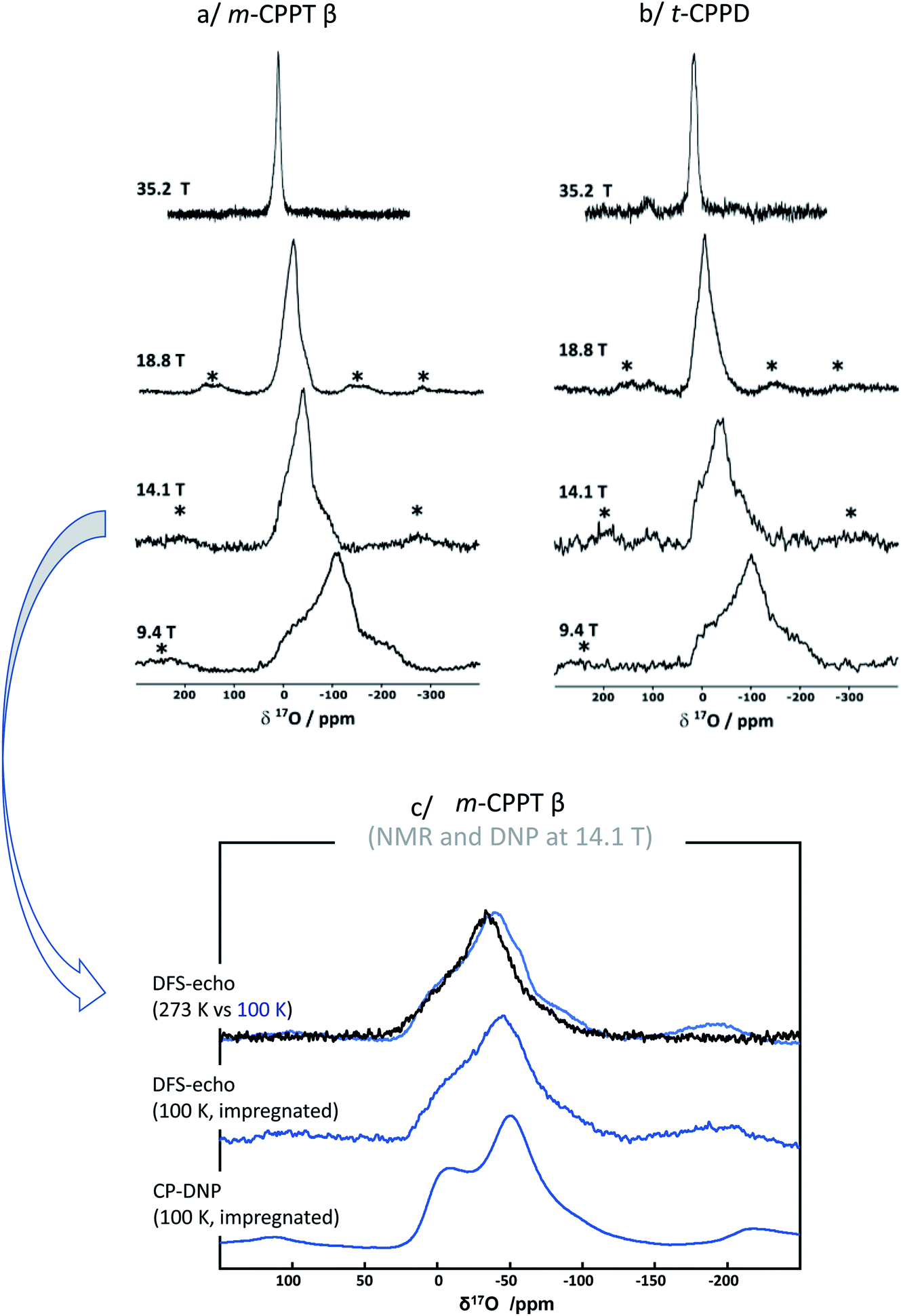 | ||
| Fig. 4 17O MAS NMR spectra of (a) m-CPPT β and (b) t-CPPD after 17O-labelling using BM, recorded at different magnetic fields (9.4, 14.1, 18.8 and 35.2 T). Zoom into the lineshapes recorded at 35.2 T is provided in the ESI (Fig. S3†). (c) Low-temperature 17O MAS NMR and DNP-enhanced NMR analyses of the m-CPPT β phase at 14.1 T (see experimental section for details). “*” symbols correspond to spinning sidebands. | ||
To further analyse the 17O MAS NMR spectra, ab initio calculations of the 17O NMR parameters were performed using the GIPAW-DFT method (see Table S2, ESI†). For each structure, the 17O NMR parameters of the crystallographically inequivalent water molecules in the crystal structure were calculated (calculations being performed at 0 K), and the corresponding 17O MAS NMR spectra were simulated, and compared to the experimental ones. For the t-CPPD phase (see ESI Fig. S4†), the overall lineshape calculated for the water molecules reproduces relatively well the experimental one, especially when looking at the breadth of the signal (which mainly arises from the second order quadrupolar broadening). The 2 water sites were not resolved at this stage. Further analyses using Multiple-Quantum Magic Angle Spinning (MQMAS) NMR sequences would be needed for this,7 which is beyond the scope of the present work. Here, only an average isotropic shift δiso and quadrupolar parameter PQ were determined for the 2 sites, by considering the change in peak maximum position as a function of the magnetic field (see ESI, Fig. S5†). Nevertheless, the changes in 17O linewidth as a function of the magnetic field reflect the presence of more than one water environment in the material. It is important, however, to highlight that the experimental 17O MAS NMR spectrum of enriched t-CPPD is more similar to the one calculated for the t-CPPD phase, rather than the m-CPPD polymorph (see ESI Fig. S4†), suggesting that the enriched water is mainly located in the core of the former compound (rather than in the less abundant m-CPPD by-product which is formed during milling).
Regarding the m-CPPT β phase (see ESI, Fig. S6†), the overall experimental lineshape was found to differ more significantly compared to the DFT-calculated one. First, it should be noted that the four crystallographically distinct water sites of m-CPPT β could not be resolved using 1D MAS experiments, nor 2D MQMAS experiments (data not shown). At this stage, only an average isotropic shift δiso and quadrupolar parameter PQ were determined for the water sites, by considering the change in peak maximum position as a function of the magnetic field (see ESI, Fig. S7†). However, it is worth noting that despite the lack of resolution, the evolution of the linewidth as a function of the magnetic field reflects the presence of more than one water site, and preliminary evidence of a higher frequency water resonance can even be noticed at the ultra-high magnetic field (35.2 T data, high-frequency tailing – see orange arrow in Fig. S3a†). The presence of such a signal would be consistent with the GIPAW-DFT calculations, which show a distinct high chemical shift water site (see OW1 in Table S2†). Second, it should be highlighted that the experimental 17O NMR spectrum of m-CPPT β is clearly narrower than the one calculated by DFT. Such difference can be indicative of rapid reorientations of the water molecules within the crystal structure at room temperature, which would lead to a partial averaging of the apparent quadrupolar coupling constant in 17O NMR, leading to a difference with the DFT results (as calculations are performed at 0 K). Considering the crystal structure of m-CPPT β (Fig. 1), the observation of such water dynamics at room temperature is actually not fully surprising. Indeed, the water molecules in this structure are all within the same interlayer space (water layer thickness ∼4 Å), where they interact with each other through hydrogen-bonding, and are capable of easily changing their local orientation. After ball-milling, the sheet-like morphology of the crystallites is maintained (as shown by SEM, Fig. 2b), and the crystal structure is preserved (as shown by XRD, Fig. 2b), which implies that the enriched water molecules are most likely also present in the hydrated layers and prone to similar motions. As a matter of fact, even after cooling the sample down to 100 K, only a slight broadening of the 17O resonance is observed in NMR, showing that local movements of the water molecules in the interlayer space are still present (see Fig. 4c, top spectra recorded at 273 vs. 100 K).
The 17O NMR analyses also revealed that the 17O-enrichment level was significantly different between the m-CPPT β and t-CPPD phases, despite both samples having been labelled using similar milling conditions. Indeed, 17O MAS NMR spectra of the m-CPPT β phase were systematically obtained with better sensitivity than for the t-CPPD phase, a difference which could not be accounted for by simply considering the difference in the mass percentage of H2O between both phases (see ESI, Fig. S8,† for a quantitative comparison). The comparison of the crystal structures of the m-CPPT β and t-CPPD phases can explain why such differences in labelling rate are observed by 17O NMR. In the former case, water molecules are all present within a layer, with notably “weak” hydrogen-bonding interactions between them, with one water molecule not coordinated to Ca2+. For t-CPPD, however, no such layered arrangement is observed, and the water molecules appear to be in a more constrained local environment within the crystal structure, with all water molecules coordinated to Ca2+ (see ESI, Fig. S9†). Consequently, the 17O labelling of the water molecules in the m-CPPT β structure would be more straightforward, as the enriched water molecules could enter the crystal structure by simple diffusion inside the hydrated layers. In contrast, in the case of the t-CPPD phase, larger structural changes involving partial dissolution/recrystallisation of the starting material would be needed for the enriched water to enter the crystal structure. This could also explain the partial formation of the m-CPPD by-product, with some of the calcium and pyrophosphate ions adopting a different arrangement upon recrystallisation. To further test this hypothesis, an attempt to enrich both materials by simple “shaking” in the presence of 17O-enriched water (but in the absence of milling beads) was performed, such protocol having been previously tested for COM as well.7 Only in the case of the m-CPPT β phase was this alternate procedure found to be successful, confirming that the labelling is more straightforward to achieve for this phase, due to its layered structure.
Overall, the different characterisations performed on the 17O-enriched phases, notably using solid state NMR, enable to propose two different mechanisms by which water can react with hydrated biominerals, when performing the liquid-assisted grinding under gentle milling conditions like those described here (5 minutes BM at 25 Hz, using 1 PTFE coated ball). On the one hand, H217O can enter the crystal structure by partial dissolution/recrystallisation of the hydrated biomineral. This requires rather significant changes at the surface/interface of the crystallites, and can possibly lead to the formation of minority side-products during this process. This is what was observed here for t-CPPD, with both significant changes in crystallite morphology after ball-milling (Fig. 2c), and the formation of small amounts of m-CPPD polymorph (Fig. 3). On the other hand, H217O can enter the crystal structure by diffusion, provided that the crystal structure exhibits continuous domains for water incorporation. In such a situation, no complete change in particle morphology is needed for the water to penetrate the crystal structure. The observations made here for m-CPPT β suggest that diffusion of labelled water is likely to occur within the hydrated layers easily. Indeed, SEM images show that the sheet-like morphology of the crystallites is essentially preserved after milling. Moreover, 17O-labelling could also be achieved in only 20 s of milling (Fig. S7b†), or even in the absence of milling (shaking experiment), which strongly points to the fact that significant surface reconstruction upon ball-milling is not needed for this material to become labelled in 17O, and that diffusion is a likely mechanism. It is worth noting that in our former work on COM, a dissolution/recrystallisation process had been proposed (notably based on mass-spectrometry analyses),7 and this is in line with the fact that the calcium oxalate monohydrate structure does not exhibit any water layers nor water channels allowing for diffusion.
Interestingly, for all the biominerals enriched so far (t-CPPD, m-CPPT β, COM and COD), no formation of a biomineral exhibiting a higher hydration level was observed, despite the addition of stoichiometric amounts of enriched water in the LAG process. For example, in our previous work on COM, the amount of enriched water added could have led to the formation of the dihydrate form COD, but no trace of this phase was observed. Similarly, here, in the case of t-CPPD, the amount of enriched water added could have led to the formation of a tetrahydrate form like m-CPPT β during the milling, but only minute conversion of t-CPPD into the m-CPPD was observed experimentally. Attempts to enable further conversion into a tetrahydrate form by increasing the energy input during the milling process were performed in the latter case (15 minutes BM at 25 Hz, using 2 stainless-steel balls), but led to a complete amorphisation of the Ca-pyrophosphate, as demonstrated by pXRD. Although such observations may not be generalisable to other hydrated biominerals, they show that the use of several characterisation tools, including multinuclear NMR, are needed to identify the labelled products and rationalise the labelling mechanism of hydrates.
Conclusion
Using liquid-assisted grinding in presence of 17O-labeled water, two hydrated Ca-pyrophosphate phases, namely t-CPPD and m-CPPT β, were enriched in 17O. Several characterisations were carried out on the enriched materials, including by 17O NMR analyses at different magnetic fields (up to 35.2 T), and using DNP. Two different enrichment mechanisms were proposed: dissolution/recrystallisation or diffusion of the enriched water inside the crystal lattice. Based on the different analyses performed, and the local environments of the water molecules within the two crystal structures, the former was proposed to be predominant for t-CPPD, while the latter for m-CPPT β. More generally speaking, similar analyses could help understand in detail how liquid-grinding agents like water can interact or react with other crystal structures. From a more practical perspective, the possibility to enrich in 17O biominerals using only microlitre amounts of expensive 17O-enriched water makes LAG a highly attractive approach for future studies on other hydrated biominerals, for which the local environment of water molecules still deserves investigation, as it is the case for canaphite (CaNa2P2O7·4H2O),23 octacalcium phosphate (OCP, Ca8(HPO4)2(PO4)4·5H2O),24 and calcium carbonate hemihydrate (CaCO3·0.5H2O).25 Further work in the case of canaphite has been initiated, which will be reported in due course. Lastly, it is worth highlighting that beyond the 17O isotopic labelling, similar synthetic procedures would also be worth performing with D2O (instead of H217O), as 2H NMR spectroscopy can provide valuable details into the local movements of water molecules within crystal structures and their characteristic frequencies.7Author contributions
IG, NDJ, TXM and DL carried out the isotopic enrichment experiments, while CC synthesised the hydrated Ca-pyrophosphate precursors. IG and DL carried out the majority of the characterisations by pXRD and solid-state NMR. JT participated in the experiments recorded at 18.8 T in Lille. CB, DL, XW, IH and ZG performed experiments at 35.2 T in Tallahassee. FMV performed the low-temperature 17O NMR and DNP analyses in Tallahassee. IG and CG carried out GIPAW DFT computations. IG and DL wrote the initial draft of the manuscript, and all authors contributed to the final preparation of the article.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreement no 772204; 2017 ERCCOG, MISOTOP project), and from the Agence Nationale de la Recherche (ANR grants PyVerres and CaPyrosis). DFT calculations were performed using HPC resources from GENCI-IDRIS (grants 097535, 2020-A0090807394, and 2021-A0110807394). Powder X-ray diffraction and SEM characterisations were performed with the support of the local Balard Plateforme d'Analyses et de Caractérisation (PAC Balard), and of Dominique Granier and Bertrand Rebière, respectively. Financial support from the IR-RMN-THC FR 3050 CNRS for conducting part of the NMR research at the UCCS facility in Lille is gratefully acknowledged. A portion of this work was also performed at the National High Magnetic Field Laboratory, which is supported by the National Science Foundation Cooperative Agreement No. DMR-1644779 and the State of Florida. The MAS-DNP instrument is supported by the NIH P41 GM122698 and NIH S10 OD018519. This project was partially supported by the European Union's Horizon 2020 research and innovation programme under Grant Agreement No. 101008500 (PANACEA).References
- C. Leroy, T.-X. Métro and D. Laurencin, The expanding frontier between mechanochemistry & solid state NMR: special focus on inorganic components of materials, in Comprehensive inorganic chemistry III, Elsevier, 2023, CH 00136, accepted Search PubMed.
- E. F. Baxter, T. D. Bennett, A. B. Cairns, N. J. Brownbill, A. L. Goodwin, D. A. Keen, P. A. Chater, F. Blanc and A. K. Cheetham, A Comparison of the Amorphization of Zeolitic Imidazolate Frameworks (ZIFs) and Aluminosilicate Zeolites by Ball-Milling, Dalton Trans., 2016, 45, 4258–4268 RSC.
- (a) M. Schmücker, H. Schneider and K. J. MacKenzie, Mechanical Amorphization of Mullite and Thermal Recrystallization, J. Non-Cryst. Solids, 1998, 226, 99–104 CrossRef; (b) S. E. Ashbrook, K. J. D. MacKenzie and S. Wimperis, 27Al Multiple-Quantum MAS NMR of Mechanically Treated Bayerite (α-Al(OH)3) and Silica Mixtures, Solid State Nucl. Magn. Reson., 2001, 20, 87–99 CrossRef CAS PubMed.
- (a) S. E. Ashbrook and M. E. Smith, Solid state 17O NMR—an introduction to the background principles and applications to inorganic materials, Chem. Soc. Rev., 2006, 35, 718–735 RSC; (b) G. Wu, 17O NMR studies of organic and biological molecules in aqueous solution and in the solid state, Prog. Nucl. Magn. Reson. Spectrosc., 2019, 114–115, 135–191 CrossRef CAS PubMed.
- (a) C. H. Chen, E. Gaillard, F. Mentink-Vigier, K. Chen, Z. Gan, P. Gaveau, B. Rebière, R. Berthelot, P. Florian, C. Bonhomme, M. E. Smith, T.-X. Métro, B. Alonso and D. Laurencin, Direct isotopic labeling of oxides using mechanochemistry, Inorg. Chem., 2020, 59, 13050–13066 CrossRef CAS PubMed; (b) C.-H. Chen, F. Mentink-Vigier, J. Trébosc, I. Goldberga, P. Gaveau, E. Thomassot, D. Iuga, M. E. Smith, K. Chen, Z. Gan, N. Fabregue, T.-X. Métro, B. Alonso and D. Laurencin, Labeling and Probing the Silica Surface Using Mechanochemistry and 17O NMR Spectroscopy, Chem.–Eur. J., 2021, 27, 12574–12588 CrossRef CAS.
- C. Leroy, T.-X. Métro, I. Hung, Z. Gan, C. Gervais and D. Laurencin, From operando Raman mechanochemistry to “NMR crystallography”: understanding the structures and interconversion of Zn-terephthalate networks using selective 17O-labelling, Chem. Mater., 2022, 34, 2292–2312 CrossRef CAS.
- I. Goldberga, N. Patris, C.-H. Chen, E. Thomassot, J. Trébosc, I. Hung, Z. Gan, D. Berthomieu, T.-X. Métro, C. Bonhomme, C. Gervais and D. Laurencin, First Direct Insight into the Local Environment and Dynamics of Water Molecules in the Whewellite Mineral Phase: Mechanochemical Isotopic Enrichment and High-Resolution 17O and 2H NMR Analyses, J. Phys. Chem. C, 2022, 126, 12044–12059 CrossRef CAS.
- J. M. Griffin, A. J. Berry, D. J. Frost, S. Wimperis and S. E. Ashbrook, Water in the Earth's mantle: a solid-state NMR study of hydrous wadsleyite, Chem. Sci., 2013, 4, 1523–1538 RSC.
- (a) P. Gras, Etude physico-chimique et structurale de pyrophosphates de calcium hydratés: application aux microcalcifications associées à l'arthrose, PhD thesis, Université de Toulouse, France, 2014 Search PubMed; (b) P. Gras, C. Rey, O. Marsan, S. Sarda and C. Combes, Synthesis and Characterisation of Hydrated Calcium Pyrophosphate Phases of Biological Interest, Eur. J. Inorg. Chem., 2013, 5886–5895 CrossRef CAS; (c) P. Gras, A. Baker, C. Combes, C. Rey, S. Sarda, A. J. Wright, M. E. Smith, J. V. Hanna, C. Gervais, D. Laurencin and C. Bonhomme, From crystalline to amorphous calcium pyrophosphates: a solid state nuclear magnetic resonance perspective, Acta Biomater., 2016, 31, 348–357 CrossRef CAS PubMed; (d) P. Gras, S. Teychené, C. Rey, C. Charvillat, B. Biscans, S. Sarda and C. Combes, Crystallisation of a highly metastable hydrated calcium pyrophosphate phase, CrystEngComm, 2013, 15, 2294–2300 RSC.
- (a) B. M. Fung, A. K. Khitrin and K. Ermolaev, An improved broadband decoupling sequence for liquid crystals and solids, J. Magn. Reson., 2000, 142, 97–101 CrossRef CAS PubMed; (b) G. Comellas, J. J. Lopez, A. J. Nieuwkoop, L. R. Lemkau and C. M. Rienstra, Straightforward, Effective Calibration of SPINAL-64 Decoupling Results in the Enhancement of Sensitivity and Resolution of Biomolecular Solid-State NMR, J. Magn. Reson., 2011, 209, 131–135 CrossRef CAS.
- (a) F. Fayon, D. Massiot, M. H. Levitt, J. J. Titman, D. H. Gregory, L. Duma, L. Emsley and S. P. Brown, Through-space contributions to two-dimensional double-quantum J correlation NMR spectra of magic-angle-spinning solids, J. Chem. Phys., 2005, 122, 194313 CrossRef; (b) F. Fayon, G. Le Saout, L. Emsley and D. Massiot, Through-bond phosphorus–phosphorus connectivities in crystalline and disordered phosphates by solid-state NMR., Chem. Commun., 2002, 1702–1703 RSC.
- A. P. M. Kentgens and R. Verhagen, Advantages of Double Frequency Sweeps in Static, MAS and MQMAS NMR of Spin I=3/2 Nuclei, Chem. Phys. Lett., 1999, 300, 435–443 CrossRef CAS.
- T. Dubroca, A. N. Smith, K. J. Pike, S. Froud, R. Wylde, B. Trociewitz, J. E. McKay, F. Mentink-Vigier, J. van Tol, S. Wi, W. W. Brey, J. R. Long, L. Frydman and S. Hill, A quasi-optical and corrugated waveguide microwave transmission system for simultaneous dynamic nuclear polarization NMR on two separate 14.1 T spectrometers, J. Magn. Reson., 2018, 289, 35–44 CrossRef CAS PubMed.
- Z. Gan, I. Hung, X. Wang, J. Paulino, G. Wu, I. M. Litvak, P. L. Gor'kov, W. W. Brey, P. Lendi and J. L. Schiano, et al., NMR Spectroscopy up to 35.2 T Using a Series-Connected Hybrid Magnet, J. Magn. Reson., 2017, 284, 125–136 CrossRef CAS.
- C. Sauvée, M. Rosay, G. Casano, F. Aussenac, R. T. Weber, O. Ouari and P. Tordo, Highly Efficient, Water-Soluble Polarizing Agents for Dynamic Nuclear Polarization at High Frequency, Angew. Chem., Int. Ed., 2013, 52, 10858–10861 CrossRef.
- G. Kresse and J. Hafner, Ab Initio Molecular Dynamics for Liquid Metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef PubMed.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni and I. Dabo, et al., QUANTUM ESPRESSO: A Modular and Open-Source Software Project for Quantum Simulations of Materials, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef PubMed.
- N. Troullier and J. L. Martins, Efficient Pseudopotentials for Plane-Wave Calculations, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 43, 1993–2006 CrossRef PubMed.
- L. Kleinman and D. M. Bylander, Efficacious Form for Model Pseudopotentials, Phys. Rev. Lett., 1982, 48, 1425–1428 CrossRef.
- C. J. Pickard and F. Mauri, All-Electron Magnetic Response with Pseudopotentials: NMR Chemical Shifts, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 245101 CrossRef.
- P. Pyykkö, Year-2017 Nuclear Quadrupole Moments, Mol. Phys., 2018, 116, 1328–1338 CrossRef.
- L. Mayen, N. D. Jensen, M. Desbord, D. Laurencin, C. Gervais, C. Bonhomme, M. E. Smith, F. Porcher, E. Elkaim, C. Charvillat, P. Gras, C. Rey, J. Soulié and C. Combes, Advances in the synthesis and structure of α-canaphite: a multitool and multiscale study, CrystEngComm, 2020, 22, 3130–3143 RSC.
- (a) E. Davies, M. J. Duer, S. E. Ashbrook and J. M. Griffin, Applications of NMR Crystallography to Problems in Biomineralization: Refinement of the Crystal Structure and 31P Solid-State NMR Spectral Assignment of Octacalcium Phosphate, J. Am. Chem. Soc., 2012, 134, 12508–12515 CrossRef CAS PubMed; (b) D. Laurencin, Y. Li, M. Duer, D. Iuga, C. Gervais and C. Bonhomme, A 43Ca NMR perspective on octacalcium phosphate and its hybrid derivatives, Magn. Reson. Chem., 2021, 59, 1048–1061 CrossRef CAS PubMed.
- Z. Zou, W. J. E. M. Habraken, G. Matveeva, A. C. S. Jensen, L. Bertinetti, M. A. Hood, C.-Y. Sun, P. U. P. A. Gilbert, I. Polishchuk, B. Pokroy, J. Mahamid, Y. Politi, S. Weiner, P. Werner, S. Bette, R. Dinnebier, U. Kolb, E. Zolotoyabko and P. Fratzl, A hydrated crystalline calcium carbonate phase: Calcium carbonate hemihydrate, Science, 2019, 363, 396–400 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Acquisition conditions of all ssNMR spectra. GIPAW-DFT calculated NMR parameters for the water sites, and simulated 17O NMR spectra. See https://doi.org/10.1039/d2fd00127f |
| This journal is © The Royal Society of Chemistry 2023 |