 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSite-selective methylene C–H oxidation of an alkyl diamine enabled by supramolecular recognition using a bioinspired manganese catalyst†
Arnau
Vicens
a,
Laia
Vicens
 a,
Giorgio
Olivo
*b,
Osvaldo
Lanzalunga
b,
Stefano
Di Stefano
b and
Miquel
Costas
*a
a,
Giorgio
Olivo
*b,
Osvaldo
Lanzalunga
b,
Stefano
Di Stefano
b and
Miquel
Costas
*a
aInstitut de Química Computacional i Catàlisi (IQCC) and Departament de Química, Universitat de Girona, Campus de Montilivi, 17071 Girona, Spain. E-mail: miquel.costas@udg.edu
bDipartimento di Chimica and Istituto CNR di Metodologie Chimiche (IMC-CNR), Sezione Meccanismi di Reazione, Sapienza Università di Roma, P.le A. Moro 5, I-00185 Rome, Italy. E-mail: giorgio.olivo@uniroma1.it
First published on 6th January 2023
Abstract
Site-selective oxidation of aliphatic C–H bonds is a powerful synthetic tool because it enables rapid build-up of product complexity and diversity from simple precursors. Besides the poor reactivity of alkyl C–H bonds, the main challenge in this reaction consists in differentiating between the multiple similar sites present in most organic molecules. Herein, a manganese oxidation catalyst equipped with two 18-benzo-6-crown ether receptors has been employed in the oxidation of the long chain tetradecane-1,14-diamine. 1H-NMR studies evidence simultaneous binding of the two protonated amine moieties to the crown ether receptors. This recognition has been used to pursue site-selective oxidation of a methylenic site, using hydrogen peroxide as oxidant in the presence of carboxylic acids as co-ligands. Excellent site-selectivity towards the central methylenic sites (C6 and C7) is observed, overcoming selectivity parameters derived from polar deactivation by simple amine protonation and selectivity observed in the oxidation of related monoprotonated amines.
Introduction
Selective functionalization of alkyl C–H bonds is one of the most challenging reactions in organic chemistry because of the poorly reactive character of these bonds.1 Moreover, organic molecules commonly have many methylenic site with similar electronic and steric properties, which make them difficult to be differentiated. Governing site-selectivity is especially challenging in the oxidation of long aliphatic chains, where methylenic site are nearly equivalent from a reactivity perspective. In these cases, C–H functionalization reactions usually proceed to yield a statistical distribution of products. However, site-selectivity has been pursued by using bulky catalysts that disfavor oxidation of internal sites that are sterically more demanding,2 or via intramolecular reactions governed by directing groups.3An appealing strategy to pursue site-selectivity is the use of weak interactions, akin to those operating in enzymatic sites.4 Supramolecular interactions may be used to govern the access and/or positioning of the substrate with respect to the catalytic site, promoting the reactivity of specific well-oriented positions of the substrate.5 One strategy consists in using supramolecular hosts that embed the catalyst and only permit access to substrate sites readily accessible or with specific 3D structure.6 Alternatively, catalysts may be equipped with a receptor that recognizes specific functional groups in the substrate leading to an exposure of specific sites to the reactive center. A critical aspect of this strategy is the use of weak interactions that ensures reversible binding of the substrate to the catalyst, enabling catalyst turnover and reducing product inhibition. Pioneering examples of the use of supramolecular receptors to govern site-selectivity in C–H oxidation have been described by Breslow,7 and more recently by Crabtree and Brudvig,8 Bach9 and Tiefenbacher.10
Recently, we reported a Mn catalyst based on a N2Py2 ligand in which 18-benzocrown-6-ether receptors ([Mn(CF3SO3)2(CRpdp)], MnCRpdp)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 11 that have high affinity towards protonated primary amines were attached (Fig. 1A). A notable increase in selectivity towards C8 and C9 methylenic sites is achieved as the protonated amine is recognized by the crown ether (Fig. 1B).12
11 that have high affinity towards protonated primary amines were attached (Fig. 1A). A notable increase in selectivity towards C8 and C9 methylenic sites is achieved as the protonated amine is recognized by the crown ether (Fig. 1B).12
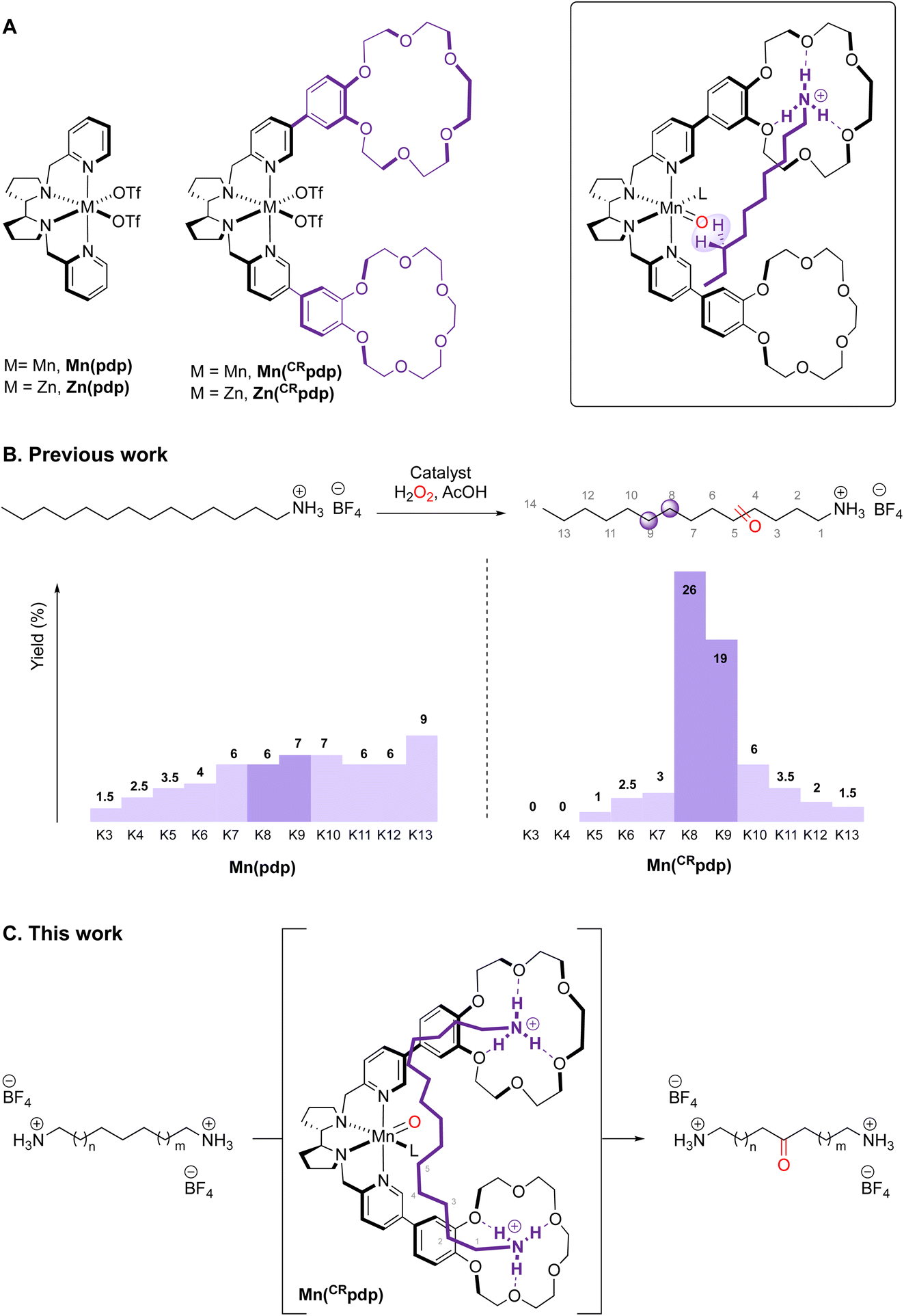 | ||
| Fig. 1 (A) Catalyst with and without 18-benzocrown-6-ether receptors and their interaction with protonated amines. (B) Previous work on the oxidation of linear protonated amines. (C) This work. | ||
Building on these precedents, the aim of this work is to explore site-selective C–H oxidations on the aliphatic chain of a protonated diamine that can bind simultaneously to the two crown ether receptors of a single catalyst. It was envisioned that, in such a case, the reduction of the degrees of freedom of the substrate upon binding to the catalyst will translate into an enhanced site-selectivity. Towards this end, the binding of the long-chain 1,14-tetradecanediamonium tetrafluoroborate (S1), selected as substrate, to Zn(CRpdp), the Zn analogue of the manganese catalyst Mn(CRpdp), was explored by NMR. Oxidation of S1 with Mn(CRpdp), H2O2 and a carboxylic acid was then studied. Compared with the previously described oxidation of monoamine substrates,11 the current reactions evidence substantially improved site-selectivities. Comparison with values obtained with a simple Mn(pdp) catalyst13 permits one to distinguish the contributions to selectivity derived from supramolecular recognition from those due to polar effects resulting from the repulsion between the ammonium groups and the electrophilic catalyst.
Results and discussion
The recently described catalysts Mn(CRpdp)11 and its Zn analogue Zn(CRpdp)14 were employed in the study. Catalytic oxidation of S1 was performed using Mn(CRpdp) as the catalyst. In addition, control experiments were done with Mn(pdp), the complex that has the same structure as Mn(CRpdp) but lacks the supramolecular 18-benzocrown-6-ether receptors.
1H-NMR studies of the binding of S1 to Zn(CRpdp)
The diamagnetic Zn complex Zn(CRpdp) shares the same topology and first coordination sphere of the metal as in Mn(CRpdp), and, therefore, it is envisioned as a suitable probe to characterize the substrate–catalyst binding by 1H-NMR.13Titration of 1.0 mM Zn(CRpdp) with S1 was followed by 1H-NMR (CD3CN, 25 °C). Focusing on the region between 3.3–4.6 ppm (Fig. 2A), a downfield shift of the crown ether signals was observed upon addition of S1, suggesting that the protonated diamine moieties interact with the crown ether. Moreover, the observed saturation profile, which reaches a plateau after addition of 1.0–1.2 molar equivalents of S1, indicates a 1:1 host–guest stoichiometry (Kass > 103 M−1, Fig. 2B).
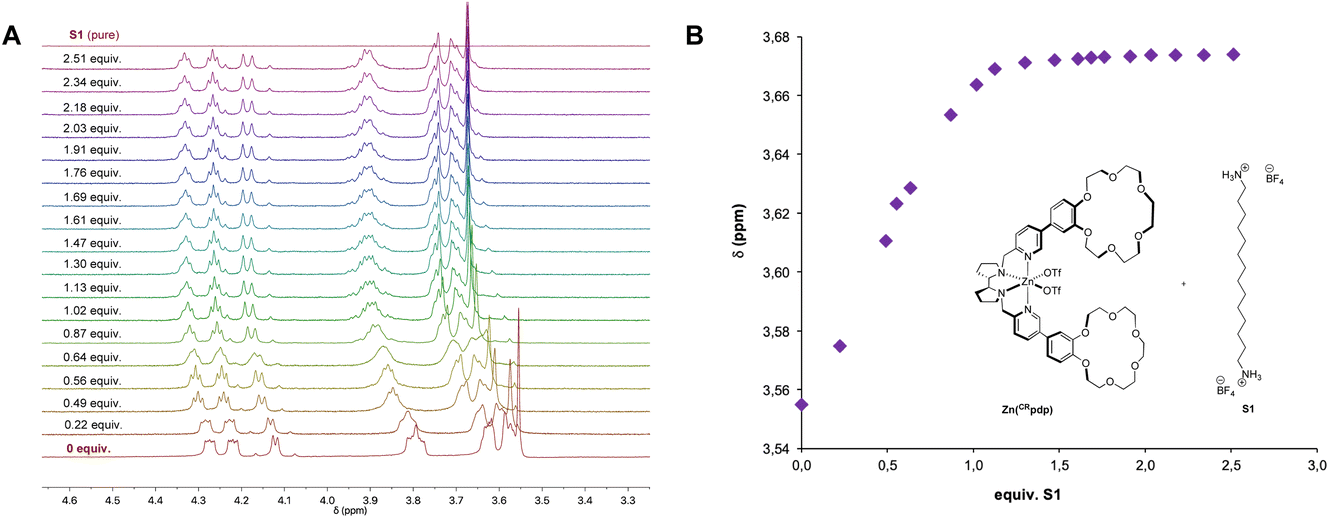 | ||
| Fig. 2 Binding of S1 to Zn(CRpdp) (1 mM) in CD3CN at 25 °C. (A) Enlargement of the region between 3.3 and 4.6 ppm, corresponding to the crown ether signals. (B) Titration curve. | ||
Two binding modalities can be considered for a 1:1 stoichiometry between the substrate and the catalyst. The first possibility is that a diamine molecule may be bound to a single molecule of catalyst, each end of the substrate interacting with one of the two crown ether receptors of the same catalyst molecule (Fig. 3A, left). A second option consistent with the stoichiometry is that each diamine binds to two crown ether moieties that belong to different catalyst molecules, which would result in the formation of cyclo-oligomeric species, the dimer being the simplest possibility (Fig. 3A, right).
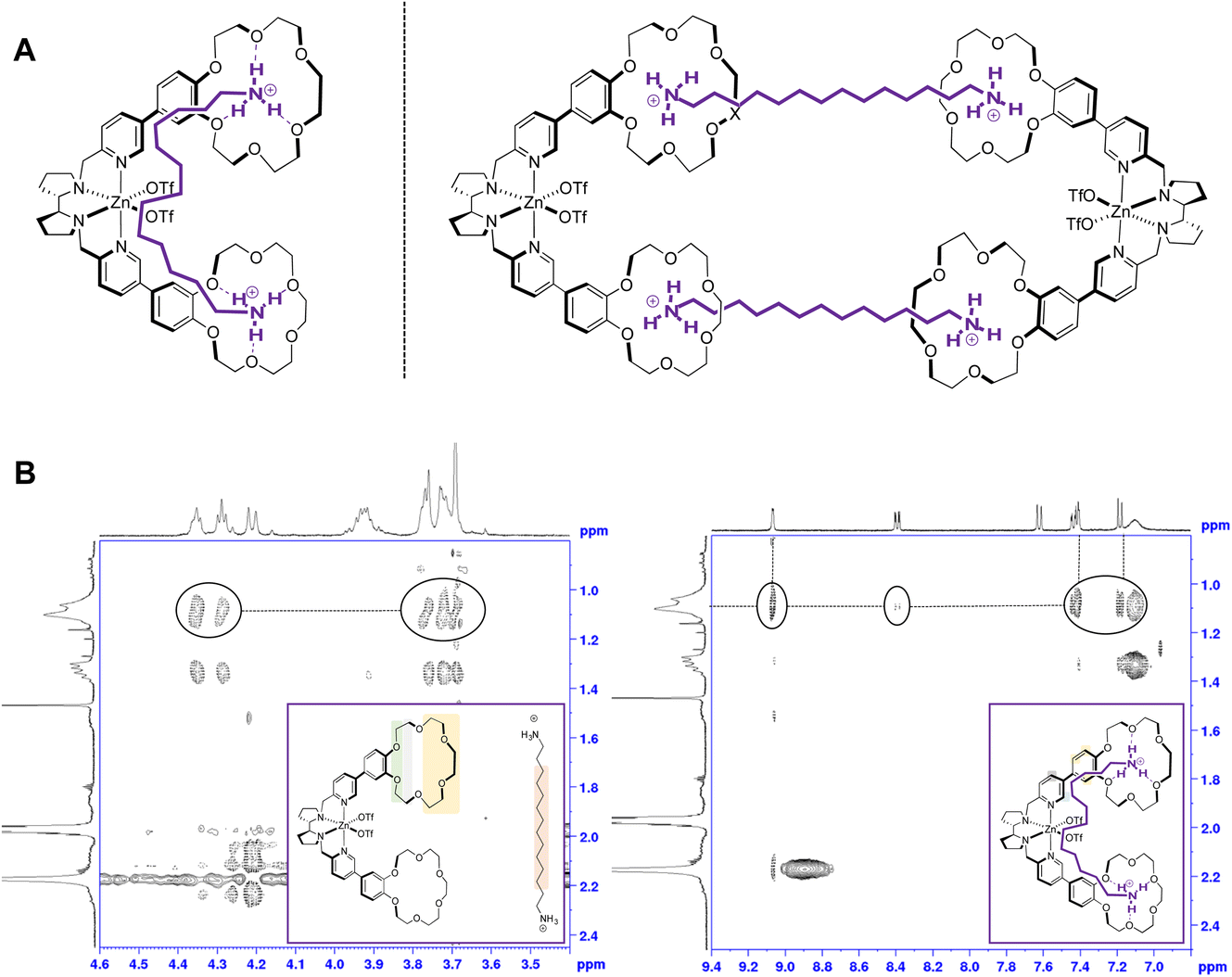 | ||
| Fig. 3 (A) Possible 1:1 stoichiometries for Zn(CRpdp):S1 binding. (B) NOESY of the 1:1 mixture of Zn(CRpdp):S1. (Left): enlargement of the aliphatic region of the crown ether catalyst (3.5–4.5 ppm) vs.S1 signals (0.8–2.5 ppm). (Right): enlargement of the aromatic region of the catalyst (7.0–9.4 ppm) vs.S1 signals (0.8–2.5 ppm). | ||
To understand how the substrate binds to the catalyst, two-dimensional NMR NOESY experiments were carried out with a 1:1 mixture of Zn(CRpdp):S1 (Fig. 3B). First, in the aliphatic region of the crown ether catalyst, an intermolecular NOESY correlation between the signals of the crown ether (3.5–4.5 ppm) and those of the substrate was observed (Fig. 3B, left), which implies proximity between the diamine and the crown ether moieties of the complex, in accordance with the adduct geometry observed with monoamines.14 Second and more interestingly, a clear NOESY correlation between the aliphatic signals of the substrate and the α-protons of the pyridines of the ligand was observed, as well as with the protons of the phenyl ring of the benzocrown. A small correlation was also observed with the γ-proton of the pyridine rings. These correlations are only consistent with the 1:1 model depicted in Fig. 3A (left), in which the protonated diamine is located close to the aromatic parts of Zn(CRpdp), and are incompatible with the 2:2 adduct on the right. The 1:1 stoichiometry, which therefore prevails on all other n:n ones (n > 1), is consistent with the desired recognition-driven site-selective oxidation.
A control experiment with Zn(pdp), devoid of the crown ether, was also performed following an analogous procedure. In this case, no shift in either the signals of the catalyst nor the signals of S1 was detected, suggesting the absence of any interaction. A NOESY experiment on the 1:1 mixture of Zn(pdp):S1 did not show any intermolecular correlation, which confirms the expected lack of binding between the substrate and the complex.
Catalytic oxidation of S1
Once the desired binding geometry between S1 and the supramolecular catalyst was confirmed, oxidation of the substrate was investigated. Catalytic oxidations were performed adopting the following standard conditions: 1 mol% of catalyst, 1 equiv. of S1 (0.1 M), 10 equiv. of H2O2 delivered by a syringe pump over 30 minutes, and 22 equiv. of AcOH at 0 °C.11 Conversions and yields were determined by 1H-NMR (Fig. 4A).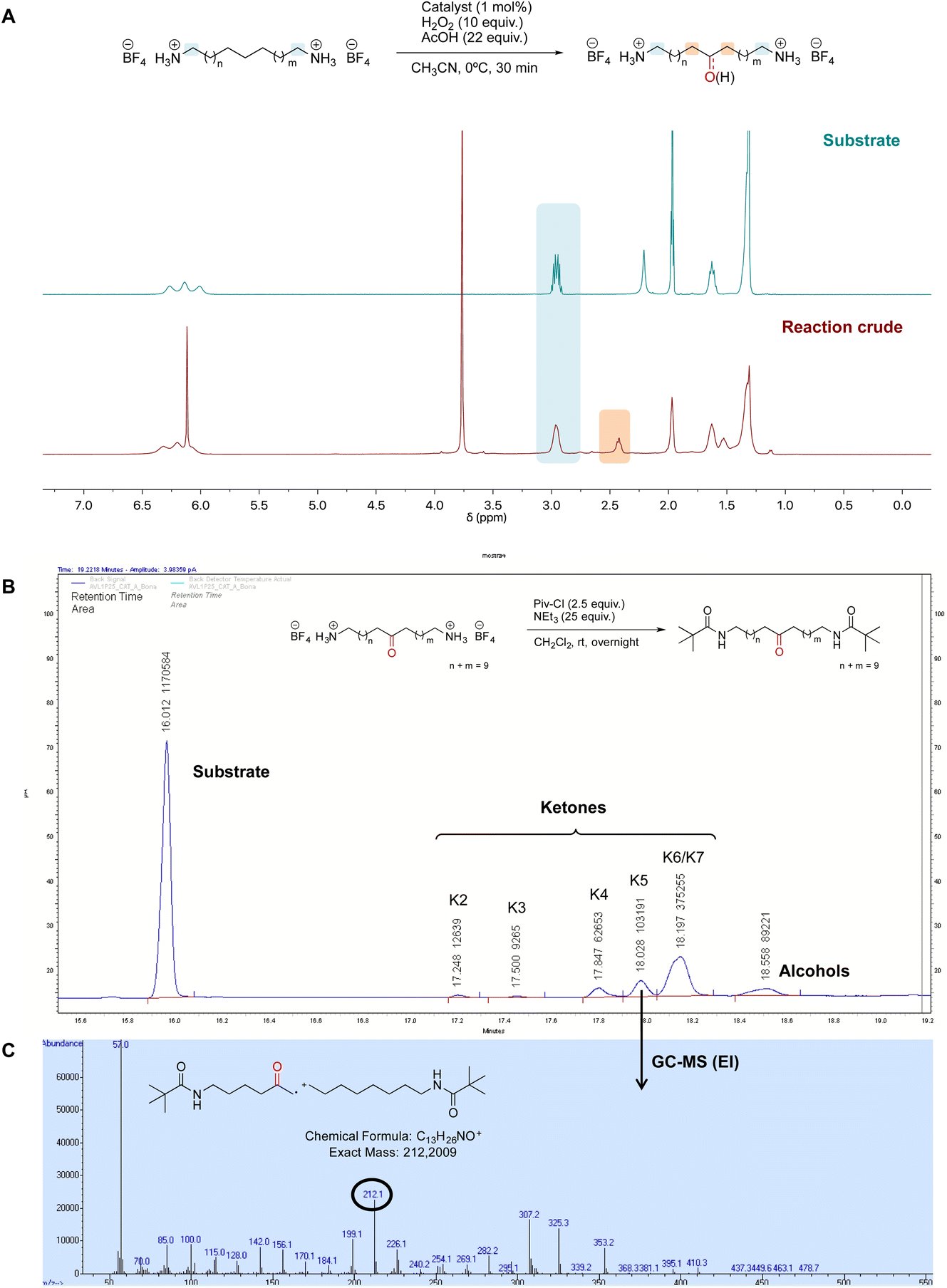 | ||
| Fig. 4 Experimental details for oxidation of S1. (A) NMR spectrum of S1 before (top) and after the oxidation (bottom; sharp peaks at 3.7 and 6.1 ppm are due to 1,3,5-trimethoxybenzene internal standard). (B) GC chromatogram of the oxidation of S1 with the Mn(pdp) catalyst analyzed after derivatization of products. (C) GC-MS (EI) of the peak at 18.0 min, showing the fragmentation of product K5 as a typical example of fragmentation. | ||
Oxidation of the different methylenic sites of S1 produced a mixture of isomeric alcohol (OH) and ketone (K) products differing in the position of the oxygenated function (i.e., K2 refers to the ketone derived from oxidation of the C2 site, etc.). Site-selectivity (the distribution of the isomeric oxygenated products) was analyzed by GC after derivatization of the products by the preparation of the corresponding dipivalamides (Fig. 4B). The oxidation site was assigned by analyzing the fragmentation of the ketone products in the EI-MS spectra (Fig. 4C).11
The results obtained are shown in Table 1. For each reaction, the analysis of site-selectivity was repeated after Jones oxidation of the reaction mixture to convert all the alcohols into ketones (Table 1, footnote d).
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Catalyst [x mol%] | Conv.a (%) | Yield Ka (%) | Yield OHb (%) | K2c | K3c | K4c | K5c | K6 + K7c |
| a Conversions and yields determined by 1H-NMR using 1,3,5-trimethoxybenzene as the internal standard. b Estimated by GC. c Normalized site-selectivity (%) determined by GC after derivatization. d Values obtained after oxidation of the mixture with Jones reagent. | ||||||||
| — | 4 | — | — | — | — | — | — | — |
| Mn(pdp) [1 mol%] | 45 | 26 | 5 | 2 | 2 | 11 | 18 | 67 |
| 6d | 1d | 14d | 17d | 62d | ||||
| Mn(CRpdp) [1 mol%] | 38 | 25 | 10 | 0 | 2 | 7 | 6 | 84 |
| 2d | 3d | 5d | 10d | 81 | ||||
| Mn(CRpdp) [3 mol%] | 55 | 43 | 7 | 0 | 1 | 6 | 7 | 86 |
| 0d | 2d | 2d | 8d | 88 | ||||
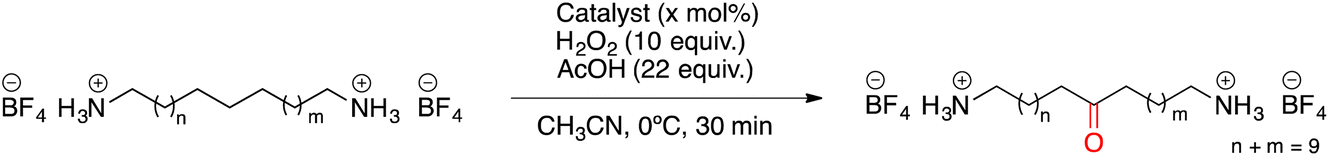
When using Mn(pdp) as the catalyst, a 26% combined yield of ketones and 5% of alcohols was obtained, with a selectivity for the central C6 and C7 sites of 62%. Significant amounts of K4 and K5 were also obtained. The origin of this selectivity is likely due to the polar deactivation exerted by the protonated amine moieties, which decreases as the methylenic site moves away from the NH3+. In contrast, when the supramolecular catalyst Mn(CRpdp) was used, the total yield of oxidation products was slightly increased, the mass balance of the reaction substantially improved (35% combined yield/38% substrate conversion) and, most remarkably, the selectivity towards K6 + K7 increased up to 81%. This means that supramolecular recognition plays an important role as the selectivity increases from 62% to 81% compared to the reaction in the absence of supramolecular recognition. With higher catalyst loading (3 mol%), the oxidation yield and the selectivity for C6 and C7 sites increased up to 43% and, remarkably, 88%, respectively. A notable finding concerns the relative selectivity for C4 and C5 oxidation. In the absence of the recognition, the two positions are nearly equally reactive. In contrast, in the presence of the crown ether moieties, oxidation at C4 is substantially prevented, since such a position is presumably placed far away from the reactive metal-oxo in the catalyst–substrate complex.
Effect of carboxylic acid co-catalyst
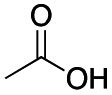
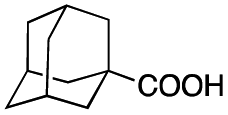
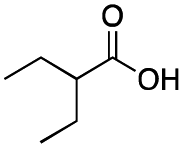
Carboxylic acids are known to be crucial co-ligands in oxidation reactions, and different examples in the literature showed that modulation of their structure may affect the selectivity of the oxidation reactions.2c The active species responsible for the C–H oxidation with this specific type of catalyst is presumably a MnV(O)(carboxylate) intermediate,15 and therefore the nature of the carboxylate ligand impacts on the structure of the C–H cleaving species. In fact, use of different carboxylic acid co-ligands allowed fine-tuning of site- and enantio-selectivity of C–H oxidation reactions catalysed by the same Mn catalyst. Along this line, we explored replacement of acetic acid with other more structured acids in order to pursue an improvement of the selectivity for the central C6 and C7 sites (Table 2).16 All these experiments were performed with 1 mol% of catalyst Mn(CRpdp).|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | R-COOH | Conv.a (%) | Yield Ka (%) | Yield OHb (%) | K2c | K3c | K4c | K5c | K6 + K7c |
| a Conversions and yields determined by 1H-NMR using 1,3,5-trimethozybenzene as the internal standard. b Estimated by GC. c Normalized site-selectivity (%) determined by GC after derivatization.. | |||||||||
| 1 |
|
38 | 25 | 10 | 0 | 2 | 7 | 6 | 84 |
| 2 |
|
— | — | — | — | — | — | — | — |
| 3 |
|
32 | 18 | 7 | 0 | 6 | 18 | 1 | 75 |
| 4 |
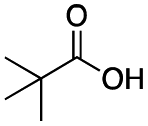
|
35 | 24 | 4 | 0 | 1 | 6 | 2 | 91 |
| 5 |
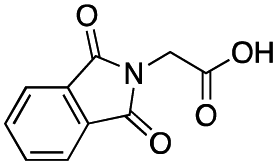
|
— | — | — | — | — | — | — | — |
| 6 |
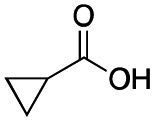
|
35 | 31 | 2 | 0 | 4 | 9 | 3 | 84 |
| 7 |
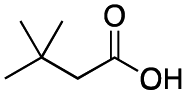
|
36 | 31 | 5 | 0 | 8 | 6 | 2 | 84 |
| 8 |
|
— | — | — | — | — | — | — | — |
| 9 |
|
51 | 42 | 4 | 0 | 3 | 5 | 1 | 91 |
| 10 |
|
55 | 38 | 3 | 0 | 2 | 5 | 1 | 92 |
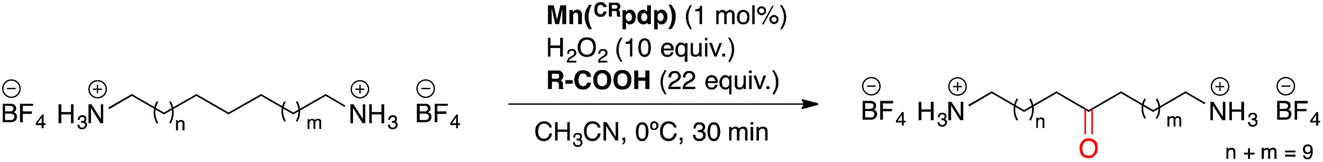
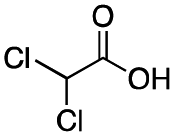
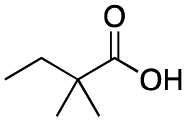
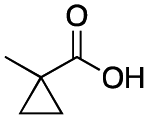
As shown in Table 2, all the tested carboxylic acids were effective co-catalysts, with the exception of 1-adamantane carboxylic acid (entry 2), Phth-Gly-OH (entry 5) and dichloroacetic acid (entry 8) that delivered no oxidation products. Cyclopropyl carboxylic acid (entry 6) and 3,3-dimethylbutyric carboxylic acid (entry 7) gave similar selectivity to acetic acid (entry 1), while 2-ethylbutyric carboxylic acid led to slightly lower values (entry 3). More interestingly, a series of bulky carboxylic acids improved site-selectivity for central C6 and C7 oxidation. For example, pivalic acid (entry 4), 2,2-dimethylcarboxylic acid (entry 9) and 1-methylcyclopropane carboxylic acid (entry 10) raised the K6/K7 selectivity up to 91–92%. In these three cases, an α,α,α-trisubstituted carboxylic acid was used, indicating that substantial steric hindrance close to the metal seems to be an important characteristic to increase the selectivity. This effect is also evident in control experiments carried out using Mn(pdp) as the catalyst where the selectivities increased compared to acetic acid (Table 3). However, the presence of recognition units in the supramolecular catalyst always led to a site-selectivity improvement with respect to Mn(pdp).
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| R-COOH | Catalyst | Conv.a (%) | Yield Ka (%) | Yield OHb (%) | K2c | K3c | K4c | K5c | K6 + K7c |
| a Conversions and yields determined by 1H-NMR using 1,3,5-trimethozybenzene as the internal standard. b Estimated by GC. c Normalized site-selectivity (%) determined by GC after derivatization. | |||||||||
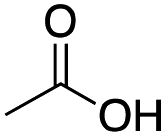
|
Mn(pdp) | 45 | 26 | 5 | 2 | 2 | 11 | 18 | 67 |
| Mn(CRpdp) | 38 | 25 | 10 | 0 | 2 | 7 | 6 | 84 | |

|
Mn(pdp) | 62 | 34 | 0 | 2 | 0 | 12 | 3 | 82 |
| Mn(CRpdp) | 35 | 24 | 4 | 0 | 1 | 6 | 2 | 91 | |
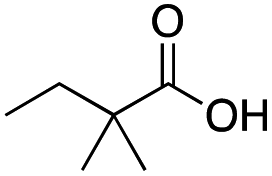
|
Mn(pdp) | 37 | 27 | 4 | 0 | 0 | 11 | 12 | 77 |
| Mn(CRpdp) | 51 | 42 | 4 | 0 | 3 | 5 | 1 | 91 | |
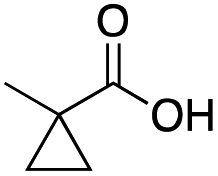
|
Mn(pdp) | 40 | 15 | 6 | 0 | 0 | 15 | 6 | 79 |
| Mn(CRpdp) | 55 | 38 | 3 | 0 | 2 | 5 | 1 | 92 | |
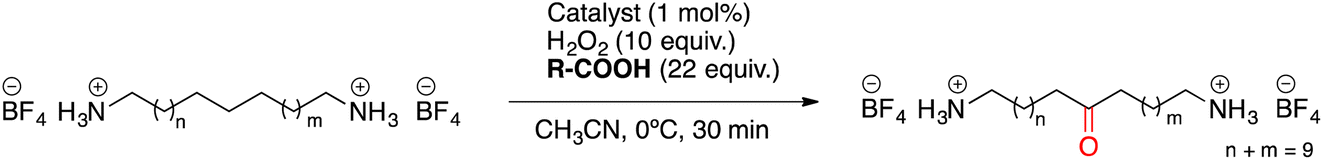
These results can be compared with those reported by Breslow's group17 and, more recently, those described by Higuchi and co-workers,18 in the oxidation of a long linear α,ω-disubstituted alkyl chain (1,12 dicarboxylate17 or 1,14 diimide derivative18) that undergoes a two-point binding to an oxidation catalyst via supramolecular interactions (ion pairing16 or hydrogen bonding18). The oxidation chemistry differs in these cases. Breslow studied a photoinduced stoichiometric oxidation with benzophenone derivatives, while Higuchi's group used a Ru porphyrin and 2,6-dichloro-pyridine N-oxide oxidant. In both reports, up to >90% selectivity for the oxidation of the two central methylenic sites was obtained in the best cases, which compares well with the present findings. Such selectivities are higher than those previously obtained with singly bound alkyl chain monoamines (up to 81% selectivity for the two favoured methylenes).10,11 Under oxidation conditions, catalyst Mn(CRpdp) is expected to be almost fully saturated (>98%) with both diammonium S1 and monoammonium guests. Hence, the increased selectivity observed with diammonium S1 can be ascribed to an improved pre-organization of the catalyst–substrate adduct that arises from the higher rigidity associated with the two-point binding of S1.
Conclusion
In summary, we have demonstrated the capability of a bioinspired manganese catalyst bearing two benzocrown ether receptors for binding a diprotonated diamine. The binding mainly occurs in a 1:1 stoichiometry, with each protonated amine of the substrate being recognized by one of the two crown ether receptors of the catalyst, resulting in a well-defined orientation of the substrate towards the metal center. In the catalytic oxidation of a diammonium substrate, such binding allows an increase of selectivity for the central C6 and C7 positions up to 92% using bulky carboxylic acids as co-ligands.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Spanish Ministry of Science, Innovation, and Universities (PID2021-129036NB-I00 to M. C. and PhD grant FPU16/04231 to L.V.), and Generalitat de Catalunya (ICREA Academia Award to M.C.). G.O. acknowledges project S-ReCHOx from SCI and Elsevier for funding. We acknowledge STR of UdG for experimental support.References
- T. Newhouse and P. S. Baran, Angew. Chem., Int. Ed., 2011, 50, 3362 CrossRef CAS PubMed.
- (a) M. C. White and J. Zhao, J. Am. Chem. Soc., 2018, 140, 13988 CrossRef CAS PubMed; (b) W. Sun and Q. Sun, Acc. Chem. Res., 2019, 52, 2370 CrossRef CAS PubMed; (c) L. Vicens, G. Olivo and M. Costas, ACS Catal., 2020, 10, 8611 CrossRef CAS.
- (a) T. Brückl, R. D. Baxter, Y. Ishihara and P. S. Baran, Acc. Chem. Res., 2012, 45, 826 CrossRef PubMed; (b) S. Bhadra and H. Yamamoto, Chem. Rev., 2018, 118, 3391 CrossRef CAS PubMed.
- (a) J. C. Lewis, P. S. Coelho and F. H. Arnold, Chem. Soc. Rev., 2011, 40, 2003 RSC; (b) R. Fasan, ACS Catal., 2012, 2, 647 CrossRef CAS; (c) S. Jena, J. Dutta, K. D. Tulsiyan, A. K. Sahu, S. S. Choudhury and H. S. Biswal, Chem. Soc. Rev., 2022, 51, 4261 RSC.
- (a) P. Dydio and J. N. H. Reek, Chem. Sci., 2014, 5, 2135 RSC; (b) H. J. Davis and R. J. Phipps, Chem. Sci., 2017, 8, 864 RSC; (c) D. Vidal, G. Olivo and M. Costas, Chem.–Eur. J., 2018, 24, 5042 CrossRef CAS PubMed; (d) G. Olivo, G. Capocasa, D. Del Giudice, O. Lanzalunga and S. Di Stefano, Chem. Soc. Rev., 2021, 50, 7681 RSC; (e) S. Pachisia and R. Gupta, Dalton Trans., 2021, 50, 14951 RSC; (f) Y. Jiao, X.-Y. Chen and J. F. Stoddart, Chem, 2022, 8, 414 CrossRef CAS.
- (a) M. Guitet, P. Zhang, F. Marcelo, C. Tugny, J. Jiménez-Barbero, O. Buriez, C. Amatore, V. Mouriès-Mansuy, J.-P. Goddard, L. Fensterbank, Y. Zhang, S. Roland, M. Ménand and M. Sollogoub, Angew. Chem., Int. Ed., 2013, 52, 7213 CrossRef CAS PubMed; (b) C. García-Simón, R. Gramage-Doria, S. Raoufmoghaddam, T. Parella, M. Costas, X. Ribas and J. N. H. Reek, J. Am. Chem. Soc., 2015, 137, 2680 CrossRef PubMed; (c) T. A. Bender, R. G. Bergman, K. N. Raymond and F. D. Toste, J. Am. Chem. Soc., 2019, 141, 11806 CrossRef CAS PubMed.
- (a) R. Breslow, Y. Huang, X. Zhang and J. Yang, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 11156 CrossRef CAS PubMed; (b) R. Breslow, X. Zhang and Y. Huang, J. Am. Chem. Soc., 1997, 119, 4535 CrossRef CAS; (c) J. Yang, B. Gabriele, S. Belvedere, Y. Huang and R. Breslow, J. Org. Chem., 2002, 67, 5057 CrossRef CAS PubMed.
- S. Das, C. D. Incarvito, R. H. Crabtree and G. W. Brudvig, Science, 2006, 312, 1941 CrossRef CAS PubMed.
- (a) F. Burg, M. Gicquel, S. Breitenlechner, A. Pöthig and T. Bach, Angew. Chem., Int. Ed., 2018, 57, 2953 CrossRef CAS PubMed; (b) F. Burg, S. Breitenlechner, C. Jandl and T. Bach, Chem. Sci., 2020, 11, 2121 RSC; (c) F. Burg, C. Buchelt, N. M. Kreienborg, C. Merten and T. Bach, Org. Lett., 2021, 23, 1829 CrossRef CAS PubMed.
- (a) M. Knezevic, M. Heilmann, G. M. Piccini and K. Tiefenbacher, Angew. Chem., Int. Ed., 2020, 59, 12387 CrossRef CAS PubMed; (b) M. Knezevic and K. Tiefenbacher, Chem.–Eur. J., 2022, e202203480 Search PubMed.
- (a) G. Olivo, G. Farinelli, A. Barbieri, O. Lanzalunga, S. Di Stefano and M. Costas, Angew. Chem., Int. Ed., 2017, 56, 16347 CrossRef CAS PubMed; (b) G. Olivo, G. Capocasa, O. Lanzalunga, S. Di Stefano and M. Costas, Chem. Commun., 2019, 55, 917 RSC.
- S. Di Stefano, G. Capocasa and L. Mandolini, Eur. J. Org. Chem., 2020, 2020, 3340 CrossRef CAS.
- R. V. Ottenbacher, D. G. Samsonenko, E. P. Talsi and K. P. Bryliakov, Org. Lett., 2012, 14, 4310 CrossRef CAS PubMed.
- (a) G. Olivo, G. Capocasa, B. Ticconi, O. Lanzalunga, S. Di Stefano and M. Costas, Angew. Chem., Int. Ed., 2020, 59, 12703 CrossRef CAS PubMed; (b) L. Vicens, G. Olivo and M. Costas, Angew. Chem., Int. Ed., 2022, 61, e202114932 CrossRef CAS PubMed.
- (a) R. V. Ottenbacher, E. P. Talsi and K. P. Bryliakov, ACS Catal., 2015, 5, 39 CrossRef CAS; (b) X.-X. Li, M. Guo, B. Qiu, K.-B. Cho, W. Sun and W. Nam, Inorg. Chem., 2019, 58, 14842 CrossRef CAS PubMed.
- (a) M. Milan, M. Bietti and M. Costas, ACS Cent. Sci., 2017, 3, 196 CrossRef CAS PubMed; (b) E. P. Talsi, D. G. Samsonenko, R. V Ottenbacher and K. P. Bryliakov, ChemCatChem, 2017, 9, 4580–4586 CrossRef CAS; (c) B. Qiu, D. Xu, Q. Sun, C. Miao, Y.-M. Lee, X.-X. Li, W. Nam and W. Sun, ACS Catal., 2018, 8, 2479–2487 CrossRef CAS; (d) M. Galeotti, L. Vicens, M. Salamone, M. Costas and M. Bietti, J. Am. Chem. Soc., 2022, 144, 7391 CrossRef CAS PubMed.
- R. Breslow, R. Rajagopalan and J. Schwarz, J. Am. Chem. Soc., 1981, 103, 2905 CrossRef CAS.
- S. Teramae, A. Kito, T. Shingaki, Y. Hamaguchi, Y. Yano, T. Nakayama, Y. Kobayashi, N. Kato, N. Umezawa, Y. Hisamatsu, T. Nagano and T. Higuchi, Chem. Commun., 2019, 55, 8378 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2fd00177b |
| This journal is © The Royal Society of Chemistry 2023 |