 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ultrafast electronic, infrared, and X-ray absorption spectroscopy study of Cu(I) phosphine diimine complexes†
Martin V.
Appleby
 a,
Rory A.
Cowin
a,
Iona I.
Ivalo
a,
Samantha L.
Peralta-Arriaga
a,
Craig C.
Robertson
a,
Stuart
Bartlett
b,
Ann
Fitzpatrick
b,
Andrew
Dent
b,
Gabriel
Karras
b,
Sofia
Diaz-Moreno
b,
Dimitri
Chekulaev
a and
Julia. A.
Weinstein
*a
a,
Rory A.
Cowin
a,
Iona I.
Ivalo
a,
Samantha L.
Peralta-Arriaga
a,
Craig C.
Robertson
a,
Stuart
Bartlett
b,
Ann
Fitzpatrick
b,
Andrew
Dent
b,
Gabriel
Karras
b,
Sofia
Diaz-Moreno
b,
Dimitri
Chekulaev
a and
Julia. A.
Weinstein
*a
aDepartment of Chemistry, The University of Sheffield, Sheffield, S3 7HF, UK. E-mail: julia.weinstein@sheffield.ac.uk
bDiamond Light Source, Rutherford Appleton Laboratory, Harwell Science and Innovation Campus, Didcot, Oxfordshire OX11 0DE, UK
First published on 7th July 2023
Abstract
The study aims to understand the role of the transient bonding in the interplay between the structural and electronic changes in heteroleptic Cu(I) diimine diphosphine complexes. This is an emerging class of photosensitisers which absorb in the red region of the spectrum, whilst retaining a sufficiently long excited state lifetime. Here, the dynamics of these complexes are explored by transient absorption (TA) and time-resolved infrared (TRIR) spectroscopy, which reveal ultrafast intersystem crossing and structural distortion occurring. Two potential mechanisms affecting excited state decay in these complexes involve a transient formation of a solvent adduct, made possible by the opening up of the Cu coordination centre in the excited state due to structural distortion, and by a transient coordination of the O-atom of the phosphine ligand to the copper center. X-ray absorption studies of the ground electronic state have been conducted as a prerequisite for the upcoming X-ray spectroscopy studies which will directly determine structural dynamics. The potential for these complexes to be used in bimolecular applications is confirmed by a significant yield of singlet oxygen production.
Introduction
Transition metal complexes have been at the forefront of light-driven applications in synthesis and catalysis for decades, owing to their intense absorption of visible light, and comparatively long – hundreds of nanoseconds or more – lifetime of the lowest-lying excited state, which enables efficient bimolecular reactions. The reason for the long excited state lifetime if compared to organic molecules is the triplet nature of the lowest excited state, which is populated due to an efficient intersystem crossing (ISC) process in the originally populated singlet excited state, the process greatly enhanced by the presence of a heavy atom, and the ensued spin–orbit coupling. Thus, transition metal complexes act as efficient photosensitisers in catalysis,1 and as photosensitisers of reactive oxygen species in light-driven antimicrobial and anticancer (PDT) applications.2 The overwhelming majority of such photosensitisers to date are based on Noble metals (e.g. Pt, Re, Ru), but there has been substantial interest in moving towards the more abundant first-row transition metals such as copper and iron. However, complexes of these lighter elements typically possess low-lying metal-centred dd-states that provide an efficient decay pathway from the excited state which limit their use as photosensitisers or photocatalysts. By designing complexes with low energy Metal–Ligand Charge Transfer (MLCT) states, one can bypass the dd deactivation pathway.Homoleptic Cu(I) diimine complexes, [Cu(NN)2]+ possess sufficiently low-energy MLCT states, but MLCT states are usually too short-lived, in the sub-nanoseconds domain, unless bulky substituents are introduced next to coordinating N-atoms. The reason for the short lifetime is that excitation into an MLCT state changes the electronic configuration of Cu from nominally d10 to d9, causing large structural changes from a pseudo-tetrahedral geometry in the ground state to a pseudo-square-planar geometry, which provides yet another efficient deactivation pathway.3,4 Heteroleptic Cu(I) complexes, such as phosphine diimine complexes, [Cu(PP)(NN)]+, can benefit from the tuneable electronic properties of the diimine acceptor ligand while extending the lifetime of the MLCT state by inhibiting the excited state distortion using a bulky phosphine co-ligand.5 For example, Cu(I) xantphos diimine complexes have excited state lifetimes in the nano to microsecond range (10–100 times longer than the homoleptic diimine complexes) which make them suitable for applications relying on bimolecular processes.6,7 We have recently shown that Cu(I) heteroleptic complexes consisting of a phen based ligand (dmp) and phosphine ligand (xantphos = 4,5-bis(diphenylphosphino)-9,9-dimethyl-xanthene) produce singlet oxygen in high yields and are capable of killing both Gram positive and Gram negative bacteria.8 However, these complexes are limited in use due to limited UV-vis absorption which does not make the most of the solar spectrum, vital for sunlight-driven applications.
Whilst heteroleptic Cu(I) phenanthroline complexes have shown strong absorption of light below 400 nm, many applications require compounds and photosensitisers which absorb in the visible range. The lowest absorption band in these complexes is due to a metal-to-ligand charge transfer (MLCT) transition to the diimine ligand. Introducing a more electron-withdrawing diimine ligand such as 2,2′-biquinoline (biq) and its derivatives (Ered = −1.46,9vs. 2,9-dimethyl-1,10-phenanthroline (dmp), Ered = −2.11)7 may shift the MLCT absorption towards lower energies.
Herein, the excited state dynamics of a series of Cu(I) 4,4′-(CO(O)Et)2-2,2′-biquinoline (deebq) complexes are studied. The biquinoline ligand, deebq, has electron-withdrawing ester groups, which further shift the MLCT absorption band to lower energies, and enable future immobilisation of the complexes on surfaces using –COOH as an anchoring group, for which the ester is a suitable proxy for spectroscopic studies. Surface immobilisation improves reusability of the photosensitiser and prevents leaching of the complex from a support, both are of specific interest in applications for water purification and H2 production. Earth-abundant photosensitisers absorbing in the red region and achieving a good yield of singlet oxygen not only will lend themselves to sun-driven applications but also photodynamic therapy (PDT), and photo-driven synthetic applications.
The heteroleptic diphosphine diimine complexes discussed in this work (Fig. 1) include [Cu(xantphos)(deebq)]BF4 (D2) where xantphos = 4,5-bis(diphenylphosphino)-9,9-dimethyl-xanthene, and [Cu(DPEphos)(deebq)]BF4 (D3) where DPEphos = bis[(2-diphenyl-phosphino)phenyl] ether, a more flexible analog of xantphos. The homoleptic complex [Cu(deebq)2]BF4 (D1) was also studied for comparison. D2 and D3 have previously been reported for use in white-light emitting electrochemical cells.10
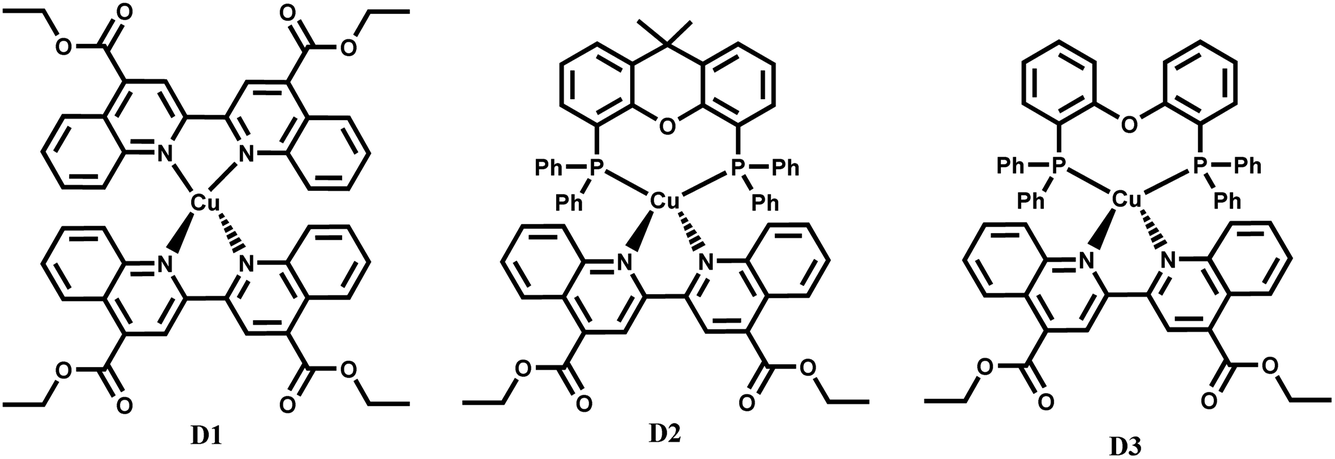 | ||
| Fig. 1 Structures of the Cu(I) complexes studied: [Cu(deebq)2]BF4 (D1), [Cu(xantphos)(deebq)]BF4 (D2) and [Cu(DPEphos)(deebq)]BF4 (D3). | ||
We aim to further understand the interplay between the structural and electronic changes in heteroleptic Cu(I) diimine complexes that will inform future design of more efficient photosensitisers. Two potential mechanisms affecting MLCT excited state decay involve a transient formation of a solvent adduct, made possible by the opening up of the Cu coordination centre in the excited state due to structural distortion, and a coordination of the O-atom on the phosphine ligand to the copper center. Here, the dynamics of these complexes are explored by transient absorption (TA) and time-resolved infrared (TRIR) spectroscopy; X-ray absorption studies of the ground electronic state have been conducted as a prerequisite for the upcoming X-ray spectroscopy studies which will directly determine structural dynamics. The potential for these complexes to be used in bimolecular applications is evaluated by determining the yield of singlet oxygen production.
Experimental
Materials and methods
Reagents were obtained from commercial sources and used without further purification unless stated otherwise. These included starting materials of [Cu(CH3CN)4](BF4), 4,4′-dicarboxylic-acid-2,2′-biquinoline (dcbq), bis[(2-diphenylphosphino)phenyl] ether (DPEphos) and 4,5-bis(diphenylphosphino)-9,9-dimethyl-xanthene (xantphos). All solvents were of spectroscopic grade or higher, and used as received from commercial suppliers, unless otherwise stated. Dry solvents were acquired from the University of Sheffield Grubbs solvent drying system. Synthetic procedures and analysis of purity of D1–D3 are presented in the ESI.†1H NMR spectroscopy
1H NMR spectra were recorded using a 400 MHz Bruker Avance 400 spectrometer equipped with a 5 mm BBFO SmartProbe. The complexes were dissolved in spectroscopic grade deuterated chloroform (CDCl3). 1H spectra were calibrated against the residual solvent peak.11 Heteronuclear NMR were referenced to the 2H lock signal of the deuterated solvent.Mass spectrometry
All mass spectra were recorded by the University of Sheffield mass spectrometry service. Positive electrospray ionisation (ES+) mass spectra were recorded using a VG AutoSpec magnetic sector instrument.Steady state X-ray diffraction
Steady state X-ray diffraction crystallography experiments were carried out in the University of Sheffield Department of Chemistry X-ray crystallography laboratory. Intensity data was collected at 100 K on a Bruker D8 Venture diffractometer equipped with a Photon 100 CMOS detector using a CuKα microfocus X-ray source from a crystal mounted in Fomblin oil on a MiTiGen microloop and cooled in a stream of cold N2. Data were corrected for absorption using empirical methods (SADABS12) based upon symmetry equivalent reflections combined with measurements at different azimuthal angles.13 The crystal structures were solved and refined against F2 values using ShelXT14 for solution and ShelXL15 for refinement, accessed via the Olex2 program.16 Non-hydrogen atoms were refined anisotropically. Hydrogen atoms were placed in calculated positions with idealized geometries and then refined by employing a riding model and isotropic displacement parameters.Steady state UV-vis, FTIR and emission spectroscopy
The UV-vis spectra were recorded using a Cary 50 Bio Spectrometer on sample solutions measured in a quartz cuvette with a path length of 1 cm. Emission and excitation spectra were collected using a Horiba Jobin Yvon Fluromax-4 spectrofluorometer. Emission lifetimes were measured with an Edinburgh Instruments mini-τ set-up using 445 nm diode as the excitation source, under aerated conditions. FTIR spectra were recorded with a PerkinElmer Spectrum One instrument with a resolution of 2–4 cm−1. Samples were recorded in a Harrick Cell with CaF2 windows (2 mm thickness) with a typical path length of 500–650 μm.Quantum yield of singlet oxygen production
Singlet oxygen was detected through measurement of the singlet oxygen emission band at ∼1275 nm. The complexes in solution were then excited by the third harmonic of a Q-Switch Nd:YAG laser (λ = 355 nm, ∼8 ns pulse length, laser model LS-1231M from LOTISII). The time-resolved signal of 1O2 luminescence at 1275 nm was detected by a liquid nitrogen cooled InGaAs photodiode of Ø3 mm active area (J22D-M204-R03M-60-1.7, Judson Technologies). The output from the photodiode was coupled into a low-noise current amplifier (DLPCA-200, FEMTO Messtechnik GmbH). The amplifier output signal was recorded with a digital oscilloscope (TDS 3032B Tektronix) and transferred to a computer. To selectively detect the 1O2 emission, the high-contrast bandpass optical filter (1277 nm centre wavelength, 28 nm FWHM, custom-made by Izovac, Belarus) was fitted in front of the InGaAs photodiode. The quantum yield of singlet oxygen production (ϕ1O2) is determined by comparing the initial amplitude of the emission signal of 1O2 generated when irradiating the air-equilibrated solution of complex and that of the standard (perinapthenone, ϕ1O2 ≈ 100% (DCM)).17 The emission lifetime for 1O2 sensitised by the complex and the standard must be similar (within the range 80–100 μs DCM)18,19 to confirm that 1O2 does not react with the photosensitiser in its ground state. The optical densities of the complex and a standard were matched at 355 nm, and the same solvent was used for both compounds. The experiments were performed at a series of excitation energies ranging from 10 μJ to 100 μJ per pulse. A correction was applied to the calculated initial intensities to account for small discrepancies in the optical density of the compound and standard solutions at 355 nm.Steady-state (or ground state) X-ray absorption spectroscopy (EXAFS)
Steady state (or ground state) EXAFS experiments were carried out at the I18 beamline at Diamond Light Source (DLS). The EXAFS region was scanned with varying step sizes of 0.5–1 eV between 8777 and 9527 eV, each energy point was measured for 4 s. The X-ray spot-size was 40 μm2. Samples were prepared in 50 mL of MeCN to a concentration of ca. 2 mM. The sample was supplied using a jet, with a diameter of 400 μm, operating at a flow rate of 25 mL min−1. The XAFS were calibrated to a Cu reference foil, and the edge energies were set as the zero intercept of the second derivative. The XAFS data was analysed using the Demeter software package, a comprehensive system for processing and analysing X-ray Absorption Spectroscopy data.20Theoretical calculations
Calculations were performed using the ORCA quantum chemistry software package, version 4.0.1.21,22 Calculations were carried out on the University of Sheffield’s Sheffield Advanced Research Computer (ShARC). Molecular structures were optimised by DFT, using a B3LYP hybrid functional, the RIJCOSX approximation, and the Douglas–Kroll–Hess (DKH3) relativistic approximation. For Cu, the Sapporo-DKH3-TZP-2012 basis set was used; For all other atoms, the relativistically adjusted Karlsruhe basis sets, DKH-def2-SVP, and the SARC/J auxiliary basis set were used. XAS calculations were carried out using the same level of theory. A 2.5 eV broadening was applied to all calculated XAS spectra.Ultrafast pump–probe spectroscopy
Transient absorption (TA) and time-resolved infrared (TRIR) measurements were carried out at the Lord Porter Laser Laboratory, University of Sheffield. A Ti:Sapphire regenerative amplifier (Spitfire ACE PA-40, Spectra-Physics) provided 800 nm pulses (40 fs FWHM, 10 kHz, 1.2 mJ). The amplifier was seeded by 800 nm pulses (25 fs FWHM, 84 MHz) generated by a Ti:Sapphire oscillator (Mai Tai, Spectra-Physics). Both amplification stages of the Spitfire ACE were pumped by two Nd:YLF lasers (Empower, Spectra-Physics). The 800 nm pulses were split between an 800 nm line, two UV-to-mid-IR commercial OPAs (TOPAS, Light Conversion) and doubler/tripler (TimePlate, Photop Technologies).Femtosecond transient absorption (TA) spectroscopy
400 nm (40 fs FWHM, 10 kHz) pulses for excitation were generated by doubling a portion of the 800 nm output, from the same Ti:Sapphire amplifier, in a β-barium borate crystal (BBO) within a commercially available doubler/tripler (TimePlate, Photop Technologies). Alternatively, tuneable pump pulses (260–800 nm, 10 nm FWHM) were generated with a commercial OPA (TOPAS, Light Conversion) also pumped by the same amplifier (Helios, Ultrafast Systems). The white light supercontinuum probe (∼340–790 nm) was generated by focusing ∼2% (∼1 μJ) of the 800 nm from the Ti:Sapphire amplifier output into a translating CaF2 crystal (∼3 mm thickness). The probe is focussed onto the sample position to a spot size of 120 μm diameter. The relative polarisation of the pump was set to magic angle (54.7°) relative to the probe polarisation in the measurements. The pump was focussed onto the sample to a spot size of ∼250 μm diameter. The pump was chopped using a mechanical chopper running at 2.5 kHz. The probe was collimated and focussed on to a CMOS sensor, with a spectral resolution of 0.5 nm, operating at 5 kHz.Samples were prepared in quartz cuvettes with a pathlength of 1 or 2 mm. The samples were stirred using a magnetic stirrer to minimise photodegradation. Steady-state UV-vis spectra were measured before and after TA measurements to confirm no photodegradation had occurred. The optical density of the sample at the excitation wavelength is specified for each experiment. Over the probe spectral region, the optical density of the sample was kept below 1. Experiments consisted of averaging 1 s at each time point for 5 scans. Temporal chirp was obtained from measuring and fitting the solvent response. This was applied to the raw data to remove contributions from the chirp and solvent response. Chirp correction and background subtraction was performed in Surface Xplorer (Ultrafast Systems). Global lifetime analysis was carried out in MATLAB R2022a.
Time-resolved infrared (TRIR) spectroscopy
The Ti:Sapphire regenerative amplifier (Spitfire ACE PA-40, Spectra-Physics) was used to pump a commercial optical parametric amplifier (TOPAS, Light Conversion), which provided tuneable mid-IR pulses (2300–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm−1, typical FWHM of ∼280 cm−1) by difference frequency generation in AgS. After the optical parametric amplifier (OPA), the probe beam passed through a Ge long pass filter and was collimated to a beam diameter of 10 cm in a two-mirror telescope. A wire-grid polariser (Thorlabs) was used in conjunction with a tuneable λ/2 wave plate (AlphasLas) to set the probe polarisation (to be s-polarised/perpendicular). Before the sample, the probe was split into a probe and reference beam using a 50:50 Ge beam splitter. The tuneable pump pulses (260–800 nm, FWHM 10 nm) were generated with a second commercial OPA (TOPAS, Light Conversion) also pumped by the Ti:Sapphire regenerative amplifier. A mechanical delay-stage (Thorlabs) is used to set the delay time between the pump and probe pulses up to 4 ns, by extending the pump path in relation to the probe. The pump repetition rate was set to 5 kHz using an optical chopper (Thorlabs). The power of the pump pulses was attenuated before the sample using a variable attenuation neutral density filter wheel. The polarisation of the pump was set to magic angle (54.7°) relative to the probe polarisation using a tuneable λ/2 wave plate (Thorlabs). The IR probe (∼1 mW) and reference (∼1 mW) were focussed onto the sample position using a gold off-axis parabolic mirror (f = 211 mm) to a spot size of ∼300 × 300 μm. The UV-vis pump was focussed onto the probe position at the sample using an aluminium 90° off-axis parabolic mirror (f = 100 mm) to a spot-size of ∼400 × 350 μm. The sample was mounted on a mechanical z stage to adjust the sample position in the focal point of the beam. After the probe and reference pass through the sample, the beams are collected on another parabolic mirror. The reference has an independent mirror path to control both beams into the detector.
000 cm−1, typical FWHM of ∼280 cm−1) by difference frequency generation in AgS. After the optical parametric amplifier (OPA), the probe beam passed through a Ge long pass filter and was collimated to a beam diameter of 10 cm in a two-mirror telescope. A wire-grid polariser (Thorlabs) was used in conjunction with a tuneable λ/2 wave plate (AlphasLas) to set the probe polarisation (to be s-polarised/perpendicular). Before the sample, the probe was split into a probe and reference beam using a 50:50 Ge beam splitter. The tuneable pump pulses (260–800 nm, FWHM 10 nm) were generated with a second commercial OPA (TOPAS, Light Conversion) also pumped by the Ti:Sapphire regenerative amplifier. A mechanical delay-stage (Thorlabs) is used to set the delay time between the pump and probe pulses up to 4 ns, by extending the pump path in relation to the probe. The pump repetition rate was set to 5 kHz using an optical chopper (Thorlabs). The power of the pump pulses was attenuated before the sample using a variable attenuation neutral density filter wheel. The polarisation of the pump was set to magic angle (54.7°) relative to the probe polarisation using a tuneable λ/2 wave plate (Thorlabs). The IR probe (∼1 mW) and reference (∼1 mW) were focussed onto the sample position using a gold off-axis parabolic mirror (f = 211 mm) to a spot size of ∼300 × 300 μm. The UV-vis pump was focussed onto the probe position at the sample using an aluminium 90° off-axis parabolic mirror (f = 100 mm) to a spot-size of ∼400 × 350 μm. The sample was mounted on a mechanical z stage to adjust the sample position in the focal point of the beam. After the probe and reference pass through the sample, the beams are collected on another parabolic mirror. The reference has an independent mirror path to control both beams into the detector.
The beams are split by an additional 50:50 Ge beam splitter. The split beams were focussed into two identical spectrometers (f = 320 mm, Horiba iHR320), with either 50 g mm−1 or 100 g mm−1 gratings which gave a spectral resolution of 6 and 3 cm−1 respectively. The probe and reference spectra were recorded employing two liquid nitrogen cooled HgCdTe (MCT) array detectors (Infrared Systems). Each detector had 128 pixels split into 2 lines; 96 pixels for the probe and 32 for the reference. Home-built software (LabVIEW) was used for data collection and processing the raw spectra to pump–probe TRIR spectra. A multichannel referencing scheme23,24 was used to improve the signal-to-noise ratio. Measurements were calculated from the average of 5 scans with 2 s averaged per time point. Samples were prepared in a Harrick Scientific IR cell with 2 mm CaF2 windows. The path length was adjusted using a PFTE spacer (650 μm). Samples were raster-scanned at the sample position using a motorised x–y stage to minimise photodegradation. In addition to the raster-scan, samples were also flowed through the Harrick cells using a peristaltic pump (ColePalmer, Teflon loop). The photostability of the sample was checked by recording the UV-vis spectra before and after laser exposure. Global Lifetime Analysis was performed using MATLAB R2022a. Additional TRIR studies were performed at the Laser for Science Facility, Rutherford Appleton Laboratory.
Results and discussion
UV-vis absorption and emission spectroscopy
The complexes exhibit high energy absorption at <400 nm attributed to π–π* transitions of the diimine and phosphine ligands, the full spectra are given in Fig. S1–S3.† All complexes studied exhibit a broad absorption band in the range >400 nm, with the extinction coefficient of the order of ∼1 × 104 M−1 cm−1 for homoleptic complexes25 and ∼3 × 103 M−1 cm−1 for heteroleptic complexes,9,26 characteristic of a CT transition. The MLCT bands in all complexes studied show slight solvatochromism, Fig. S1–S3 and Table S1,† confirming the charge-transfer nature of this absorption band. The homoleptic D1 complex exhibits the most red-shifted MLCT absorption bands at 577–581 nm, with a broad, weaker band at ca. 640 nm. On the basis of calculations reported in the literature for [Cu(phen)2]+, it could be suggested that the main MLCT absorption band at 577–581 nm corresponds to S0 → S3 transition, and the lower-energy band to the symmetry-forbidden S0 → S1/S2 transition. The MLCT absorption band in the heteroleptic complexes D2 and D3 corresponds to the S0 → S1 transition,27 and is blue-shifted with respect to the homoleptic complex, Fig. 2, similar to what has been reported for heteroleptic phosphine phenanthroline Cu(I) complexes.28 The MLCT absorption band of D3, the DPEphos containing complex, is slightly shifted to higher energy with respect to D2, which has the more rigid xantphos ligand. Finally, an additional absorption band at longer wavelengths may appear in the spectra for D2 due to formation of a very small amount of D1 in the “ligand scrambling” process.10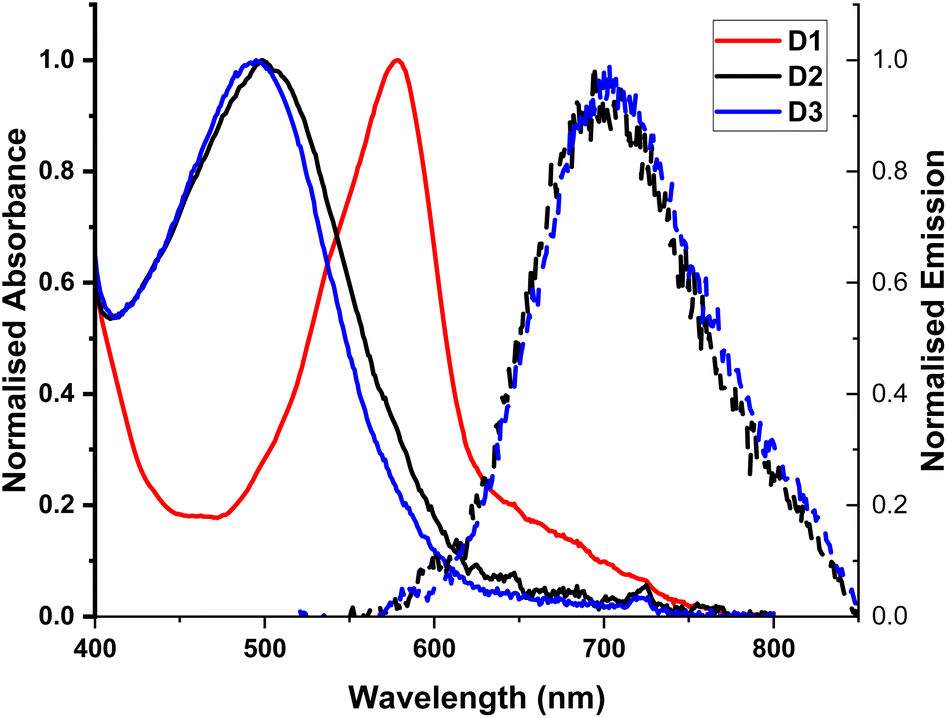 | ||
| Fig. 2 UV-Vis absorption spectra of complexes D1–D3 in DCM, normalised to the maxima of the MLCT transition band. Emission recorded for D2 and D3 following excitation at the absorption maxima. | ||
The photophysical properties of the Cu(I) deebq complexes are summarised in Table 1. The homoleptic complex D1 is non-emissive in DCM Fig. 2. The deebq complexes, D2 and D3, show a main emission band at 720 and 725 nm respectively. The emission maximum in deebq complexes is red-shifted relative to the complexes with unsubstituted biq ligands by ∼25 nm (∼610 cm−1).9,29 Both D2 and D3 exhibit emission lifetimes of the order of tens of nanoseconds. A slightly longer excited state lifetime for D3 in comparison to D2 (82 ns vs. 52 ns, in aerated DCM) could be due to the increased flexibility of the DPEphos not only restricting the structural reorganisation in the excited state, but also inhibiting solvent interactions more effectively than xantphos.
| Complex | λ abs /nm | λ em /nm | τ air , /ns | τ 1(TA) /ps | τ 2(TA)/ps | τ 3(TA)/ps | τ 1(TRIR)/ps | τ 2(TRIR)/ps | ϕ 1O2 |
|---|---|---|---|---|---|---|---|---|---|
| a In aerated solution. b Excitation wavelength 445 nm. c Quantum yield of singlet oxygen calculated with respect to perinapthenone (assumed ϕPN ≈ 1 in DCM).17 d A component with the lifetime significantly exceeding the instrument-limited time window of 7 ns. e Additional ultrafast component of <0.2 ps is present in all TA data. | |||||||||
| D1 | 540, 580, 617 | — | — | 1.7 | 6.1 | 1070 | <0.2 | 1000 | — |
| D2 | 500 | 720 (698) | 52 ± 7 | 0.9 | 15 | Infd | <0.2 | Infd | 0.13 ± 0.05 |
| D3 | 495 | 725 (703) | 89 ± 5 | 0.7 | 14 | Infd | <0.2 | Infd | 0.09 ± 0.05 |
Crystallography
Single crystals of complexes D1–D3 were grown via vapour diffusion of DCM/diethyl-ether (Table 2 and Fig. 3). Similarly to other homoleptic and heteroleptic Cu(I) complexes,30D1–D3 exhibit a distorted tetrahedral structure; the ∠PCuN angles for complex D2 that contains the most rigid ligand xantphos are closest to the ideal angle of 109.5°. The distortion in complexes D2 and D3 from the ideal tetrahedral geometry around the metal center is also shown by the value of the dihedral angle between the planes N1–Cu–N2 and P1–Cu–P2, 92.94° and 92.81°, respectively. Complex D1 shows the largest distortion from the tetrahedral geometry, with a dihedral angle N1–Cu–N2/N3–Cu–N4 of 82.6° (the dihedral angle of ∼80° was reported for a similar complex [Cu(4,4′-(COOH)2-biq)2]+).31 The two heteroleptic complexes, D2 and D3, display structural disorder. D3 displays two distinct conformations which coexist in a ratio of 70:30 that differ in the twist of the DPEphos ligand (Fig. 3), and the distance between the oxygen atom of the DPEphos ligand and the Cu centre (3.21 vs. 3.15 Å).
| Complexes | D2 | D3 | D1 | |
|---|---|---|---|---|
| Bond lengths (Å) | ||||
| Cu–N1 | 2.085(5) | 2.097(5) | 2.019(4) | |
| Cu–N2 | 2.087(5) | 2.058(5) | 2.016(5) | |
| Cu–P1 | 2.2664(17) | 2.2344(18) | Cu–N3 | 2.015(5) |
| Cu–P2 | 2.2839(18) | 2.3222(18) | Cu–N4 | 2.040(5) |
| Cu–O1 | 3.170(4) | 3.152(12) | — | |
|
||||
| Dihedral angle | ||||
| N1–Cu–N2/P1–Cu–P2 | 92.94° | 92.81° | N1–Cu–N2/N3–Cu–N4 | 82.6° |
|
||||
| Bond angles (°) | ||||
| N1–Cu–N2 | 79.7(2) | 79.2(2) | N1–Cu–N2 | 80.76(18) |
| P1–Cu–P2 | 116.74(8) | 116.80(6) | N3–Cu–N4 | 81.66(19) |
| P1–Cu–N1 | 114.45(14) | 124.95(15) | N1–Cu–N3 | 124.12(19) |
| P1–Cu–N2 | 119.49(15) | 127.37(15) | N2–Cu–N3 | 132.49(19) |
| P2–Cu–N1 | 111.63(14) | 97.59(14) | N1–Cu–N4 | 123.94(18) |
| P2–Cu–N2 | 109.25(14) | 102.40(15) | N2–Cu–N4 | 120.00(19) |
 | ||
| Fig. 3 Crystal structures of complexes D1, D2 and D3 obtained by single-crystal X-ray crystallography. | ||
Steady-state X-ray absorption spectroscopy
The experimental Cu K-edge XANES spectra measured for complexes D1–D3 in MeCN are shown in Fig. 4. The heteroleptic Cu(I) complexes, D2 and D3, display an edge feature at 8982.6 eV which is assigned to the 1s → 4pz transition. This feature is less prominent in the homoleptic complex, D1. It has been observed that the 1s → 4pz edge feature is heavily affected by the Cu(I) coordination environment as the 4pz orbital is easily perturbed.32–34 The heteroleptic complexes exhibit a second edge feature at ∼8988.6 eV which is most prominent in complex D3 and appears to be absent in the spectrum for the homoleptic complex, D1. An additional edge feature is present in the spectrum for complex D1 at 8985.6 eV which is seemingly absent from the heteroleptic complexes. The maximum of the rising edge peak is centred around 8994.0 eV for D2 and D3, and at ∼8995 eV for D1. | ||
| Fig. 4 (a) Experimental Cu K-edge XANES spectra for D1 (black), D2 (red) and D3 (blue) in MeCN solution at r.t. (b–d) Experimental XANES (blue) and results of preliminary calculations of XANES (black) of complexes D1–D3. The energy axis for the calculated spectra has been shifted upwards by 98.25 eV so that the edge corresponds to the edge of the experimental data. Experimental data are average of multiple runs, see Fig. S17† for the data from individual runs. | ||
Ground state XAFS spectra for each complex in the k-space and R-space are displayed in Fig. 5. The results from the best fits are shown in Table 3. The scattering paths described are the single scattering paths to the N, P and C atoms closest to the Cu centre. The fits show that the average Cu–N bond length is shorter (2.01 Å) for the homoleptic complex D1 than the two heteroleptic complexes, D2 and D3 (2.11 Å and 2.12 Å respectively). The obtained Cu–N and Cu–P distances for D1–D3 in MeCN are similar to those obtained in the single crystal (Table 2). The average Cu–N bond length for D1 is slightly larger than for the similar complex [Cu(biq(COOH)2)2]+, 1.98 Å.35 The average Cu–N and Cu–P bond lengths for D2 and D3 are also comparable to those reported for other heteroleptic Cu(I) complexes such as [Cu(xantphos)(2,9-(OMe)2-1,10-phenanthroline)], 2.08 Å (Cu–N) and 2.28 Å (Cu–P).30
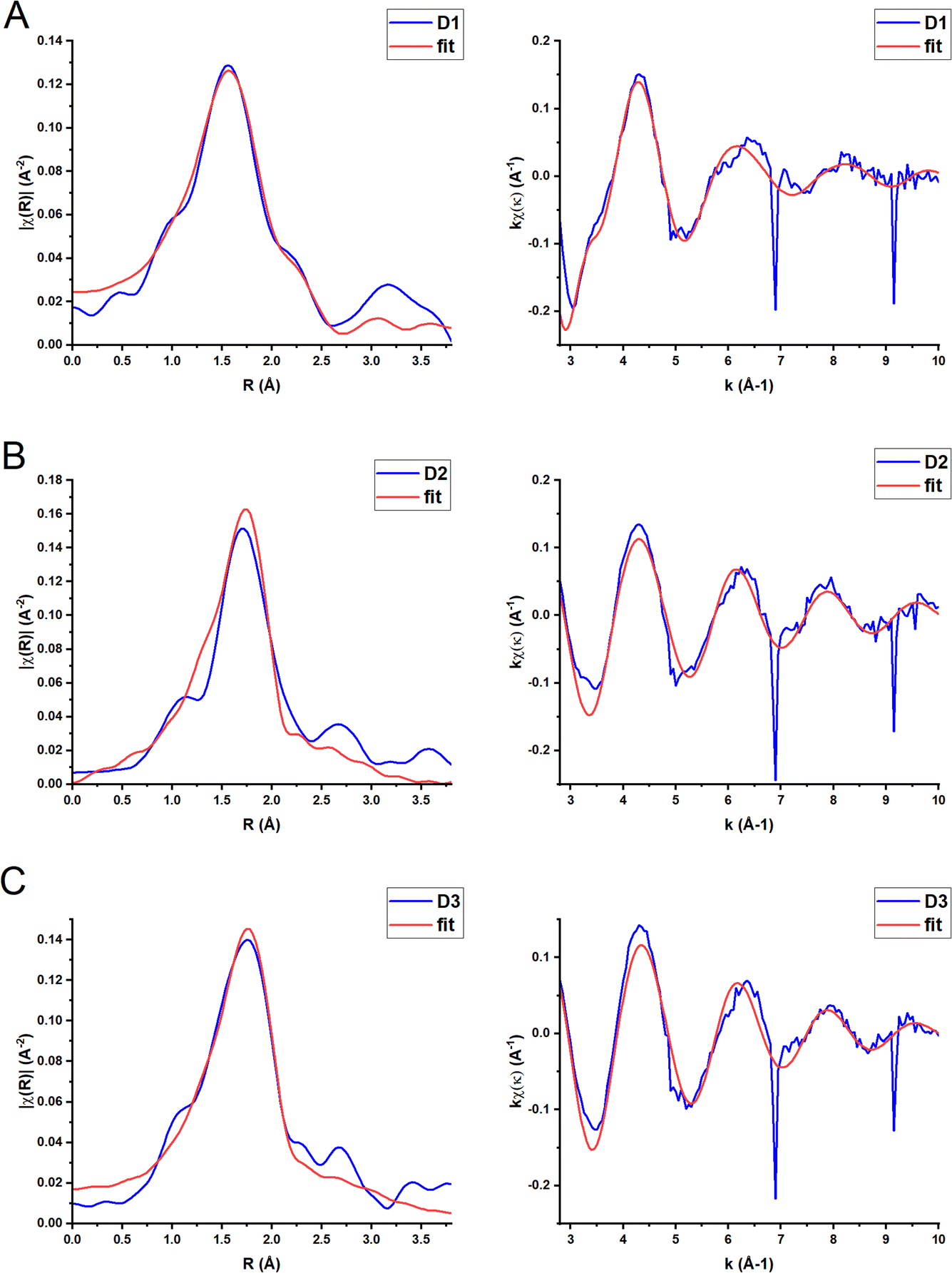 | ||
| Fig. 5 Fourier transform (left) and EXAFS extracted signal (right) for complexes D1 (A), D2 (B) and D3 (C) in MeCN solution (k weighting = 1) (blue line). The red lines represent the best fit to the data using the parameters included in Table 3. | ||
a
| Complex | Paths | Fitted distance (Å) | Fitted Debye–Waller (Å2) |
|---|---|---|---|
| a D1: S02 = 0.8, E0 = 6(2) eV, k-weight = 1, k-range = 3–10 Å, R-range = 1–3.5 Å. R-Factor = 0.02. D2: S02 = 0.8, E0 = 7(3) eV, k-weight = 1, k-range = 2.7–12 Å, R-range = 1.35–3 Å. R-Factor = 0.06. D3. S02 = 0.8, E0 = 7(3) eV, k-weight = 1, k-range = 2.7–12 Å, R-range = 1.35–3 Å. R-Factor = 0.06. | |||
| D1 | 4 Cu–N | 2.01(2) | 0.009(1) |
| 4 Cu–C | 3.10(2) | 0.005(2) | |
| 4 Cu–C | 3.32(2) | 0.005(2) | |
| D2 | 2 Cu–N | 2.11(4) | 0.002(6) |
| 2 Cu–P | 2.26(3) | 0.014(11) | |
| D3 | 2 Cu–N | 2.12(3) | 0.006(6) |
| 2 Cu–P | 2.27(2) | 0.013(7) | |
For D1 the calculated XANES spectrum (Fig. 4b) only roughly reproduces the edge shoulder at 8985 eV. Importantly, although the calculated spectrum underestimates the strength of the edge feature at 8982 eV, it does predict the pre-edge shoulder at 8978 eV associated with the low-lying, mixed metal–ligand states. The XANES spectra of D2 and D3 are more accurately predicted, including the first prominent edge feature, but still fails to predict the absorption maxima. The modelling of the 8985 eV edge feature in D2 and D3 but not D1 suggests that these transitions are fully allowed in the heteroleptic complexes, but are only enabled by spin–orbit coupling in the homoleptic D1 complex. Similarly, the calculations suggest that the absorption maxima at ∼8994 eV is highly dependent on a strong spin–orbit coupling interaction between the copper centre and the ligand. This would increase the absorption cross section for the higher energy, more ligand-centred virtual states, creating the broad absorption pattern seen at these energies. The calculated bond length of the Cu–N bonds in D1 (Table S25†) correspond well with the experimental results obtained in solution by EXAFS and by X-ray diffraction in the solid state. For D2 and D3, however, the calculated bond lengths (Table S25†) for Cu–N are smaller, and the calculated Cu–P bond lengths are significantly larger (∼2.5 Å vs. ∼2.3 Å) than the experimental values. The calculated dihedral angle for complex D3 is in good agreement with the crystallographic measurements, however it is underestimated for complexes D1 and D2.
Transient absorption spectroscopy
Transient absorption spectra of the heteroleptic (D2 and D3) and homoleptic (D1) complexes in DCM following excitation at 500 and 525 nm, respectively, are shown in Fig. 6; species-associated spectra (SAS) and decay-associated spectra (DAS) obtained by global analysis are given in the ESI, Fig. S4–S6.† The TA spectrum of D1 at early times shows a broad transient, featuring peaks at 395 nm, 419 nm and 476 nm along with the ground state bleach (GSB) centred at 579 nm. Over the first 5–10 ps, the transient bands at 419 and 476 nm decay, revealing a peak at 456 nm. The peaks at 393 nm and 456 nm, and a broad weak transient extending beyond 700 nm fully decay over the experimental time window (7 ns). The transient bands are characteristic of biquinoline-type radical anions.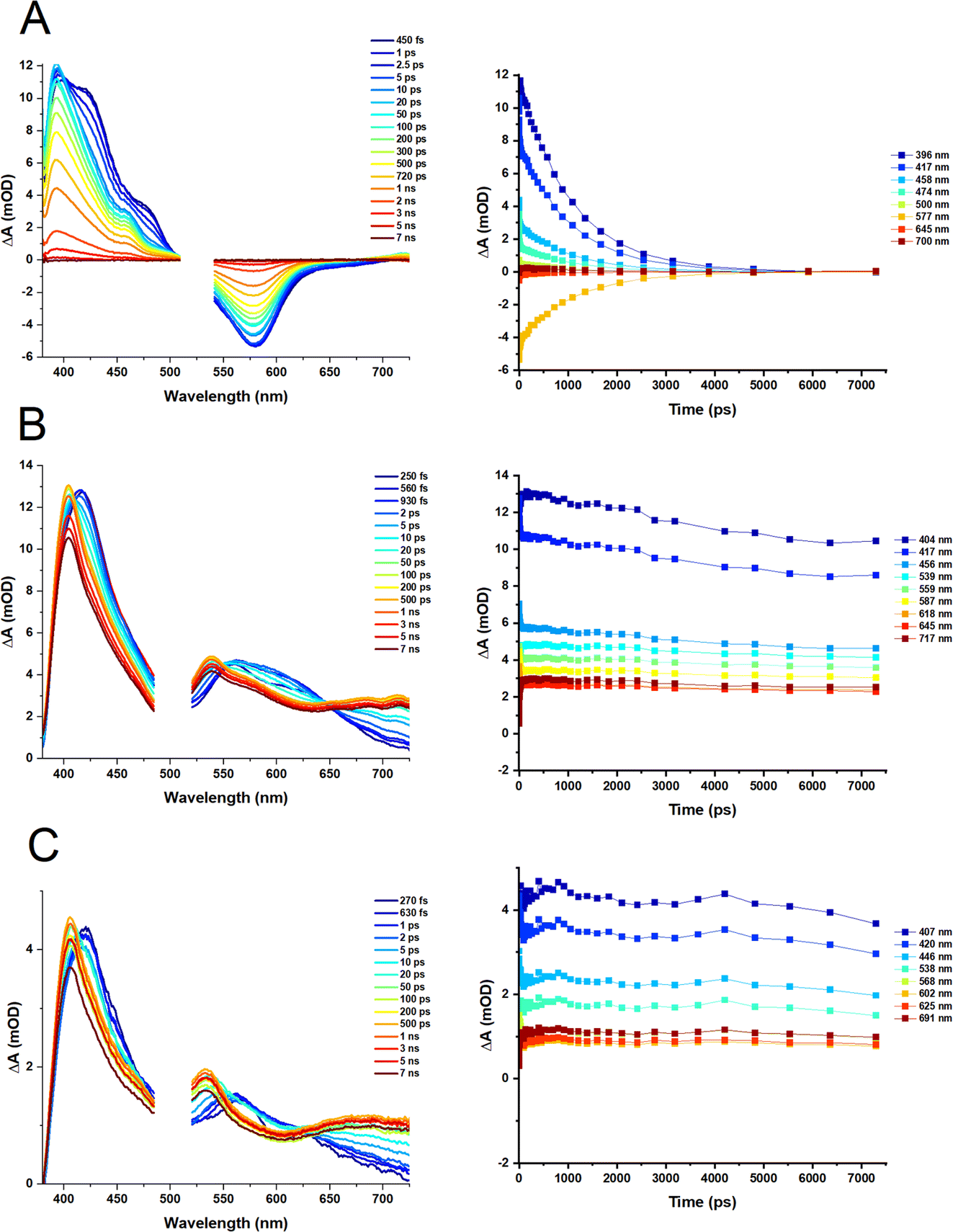 | ||
| Fig. 6 Fs-TA of (A) D1, (B) D2 and (C) D3 in DCM. Fs-TA for D2 and D3 was recorded following excitation at 500 nm (2 mW, OD@500 nm = 0.38 (D2) and 0.2 (D3)). Fs-TA for D1 was recorded following photoexcitation at 525 nm (2 mW, OD@525 nm = 0.2). The spectra were collected with the pump set to magic angle (54.7°) with respect to the probe. The spectra have been cropped around the pump excitation wavelength to remove the pump scatter. The time delay between the pump and probe for each spectrum is shown in the legend. Kinetics for each complex are shown corresponding wavelength is shown in the legend. | ||
For D2 and D3, initially, transients at ∼420, 461, 560, and 627 nm are observed, with the latter becoming obscured by a broader peak, 560 nm, by 1 ps. The GSB is obscured by the pump scatter and electronic offset but can be partially observed at ∼520 nm. A dip in the spectra at ∼600 nm observed at early times (∼250 fs, which could be potentially assigned to stimulated emission) disappears by 1 ps. The transient at 420 nm shifts to ∼404 nm by ca. 100 ps, and remains unchanged within the temporal limit of the experiment (7 ns). The peak at 560 nm decays over this timescale and a new peak at 538 nm grows in, appearing as a blue shift in the TA spectra which may also be assigned to the decay of stimulated emission. An additional transient >650 nm grows in by 100 ps, and persists.
The results of the global analysis of the transient data for each complex and the corresponding time components are listed in Table 1. The dynamics of the homoleptic complex D1 requires four time constants to describe the fs-TA satisfactory, of sub-200 fs (IRF), 1.7 ps, 6.1 ps and 1070 ns, Fig. 6A. Structural distortion in a singlet state (S1) of [Cu(NN)2]+ is usually observed on the sub-ps timescale.36 The observed 1.7 ps process could relate to either geometry change in an Sn state, or to the flattening distortion in a T13MLCT state, populated via an ultrafast (not detected) intersystem crossing (ISC) from an initially populated S2 state. The decay component of 6.1 ps is assigned to ISC from the flattened S1 state to the flattened T1 state which is similar to the 10 ps values reported for homoleptic [Cu(dmp)2]+ complexes.37
Overall, it appears that there is a branched (rather than a consecutive) decay process of the initially populated S2 state in D1 that leads to the final T1, 3MLCT state. The T1 state has a lifetime of 1.1 ns in DCM, which is longer than [Cu(phen)2]+ (143 ps),36,37 but shorter than the 54 ns lifetime of the [Cu(dmp)2]+ complex.
For complexes D2 and D3, four components are required to fit the TA data (Fig. 6B and C). The sub-200 fs, 900 fs (D2) and 700 fs (D3) decay components, could be assigned to the structural distortion which is similar to the rate of structural distortion in [Cu(xantphos)(NN)]+ bearing bulky NN = derivatives of 2,9-dimethyl-1,10-phenanthroline.38 The slight decrease of the sub-ps component from D2 (900 fs) to D3 (700 fs), could be due to the increased flexibility of the DPEphos ligand compared to the xantphos ligand. The component, 14–15 ps for both complexes, is tentatively assigned to ISC corresponding to the blue shift of the 420 nm to 404 nm and the appearance of the peak at 538 nm in the TA spectra.
Fourier-transform (FTIR) and time-resolved infrared (TRIR) spectroscopy
FTIR spectra for D1–D3 recorded between 1300 and 1850 cm−1, Fig. 7, display a band at 1728 cm−1 (![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) (C
(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) of the ester group), and multiple bands of the coordinated biq-ligand, at 1357, 1375.5, 1512, 1566, 1575 and 1590.5 cm−1.39
O) of the ester group), and multiple bands of the coordinated biq-ligand, at 1357, 1375.5, 1512, 1566, 1575 and 1590.5 cm−1.39
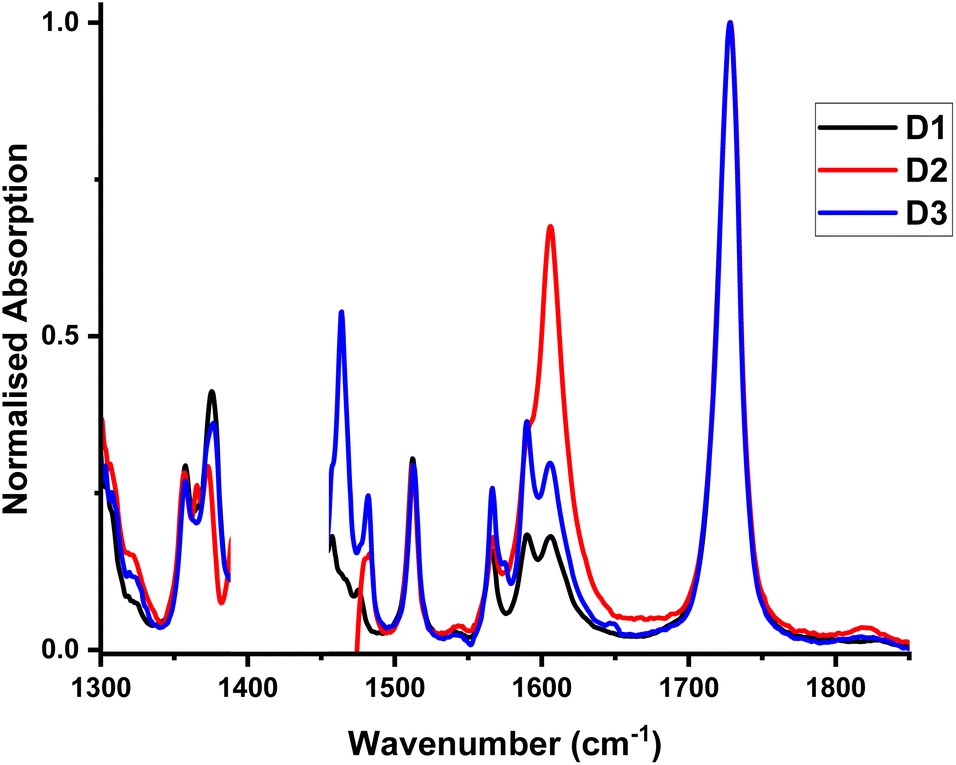 | ||
| Fig. 7 FTIR spectra of complexes D1–D3 in DCM. The peak at 1606 cm−1 is due to water absorption. | ||
To further investigate the excited state dynamics of these complexes, TRIR was recorded Fig. 8, S7 to S10,† following excitation with 40 fs, 525 nm (D1) and 500 nm (D2 and D3) laser pulses in DCM. Upon excitation, bleach of the ground state absorbencies of several of the biq bands are observed, with intense transient bands formed across the entire region. The ν(CO) stretching vibration of the ester group at ca. 1728 cm−1 is bleached, and a transient at lower energies is observed, at ca. 1700 cm−1. This is consistent with electron density increase on the diimine ligand, thus confirming the MLCT nature of the lowest excited state.
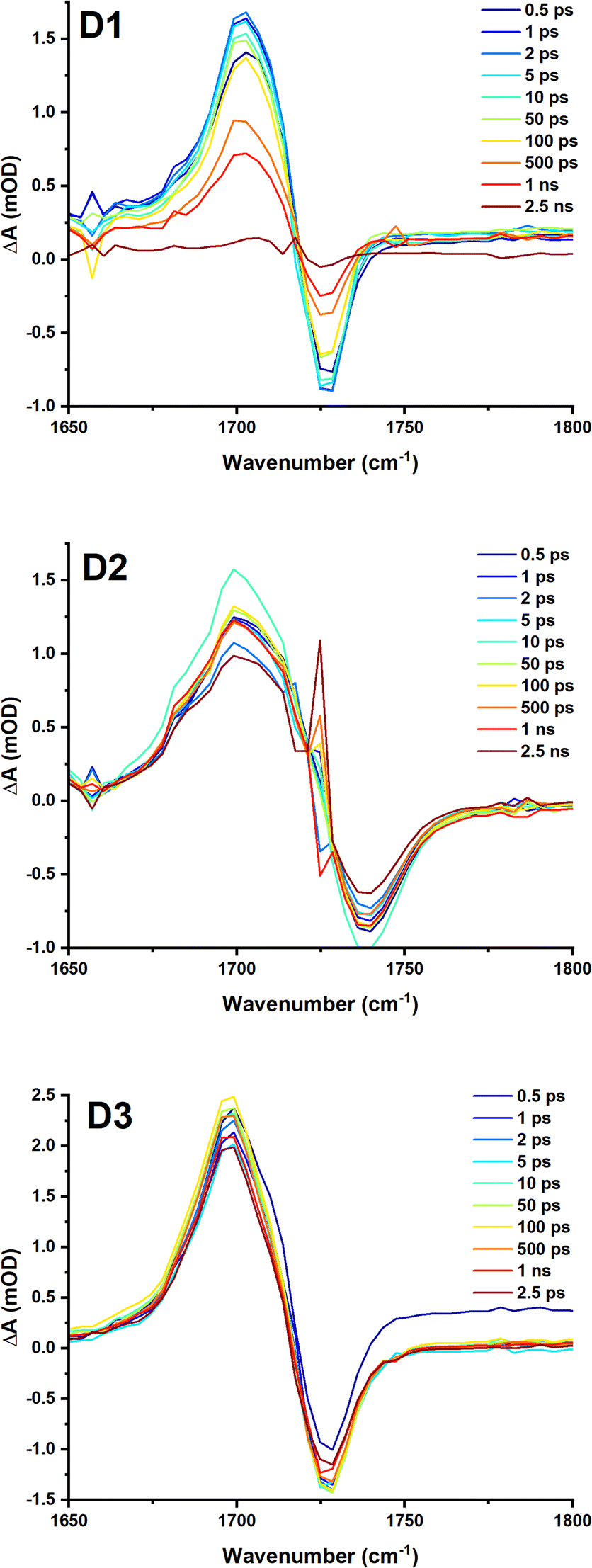 | ||
| Fig. 8 TRIR spectra of D1, D2 and D3 in DCM following excitation at 525, 500 and 500 nm respectively (2 mW, OD@λex = ∼0.6), collected with the pump set to magic angle (54.7°) with respect to the probe. The time delay between the pump and probe for each spectrum is shown in the legend. Kinetic traces are shown in the ESI.† | ||
For complex D1, global analysis revealed two decay components for the region between 1650 cm−1 and 1760 cm−1, corresponding to the (CO) of the ester group (Fig. 8) including a sub-ps component likely due to vibrational cooling, and 1 ns.
Global analysis of the data D2–D3 Fig. S8 and S11,† yields 2 components: a long component beyond the timescale of the experiment of 6 ns, and sub-ps component, likely vibrational cooling which would not be easily observed in the TA. This component corresponds to a small increase in the peak intensity at 1710 cm−1, the bleach also increases in intensity, meanwhile the peak of the transient shifts from 1699 cm−1 to 1702 cm−1 (the apparent position of the bleach shifts from 1733 cm−1 to 1728 cm−1). This is a little faster than the typical timescale of vibrational relaxation of the (CO) of ester groups.40,41
Overall, the dynamics of the excited states for D1–D3 elucidated from the transient absorption and TRIR data indicate at least three components of the MLCT excited state decay, including a structural change, an ISC, and the final decay to the ground state in the process of charge recombination.
Conclusions
Ultrafast dynamics of two heteroleptic diimine diphosphine complexes of Cu(I) bearing diester-bisquinoline (deebq) diimine ligand, and two diphosphine ligands, a xantphos, and a less rigid DPEphos, have been investigated, and compared to the homoleptic [Cu(deebq)2]+ complex. The two heteroleptic compounds absorb intensely in the red region of the spectrum owing to an MLCT transition, whilst retaining the comparatively long excited state lifetime, a combination of properties desirable in photosensitisers for catalysis, synthesis, or cell killing. Preliminary dynamics of D1–D3 complexes were described on the basis of the results of fs-TA and TRIR in DCM.For the homoleptic complex D1, a 200 fs component observed is comparable to the rate of the flattening distortion in [Cu(phen)2]+.36,37,42 A second decay component of 1.7 ps for D1 was tentatively assigned to IC; however this component was not observed in TRIR, and one may assume that this is a process that does not affect electron density on the ester groups of the diimine ligand and hence is associated only with the structural change around the metal center. A 6.1 ps component was tentatively assigned to a combination of intersystem crossing, ISC, and vibrational cooling due to the blue shift in the TA spectrum and due to the similar magnitude of the component to the rate of ISC observed for other homoleptic Cu(I) complexes (9–13 ps)−1.37 The final state, here assigned to 3MLCT T1, decays over ∼1–1.1 ns as observed in both TA and TRIR data.
For the heteroleptic complexes, D2 and D3 a structural distortion of the S1 state occurs over 700–900 fs which is followed by ISC to the flattened T1 state over 14–15 ps. The final T1 state is long-lived, exhibiting lifetimes of 52 and 89 ns (D2 and D3). The propensity of the compounds D2–D3 to bimolecular reactions is demonstrated by a high yield of singlet oxygen photosensitisation (9–13%).
One of the mechanisms of excited state quenching in the nascent MLCT excited state in Cu(I) complexes could be a non-covalent, transient interaction between the solvent molecules and the copper center due to structural distortion from pseudo-tetrahedral to pseudo-square planar configuration that makes the metal centre more accessible. A process that may influence and/or prevent solvent coordination may involve transient coordination of the –O-atom of the phosphine ligand to the copper center. Ground state X-ray absorption results from D1–D3 in solution reveal the local structure around the metal center. Preliminary DFT calculations identified the geometry of the ground states of D1–D3 which correspond well to the parameters obtained from the EXAFS modelling. Building on the ground state EXAFS data, and the excited state dynamics resolved by optical spectroscopies, the next step will be an investigation of structural changes and transient coordination of solvent and O-atom to the copper center in the excited state of these promising photosensitisers using excited state X-ray spectroscopies.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the EPSRC (capital equipment award to Lord Porter Laser Laboratory and the PORTO project), the Grantham Centre for Sustainable Futures (M. V. A.), CONACYT (S. L. P.-A.), UK XFEL Physical Sciences Hub (R. A. C.), the EPSRC MPB project (I. I. I.) the University of Sheffield for support (and publication Scholarship for M. V. A.). We also thank Diamond Light Source, STFC and the staff of I18 for beam time at the facility. Finally, we thank Dr I. Sazanovich, STFC, for collaboration, and Dr R. J. Fernández-Terán for the use of his analysis scripts.Notes and references
- J. Twilton, C. C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem., 2017, 1, 0052 CrossRef CAS.
- L. K. McKenzie, H. E. Bryant and J. A. Weinstein, Coord. Chem. Rev., 2019, 379, 2–29 CrossRef CAS.
- D. R. McMillin, J. R. Kirchhoff and K. V. Goodwin, Coord. Chem. Rev., 1985, 64, 83–92 CrossRef CAS.
- T. Katayama, T.-K. Choi, D. Khakhulin, A. O. Dohn, C. J. Milne, G. Vankó, Z. Németh, F. A. Lima, J. Szlachetko, T. Sato, S. Nozawa, S.-I. Adachi, M. Yabashi, T. J. Penfold, W. Gawelda and G. Levi, Atomic-scale observation of solvent reorganization influencing photoinduced structural dynamics in a copper complex photosensitizer, Chem. Sci., 2023, 14, 2572–2584 RSC.
- Y. Zhang, M. Schulz, M. Wächtler, M. Karnahl and B. Dietzek, Coord. Chem. Rev., 2018, 356, 127–146 CrossRef CAS.
- D. Moonshiram, P. Garrido-Barros, C. Gimbert-Suriñach, A. Picón, C. Liu, X. Zhang, M. Karnahl and A. Llobet, Chem.–Eur. J., 2018, 24, 6464–6472 CrossRef CAS PubMed.
- Y. Zhang, P. Traber, L. Zedler, S. Kupfer, S. Gräfe, M. Schulz, W. Frey, M. Karnahl and B. Dietzek, Phys. Chem. Chem. Phys., 2018, 20, 24843–24857 RSC.
- M. V. Appleby, P. G. Walker, D. Pritchard, S. van Meurs, C. M. Booth, C. Robertson, M. D. Ward, D. J. Kelly and J. A. Weinstein, Mater. Adv., 2020, 1, 3417–3427 RSC.
- B. J. McCullough, B. J. Neyhouse, B. R. Schrage, D. T. Reed, A. J. Osinski, C. J. Ziegler and T. A. White, Inorg. Chem., 2018, 57, 2865–2875 CrossRef CAS PubMed.
- E. Fresta, M. D. Weber, J. Fernandez-Cestau and R. D. Costa, Adv. Opt. Mater., 2019, 7, 1900830 CrossRef CAS.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- Bruker, SADABS, Bruker Axs Inc., Madison, Wisconsin, USA, 2016 Search PubMed.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3–10 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- R. Schmidt, C. Tanielian, R. Dunsbach and C. Wolff, J. Photochem. Photobiol., A, 1994, 79, 11–17 CrossRef CAS.
- M. Bregnhøj, M. Westberg, F. Jensen and P. R. Ogilby, Phys. Chem. Chem. Phys., 2016, 18, 22946–22961 RSC.
- N. Epelde-Elezcano, V. Martínez-Martínez, E. Peña-Cabrera, C. F.A. Gómez-Durán, I. López Arbeloa and S. Lacombe, RSC Adv., 2016, 6, 41991–41998, 10.1039/c6ra05820e.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327 Search PubMed.
- F. Neese and J. Wiley, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- Y. Feng, I. Vinogradov and N.-H. Ge, Opt. Express, 2019, 27(15), 20323–20346 CrossRef CAS PubMed.
- Y. Feng, I. Vinogradov, N.-H. Ge, W. S. Chan and T. K. Yee, Opt. Express, 2017, 25(21), 26262–26279 CrossRef CAS PubMed.
- K. A. Wills, H. J. Mandujano-Ramírez, G. Merino, D. Mattia, T. Hewat, N. Robertson, G. Oskam, M. D. Jones, S. E. Lewis and P. J. Cameron, RSC Adv., 2013, 3, 23361–23369 RSC.
- N. Arnosti, M. Meyer, A. Prescimone, E. C. Constable and C. E. Housecroft, Crystals, 2021, 11, 185, DOI:10.3390/cryst11020185.
- K. Kubiček, S. Thekku Veedu, D. Storozhuk, R. Kia and S. Techert, Polyhedron, 2017, 124, 166–176 CrossRef.
- S. M. Kuang, D. G. Cuttell, D. R. McMillin, P. E. Fanwick and R. A. Walton, Inorg. Chem., 2002, 41, 3313–3322 CrossRef CAS PubMed.
- S. Saeedi, C. Xue, B. J. McCullough, S. E. Roe, B. J. Neyhouse and T. A. White, ACS Appl. Energy Mater., 2019, 2, 131–143 CrossRef CAS.
- M. Rentschler, S. Iglesias, M. A. Schmid, C. Liu, S. Tschierlei, W. Frey, X. Zhang, M. Karnahl and D. Moonshiram, Chem.–Eur. J., 2020, 26, 9527–9536 CrossRef CAS PubMed.
- M. K. Eggleston, P. E. Fanwick, A. J. Pallenberg and D. R. McMillin, Inorg. Chem., 1997, 36, 4007–4010 CrossRef CAS.
- J. V. Lockard, S. Kabehie, J. I. Zink, G. Smolentsev, A. Soldatov and L. X. Chen, J. Phys. Chem. B, 2010, 114, 14521–14527 CrossRef CAS PubMed.
- L. Kohler, D. Hayes, J. Hong, T. J. Carter, M. L. Shelby, K. A. Fransted, L. X. Chen and K. L. Mulfort, Dalton Trans., 2016, 45, 9871–9883 RSC.
- L.-S. Kau, D. J. Spira-Solomon, J. E. Penner-Hahn, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 1987, 109, 6433–6442 CrossRef CAS.
- K. A. Fransted, N. E. Jackson, R. Zong, M. W. Mara, J. Huang, M. R. Harpham, M. L. Shelby, R. P. Thummel and L. X. Chen, J. Phys. Chem. A, 2014, 118, 10497–10506 CrossRef CAS PubMed.
- L. Hua, M. Iwamura, S. Takeuchi and T. Tahara, Phys. Chem. Chem. Phys., 2015, 17, 2067–2077 RSC.
- M. Iwamura, S. Takeuchi and T. Tahara, Acc. Chem. Res., 2015, 48, 782–791 CrossRef CAS PubMed.
- S. Tschierlei, M. Karnahl, N. Rockstroh, H. Junge, M. Beller and S. Lochbrunner, ChemPhysChem, 2014, 15, 3709–3713 CrossRef CAS PubMed.
- M. Trpkovska, B. Šoptrajanov and L. Pejov, J. Mol. Struct., 2003, 654, 21–26 CrossRef CAS.
- I. V. Sazanovich, J. Best, P. A. Scattergood, M. Towrie, S. A. Tikhomirov, O. V. Bouganov, A. J. H. M. Meijer and J. A. Weinstein, Phys. Chem. Chem. Phys., 2014, 16, 25775–25788 RSC.
- J. Dyer, D. C. Grills, P. Matousek, A. W. Parker, M. Towrie, J. A. Weinstein and M. W. George, Chem. Commun., 2002, 872–873 RSC.
- M. Iwamura, S. Takeuchi and T. Tahara, Phys. Chem. Chem. Phys., 2014, 16, 4143–4154 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2239630–2239632. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3fd00027c |
| This journal is © The Royal Society of Chemistry 2023 |